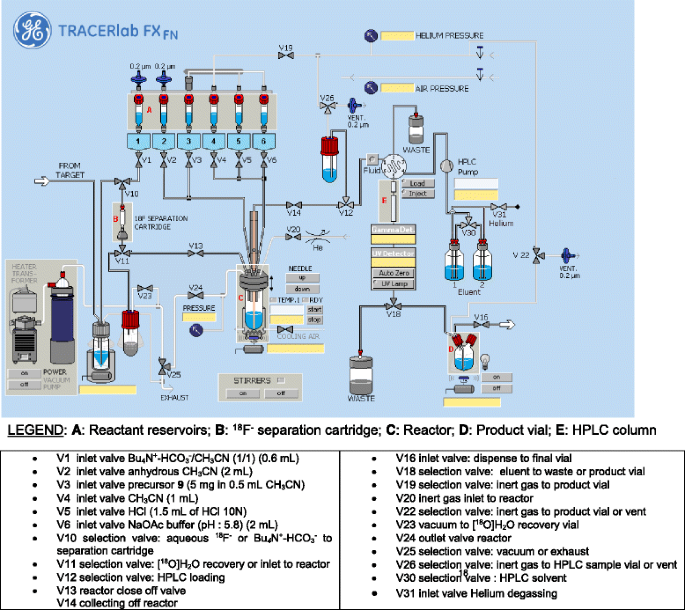
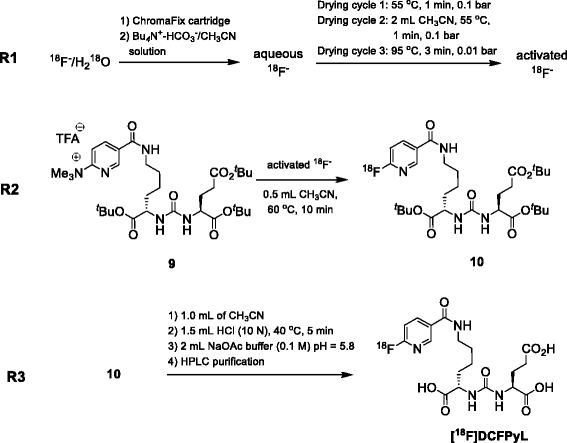
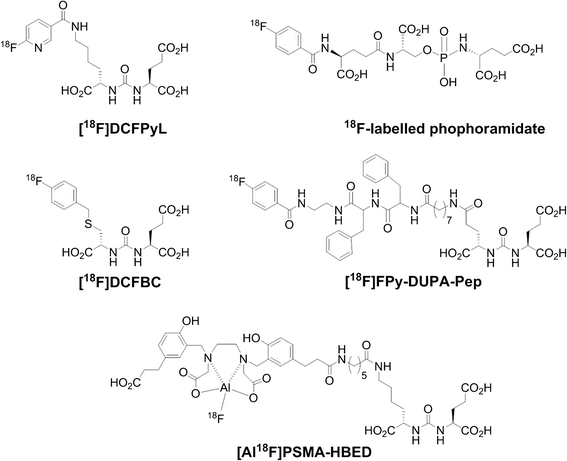
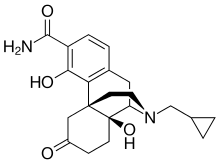
Dostarlimab, sold under the brand name Jemperli, is a monoclonal antibody medication used for the treatment of endometrial cancer.[1][2][3][4]
The most common adverse reactions (≥20%) were fatigue/asthenia, nausea, diarrhea, anemia, and constipation.[1][2] The most common grade 3 or 4 adverse reactions (≥2%) were anemia and transaminases increased.[1][2]
Dostarlimab is a programmed death receptor-1 (PD-1)–blocking antibody.[1][2]
Dostarlimab was approved for medical use in the United States in April 2021.[1][2][5]
NAME
DOSAGE
STRENGTH
ROUTE
LABELLER
MARKETING START
MARKETING END
Jemperli
Injection
50 mg/1mL
Intravenous
GlaxoSmithKline LLC
2021-04-22
Not applicable
Medical uses
Dostarlimab is indicated for the treatment of adults with mismatch repair deficient (dMMR) recurrent or advanced endometrial cancer, as determined by an FDA-approved test, that has progressed on or following prior treatment with a platinum-containing regimen.[1][2]
On April 22, 2021, the Food and Drug Administration granted accelerated approval to dostarlimab-gxly (Jemperli, GlaxoSmithKline LLC) for adult patients with mismatch repair deficient (dMMR) recurrent or advanced endometrial cancer, as determined by an FDA-approved test, that has progressed on or following a prior platinum-containing regimen.
Efficacy was evaluated based on cohort (A1) in GARNET Trial (NCT02715284), a multicenter, multicohort, open-label trial in patients with advanced solid tumors. The efficacy population consisted of 71 patients with dMMR recurrent or advanced endometrial cancer who progressed on or after a platinum-containing regimen. Patients received dostarlimab-gxly, 500 mg intravenously, every 3 weeks for 4 doses followed by 1,000 mg intravenously every 6 weeks.
The main efficacy endpoints were overall response rate (ORR) and duration of response (DOR), as assessed by blinded independent central review (BICR) according to RECIST 1.1. Confirmed ORR was 42.3% (95% CI: 30.6%, 54.6%). The complete response rate was 12.7% and partial response rate was 29.6%. Median DOR was not reached, with 93.3% of patients having durations ≥6 months (range: 2.6 to 22.4 months, ongoing at last assessment).
Serious adverse reactions occurred in 34% of patients receiving dostarlimab-gxly. Serious adverse reactions in >2% of patients included sepsis , acute kidney injury , urinary tract infection , abdominal pain , and pyrexia . The most common adverse reactions (≥20%) were fatigue/asthenia, nausea, diarrhea, anemia, and constipation. The most common grade 3 or 4 adverse reactions (≥2%) were anemia and transaminases increased. Immune-mediated adverse reactions can occur including pneumonitis, colitis, hepatitis, endocrinopathies, and nephritis.
The recommended dostarlimab-gxly dose and schedule (doses 1 through 4) is 500 mg every 3 weeks. Subsequent dosing, beginning 3 weeks after dose 4, is 1,000 mg every 6 weeks until disease progression or unacceptable toxicity. Dostarlimab-gxly should be administered as an intravenous infusion over 30 minutes.
This indication is approved under accelerated approval based on tumor response rate and durability of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory trial(s).
FDA also approved the VENTANA MMR RxDx Panel as a companion diagnostic device for selecting endometrial cancer patients for treatment with dostarlimab-gxly.
This review used the Real-Time Oncology Review (RTOR) pilot program, which streamlined data submission prior to the filing of the entire clinical application, and the Assessment Aid, a voluntary submission from the applicant to facilitate the FDA’s assessment.
Serious adverse reactions in >2% of patients included sepsis, acute kidney injury, urinary tract infection, abdominal pain, and pyrexia.[1][2]
Immune-mediated adverse reactions can occur including pneumonitis, colitis, hepatitis, endocrinopathies, and nephritis.[1][2]
History
Like several other available and experimental monoclonal antibodies, it is a PD-1 inhibitor. As of 2020, it is undergoing Phase I/II and Phase III clinical trials.[6][7][8] The manufacturer, Tesaro, announced prelimary successful results from the Phase I/II GARNET study.[6][9][10]
In 2020, the GARNET study announced that Dostarlimab was demonstrating potential to treat a subset of women with recurrent or advanced endometrial cancer.[11]
April 2021, Dostarlimab is approved for the treatment of recurrent or advanced endometrial cancer with deficient mismatch repair (dMMR), which are genetic anomalies abnormalities that disrupt DNA repair.[12]
On April 22, 2021, the Food and Drug Administration granted accelerated approval to dostarlimab-gxly (Jemperli, GlaxoSmithKline LLC).[1] Efficacy was evaluated based on cohort (A1) in GARNET Trial (NCT02715284), a multicenter, multicohort, open-label trial in patients with advanced solid tumors.[1]
Society and culture
Legal status
On 25 February 2021, the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) adopted a positive opinion, recommending the granting of a conditional marketing authorization for the medicinal product Jemperli, intended for the treatment of certain types of recurrent or advanced endometrial cancer.[13] The applicant for this medicinal product is GlaxoSmithKline (Ireland) Limited.[13]
^ Jump up to:ab Clinical trial number NCT02715284 for “A Phase 1 Dose Escalation and Cohort Expansion Study of TSR-042, an Anti-PD-1 Monoclonal Antibody, in Patients With Advanced Solid Tumors (GARNET)” at ClinicalTrials.gov
^ Clinical trial number NCT03981796 for “A Study of Dostarlimab (TSR-042) Plus Carboplatin-paclitaxel Versus Placebo Plus Carboplatin-paclitaxel in Patients With Recurrent or Primary Advanced Endometrial Cancer (RUBY)” at ClinicalTrials.gov
^ Clinical trial number NCT03602859 for “A Phase 3 Comparison of Platinum-Based Therapy With TSR-042 and Niraparib Versus Standard of Care Platinum-Based Therapy as First-Line Treatment of Stage III or IV Nonmucinous Epithelial Ovarian Cancer (FIRST)” at ClinicalTrials.gov
“Dostarlimab”. Drug Information Portal. U.S. National Library of Medicine.
Clinical trial number NCT02715284 for “Study of TSR-042, an Anti-programmed Cell Death-1 Receptor (PD-1) Monoclonal Antibody, in Participants With Advanced Solid Tumors (GARNET)” at ClinicalTrials.gov
Kaplon H, Muralidharan M, Schneider Z, Reichert JM: Antibodies to watch in 2020. MAbs. 2020 Jan-Dec;12(1):1703531. doi: 10.1080/19420862.2019.1703531. [Article]
Temrikar ZH, Suryawanshi S, Meibohm B: Pharmacokinetics and Clinical Pharmacology of Monoclonal Antibodies in Pediatric Patients. Paediatr Drugs. 2020 Apr;22(2):199-216. doi: 10.1007/s40272-020-00382-7. [Article]
Green AK, Feinberg J, Makker V: A Review of Immune Checkpoint Blockade Therapy in Endometrial Cancer. Am Soc Clin Oncol Educ Book. 2020 Mar;40:1-7. doi: 10.1200/EDBK_280503. [Article]
Deshpande M, Romanski PA, Rosenwaks Z, Gerhardt J: Gynecological Cancers Caused by Deficient Mismatch Repair and Microsatellite Instability. Cancers (Basel). 2020 Nov 10;12(11). pii: cancers12113319. doi: 10.3390/cancers12113319. [Article]
FDA Approved Drug Products: Jemperli (dostarlimab-gxly) for intravenous injection [Link]
FDA News Release: FDA grants accelerated approval to dostarlimab-gxly for dMMR endometrial cancer [Link]
Statement on a Nonproprietary Name Adopted by the USAN Council: Dostarlimab [Link]
ZyCoV-D is a genetically engineered DNA plasmid based vaccine encoding for the membrane proteins of the virus. The clinical trials to study the immunogenicity, and safety of the vaccine, will administer three doses at an interval of 28 days in 1048 individuals.
The ZYCOV-D vaccine candidate was developed by Cadila Healthcare Ltd. based in India1. The vaccine was developed using a DNA vaccine platform with a non-replicating and non-integrating plasmid carrying the gene of interest3. Once the plasmid DNA is introduced into host cells and the viral protein is translated, it elicits a strong immune response, stimulating the humoral and cellular components of the immune system3. The DNA vaccine platform offers minimal biosafety requirements, more improved vaccine stability, and lower cold chain requirements3. Phase I clinical trials of this vaccine candidate were completed in July 2020, with the company reporting successful dosing and tolerance1,2. As of August, 2020 the candidate is in Phase II clinical trials1.
In February 2020, Cadila Healthcare decided to develop a DNA plasmid based COVID-19 vaccine at their Vaccine Technology Centre (VTC) in Ahmedabad.[1] The vaccine candidate was able to pass the pre-clinical trials on animal models successfully. A report of the study was made available via bioRxiv.[2] Thereafter, human trials for Phase I and II were approved by the regulator.[3]
The Phase II trials of the vaccine candidate were conducted in over 1,000 volunteers as part of the adaptive Phase I/II multi-centric, dose escalation, randomised, double-blind placebo controlled method.[4][5]
Phase III trials
In November 2020, the company announced it would test the vaccine candidate on 30,000 patients in Phase III trials.[6] The vaccine would be given out in three doses at five sites across four cities of India.[7] In January 2021, the Drugs Controller General of India (DCGI) granted permission to conduct the Phase III clinical trials for 28,216 Indian participants.[8][9]
In April 2021, the company reported that they expected to have initial data for the Phase III trials by May 2021.[10]
Production
On 23 April 2021, production of the ZyCoV-D vaccine was started, with a yearly capacity of 240 million doses. It is expected to get emergency use authorization in May or June.[11]
^ Dey A, Rajanathan C, Chandra H, Pericherla HP, Kumar S, Choonia HS, et al. (26 January 2021). “Immunogenic Potential of DNA Vaccine candidate, ZyCoV-D against SARS-CoV-2 in Animal Models”. bioRxiv: 2021.01.26.428240. doi:10.1101/2021.01.26.428240. S2CID231777527.
In an exclusive interview with India Today TV, Managing Director of Zydus Cadila Dr Sharvil Patel said the company’s Covid vaccine candidate ZyCoV-D against the Covid-19 infection is very close to getting approved in India. They are likely to apply for emergency use authorisation this month.
Ahmedabad-based pharmaceutical company Zydus Cadila is likely to submit the application for emergency use authorisation of its Covid-19 vaccine candidate ‘ZyCoV-D’ in India this month. The company is confident that the vaccine will be approved in May itself. The company plants to produce one crore doses of its ‘painless’ Covid-19 vaccine per month.
If approved, ZyCoV-D will be the fourth vaccine to be used in India’s Covid-19 vaccination drive. Made in India, the company plans to ramp up the vaccine’s production to 3-4 crore doses per month and is already in talks with two other manufacturing companies for the same
Although the vaccine should ideally be stored between 2 and 8 degrees Celsius, it remains stable even at room temperature conditions at 25 degrees Celsius. It is easy to administer, the developers said, and will be administered via intradermal injection.
If approved for emergency use, ZyCoV-D could help India fill the vacuum of vaccine doses currently being experienced in the country’s immunisation drive.
In an exclusive interview with India Today TV, Sharvil Patel sheds details on all aspects of the Covid-19 vaccine ZyCoV-D.
When asked the status of Covid vaccine candidate ZyCoV-D and when exactly Zydus Cadila would apply for emergency use authorisation in India, Dr Sharvil Patel said the vaccine was getting very close to getting approved in the country.
“I am very happy to say that India’s first indigenously developed DNA vaccine candidate against Covid, which is our ZyCoV-D, is getting very close to approval,” he said.
“We have almost completed all our recruitment for the clinical trials. We have, by far, recruited the largest number of patients for a Covid vaccine trial in India. The number of volunteers who have been vaccinated as a part of the trial is 28,000,” Sharvil Patel said.
Sharvil Patel also said that his company has also included children in the 12-17 age group for the vaccine trials.
He said, “The recruitment holds very important milestones in terms of cohorts because not only have we included the elderly and those with co-morbidities, but also children in the age group of 12 to 17 years.”
Sharvil Patel said as soon as the efficacy data is obtained, Sydus Cadila will file for emergency use authorisation. As soon as the approval is granted, Zydus Cadila will start production of Covid-19 vaccines from July, he said.
“We hope to see our efficacy data in the middle of May. As soon as we see strong efficacy which correlates to the vaccine’s strong immunogenicity in Phase 2, we will file for emergency use authorization. We hope to produce a good quantity of the vaccine from July onwards to make sure it is available to the people. That is the need of the hour right now,” Sharvil Patel said.
He said by May the company will be in a position to talk to the regulators about the restricted use of the Covid-19 vaccine. “The regulatory process is a rolling one. I believe the regulators look at the data in a short period of time,” Sharvil Patel said.
“We have submitted a lot of data already so that it will aid the regulators once we provide them with the efficacy results. We are, hence, expecting to get the approval in May itself,” Sharvil Patel said.
Loncastuximab tesirine-lpyl is a CD19-directed antibody and alkylating agent conjugate, consisting of a humanized IgG1 kappa monoclonal antibody conjugated to SG3199, a pyrrolobenzodiazepine (PBD) dimer cytotoxic alkylating agent, through a protease-cleavable valine–alanine linker. SG3199 attached to the linker is designated as SG3249, also known as tesirine.
Loncastuximab tesirine-lpyl has an approximate molecular weight of 151 kDa. An average of 2.3 molecules of SG3249 are attached to each antibody molecule. Loncastuximab tesirine-lpyl is produced by chemical conjugation of the antibody and small molecule components. The antibody is produced by mammalian (Chinese hamster ovary) cells, and the small molecule components are produced by chemical synthesis.
ZYNLONTA (loncastuximab tesirine-lpyl) for injection is supplied as a sterile, white to off-white, preservative-free, lyophilized powder, which has a cake-like appearance, for intravenous infusion after reconstitution and dilution. Each single-dose vial delivers 10 mg of loncastuximab tesirine-lpyl, L-histidine (2.8 mg), L-histidine monohydrochloride (4.6 mg), polysorbate 20 (0.4 mg), and sucrose (119.8 mg). After reconstitution with 2.2 mL Sterile Water for Injection, USP, the final concentration is 5 mg/mL with a pH of approximately 6.0.
Loncastuximab tesirine , sold under the brand name Zynlonta, is used for the treatment of large B-cell lymphoma. It is an antibody-drug conjugate (ADC) composed of a humanized antibody targeting the protein CD19, which is expressed in a wide range of B cell hematological tumors.[2] The experimental drug, developed by ADC Therapeutics is being tested in clinical trials for the treatment of B-cell non-Hodgkin lymphoma (NHL) and B-cell acute lymphoblastic leukemia (ALL).
On April 23, 2021, the Food and Drug Administration granted accelerated approval to loncastuximab tesirine-lpyl (Zynlonta, ADC Therapeutics SA), a CD19-directed antibody and alkylating agent conjugate, for adult patients with relapsed or refractory large B-cell lymphoma after two or more lines of systemic therapy, including diffuse large B-cell lymphoma (DLBCL) not otherwise specified, DLBCL arising from low grade lymphoma, and high-grade B-cell lymphoma.
Approval was based on LOTIS-2 (NCT03589469), an open-label, single-arm trial in 145 adult patients with relapsed or refractory DLBCL or high-grade B-cell lymphoma after at least two prior systemic regimens. Patients received loncastuximab tesirine-lpyl 0.15 mg/kg every 3 weeks for 2 cycles, then 0.075 mg/kg every 3 weeks for subsequent cycles. Patients received treatment until progressive disease or unacceptable toxicity.
The main efficacy outcome measure was overall response rate (ORR), as assessed by an independent review committee using Lugano 2014 criteria. The ORR was 48.3% (95% CI: 39.9, 56.7) with a complete response rate of 24.1% (95% CI: 17.4, 31.9). After a median follow-up of 7.3 months, median response duration was 10.3 months (95% CI: 6.9, NE). Of the 70 patients who achieved objective responses, 36% were censored for response duration prior to 3 months.
Most common (≥20%) adverse reactions in patients receiving loncastuximab tesirine-lpyl, including laboratory abnormalities, are thrombocytopenia, increased gamma-glutamyltransferase, neutropenia, anemia, hyperglycemia, transaminase elevation, fatigue, hypoalbuminemia, rash, edema, nausea, and musculoskeletal pain.
The prescribing information provides warnings and precautions for adverse reactions including edema and effusions, myelosuppression, infections, and cutaneous reactions.
The recommended loncastuximab tesirine-lpyl dosage is 0.15 mg/kg every 3 weeks for 2 cycles, then 0.075 mg/kg every 3 weeks for subsequent cycles, by intravenous infusion over 30 minutes on day 1 of each cycle (every 3 weeks). Patients should be premedicated with dexamethasone 4 mg orally or intravenously twice daily for 3 days beginning the day before loncastuximab tesirine-lpyl.
Technology
The humanized monoclonal antibody is stochastically conjugated via a valine-alanine cleavable, maleimide linker to a cytotoxic (anticancer) pyrrolobenzodiazepine (PBD) dimer. The antibody binds to CD19, a protein which is highly expressed on the surface of B-cell hematological tumors[3] including certain forms of lymphomas and leukemias. After binding to the tumor cells the antibody is internalized, the cytotoxic drug PBD is released and the cancer cells are killed. PBD dimers are generated out of PBD monomers, a class of natural products produced by various actinomycetes. PBD dimers work by crosslinking specific sites of the DNA, blocking the cancer cells’ division that cause the cells to die. As a class of DNA-crosslinking agents they are significantly more potent than systemic chemotherapeutic drugs.[4]
Clinical trials
Two phase I trials are evaluating the drug in patients with relapsed or refractory B-cell non-Hodgkin’s lymphoma and relapsed or refractory B-cell acute lymphoblastic leukemia.[5] At the 14th International Conference on Malignant Lymphoma interim results from a Phase I, open-label, dose-escalating study designed to evaluate the treatment of loncastuximab tesirine in relapsed or refractory non-Hodgkin’s lymphoma were presented.[6] Among the patients enrolled at the time of the data cutoff the overall response rate was 61% in the total patient population (42% complete response and 19% partial response) and in patients with relapsing or refractory diffuse large B-cell lymphoma (DLBCL) the overall response rate was 57% (43% complete response and 14% partial response).[7][8]
Orphan drug designation
Loncastuximab tesirine was granted Orphan Drug Designation by the U.S. Food and Drug Administration (FDA) for the treatment of diffuse large B-cell lymphoma and mantle cell lymphoma.[9]
An autologous T lymphocyte-enriched cell transduced ex vivo with an anti-BCMA CAR lentiviral vector encoding a chimeric antigen receptor CAR, comprising a CD8 hinge and TM domain, 4-1BB costimulatory domain and CD3ζ signaling domain, targeting human B cell maturation antigen for cancer immunotherapy (Celgene Corp., NJ)
Dendritic cells (DCs) are antigen-presenting cells (APCs) that process antigens and display them to other cells of the immune system. Specifically, dendritic cells are capable of capturing and presenting antigens on their surfaces to activate T cells such as cytotoxic T cells (CTLs). Further, activated dendritic cells are capable of recruiting additional immune cells such as macrophages, eosinophils, natural killer cells, and T cells such as natural killer T cells.
Despite major advances in cancer treatment, cancer remains one of the leading causes of death globally. Hurdles in designing effective therapies include cancer immune evasion, in which cancer cells escape destructive immunity, as well as the toxicity of many conventional cancer treatments such as radiation therapy and chemotherapy, which significantly impacts a patient’s ability to tolerate the therapy and/or impacts the efficacy of the treatment.
Given the important role of dendritic cells in immunity, derailed dendritic cell functions have been implicated in diseases such as cancer and autoimmune diseases. For example, cancer cells may evade immune detection and destruction by crippling dendritic cell functionality through prevention of dendritic cell recruitment and activation. In addition, dendritic cells have been found in the brain during central nervous system inflammation and may be involved in the pathogenesis of autoimmune diseases in the brain.
One mechanism by which cancers evade immune detection and destruction is by crippling dendritic cell functionality through prevention of dendritic cell (DC) recruitment and activation. Accordingly, there remains a need for cancer therapies that can effectively derail tumor evasion and enhance anti-tumor immunity as mediated, for example, by dendritic cells.
ABECMA is a BCMA-directed genetically modified autologousT cellimmunotherapy product consisting of a patientâ€s own T cells that are harvested and genetically modified ex vivo through transduction with an anti-BCMA02 chimeric antigen receptor (CAR) lentiviral vector (LVV). Autologous T cells transduced with the anti-BCMA02 CAR LVV express the anti-BCMA CAR on the T cell surface. The CAR is comprised of a murine extracellular single-chain variable fragment (scFv) specific for recognizing B cell maturation antigen (BCMA) followed by a human CD8α hinge and transmembrane domain fused to the T cell cytoplasmic signaling domains of CD137 (4-1BB) and CD3ζ chain, in tandem. Binding of ABECMA to BCMA-expressing target cells leads to signaling initiated by CD3ζ and 4-1BB domains, and subsequent CAR-positive T cell activation. Antigen-specific activation of ABECMA results in CAR-positive T cell proliferation, cytokine secretion, and subsequent cytolytic killing of BCMA-expressing cells.
ABECMA is prepared from the patientâ€s peripheral blood mononuclear cells (PBMCs), which are obtained via a standard leukapheresis procedure. The mononuclear cells are enriched for T cells, through activation with anti-CD3 and anti-CD28 antibodies in the presence of IL-2, which are then transduced with the replication-incompetent lentiviral vector containing the anti-BCMA CAR transgene. The transduced T cells are expanded in cell culture, washed, formulated into a suspension, and cryopreserved. The product must pass a sterility test before release for shipping as a frozen suspension in one or more patient-specific infusion bag(s). The product is thawed prior to infusion back into the patient [see DOSAGE AND ADMINISTRATION and HOW SUPPLIED/Storage And Handling].
The ABECMA formulation contains 50% Plasma-Lyte A and 50% CryoStor® CS10, resulting in a final DMSO concentration of 5%.
FDA approves idecabtagene vicleucel for multiple myeloma
On March 26, 2021, the Food and Drug Administration approved idecabtagene vicleucel (Abecma, Bristol Myers Squibb) for the treatment of adult patients with relapsed or refractory multiple myeloma after four or more prior lines of therapy, including an immunomodulatory agent, a proteasome inhibitor, and an anti-CD38 monoclonal antibody. This is the first FDA-approved cell-based gene therapy for multiple myeloma.
Idecabtagene vicleucel is a B-cell maturation antigen (BCMA)-directed genetically modified autologous chimeric antigen receptor (CAR) T-cell therapy. Each dose is customized using a patient’s own T-cells, which are collected and genetically modified, and infused back into the patient.
Safety and efficacy were evaluated in a multicenter study of 127 patients with relapsed and refractory multiple myeloma who received at least three prior lines of antimyeloma therapies; 88% had received four or more prior lines of therapies. Efficacy was evaluated in 100 patients who received idecabtagene vicleucel in the dose range of 300 to 460 x 106 CAR-positive T cells. Efficacy was established based on overall response rate (ORR), complete response (CR) rate, and duration of response (DOR), as evaluated by an Independent Response committee using the International Myeloma Working Group Uniform Response Criteria for Multiple Myeloma.
The ORR was 72% (95% CI: 62%, 81%) and CR rate was 28% (95% CI 19%, 38%). An estimated 65% of patients who achieved CR remained in CR for at least 12 months.
The idecabtagene vicleucel label carries a boxed warning for cytokine release syndrome (CRS), neurologic toxicities, hemophagocytic lymphohistiocytosis/ macrophage activation syndrome, and prolonged cytopenias. The most common side effects of idecabtagene vicleucel include CRS, infections, fatigue, musculoskeletal pain, and hypogammaglobulinemia.
Idecabtagene vicleucel is approved with a risk evaluation and mitigation strategy requiring that healthcare facilities that dispense the therapy must be specially certified to recognize and manage CRS and nervous system toxicities. To evaluate long-term safety, the FDA is requiring the manufacturer to conduct a post-marketing observational study involving patients treated with idecabtagene vicleucel.
FDA D.I.S.C.O. Burst Edition: FDA approval of ABECMA (idecabtagene vicleucel) the first FDA approved cell-based gene therapy for the treatment of adult patients with relapsed or refractory multiple myeloma
Welcome back to the D.I.S.C.O., FDA’s Drug Information Soundcast in Clinical Oncology, Burst Edition, brought to you by FDA’s Division of Drug Information in partnership with FDA’s Oncology Center of Excellence. Today we have another quick update on a recent FDA cancer therapeutic approval.
On March 26, 2021, the FDA approved idecabtagene vicleucel (brand name Abecma) for the treatment of adult patients with relapsed or refractory multiple myeloma after four or more prior lines of therapy, including an immunomodulatory agent, a proteasome inhibitor, and an anti-CD38 monoclonal antibody. This is the first FDA-approved cell-based gene therapy for multiple myeloma.
Idecabtagene vicleucel is a B-cell maturation antigen-directed genetically modified autologous chimeric antigen receptor T-cell therapy. Each dose is customized using a patient’s own T-cells, which are collected and genetically modified, and infused back into the patient.
Safety and efficacy were evaluated in a multicenter study of 127 patients with relapsed and refractory multiple myeloma who received at least three prior lines of antimyeloma therapies, 88% of whom had received four or more prior lines of therapies. Efficacy was evaluated in 100 patients who received idecabtagene vicleucel and was established based on overall response rate, complete response rate, and duration of response, as evaluated by an Independent Response committee using the International Myeloma Working Group Uniform Response Criteria for Multiple Myeloma.
The overall response rate was 72% and complete response rate was 28%. An estimated 65% of patients who achieved complete response remained in complete response for at least 12 months.
The idecabtagene vicleucel label carries a boxed warning for cytokine release syndrome, neurologic toxicities, hemophagocytic lymphohistiocytosis/ macrophage activation syndrome, and prolonged cytopenias. Idecabtagene vicleucel is approved with a risk evaluation and mitigation strategy requiring that healthcare facilities dispensing the therapy must be specially certified to recognize and manage cytokine release syndrome and nervous system toxicities. To evaluate long-term safety, the FDA is requiring the manufacturer to conduct a post-marketing observational study involving patients treated with idecabtagene vicleucel.
Full prescribing information for this approval can be found on the web at www.fda.gov, with key word search “Approved Cellular and Gene Therapy Products”.
Health care professionals should report serious adverse events to FDA’s MedWatch Reporting System at www.fda.gov/medwatch.
In various aspects, the present invention relates to XCR1 binding agents having at least one targeting moiety that specifically binds to XCR1. In various embodiments, these XCR1 binding agents bind to, but do not functionally modulate ( e.g . partially or fully neutralize) XCR1. Therefore, in various embodiments, the present XCR1 binding agents have use in, for instance, directly or indirectly recruiting a XCR1-expressing cell to a site of interest while still allowing the XCR1-expressing cell to signal via XCR1 (i.e. the binding of the XCR1 binding agent does not reduce or eliminate XCR1 signaling at the site of interest). In various embodiments, the XCR-1 binding agent functionally modulates XCR1. In an embodiment, the targeting moiety is a single domain antibody (e.g. VHH, HUMABODY, scFv, on antibody). In various embodiments, the XCR1 binding agent further comprises a signaling agent, e.g., without limitation, an interferon, an interleukin, and a tumor necrosis factor, that may be modified to attenuate activity. In various embodiments, the XCR1 binding agent comprises additional targeting moieties that bind to other targets (e.g. antigens, receptor) of interest. In an embodiment, the other targets (e.g. antigens, receptor) of interest are present on tumor cells. In another embodiment, the other targets (e.g. antigens, receptor) of interest are present on immune cells. In some embodiments, the present XCR1 binding agent may directly or indirectly recruit an immune cell (e.g. a dendritic cell) to a site of action (such as, by way of non-limiting example, the tumor microenvironment). In some embodiments, the present XCR1 binding agent facilitates the presentation of antigens (e.g., tumor antigens) by dendritic cells.
In various embodiments, the present XCR binding agent or targeting moiety of the present chimeric proteins comprises the heavy chain of SEQ ID NO: 223 and/or the light chain of SEQ ID NO: 224, or a variant thereof (e.g. an amino acid sequence having at least about 90%, or at least about 93%, at least about 95%, at least about 97%, at least about 98%, at least about 99%, identity with SEQ ID NO: 223 and/or SEQ ID NO: 224).
In various embodiments, the present XCR binding agent or targeting moiety of the present chimeric proteins comprises a heavy chain CDR 1 of SHNLH (SEQ ID NO: 225), heavy chain CDR 2 of AIYPGNGNTAYNQKFKG (SEQ ID NO: 226), and heavy chain CDR 3 of WGSVVGDWYFDV (SEQ ID NO: 227) and/or a light chain CDR 1 of RSSLGLVHRNGNTYLH (SEQ ID NO: 228), light chain CDR 2 of KVSHRFS (SEQ ID NO: 229), and light chain CDR 3 of SQSTFIVPWT (SEQ ID NO: 230), or a variant thereof (e.g. with four or fewer amino acid substitutions, or with three or fewer amino acid substitutions, or with two or fewer amino acid substitutions, or with one amino acid substitution).
In various embodiments, the present XCR binding agent or targeting moiety of the present chimeric proteins comprises a heavy chain CDR 1 of SHNLH (SEQ ID NO: 225), heavy chain CDR 2 of AIYPGNGNTAYNQKFKG (SEQ ID NO: 226), and heavy chain CDR 3 of WGSVVGDWYFDV (SEQ ID NO: 227).
Illustrative Disease Modifying Therapies
EXAMPLES
Example 1. Identification and Characterization of Human XCR1 Ab AFNs
As used in this Example and associated figures,“AFN” is a chimera of the anti-Xcr1 5G7 antibody and human IFNa2 with an R149A mutation.
AFNs were made based on the 5G7 anti-hXcr1 Ab using the intact (full) Ab or a scFv format.
DWMTQTPLSLPVTLGNQASIFCRSSLGLVHRNGNTYLHWYLQKPGQSPKLLIYKVSHRFSGVPDRFSGSGSGT DFTLKISRVEAEDLGVYFCSQSTHVPWTFGGGTKLEIKRTVAAPSVFIFPPSDEQLKSGTASWCLLNNFYPREAK VQWKVDNALQSGNSQESVTEQDSKDSTYSLSSTLTLSKADYEKHKVYACEVTHQGLSSPVTKSFNRGEC (SEQ ID NO: 224)
5G7 Heavy chain CDR 1 is SHNLH (SEQ ID NO: 225), Heavy chain CDR 2 is AIYPGNGNTAYNQKFKG (SEQ ID NO: 226), Heavy chain CDR 3 is WGSVVGDWYFDV (SEQ ID NO: 227). 5G7 Light chain CDR 1 is RSSLGLVHRNGNTYLH (SEQ ID NO: 228), Light chain CDR 2 is KVSHRFS (SEQ ID NO: 229), and Light chain CDR 3 is SQSTHVPWT (SEQ ID NO: 230).
The sequence of hulFNa2(R149A) is:
CDLPQTHSLGSRRTLMLLAQMRKISLFSCLKDRHDFGFPQEEFGNQFQKAETIPVLHEMIQQIFNLFSTKDSSAA WDETLLDKFYTELYQQLNDLEACVIQGVGVTETPLMKEDSILAVRKYFQRITLYLKEKKYSPCAWEVVRAEIMASF SLSTNLQESLRSKE (SEQ ID NO: 231).
In case of the intact Ab AFN, the 5G7 Ab heavy chain was fused to h I FN a2_R149A (human IFNal with a R149A mutation) via a flexible (GGS)2oG-linker and co-expressed with the 5G7 Ab light chain (sequences shown below). 5G7 scFv-AFN was constructed by linking the Ab VL and VH domains via a (GGGS)4 linker and followed by a (GGS)2o-linker and the sequence encoding hlFNa2_R149A. Recombinant proteins, cloned in the pcDNA3.4 expression-vector, were produced in ExpiCHO cells (Thermo Fisher Scientific) and purified on HisPUR spin plates (Thermo Fisher Scientific) according to the manufacturer’s instructions.
To test binding of the AFNs, parental HL1 16 and HL1 16 cells stably expressing hXcrl (HL116-hXcr1) were incubated with a serial dilution AFN for two hours at 4°C. Binding was detected using THE HIS antibody-FITC (GenScript) and measured on a MACSQuant X instrument (Miltenyi Biotec) and analysed using the FlowLogic software (Miltenyi Biotec). Data in Figures 1A and 1 B clearly show that both 5G7 Ab-AFN and 5G7 scFv bind specifically to hXcrl expressing cells.
Biological activity was measured on parental HL1 16 cells (an IFN responsive cell-line stably transfected with a p6-16 luciferase reporter) and the derived HL116-hXcr1 cells. Cells were seeded overnight and stimulated for 6 hours with a serial dilution 5G7 AFNs. Luciferase activity was measured on an EnSight Multimode Plate Reader (Perkin Elmer). Data in Figures 2A and 2B clearly illustrate that 5G7 AFNs, in the intact Ab format or as scFv, are clearly more active on cells expressing hXcrl compared to parental cells, illustrating that it is possible to restore signaling of an IFNa2 mutant by specific targeting to hXcrl .
Example 2. Identification and Characterization of Mouse Xcr1 Ab AFNs
As used in this Example and associated figures,“AFN” is a chimera of the anti-Xcr1 MAARX10 antibody and human IFNa2 with Q124R mutation.
Similar to the anti-human Xcr1 Ab, AFNs based on the MARX10 anti-mouse Xcr1 Ab were made, as intact Ab or as scFv. In case of the intact Ab AFN, the MARX10 Ab heavy chain was fused to hlFNa2_Q124R (human IFNa2 with Q124R mutation) via a flexible (GGS)2oG-linker and co-expressed with the MARX10 Ab light chain. scFv-AFN was constructed by linking the Ab VL and VH domains, in VH-VL (scFv(1 )) or VL-VH (scFv(2)) orientation, via a (GGGS)4 linker and followed by a (GGS)2o-linker and h I FN a2_Q 124R.
Selectivity of AFNs (produced and purified as described above for the human Xcr1 Ab AFNs) was tested by comparing binding at 2.5 pg/ml to MOCK or mouse Xcr1 transfected Hek293T cells. Binding was detected using THE HIS antibody-FITC (GenScript) and measured on a MACSQuant X instrument (Miltenyi Biotec) and analysed using the FlowLogic software (Miltenyi Biotec). Data in Figure 3 clearly show that all three specifically bind to mXcrl expressing cells.
///////////Idecabtagene vicleucel, breakthrough therapy designation, orphan drug designation, FDA 2021, APPROVALS 2021, Bb2121, Bb , ABECMA
Manufacturer: Celgene Corporation, a Bristol-Myers Squibb Company Indications:
Treatment of adult patients with relapsed or refractory multiple myeloma after four or more prior lines of therapy including an immunomodulatory agent, a proteasome inhibitor, and an anti-CD38 monoclonal antibody.
1H-1,2,3-Triazolium, 3-(((2S,3S,5R)-2-carboxy-3-methyl-4,4-dioxido-7-oxo-4-thia-1-azabicyclo(3.2.0)hept-3-yl)methyl)-1-methyl-, inner salt
Enmetazobactam
The Board of directors of Orchid Pharma Ltd has announced that the company had developed a new molecule known as OCID-5090, which was licensed to a company named Allecra Therapeutics, this molecule was undergoing the clinical trials and the company is happy to announce that the molecule has cleared the Phase 3 clinical trials.
Allecra Therapeutics would now either directly or through out license file for NDA of this molecule. Allecra has already out licensed the product to Haini Pharmaceuticals, China for the Chinese Territory at a value of $78mn plus royalties.
As per the IP Agreement between Orchid Pharma Limited and Allecra Therapeutics, Orchid is entitled to receive a Royalty of 6-8% on the worldwide sales of the product. Therefore, once the molecule is commercialised, Orchid can expect a regular stream of Royalty from Allecra. Further, the rights to develop and commercialise the molecule in India (which is under patent protection) remain with Orchid Pharma Limited, and the company is evaluating the various options to commercialise the product.
Orchid had developed a new molecule known as OCID-5090, which was licensed to a company named Allecra Therapeutics, this molecule was undergoing the clinical trials and the molecule has cleared the Phase 3 clinical trials.
Allecra Therapeutics would now either directly or through out license file for NDA of this molecule. Allecra has already out licensed the product to Haini Pharmaceuticals, China for the Chinese Territory at a value of $78mn plus royalties.
As per the IP Agreement between Orchid Pharma Limited and Allecra Therapeutics, Orchid is entitled to receive a Royalty of 6-8% on the worldwide sales of the product. Therefore, once the molecule is commercialised, Orchid can expect a regular stream of Royalty from Allecra. Further, the rights to develop and commercialise the molecule in India (which is under patent protection) remain with Orchid Pharma Limited, and the company is evaluating the various options to commercialise the product.
[0051]To a suspension of (2S,3S,5R)-3-methyl-7-oxo-3-(1H-1,2,3-triazol-1-ylmethyl)-4-thia-1-azabicyclo-[3.2.0]heptane-2-carboxylic acid 4,4-dioxide (25 g) in acetone (100 mL) at 25-30° C. was added slowly N,O-bis(silylacetamide) (18.6 g) with stirring. The reaction mixture was stirred at this temperature (25-30° C.) for 15-20 min. To the clear solution obtained, methyl iodide (100 mL) was added over a period of 15 min. and stirred at 25-30 min. for 24 h. The precipitated solid was separated by filtration and washed with acetone (25 mL). Wet weight of the solid obtained was 30 g.
[0052]The above wet solid was stirred with purified water (300 mL) at 10-15° C. for 2.5 h. To the resulted reaction mixture was added sodium thiosulfate (0.1 g) and stirred at 10-15° C. for 10-15 min. To the reaction mixture, dichloromethane (300 mL) was added, stirred and the organic layer separated. The aqueous layer was washed with a solution of Amberlite LA-2 resin (5% solution in dichloromethane twice, followed by dichloromethane twice. To the aqueous solution, activated carbon (1 g) was added, stirred for 15 min, filtered and washed with purified water (25 mL). The solution was filtered and lyophilized to get the title compound in pure form (10 g). 1H NMR (400 MHz, DMSO) δ ppm: 1.39 (s, 3H), 3.14 (dd, J=16.0, 1.3 Hz, 1H), 3.55 (dd, J=16.0, 4.2 Hz, 1H), 3.97 (s, 1H), 4.34 (s, 3H), 5.05 (dd, J=4.2, 1.3 Hz, 1H), 5.29 (d, J=14.7 Hz, 1H), 5.42 (d, J=14.7 Hz, 1H), 8.91 (d, J=1.3 Hz, 1H), 8.99 (d, J=1.3 Hz, 1H). Mass m/z: M+1 peak at 315. Alternatively the solution could be subjected to spray-drying to yield the title compound.
Synthesis of (2535.5R)-3-methyl-3-((3-methyl-lH-1.2 -triazol-3-ium-l-yl)methvn-7-oxo-4-thia-l-azabicyclor3.2.01heptane-2-carboxylate 4,4-dioxide (4),
Compound (4) was prepared according to Scheme 2.
Scheme 2
i) Ν,Ο-bis-trimethylsilylacetamide, CH2CI2; ii) CH3OTf; iii) Na 2-ethylhexanoate
In a round bottom flask under nitrogen flow 100 g of Tazobactam acid (1) and 500 mL of Dichloromethane are loaded. The temperature is adjusted to +30/35°C then 37 g of Ν,Ο-Bis(trimethylsilyl) acetamide are loaded in 15-20 minutes maintaining the temperature to +35/42°C. The mixture is heated to reflux (+40/42°C) for 60 minutes. If the solution is not clear, N,0-Bis(trimethylsilyl) acetamide is loaded in small portions (0,5-1.0 g each) waiting 15 minutes every time till a clear solution containing intermediate (2) is obtained. 0.55 moles of N,0-Bis(trimethylsilyl) acetamide is used, with further 0.1-0.2 equivalents being added if the reaction is not complete.
Then the temperature is cooled down to 0/+5°C and 70 g of Methyl trifluoromethanesulfonate are loaded in 60-90 minutes maintaining the temperature at 0/+5°C. After 30 minutes the reaction is monitored by HPLC to control the disappearance of intermediate (2) and formation of intermediate (3). The reaction is monitored every 30 minutes until completion.
In a round bottom flask, under nitrogen, are loaded 500 mL of Ethanol and 55 g of Sodium 2-Ethylhexanoate and the temperature is adjusted to +20/25°C, then the reaction solution containing intermediate (3) is added in 60-90 minutes maintaining the temperature of +20/25 °C under vigorous stirring. The suspension is stirred for 30 minutes then is filtered and washed with 300 mL of Ethanol followed by 500 mL of Dichloromethane under nitrogen. The crude product (4) is dried under nitrogen flow till constant weight (150 g) is obtained. The crude product compound (4) was isolated as a solid product (HPLC assay = 70%, yield = 80%).
Purification of (2tS’,3^5^)-3-methyl-3-((3-methyl-lH-l,2,3-triazol-3-ium-l-yl)methyl)-7-oxo-4-thia-l-azabicyclor3.2.01heptane-2-carboxylate 4,4-dioxide (4)
In a round bottom flask 800 mL of Dimethylformamide are loaded, the temperature is adjusted to +20/25°C then crude Compound 4 (150g) obtained above is loaded using 100 mL of Dimethylformamide to facilitate the transfer. The mixture is stirred for 5 minutes and a solution is obtained, then and after a few minutes crystallization takes place. The suspension is stirred for about 3 hours, then is cooled to 0/+5°C and stirred for another 3 hours.
The solid is filtered and washed with 300 mL of Dimethylformamide pre-cooled to 0/+5°C. Compound 4 is then suspended in 700 mL of Ethyl acetate and the temperature is adjusted to +40/45°C. The suspension is stirred for 30 minutes then the solid is filtered and washed with 150 mL of Ethyl acetate pre-heated to +40/45°C. The suspension with
Ethyl acetate is repeated twice. Finally Compound 4 is dried under vacuum at +40°C till constant weight is achieved (66 g, HPLC assay = 99%, yield = 76%).
Compound 4 Sterile filtration and recrystallization Procedure
In a round bottom flask 350 mL of Methanol are loaded, the temperature is adjusted to +30/35°C then 100 g of Compound 4 are loaded and finally the flask is washed with 60 mL of Methanol. After 5-10 minutes a solution is obtained. The solution is diluted with 330 mL of acetone adjusting the temperature to +20/+25°C. The obtained solution is treated with 2,2 g of charcoal for 20 minutes then filtered using a 0.22microM filter and the filter is washed with a mixture of 13 mL of Methanol and 110 mL of Acetone. The temperature of the solution is adjusted to +30/35°C and under vigorous stirring 830 mL of Acetone are loaded in about 15-20 minutes. After stirring for 60 minutes at temperature of +30/35°C 1170 mL of Acetone are loaded in 45-60 minutes. Then the temperature is adjusted to +20/25 °C in about 30-60 minutes and maintained for 30 minutes. The obtained crystalline solid is filtered and washed with 430 mL of Acetone. Finally the product is dried under vacuum at +40°C till constant weight is achieved (83 g of Compound 4) are obtained with an HPLC assay = 98-99%, yield =t 80%).
Mr. Ram Gopal Agarwal
Chairman and Non-Executive Director
Mr. Ram Gopal Agarwal is Founder Chairman of Dhanuka Group.
He is a decisive and action oriented visionary who took over a sick pesticide Company named Northern Mineral Pvt. Ltd. in 1980 and transformed it today into a Rs 1000 Crore organization called Dhanuka Agritech Ltd.
His deep commitment and inspiring leadership in initial turbulent days is an example worth inculcating and his passion to contribute to Indian Agriculture is commendable.
His ability to prioritize and deal effectively with a number of tasks simultaneously reinforced with the skills to make effective decisions, has metamorphosed the business venture into one of the fastest growing Agrochemical Company in India which has thrice been rated as ‘Best under a Billion Company’ by Forbes Magazine.
In order to achieve his aspiration of “Transforming India through Agriculture” he has dedicated himself to bring changes in Agrochemicals Industry and the farming community. His contribution for adopting newer farming techniques at the grass root level, judicious use of agro chemicals in farming and imparting knowledge through his nationwide network of distributors and Dhanuka Doctors in field has resulted in the overall prosperity of farmers.
Mr. Ram Gopal Agarwal has been the past Chairman of CCFI, (Crop Care Federation of India) the apex Chamber of all Indian Agrochemical majors. He is also Chairman of Advisory Committee of AGRO Chemicals Federation of India.
Mr. Ram Gopal Agarwal, Group Chairman, has been bestowed with many Awards for his tremendous contribution in Agro Industry like “Life Time Achievement Award” by Agri Business Summit and Agri Awards 2019, “Distinguished Contribution to Indian Agrochemicals Industry” during India Chem 2016 International Conference organised by FICCI etc.
Mr. Manish Dhanuka
Managing Director
Mr. Manish Dhanuka is the Director of Orchid Pharma Limited; he has the vision to rejuvenate Orchid Pharma Ltd. and take it on a fruitful path. His wide-ranging experience of handling operations, commercial, marketing and finance in the manufacturing industry provides for his analytical and decision-making skills facilitating the restoration of the company to its glorious past and to achieve even greater heights.
He excels in creating economical Pharmaceutical technologies and accelerated evaluation process for improving healthcare. Experience of 25 years in research, evaluation, and teaching in the pharmaceutical industry equips him with the expertise in innovative pharmaceutical technologies…
He holds a B.Tech in Chemical Engineering from IIT, New Delhi, and M.S in Chemical Engineering from the University of Akron, USA.
Before establishing Dhanuka Laboratories Ltd. in 1993, he began his career at Ranbaxy Labs Ltd. in New Delhi and worked there for 5 years. His vision and strategy to grow the Pharmaceutical industry in the Indian sub-continent, have helped the Dhanuka Group of companies enhance its Bulk Drugs manufacturing arm exponentially. He spearheaded the acquisition of Synmedic Laboratories in the year 2013 which is involved in pharmaceutical formulations. This entrepreneurial vigor enabled him to take over the operations of Orchid Pharma Ltd. in March 2020.
Outside of work, he likes to travel for wildlife adventures.
CLIP
Orchid Chemicals & Pharmaceuticals, or Orchid Pharma since its recent name change in 2015, was established in 1992 in Chennai to manufacture antibiotics, and entered drug discovery in 2001 with projects in the areas of anti-infectives and treatments for pain.32, 197 In 2002, the company engaged in a joint venture to develop US-based firm Bexel Biotechnology’s BLX-1002, an oral, non-PPAR AMPK activator for the treatment of diabetes,198 later repositioned for NASH (2012), but no further progress has been reported recently.197 In 2008, Orchid invested in Diakron Pharmaceuticals, a US-based company that had an exclusive license to MSD′s investigational oral anticoagulant drug, a direct thrombin inhibitor later known as DPOC-4088 (or DP-4088),199 which reached Phase 1 clinical studies in Europe in 2012 (Supporting Information Table 6b, entries 5–6).200 The company’s own internal discovery efforts had a broad therapeutic focus, covering infectious diseases, inflammation, pain, oncology, metabolic disorders, and CNS diseases. OCID-2987,197, 201 a PDE4 inhibitor for the treatment of inflammatory disorders such as COPD, completed successfully Phase 1 studies in Europe in 2012, and OCID-4681 29,202, 203 a histone deacetylase (HDAC) inhibitor for cancer had received approval in 2011 for Phase 1 studies for solid tumors in India, but we assume both have been abandoned, as cancer and inflammation are not mentioned in the company’s latest annual reports.197 Two additional compounds were abandoned at the preclinical stage: OCID-5005, a STAT-3/IL-6 inhibitor for oncology, and a unnamed Th1/Th2 cytokine synthesis inhibitor for inflammation (Supporting Information Table 2a, entries 134–138).197 Financial issues led Orchid, as of 2009, to sell parts of its business to Hospira (now part of Pfizer). As a consequence, no progress has been reported on its discovery programs since 2010, and no further NCE patent application has been published since 2012. However, in 2013 Orchid licensed its broad-spectrum β-lactamase inhibitor OCID-5090, a zwitterionic N-methylated tazobactam derivative, to the German Allecra Therapeutics for a 20 % stake in the company, for use in combination with antibiotics to treat multidrug-resistant gram negative bacteria.204–207 Allecra’s lead compound AAI202, a combination of cefepime and AAI101/OCID-5090 30, is currently in Phase 1 studies in France.208, 209
Dr. Gopalan is a synthetic organic chemist with extensive experience in the field of drug discovery and development. After completing his PhD from University of Madras, he went to Harvard University where he worked with the Nobel Laureate, Prof. E.J. Corey, as a post-doctoral fellow. Subsequent to this he joined Syntex Research Inc. in California to work on the synthesis of unnatural amino acids. After a year, he moved to Bristol-Meyers Squibb, Princeton, New Jersey, to contribute to their program on novel antibiotics and ACE inhibitors. Dr. Gopalan then moved back to India in 1982 to join the Drug Discovery Research Division of Boots Pharmaceuticals (India) Ltd. in Mumbai. Over his decade long stint there he contributed extensively to their drug discovery program, and one of the product candidates that he developed went up to Phase-2 clinical trials in both USA and UK. He then moved to Sun Pharma Advanced Research Center as Vice-President and, after a year, took up the position as General Manager at Glaxo (India) Ltd. in 1993. Here, he worked in a broad range of areas that included process development, synthesis of impurities of APIs, and generation of small molecule libraries to support drug discovery efforts to Glaxo, France. In 1999 he took over as Senior Vice President of the Drug Discovery Chemistry Division of Glenmark Pharmaceuticals Ltd. where he was involved in the design and development of inhibitors for PDE IV and DPP IV, as well as agonists for CB2. After a 6-year stint at Glenmark, Dr. Gopalan joined Matrix Laboratories Ltd. as CSO and Executive Vice-President, where he successfully helped to develop novel and selective inhibitors for PDE4 and DPP4. Five years later he became CSO and Executive Director of Orchid Pharmaceuticals Ltd in Chennai. He served in this capacity for close to a decade, contributing extensively to drug design and development in the broad segments of oncology, anti-infectives, and anti-inflammatory & metabolic disorders. Since 2017, Dr. Gopalan has been associated with CSIR-Indian Institute of Chemical Technology as a Scientific Advisor.
Dr. Gopalan’s illustrious career is endowed with numerous successes. He has been inventor, or co-inventor, of several drugs or candidate drugs. These include the novel potassium channel blockers BTS-67582 (BTI-2927) for tpe-2 diabetes, the PDE IV inhibitors Oglemilast (COPD) and Revamilast (RA); DPP IV inhibitor Melogliptin; a selective Cannaboid-2 agonist Tedalinib (Neuropathic pain); a Beta lactamase inhibitor Enmetazobactum (OCID-5090); OCID-18034 (an inhibitor of KPC enzyme); and OCID-18174 (an inhibitor of P. arugenosa). Most of these compounds were out-licensed to major international pharmaceutical companies such as Forest Laboratories Inc. USA, Teijin of Japan, Merck KGaA of Germany, Allecra of Switzerland, and Merck & Co. USA. Dr.Gopalan has 34 publications in National and International Journals, has contributed a Chapter,Co-authored with Professor K.K.Balasubramanian (IITM) on Applications of Click Chemistry in Drug Discovery and Development in a Book on Click reaction in Organic Synthesis, published by Wiley-VCH VERLAG GmbH &Co,KGaA, Weinheim,Germany,Chapter 2, p 25-70,2016, edited by Prof. S. Chandrasekharan (IISc,Bangalore) & 51 Patents.
Commensurate with his achievements, Dr. Gopalan has also received many awards. The more prominent of these include Inventor’s award by Glenmark (2004), Ranbaxy Science Foundation Award in Pharmaceutical Sciences (2005), and the Lifetime Achievement Award in the Field of Chemistry from Vels University (2011).
Today, the U.S. Food and Drug Administration issued an emergency use authorization (EUA) for casirivimab and imdevimab to be administered together for the treatment of mild to moderate COVID-19 in adults and pediatric patients (12 years of age or older weighing at least 40 kilograms [about 88 pounds]) with positive results of direct SARS-CoV-2 viral testing and who are at high risk for progressing to severe COVID-19. This includes those who are 65 years of age or older or who have certain chronic medical conditions.
In a clinical trial of patients with COVID-19, casirivimab and imdevimab, administered together, were shown to reduce COVID-19-related hospitalization or emergency room visits in patients at high risk for disease progression within 28 days after treatment when compared to placebo. The safety and effectiveness of this investigational therapy for use in the treatment of COVID-19 continues to be evaluated.
Casirivimab and imdevimab must be administered together by intravenous (IV) infusion.
Casirivimab and imdevimab are not authorized for patients who are hospitalized due to COVID-19 or require oxygen therapy due to COVID-19. A benefit of casirivimab and imdevimab treatment has not been shown in patients hospitalized due to COVID-19. Monoclonal antibodies, such as casirivimab and imdevimab, may be associated with worse clinical outcomes when administered to hospitalized patients with COVID-19 requiring high flow oxygen or mechanical ventilation.
“The FDA remains committed to advancing the nation’s public health during this unprecedented pandemic. Authorizing these monoclonal antibody therapies may help outpatients avoid hospitalization and alleviate the burden on our health care system,” said FDA Commissioner Stephen M. Hahn, M.D. “As part of our Coronavirus Treatment Acceleration Program, the FDA uses every possible pathway to make new treatments available to patients as quickly as possible while continuing to study the safety and effectiveness of these treatments.”
Monoclonal antibodies are laboratory-made proteins that mimic the immune system’s ability to fight off harmful pathogens such as viruses. Casirivimab and imdevimab are monoclonal antibodies that are specifically directed against the spike protein of SARS-CoV-2, designed to block the virus’ attachment and entry into human cells.
“The emergency authorization of these monoclonal antibodies administered together offers health care providers another tool in combating the pandemic,” said Patrizia Cavazzoni, M.D., acting director of the FDA’s Center for Drug Evaluation and Research. “We will continue to facilitate the development, evaluation and availability of COVID-19 therapies.”
The issuance of an EUA is different than an FDA approval. In determining whether to issue an EUA, the FDA evaluates the totality of available scientific evidence and carefully balances any known or potential risks with any known or potential benefits of the product for use during an emergency. Based on the FDA’s review of the totality of the scientific evidence available, the agency has determined that it is reasonable to believe that casirivimab and imdevimab administered together may be effective in treating patients with mild or moderate COVID-19. When used to treat COVID-19 for the authorized population, the known and potential benefits of these antibodies outweigh the known and potential risks. There are no adequate, approved and available alternative treatments to casirivimab and imdevimab administered together for the authorized population.
The data supporting this EUA for casirivimab and imdevimab are based on a randomized, double-blind, placebo-controlled clinical trial in 799 non-hospitalized adults with mild to moderate COVID-19 symptoms. Of these patients, 266 received a single intravenous infusion of 2,400 milligrams casirivimab and imdevimab (1,200 mg of each), 267 received 8,000 mg casirivimab and imdevimab (4,000 mg of each), and 266 received a placebo, within three days of obtaining a positive SARS-CoV-2 viral test.
The prespecified primary endpoint for the trial was time-weighted average change in viral load from baseline. Viral load reduction in patients treated with casirivimab and imdevimab was larger than in patients treated with placebo at day seven. However, the most important evidence that casirivimab and imdevimab administered together may be effective came from the predefined secondary endpoint of medically attended visits related to COVID-19, particularly hospitalizations and emergency room visits within 28 days after treatment. For patients at high risk for disease progression, hospitalizations and emergency room visits occurred in 3% of casirivimab and imdevimab-treated patients on average compared to 9% in placebo-treated patients. The effects on viral load, reduction in hospitalizations and ER visits were similar in patients receiving either of the two casirivimab and imdevimab doses.
Under the EUA, fact sheets that provide important information about using casirivimab and imdevimab administered together in treating COVID-19 as authorized must be made available to health care providers and to patients and caregivers. These fact sheets include dosing instructions, potential side effects and drug interactions. Possible side effects of casirivimab and imdevimab include: anaphylaxis and infusion-related reactions, fever, chills, hives, itching and flushing.
The EUA was issued to Regeneron Pharmaceuticals Inc.
The FDA, an agency within the U.S. Department of Health and Human Services, protects the public health by assuring the safety, effectiveness, and security of human and veterinary drugs, vaccines and other biological products for human use, and medical devices. The agency also is responsible for the safety and security of our nation’s food supply, cosmetics, dietary supplements, products that give off electronic radiation, and for regulating tobacco products.
In a clinical trial of people with COVID-19, casirivimab and imdevimab, administered together, were shown to reduce COVID-19-related hospitalization or emergency room visits in people at high risk for disease progression within 28 days after treatment when compared to placebo.[2] The safety and effectiveness of this investigational therapy for use in the treatment of COVID-19 continues to be evaluated.[2]
The data supporting the emergency use authorization (EUA) for casirivimab and imdevimab are based on a randomized, double-blind, placebo-controlled clinical trial in 799 non-hospitalized adults with mild to moderate COVID-19 symptoms.[2] Of these participants, 266 received a single intravenous infusion of 2,400 milligrams casirivimab and imdevimab (1,200 mg of each), 267 received 8,000 mg casirivimab and imdevimab (4,000 mg of each), and 266 received a placebo, within three days of obtaining a positive SARS-CoV-2 viral test.[2]
The prespecified primary endpoint for the trial was time-weighted average change in viral load from baseline.[2] Viral load reduction in participants treated with casirivimab and imdevimab was larger than in participants treated with placebo at day seven.[2] However, the most important evidence that casirivimab and imdevimab administered together may be effective came from the predefined secondary endpoint of medically attended visits related to COVID-19, particularly hospitalizations and emergency room visits within 28 days after treatment.[2] For participants at high risk for disease progression, hospitalizations and emergency room visits occurred in 3% of casirivimab and imdevimab-treated participants on average compared to 9% in placebo-treated participants.[2] The effects on viral load, reduction in hospitalizations and ER visits were similar in participants receiving either of the two casirivimab and imdevimab doses.[2]
As of September 2020, REGEN-COV is being evaluated as part of the RECOVERY Trial.[8]
On 12 April 2021, Roche and Regeneron announced that the Phase III clinical trial REGN-COV 2069 met both primary and secondary endpoints, reducing risk of infection by 81% for the non-infected patients, and reducing time-to-resolution of symptoms for symptomatic patients to one week vs. three weeks in the placebo group.[9]
Authorization
On 21 November 2020, the U.S. Food and Drug Administration (FDA) issued an emergency use authorization (EUA) for casirivimab and imdevimab to be administered together for the treatment of mild to moderate COVID-19 in people twelve years of age or older weighing at least 40 kilograms (88 lb) with positive results of direct SARS-CoV-2 viral testing and who are at high risk for progressing to severe COVID-19.[2][10][11] This includes those who are 65 years of age or older or who have certain chronic medical conditions.[2] Casirivimab and imdevimab must be administered together by intravenous (IV) infusion.[2]
Casirivimab and imdevimab are not authorized for people who are hospitalized due to COVID-19 or require oxygen therapy due to COVID-19.[2] A benefit of casirivimab and imdevimab treatment has not been shown in people hospitalized due to COVID-19.[2] Monoclonal antibodies, such as casirivimab and imdevimab, may be associated with worse clinical outcomes when administered to hospitalized people with COVID-19 requiring high flow oxygen or mechanical ventilation.[2]
The EUA was issued to Regeneron Pharmaceuticals Inc.[2][10][12]
On 1 February 2021, the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) started a rolling review of data on the REGN‑COV2 antibody combination (casirivimab/imdevimab), which is being co-developed by Regeneron Pharmaceuticals, Inc. and F. Hoffman-La Roche, Ltd (Roche) for the treatment and prevention of COVID‑19.[13][14] In February 2021, the CHMP concluded that the combination, also known as REGN-COV2, can be used for the treatment of confirmed COVID-19 in people who do not require supplemental oxygen and who are at high risk of progressing to severe COVID-19.[15]
The Central Drugs Standards Control Organisation (CDSCO) in India, on 5 May 2021, granted an Emergency Use Authorisation to Roche (Genentech)[16] and Regeneron[17] for use of the casirivimab/imdevimab cocktail in the country. The announcement came in light of the second wave of the COVID-19 pandemic in India. Roche India maintains partnership with Cipla, thereby permitting the latter to market the drug in the country.[18]
Deployment
Although Regeneron is headquartered in Tarrytown, New York (near New York City), REGEN-COV is manufactured at the company’s primary U.S. manufacturing facility in Rensselaer, New York (near the state capital at Albany).[19] In September 2020, to free up manufacturing capacity for REGEN-COV, Regeneron began to shift production of its existing products from Rensselaer to the Irish city of Limerick.[20]
Regeneron has a deal in place with Roche (Genentech)[21]to manufacture and market REGEN-COV outside the United States.[10][22]
On 2 October 2020, Regeneron Pharmaceuticals announced that US President Donald Trump had received “a single 8 gram dose of REGN-COV2” after testing positive for SARS-CoV-2.[23][24] The drug was provided by the company in response to a “compassionate use” (temporary authorization for use) request from the president’s physicians.[23]
Monoclonal antibody Treatment and prophylaxis of SARS-CoV-2 infection
ANTIVIRAL
SARS-CoV-2 spike glycoprotein
REGN 10987
RG 6412
Fact Sheet – US Food and Drug Administration
https://www.fda.gov › media › download PDFBenefit of treatment with casirivimab and imdevimab has not been observed in patients hospitalized due to COVID-19. Monoclonal antibodies, such as casirivimab.
In a clinical trial of people with COVID-19, casirivimab and imdevimab, administered together, were shown to reduce COVID-19-related hospitalization or emergency room visits in people at high risk for disease progression within 28 days after treatment when compared to placebo.[2] The safety and effectiveness of this investigational therapy for use in the treatment of COVID-19 continues to be evaluated.[2]
The data supporting the emergency use authorization (EUA) for casirivimab and imdevimab are based on a randomized, double-blind, placebo-controlled clinical trial in 799 non-hospitalized adults with mild to moderate COVID-19 symptoms.[2] Of these participants, 266 received a single intravenous infusion of 2,400 milligrams casirivimab and imdevimab (1,200 mg of each), 267 received 8,000 mg casirivimab and imdevimab (4,000 mg of each), and 266 received a placebo, within three days of obtaining a positive SARS-CoV-2 viral test.[2]
The prespecified primary endpoint for the trial was time-weighted average change in viral load from baseline.[2] Viral load reduction in participants treated with casirivimab and imdevimab was larger than in participants treated with placebo at day seven.[2] However, the most important evidence that casirivimab and imdevimab administered together may be effective came from the predefined secondary endpoint of medically attended visits related to COVID-19, particularly hospitalizations and emergency room visits within 28 days after treatment.[2] For participants at high risk for disease progression, hospitalizations and emergency room visits occurred in 3% of casirivimab and imdevimab-treated participants on average compared to 9% in placebo-treated participants.[2] The effects on viral load, reduction in hospitalizations and ER visits were similar in participants receiving either of the two casirivimab and imdevimab doses.[2]
As of September 2020, REGEN-COV is being evaluated as part of the RECOVERY Trial.[8]
On 12 April 2021, Roche and Regeneron announced that the Phase III clinical trial REGN-COV 2069 met both primary and secondary endpoints, reducing risk of infection by 81% for the non-infected patients, and reducing time-to-resolution of symptoms for symptomatic patients to one week vs. three weeks in the placebo group.[9]
Authorization
On 21 November 2020, the U.S. Food and Drug Administration (FDA) issued an emergency use authorization (EUA) for casirivimab and imdevimab to be administered together for the treatment of mild to moderate COVID-19 in people twelve years of age or older weighing at least 40 kilograms (88 lb) with positive results of direct SARS-CoV-2 viral testing and who are at high risk for progressing to severe COVID-19.[2][10][11] This includes those who are 65 years of age or older or who have certain chronic medical conditions.[2] Casirivimab and imdevimab must be administered together by intravenous (IV) infusion.[2]
Casirivimab and imdevimab are not authorized for people who are hospitalized due to COVID-19 or require oxygen therapy due to COVID-19.[2] A benefit of casirivimab and imdevimab treatment has not been shown in people hospitalized due to COVID-19.[2] Monoclonal antibodies, such as casirivimab and imdevimab, may be associated with worse clinical outcomes when administered to hospitalized people with COVID-19 requiring high flow oxygen or mechanical ventilation.[2]
The EUA was issued to Regeneron Pharmaceuticals Inc.[2][10][12]
On 1 February 2021, the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) started a rolling review of data on the REGN‑COV2 antibody combination (casirivimab/imdevimab), which is being co-developed by Regeneron Pharmaceuticals, Inc. and F. Hoffman-La Roche, Ltd (Roche) for the treatment and prevention of COVID‑19.[13][14] In February 2021, the CHMP concluded that the combination, also known as REGN-COV2, can be used for the treatment of confirmed COVID-19 in people who do not require supplemental oxygen and who are at high risk of progressing to severe COVID-19.[15]
The Central Drugs Standards Control Organisation (CDSCO) in India, on 5 May 2021, granted an Emergency Use Authorisation to Roche (Genentech)[16] and Regeneron[17] for use of the casirivimab/imdevimab cocktail in the country. The announcement came in light of the second wave of the COVID-19 pandemic in India. Roche India maintains partnership with Cipla, thereby permitting the latter to market the drug in the country.[18]
Deployment
Although Regeneron is headquartered in Tarrytown, New York (near New York City), REGEN-COV is manufactured at the company’s primary U.S. manufacturing facility in Rensselaer, New York (near the state capital at Albany).[19] In September 2020, to free up manufacturing capacity for REGEN-COV, Regeneron began to shift production of its existing products from Rensselaer to the Irish city of Limerick.[20]
Regeneron has a deal in place with Roche (Genentech)[21]to manufacture and market REGEN-COV outside the United States.[10][22]
On 2 October 2020, Regeneron Pharmaceuticals announced that US President Donald Trump had received “a single 8 gram dose of REGN-COV2” after testing positive for SARS-CoV-2.[23][24] The drug was provided by the company in response to a “compassionate use” (temporary authorization for use) request from the president’s physicians.[23]
Safe and effective vaccines are needed to end the COVID-19 pandemic caused by SARS-CoV-2. Here we report the preclinical development of a lipid nanoparticle (LNP) formulated SARS-CoV-2 mRNA vaccine, PTX-COVID19-B. PTX-COVID19-B was chosen among three candidates after the initial mouse vaccination results showed that it elicited the strongest neutralizing antibody response against SARS-CoV-2. Further tests in mice and hamsters indicated that PTX-COVID19-B induced robust humoral and cellular immune responses and completely protected the vaccinated animals from SARS-CoV-2 infection in the lung. Studies in hamsters also showed that PTX-COVID19-B protected the upper respiratory tract from SARS-CoV-2 infection. Mouse immune sera elicited by PTX-COVID19-B vaccination were able to neutralize SARS-CoV-2 variants of concern (VOCs), including the B.1.1.7, B.1.351 and P.1 lineages. No adverse effects were induced by PTX-COVID19-B in both mice and hamsters. These preclinical results indicate that PTX-COVID19-B is safe and effective. Based on these results, PTX-COVID19-B was authorized by Health Canada to enter clinical trials in December 2020 with a phase 1 clinical trial ongoing (ClinicalTrials.gov number: NCT04765436).
PTX-COVID19-B is a messenger RNA (mRNA)-based COVID-19 vaccine, a vaccine for the prevention of the COVID-19 disease caused by an infection of the SARS-CoV-2coronavirus, created by Providence Therapeutics—a private Canadian drug company co-founded by Calgary, Alberta-based businessman Brad T. Sorenson and San Francisco-based Eric Marcusson.[1] in 2013. A team of eighteen working out of Sunnybrook Research Institute in Toronto, Ontario developed PTX-COVID19-B[2] in less than four weeks, according to the Calgary Herald.[3] Human trials with sixty volunteers began on January 26, 2021 in Toronto.[4][5][6]
Providence, which has no manufacturing facilities, partnered with Calgary-based Northern mRNA—the “anchor tenant” in their future manufacturing facilities pending financing.[2]
On 30 April 2021, Sorenson announced that Providence Therapeutics would be leaving Canada and any vaccine that it developed would not be manufactured in Canada.[2]
Overview
Providence Therapeutics Holdings Inc. was co-founded in Toronto, Ontario[7][8] by Calgary, Alberta-based businessman Brad T. Sorenson and San Francisco-based Eric Marcusson Ph.D, who was also the Chief Scientific Officer.[9][3]
PTX-COVID19-B is a messenger RNA (mRNA)-based COVID-19 vaccine. In an interview with CTV news, Sorenson said they were “building some of the important building blocks for the messenger RNA … that provides instructions to cells … to build proteins that may treat or prevent disease”.
As of January 2021, Northern RNA’s Calgary lab was proposed as the site where manufacturing of PTX-COVID19-B would take place.[10] Providence Therapeutics’ partner, Northern RNA, which located at 421 7 Avenue SW in Calgary, has been described as Providence Therapeutics northern division.[7][8]
A February 2021 Manitoba government press release said that the Winnipeg-based Emergent BioSolutions would be manufacturing the vaccine.[11]
Human trials
Phase 1
Human trials began on January 26, 2021 with 60 volunteers between the ages of 18 to 65 in Toronto.[12][13][3] Of these, 15 would receive a placebo and 3 groups of 15 would receive different doses of the vaccine.[10] The volunteers will be monitored for 13 months. The company said that enough data would be available in May which could result in a Phase 2 clinical testing beginning soon after that, pending regulatory approval. If the results of a subsequent larger human trial are positive, the vaccine could enter a commercialization phase in 2022.[14] The Phase 1 clinical trial lead was Piyush Patel. At the 29 April meeting with the House of Commons, Sorenson estimated that PTX-COVID19-B could be approved by Health Canada by “January or February 2022”.[15]:8
Provincial funding
Shortly after the first human trials on PTX-COVID19-B began in late January, on 11 February 2021, Manitoba Premier Brian Pallister announced a “term sheet” between the province and Providence Therapeutics through which Manitoba would receive 2 million doses of PTX-COVID19-B pending its approval by Health Canada.[11] The term sheet includes “best-price guarantee” PTX-COVID19-B.[13] According to a provincial statement released by the Manitoba government, pending approval of the vaccine, the actual manufacturing would take place in Winnipeg by Emergent BioSolutions.[11] Pallister said that, “Building a secure, made-in-Canada vaccine supply will put Canadians at the head of the line to get a COVID vaccine, where we belong.”[11] The down payment would be 20% with a subsequent 40% to be paid when the vaccine was approved by Health Canada; the balance would be paid on delivery of the doses.[13] Specifics about the contract were released in April 2021: the total cost was estimated as CAD $36 million and the agreement included a clause for a non-refundable advance payment of CAD $7.2 million.[2] Sorenson made this comment to Global News: “Under no circumstances is Manitoba going to be on the hook for $7.2 million unless they get real value out of it”.
Federal funding
Canada’s National Research Council (NRC) provided Providence Therapeutics with CAD $5 million for the launch of January 2021 first phase of PTX-COVID19-B clinical trials.[2]
As part of the federal government’s “next generation manufacturing supercluster” program, Providence and Northern mRNA had also been “cleared to access up to $5 million” towards the manufacturing start up process, according to a federal government spokesperson.[2]
The CBC report in late April 2021 also stated that “it could be eligible for a slice of $113 million in additional funding from the National Research Council of Canada Industrial Research Assistance Program”. The federal government had provided funding to some other companies in Canada that were also working to develop a COVID-19 vaccine.[2]
Sorenson as Providence Therapeutics CEO posted an open letter to Prime Minister Justin Trudeau, in which he requested $CDN 150 million upfront to be used to pay for clinical trial and material costs.[16][9]
On 29 April 2021, Sorenson appeared before the House of Commons standing committee on international trade, to ask the Minister of Procurement, Anita Anand, to consider PTX-COVID19-B as an alternative to Moderna and Pfizer for the “2022 booster vaccines”.[15] Sorenson said that the NRC had approached Providence Therapeutics in 2020 after the company had announced their Phase I trial PTX-COVID19-B. Sorenson told the Standing Committee that, “We’ve had really good dialogue ever since phase I started. That process has gone on. That started probably [in February], as we geared up to conclude our phase I trial and release data. Although the NRC is capped at $10 million, which is certainly not sufficient to carry out phase II and phase III trials, the NRC has, through the bureaucracy, elevated us back up to the strategic innovation fund. That occurred about three weeks ago. We’re now working with the strategic innovation fund.”[15]:7
He later said that no reply had been received from the government.[17]
In a meeting with the federal COVID-19 vaccine task force and Sorenson, task force members expressed concerns that “Providence might not be able to scale up production fast enough”.[2]
Clinical trials
PTX-COVID19-B, an mRNA Humoral Vaccine, is Intended for Prevention of COVID-19 in a General Population. This Study is Designed to Evaluate Safety, Tolerability, and Immunogenicity of PTX-COVID19-B Vaccine in Healthy Seronegative Adults Aged 18-64… https://clinicaltrials.gov/ct2/show/NCT04765436
Biological E., will run a clinical trial of Providence’s vaccine in India and seek emergency use approval for it, the company said in a statement
Hyderabad-based Biological E said on Tuesday it has entered into a licensing agreement with Providence Therapeutics Holdings to manufacture the Canadian company’s mRNA COVID-19 vaccine in India.
Biological E., which also has a separate deal to produce about 600 million doses of Johnson & Johnson’s COVID-19 shot annually, will run a clinical trial of Providence’s vaccine in India and seek emergency use approval for it, the company said in a statement.
Providence will sell up to 30 million doses of its mRNA vaccine, PTX-COVID19-B, to Biological E, and will also provide the necessary technology transfer of the shot, with a minimum production capacity of 600 million doses in 2022 and a target capacity of 1 billion doses.
Financial details of the transaction were not disclosed.
India has been struggling with a devastating second wave of the pandemic and has managed to fully vaccinate only about 3% of its population. On Monday, the Serum Institute of India said it will increase production of AstraZeneca’s shot by nearly 40% in June, a step towards bridging the shortfall in the country.
“The mRNA platform has emerged as the front runner in delivering the first vaccines for emergency use to combat the COVID-19 pandemic,” said Mahima Datla, Biological E.’s managing director.
Messenger ribonucleic acid (mRNA) vaccines prompt the body to make a protein that is part of the virus, triggering an immune response. US companies Pfizer and Moderna use mRNA technology in their COVID-19 shots.
The drug regulator has approved clinical trials of another mRNA vaccine developed by local firm Gennova Biopharmaceuticals, and the government has said it will fund the studies.
Providence Therapeutics Announces Very Favorable Interim Phase 1 Trial Data for PTX-COVID19-B, its mRNA Vaccine Against COVID-19
CALGARY, AB, May 12, 2021 / – Providence Therapeutics Holdings Inc. (“Providence”) announced today very favorable interim clinical data of PTX-COVID19-B, its vaccine candidate against SARS-CoV-2 (“COVID-19”), from its Phase 1 study entitled “PRO-CL-001, A Phase 1, First-in-Human, Observer-Blinded, Randomized, Placebo Controlled, Ascending Dose Study to Evaluate the Safety, Tolerability, and Immunogenicity of PTX-COVID19-B Vaccine in Healthy Seronegative Adults Aged 18-64” (the “Phase 1 Study”), which found that PTX-COVID19-B met Providence’s target results for safety, tolerability, and immunogenicity in the participants of the Phase 1 Study.
Highlights from Providence Therapeutics’ “Phase 1 Study”:
PTX-COVID19-B was generally safe and well tolerated
PTX-COVID19-B exhibited strong virus neutralization capability across the 16µg, 40µg and 100µg dose cohorts
PTX-COVID19-B 40µg dose was selected for Phase 2 study
PTX-COVID19-B will be evaluated in additional Phase 1 population cohorts
The Phase 1 Study was designed with dose-escalations and was performed in seronegative adult subjects without evidence of recent exposure to COVID-19. The subjects were randomized to receive either the PTX-COVID19-B vaccine or a placebo in a 3:1 ratio. A total of 60 subjects participated in the Phase 1 Study.
The overall results of the Phase 1 Study are that PTX-COVID19-B was safe and well tolerated at the three dose levels of 16µg, 40µg and 100µg. Adverse events identified in the Phase 1 Study were generally mild to moderate in severity, self-resolving and transient. There were no serious adverse events reported in the Phase 1 Study. The most common adverse event reported in the Phase 1 Study was redness and pain at the injection site. Systemic reactions reported in the Phase 1 Study were generally mild to moderate and well tolerated with headache being the most common reaction reported. The reported adverse events of the Phase 1 Study were in line with the expectations of management of Providence as they compare very favorably to the adverse events data published on other mRNA vaccines for COVID-19 that have been approved for use by various health authorities around the world.
Based on the results of the Phase 1 Study, Providence intends to use a 40µg dose for the Phase 2 study of PTX-COVID19-B that is anticipated to be initiated in June 2021. Additional Phase 1 studies in adolescent and elderly populations are also planned to be undertaken by Providence.
PTX-COVID19-B vaccination induced high anti-S IgG antibodies:
Participants in the Phase 1 Study were vaccinated on day zero and day twenty-eight. Plasma samples were collected on day 1, day 8, day 28 (prior to the participant receiving the second dose), and day 42 to determine levels of IgG anti-S protein using electrochemiluminescence (“ECL”) assays from Meso Scale Discovery (“MSD”). Study participants in all three vaccine dose cohorts of the Phase 1 Study developed a strong IgG antibody response against Spike protein that was detected by day 28 and enhanced by day 42. No antibodies against S protein were detected in participants in the Phase 1 Study injected with placebo. The highest levels of antibodies were found in the 40 and 100 µg doses. By day 42, PTX-COVID19-B vaccinated participants had more than one log higher antibody levels than convalescent subjects-plasma (indicated in the dotted line) which was evaluated in the same assay.
Based on the interim data of the Phase 1 Study, the level of antibodies produced in participants by PTX-COVID19-B compare favorably to the levels of antibodies produced by other mRNA vaccines that have been approved for use against COVID-19 based on the recently published report from Stanford University, where IgG responses in individuals vaccinated with the COVID-19 mRNA vaccine compared to COVID-19 infected patients were evaluated[1].
PTX-COVID19-B vaccination induced high neutralizing antibody levels:
Neutralizing activity from the Phase 1 Study participants’ plasma was evaluated by S-ACE2 MSD assay. The results indicate that the antibodies block the interaction between S protein with the ACE2 receptor and the decrease in ECL signal is used to calculate percentage inhibition of the plasma at the same dilution. All participants in the Phase 1 Study from the 16, 40 and 100 µg dose levels showed blocking activity by day 28 and all of them reached 100% blocking activity by day 42 with samples diluted 1:100 or greater. Moreover, the quantification of the antibody levels in ng/mL with a reference standard showed that all participants in the Phase 1 Study produced neutralizing antibodies by day 28 with the first immunization and increase ten-fold by day 42, two weeks after the administration of the second dose. These results indicate that PTX-COVID19-B induced a strong neutralizing antibody response which compares very favorably to the published results of other mRNA vaccines. Further studies are being conducted by Providence to determine neutralization activity using a pseudo-virus assay.
Providence intends to advance a Phase 2 clinical trial in early June 2021, with multiple trial sites in Canada. The Phase 2 clinical trial is anticipated to be structured as a comparator trial using Pfizer/BioNTech vaccine as the positive control.
About Providence Therapeutics
Providence is a leading Canadian clinical stage biotechnology company pioneering mRNA therapeutics and vaccines with operations in Calgary, Alberta and Toronto, Ontario. In response to a worldwide need for a COVID-19 vaccine, Providence expanded its focus beyond oncology therapies and devoted its energy and resources to develop a world-class mRNA vaccine for COVID-19. Providence is focused on serving the needs of Canada, and other countries that may be underserved by large pharmaceutical programs. For more information, please visit providencetherapeutics.com.
^ Clinical trial number NCT04765436 for “PTX-COVID19-B, an mRNA Humoral Vaccine, is Intended for Prevention of COVID-19 in a General Population. This Study is Designed to Evaluate Safety, Tolerability, and Immunogenicity of PTX-COVID19-B Vaccine in Healthy Seronegative Adults Aged 18-64” at ClinicalTrials.gov
Antifungal, Cell wall biosynthesis inhibitor, Treatment of invasive fungal infections due to Candida spp. or Aspergillus spp., vulvovaginal candidiasis
Mechanism of ActionBeta-1,3-D glucan synthetase inhibitors
Orphan Drug StatusYes – Invasive bronchopulmonary aspergillosis; Candidiasis
RegisteredVulvovaginal candidiasis
Phase IIICandidiasis
Phase IIInvasive bronchopulmonary aspergillosis
Phase IUnspecified
PreclinicalPneumocystis pneumonia
01 Jun 2021Registered for Vulvovaginal candidiasis (In adolescents, In children, In the elderly, In adults) in USA (PO)
01 May 2021Ibrexafungerp – SCYNEXIS receives Qualified Infectious Disease Product status for Vulvovaginal candidiasis (Recurrent, Prevention) in USA
30 Apr 2021Efficacy data from phase III VANISH-303 and VANISH-306 trials in Vulvovaginal Candidiasis presented at the 2021 American College of Obstetricians and Gynecologists Annual Meeting (ACOG-2021)
We previously reported medicinal chemistry efforts that identified MK-5204, an orally efficacious β-1,3-glucan synthesis inhibitor derived from the natural product enfumafungin. Further extensive optimization of the C2 triazole substituent identified 4-pyridyl as the preferred replacement for the carboxamide of MK-5204, leading to improvements in antifungal activity in the presence of serum, and increased oral exposure. Reoptimizing the aminoether at C3 in the presence of this newly discovered C2 substituent, confirmed that the (R) t-butyl, methyl aminoether of MK-5204 provided the best balance of these two key parameters, culminating in the discovery of ibrexafungerp, which is currently in phase III clinical trials. Ibrexafungerp displayed significantly improved oral efficacy in murine infection models, making it a superior candidate for clinical development as an oral treatment for Candida and Aspergillus infections.
“Ibrexafungerp”. Drug Information Portal. U.S. National Library of Medicine.
Clinical trial number NCT03734991 for “Efficacy and Safety of Oral Ibrexafungerp (SCY-078) vs. Placebo in Subjects With Acute Vulvovaginal Candidiasis (VANISH 303)” at ClinicalTrials.gov
Clinical trial number NCT03987620 for “Efficacy and Safety of Oral Ibrexafungerp (SCY-078) vs. Placebo in Subjects With Acute Vulvovaginal Candidiasis (Vanish 306)” at ClinicalTrials.gov
Ibrexafungerp, also known as SCY-078 or MK-3118, is a novel enfumafungin derivative oral triterpene antifungal approved for the treatment of vulvovaginal candidiasis (VVC), also known as a vaginal yeast infection.1,9 It was developed out of a need to treat fungal infections that may have become resistant to echinocandins or azole antifungals.1 Ibrexafungerp is orally bioavailable compared to the echinocandins caspofungin, micafungin, and anidulafungin; which can only be administered parenterally.1,2 Similar to echinocandins, ibrexafungerp targets the fungal β-1,3-glucan synthase, which is not present in humans, limiting the chance of renal or hepatic toxicity.6,9
Ibrexafungerp was granted FDA approval on 1 June 2021.9
β-1,3-glucan synthase is composed of a catalytic subunit, FKS1 or FKS2, and a GTP-binding regulatory subunit, Rho1.5,6 This synthase is involved in the synthesis of β-1,3-glucan, a fungal cell wall component.6
Ibrexafungerp acts similarly to the echinocandin antifungals, by inhibiting the synthesis of β-1,3-glucan synthase.1,9 While echinocandins bind to the FKS1 domain of β-1,3-glucan synthase, enfumafungin and its derivatives bind at an alternate site which allows them to maintain their activity against fungal infections that are resistant to echinocandins.3,4
Ibrexafungerp has been shown in animal studies to distribute well to vaginal tissue, making it a favourable treatment for vulvovaginal candidiasis.4
Wring SA, Randolph R, Park S, Abruzzo G, Chen Q, Flattery A, Garrett G, Peel M, Outcalt R, Powell K, Trucksis M, Angulo D, Borroto-Esoda K: Preclinical Pharmacokinetics and Pharmacodynamic Target of SCY-078, a First-in-Class Orally Active Antifungal Glucan Synthesis Inhibitor, in Murine Models of Disseminated Candidiasis. Antimicrob Agents Chemother. 2017 Mar 24;61(4). pii: AAC.02068-16. doi: 10.1128/AAC.02068-16. Print 2017 Apr. [Article]
Hector RF, Bierer DE: New beta-glucan inhibitors as antifungal drugs. Expert Opin Ther Pat. 2011 Oct;21(10):1597-610. doi: 10.1517/13543776.2011.603899. Epub 2011 Jul 25. [Article]
Kuhnert E, Li Y, Lan N, Yue Q, Chen L, Cox RJ, An Z, Yokoyama K, Bills GF: Enfumafungin synthase represents a novel lineage of fungal triterpene cyclases. Environ Microbiol. 2018 Sep;20(9):3325-3342. doi: 10.1111/1462-2920.14333. Epub 2018 Sep 13. [Article]
Larkin EL, Long L, Isham N, Borroto-Esoda K, Barat S, Angulo D, Wring S, Ghannoum M: A Novel 1,3-Beta-d-Glucan Inhibitor, Ibrexafungerp (Formerly SCY-078), Shows Potent Activity in the Lower pH Environment of Vulvovaginitis. Antimicrob Agents Chemother. 2019 Apr 25;63(5). pii: AAC.02611-18. doi: 10.1128/AAC.02611-18. Print 2019 May. [Article]
Ha YS, Covert SF, Momany M: FsFKS1, the 1,3-beta-glucan synthase from the caspofungin-resistant fungus Fusarium solani. Eukaryot Cell. 2006 Jul;5(7):1036-42. doi: 10.1128/EC.00030-06. [Article]
Perlin DS: Mechanisms of echinocandin antifungal drug resistance. Ann N Y Acad Sci. 2015 Sep;1354:1-11. doi: 10.1111/nyas.12831. Epub 2015 Jul 17. [Article]
Wring S, Murphy G, Atiee G, Corr C, Hyman M, Willett M, Angulo D: Clinical Pharmacokinetics and Drug-Drug Interaction Potential for Coadministered SCY-078, an Oral Fungicidal Glucan Synthase Inhibitor, and Tacrolimus. Clin Pharmacol Drug Dev. 2019 Jan;8(1):60-69. doi: 10.1002/cpdd.588. Epub 2018 Jun 27. [Article]
Ghannoum M, Arendrup MC, Chaturvedi VP, Lockhart SR, McCormick TS, Chaturvedi S, Berkow EL, Juneja D, Tarai B, Azie N, Angulo D, Walsh TJ: Ibrexafungerp: A Novel Oral Triterpenoid Antifungal in Development for the Treatment of Candida auris Infections. Antibiotics (Basel). 2020 Aug 25;9(9). pii: antibiotics9090539. doi: 10.3390/antibiotics9090539. [Article]
FDA Approved Drug Products: Brexafemme (Ibrexafungerp) Oral Tablet [Link]
Corbevax is a “recombinant protein sub-unit” vaccine, which means it is made up of a specific part of SARS-CoV-2 — the spike protein on the virus’s surface.
The spike protein allows the virus to enter the cells in the body so that it can replicate and cause disease. However, when this protein alone is given to the body, it is not expected to be harmful as the rest of the virus is absent. The body is expected to develop an immune response against the injected spike protein. Therefore, when the real virus attempts to infect the body, it will already have an immune response ready that will make it unlikely for the person to fall severely ill.
Although this technology has been used for decades to make hepatitis B vaccines, Corbevax will be among the first Covid-19 vaccines to use this platform. Novavax has also developed a protein-based vaccine, which is still waiting for emergency use authorisation from various regulators.
How Corbevax was made
While it is indigenously produced, Corbevax’s beginnings can be traced to the Baylor College of Medicine’s National School of Tropical Medicine. The School had been working on recombinant protein vaccines for coronaviruses SARS and MERS for a decade.
“We knew all the techniques required to produce a recombinant protein (vaccine) for coronaviruses at high levels of efficiency and integrity,” said Dr Peter Hotez, Professor and Dean at the School.
When the genetic sequence for SARS-CoV-2 was made available in February 2020, researchers at the School pulled out the sequence for the gene for the spike protein, and worked on cloning and engineering it. The gene was then put into yeast, so that it could manufacture and release copies of the protein. “It’s actually similar to the production of beer. Instead of releasing alcohol, in this case, the yeast is releasing the recombinant protein,” Dr Hotez said.
After this, the protein was purified to remove any remnants of the yeast “to make it pristine”. Then, the vaccine was formulated using an adjuvant to better stimulate the immune response.
Most of these ingredients are cheap and easy to find.
In August, BCM transferred its production cell bank for this vaccine to Biological E, so that the Hyderabad-based company could take the candidate through trials. The vaccine has received approval for phase 3 trials, which the government expects will be over by July.
Biological E is also expected to scale up production for the world.
How Corbevax is different
Other Covid-19 vaccines approved so far are either mRNA vaccines (Pfizer and Moderna), viral vector vaccines (AstraZeneca-Oxford/Covishield, Johnson & Johnson and Sputnik V) or inactivated vaccines (Covaxin, Sinovac-CoronaVac and Sinopharm’s SARS-CoV-2 Vaccine–Vero Cell).
Inactivated vaccines, which include killed particles of the whole SARS-CoV-2 virus, attempt to target the entire structure of the virus. On the other hand, Corbevax, like the mRNA and viral vector Covid-19 vaccines, targets only the spike protein, but in a different way.
Viral vector and mRNA and vaccines use a code to induce our cells to make the spike proteins against which the body have to build immunity. “In this case (Corbevax), we’re actually giving the protein,” said Dr Hotez.
Like most other Covid-19 vaccines, Corbevax is administered in two doses. However, as it is made using a low-cost platform, it is also expected to be among the cheapest available in the country.
Why Corbevax matters
This is the first time the Indian government has placed an order for a vaccine that has not received emergency use authorisation, paying Rs 1,500 crore in advance to block an order that could vaccinate 15 crore Indian citizens. The Centre has provided major pre-clinical and clinical trial support towards the vaccine’s development, including a grant-in-aid of Rs 100 crore from the Department of Biotechnology.
A major reason for India placing such a big order is the difficulties it is facing in enhancing vaccine supplies. While the US, UK and the EU had made advance payments and at-risk investments into vaccines like Pfizer, AstraZeneca and Moderna, India waited until after its first two vaccines were approved before placing limited orders. Even after the government eased regulatory requirements for foreign vaccines, it did not receive a speedy response from companies like Pfizer and Moderna, their supplies already blocked through orders from other countries. India is currently in negotiations for a limited supply of Pfizer’s vaccine, and expecting to secure up to two billion doses of Covid vaccines by December this year. Given the ease with which it can be mass produced, Corbevax could make up a sizeable portion of this expected supply.
Biological E, the manufacturer of Corbevax
Biological E, headquartered in Hyderabad, was founded by Dr D V K Raju in 1953 as a biological products company that pioneered the production of heparin in India. By 1962, it forayed into the vaccines space, producing DPT vaccines on a large-scale. Today, it is among the major vaccine makers in India and, by its own claim, the “largest” tetanus vaccine producer in the world.
It has seven WHO-prequalified shots, including a five-in-one vaccine against diphtheria, tetanus, pertussis, hepatitis B and haemophilus influenza type-b infections. Its vaccines are supplied to over 100 countries and it has supplied more than two billion doses in the last 10 years alone.
Since 2013, the company has been under the management of Mahima Datla — the third generation of the founding family. During her time as managing director, the company has received WHO prequalification of its Japanese encephalitis, DTwP and Td as well as measles and rubella vaccines and also commenced commercial operations in the US.
In phase I clinical trial was carried to evaluate the safety and immunogenicity of the vaccine candidate in about 360 participants.[5]The phase II concluded in April 2021.[6][7]
Phase III trials
In April 2021, the Drugs Controller General of India permitted the vaccine candidate to start phase III clinical trials. A total of 1,268 healthy participants between the age of 18 and 80 years to be selected from 15 sites across India for the trial and intended to be part of a larger global Phase III study.[8][7]
Manufacturing and Orders
In April 2021, the U.S. International Development Finance Corporation (DFC) announced that it would fund the expansion of BioE’s manufacturing capabilities, so that it could produce at least 1 billion doses by end of 2022.[9]
31 May 2021Clinical development is ongoing in Bladder cancer (QED Therapeutics pipeline, May 2020)
28 May 2021Registered for Cholangiocarcinoma (Second-line therapy or greater, Metastatic disease, Inoperable/Unresectable, Late-stage disease) in USA (PO) – First global approval (under Project Orbis using RTOR program)
28 May 2021Efficacy and safety data from a phase II trial in Cholangiocarcinoma released by QED Therapeutics
Infigratinib is a pan-fibroblast growth factor receptor (FGFR) kinase inhibitor. By inhibiting the FGFR pathway, which is often aberrated in cancers such as cholangiocarcinoma, infigratinib suppresses tumour growth.1 Cholangiocarcinoma is the most common primary malignancy affecting the biliary tract and the second most common primary hepatic malignancy.2 Infitratinib is a pan-FGFR inhibitor, as it is an ATP-competitive inhibitor of all four FGFR receptor subtypes.1
On May 28, 2021, the FDA granted accelerated approval to infigratinib – under the market name Truseltiq – for the treatment of previously treated, unresectable locally advanced or metastatic cholangiocarcinoma in adults with a fibroblast growth factor receptor 2 (FGFR2) fusion or another rearrangement as detected by an FDA-approved test.5 This approval follows pemigatinib, another FGFR inhibitor approved by the FDA for the same therapeutic indication.
Infigratinib is indicated for the treatment of previously treated, unresectable locally advanced or metastatic cholangiocarcinoma in adults with a fibroblast growth factor receptor 2 (FGFR2) fusion or another rearrangement as detected by an FDA-approved test.4
Medical uses
Infigratinib is indicated for the treatment of adults with previously treated, unresectable locally advanced or metastatic cholangiocarcinoma (bile duct cancer) with a fibroblast growth factor receptor 2 (FGFR2) fusion or other rearrangement as detected by an FDA-approved test.[1]
PAPER
Journal of Medicinal Chemistry (2011), 54(20), 7066-7083.
A novel series of N-aryl-N′-pyrimidin-4-yl ureas has been optimized to afford potent and selective inhibitors of the fibroblast growth factor receptor tyrosine kinases 1, 2, and 3 by rationally designing the substitution pattern of the aryl ring. On the basis of its in vitro profile, compound 1h (NVP-BGJ398) was selected for in vivo evaluation and showed significant antitumor activity in RT112 bladder cancer xenografts models overexpressing wild-type FGFR3. These results support the potential therapeutic use of 1h as a new anticancer agent.
In 2018, it was estimated that 150,350 new patients would be diagnosed with urinary system cancer: 81,190 urinary bladder; 65,340 kidney and renal pelvis; and, 3,820 ureter and other urinary organs. Excluding non-urothelial kidney cancers, approximately 5 to 10% of all urothelial carcinomas are upper tract urothelial carcinomas (UTUC). The incidence of UTUC is 2 to 3 times greater in men than women (Siegel et al, 2018; Roupret et al, 2015).
[0003] In contrast to invasive urinary bladder cancer (UCB), UTUC has a more aggressive clinical course. At the time of diagnosis, 60% of patients with UTUC have invasive cancer compared to 15% to 25% of patients with UCB (Roupret et al, 2015; Margulis et al., 2009). Thirty-six percent have regional disease and 9% distant disease (Raman et al., 2010). A large retrospective review of 1363 patients with UTUC who underwent radical nephroureterectomy (RNU) at 12 centers demonstrated that 28% of the total population had recurrence after RNU (Margulis et al, 2009).
[0004] To reduce the morbidity and mortality in patients with UTUC, neoadjuvant or adjuvant treatment is needed. The POUT study, a large randomized trial in UTUC supports the use of standard-of-care adjuvant cisplatin-based chemotherapy (Birtle et al., 2020). Because many patients with UTUC will have one remaining kidney following RNU and frequently have other significant co-morbid conditions, cisplatin-based therapy is not well tolerated (NCCN Guidelines Version 3, 2018). Renal function before and after RNU greatly limits the number of patients with UTUC who are eligible for platinum-based neoadjuvant or adjuvant therapy. Therefore, targeted therapies are needed for treating UTUC (Lane et al., 2010).
[0005] Despite demonstrated survival benefit for neoadjuvant treatment of invasive UCB, many patients with invasive UCB are unlikely to receive (neo)adjuvant cisplatin-based chemotherapy, due in part to cisplatin ineligibility (Porter et al., 2011). In addition, residual disease following neoadjuvant therapy is associated with a poor prognosis (Grossman et al, 2003). Therefore,
there remains an unmet need for a substantial proportion of patients with invasive UCB who are ineligible or refuse to receive cisplatin-based adjuvant chemotherapy or who have residual disease following neoadjuvant therapy.
Infigratinib, as depicted in formula (I), is a selective and ATP-competitive pan-fibroblast growth factor receptor (FGFR) kinase inhibitor, also known as 3-(2,6-dichloro-3,5-dimethoxyphenyl)- 1 – { 6- [4-(4-ethyl- 1 -piperazin- 1 -yljphenylamino] -pyrimidinyl-4-yl } – 1 -methylurea. Infigratinib selectively inhibits the kinase activity of FGFR1, FGFR2, FGFR3, and
FGFR4.
PATENT
WO 2011071821
https://patents.google.com/patent/WO2011071821A1/en3-(2,6-Dichloro-3,5-dimethoxy-phenyl)-l-{6-[4-(4-ethyl-piperazin-l-yl)- phenylamino]-pyrimidin-4-yl}-l -methyl-urea (described in USSN 11/570983, filed June 23, 2005, and incorporated by reference in its entirety herein) has the structure of Formula I:
The compound of Formula I is a protein kinase inhibitor and is useful in the treatment of proliferative diseases mediated by protein kinases. In particular, the compound of Formula I inhibits FGFR1, FGFR2, FGFR3, FGFR4, KDR, HER1, HER2, Bcr-Abl, Tie2, and Ret kinases. It is therefore useful in the treatment of cancers including AML, melanocyte neoplasia, breast cancer, colon cancer, lung cancer (especially small-cell lung cancer), cancer of the prostate or Kaposi’s sarcoma.[0003] It is well known that the crystalline form of the active pharmaceutical ingredient (API) of a particular drug is often an important determinant of the drug’s ease of preparation, hygroscopicity, stability, solubility, storage stability, ease of formulation, rate of dissolution in gastrointestinal fluids and in vivo bioavailability. Crystalline forms occur where the same composition of matter crystallizes in a different lattice arrangement resulting in different thermodynamic properties and stabilities specific to the particular crystalline form.Crystalline forms may also include different hydrates or solvates of the same compound. In deciding which form is preferable, the numerous properties of the forms are compared and the preferred form chosen based on the many physical property variables. It is entirely possible that one form can be preferable in some circumstances where certain aspects such as ease of preparation, stability, etc. are deemed to be critical. In other situations, a different form may be preferred for greater dissolution rate and/or superior bioavailability. It is not yet possible to predict whether a particular compound or salt of a compound will form polymorphs, whether any such polymorphs will be suitable for commercial use in a therapeutic composition, or which polymorphs will display such desirable properties.Example 2: Manufacture of the Free Base of the Compound of Formula I
IA. N- [4-(4-ethyl-piperazin- 1 -yl)-phenyl] -N’ -methyl-pyrimidine-4,6-diamine[0077] A mixture of 4-(4-ethylpiperazin-l-yl)-aniline (1 g, 4.88 mmol), (6-chloro- pyrimidin-4-yl)-methyl-amine (1.81 g, 12.68 mmol, 1.3 eq.), and 4N HC1 in dioxane (15 ml) is heated in a sealed tube to 150°C for 5h. The reaction mixture is concentrated, diluted with DCM and a saturated aqueous solution of sodium bicarbonate. The aqueous layer is separated and extracted with DCM. The organic phase is washed with brine, dried (sodium sulfate), filtered and concentrated. Purification of the residue by silica gel column chromatography (DCM/MeOH, 93:7) followed by trituration in diethyl ether affords the title compound as a white solid: ESI-MS: 313.2 [MH]+; tR= 1.10 min (gradient J); TLC: Rf = 0.21 (DCM/MeOH, 93:7).B. 4-(4-Ethylpiperazin- 1 -yl)-aniline[0078] A suspension of l-ethyl-4-(4-nitro-phenyl)-piperazine (6.2 g, 26.35 mmol) and Raney Nickel (2 g) in MeOH (120 mL) is stirred for 7 h at RT, under a hydrogen atmosphere. The reaction mixture is filtered through a pad of celite and concentrated to afford 5.3 g of the title compound as a violet solid: ESI-MS: 206.1 [MH]+; TLC: Rf = 0.15 (DCM/MeOH + 1 % NH3aq, 9:l).C. 1 -Ethyl-4-(4-nitro-phenyl)-piperazine[0079] A mixture of l-bromo-4-nitrobenzene (6 g, 29.7 mmol) and 1-ethylpiperazine (7.6 ml, 59.4 mmol, 2 eq.) is heated to 80°C for 15h. After cooling to RT, the reaction mixture is diluted with water and DCM/MeOH, 9:1. The aqueous layer is separated and extracted with DCM/MeOH, 9:1. The organic phase is washed with brine, dried (sodium sulfate), filtered and concentrated. Purification of the residue by silica gel column chromatography(DCM MeOH + 1 % NH3aq, 9:1) affords 6.2 g of the title compound as a yellow solid: ESI- MS: 236.0 [MH]+; tR= 2.35 min (purity: 100%, gradient J); TLC: Rf = 0.50 (DCM/MeOH + 1 % NH3aq, 9:1).D. (6-chloro-pyrimidin-4-yl)-methyl-amine[0080] This material was prepared by a modified procedure published in the literature (J. Appl. Chem. 1955, 5, 358): To a suspension of commercially available 4,6- dichloropyrimidine (20 g, 131.6 mmol, 1.0 eq.) in isopropanol (60 ml) is added 33% methylamine in ethanol (40.1 ml, 328.9 mmol, 2.5 eq.) at such a rate that the internal temperature does not rise above 50°C. After completion of the addition the reaction mixture was stirred for lh at room temperature. Then, water (50 ml) is added and the suspension formed is chilled in an ice bath to 5°C. The precipitated product is filtered off, washed with cold isopropanol/water 2:1 (45 ml) and water. The collected material is vacuum dried over night at 45°C to afford the title compound as colorless powder: tR = 3.57 min (purity: >99%, gradient A), ESI-MS: 144.3 / 146.2 [MH]+.E. (3-(2,6-Dichloro-3,5-dimethoxy-phenyl)-l-{6-[4-(4-ethyl-piperazin-l-yl)-phenylamino]- pyrimidin-4-yl} – 1 -methyl-urea)[0081] The title compound was prepared by adding 2,6-dichloro-3,5-dimethoxyphenyl- isocyanate (1.25 eq.) to a solution of N-[4-(4-ethyl-piperazin-l-yl)-phenyl]-N’-methyl- pyrimidine-4,6-diamine (2.39 g, 7.7 mmol, 1 eq.) in toluene and stirring the reaction mixture for 1.5h at reflux. Purification of the crude product by silica gel column chromatography (DCM MeOH + 1 % NH3aq, 95:5) affords the title compound as a white solid: ESI-MS: 560.0 / 561.9 [MHf; tR= 3.54 min (purity: 100%, gradient J); TLC: Rf = 0.28 (DCM/MeOH + 1 % NH3aq, 95:5). Analysis: C26H3iN703Cl2, calc. C 55.72% H 5.57% N 17.49% O 8.56% CI 12.65%; found C 55.96% H 5.84% N 17.17% O 8.46% CI 12.57%. The title compound was characterized by XRPD, thermal and other methods as described below. Example 3: Manufacture of the Monophosphoric Acid Salt Form A of the Compound of Formula I.[0082] To a round bottom flask was added 3-(2,6-dichloro-3,5-dimethoxyphenyl)-l-{6-[4- (4-ethylpiperazin-l-yl)phenylamino]-pyrimidine-4-yl}-l -methyl-urea (134 g, 240 mmol) and IPA (2000 ml). The suspension was stirred and heated to 50°C and a solution of phosphoric acid (73.5 g, 750 mmol) in water (2000 ml) added to it portions. The mixture was stirred at 60°C for 30 min. and filtered through a polypropylene pad. The pad was washed with warm IP A/water (1:1, 200 ml) and the filtrates were combined. To this clear solution, IPA (6000 ml) was added and the mixture was stirred under reflux for 20 min, cooled slowly to room temperature (25° C), and stirred for 24 hours. The white salt product was collected by filtration, washed with IPA (2 χ 500 ml) and dried in the oven at 60° C under reduced pressure for two days to provide the phosphate salt (form A) 110 g. Yield 70%. Purity >98% by HPLC. Analysis: C26H34 707C12P, calc. C 47.42% H 5.20% N 14.89% O 17.01% CI 10.77% P 4.70%; found C 47.40% H 5.11% N 14.71% O 17.18% CI 10.73% P 4.87%. The title compound was characterized by XRPD, thermal and other methods as described below.
^ Botrus G, Raman P, Oliver T, Bekaii-Saab T (April 2021). “Infigratinib (BGJ398): an investigational agent for the treatment of FGFR-altered intrahepatic cholangiocarcinoma”. Expert Opinion on Investigational Drugs. 30 (4): 309–316. doi:10.1080/13543784.2021.1864320. PMID33307867.
Anti-inflammatory, Farnesoid X receptor (FXR) agonist
Comment
Treatment of non-alcoholic steatohepatitis
Novartis is developing tropifexor, a non-bile acid farnesoid X receptor agonist, and its analog LJP-305, for treating NASH, PBC, liver fibrosis, bile acid diarrhea and non-alcoholic fatty liver disease. In June 2021, this drug was reported to be in phase 2 clinical development.
Nonalcoholic steatohepatitis (NASH) is a liver disease that is becoming more prevalent as worldwide obesity and type 2 diabetes increase. It is characterized by accumulation of fat in the liver, inflammation, hepatocyte ballooning, and fibrosis.
Another liver disease, primary biliary cholangitis (PBC), is a cholestatic condition in which bile flow from the liver to the intestine is reduced or interrupted. It is thought to be autoimmune.
PBC is associated with decreased expression of the farnesoid X receptor (FXR), a ligand-activated nuclear receptor that is highly expressed in the liver and other organs. FXR is a key regulator of bile acid production, conjugation, and transport. FXR activation also suppresses lipogenesis; thus, it has been proposed as a treatment for NASH.
Recently, David C. Tully and colleagues at the Genomics Institute of the Novartis Research Foundation (San Diego) and the Novartis Institutes for Biomedical Research (Emeryville, CA) discovered tropifexor, a highly potent FXR agonist. They began by replacing an indole group in an existing partial FXR agonist with a 2-substituted benzothiazole-6-carboxylic acid, a change that resulted in a dramatic increase in potency. Further changes, including optimization of the benzothiazole substituent, resulted in more potent, orally bioavailable tropifexor.
Rats treated orally with tropifexor (0.03 to 1 mg/kg) showed an upregulation of the FXR target genes, BSEP and SHP, and a down-regulation of CYP8B1. Its EC50 for FXR is between 0.2 and 0.26 nM depending on the biochemical assay.
The patent which covers tropifexor and related compounds was published in 2010.[3]
Novel, stable crystalline polymorphic form II of tropifexor , useful for treating non-alcoholic steatohepatitis (NASH), fatty liver and primary biliary cholangitis (PBC).Tropifexor was originally developed by Novartis and then licensed to Pfizer for cooperative development. It is a non-steroidal FXR (farnesoid receptor) agonist, currently in clinical phase II of indications for NASH (non-alcoholic steatohepatitis), fatty liver and primary biliary cholangitis. The structure of Tropifexor is shown in the following formula (1):
Drug polymorphism is a common phenomenon in drug development and an important factor affecting drug quality. Different crystal forms of the same drug may have significant differences in physical and chemical properties such as appearance, fluidity, solubility, storage stability, bioavailability, etc., and there may be great differences, which will affect the storage transfer, application, stability, and efficacy of the drug In order to obtain an effective crystal form that is conducive to production or pharmaceutical preparations, it is necessary to conduct a comprehensive investigation of the crystallization behavior of the drug to obtain a crystal form that meets the production requirements. At present, there is no literature that discloses the crystal form of Tropifexor, and there is no related literature report. The present invention obtains a new crystal form of the compound through a large number of experimental studies on the Tropifexor compound. The new crystal form has the advantages of high solubility, good stability, low moisture absorption, simple preparation process and easy operation, etc., and has excellent properties in industrial production. Superiority.Example 1 Preparation method of Tropifexor crystal form II[0049]After mixing 60.3 mg of Tropifexor and p-aminobenzoic acid (13.7 mg), they were added to ethanol (3.0 ml), stirred at 27° C. to obtain a clear solution, and then allowed to stand at room temperature for about 2 days to precipitate a solid product. It was filtered with suction and placed in a drying box at 50°C and vacuum dried to constant weight to obtain 51.3 mg of solid powder. The obtained crystal was detected by XPRD and confirmed to be Tropifexor crystal form II; its X-ray powder diffraction pattern was basically consistent with Fig. 1, its DSC pattern was basically the same as Fig. 2, and its TGA pattern was basically the same as Fig. 3.[0050]Example 2 Preparation method of Tropifexor crystal form II[0051]After mixing 60.3 mg of Tropifexor and p-hydroxybenzoic acid (13.8 mg), they were added to ethanol (3.0 ml), stirred at 27° C. to obtain a clear solution, and then allowed to stand at room temperature for about 2 days to precipitate a solid product. It was filtered with suction and placed in a drying box at 50°C and vacuum dried to constant weight to obtain 48.5 mg of solid powder. The obtained crystal was detected by XPRD and confirmed to be Tropifexor crystal form II; its X-ray powder diffraction pattern was basically consistent with Fig. 1, its DSC pattern was basically the same as Fig. 2, and its TGA pattern was basically the same as Fig. 3.[0052]Example 3 Preparation method of Tropifexor crystal form II[0053]After mixing 60.3 mg of Tropifexor and salicylic acid (13.8 mg), they were added to ethanol (3.0 ml), stirred at 27°C to obtain a clear solution, and then allowed to stand at room temperature for about 2 days to precipitate a solid product. Filter with suction and place in a drying box at 50°C and vacuum dry to constant weight to obtain 50.0 mg of solid powder. The obtained crystal was detected by XPRD and confirmed to be Tropifexor crystal form II; its X-ray powder diffraction pattern was basically consistent with Fig. 1, its DSC pattern was basically the same as Fig. 2, and its TGA pattern was basically the same as Fig. 3.[0054]Example 4 Preparation method of Tropifexor crystal form II[0055]After mixing 60.3 mg of Tropifexor and 2,4-dihydroxybenzoic acid (15.4 mg), they were added to ethanol (3.0 ml), stirred at 27°C to obtain a clear solution, and then allowed to stand at room temperature for about 2 days to precipitate a solid product. It was filtered with suction and placed in a drying box at 50°C and vacuum dried to constant weight to obtain 49.5 mg of solid powder. The obtained crystal was detected by XPRD and confirmed to be Tropifexor crystal form II; its X-ray powder diffraction pattern was basically consistent with Fig. 1, its DSC pattern was basically the same as Fig. 2, and its TGA pattern was basically the same as Fig. 3.
claiming crystalline polymorphic form I of tropifexor,Example 1 Preparation method of Tropifexor crystal form I 50.0 mg of Tropifexor was added to ethanol (1.0 ml), heated to 60° C. and stirred to obtain a clear solution, and then water (3 ml) was added dropwise to the Tropifexor solution. Stir and precipitate solid product. It was filtered with suction and placed in a drying box at 50°C and vacuum dried to constant weight to obtain 38.5 mg of solid powder. The obtained crystal was detected by XPRD and confirmed to be Tropifexor crystal form I; its X-ray powder diffraction pattern was basically consistent with Figure 1, its DSC pattern was basically consistent with Figure 2, and its TGA pattern was basically consistent with Figure 3
Methyl 2-[(1 R,3r,5S)-3-(i5-cvclopropyl-3-r2-(trifluoromethoxy)phenyll-1 ,2-oxazol-4- yl}methoxy)-8-azabicvcloi3.2.1 loctan-8-yll-4-fluoro-1 ,3-benzothiazole-6-carboxylate (1 -1 A). Into a 25-mL round-bottom flask equipped with a stir bar was added sequentially 4-(((1 R,3r,5S)- 8-azabicyclo[3.2.1 ]octan-3-yloxy)methyl)-5-cyclopropyl-3-(2-(trifluoromethoxy)phenyl)isoxazole (1 .29 mmol), N,N-dimethylacetamide (3.6 mL), cesium carbonate (3.31 mmol), and methyl 2- bromo-4-fluorobenzo[d]thiazole-6-carboxylate (3.87 mmol). After stirring the resulting slurry at room temperature for 10 minutes, the mixture was then warmed to 60 °C and stirred for 1 h. The reaction slurry was allowed to cool to room temperature, and was diluted with 200 mL of ethyl acetate and washed with water (3 χ 30 mL). The organic extracts were concentrated under vacuum and directly purified using normal phase silica gel chromatography (40 g silica column) with a 15 min gradient of 10 % to 60 % ethyl acetate/hexanes. Desired fractions were concentrated in vacuo, and the resulting residue crystallized upon standing to give methyl 2- [(1 R,3r,5S)-3-({5-cyclopropyl-3-[2-(trifluoromethoxy)phenyl]-1 ,2-oxazol-4-yl}methoxy)-8- azabicyclo[3.2.1 ]octan-8-yl]-4-fluoro-1 ,3-benzothiazole-6-carboxylate (1-1 A) as a white crystalline solid. MS (m/z) : 618.2 (M+1 ).
2-r(1 R,3r,5S)-3-(i5-cvclopropyl-3-r2-(trifluoromethoxy)phenyll-1 ,2-oxazol-4-yl}methoxy)- 8-azabicvcloi3.2.1 loctan-8-yll-4-fluoro-1 ,3-benzothiazole-6-carboxylic acid (1 -1 B). To a 25-mL round-bottom flask equipped with a stir bar was added the ester (0.89 mmol), THF (4 mL),
MeOH (2 mL), and 3 N aqueous KOH solution (1 mL, 3 mmol). The resulting homogenous solution was stirred for 1 hour at 70 °C, cooled to room temperature, and then quenched with AcOH (roughly 0.2 mL of glacial acetic, 3 mmol) until pH=6 was achieved (Whatman class pH strip paper). At this time the reaction was diluted with ethyl acetate (40 mL) and washed with water (3 5 mL). The ethyl acetate fraction was concentrated under vacuum to give to an oily residue. To the resulting oil was then added MeOH (6 mL). The oil quickly dissolved, then immediately began to crystallize. Upon standing for 2.5 hrs, the mother liquor was withdrawn and crystals washed (3 x 2 mL of ice cold MeOH). The crystals were dried via vacuum (10 mm Hg pressure at 45 °C overnight) and then recrystallized from acetonitrile, filtered, and dried under vacuum to give 2-[(1 R,3r,5S)-3-({5-cyclopropyl-3-[2-(trifluoromethoxy)phenyl]-1 ,2-oxazol-4-yl}methoxy)-8-azabicyclo[3.2.1 ]octan-8-yl]-4-fluoro-1 ,3-benzothiazole-6-carboxylic acid (1 -1 B). 2-[(1 R,3r,5S)-3-({5-cyclopropyl-3-[2-(trifluoromethyl)phenyl]-1 ,2-oxazol-4-yl}methoxy)-8-azabicyclo[3.2.1 ]octan-8-yl]-4-fluoro-1 ,3-benzothiazole-6-carboxylic acid (1 -2B).
Examples 1 -2A and the corresponding acid 1 -2B can be prepared following the same procedures, from the reaction of intermediate 4-((8-azabicyclo[3.2.1 ]octan-3-yloxy)methyl)-5-cyclopropyl-3-(2-(trifluoromethyl)phenyl)isoxazole.
PAPER
European journal of medicinal chemistry (2021), 209, 112910
Farnesoid X receptor (FXR) agonists are emerging as potential therapeutics for the treatment of various metabolic diseases, as they display multiple effects on bile acid, lipid, and glucose homeostasis. Although the steroidal obeticholic acid, a full FXR agonist, was recently approved, several side effects probably due to insufficient pharmacological selectivity impede its further clinical application. Activating FXR in a partial manner is therefore crucial in the development of novel FXR modulators. Our efforts focusing on isoxazole-type FXR agonists, common nonsteroidal agonists for FXR, led to the discovery a series of novel FXR agonists bearing aryl urea moieties through structural simplification of LJN452 (phase 2). Encouragingly, compound 11k was discovered as a potent FXR agonist which exhibited similar FXR agonism potency but lower maximum efficacy compared to full agonists GW4064 and LJN452 in cell-based FXR transactivation assay. Extensive in vitro evaluation further confirmed partial efficacy of 11k in cellular FXR-dependent gene modulation, and revealed its lipid-reducing activity. More importantly, orally administration of 11k in mice exhibited desirable pharmacokinetic characters resulting in promising in vivo FXR agonistic activity.
^ Clinical trial number NCT03517540 for “Safety, Tolerability, and Efficacy of a Combination Treatment of Tropifexor (LJN452) and Cenicriviroc (CVC) in Adult Patients With Nonalcoholic Steatohepatitis (NASH) and Liver Fibrosis. (TANDEM)” at ClinicalTrials.gov
^WO Application Filing 2012087519, Alper PB, Chianelli D, Mutnick D, Vincent P, Tully DC, “Compositions and methods for modulating fxr”, published 2012-06-28, assigned to Genomics Institute of the Novartis Research Foundation. Retrieved 17 May 2019.
Sotorasib is an inhibitor of the RAS GTPase family. The molecular formula is C30H30F2N6O3, and the molecular weight is 560.6 g/mol. The chemical name of sotorasib is 6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-(1M)-1-[4-methyl-2-(propan-2-yl)pyridin-3-yl]-4-[(2S)-2-methyl-4-(prop-2enoyl) piperazin-1-yl]pyrido[2,3-d]pyrimidin-2(1H)-one. The chemical structure of sotorasib is shown below:
Sotorasib has pKa values of 8.06 and 4.56. The solubility of sotorasib in the aqueous media decreases over the range pH 1.2 to 6.8 from 1.3 mg/mL to 0.03 mg/mL.
LUMAKRAS is supplied as film-coated tablets for oral use containing 120 mg of sotorasib. Inactive ingredients in the tablet core are microcrystalline cellulose, lactose monohydrate, croscarmellose sodium, and magnesium stearate. The film coating material consists of polyvinyl alcohol, titanium dioxide, polyethylene glycol, talc, and iron oxide yellow.
FDA grants accelerated approval to sotorasib for KRAS G12C mutated NSCLC
On May 28, 2021, the Food and Drug Administration granted accelerated approval to sotorasib (Lumakras, Amgen, Inc.), a RAS GTPase family inhibitor, for adult patients with KRAS G12C ‑mutated locally advanced or metastatic non-small cell lung cancer (NSCLC), as determined by an FDA ‑approved test, who have received at least one prior systemic therapy.
FDA also approved the QIAGEN therascreen® KRAS RGQ PCR kit (tissue) and the Guardant360® CDx (plasma) as companion diagnostics for Lumakras. If no mutation is detected in a plasma specimen, the tumor tissue should be tested.
Approval was based on CodeBreaK 100, a multicenter, single-arm, open label clinical trial (NCT03600883) which included patients with locally advanced or metastatic NSCLC with KRAS G12C mutations. Efficacy was evaluated in 124 patients whose disease had progressed on or after at least one prior systemic therapy. Patients received sotorasib 960 mg orally daily until disease progression or unacceptable toxicity.
The main efficacy outcome measures were objective response rate (ORR) according to RECIST 1.1, as evaluated by blinded independent central review and response duration. The ORR was 36% (95% CI: 28%, 45%) with a median response duration of 10 months (range 1.3+, 11.1).
The most common adverse reactions (≥ 20%) were diarrhea, musculoskeletal pain, nausea, fatigue, hepatotoxicity, and cough. The most common laboratory abnormalities (≥ 25%) were decreased lymphocytes, decreased hemoglobin, increased aspartate aminotransferase, increased alanine aminotransferase, decreased calcium, increased alkaline phosphatase, increased urine protein, and decreased sodium.
The recommended sotorasib dose is 960 mg orally once daily with or without food.
The approved 960 mg dose is based on available clinical data, as well as pharmacokinetic and pharmacodynamic modeling that support the approved dose. As part of the evaluation for this accelerated approval, FDA is requiring a postmarketing trial to investigate whether a lower dose will have a similar clinical effect.
This indication is approved under accelerated approval based on overall response rate and duration of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory trial(s).
This review was conducted under Project Orbis, an initiative of the FDA Oncology Center of Excellence. Project Orbis provides a framework for concurrent submission and review of oncology drugs among international partners. For this review, FDA collaborated with the Australian Therapeutic Goods Administration (TGA), the Brazilian Health Regulatory Agency (ANVISA), Health Canada, and the United Kingdom Medicines and Healthcare products Regulatory Agency (MHRA). The application reviews are ongoing at the other regulatory agencies.
This review used the Real-Time Oncology Review (RTOR) pilot program, which streamlined data submission prior to the filing of the entire clinical application, the Assessment Aid, and the Product Quality Assessment Aid (PQAA), voluntary submissions from the applicant to facilitate the FDA’s assessment. The FDA approved this application approximately 10 weeks ahead of the FDA goal date.
The most common side effects include diarrhea, musculoskeletal pain, nausea, fatigue, liver damage and cough.[1][2]
Sotorasib is an inhibitor of the RAS GTPase family.[1]
Sotorasib is the first approved targeted therapy for tumors with any KRAS mutation, which accounts for approximately 25% of mutations in non-small cell lung cancers.[2] KRAS G12C mutations represent about 13% of mutations in non-small cell lung cancers.[2] Sotorasib was approved for medical use in the United States in May 2021.[2][5]
Sotorasib is an experimental KRAS inhibitor being investigated for the treatment of KRAS G12C mutant non small cell lung cancer, colorectal cancer, and appendix cancer.
Sotorasib, also known as AMG-510, is an acrylamide derived KRAS inhibitor developed by Amgen.1,3 It is indicated in the treatment of adult patients with KRAS G12C mutant non small cell lung cancer.6 This mutation makes up >50% of all KRAS mutations.2 Mutant KRAS discovered in 1982 but was not considered a druggable target until the mid-2010s.5 It is the first experimental KRAS inhibitor.1
The drug MRTX849 is also currently being developed and has the same target.1
Sotorasib was granted FDA approval on 28 May 2021.6
Medical uses
Sotorasib is indicated for the treatment of adults with KRAS G12C-mutated locally advanced or metastatic non-small cell lung cancer (NSCLC), as determined by an FDA-approved test, who have received at least one prior systemic therapy.[1][2]
Clinical development
Sotorasib is being developed by Amgen. Phase I clinical trials were completed in 2020.[6][7][8] In December 2019, it was approved to begin Phase II clinical trials.[9]
Because the G12C KRAS mutation is relatively common in some cancer types, 14% of non-small-cell lung cancer adenocarcinoma patients and 5% of colorectal cancer patients,[10] and sotorasib is the first drug candidate to target this mutation, there have been high expectations for the drug.[10][11][12] The Food and Drug Administration has granted a fast track designation to sotorasib for the treatment of metastatic non-small-cell lung carcinoma with the G12C KRAS mutation.[13]
Chemistry and pharmacology
Sotorasib can exist in either of two atropisomeric forms and one is more active than the other.[10] It selectively forms an irreversible covalent bond to the sulfur atom in the cysteine residue that is present in the mutated form of KRAS, but not in the normal form.[10]
History
Researchers evaluated the efficacy of sotorasib in a study of 124 participants with locally advanced or metastatic KRAS G12C-mutated non-small cell lung cancer with disease progression after receiving an immune checkpoint inhibitor and/or platinum-based chemotherapy.[2] The major outcomes measured were objective response rate (proportion of participants whose tumor is destroyed or reduced) and duration of response.[2] The objective response rate was 36% and 58% of those participants had a duration of response of six months or longer.[2]
KRAS is the most frequently mutated oncogene in cancer and encodes a key signalling protein in tumours1,2. The KRAS(G12C) mutant has a cysteine residue that has been exploited to design covalent inhibitors that have promising preclinical activity3,4,5. Here we optimized a series of inhibitors, using novel binding interactions to markedly enhance their potency and selectivity. Our efforts have led to the discovery of AMG 510, which is, to our knowledge, the first KRAS(G12C) inhibitor in clinical development. In preclinical analyses, treatment with AMG 510 led to the regression of KRASG12C tumours and improved the anti-tumour efficacy of chemotherapy and targeted agents. In immune-competent mice, treatment with AMG 510 resulted in a pro-inflammatory tumour microenvironment and produced durable cures alone as well as in combination with immune-checkpoint inhibitors. Cured mice rejected the growth of isogenic KRASG12D tumours, which suggests adaptive immunity against shared antigens. Furthermore, in clinical trials, AMG 510 demonstrated anti-tumour activity in the first dosing cohorts and represents a potentially transformative therapy for patients for whom effective treatments are lacking.
Paper
Scientific Reports (2020), 10(1), 11992
PAPER
European journal of medicinal chemistry (2021), 213, 113082.
KRAS is the most commonly altered oncogene of the RAS family, especially the G12C mutant (KRASG12C), which has been a promising drug target for many cancers. On the basis of the bicyclic pyridopyrimidinone framework of the first-in-class clinical KRASG12C inhibitor AMG510, a scaffold hopping strategy was conducted including a F–OH cyclization approach and a pyridinyl N-atom working approach leading to new tetracyclic and bicyclic analogues. Compound 26a was identified possessing binding potency of 1.87 μM against KRASG12C and cell growth inhibition of 0.79 μM in MIA PaCa-2 pancreatic cancer cells. Treatment of 26a with NCI–H358 cells resulted in down-regulation of KRAS-GTP levels and reduction of phosphorylation of downstream ERK and AKT dose-dependently. Molecular docking suggested that the fluorophenol moiety of 26a occupies a hydrophobic pocket region thus forming hydrogen bonding to Arg68. These results will be useful to guide further structural modification.
PAPER
Journal of Medicinal Chemistry (2020), 63(1), 52-65.
KRASG12C has emerged as a promising target in the treatment of solid tumors. Covalent inhibitors targeting the mutant cysteine-12 residue have been shown to disrupt signaling by this long-“undruggable” target; however clinically viable inhibitors have yet to be identified. Here, we report efforts to exploit a cryptic pocket (H95/Y96/Q99) we identified in KRASG12C to identify inhibitors suitable for clinical development. Structure-based design efforts leading to the identification of a novel quinazolinone scaffold are described, along with optimization efforts that overcame a configurational stability issue arising from restricted rotation about an axially chiral biaryl bond. Biopharmaceutical optimization of the resulting leads culminated in the identification of AMG 510, a highly potent, selective, and well-tolerated KRASG12C inhibitor currently in phase I clinical trials (NCT03600883).
The present disclosure relates to an improved, efficient, scalable process to prepare intermediate compounds, such as compound of Formula 6A, having the structure,
useful for the synthesis of compounds for the treatment of KRAS G12C mutated cancers.
BACKGROUND
[0003] KRAS gene mutations are common in pancreatic cancer, lung adenocarcinoma, colorectal cancer, gall bladder cancer, thyroid cancer, and bile duct cancer. KRAS mutations are also observed in about 25% of patients with NSCLC, and some studies have indicated that KRAS mutations are a negative prognostic factor in patients with NSCLC. Recently, V-Ki-ras2 Kirsten rat sarcoma viral oncogene homolog (KRAS) mutations have been found to confer resistance to epidermal growth factor receptor (EGFR) targeted therapies in colorectal cancer; accordingly, the mutational status of KRAS can provide important information prior to the prescription of TKI therapy. Taken together, there is a need for new medical treatments for patients with pancreatic cancer, lung adenocarcinoma, or colorectal cancer, especially those who have been diagnosed to have such cancers characterized by a KRAS mutation, and including those who have progressed after chemotherapy.
Related Synthetic Processes
[0126] The following intermediate compounds of 6-Fluoro-7-(2-fluoro-6-hydroxyphenyl)-1-(4-methyl-2-(2-propanyl)-3-pyridinyl)-4-((2S)-2-methyl-4-(2-propenoyl)-1-piperazinyl)pyrido[2,3-d]pyrimidin-2(1H)-one are representative examples of the disclosure and are not intended to be construed as limiting the scope of the present invention.
[0127] A synthesis of Compound 9 and the relevant intermediates is described in U.S. Serial No.15/984,855, filed May 21, 2018 (U.S. Publication No.2018/0334454, November 22, 2018) which claims priority to and the benefit claims the benefit of U.S. Provisional Application No.62/509,629, filed on May 22, 2017, both of which are incorporated herein by reference in their entireties for all purposes. 6-Fluoro-7-(2-fluoro-6-hydroxyphenyl)-1-(4-methyl-2-(2-propanyl)-3-pyridinyl)-4-((2S)-2-methyl-4-(2-propenoyl)-1-piperazinyl)pyrido[2,3-d]pyrimidin-2(1H)-one was prepared using the following process, in which the isomers of the final product were isolated via chiral chromatography.
[0128] Step 1: 2,6-Dichloro-5-fluoronicotinamide (Intermediate S). To a mixture of 2,6-dichloro-5-fluoro-nicotinic acid (4.0 g, 19.1 mmol, AstaTech Inc., Bristol, PA) in dichloromethane (48 mL) was added oxalyl chloride (2M solution in DCM, 11.9 mL, 23.8 mmol), followed by a catalytic amount of DMF (0.05 mL). The reaction was stirred at room temperature overnight and then was concentrated. The residue was dissolved in 1,4-dioxane (48 mL) and cooled to 0 °C. Ammonium hydroxide solution (28.0-30% NH3 basis, 3.6 mL, 28.6 mmol) was added slowly via syringe. The resulting mixture was stirred at 0 °C for 30 min and then was concentrated. The residue was diluted with a 1:1 mixture of EtOAc/Heptane and agitated for 5 min, then was filtered. The filtered solids were discarded, and the remaining mother liquor was partially concentrated to half volume and filtered. The filtered solids were washed with heptane and dried in a reduced-pressure oven (45 °C) overnight to provide 2,6-dichloro-5-fluoronicotinamide. 1H NMR (400 MHz, DMSO-d6) δ ppm 8.23 (d, J = 7.9 Hz, 1 H) 8.09 (br s, 1 H) 7.93 (br s, 1 H). m/z (ESI, +ve ion): 210.9 (M+H)+.
[0129] Step 2: 2,6-Dichloro-5-fluoro-N-((2-isopropyl-4-methylpyridin-3-yl)carbamoyl)nicotinamide. To an ice-cooled slurry of 2,6-dichloro-5-fluoronicotinamide (Intermediate S, 5.0 g, 23.9 mmol) in THF (20 mL) was added oxalyl chloride (2 M solution in DCM, 14.4 mL, 28.8 mmol) slowly via syringe. The resulting mixture was heated at 75 °C for 1 h, then heating was stopped, and the reaction was concentrated to half volume. After cooling to 0 °C, THF (20 mL) was added, followed by a solution of 2-isopropyl-4-methylpyridin-3-amine (Intermediate R, 3.59 g, 23.92 mmol) in THF (10 mL), dropwise via cannula. The resulting mixture was stirred at 0 °C for 1 h and then was quenched with a 1:1 mixture of brine and saturated aqueous ammonium chloride. The mixture was extracted with EtOAc (3x) and the combined organic layers were dried over anhydrous sodium sulfate and concentrated to provide 2,6-dichloro-5-fluoro-N-((2-isopropyl-4-methylpyridin-3-yl)carbamoyl)nicotinamide. This material was used without further purification in the following step. m/z (ESI, +ve ion): 385.1(M+H)+.
[0130] Step 3: 7-Chloro-6-fluoro-1-(2-isopropyl-4-methylpyridin-3-yl)pyrido[2,3-d]pyrimidine-2,4(1H,3H)-dione. To an ice-cooled solution of 2,6-dichloro-5-fluoro-N-((2-isopropyl-4-methylpyridin-3-yl)carbamoyl)nicotinamide (9.2 g, 24.0 mmol) in THF (40 mL) was added KHMDS (1 M solution in THF, 50.2 mL, 50.2 mmol) slowly via syringe. The ice bath was removed and the resulting mixture was stirred for 40 min at room temperature. The reaction was quenched with saturated aqueous ammonium chloride and extracted with EtOAc (3x). The combined organic layers were dried over anhydrous sodium sulfate and concentrated. The residue was purified by silica gel chromatography (eluent: 0-50% 3:1 EtOAc-EtOH/heptane) to provide 7-chloro-6-fluoro-1-(2-isopropyl-4-methylpyridin-3-yl)pyrido[2,3-d]pyrimidine-2,4(1H,3H)-dione.1H NMR (400 MHz, DMSO-d6) δ ppm 12.27 (br s, 1H), 8.48-8.55 (m, 2 H), 7.29 (d, J = 4.8 Hz, 1 H), 2.87 (quin, J = 6.6 Hz, 1 H), 1.99-2.06 (m, 3 H), 1.09 (d, J = 6.6 Hz, 3 H), 1.01 (d, J = 6.6 Hz, 3 H).19F NMR (376 MHz, DMSO-d6) δ: -126.90 (s, 1 F). m/z (ESI, +ve ion): 349.1 (M+H)+.
[0131] Step 4: 4,7-Dichloro-6-fluoro-1-(2-isopropyl-4-methylpyridin-3-yl)pyrido[2,3-d]pyrimidin-2(1H)-one. To a solution of 7-chloro-6-fluoro-1-(2-isopropyl-4-methylpyridin-3-yl)pyrido[2,3-d]pyrimidine-2,4(1H,3H)-dione (4.7 g, 13.5 mmol) and DIPEA (3.5 mL, 20.2 mmol) in acetonitrile (20 mL) was added phosphorus oxychloride (1.63 mL, 17.5 mmol), dropwise via syringe. The resulting mixture was heated at 80 °C for 1 h, and then was cooled to room temperature and concentrated to provide 4,7-dichloro-6-fluoro-1-(2-isopropyl-4-methylpyridin-3-yl)pyrido[2,3-d]pyrimidin-2(1H)-one. This material was used without further purification in the following step. m/z (ESI, +ve ion): 367.1 (M+H)+.
[0132] Step 5: (S)-tert-Butyl 4-(7-chloro-6-fluoro-1-(2-isopropyl-4-methylpyridin-3-yl)-2-oxo-1,2-dihydropyrido[2,3-d]pyrimidin-4-yl)-3-methylpiperazine-1-carboxylate. To an ice-cooled solution of 4,7-dichloro-6-fluoro-1-(2-isopropyl-4-methylpyridin-3-yl)pyrido[2,3-d]pyrimidin-2(1H)-one (13.5 mmol) in acetonitrile (20 mL) was added DIPEA (7.1 mL, 40.3 mmol), followed by (S)-4-N-Boc-2-methyl piperazine (3.23 g, 16.1 mmol, Combi-Blocks, Inc., San Diego, CA, USA). The resulting mixture was warmed to room temperature and stirred for 1 h, then was diluted with cold saturated aqueous sodium bicarbonate solution (200 mL) and EtOAc (300 mL). The mixture was stirred for an additional 5 min, the layers were separated, and the aqueous layer was extracted with more EtOAc (1x). The combined organic layers were dried over anhydrous sodium sulfate and concentrated. The residue was purified by silica gel chromatography (eluent: 0-50% EtOAc/heptane) to provide (S)-tert-butyl 4-(7-chloro-6-fluoro-1-(2-isopropyl-4-methylpyridin-3-yl)-2-oxo-1,2-dihydropyrido[2,3-d]pyrimidin-4-yl)-3-methylpiperazine-1-carboxylate. m/z (ESI, +ve ion): 531.2 (M+H)+.
[0133] Step 6: (3S)-tert-Butyl 4-(6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-1-(2-isopropyl-4-methylpyridin-3-yl)-2-oxo-1,2-dihydropyrido[2,3-d]pyrimidin-4-yl)-3-methylpiperazine-1-carboxylate. A mixture of (S)-tert-butyl 4-(7-chloro-6-fluoro-1-(2-isopropyl-4-methylpyridin-3-yl)-2-oxo-1,2-dihydropyrido[2,3-d]pyrimidin-4-yl)-3-methylpiperazine-1-carboxylate (4.3 g, 8.1 mmol), potassium trifluoro(2-fluoro-6-hydroxyphenyl)borate (Intermediate Q, 2.9 g, 10.5 mmol), potassium acetate (3.2 g, 32.4 mmol) and [1,1′-bis(diphenylphosphino)ferrocene]dichloropalladium(II), complex with dichloromethane (661 mg, 0.81 mmol) in 1,4-dioxane (80 mL) was degassed with nitrogen for 1 min. De-oxygenated water (14 mL) was added, and the resulting mixture was heated at 90 °C for 1 h. The reaction was allowed to cool to room temperature, quenched with half-saturated aqueous sodium bicarbonate, and extracted with EtOAc (2x) and DCM (1x). The combined organic layers were dried over anhydrous sodium sulfate and concentrated. The residue was purified by silica gel chromatography (eluent: 0-60% 3:1 EtOAc-EtOH/heptane) to provide (3S)-tert-butyl 4-(6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-1-(2-isopropyl-4-methylpyridin-3-yl)-2-oxo-1,2-dihydropyrido[2,3-d]pyrimidin-4-yl)-3-methylpiperazine-1-carboxylate.1H NMR (400 MHz, DMSO-d6) δ ppm 10.19 (br s, 1 H), 8.38 (d, J = 5.0 Hz, 1 H), 8.26 (dd, J = 12.5, 9.2 Hz, 1 H), 7.23-7.28 (m, 1 H), 7.18 (d, J = 5.0 Hz, 1 H), 6.72 (d, J = 8.0 Hz, 1 H), 6.68 (t, J = 8.9 Hz, 1 H), 4.77-4.98 (m, 1 H), 4.24 (br t, J = 14.2 Hz, 1 H), 3.93-4.08 (m, 1 H), 3.84 (br d, J=12.9 Hz, 1 H), 3.52-3.75 (m, 1 H), 3.07-3.28 (m, 1 H), 2.62-2.74 (m, 1 H), 1.86-1.93 (m, 3 H), 1.43-1.48 (m, 9 H), 1.35 (dd, J = 10.8, 6.8 Hz, 3 H), 1.26-1.32 (m, 1 H), 1.07 (dd, J = 6.6, 1.7 Hz, 3 H), 0.93 (dd, J = 6.6, 2.1 Hz, 3 H).19F NMR (376 MHz, DMSO-d6) δ: -115.65 (s, 1 F), -128.62 (s, 1 F). m/z (ESI, +ve ion): 607.3 (M+H)+.
[0134] Step 7: 6-Fluoro-7-(2-fluoro-6-hydroxyphenyl)-1-(4-methyl-2-(2-propanyl)-3-pyridinyl)-4-((2S)-2-methyl-4-(2-propenoyl)-1-piperazinyl)pyrido[2,3-d]pyrimidin-2(1H)-one. Trifluoroacetic acid (25 mL, 324 mmol) was added to a solution of (3S)-tert-butyl 4-(6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-1-(2-isopropyl-4-methylpyridin-3-yl)-2-oxo-1,2-dihydropyrido[2,3-d]pyrimidin-4-yl)-3-methylpiperazine-1-carboxylate (6.3 g, 10.4 mmol) in DCM (30 mL). The resulting mixture was stirred at room temperature for 1 h and then was concentrated. The residue was dissolved in DCM (30 mL), cooled to 0 °C, and sequentially treated with DIPEA (7.3 mL, 41.7 mmol) and a solution of acryloyl chloride (0.849 mL, 10.4 mmol) in DCM (3 mL; added dropwise via syringe). The reaction was stirred at 0 °C for 10 min, then was quenched with half-saturated aqueous sodium bicarbonate and extracted with DCM (2x). The combined organic layers were dried over anhydrous sodium sulfate and concentrated. The residue was purified by silica gel chromatography (eluent: 0-100% 3:1 EtOAc-EtOH/heptane) to provide 6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-1-(4-methyl-2-(2-propanyl)-3-pyridinyl)-4-((2S)-2-methyl-4-(2-propenoyl)-1-piperazinyl)pyrido[2,3-d]pyrimidin-2(1H)-one.1H NMR (400 MHz, DMSO-d6) δ ppm 10.20 (s, 1 H), 8.39 (d, J = 4.8 Hz, 1 H), 8.24-8.34 (m, 1 H), 7.23-7.32 (m, 1 H), 7.19 (d, J = 5.0 Hz, 1 H), 6.87 (td, J = 16.3, 11.0 Hz, 1 H), 6.74 (d, J = 8.6 Hz, 1 H), 6.69 (t, J = 8.6 Hz, 1 H), 6.21 (br d, J = 16.2 Hz, 1 H), 5.74-5.80 (m, 1 H), 4.91 (br s, 1 H), 4.23-4.45 (m, 2 H), 3.97-4.21 (m, 1 H), 3.44-3.79 (m, 2 H), 3.11-3.31 (m, 1 H), 2.67-2.77 (m, 1 H), 1.91 (s, 3 H), 1.35 (d, J = 6.8 Hz, 3 H), 1.08 (d, J = 6.6 Hz, 3 H), 0.94 (d, J = 6.8 Hz, 3 H).19F NMR (376 MHz, DMSO-d6) δ ppm -115.64 (s, 1 F), -128.63 (s, 1 F). m/z (ESI, +ve ion): 561.2 (M+H)+.
[0135] Another synthesis of Compound 9 and the relevant intermediates was described in a U.S. provisional patent application filed November 16, 2018, which is incorporated herein by reference in its entirety for all purposes.
Representative Synthetic Processes
[0136] The present disclosure comprises the following steps wherein the synthesis and utilization of the boroxine intermediate is a novel and inventive step in the manufacture of AMG 510 (Compound 9):
Raw Materials
Step la
[0137] To a solution of 2,6-dichloro-5-fluoro-3-pyridinecarboxylic acid (25kg; 119. lmol) in dichloromethane (167kg) and DMF (592g) was added Oxalyl chloride (18.9kg; 148.9mol) while maintaining an internal temp between 15-20 °C. Additional dichloromethane (33kg) was added as a rinse and the reaction mixture stirred for 2h. The reaction mixture is cooled then quenched with ammonium hydroxide (40.2L; 595.5mol) while maintaining internal temperature 0 ± 10°C. The resulting slurry was stirred for 90min then the product collected by filtration. The filtered solids were washed with DI water (3X 87L) and dried to provide 2,6-dichloro-5-fluoronicotinamide (Compound 1).
Step 1b
[0138] In reactor A, a solution of 2,6-dichloro-5-fluoronicotinamide (Compound 1) (16.27kg; 77.8mol) in dichloromethane (359.5kg) was added oxalyl chloride (11.9kg;
93.8mol) while maintaining temp ≤ 25°C for 75min. The resulting solution was then headed to 40°C ± 3°C and aged for 3h. Using vacuum, the solution was distilled to remove dichloromethane until the solution was below the agitator. Dichloromethane (300 kg) was then added and the mixture cooled to 0 ± 5°C. To a clean, dry reactor (reactor B) was added,2-isopropyl-4-methylpyridin-3-amine (ANILINE Compound 2A) (12.9kg; 85.9mol) followed by dichloromethane (102.6 kg). The ANILINE solution was azeodried via vacuum distillation while maintaining an internal temperature between 20-25 °), replacing with additional dichloromethane until the solution was dry by KF analysis (limit ≤ 0.05%). The solution volume was adjusted to approx. 23L volume with dichloromethane. The dried ANILINE solution was then added to reactor A while maintaining an internal temperature of 0 ± 5°C throughout the addition. The mixture was then heated to 23 °C and aged for 1h. the solution was polish filtered into a clean reactor to afford 2,6-dichloro-5-fluoro-N-((2- isopropyl-4-methylpyridin-3-yl)carbamoyl)nicotinamide (Compound 3) as a solution in DCM and used directly in the next step.
Step 2
[0139] A dichloromethane solution of 2,6-dichloro-5-fluoro-N-{[4-methyl-2-(propan-2- yl)pyridin-3-yl]carbamoyl}pyridine-3-carboxamide (UREA (Compound 3)) (15kg contained; 38.9mol) was solvent exchanged into 2-MeTHF using vacuum distillation while maintaining internal temperature of 20-25 °C. The reactor volume was adjusted to 40L and then
additional 2-MeTHF was charged (105.4 kg). Sodium t-butoxide was added (9.4 kg;
97.8mol) while maintaining 5-10 °C. The contents where warmed to 23 °C and stirred for 3h. The contents where then cooled to 0-5C and ammonium chloride added (23.0kg; 430mol) as a solution in 60L of DI water. The mixture was warmed to 20 C and DI water added (15L) and further aged for 30min. Agitation was stopped and the layers separated. The aqueous layer was removed and to the organic layer was added DI water(81.7L). A mixture of conc HCl (1.5kg) and water (9L) was prepared then added to the reactor slowly until pH measured between 4-5. The layers were separated, and the aqueous layer back extracted using 2-MeTHF (42.2kg). The two organic layers combined and washed with a 10% citric acid solution (75kg) followed by a mixture of water (81.7L) and saturated NaCl (19.8 kg). The organic layer was then washed with saturated sodium bicarbonate (75kg) repeating if necessary to achieve a target pH of ≥ 7.0 of the aqueous. The organic layer was washed again with brine (54.7kg) and then dried over magnesium sulfate (5kg). The mixture was filtered to remove magnesium sulfate rinsing the filtered bed with 2-MeTHF (49.2 kg). The combined filtrate and washes where distilled using vacuum to 40L volume. The concentrated solution was heated to 55 °C and heptane (10-12kg) slowly added until cloud point. The solution was cooled to 23 °C over 2h then heptane (27.3 kg) was added over 2h. The product slurry was aged for 3h at 20-25 °C then filtered and washed with a mixture of 2-MeTHF (2.8kg) and heptane (9kg). The product was dried using nitrogen and vacuum to afford solid 7-chloro-6-fluoro-1-(2-isopropyl-4-methylpyridin-3-yl)pyrido[2,3-d]pyrimidine-2,4(1H,3H)-dione (rac-DIONE (Compound 4)).
Step 3
[0140] To a vessel, an agitated suspension of Compound 4, (1.0 eq.) in 2- methylterahydrofuran (7.0 L/kg) was added (+)-2,3-dibenzoyl-D-tartaric acid (2.0 eq.) under an atmosphere of nitrogen. 2-MeTHF is chiral, but it is used as a racemic mixture. The different enantiomers of 2-MeTHF are incorporated randomly into the co-crystal. The resulting suspension was warmed to 75°C and aged at 75°C until full dissolution was observed (< 30 mins.). The resulting solution was polish filtered at 75°C into a secondary vessel. To the polish filtered solution was charged n-Heptane (2.0 L/kg) at a rate that maintained the internal temperature above 65°C. The solution was then cooled to 60°C, seeded with crystals (0.01 kg/kg) and allowed to age for 30 minutes. The resulting suspension was cooled to 20°C over 4 hours and then sampled for chiral purity analysis by HPLC. To the suspension, n-Heptane (3.0 L/kg) was charged and then aged for 4 hours at 20°C under an atmosphere of nitrogen. The suspension was filtered, and the isolated solids were washed two times with (2:1) n-Heptane:2-methyltetrahydrofuran (3.0 L/kg). The material was dried with nitrogen and vacuum to afford M-Dione:DBTA: Me-THF complex (Compound 4a).
Step 4
[0141] To vessel A, a suspension of disodium hydrogen phosphate (21.1 kg, 2.0 equiv) in DI water (296.8 L, 6.3 L/kg) was agitated until dissolution was observed (≥ 30 min.). To vessel B, a suspension of the M-Dione:DBTA: Me-THF complex (Composition 4a)[46.9 kg (25.9 kg corrected for M-dione, 1.0 equiv.)] in methyl tert-butyl ether (517.8 L, 11.0 L/kg) was agitated for 15 to 30 minutes. The resulting solution from vessel A was added to vessel B, and then the mixture was agitated for more than 3 hours. The agitation was stopped, and the biphasic mixture was left to separate for more than 30 minutes. The lower aqueous phase was removed and then back extracted with methyl tert-butyl ether (77.7 L, 1.7 L/kg). The organic phases were combined in vessel B and dried with magnesium sulfate (24.8 kg, 0.529 kg/kg). The resulting suspension from vessel B was agitated for more than three hours and then filtered into vessel C. To vessel B, a methyl tert-butyl ether (46.9 L, 1.0 L/kg) rinse was charged and then filtered into vessel C. The contents of vessel C were cooled to 10 °C and then distilled under vacuum while slowly being warmed to 35°C. Distillation was continued until 320-350 kg (6.8-7.5 kg/kg) of methyl tert-butyl ether was collected. After cooling the contents of vessel C to 20°C, n-Heptane (278.7 L, 5.9 L/kg) was charged over one hour and then distilled under vacuum while slowly being warmed to 35°C. Distillation was continued until a 190-200 kg (4.1-4.3 kg/kg) mixture of methyl tert-butyl ether and n-Heptane was collected. After cooling the contents of vessel C to 20°C, n-Heptane (278.7 L, 5.9 L/kg) was charged a second time over one hour and then distilled under vacuum while slowly being warmed to 35°C. Distillation was continued until a 190-200 kg (4.1-4.3 kg/kg) mixture of methyl tert-butyl ether and n-Heptane was collected. After cooling the contents of vessel C to 20°C, n-Heptane (195.9 L, 4.2 L/kg) was charged a third time over one hour and then sampled for solvent composition by GC analysis. The vessel C suspension continued to agitate for more than one hour. The suspension was filtered, and then washed with a n-Heptane (68.6 L, 1.5 L/kg) rinse from vessel C. The isolated solids were dried at 50°C, and a sample was submitted for stock suitability. Afforded 7-chloro-6-fluoro-(1M)-1-[4-methyl-2-(propan-2-yl)pyridin-3-yl]pyrido[2,3-d]pyrimidine-2,4(1H,3H)-dione (M-DIONE) Compound 5M.
[0142] The first-generation process highlighted above has been successfully scaled on 200+ kg of rac-dione starting material (Compound 4). In this process, seeding the crystallization with the thermodynamically-stable rac-dione crystal form (which exhibits low solubility) would cause a batch failure. Based on our subsequent studies, we found that increasing the DBTA equivalents and lowering the seed temperature by adjusting heptane
charge schedule improves robustness of the process. The improved process is resistant to the presence of the thermodynamically-stable rac-dione crystal form and promotes successful separation of atropisomers. Subsequent batches will incorporate the improved process for large scale manufacture.
Step 5
Note: All L/kg amounts are relative to M-Dione input; All equiv. amounts are relative to M-Dione input after adjusted by potency.
[0143] M-Dione (Compound 5M, 1.0 equiv.) and Toluene-1 (10.0 L/kg) was charged to Vessel A. The resulting solution was dried by azeotropic distillation under vacuum at 45 °C until 5.0 L/kg of solvents has been removed. The contents of Vessel A were then cooled to 20 °C.
[0144] Vessel C was charged with Toluene-3 (4.5 L/kg), Phosphoryl chloride (1.5 equiv.) and N,N-Diisopropylethylamine-1 (2.0 equiv.) while maintaining the internal temperature below 20 ± 5 °C.
Upon finishing charging, Vessel C was warmed to 30 ± 5 °C. The contents of Vessel A were then transferred to Vessel C over 4 hours while maintaining the internal temperature at 30 ± 5°C. Vessel A was rinsed with Toluene-2 (0.5 L/kg) and transferred to Vessel C. The contents of Vessel C were agitated at 30°C for an additional 3 hours. The contents of Vessel C were cooled to 20 ± 5 °C. A solution of (s)-1-boc-3-methylpiperazine (1.2 equiv.), N,N-Diisopropylethylamine-2 (1.2 equiv.) in isopropyl acetate-1 (1.0 L/kg) was prepared in Vessel D. The solution of Vessel D was charged to vessel C while maintaining a batch temperature of 20 ± 5 °C (Note: Exotherm is observed). Upon the end of transfer, Vessel D was rinsed with additional dichloromethane (1.0 L/kg) and transferred to Vessel C. The contents of Vessel C were agitated for an additional 60 minutes at 20 °C. A solution of sodium bicarbonate [water-1 (15.0 L/kg + Sodium bicarbonate (4.5 equiv.)] was then charged into Vessel C over an hour while maintaining an internal temperature at 20 ± 5 °C throughout the addition. The contents of Vessel C were agitated for at least 12 hours at which point the Pipazoline (Compound 6) product was isolated by filtration in an agitated filter dryer. The cake was washed with water-2 and -3 (5.0 L/kg x 2 times, agitating each wash for 15 minutes) and isopropyl acetate-2 and 3 (5.0 L/kg x 2 times, agitating each wash for 15 min). The cake as dried under nitrogen for 12 hours.
Acetone Re-slurry (Optional):
[0145] Pipazoline (Compound 6) and acetone (10.0 L/kg) were charged to Vessel E. The suspension was heated to 50 °C for 2 hours. Water-4 (10.0 L/kg) was charged into Vessel E over 1 hour. Upon completion of water addition, the mixture was cooled to 20 °C over 1 hour. The contents of Vessel E were filtered to isolate the product, washing the cake with 1:1 acetone/water mixture (5.0 L/kg). The cake was dried under nitrogen for 12 hours.
Step 6
General Note: All equivalents and volumes are reported in reference to Pipazoline input
Note: All L/kg and kg/kg amounts are relative to Pipazoline input
[0146] Reactor A is charged with Pipazoline (Compound 6, 1.0 equiv), degassed 2- MeTHF (9.0 L/kg) and a solution of potassium acetate (2.0 equiv) in degassed water (6.5 L/kg). The resulting mixture is warmed to 75 ± 5 °C and then, charge a slurry of
Pd(dpePhos)Cl2 (0.003 equiv) in 2-MeTHF (0.5 L/kg). Within 2 h of catalyst charge, a solution of freshly prepared Boroxine (Compound 6A, 0.5 equiv) in wet degassed 2-MeTHF (4.0 L/kg, KF > 4.0%) is charged over the course of >1 hour, but < 2 hours, rinsing with an additional portion of wet 2-MeTHF (0.5 L/kg) after addition is complete. After reaction completion ( <0.15 area % Pipazoline remaining, typically <1 h after boroxine addition is complete), 0.2 wt% (0.002 kg/kg) of Biaryl seed is added as a slurry in 0.02 L/kg wet 2- MeTHF, and the resulting seed bed is aged for > 60 min. Heptane (5.0 L/kg) is added over 2 hours at 75 ± 5 °C. The batch is then cooled to 20 ± 5 °C over 2 hours and aged for an additional 2 h. The slurry is then filtered and cake washed with 1 x 5.0L/kg water, 1 x 5.0L/kg 1:1 iPrOH:water followed by 1 x 5.0 L/kg 1:1 iPrOH:heptane (resuspension wash: the cake is resuspended by agitator and allow to set before filtering) . The cake (Biaryl, Compound 7) is then dried under vacuum with a nitrogen sweep.
Note: If the reaction stalls, an additional charge of catalyst and boroxine is required
Step 7 Charcoal Filtration for Pd removal
General Note: All equivalents and volumes are reported in reference to crude Biaryl input
Note: All L/kg and kg/kg amounts are relative to crude Biaryl input
[0147] In a clean Vessel A, charge crude Biaryl (1 equiv) and charge DCM (10 L/kg). Agitate content for > 60 minutes at 22 ± 5 °C, observing dissolution. Pass crude Biaryl from Vessel A, through a bag filter and carbon filters at a flux ≤ 3 L2/min/m and collect filtrate in clean Vessel B. Charge DCM rinse (1 L/kg) to Vessel A, and through carbon filters to collect in vessel B.
[0148] From filtrate in Vessel B, pull a solution sample for IPC Pd content. Sample is concentrated to solid and analyzed by ICP-MS. IPC: Pd ≤ 25 ppm with respect to Biaryl. a. If Pd content is greater than 25 ppm with respect to Biaryl on first or second IPC sample, pass solution through carbon filter a second time at ≤ 3 L2/min/m2, rinsing with 1 L/kg DCM; sample filtrate for IPC.
b. If Pd content remains greater than 25 ppm after third IPC, install and condition fresh carbon discs. Pass Biaryl filtrate through refreshed carbon filter, washing with 1 L/kg DCM. Sample for IPC.
[0149] Distill and refill to appropriate concentration. Prepare for distillation of recovered filtrate by concentrating to ≤ 4 L/kg DCM, and recharge to reach 5.25 ± 0.25 L/kg DCM prior to moving into Step 7 Boc-deprotection reaction.
Step 7
General Note: All equivalents and volumes are reported in reference to crude Biaryl input
Note: All L/kg and kg/kg amounts are relative to Biaryl input
[0150] To Reactor A was added: tert-butyl (3S)-4-{6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-(1M)-1-[4-methyl-2-(propan-2-yl)pyridin-3-yl]-2-oxo-1,2-dihydropyrido[2,3-d]pyrimidin-4-yl}-3-methylpiperazine-1-carboxylate (Biaryl) (1.0 equiv), dichloromethane (5.0 L/kg), and the TFA (15.0 equiv, 1.9 L/kg) is charged slowly to maintain the internal temperature at 20 ± 5 °C. The reaction was stirred for 4 h at 20 ± 5 °C.
[0151] To Reactor B was added: potassium carbonate (18.0 equiv), water (20.0 L/kg), and NMP (1.0) to form a homogenous solution. While agitating at the maximum acceptable rate for the equipment, the reaction mixture in A was transferred into the potassium carbonate solution in B over 30 minutes (~ 0.24 L/kg/min rate). The mixture was stirred at 20 ± 5 °C for an additional 12 h.
[0152] The resulting slurry was filtered and rinsed with water (2 x 10 L/kg). The wet cake was dried for 24 h to give 6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-4-[(2S)-2-methylpiperazin- 1-yl]-(1M)-1-[4-methyl-2-(propan-2-yl)pyridin-3-yl]pyrido[2,3-d]pyrimidin-2(1H)-one (Des- Boc, Compound 8).
Step 8
Note: All L/kg and kg/kg amounts are relative to Des-Boc input
[0153] Des-Boc (Compound 8, 1.0 equiv) and NMP (4.2 L/kg) are charged to Vessel A under nitrogen, charge the TFA (1.0 equiv.) slowly to maintain the Tr <25 °C. The mixture is aged at 25 °C until full dissolution is observed (about 0.5 hour). The solution is then polish filtered through a 0.45 micron filter into Vessel B, washing with a NMP (0.8 L/kg). The filtrate and wash are combined, and then cooled to 0 °C. To the resulting solution, Acryloyl Chloride (1.3 equiv.) is added while maintaining temperature < 10 C. The reaction mixture is then aged at 5 ±5°C until completed by IPC (ca.1.5 hrs).
Preparation of Aqueous Disodium Phosphate Quench:
[0154] Disodium Phosphate (3.0 equiv) and Water (15.0 L/kg) are charged to Vessel C. The mixture is aged at 25 °C until full dissolution is observed. The solution is warmed to 45 ±5°C. A seed slurry of AMG 510 (0.005 equiv.) in Water (0.4 L/kg) is prepared and added to Vessel C while maintaining temperature at 45 ±5°C.
[0155] The reaction mixture in Vessel B is transferred to Vessel C (quench solution) while maintaining temperature at 45 ±5°C (ca.1 hrs). Vessel B is washed with a portion of NMP (0.5 L/kg). The product slurry is aged for 2 hrs at 45 ±5°C, cooled to 20 °C over 3 hrs, aged at 20 °C for a minimum of 12 hrs, filtered and washed with Water (2 x 10.0 L/kg). The product is dried using nitrogen and vacuum to afford Crude AMG 510 (Compound 9A).
Step 9
General Note: All equivalents and volumes are reported in reference to crude AMG 510 input
Note: All L/kg and kg/kg amounts are relative to Crude AMG 510 input
[0156] Reactor A was charged with 6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-(1M)-1-[4- methyl-2-(propan-2-yl)pyridin-3-yl]-4-[(2S)-2-methyl-4-(prop-2-enoyl)piperazin-1- yl]pyrido[2,3-d]pyrimidin-2(1H)-one (Crude AMG 510) (1.0 equiv), ethanol (7.5 L/kg), and water (1.9 L/kg). The mixture heated to 75 °C and polish filtered into a clean Reactor B. The solution was cool to 45 °C and seeded with authentic milled AMG 510 seed (0.015 േ 0.005
1 Seed performs best when reduced in particle size via milling or with other type of mechanical grinding if mill is not available (mortar/ pestle). Actual seed utilized will be based on seed availability. 1.0- 2.0% is seed is target amount.
kg/kg); the resulting slurry was aged for 30 min. Water (15.0 L/kg) was added over 5h while maintaining an internal temperature > 40 °C; the mixture was aged for an additional 2h.
[0157] The mixture was cooled to 20 °C over 3 hours and aged for 8h, after which the solid was collected by filtration and washed using a mixture of ethanol (2.5 L/kg) and water (5.0 L/kg). The solid was dried using vacuum and nitrogen to obtain 6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-(1M)-1-[4-methyl-2-(propan-2-yl)pyridin-3-yl]-4-[(2S)-2-methyl-4-(prop-2-enoyl)piperazin-1-yl]pyrido[2,3-d]pyrimidin-2(1H)-one (AMG 510, Compound 9).
Compound 6A Boroxine Synthesis:
Lithiation/borylation
[0158] Reactor A was charged with THF (6 vol), a secondary amine base, Diisopropylamine (1.4 equiv), and a catalyst, such as triethylamine hydrochloride (0.01 equiv.). The resulting solution was cooled to -70 °C and a first base, n-BuLi (2.5 M in hexane, 1.5 equiv) was slowly added. After addition is complete, a solution of 3-fluoroanisole (1.0 equiv) in THF (6 vol) was added slowly and kept at -70 °C for 5 min. Concurrently or subsequently, a reagent, B(EtO)3 (2.0 equiv), was added slowly and kept at -70 °C for 10 min. The reaction mixture was quenched with an acid, 2N HCl. The quenched reaction mixture was extracted with MTBE (3 x 4 vol). The combined organic phases were concentrated to 1.5-3 total volumes. Heptane (7-9 vol) was added drop-wise and the mixture was cooled to 0-10 °C and stirred for 3 h. The mixture was filtrated and rinsed with heptane (1.5 vol). The solid was dried under nitrogen at < 30 °C to afford (2-fluoro-6-methoxyphenyl)boronic acid.
Demethylation:
Note: All L/kg and kg/kg amounts are relative to (2-fluoro-6-methoxyphenyl)boronic acid input
[0159] To a reactor, charge dichloromethane (solvent, 4.0 L/kg) and an acid, BBr3 (1.2 equiv), and cool to -20 °C. To this solution, a suspension of (2-fluoro-6-methoxyphenyl)boronic acid (1.0 equiv) in dichloromethane (4.0 L/kg) was added into the BBr3/DCM mixture while keeping temperature -15 to -25 °C. The reaction was allowed to proceed for approximately 2 hours while monitored by HPLC [≤1% (2-fluoro-6-methoxyphenyl)boronic acid] before reverse quenching into water (3.0 L/kg). The precipitated solid was then isolated by filtration and slurried with water (3.0 L/kg) on the filter prior to deliquoring. The filtrates were adjusted to pH 4-6 by the addition of sodium bicarbonate. The bottom organic phase was separated and the resulting aqueous layer was washed with dichloromethane (solvent, 5.0 Vol) and adjusted to pH = 1 by addition of concentrated hydrochloric acid. The resulting solids were isolated by filtration, washing the cake with water (2 x 5.0 L/kg)
Purification via Reslurry (required)
[0160] The combined crude solids were charged into a reactor and slurried with 5% EtOH/water (5.0 L/kg) at 20 °C for >1 h. The purified product was then isolated by filtration and rinsed with water (2 x 3 L/kg) before drying on the filter at < 30 °C to with nitrogen/vacuum to afford 2,2′,2”-(1,3,5,2,4,6-trioxatriborinane-2,4,6-triyl)tris(3-fluorophenol) (Boroxine, Compound 6A).
^ Clinical trial number NCT03600883 for “A Phase 1/2, Study Evaluating the Safety, Tolerability, PK, and Efficacy of AMG 510 in Subjects With Solid Tumors With a Specific KRAS Mutation ” at ClinicalTrials.gov
“Sotorasib”. Drug Information Portal. U.S. National Library of Medicine.
Clinical trial number NCT03600883 for “A Phase 1/2, Study Evaluating the Safety, Tolerability, PK, and Efficacy of AMG 510 in Subjects With Solid Tumors With a Specific KRAS Mutation (CodeBreaK 100)” at ClinicalTrials.gov
Sotorasib is an inhibitor of the RAS GTPase family. The molecular formula is C30H30F2N6O3, and the molecular weight is 560.6 g/mol. The chemical name of sotorasib is 6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-(1M)-1-[4-methyl-2-(propan-2-yl)pyridin-3-yl]-4-[(2S)-2-methyl-4-(prop-2enoyl) piperazin-1-yl]pyrido[2,3-d]pyrimidin-2(1H)-one. The chemical structure of sotorasib is shown below:
Sotorasib has pKa values of 8.06 and 4.56. The solubility of sotorasib in the aqueous media decreases over the range pH 1.2 to 6.8 from 1.3 mg/mL to 0.03 mg/mL.
LUMAKRAS is supplied as film-coated tablets for oral use containing 120 mg of sotorasib. Inactive ingredients in the tablet core are microcrystalline cellulose, lactose monohydrate, croscarmellose sodium, and magnesium stearate. The film coating material consists of polyvinyl alcohol, titanium dioxide, polyethylene glycol, talc, and iron oxide yellow.
FDA grants accelerated approval to sotorasib for KRAS G12C mutated NSCLC
On May 28, 2021, the Food and Drug Administration granted accelerated approval to sotorasib (Lumakras, Amgen, Inc.), a RAS GTPase family inhibitor, for adult patients with KRAS G12C ‑mutated locally advanced or metastatic non-small cell lung cancer (NSCLC), as determined by an FDA ‑approved test, who have received at least one prior systemic therapy.
FDA also approved the QIAGEN therascreen® KRAS RGQ PCR kit (tissue) and the Guardant360® CDx (plasma) as companion diagnostics for Lumakras. If no mutation is detected in a plasma specimen, the tumor tissue should be tested.
Approval was based on CodeBreaK 100, a multicenter, single-arm, open label clinical trial (NCT03600883) which included patients with locally advanced or metastatic NSCLC with KRAS G12C mutations. Efficacy was evaluated in 124 patients whose disease had progressed on or after at least one prior systemic therapy. Patients received sotorasib 960 mg orally daily until disease progression or unacceptable toxicity.
The main efficacy outcome measures were objective response rate (ORR) according to RECIST 1.1, as evaluated by blinded independent central review and response duration. The ORR was 36% (95% CI: 28%, 45%) with a median response duration of 10 months (range 1.3+, 11.1).
The most common adverse reactions (≥ 20%) were diarrhea, musculoskeletal pain, nausea, fatigue, hepatotoxicity, and cough. The most common laboratory abnormalities (≥ 25%) were decreased lymphocytes, decreased hemoglobin, increased aspartate aminotransferase, increased alanine aminotransferase, decreased calcium, increased alkaline phosphatase, increased urine protein, and decreased sodium.
The recommended sotorasib dose is 960 mg orally once daily with or without food.
The approved 960 mg dose is based on available clinical data, as well as pharmacokinetic and pharmacodynamic modeling that support the approved dose. As part of the evaluation for this accelerated approval, FDA is requiring a postmarketing trial to investigate whether a lower dose will have a similar clinical effect.
This indication is approved under accelerated approval based on overall response rate and duration of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory trial(s).
This review was conducted under Project Orbis, an initiative of the FDA Oncology Center of Excellence. Project Orbis provides a framework for concurrent submission and review of oncology drugs among international partners. For this review, FDA collaborated with the Australian Therapeutic Goods Administration (TGA), the Brazilian Health Regulatory Agency (ANVISA), Health Canada, and the United Kingdom Medicines and Healthcare products Regulatory Agency (MHRA). The application reviews are ongoing at the other regulatory agencies.
This review used the Real-Time Oncology Review (RTOR) pilot program, which streamlined data submission prior to the filing of the entire clinical application, the Assessment Aid, and the Product Quality Assessment Aid (PQAA), voluntary submissions from the applicant to facilitate the FDA’s assessment. The FDA approved this application approximately 10 weeks ahead of the FDA goal date.
The most common side effects include diarrhea, musculoskeletal pain, nausea, fatigue, liver damage and cough.[1][2]
Sotorasib is an inhibitor of the RAS GTPase family.[1]
Sotorasib is the first approved targeted therapy for tumors with any KRAS mutation, which accounts for approximately 25% of mutations in non-small cell lung cancers.[2] KRAS G12C mutations represent about 13% of mutations in non-small cell lung cancers.[2] Sotorasib was approved for medical use in the United States in May 2021.[2][5]
Sotorasib is an experimental KRAS inhibitor being investigated for the treatment of KRAS G12C mutant non small cell lung cancer, colorectal cancer, and appendix cancer.
Sotorasib, also known as AMG-510, is an acrylamide derived KRAS inhibitor developed by Amgen.1,3 It is indicated in the treatment of adult patients with KRAS G12C mutant non small cell lung cancer.6 This mutation makes up >50% of all KRAS mutations.2 Mutant KRAS discovered in 1982 but was not considered a druggable target until the mid-2010s.5 It is the first experimental KRAS inhibitor.1
The drug MRTX849 is also currently being developed and has the same target.1
Sotorasib was granted FDA approval on 28 May 2021.6
Medical uses
Sotorasib is indicated for the treatment of adults with KRAS G12C-mutated locally advanced or metastatic non-small cell lung cancer (NSCLC), as determined by an FDA-approved test, who have received at least one prior systemic therapy.[1][2]
Clinical development
Sotorasib is being developed by Amgen. Phase I clinical trials were completed in 2020.[6][7][8] In December 2019, it was approved to begin Phase II clinical trials.[9]
Because the G12C KRAS mutation is relatively common in some cancer types, 14% of non-small-cell lung cancer adenocarcinoma patients and 5% of colorectal cancer patients,[10] and sotorasib is the first drug candidate to target this mutation, there have been high expectations for the drug.[10][11][12] The Food and Drug Administration has granted a fast track designation to sotorasib for the treatment of metastatic non-small-cell lung carcinoma with the G12C KRAS mutation.[13]
Chemistry and pharmacology
Sotorasib can exist in either of two atropisomeric forms and one is more active than the other.[10] It selectively forms an irreversible covalent bond to the sulfur atom in the cysteine residue that is present in the mutated form of KRAS, but not in the normal form.[10]
History
Researchers evaluated the efficacy of sotorasib in a study of 124 participants with locally advanced or metastatic KRAS G12C-mutated non-small cell lung cancer with disease progression after receiving an immune checkpoint inhibitor and/or platinum-based chemotherapy.[2] The major outcomes measured were objective response rate (proportion of participants whose tumor is destroyed or reduced) and duration of response.[2] The objective response rate was 36% and 58% of those participants had a duration of response of six months or longer.[2]
KRAS is the most frequently mutated oncogene in cancer and encodes a key signalling protein in tumours1,2. The KRAS(G12C) mutant has a cysteine residue that has been exploited to design covalent inhibitors that have promising preclinical activity3,4,5. Here we optimized a series of inhibitors, using novel binding interactions to markedly enhance their potency and selectivity. Our efforts have led to the discovery of AMG 510, which is, to our knowledge, the first KRAS(G12C) inhibitor in clinical development. In preclinical analyses, treatment with AMG 510 led to the regression of KRASG12C tumours and improved the anti-tumour efficacy of chemotherapy and targeted agents. In immune-competent mice, treatment with AMG 510 resulted in a pro-inflammatory tumour microenvironment and produced durable cures alone as well as in combination with immune-checkpoint inhibitors. Cured mice rejected the growth of isogenic KRASG12D tumours, which suggests adaptive immunity against shared antigens. Furthermore, in clinical trials, AMG 510 demonstrated anti-tumour activity in the first dosing cohorts and represents a potentially transformative therapy for patients for whom effective treatments are lacking.
Paper
Scientific Reports (2020), 10(1), 11992
PAPER
European journal of medicinal chemistry (2021), 213, 113082.
KRAS is the most commonly altered oncogene of the RAS family, especially the G12C mutant (KRASG12C), which has been a promising drug target for many cancers. On the basis of the bicyclic pyridopyrimidinone framework of the first-in-class clinical KRASG12C inhibitor AMG510, a scaffold hopping strategy was conducted including a F–OH cyclization approach and a pyridinyl N-atom working approach leading to new tetracyclic and bicyclic analogues. Compound 26a was identified possessing binding potency of 1.87 μM against KRASG12C and cell growth inhibition of 0.79 μM in MIA PaCa-2 pancreatic cancer cells. Treatment of 26a with NCI–H358 cells resulted in down-regulation of KRAS-GTP levels and reduction of phosphorylation of downstream ERK and AKT dose-dependently. Molecular docking suggested that the fluorophenol moiety of 26a occupies a hydrophobic pocket region thus forming hydrogen bonding to Arg68. These results will be useful to guide further structural modification.
PAPER
Journal of Medicinal Chemistry (2020), 63(1), 52-65.
KRASG12C has emerged as a promising target in the treatment of solid tumors. Covalent inhibitors targeting the mutant cysteine-12 residue have been shown to disrupt signaling by this long-“undruggable” target; however clinically viable inhibitors have yet to be identified. Here, we report efforts to exploit a cryptic pocket (H95/Y96/Q99) we identified in KRASG12C to identify inhibitors suitable for clinical development. Structure-based design efforts leading to the identification of a novel quinazolinone scaffold are described, along with optimization efforts that overcame a configurational stability issue arising from restricted rotation about an axially chiral biaryl bond. Biopharmaceutical optimization of the resulting leads culminated in the identification of AMG 510, a highly potent, selective, and well-tolerated KRASG12C inhibitor currently in phase I clinical trials (NCT03600883).
The present disclosure relates to an improved, efficient, scalable process to prepare intermediate compounds, such as compound of Formula 6A, having the structure,
useful for the synthesis of compounds for the treatment of KRAS G12C mutated cancers.
BACKGROUND
[0003] KRAS gene mutations are common in pancreatic cancer, lung adenocarcinoma, colorectal cancer, gall bladder cancer, thyroid cancer, and bile duct cancer. KRAS mutations are also observed in about 25% of patients with NSCLC, and some studies have indicated that KRAS mutations are a negative prognostic factor in patients with NSCLC. Recently, V-Ki-ras2 Kirsten rat sarcoma viral oncogene homolog (KRAS) mutations have been found to confer resistance to epidermal growth factor receptor (EGFR) targeted therapies in colorectal cancer; accordingly, the mutational status of KRAS can provide important information prior to the prescription of TKI therapy. Taken together, there is a need for new medical treatments for patients with pancreatic cancer, lung adenocarcinoma, or colorectal cancer, especially those who have been diagnosed to have such cancers characterized by a KRAS mutation, and including those who have progressed after chemotherapy.
Related Synthetic Processes
[0126] The following intermediate compounds of 6-Fluoro-7-(2-fluoro-6-hydroxyphenyl)-1-(4-methyl-2-(2-propanyl)-3-pyridinyl)-4-((2S)-2-methyl-4-(2-propenoyl)-1-piperazinyl)pyrido[2,3-d]pyrimidin-2(1H)-one are representative examples of the disclosure and are not intended to be construed as limiting the scope of the present invention.
[0127] A synthesis of Compound 9 and the relevant intermediates is described in U.S. Serial No.15/984,855, filed May 21, 2018 (U.S. Publication No.2018/0334454, November 22, 2018) which claims priority to and the benefit claims the benefit of U.S. Provisional Application No.62/509,629, filed on May 22, 2017, both of which are incorporated herein by reference in their entireties for all purposes. 6-Fluoro-7-(2-fluoro-6-hydroxyphenyl)-1-(4-methyl-2-(2-propanyl)-3-pyridinyl)-4-((2S)-2-methyl-4-(2-propenoyl)-1-piperazinyl)pyrido[2,3-d]pyrimidin-2(1H)-one was prepared using the following process, in which the isomers of the final product were isolated via chiral chromatography.
[0128] Step 1: 2,6-Dichloro-5-fluoronicotinamide (Intermediate S). To a mixture of 2,6-dichloro-5-fluoro-nicotinic acid (4.0 g, 19.1 mmol, AstaTech Inc., Bristol, PA) in dichloromethane (48 mL) was added oxalyl chloride (2M solution in DCM, 11.9 mL, 23.8 mmol), followed by a catalytic amount of DMF (0.05 mL). The reaction was stirred at room temperature overnight and then was concentrated. The residue was dissolved in 1,4-dioxane (48 mL) and cooled to 0 °C. Ammonium hydroxide solution (28.0-30% NH3 basis, 3.6 mL, 28.6 mmol) was added slowly via syringe. The resulting mixture was stirred at 0 °C for 30 min and then was concentrated. The residue was diluted with a 1:1 mixture of EtOAc/Heptane and agitated for 5 min, then was filtered. The filtered solids were discarded, and the remaining mother liquor was partially concentrated to half volume and filtered. The filtered solids were washed with heptane and dried in a reduced-pressure oven (45 °C) overnight to provide 2,6-dichloro-5-fluoronicotinamide. 1H NMR (400 MHz, DMSO-d6) δ ppm 8.23 (d, J = 7.9 Hz, 1 H) 8.09 (br s, 1 H) 7.93 (br s, 1 H). m/z (ESI, +ve ion): 210.9 (M+H)+.
[0129] Step 2: 2,6-Dichloro-5-fluoro-N-((2-isopropyl-4-methylpyridin-3-yl)carbamoyl)nicotinamide. To an ice-cooled slurry of 2,6-dichloro-5-fluoronicotinamide (Intermediate S, 5.0 g, 23.9 mmol) in THF (20 mL) was added oxalyl chloride (2 M solution in DCM, 14.4 mL, 28.8 mmol) slowly via syringe. The resulting mixture was heated at 75 °C for 1 h, then heating was stopped, and the reaction was concentrated to half volume. After cooling to 0 °C, THF (20 mL) was added, followed by a solution of 2-isopropyl-4-methylpyridin-3-amine (Intermediate R, 3.59 g, 23.92 mmol) in THF (10 mL), dropwise via cannula. The resulting mixture was stirred at 0 °C for 1 h and then was quenched with a 1:1 mixture of brine and saturated aqueous ammonium chloride. The mixture was extracted with EtOAc (3x) and the combined organic layers were dried over anhydrous sodium sulfate and concentrated to provide 2,6-dichloro-5-fluoro-N-((2-isopropyl-4-methylpyridin-3-yl)carbamoyl)nicotinamide. This material was used without further purification in the following step. m/z (ESI, +ve ion): 385.1(M+H)+.
[0130] Step 3: 7-Chloro-6-fluoro-1-(2-isopropyl-4-methylpyridin-3-yl)pyrido[2,3-d]pyrimidine-2,4(1H,3H)-dione. To an ice-cooled solution of 2,6-dichloro-5-fluoro-N-((2-isopropyl-4-methylpyridin-3-yl)carbamoyl)nicotinamide (9.2 g, 24.0 mmol) in THF (40 mL) was added KHMDS (1 M solution in THF, 50.2 mL, 50.2 mmol) slowly via syringe. The ice bath was removed and the resulting mixture was stirred for 40 min at room temperature. The reaction was quenched with saturated aqueous ammonium chloride and extracted with EtOAc (3x). The combined organic layers were dried over anhydrous sodium sulfate and concentrated. The residue was purified by silica gel chromatography (eluent: 0-50% 3:1 EtOAc-EtOH/heptane) to provide 7-chloro-6-fluoro-1-(2-isopropyl-4-methylpyridin-3-yl)pyrido[2,3-d]pyrimidine-2,4(1H,3H)-dione.1H NMR (400 MHz, DMSO-d6) δ ppm 12.27 (br s, 1H), 8.48-8.55 (m, 2 H), 7.29 (d, J = 4.8 Hz, 1 H), 2.87 (quin, J = 6.6 Hz, 1 H), 1.99-2.06 (m, 3 H), 1.09 (d, J = 6.6 Hz, 3 H), 1.01 (d, J = 6.6 Hz, 3 H).19F NMR (376 MHz, DMSO-d6) δ: -126.90 (s, 1 F). m/z (ESI, +ve ion): 349.1 (M+H)+.
[0131] Step 4: 4,7-Dichloro-6-fluoro-1-(2-isopropyl-4-methylpyridin-3-yl)pyrido[2,3-d]pyrimidin-2(1H)-one. To a solution of 7-chloro-6-fluoro-1-(2-isopropyl-4-methylpyridin-3-yl)pyrido[2,3-d]pyrimidine-2,4(1H,3H)-dione (4.7 g, 13.5 mmol) and DIPEA (3.5 mL, 20.2 mmol) in acetonitrile (20 mL) was added phosphorus oxychloride (1.63 mL, 17.5 mmol), dropwise via syringe. The resulting mixture was heated at 80 °C for 1 h, and then was cooled to room temperature and concentrated to provide 4,7-dichloro-6-fluoro-1-(2-isopropyl-4-methylpyridin-3-yl)pyrido[2,3-d]pyrimidin-2(1H)-one. This material was used without further purification in the following step. m/z (ESI, +ve ion): 367.1 (M+H)+.
[0132] Step 5: (S)-tert-Butyl 4-(7-chloro-6-fluoro-1-(2-isopropyl-4-methylpyridin-3-yl)-2-oxo-1,2-dihydropyrido[2,3-d]pyrimidin-4-yl)-3-methylpiperazine-1-carboxylate. To an ice-cooled solution of 4,7-dichloro-6-fluoro-1-(2-isopropyl-4-methylpyridin-3-yl)pyrido[2,3-d]pyrimidin-2(1H)-one (13.5 mmol) in acetonitrile (20 mL) was added DIPEA (7.1 mL, 40.3 mmol), followed by (S)-4-N-Boc-2-methyl piperazine (3.23 g, 16.1 mmol, Combi-Blocks, Inc., San Diego, CA, USA). The resulting mixture was warmed to room temperature and stirred for 1 h, then was diluted with cold saturated aqueous sodium bicarbonate solution (200 mL) and EtOAc (300 mL). The mixture was stirred for an additional 5 min, the layers were separated, and the aqueous layer was extracted with more EtOAc (1x). The combined organic layers were dried over anhydrous sodium sulfate and concentrated. The residue was purified by silica gel chromatography (eluent: 0-50% EtOAc/heptane) to provide (S)-tert-butyl 4-(7-chloro-6-fluoro-1-(2-isopropyl-4-methylpyridin-3-yl)-2-oxo-1,2-dihydropyrido[2,3-d]pyrimidin-4-yl)-3-methylpiperazine-1-carboxylate. m/z (ESI, +ve ion): 531.2 (M+H)+.
[0133] Step 6: (3S)-tert-Butyl 4-(6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-1-(2-isopropyl-4-methylpyridin-3-yl)-2-oxo-1,2-dihydropyrido[2,3-d]pyrimidin-4-yl)-3-methylpiperazine-1-carboxylate. A mixture of (S)-tert-butyl 4-(7-chloro-6-fluoro-1-(2-isopropyl-4-methylpyridin-3-yl)-2-oxo-1,2-dihydropyrido[2,3-d]pyrimidin-4-yl)-3-methylpiperazine-1-carboxylate (4.3 g, 8.1 mmol), potassium trifluoro(2-fluoro-6-hydroxyphenyl)borate (Intermediate Q, 2.9 g, 10.5 mmol), potassium acetate (3.2 g, 32.4 mmol) and [1,1′-bis(diphenylphosphino)ferrocene]dichloropalladium(II), complex with dichloromethane (661 mg, 0.81 mmol) in 1,4-dioxane (80 mL) was degassed with nitrogen for 1 min. De-oxygenated water (14 mL) was added, and the resulting mixture was heated at 90 °C for 1 h. The reaction was allowed to cool to room temperature, quenched with half-saturated aqueous sodium bicarbonate, and extracted with EtOAc (2x) and DCM (1x). The combined organic layers were dried over anhydrous sodium sulfate and concentrated. The residue was purified by silica gel chromatography (eluent: 0-60% 3:1 EtOAc-EtOH/heptane) to provide (3S)-tert-butyl 4-(6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-1-(2-isopropyl-4-methylpyridin-3-yl)-2-oxo-1,2-dihydropyrido[2,3-d]pyrimidin-4-yl)-3-methylpiperazine-1-carboxylate.1H NMR (400 MHz, DMSO-d6) δ ppm 10.19 (br s, 1 H), 8.38 (d, J = 5.0 Hz, 1 H), 8.26 (dd, J = 12.5, 9.2 Hz, 1 H), 7.23-7.28 (m, 1 H), 7.18 (d, J = 5.0 Hz, 1 H), 6.72 (d, J = 8.0 Hz, 1 H), 6.68 (t, J = 8.9 Hz, 1 H), 4.77-4.98 (m, 1 H), 4.24 (br t, J = 14.2 Hz, 1 H), 3.93-4.08 (m, 1 H), 3.84 (br d, J=12.9 Hz, 1 H), 3.52-3.75 (m, 1 H), 3.07-3.28 (m, 1 H), 2.62-2.74 (m, 1 H), 1.86-1.93 (m, 3 H), 1.43-1.48 (m, 9 H), 1.35 (dd, J = 10.8, 6.8 Hz, 3 H), 1.26-1.32 (m, 1 H), 1.07 (dd, J = 6.6, 1.7 Hz, 3 H), 0.93 (dd, J = 6.6, 2.1 Hz, 3 H).19F NMR (376 MHz, DMSO-d6) δ: -115.65 (s, 1 F), -128.62 (s, 1 F). m/z (ESI, +ve ion): 607.3 (M+H)+.
[0134] Step 7: 6-Fluoro-7-(2-fluoro-6-hydroxyphenyl)-1-(4-methyl-2-(2-propanyl)-3-pyridinyl)-4-((2S)-2-methyl-4-(2-propenoyl)-1-piperazinyl)pyrido[2,3-d]pyrimidin-2(1H)-one. Trifluoroacetic acid (25 mL, 324 mmol) was added to a solution of (3S)-tert-butyl 4-(6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-1-(2-isopropyl-4-methylpyridin-3-yl)-2-oxo-1,2-dihydropyrido[2,3-d]pyrimidin-4-yl)-3-methylpiperazine-1-carboxylate (6.3 g, 10.4 mmol) in DCM (30 mL). The resulting mixture was stirred at room temperature for 1 h and then was concentrated. The residue was dissolved in DCM (30 mL), cooled to 0 °C, and sequentially treated with DIPEA (7.3 mL, 41.7 mmol) and a solution of acryloyl chloride (0.849 mL, 10.4 mmol) in DCM (3 mL; added dropwise via syringe). The reaction was stirred at 0 °C for 10 min, then was quenched with half-saturated aqueous sodium bicarbonate and extracted with DCM (2x). The combined organic layers were dried over anhydrous sodium sulfate and concentrated. The residue was purified by silica gel chromatography (eluent: 0-100% 3:1 EtOAc-EtOH/heptane) to provide 6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-1-(4-methyl-2-(2-propanyl)-3-pyridinyl)-4-((2S)-2-methyl-4-(2-propenoyl)-1-piperazinyl)pyrido[2,3-d]pyrimidin-2(1H)-one.1H NMR (400 MHz, DMSO-d6) δ ppm 10.20 (s, 1 H), 8.39 (d, J = 4.8 Hz, 1 H), 8.24-8.34 (m, 1 H), 7.23-7.32 (m, 1 H), 7.19 (d, J = 5.0 Hz, 1 H), 6.87 (td, J = 16.3, 11.0 Hz, 1 H), 6.74 (d, J = 8.6 Hz, 1 H), 6.69 (t, J = 8.6 Hz, 1 H), 6.21 (br d, J = 16.2 Hz, 1 H), 5.74-5.80 (m, 1 H), 4.91 (br s, 1 H), 4.23-4.45 (m, 2 H), 3.97-4.21 (m, 1 H), 3.44-3.79 (m, 2 H), 3.11-3.31 (m, 1 H), 2.67-2.77 (m, 1 H), 1.91 (s, 3 H), 1.35 (d, J = 6.8 Hz, 3 H), 1.08 (d, J = 6.6 Hz, 3 H), 0.94 (d, J = 6.8 Hz, 3 H).19F NMR (376 MHz, DMSO-d6) δ ppm -115.64 (s, 1 F), -128.63 (s, 1 F). m/z (ESI, +ve ion): 561.2 (M+H)+.
[0135] Another synthesis of Compound 9 and the relevant intermediates was described in a U.S. provisional patent application filed November 16, 2018, which is incorporated herein by reference in its entirety for all purposes.
Representative Synthetic Processes
[0136] The present disclosure comprises the following steps wherein the synthesis and utilization of the boroxine intermediate is a novel and inventive step in the manufacture of AMG 510 (Compound 9):
Raw Materials
Step la
[0137] To a solution of 2,6-dichloro-5-fluoro-3-pyridinecarboxylic acid (25kg; 119. lmol) in dichloromethane (167kg) and DMF (592g) was added Oxalyl chloride (18.9kg; 148.9mol) while maintaining an internal temp between 15-20 °C. Additional dichloromethane (33kg) was added as a rinse and the reaction mixture stirred for 2h. The reaction mixture is cooled then quenched with ammonium hydroxide (40.2L; 595.5mol) while maintaining internal temperature 0 ± 10°C. The resulting slurry was stirred for 90min then the product collected by filtration. The filtered solids were washed with DI water (3X 87L) and dried to provide 2,6-dichloro-5-fluoronicotinamide (Compound 1).
Step 1b
[0138] In reactor A, a solution of 2,6-dichloro-5-fluoronicotinamide (Compound 1) (16.27kg; 77.8mol) in dichloromethane (359.5kg) was added oxalyl chloride (11.9kg;
93.8mol) while maintaining temp ≤ 25°C for 75min. The resulting solution was then headed to 40°C ± 3°C and aged for 3h. Using vacuum, the solution was distilled to remove dichloromethane until the solution was below the agitator. Dichloromethane (300 kg) was then added and the mixture cooled to 0 ± 5°C. To a clean, dry reactor (reactor B) was added,2-isopropyl-4-methylpyridin-3-amine (ANILINE Compound 2A) (12.9kg; 85.9mol) followed by dichloromethane (102.6 kg). The ANILINE solution was azeodried via vacuum distillation while maintaining an internal temperature between 20-25 °), replacing with additional dichloromethane until the solution was dry by KF analysis (limit ≤ 0.05%). The solution volume was adjusted to approx. 23L volume with dichloromethane. The dried ANILINE solution was then added to reactor A while maintaining an internal temperature of 0 ± 5°C throughout the addition. The mixture was then heated to 23 °C and aged for 1h. the solution was polish filtered into a clean reactor to afford 2,6-dichloro-5-fluoro-N-((2- isopropyl-4-methylpyridin-3-yl)carbamoyl)nicotinamide (Compound 3) as a solution in DCM and used directly in the next step.
Step 2
[0139] A dichloromethane solution of 2,6-dichloro-5-fluoro-N-{[4-methyl-2-(propan-2- yl)pyridin-3-yl]carbamoyl}pyridine-3-carboxamide (UREA (Compound 3)) (15kg contained; 38.9mol) was solvent exchanged into 2-MeTHF using vacuum distillation while maintaining internal temperature of 20-25 °C. The reactor volume was adjusted to 40L and then
additional 2-MeTHF was charged (105.4 kg). Sodium t-butoxide was added (9.4 kg;
97.8mol) while maintaining 5-10 °C. The contents where warmed to 23 °C and stirred for 3h. The contents where then cooled to 0-5C and ammonium chloride added (23.0kg; 430mol) as a solution in 60L of DI water. The mixture was warmed to 20 C and DI water added (15L) and further aged for 30min. Agitation was stopped and the layers separated. The aqueous layer was removed and to the organic layer was added DI water(81.7L). A mixture of conc HCl (1.5kg) and water (9L) was prepared then added to the reactor slowly until pH measured between 4-5. The layers were separated, and the aqueous layer back extracted using 2-MeTHF (42.2kg). The two organic layers combined and washed with a 10% citric acid solution (75kg) followed by a mixture of water (81.7L) and saturated NaCl (19.8 kg). The organic layer was then washed with saturated sodium bicarbonate (75kg) repeating if necessary to achieve a target pH of ≥ 7.0 of the aqueous. The organic layer was washed again with brine (54.7kg) and then dried over magnesium sulfate (5kg). The mixture was filtered to remove magnesium sulfate rinsing the filtered bed with 2-MeTHF (49.2 kg). The combined filtrate and washes where distilled using vacuum to 40L volume. The concentrated solution was heated to 55 °C and heptane (10-12kg) slowly added until cloud point. The solution was cooled to 23 °C over 2h then heptane (27.3 kg) was added over 2h. The product slurry was aged for 3h at 20-25 °C then filtered and washed with a mixture of 2-MeTHF (2.8kg) and heptane (9kg). The product was dried using nitrogen and vacuum to afford solid 7-chloro-6-fluoro-1-(2-isopropyl-4-methylpyridin-3-yl)pyrido[2,3-d]pyrimidine-2,4(1H,3H)-dione (rac-DIONE (Compound 4)).
Step 3
[0140] To a vessel, an agitated suspension of Compound 4, (1.0 eq.) in 2- methylterahydrofuran (7.0 L/kg) was added (+)-2,3-dibenzoyl-D-tartaric acid (2.0 eq.) under an atmosphere of nitrogen. 2-MeTHF is chiral, but it is used as a racemic mixture. The different enantiomers of 2-MeTHF are incorporated randomly into the co-crystal. The resulting suspension was warmed to 75°C and aged at 75°C until full dissolution was observed (< 30 mins.). The resulting solution was polish filtered at 75°C into a secondary vessel. To the polish filtered solution was charged n-Heptane (2.0 L/kg) at a rate that maintained the internal temperature above 65°C. The solution was then cooled to 60°C, seeded with crystals (0.01 kg/kg) and allowed to age for 30 minutes. The resulting suspension was cooled to 20°C over 4 hours and then sampled for chiral purity analysis by HPLC. To the suspension, n-Heptane (3.0 L/kg) was charged and then aged for 4 hours at 20°C under an atmosphere of nitrogen. The suspension was filtered, and the isolated solids were washed two times with (2:1) n-Heptane:2-methyltetrahydrofuran (3.0 L/kg). The material was dried with nitrogen and vacuum to afford M-Dione:DBTA: Me-THF complex (Compound 4a).
Step 4
[0141] To vessel A, a suspension of disodium hydrogen phosphate (21.1 kg, 2.0 equiv) in DI water (296.8 L, 6.3 L/kg) was agitated until dissolution was observed (≥ 30 min.). To vessel B, a suspension of the M-Dione:DBTA: Me-THF complex (Composition 4a)[46.9 kg (25.9 kg corrected for M-dione, 1.0 equiv.)] in methyl tert-butyl ether (517.8 L, 11.0 L/kg) was agitated for 15 to 30 minutes. The resulting solution from vessel A was added to vessel B, and then the mixture was agitated for more than 3 hours. The agitation was stopped, and the biphasic mixture was left to separate for more than 30 minutes. The lower aqueous phase was removed and then back extracted with methyl tert-butyl ether (77.7 L, 1.7 L/kg). The organic phases were combined in vessel B and dried with magnesium sulfate (24.8 kg, 0.529 kg/kg). The resulting suspension from vessel B was agitated for more than three hours and then filtered into vessel C. To vessel B, a methyl tert-butyl ether (46.9 L, 1.0 L/kg) rinse was charged and then filtered into vessel C. The contents of vessel C were cooled to 10 °C and then distilled under vacuum while slowly being warmed to 35°C. Distillation was continued until 320-350 kg (6.8-7.5 kg/kg) of methyl tert-butyl ether was collected. After cooling the contents of vessel C to 20°C, n-Heptane (278.7 L, 5.9 L/kg) was charged over one hour and then distilled under vacuum while slowly being warmed to 35°C. Distillation was continued until a 190-200 kg (4.1-4.3 kg/kg) mixture of methyl tert-butyl ether and n-Heptane was collected. After cooling the contents of vessel C to 20°C, n-Heptane (278.7 L, 5.9 L/kg) was charged a second time over one hour and then distilled under vacuum while slowly being warmed to 35°C. Distillation was continued until a 190-200 kg (4.1-4.3 kg/kg) mixture of methyl tert-butyl ether and n-Heptane was collected. After cooling the contents of vessel C to 20°C, n-Heptane (195.9 L, 4.2 L/kg) was charged a third time over one hour and then sampled for solvent composition by GC analysis. The vessel C suspension continued to agitate for more than one hour. The suspension was filtered, and then washed with a n-Heptane (68.6 L, 1.5 L/kg) rinse from vessel C. The isolated solids were dried at 50°C, and a sample was submitted for stock suitability. Afforded 7-chloro-6-fluoro-(1M)-1-[4-methyl-2-(propan-2-yl)pyridin-3-yl]pyrido[2,3-d]pyrimidine-2,4(1H,3H)-dione (M-DIONE) Compound 5M.
[0142] The first-generation process highlighted above has been successfully scaled on 200+ kg of rac-dione starting material (Compound 4). In this process, seeding the crystallization with the thermodynamically-stable rac-dione crystal form (which exhibits low solubility) would cause a batch failure. Based on our subsequent studies, we found that increasing the DBTA equivalents and lowering the seed temperature by adjusting heptane
charge schedule improves robustness of the process. The improved process is resistant to the presence of the thermodynamically-stable rac-dione crystal form and promotes successful separation of atropisomers. Subsequent batches will incorporate the improved process for large scale manufacture.
Step 5
Note: All L/kg amounts are relative to M-Dione input; All equiv. amounts are relative to M-Dione input after adjusted by potency.
[0143] M-Dione (Compound 5M, 1.0 equiv.) and Toluene-1 (10.0 L/kg) was charged to Vessel A. The resulting solution was dried by azeotropic distillation under vacuum at 45 °C until 5.0 L/kg of solvents has been removed. The contents of Vessel A were then cooled to 20 °C.
[0144] Vessel C was charged with Toluene-3 (4.5 L/kg), Phosphoryl chloride (1.5 equiv.) and N,N-Diisopropylethylamine-1 (2.0 equiv.) while maintaining the internal temperature below 20 ± 5 °C.
Upon finishing charging, Vessel C was warmed to 30 ± 5 °C. The contents of Vessel A were then transferred to Vessel C over 4 hours while maintaining the internal temperature at 30 ± 5°C. Vessel A was rinsed with Toluene-2 (0.5 L/kg) and transferred to Vessel C. The contents of Vessel C were agitated at 30°C for an additional 3 hours. The contents of Vessel C were cooled to 20 ± 5 °C. A solution of (s)-1-boc-3-methylpiperazine (1.2 equiv.), N,N-Diisopropylethylamine-2 (1.2 equiv.) in isopropyl acetate-1 (1.0 L/kg) was prepared in Vessel D. The solution of Vessel D was charged to vessel C while maintaining a batch temperature of 20 ± 5 °C (Note: Exotherm is observed). Upon the end of transfer, Vessel D was rinsed with additional dichloromethane (1.0 L/kg) and transferred to Vessel C. The contents of Vessel C were agitated for an additional 60 minutes at 20 °C. A solution of sodium bicarbonate [water-1 (15.0 L/kg + Sodium bicarbonate (4.5 equiv.)] was then charged into Vessel C over an hour while maintaining an internal temperature at 20 ± 5 °C throughout the addition. The contents of Vessel C were agitated for at least 12 hours at which point the Pipazoline (Compound 6) product was isolated by filtration in an agitated filter dryer. The cake was washed with water-2 and -3 (5.0 L/kg x 2 times, agitating each wash for 15 minutes) and isopropyl acetate-2 and 3 (5.0 L/kg x 2 times, agitating each wash for 15 min). The cake as dried under nitrogen for 12 hours.
Acetone Re-slurry (Optional):
[0145] Pipazoline (Compound 6) and acetone (10.0 L/kg) were charged to Vessel E. The suspension was heated to 50 °C for 2 hours. Water-4 (10.0 L/kg) was charged into Vessel E over 1 hour. Upon completion of water addition, the mixture was cooled to 20 °C over 1 hour. The contents of Vessel E were filtered to isolate the product, washing the cake with 1:1 acetone/water mixture (5.0 L/kg). The cake was dried under nitrogen for 12 hours.
Step 6
General Note: All equivalents and volumes are reported in reference to Pipazoline input
Note: All L/kg and kg/kg amounts are relative to Pipazoline input
[0146] Reactor A is charged with Pipazoline (Compound 6, 1.0 equiv), degassed 2- MeTHF (9.0 L/kg) and a solution of potassium acetate (2.0 equiv) in degassed water (6.5 L/kg). The resulting mixture is warmed to 75 ± 5 °C and then, charge a slurry of
Pd(dpePhos)Cl2 (0.003 equiv) in 2-MeTHF (0.5 L/kg). Within 2 h of catalyst charge, a solution of freshly prepared Boroxine (Compound 6A, 0.5 equiv) in wet degassed 2-MeTHF (4.0 L/kg, KF > 4.0%) is charged over the course of >1 hour, but < 2 hours, rinsing with an additional portion of wet 2-MeTHF (0.5 L/kg) after addition is complete. After reaction completion ( <0.15 area % Pipazoline remaining, typically <1 h after boroxine addition is complete), 0.2 wt% (0.002 kg/kg) of Biaryl seed is added as a slurry in 0.02 L/kg wet 2- MeTHF, and the resulting seed bed is aged for > 60 min. Heptane (5.0 L/kg) is added over 2 hours at 75 ± 5 °C. The batch is then cooled to 20 ± 5 °C over 2 hours and aged for an additional 2 h. The slurry is then filtered and cake washed with 1 x 5.0L/kg water, 1 x 5.0L/kg 1:1 iPrOH:water followed by 1 x 5.0 L/kg 1:1 iPrOH:heptane (resuspension wash: the cake is resuspended by agitator and allow to set before filtering) . The cake (Biaryl, Compound 7) is then dried under vacuum with a nitrogen sweep.
Note: If the reaction stalls, an additional charge of catalyst and boroxine is required
Step 7 Charcoal Filtration for Pd removal
General Note: All equivalents and volumes are reported in reference to crude Biaryl input
Note: All L/kg and kg/kg amounts are relative to crude Biaryl input
[0147] In a clean Vessel A, charge crude Biaryl (1 equiv) and charge DCM (10 L/kg). Agitate content for > 60 minutes at 22 ± 5 °C, observing dissolution. Pass crude Biaryl from Vessel A, through a bag filter and carbon filters at a flux ≤ 3 L2/min/m and collect filtrate in clean Vessel B. Charge DCM rinse (1 L/kg) to Vessel A, and through carbon filters to collect in vessel B.
[0148] From filtrate in Vessel B, pull a solution sample for IPC Pd content. Sample is concentrated to solid and analyzed by ICP-MS. IPC: Pd ≤ 25 ppm with respect to Biaryl. a. If Pd content is greater than 25 ppm with respect to Biaryl on first or second IPC sample, pass solution through carbon filter a second time at ≤ 3 L2/min/m2, rinsing with 1 L/kg DCM; sample filtrate for IPC.
b. If Pd content remains greater than 25 ppm after third IPC, install and condition fresh carbon discs. Pass Biaryl filtrate through refreshed carbon filter, washing with 1 L/kg DCM. Sample for IPC.
[0149] Distill and refill to appropriate concentration. Prepare for distillation of recovered filtrate by concentrating to ≤ 4 L/kg DCM, and recharge to reach 5.25 ± 0.25 L/kg DCM prior to moving into Step 7 Boc-deprotection reaction.
Step 7
General Note: All equivalents and volumes are reported in reference to crude Biaryl input
Note: All L/kg and kg/kg amounts are relative to Biaryl input
[0150] To Reactor A was added: tert-butyl (3S)-4-{6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-(1M)-1-[4-methyl-2-(propan-2-yl)pyridin-3-yl]-2-oxo-1,2-dihydropyrido[2,3-d]pyrimidin-4-yl}-3-methylpiperazine-1-carboxylate (Biaryl) (1.0 equiv), dichloromethane (5.0 L/kg), and the TFA (15.0 equiv, 1.9 L/kg) is charged slowly to maintain the internal temperature at 20 ± 5 °C. The reaction was stirred for 4 h at 20 ± 5 °C.
[0151] To Reactor B was added: potassium carbonate (18.0 equiv), water (20.0 L/kg), and NMP (1.0) to form a homogenous solution. While agitating at the maximum acceptable rate for the equipment, the reaction mixture in A was transferred into the potassium carbonate solution in B over 30 minutes (~ 0.24 L/kg/min rate). The mixture was stirred at 20 ± 5 °C for an additional 12 h.
[0152] The resulting slurry was filtered and rinsed with water (2 x 10 L/kg). The wet cake was dried for 24 h to give 6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-4-[(2S)-2-methylpiperazin- 1-yl]-(1M)-1-[4-methyl-2-(propan-2-yl)pyridin-3-yl]pyrido[2,3-d]pyrimidin-2(1H)-one (Des- Boc, Compound 8).
Step 8
Note: All L/kg and kg/kg amounts are relative to Des-Boc input
[0153] Des-Boc (Compound 8, 1.0 equiv) and NMP (4.2 L/kg) are charged to Vessel A under nitrogen, charge the TFA (1.0 equiv.) slowly to maintain the Tr <25 °C. The mixture is aged at 25 °C until full dissolution is observed (about 0.5 hour). The solution is then polish filtered through a 0.45 micron filter into Vessel B, washing with a NMP (0.8 L/kg). The filtrate and wash are combined, and then cooled to 0 °C. To the resulting solution, Acryloyl Chloride (1.3 equiv.) is added while maintaining temperature < 10 C. The reaction mixture is then aged at 5 ±5°C until completed by IPC (ca.1.5 hrs).
Preparation of Aqueous Disodium Phosphate Quench:
[0154] Disodium Phosphate (3.0 equiv) and Water (15.0 L/kg) are charged to Vessel C. The mixture is aged at 25 °C until full dissolution is observed. The solution is warmed to 45 ±5°C. A seed slurry of AMG 510 (0.005 equiv.) in Water (0.4 L/kg) is prepared and added to Vessel C while maintaining temperature at 45 ±5°C.
[0155] The reaction mixture in Vessel B is transferred to Vessel C (quench solution) while maintaining temperature at 45 ±5°C (ca.1 hrs). Vessel B is washed with a portion of NMP (0.5 L/kg). The product slurry is aged for 2 hrs at 45 ±5°C, cooled to 20 °C over 3 hrs, aged at 20 °C for a minimum of 12 hrs, filtered and washed with Water (2 x 10.0 L/kg). The product is dried using nitrogen and vacuum to afford Crude AMG 510 (Compound 9A).
Step 9
General Note: All equivalents and volumes are reported in reference to crude AMG 510 input
Note: All L/kg and kg/kg amounts are relative to Crude AMG 510 input
[0156] Reactor A was charged with 6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-(1M)-1-[4- methyl-2-(propan-2-yl)pyridin-3-yl]-4-[(2S)-2-methyl-4-(prop-2-enoyl)piperazin-1- yl]pyrido[2,3-d]pyrimidin-2(1H)-one (Crude AMG 510) (1.0 equiv), ethanol (7.5 L/kg), and water (1.9 L/kg). The mixture heated to 75 °C and polish filtered into a clean Reactor B. The solution was cool to 45 °C and seeded with authentic milled AMG 510 seed (0.015 േ 0.005
1 Seed performs best when reduced in particle size via milling or with other type of mechanical grinding if mill is not available (mortar/ pestle). Actual seed utilized will be based on seed availability. 1.0- 2.0% is seed is target amount.
kg/kg); the resulting slurry was aged for 30 min. Water (15.0 L/kg) was added over 5h while maintaining an internal temperature > 40 °C; the mixture was aged for an additional 2h.
[0157] The mixture was cooled to 20 °C over 3 hours and aged for 8h, after which the solid was collected by filtration and washed using a mixture of ethanol (2.5 L/kg) and water (5.0 L/kg). The solid was dried using vacuum and nitrogen to obtain 6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-(1M)-1-[4-methyl-2-(propan-2-yl)pyridin-3-yl]-4-[(2S)-2-methyl-4-(prop-2-enoyl)piperazin-1-yl]pyrido[2,3-d]pyrimidin-2(1H)-one (AMG 510, Compound 9).
Compound 6A Boroxine Synthesis:
Lithiation/borylation
[0158] Reactor A was charged with THF (6 vol), a secondary amine base, Diisopropylamine (1.4 equiv), and a catalyst, such as triethylamine hydrochloride (0.01 equiv.). The resulting solution was cooled to -70 °C and a first base, n-BuLi (2.5 M in hexane, 1.5 equiv) was slowly added. After addition is complete, a solution of 3-fluoroanisole (1.0 equiv) in THF (6 vol) was added slowly and kept at -70 °C for 5 min. Concurrently or subsequently, a reagent, B(EtO)3 (2.0 equiv), was added slowly and kept at -70 °C for 10 min. The reaction mixture was quenched with an acid, 2N HCl. The quenched reaction mixture was extracted with MTBE (3 x 4 vol). The combined organic phases were concentrated to 1.5-3 total volumes. Heptane (7-9 vol) was added drop-wise and the mixture was cooled to 0-10 °C and stirred for 3 h. The mixture was filtrated and rinsed with heptane (1.5 vol). The solid was dried under nitrogen at < 30 °C to afford (2-fluoro-6-methoxyphenyl)boronic acid.
Demethylation:
Note: All L/kg and kg/kg amounts are relative to (2-fluoro-6-methoxyphenyl)boronic acid input
[0159] To a reactor, charge dichloromethane (solvent, 4.0 L/kg) and an acid, BBr3 (1.2 equiv), and cool to -20 °C. To this solution, a suspension of (2-fluoro-6-methoxyphenyl)boronic acid (1.0 equiv) in dichloromethane (4.0 L/kg) was added into the BBr3/DCM mixture while keeping temperature -15 to -25 °C. The reaction was allowed to proceed for approximately 2 hours while monitored by HPLC [≤1% (2-fluoro-6-methoxyphenyl)boronic acid] before reverse quenching into water (3.0 L/kg). The precipitated solid was then isolated by filtration and slurried with water (3.0 L/kg) on the filter prior to deliquoring. The filtrates were adjusted to pH 4-6 by the addition of sodium bicarbonate. The bottom organic phase was separated and the resulting aqueous layer was washed with dichloromethane (solvent, 5.0 Vol) and adjusted to pH = 1 by addition of concentrated hydrochloric acid. The resulting solids were isolated by filtration, washing the cake with water (2 x 5.0 L/kg)
Purification via Reslurry (required)
[0160] The combined crude solids were charged into a reactor and slurried with 5% EtOH/water (5.0 L/kg) at 20 °C for >1 h. The purified product was then isolated by filtration and rinsed with water (2 x 3 L/kg) before drying on the filter at < 30 °C to with nitrogen/vacuum to afford 2,2′,2”-(1,3,5,2,4,6-trioxatriborinane-2,4,6-triyl)tris(3-fluorophenol) (Boroxine, Compound 6A).
^ Clinical trial number NCT03600883 for “A Phase 1/2, Study Evaluating the Safety, Tolerability, PK, and Efficacy of AMG 510 in Subjects With Solid Tumors With a Specific KRAS Mutation ” at ClinicalTrials.gov
“Sotorasib”. Drug Information Portal. U.S. National Library of Medicine.
Clinical trial number NCT03600883 for “A Phase 1/2, Study Evaluating the Safety, Tolerability, PK, and Efficacy of AMG 510 in Subjects With Solid Tumors With a Specific KRAS Mutation (CodeBreaK 100)” at ClinicalTrials.gov
02 Jun 2021Apellis Pharmaceuticals plans a phase III trial for Glomerulonephritis in the second half of 2021
25 May 2021Top-line efficacy and safety results from the phase III PRINCE trial for Paroxysmal nocturnal haemoglobinuria released by Apellis Pharmaceuticals
18 May 2021Registered for Paroxysmal nocturnal haemoglobinuria in USA (SC) – First global approval
The most common side effects include injection-site reactions, infections, diarrhea, abdominal pain, respiratory tract infection, viral infection, and fatigue.[2]
Paroxysmal nocturnal hemoglobinuria is characterized by red blood cell destruction, anemia (red blood cells unable to carry enough oxygen to tissues), blood clots, and impaired bone marrow function (not making enough blood cells).[1]
Pegcetacoplan is the first treatment for paroxysmal nocturnal hemoglobinuria that binds to complement protein C3.[1] Pegcetacoplan was approved for medical use in the United States in May 2021.[1][3]
Pegcetacoplan is a complement inhibitor indicated in the treatment of paroxysmal nocturnal hemoglobinuria (PNH).5,7 Prior to its FDA approval, patients with PNH were typically treated with the C5 inhibiting monoclonal antibody eculizumab.5 Patients given eculizumab experienced less hemolysis caused by the membrane attack complex, but were still somewhat susceptible to hemolysis caused by C3b opsonization.5,6 Pegcetacoplan was developed out of a need for an inhibitor of complement mediated hemolysis further upstream of C5.5,6 Pegcetacoplan is a pegylated C3 inhibitor that can disrupt the processes leading to both forms of hemolysis that threaten patients with PNH.5
Pegcetacoplan was granted FDA approval on 14 May 2021.7
Medical uses
Pegcetacoplan is indicated to treat adults with paroxysmal nocturnal hemoglobinuria (PNH).[1][2]
EMPAVELI contains pegcetacoplan, a complement inhibitor. Pegcetacoplan is a symmetrical molecule comprised of two identical pentadecapeptides covalently bound to the ends of a linear 40-kiloDalton (kDa) PEG molecule. The peptide portions of pegcetacoplan contain 1-methyl-L-tryptophan (Trp(Me)) in position 4 and amino(ethoxyethoxy)acetic acid (AEEA) in position 14.
The molecular weight of pegcetacoplan is approximately 43.5 kDa. The molecular formula is C1970H3848N50O947S4. The structure of pegcetacoplan is shown below.
EMPAVELI injection is a sterile, clear, colorless to slightly yellowish aqueous solution for subcutaneous use and is supplied in a 20-mL single-dose vial. Each 1 mL of solution contains 54 mg of pegcetacoplan, 41 mg of sorbitol, 0.384 mg of glacial acetic acid, 0.490 mg of sodium acetate trihydrate, and Water for Injection USP. EMPAVELI may also contain sodium hydroxide and/or additional glacial acetic acid for adjustment to a target pH of 5.0.
FDA approves new treatment for adults with serious rare blood disease..
FDA has approved Empaveli (pegcetacoplan) injection to treat adults with paroxysmal nocturnal hemoglobinuria (PNH), a rare, life-threatening blood disease. Empaveli is the first PNH treatment that binds to compliment protein C3.
PNH is characterized by red blood cell destruction, anemia (red blood cells unable to carry enough oxygen to tissues), blood clots, and impaired bone marrow function (not making enough blood cells). The disease affects 1-1.5 people per million. Individuals are typically diagnosed around ages 35 to 40. PNH can be serious, with median survival of 10 years after diagnosis. However, some patients live for decades with only minor symptoms.
PNH is caused by gene mutations that affect red blood cells. Red blood cells in people with these mutations are defective and can be destroyed by the immune system, which causes anemia.
The effectiveness of Empaveli was evaluated in a study enrolling 80 patients with PNH and anemia who had been taking eculizumab, a treatment previously approved for PNH. Patients first completed a four-week period during which they received Empaveli 1,080 mg twice weekly in addition to eculizumab at their previous dose. After the first four weeks, patients were randomly assigned to receive either Empaveli or their current dose of eculizumab for 16 weeks.
After 16 weeks, the severity of anemia was compared in the two treatment groups on the basis of hemoglobin concentration (a laboratory measure of anemia). In both treatment groups, the average hemoglobin was 8.7 g/dL at baseline, indicating severe anemia. (Normal hemoglobin values in adult men are 14 g/dL or above; normal values in adult women are 12 g/dL or above.) During the 16 weeks of treatment, patients in the Empaveli group had an average increase in their hemoglobin of 2.4 g/dL. Meanwhile, patients in the eculizumab group had an average decrease in their hemoglobin of 1.5 g/dL.
Empaveli is available only through a restricted program under a risk evaluation and mitigation strategy. Meningococcal (a type of bacteria) infections can occur in patients taking Empaveli and can become life-threatening or fatal if not treated early. Empaveli may also predispose individuals to serious infections, especially infections caused by encapsulated bacteria. Patients should be monitored for infusion-related reactions. Empaveli can interfere with certain laboratory tests. The most common side effects are injection site reactions, infections, diarrhea, abdominal pain, respiratory tract infection, viral infection, and fatigue.
FDA granted the approval of Empaveli to Apellis Pharmaceuticals.
Adverse effects
Meningococcal (a type of bacteria) infections can occur in people taking pegcetacoplan and can become life-threatening or fatal if not treated early.[1] Pegcetacoplan may also predispose individuals to serious infections, especially infections caused by encapsulated bacteria.[1]
History
The effectiveness of pegcetacoplan was evaluated in a study enrolling 80 participants with paroxysmal nocturnal hemoglobinuria and anemia who had been taking eculizumab, a treatment previously approved for paroxysmal nocturnal hemoglobinuria.[1]
“Pegcetacoplan”. Drug Information Portal. U.S. National Library of Medicine.
Clinical trial number NCT03500549 for “Study to Evaluate the Efficacy and Safety of APL-2 in Patients With Paroxysmal Nocturnal Hemoglobinuria (PNH)” at ClinicalTrials.gov
/////////Pegcetacoplan, ペグセタコプラン , FDA 2021, APPROVALS 2021, APL-2, WHO 10743, Apellis Pharmaceuticals, Empaveli, priority review, fast track,orphan drug
On May 21, 2021, the Food and Drug Administration granted accelerated approval to amivantamab-vmjw (Rybrevant, Janssen Biotech, Inc.), a bispecific antibody directed against epidermal growth factor (EGF) and MET receptors, for adult patients with locally advanced or metastatic non-small cell lung cancer (NSCLC) with epidermal growth factor receptor (EGFR) exon 20 insertion mutations, as detected by an FDA-approved test, whose disease has progressed on or after platinum-based chemotherapy.
FDA also approved the Guardant360® CDx (Guardant Health, Inc.) as a companion diagnostic for amivantamab-vmjw.
Approval was based on CHRYSALIS, a multicenter, non-randomized, open label, multicohort clinical trial (NCT02609776) which included patients with locally advanced or metastatic NSCLC with EGFR exon 20 insertion mutations. Efficacy was evaluated in 81 patients with advanced NSCLC with EGFR exon 20 insertion mutations whose disease had progressed on or after platinum-based chemotherapy. Patients received amivantamab-vmjw once weekly for 4 weeks, then every 2 weeks thereafter until disease progression or unacceptable toxicity.
The main efficacy outcome measures were overall response rate (ORR) according to RECIST 1.1 as evaluated by blinded independent central review (BICR) and response duration. The ORR was 40% (95% CI: 29%, 51%) with a median response duration of 11.1 months (95% CI: 6.9, not evaluable).
The most common adverse reactions (≥ 20%) were rash, infusion-related reactions, paronychia, musculoskeletal pain, dyspnea, nausea, fatigue, edema, stomatitis, cough, constipation, and vomiting.
The recommended dose of amivantamab-vmjw is 1050 mg for patients with baseline body weight < 80 kg, and 1400 mg for those with body weight ≥ 80 kg, administered weekly for 4 weeks, then every 2 weeks thereafter until disease progression or unacceptable toxicity.
This indication is approved under accelerated approval based on overall response rate and duration of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials.
This review was conducted under Project Orbis, an initiative of the FDA Oncology Center of Excellence. Project Orbis provides a framework for concurrent submission and review of oncology drugs among international partners. For this review, FDA collaborated with the Brazilian Health Regulatory Agency (ANVISA) and United Kingdom’s Medicines and Healthcare products Regulatory Agency (MHRA). The application reviews are ongoing at the other regulatory agencies.
This review used the Assessment Aid, a voluntary submission from the applicant to facilitate the FDA’s assessment. The FDA approved this application 2 months ahead of the FDA goal date.
The most common side effects include rash, infusion-related reactions, skin infections around the fingernails or toenails, muscle and joint pain, shortness of breath, nausea, fatigue, swelling in the lower legs or hands or face, sores in the mouth, cough, constipation, vomiting and changes in certain blood tests.[2][3]
Amivantamab was approved for medical use in the United States in May 2021.[2][3][4][5]
Amivantamab, also known as JNJ-61186372, is an anti-EGFR-MET bispecific antibody, derived from Chinese hamster ovary cells, approved for the treatment of adult patients with locally advanced or metastatic non-small cell lung cancer (NSCLC) with epidermal growth factor receptor (EGFR) exon 20 insertion mutations, as detected by an FDA-approved test, whose disease has progressed on or after platinum-based chemotherapy.1,9 Patients with NSCLC often develop resistance to drugs that target EGFR and MET individually, so amivantamab was developed to attack both targets, reducing the chance of resistance developing.1,2 Amivantamab was found to be more effective than the EGFR inhibitor erlotinib or the MET inhibitor crizotinibin vivo.1,3 Patients with NSCLC with exon 20 insertion mutations in EGFR do not respond to tyrosine kinase inhibitors, and were generally treated with platinum-based therapy.5
Amivantamab was granted FDA approval on 21 May 2021.9
Medical uses
Amivantamab is indicated for the treatment of adults with locally advanced or metastatic non-small cell lung cancer (NSCLC) with epidermal growth factor receptor (EGFR) exon 20 insertion mutations, as detected by an FDA-approved test, whose disease has progressed on or after platinum-based chemotherapy.[3]
History
The U.S. Food and Drug Administration (FDA) approved amivantamab based on CHRYSALIS, a multicenter, non-randomized, open label, multicohort clinical trial (NCT02609776) which included participants with locally advanced or metastatic non-small cell lung cancer (NSCLC) with EGFR exon 20 insertion mutations.[3] Efficacy was evaluated in 81 participants with advanced NSCLC with EGFR exon 20 insertion mutations whose disease had progressed on or after platinum-based chemotherapy.[3]
The FDA collaborated on the review of amivantamab with the Brazilian Health Regulatory Agency (ANVISA) and the United Kingdom’s Medicines and Healthcare products Regulatory Agency (MHRA).[3] The application reviews are ongoing at the other regulatory agencies.[3]
Society and culture
Legal status
Amivantamab was approved for medical use in the United States in May 2021.[2][3][4][5] A marketing authorization application is pending in the EU.[6][7]
Amivantamab is being investigated in combination with lazertinib versus osimertinib; and in combination with carboplatin-pemetrexed chemotherapy compared to carboplatin-pemetrexed.[9][10]
). The MET parental mAbs had the F405L mutation and the EGFR parental mAbs had the K409R mutation. The IgG1 b12 arm served as isotype control and null arm to preserve the BsAb architecture. The low fucose parental mAbs were generated using proprietary cell lines. The quality of the BsAb were confirmed as being monodisperse and monomeric via size exclusion chromatography and being pure via SDS-PAGE.
Flow cytometric binding assay
Binding to cells expressing EGFR and MET (A549 [ATCC CCL-185], NCI-H1975 [ATCC, CRL-5908], and NCI-H441 [ATCC HTB-174] cells) was evaluated using flow cytometry (fluorescence-activated cell sorting [FACS]). All BsAbs and controls were diluted in FACS buffer (PBS supplemented with 1% bovine serum albumin and 0.2% sodium azide). After 1 h incubation, unbound antibodies were removed by a FACS buffer wash. The cells were then incubated with goat anti-human IgG-PE (Jackson) for FACS detection (BD FACS Canto). The mean fluorescence intensity of the cells in the live gate was plotted against antibody concentration, and the EC50 was determined by nonlinear regression fitting. Anti-EGFR zalutumumab and anti-MET 5D5 (onartuzumab) were positive controls and anti-CD20 7D8 (Genmab) was the negative control.
MET phosphorylation assay
A549 cells were incubated with 30 μg/ml of test antibody for 15 min and tested for MET phosphorylation using rabbit anti-phospho MET (Tyr1234–1235) (Cell Signaling 3129) and total MET protein using mouse anti-human MET antibody (Cell Signaling 3127). A score of 1 to 4 was given, where 1 = no visible band, 2 = slightly visible band, 3 = phosphorylation comparable with weak agonist (MET B IgG1), and 4 = phosphorylation level similar to positive controls (MET A and MET 5D5 IgG1 mAbs).
Proliferation assays
Test molecules were added to H1975, KP4 (Riken Cell bank, RCB1005), or NCI-H441 cells plated at 5000 or 10,000 (KP4) cells/well in 96-well plates. After 6 (KP4) or 7 (H1975 and NCI-H441) days of incubation at 37 °C and 5% CO2, the number of viable cells was determined using an AlamarBlue assay (Biosource DAL1100). A615 values were measured and plotted in a bar diagram.
EGFR phosphorylation assay
Approximately 106 A549 or SNU-5 cells/well were grown overnight in six-well plates and incubated for 15 min with 30 μg/ml of antibody in the absence or presence of 40 ng/ml EGF. After cell lysis, Western blots determined EGFR phosphorylation status with phospho-EGFR (Tyr1068) antibody (Cell Signaling 2234) and total EGFR protein using an anti-EGFR antibody (Cell Signaling 2232).
Expression and purification of proteins for crystallization
Human MET Sema-PSI region (residues 39–564) containing a C-terminal 8xHis tag was expressed in Tni PRO insect cells infected with recombinant baculovirus. The culture was harvested 72 h post infection, and the MET Sema-PSI protein was purified by affinity and size exclusion chromatography. Briefly, MET was captured with a Ni-NTA resin (Novagen) equilibrated in TBS, 10 mM imidazole, pH 7.4 and eluted from the column with 250 mM imidazole, TBS, pH 7.4. Fractions containing MET were identified by SDS-PAGE and loaded into a Superdex 200 column (GE Healthcare) equilibrated in 20 mM Tris, 50 mM NaCl, pH 7. The final protein concentration was determined by absorbance at 280 nm.The anti-MET Fab of amivantamab was transiently expressed in Expi293F cells. Briefly, the cells were cotransfected with separate plasmids encoding the Fab heavy and light chains at 3:1 (light:heavy chain) molar ratio following transfection kit instructions (Life Technologies). The culture was harvested 5 days post transfection, and the Fab was purified by affinity and cation exchange chromatography. Briefly, the Fab was captured with a HiTrap resin (GE Healthcare) equilibrated in PBS pH 7.2 and eluted from the column with a gradient of 30 to 300 mM imidazole in PBS pH 7.2. The eluate was buffer exchanged into 25 mM NaCl, 20 mM MES pH 6.0, bound to a Source 15S column (GE Healthcare), and eluted with a NaCl gradient in 20 mM MES pH 6.0.
Crystallization and structure determination
The amivantamab anti-MET Fab–MET Sema-PSI complex was prepared by overnight mixing of MET and Fab at a molar ratio of 1:1.3 (excess Fab) at 4 °C, while buffer exchanging to 20 mM Hepes pH 7.0. The complex was captured with a monoS 5/50 column (GE Healthcare) equilibrated in 20 mM Hepes pH 7.0 and eluted from the column with a gradient of NaCl. The complex was concentrated to 4.8 mg/ml.Crystallization trials for the Fab–MET complex were carried out with a Mosquito LCP robot (TTP LabTech) for the setup of sitting drops on 96-well plates (Corning 3550) and a Rock Imager 54 (Formulatrix) for plate storage at 20 °C and automated imaging of drops. Small crystals were initially obtained from 2 M NH4(SO4)2, 0.1 M MES pH 6.5, and they were used as seeds in next rounds of optimization. Crystals suitable for X-ray diffraction were obtained from 2.5 M sodium formate, 5% PEG 400 Da, 0.1 M Tris pH 8.5 after multiple rounds of seeding. The crystals were soaked for a few seconds in a cryoprotectant solution containing mother liquor supplemented with 20% glycerol and then flash frozen in liquid nitrogen. X-ray diffraction data were collected with a Pilatus 6M detector on beamline 17-ID at the Advanced Photon Source (Argonne National Laboratory), and the diffraction data were processed with the program HKL2000. The crystal structure of the Fab–MET complex was solved by molecular replacement with PHASER using previously solved MET Sema-PSI (PDB code 1SHY) and anti-HER3 Fab RG7116 (PDB code 4LEO) structures as search models. The structure was refined with PHENIX, and model adjustments were performed using COOT. His tags (at C-terminal of heavy chain and PSI), Fab interchain disulfide bond, heavy chain residues 133 to 139, Sema residues 303 to 309, 407, and glycan linked to N399 are disordered and not included in the structure. The Fab was numbered sequentially and Sema-PSI numbering starts at the N terminus of the signal peptide.
Epitope and paratope residues were assigned within a 4-Å contact distance cutoff using the CCP4 program CONTACT. The epitope area was calculated with the CCP4 program AREA. The buried surface area of binding residues was calculated with the program MOE (47
). Structural overlays of equivalent Cα atoms in the Sema domain (residues 40–515; PDB codes 1SHY, 4K3J, 2UZX, and 2UZY) were performed with COOT. Molecular graphics were generated with PyMol (PyMOL Molecular Graphics System, Version 1.4.1, Schrödinger, LLC) and MOE. The atomic coordinates and structure factors for the amivantamab anti-MET Fab–MET Sema-PSI complex were deposited in the RCSB PDB (accession code 6WVZ).
HCC827-HGF xenograft model
Female SCID Beige mice CB17.B6-Prkdcscid Lystbg/Crl (Charles River) bearing established subcutaneous HCC827-HGF tumors were randomized 13 days post inoculation (day 1). Individual tumor volumes ranged from 144 to 221 mm3; mean tumor volume ranged from 180 to 184 mm3. PBS and amivantamab (10 mg/kg) were dosed i.p. biweekly for 3 weeks. Crizotinib (30 mg/kg), erlotinib (25 mg/kg), crizotinib (30 mg/kg) and erlotinib (25 mg/kg), and vehicle controls (0.5% carboxymethyl cellulose in sterile water and 1% carboxymethyl cellulose in 0.1% Tween 80) were dosed daily p.o. for 3 weeks. Subcutaneous tumors were measured twice weekly as the mean tumor volume (mm3 ± standard error of the mean [SEM]). To calculate the percent tumor growth inhibition (%TGI) for group A versus group B, the tumor volumes were log transformed, where A = treated and B = control. The difference between these transformed values was taken at day 1 versus the designated day. Means were taken and converted by anti-log to numerical scale. Percentage TGIs were then calculated as (1 − A/B) × 100%. In vivo experiment was reviewed and approved by the Charles River Laboratories Institutional Animal Care and Use Committee and was done in accordance with the Guide for Care and Use of Laboratory Animals.
“Amivantamab”. Drug Information Portal. U.S. National Library of Medicine.
Clinical trial number NCT02609776 for “Study of Amivantamab, a Human Bispecific EGFR and cMet Antibody, in Participants With Advanced Non-Small Cell Lung Cancer (CHRYSALIS)” at ClinicalTrials.gov
PLAIN F 1423758-00-2 WITHOUT RADIO LABELC18 H23 F N4 O8, 441.4L-Glutamic acid, N-[[[(1S)-1-carboxy-5-[[[6-(fluoro-18F)-3-pyridinyl]carbonyl]amino]pentyl]amino]carbonyl]-2-(3-{1-carboxy-5-[(6-[18F]fluoro-pyridine-3-carbonyl) amino]-pentyl}ureido)-pentanedioic acid
For positron emission tomography imaging of prostate-specific membrane antigen-positive lesions in men with prostate cancer
For positron emission tomography (PET) of prostatespecific membrane antigen (PSMA) positive lesions in men with prostate cancer: • with suspected metastasis who are candidates for initial definitive therapy. • with suspected recurrence based on elevated serum prostate-specific antigen (PSA) level.
Originator Johns Hopkins University School of Medicine
Class Amides; Carboxylic acids; Fluorinated hydrocarbons; Imaging agents; Pyridines; Radiopharmaceutical diagnostics; Radiopharmaceuticals; Small molecules; Urea compounds
Mechanism of ActionPositron-emission tomography enhancers
Orphan Drug StatusNo
MarketedProstate cancer
28 May 2021Registered for Prostate cancer (Diagnosis) in USA (IV) – First global approval
28 May 2021Adverse events data from phase III CONDOR and phase II/III OSPREY trials in prostate cancer released by Lantheus Holdings
27 May 2021Lantheus Holdings intends to launch Fluorine-18 DCFPyL in USA at end of 2021
PYLARIFY contains fluorine 18 (F 18), radiolabeled prostate-specific membrane antigen inhibitor imaging agent. Chemically piflufolastat F 18 is 2-(3-{1-carboxy-5-[(6-[18F]fluoro-pyridine-3-carbonyl) amino]-pentyl}ureido)-pentanedioic acid. The molecular weight is 441.4 and the structural formula is:
The chiral purity of the unlabeled piflufolastat F 18 precursor is greater than 99% (S,S). PYLARIFY is a sterile, non-pyrogenic, clear, colorless solution for intravenous injection. Each milliliter contains 37 to 2,960 MBq (1 to 80 mCi) piflufolastat F 18 with ≤0.01 µg/mCi of piflufolastat at calibration time and date, and ≤ 78.9 mg ethanol in 0.9% sodium chloride injection USP. The pH of the solution is 4.5 to 7.0. PYLARIFY has a radiochemical purity of at least 95% up to 10 hours following end of synthesis, and specific activity of at least 1000 mCi/µmol at the time of administration.
PYLARIFY contains fluorine 18 (F 18), radiolabeled prostate-specific membrane antigen inhibitor imaging agent. Chemically piflufolastat F 18 is 2-(3-{1-carboxy-5-[(6-[18F]fluoro-pyridine-3-carbonyl)amino]-pentyl}ureido)-pentanedioic acid. The molecular weight is 441.4 and the structural formula is:
The chiral purity of the unlabeled piflufolastat F 18 precursor is greater than 99% (S,S).
PYLARIFY is a sterile, non-pyrogenic, clear, colorless solution for intravenous injection. Each milliliter contains 37 to 2,960 MBq (1 to 80 mCi) piflufolastat F 18 with ≤0.01 μg/mCi of piflufolastat at calibration time and date, and ≤ 78.9 mg ethanol in 0.9% sodium chloride injection USP. The pH of the solution is 4.5 to 7.0.
PYLARIFY has a radiochemical purity of at least 95% up to 10 hours following end of synthesis, and specific activity of at least 1000 mCi/μmol at the time of administration.
Physical Characteristics
PYLARIFY is radiolabeled with fluorine 18 (F 18), a cyclotron produced radionuclide that decays by positron emission to stable oxygen 18 with a half-life of 109.8 minutes. The principal photons useful for diagnostic imaging are the coincident pair of 511 keV gamma photons, resulting from the interaction of the emitted positron with an electron (Table 3).
Table 3: Principal Radiation Produced from Decay of Fluorine 18
It is an important therapeutic intervention for Covid-19 patients with cardiac co-morbidities and also for reducing proinflammatory cytokines
The Council of Scientific & Industrial Research (CSIR), and Laxai Life Sciences Pvt. Ltd. Hyderabad, have obtained approval from the Drug Controller General of India (DCGI) to undertake a two-arm phase-II clinical trial of the drug Colchicine for Covid-19 treatment.
The partner CSIR institutes in this important clinical trial are the CSIR-Indian Institute of Chemical Technology (IICT), Hyderabad and CSIR-Indian Institute of Integrative Medicine (IIIM), Jammu.
According to Ram Vishwakarma, advisor to DG-CSIR, colchicine, in combination with standard of care, will be an important therapeutic intervention for Covid-19 patients with cardiac co-morbidities and also for reducing proinflammatory cytokines, leading to faster recovery.
A number of global studies have confirmed now that cardiac complications during the course of Covid-19 infections and post-covid syndrome are leading to the loss of many lives, and it is essential to look for new or repurposed drugs.
VAMI MADDIPATLA
CHAIRMAN AND MD, LAXAI
A visionary & an entrepreneur with 17 years of experience in technology and bio-pharma industries. Founder and ex-CEO of LAXAI Pharma Ltd – a clinical data services company based in NJ, USA. Past employment: Pfizer, Wyeth Pharmaceuticals, Johnson & Johnson and Deloitte.
Vamsi provides a unique blend of operational and financial experience – along with a strong and expansive network of key influencers, industry experts and financial partners. He delivers a visionary understanding of client challenges and opportunities, and the instinctive ability to facilitate collaboration between the right people to turn strategic concepts into actionable plans – and, ultimately, into business results.
Dr S Chandrasekhar (Director CSIR-IICT, Hyderabad) and Dr. DS Reddy (Director, CSIR-IIIM, Jammu), the two partner institutes from CSIR said that they were looking forward to the outcome of this Phase II clinical efficacy trial on Colchicine, which may lead to life-saving intervention in the management of hospitalised patients.
Dr S Chandrasekhar (Director CSIR-IICT, Hyderabad)
Dr. DS Reddy (Director, CSIR-IIIM, Jammu)
India is one of the largest producers of this key drug and if successful, it will be made available to the patients at an affordable cost.
According to Ram Upadhayay, CEO, Laxai the enrollment of patients has already begun at multiple sites across India and the trial is likely to be completed in the next 8-10 weeks.
The drug can be made available to the large population of India based on the results of this trial and regulatory approval, he added.
Recent clinical studies have reported in leading medical journals about colchicine being associated with a significant reduction in the rates of recurrent pericarditis, post-pericardiotomy syndrome, and peri-procedural atrial fibrillation following cardiac surgery and atrial fibrillation ablation, according to a release.
Ram Upadhayaya, PhD
Chief Executive Officer, LAXAI
Ram Upadhayaya, CEO of Laxai Life Sciences, brings with him more than two decades of R&D experience spanning both academia and industry. A Ph. D in synthetic organic Chemistry, Ram has held key positions with leading international drug discovery organizations such as Bioimics AB Sweden, and Lupin India. Apart from his industrial background, Ram has been deeply associated with academic research. He was associated with Institute of Molecular Medicine, India as Principal Scientist as well as Uppsala University, Sweden in the capacity of Assistant Professor (Forskare). During these stints he significantly contributed to the development of novel therapeutics against infectious diseases such as AIDS and TB.
Ram has 10 international patents to his credit and has authored 25 peer reviewed publications. He is concurrently a consultant to the scientific advisory committee of the Principal Scientific Advisor, Government of India.
Raghava Reddy Kethiri, PhD, LAXAI
Chief Scientific Officer
25+ years of experience at various leadership positions in Biotech, CRO and Universities; Ex Karlsruhe Institute of Technology (KIT), Technical University of Dresden (TUD), JADO Technologies , Dresden, Germany, Jubilant Biosys, India
Delivered several leads, optimised leads and PCCs/DCs across Oncology, Pain, CNS, MD and Antibacterial therapeutics areas for global pharmaceutical companies. Co-Inventor of two clinical candidates ASN-001 ( NCT 02349139) for Metastatic Castration Resistant Prostrate Cancer & ASN-007 (NCT 03415126) for metastatic KRAS, NRAS & HRAS mutated solid tumors. Co-authored over 60 publications/patents (US/EU/Indian)
Colchicine
CAS Registry Number: 64-86-8
CAS Name:N-[(7S)-5,6,7,9-Tetrahydro-1,2,3,10-tetramethoxy-9-oxobenzo[a]heptalen-7-yl]acetamideMolecular Formula: C22H25NO6Molecular Weight: 399.44Percent Composition: C 66.15%, H 6.31%, N 3.51%, O 24.03%
Literature References: A major alkaloid of Colchicum autumnale L., Liliaceae. Extraction procedure: Chemnitius, J. Prakt. Chem. [II] 118, 29 (1928); F. E. Hamerslag, Technology and Chemistry of Alkaloids (New York, 1950) pp 66-80. Structure: Dewar, Nature155, 141 (1945); King et al.,Acta Crystallogr.5, 437 (1952); Horowitz, Ullyot, J. Am. Chem. Soc.74, 487 (1952). Crystal structure: L. Lessinger, T. N. Margulis, Acta Crystallogr.B34, 578 (1978). Total synthesis: Schreiber et al.,Helv. Chim. Acta44, 540 (1961); Van Tamelen et al.,Tetrahedron14, 8 (1961); Nakamura, Chem. Pharm. Bull.8, 843 (1960); Sunagawa et al.,ibid.9, 81 (1961); 10, 281 (1962); Scott et al.,Tetrahedron21, 3605 (1965); Woodward, Harvey Lectures, Ser. 59 (Academic Press, New York, 1965) p 31; Kotani et al.,Chem. Commun.1974, 300; D. A. Evans et al.,J. Am. Chem. Soc.103, 5813 (1981). Biosynthesis: Leete, Tetrahedron Lett.1965, 333; Battersby et al.,J. Chem. Soc.1964, 4257; Hill, Unrau, Can. J. Chem.43, 709 (1965). Tubulin-binding activity: J. M. Andreu, S. N. Timasheff, Proc. Natl. Acad. Sci. USA79, 6753 (1982). Toxicity: S. J. Rosenbloom, F. C. Ferguson, Toxicol. Appl. Pharmacol.13, 50 (1968); R. P. Beliles, ibid.23, 537 (1972). Clinical evaluations in cirrhosis of the liver: M. M. Kaplan et al.,N. Engl. J. Med.315, 1448 (1986); D. Kershenobich et al.,ibid.318, 1709 (1988). Bibliography of early literature: Eigsti, Lloydia10, 65 (1947). Monograph: O. J. Eigsti, P. Dustin, Jr., Colchicine in Agriculture, Medicine, Biology and Chemistry (Iowa State College Press, Ames, Iowa, 1955). Reviews: Fleming, Selected Organic Syntheses (John Wiley, London, 1973) pp 183-207; G. Lagrue et al.,Ann. Med. Interne132, 496-500 (1981); F. D. Malkinson, Arch. Dermatol.118, 453-457 (1982). Comprehensive description: D. K. Wyatt et al.,Anal. Profiles Drug Subs.10, 139-182 (1981). Properties: Pale yellow scales or powder, mp 142-150°. Darkens on exposure to light. Has been crystallized from ethyl acetate, pale yellow needles, mp 157°. [a]D17 -429° (c = 1.72). [a]D17 -121° (c = 0.9 in chloroform). pK at 20°: 12.35; pH of 0.5% soln: 5.9. uv max (95% ethanol): 350.5, 243 nm (log e 4.22; 4.47). One gram dissolves in 22 ml water, 220 ml ether, 100 ml benzene; freely sol in alcohol or chloroform. Practically insol in petr ether. Forms two cryst compds with chloroform, B.CHCl3 or B.2CHCl3, which do not give up their chloroform unless heated between 60 and 70° for considerable time. LD50 in rats (mg/kg): 1.6 i.v. (Rosenbloom, Ferguson); in mice (mg/kg): 4.13 i.v. (Beliles).
Melting point: mp 142-150°; mp 157°pKa: pK at 20°: 12.35; pH of 0.5% soln: 5.9Optical Rotation: [a]D17 -429° (c = 1.72); [a]D17 -121° (c = 0.9 in chloroform)Absorption maximum: uv max (95% ethanol): 350.5, 243 nm (log e 4.22; 4.47)
Toxicity data: LD50 in rats (mg/kg): 1.6 i.v. (Rosenbloom, Ferguson); in mice (mg/kg): 4.13 i.v. (Beliles)Use: In research in plant genetics (for doubling chromosomes).Therap-Cat: Gout suppressant. Treatment of Familial Mediterranean Fever.Therap-Cat-Vet: Has been used as an antineoplastic.Keywords: Antigout.
Here, we describe a concise, enantioselective, and scalable synthesis of (−)-colchicine (9.2% overall yield, >99% ee). Moreover, we have also achieved the first syntheses of (+)-demecolcinone and metacolchicine, and determined their absolute configurations. The challenging tricyclic 6-7-7 core of colchicinoids was efficiently introduced using an intramolecular oxidopyrylium-mediated [5 + 2] cycloaddition reaction. Notably, the synthesized colchicinoid 23 exhibited potent inhibitory activity toward the cell growth of human cancer cell lines (IC50 = ∼3.0 nM), and greater inhibitory activity towards microtubule assembly than colchicine, making it a promising lead in the search for novel anticancer agents.
Enantioselective total synthesis of (−)- and (+)-colchicine
The synthesis began with the transition-metal-catalyzed C–H bond functionalization of 7 with 14 (Scheme 1). Inspired by Li’s seminal work,18 we applied the strategy to compound 7. Pleasingly, after optimization, we successfully generated the N-sulfonyl imine in situ by reaction of 7 with TsNH2 (15) in the presence of anhydrous CuSO4 in THF. Furthermore, subsequent treatment of this imine with [RhCp*Cl2]2 (1 mol%), AgSbF6 (4 mol%), NaOAc (2.0 equiv.), and 14 (2.0 equiv.) at 80 °C afforded ortho-olefinated benzaldehyde 16 in good yield (90% on a 0.5 g scale; 70% on a 5.0 g scale). This modified catalytic C–H bond activation involved a transient directing group.19
Scheme 1 Enantioselective synthesis of (−)-colchicine and (+)-colchicine.
Recently one of my relatives have fallen ill and was prescribed with some colchicine. Looking at the structure of the molecule, and with nothing much to do, I decided to put my retrosynthetic skills to the test. Here is a picture of my thought process:
Is there a better way to design a synthesis for this compound using the disconnection method.
From 11b, a Birch reduction is carried out to give the qunione 10b. A rearrangement of the ketone with methanediazonium gives 9b. A dihydroxylation with a peroxy acid and subsequent addition of water gives 8b. A double dehydration reaction with sulfuric acid, coupled with the protection of the ketone with propan-1,3-diol gives the seven-membered quinone 7b. A Heck reaction (or Ullmann reaction) with 7a with a palladium catalyst yields 6. (The protection group is thereafter labelled “PG”) Friedel-Crafts acylation with ethanoyl chloride yields 5 (although on second thoughts, I should have done the acylation from 7a from the start). A Michael addition is then carried out with BuLiBuLi to lithiate the ketone to give the terminal imine 4. Since this terminal imine is unstable, a mild reducing agent converts the imine to the amine 3. The ketone is then removed by addition of dithiol and subsequently reduced by Raney nickel to form 2. Finally, a simple condensation reaction between the amine and acetic anhydride, followed by deprotection of the ketone using an acid, yields the final product colchicine, 1.
Colchicine has a narrow therapeutic index and overdosing is therefore a significant risk. Common side effects of colchicine include gastrointestinal upset, particularly at high doses.[5] Severe side effects may include low blood cells and rhabdomyolysis, and the medication can be deadly in overdose.[1] It is not clear whether colchicine is safe for use during pregnancy, but its use during breastfeeding appears to be safe.[1][6] Colchicine works by decreasing inflammation via multiple mechanisms.[7]
Colchicine, in the form of the autumn crocus (Colchicum autumnale), has been used as early as 1500 BC to treat joint swelling.[8] It was approved for medical use in the United States in 1961.[9] It is available as a generic medication in the United Kingdom.[6] In 2017, it was the 201st-most commonly prescribed medication in the United States, with more than two million prescriptions.[10][11]
Medical uses
Gout
Colchicine is an alternative for those unable to tolerate NSAIDs in gout.[12] At high doses, side effects (primarily gastrointestinal upset) limit its use.[13][14] At lower doses, it is well tolerated.[13][15][16][17] One review found low-quality evidence that low-dose colchicine (1.8 mg in one hour or 1.2 mg per day) reduced gout symptoms and pain, whereas high-dose colchicine (4.8 mg over 6 hours) was effective against pain, but caused more severe side effects, such as diarrhea, nausea or vomiting.[16]
For treating gout symptoms, colchicine is used orally with or without food, as symptoms first appear.[18] Subsequent doses may be needed if symptoms worsen.[18][16] There is preliminary evidence that daily colchicine (0.6 mg twice daily) was effective as a long-term prophylaxis when used with allopurinol to reduce the risk of increased uric acid levels and acute gout flares,[2] although adverse gastrointestinal effects may occur.[19]
Other conditions
Colchicine is also used as an anti-inflammatory agent for long-term treatment of Behçet’s disease.[20] It appears to have limited effect in relapsing polychondritis, as it may only be useful for the treatment of chondritis and mild skin symptoms.[21] It is a component of therapy for several other conditions, including pericarditis, pulmonary fibrosis, biliary cirrhosis, various vasculitides, pseudogout, spondyloarthropathies, calcinosis, scleroderma, and amyloidosis.[20][22][23] Research regarding the efficacy of colchicine in many of these diseases has not been performed.[23] It is also used in the treatment of familial Mediterranean fever,[20] in which it reduces attacks and the long-term risk of amyloidosis.[24]
Colchicine is effective for prevention of atrial fibrillation after cardiac surgery.[25] Potential applications for the anti-inflammatory effect of colchicine have been studied with regard to atherosclerosis and chronic coronary disease (e.g., stable ischemic heart disease).[26] In people with recent myocardial infarction (recent heart attack), it has been found to reduce risk of future cardiovascular events. Its clinical use may grow to include this indication.[27][28]
Colchicine is also being studied in clinical trials for possible effects on COVID-19.[29][30]
Contraindications
Long-term (prophylactic) regimens of oral colchicine are absolutely contraindicated in people with advanced kidney failure (including those on dialysis).[18] About 10-20 percent of a colchicine dose is excreted unchanged by the kidneys; it is not removed by hemodialysis. Cumulative toxicity is a high probability in this clinical setting, and a severe neuromyopathy may result. The presentation includes a progressive onset of proximal weakness, elevated creatine kinase, and sensorimotor polyneuropathy. Colchicine toxicity can be potentiated by the concomitant use of cholesterol-lowering drugs.[18]
Adverse effects
Deaths – both accidental and intentional – have resulted from overdose of colchicine.[18] Typical side effects of moderate doses may include gastrointestinal upset, diarrhea, and neutropenia.[13] High doses can also damage bone marrow, lead to anemia, and cause hair loss. All of these side effects can result from inhibition of mitosis,[31] which may include neuromuscular toxicity and rhabdomyolysis.[18]
Toxicity
According to one review, colchicine poisoning by overdose (range of acute doses of 7 to 26 mg) begins with a gastrointestinal phase occurring 10–24 hours after ingestion, followed by multiple organ dysfunction occurring 24 hours to 7 days after ingestion, after which the affected person either declines into multi-organ failure or recovers over several weeks.[32]
Colchicine can be toxic when ingested, inhaled, or absorbed in the eyes.[13] Colchicine can cause a temporary clouding of the cornea and be absorbed into the body, causing systemic toxicity. Symptoms of colchicine overdose start 2 to 24 hours after the toxic dose has been ingested and include burning in the mouth and throat, fever, vomiting, diarrhea, and abdominal pain.[18] This can cause hypovolemic shock due to extreme vascular damage and fluid loss through the gastrointestinal tract, which can be fatal.[32][33]
No specific antidote for colchicine is known, but supportive care is used in cases of overdose. In the immediate period after an overdose, monitoring for gastrointestinal symptoms, cardiac dysrhythmias, and respiratory depression is appropriate,[31] and may require gastrointestinal decontamination with activated charcoal or gastric lavage.[32][33]
Mechanism of toxicity
With overdoses, colchicine becomes toxic as an extension of its cellular mechanism of action via binding to tubulin.[32] Cells so affected undergo impaired protein assembly with reduced endocytosis, exocytosis, cellular motility, and interrupted function of heart cells, culminating in multi-organ failure.[7][32]
Epidemiology
In the United States, there are several hundred recorded cases of colchicine toxicity annually; approximately 10% of which end with serious morbidity or mortality. Many of these cases are intentional overdoses, but others were accidental; for example, if the drug was not dosed appropriately for kidney function. Most cases of colchicine toxicity occur in adults. Many of these adverse events resulted from the use of intravenous colchicine.[23]
In gout, inflammation in joints results from the precipitation of circulating uric acid, exceeding its solubility in blood and depositing as crystals of monosodium urate in and around synovial fluid and soft tissues of joints.[7] These crystal deposits cause inflammatory arthritis, which is initiated and sustained by mechanisms involving various proinflammatory mediators, such as cytokines.[7] Colchicine accumulates in white blood cells and affects them in a variety of ways: decreasing motility, mobilization (especially chemotaxis) and adhesion.[23]
Under preliminary research are various mechanisms by which colchicine may interfere with gout inflammation:
inhibits microtubule polymerization by binding to its constitutive protein, tubulin[7]
as availability of tubulin is essential to mitosis, colchicine may inhibit mitosis[7]
inhibits activation and migration of neutrophils to sites of inflammation[18]
Generally, colchicine appears to inhibit multiple proinflammatory mechanisms, while enabling increased levels of anti-inflammatory mediators.[7] Apart from inhibiting mitosis, colchicine inhibits neutrophil motility and activity, leading to a net anti-inflammatory effect, which has efficacy for inhibiting or preventing gout inflammation.[7][18]
The plant source of colchicine, the autumn crocus (Colchicum autumnale), was described for treatment of rheumatism and swelling in the Ebers Papyrus (circa 1500 BC), an Egyptian medical papyrus.[34] It is a toxic alkaloid and secondary metabolite.[13][35][18]Colchicum extract was first described as a treatment for gout in De Materia Medica by Pedanius Dioscorides, in the first century AD. Use of the bulb-like corms of Colchicum to treat gout probably dates to around 550 AD, as the “hermodactyl” recommended by Alexander of Tralles. Colchicum corms were used by the Persian physician Avicenna, and were recommended by Ambroise Paré in the 16th century, and appeared in the London Pharmacopoeia of 1618.[36][23]Colchicum use waned over time, likely due to the severe gastrointestinal side effects preparations caused. In 1763, Colchicum was recorded as a remedy for dropsy (now called edema) among other illnesses.[23]Colchicum plants were brought to North America by Benjamin Franklin, who had gout himself and had written humorous doggerel about the disease during his stint as United States Ambassador to France.[37]
Colchicine was first isolated in 1820 by the French chemists P. S. Pelletier and J. B.Caventou.[38] In 1833, P. L. Geiger purified an active ingredient, which he named colchicine.[39] It quickly became a popular remedy for gout.[23] The determination of colchicine’s structure required decades, although in 1945, Michael Dewar made an important contribution when he suggested that, among the molecule’s three rings, two were seven-member rings.[40] Its pain-relieving and anti-inflammatory effects for gout were linked to its ability to bind with tubulin.
An unintended consequence of the 2006 U.S. Food and Drug Administration (FDA) safety program called the Unapproved Drugs Initiative—through which the FDA sought more rigorous testing of efficacy and safety of colchicine and other unapproved drugs[41]—was a price increase of 2000 percent [42] for “a gout remedy so old that the ancient Greeks knew about its effects.”[42] Under Unapproved Drugs Initiative small companies like URL Pharma, a Philadelphia drugmaker, were rewarded with licenses for testing of medicines like colchicine. In 2009, the FDA reviewed a New Drug Application for colchicine submitted by URL Pharma. URL Pharma did the testing, gained FDA formal approval, and was granted rights over colchicine. With this monopoly pricing power, the price of colchicine increased.
In 2012 Asia’s biggest drugmaker, Takeda Pharmaceutical Co., acquired URL Pharma for $800 million including the rights to colchicine (brand name Colcrys) earning $1.2 billion in revenue by raising the price even more.[42]
Oral colchicine had been used for many years as an unapproved drug with no FDA-approved prescribing information, dosage recommendations, or drug interaction warnings.[43] On July 30, 2009, the FDA approved colchicine as a monotherapy for the treatment of three different indications (familial Mediterranean fever, acute gout flares, and for the prophylaxis of gout flares[43]), and gave URL Pharma a three-year marketing exclusivity agreement[44] in exchange for URL Pharma doing 17 new studies and investing $100 million into the product, of which $45 million went to the FDA for the application fee. URL Pharma raised the price from $0.09 per tablet to $4.85, and the FDA removed the older unapproved colchicine from the market in October 2010, both in oral and intravenous forms, but allowed pharmacies to buy up the older unapproved colchicine.[45]Colchicine in combination with probenecid has been FDA-approved before 1982.[44]
July 29, 2009, colchicine won FDA approval in the United States as a stand-alone drug for the treatment of acute flares of gout and familial Mediterranean fever.[46][47] It had previously been approved as an ingredient in an FDA-approved combination product for gout. The approval was based on a study in which two doses (1.2 mg and 0.6 mg) an hour apart were as effective as higher doses in combating the acute flare of gout.[17]
As a drug antedating the FDA, colchicine was sold in the United States for many years without having been reviewed by the FDA for safety and efficacy. The FDA reviewed approved colchicine for gout flares, awarding Colcrys a three-year term of market exclusivity, prohibiting generic sales, and increasing the price of the drug from $0.09 to $4.85 per tablet.[48][49][50]
Numerous consensus guidelines, and previous randomized controlled trials, had concluded that colchicine is effective for acute flares of gouty arthritis. However, as of 2006, the drug was not formally approved by the FDA, owing to the lack of a conclusive randomized control trial (RCT). Through the Unapproved Drugs Initiative, the FDA sought more rigorous testing of the efficacy and safety of colchicine and other unapproved drugs.[41] In exchange for paying for the costly testing, the FDA gave URL Pharma three years of market exclusivity for its Colcrys brand,[51] under the Hatch-Waxman Act, based in part on URL-funded research in 2007, including pharmacokinetic studies and a randomized control trial with 185 patients with acute gout.
In April 2010, an editorial in the New England Journal of Medicine said that the rewards of this legislation are not calibrated to the quality or value of the information produced, that no evidence of meaningful improvement to public health was seen, and that it would be less expensive for the FDA, the National Institutes of Health or large insurers to pay for trials themselves. Furthermore, the cost burden of this subsidy falls primarily on patients or their insurers.[52] In September 2010, the FDA ordered a halt to marketing unapproved single-ingredient oral colchicine.[53]
Colchicine patents expire on February 10, 2029.[54]
URL Pharma also received seven years of market exclusivity for Colcrys in the treatment of familial Mediterranean fever, under the Orphan Drug Law. URL Pharma then raised the price per tablet from $0.09 to $4.85 and sued to remove other versions from the market, increasing annual costs for the drug to U.S. state Medicaid programs from $1 million to $50 million. Medicare also paid significantly higher costs, making this a direct money-loser for the government. (In a similar case, thalidomide was approved in 1998 as an orphan drug for leprosy and in 2006 for multiple myeloma.)[52]
Trade names for colchicine are Colcrys or Mitigare which are manufactured as a dark– and light-blue capsule having a dose of 0.6 mg.[18][56] Colchicine is also prepared as a white, yellow, or purple pill (tablet) having a dose of 0.6 mg.[56]
Colchicine is typically prescribed to mitigate or prevent the onset of gout, or its continuing symptoms and pain, using a low-dose prescription of 0.6 to 1.2 mg per day, or a high-dose amount of up to 4.8 mg in the first 6 hours of a gout episode.[5][18][16] With an oral dose of 0.6 mg, peak blood levels occur within one to two hours.[35] For treating gout, the initial effects of colchicine occur in a window of 12 to 24 hours, with a peak within 48 to 72 hours.[18] It has a narrow therapeutic window, requiring monitoring of the subject for potential toxicity.[18] Colchicine is not a general pain relief drug, and is not used to treat pain in other disorders.[18]
Biosynthesis
According to laboratory research, the biosynthesis of colchicine involves the amino acidsphenylalanine and tyrosine as precursors. Giving radioactive phenylalanine-2-14C to C. byzantinum, another plant of the family Colchicaceae, resulted in its incorporation into colchicine.[57] However, the tropolone ring of colchicine resulted from the expansion of the tyrosine ring. Radioactive feeding experiments of C. autumnale revealed that colchicine can be synthesized biosynthetically from (S)-autumnaline. That biosynthesic pathway occurs primarily through a phenolic coupling reaction involving the intermediate isoandrocymbine. The resulting molecule undergoes O-methylation directed by S-adenosylmethionine. Two oxidation steps followed by the cleavage of the cyclopropane ring leads to the formation of the tropolone ring contained by N-formyldemecolcine. N-formyldemecolcine hydrolyzes then to generate the molecule demecolcine, which also goes through an oxidative demethylation that generates deacetylcolchicine. The molecule of colchicine appears finally after addition of acetyl-coenzyme A to deacetylcolchicine.[58][59]
Purification
Colchicine may be purified from Colchicum autumnale (autumn crocus) or Gloriosa superba (glory lily). Concentrations of colchicine in C. autumnale peak in the summer, and range from 0.1% in the flower to 0.8% in the bulb and seeds.[23]
Colchicine is widely used in plant breeding by inducing polyploidy in plant cells to produce new or improved varieties, strains and cultivars.[60] When used to induce polyploidy in plants, colchicine cream is usually applied to a growth point of the plant, such as an apical tip, shoot, or sucker. Seeds can be presoaked in a colchicine solution before planting. Since chromosome segregation is driven by microtubules, colchicine alters cellular division by inhibiting chromosome segregation during meiosis; half the resulting gametes, therefore, contain no chromosomes, while the other half contains double the usual number of chromosomes (i.e., diploid instead of haploid, as gametes usually are), and lead to embryos with double the usual number of chromosomes (i.e., tetraploid instead of diploid).[60] While this would be fatal in most higher animal cells, in plant cells it is not only usually well-tolerated, but also frequently results in larger, hardier, faster-growing, and in general more desirable plants than the normally diploid parents. For this reason, this type of genetic manipulation is frequently used in breeding plants commercially.[60]
When such a tetraploid plant is crossed with a diploid plant, the triploid offspring are usually sterile (unable to produce fertileseeds or spores), although many triploids can be propagated vegetatively. Growers of annual triploid plants not readily propagated vegetatively cannot produce a second-generation crop from the seeds (if any) of the triploid crop and need to buy triploid seed from a supplier each year. Many sterile triploid plants, including some trees, and shrubs, are becoming increasingly valued in horticulture and landscaping because they do not become invasive species and will not drop undesirable fruit and seed litter. In certain species, colchicine-induced triploidy has been used to create “seedless” fruit, such as seedless watermelons (Citrullus lanatus). Since most triploids do not produce pollen themselves, such plants usually require cross-pollination with a diploid parent to induce seedless fruit production.
The ability of colchicine to induce polyploidy can be also exploited to render infertile hybrids fertile, for example in breeding triticale (× Triticosecale) from wheat (Triticum spp.) and rye (Secale cereale). Wheat is typically tetraploid and rye diploid, with their triploid hybrid infertile; treatment of triploid triticale with colchicine gives fertile hexaploid triticale.[61]
^ Jump up to:abcd van Echteld I, Wechalekar MD, Schlesinger N, Buchbinder R, Aletaha D (August 2014). “Colchicine for acute gout”. The Cochrane Database of Systematic Reviews. 8 (8): CD006190. doi:10.1002/14651858.CD006190.pub2. PMID25123076.
^ Jump up to:ab Terkeltaub RA, Furst DE, Bennett K, Kook KA, Crockett RS, Davis MW (April 2010). “High versus low dosing of oral colchicine for early acute gout flare: Twenty-four-hour outcome of the first multicenter, randomized, double-blind, placebo-controlled, parallel-group, dose-comparison colchicine study”. Arthritis and Rheumatism. 62 (4): 1060–8. doi:10.1002/art.27327. PMID20131255.
^ Jump up to:abc Cocco G, Chu DC, Pandolfi S (December 2010). “Colchicine in clinical medicine. A guide for internists”. European Journal of Internal Medicine. 21 (6): 503–8. doi:10.1016/j.ejim.2010.09.010. PMID21111934.
^ Puéchal X, Terrier B, Mouthon L, Costedoat-Chalumeau N, Guillevin L, Le Jeunne C (March 2014). “Relapsing polychondritis”. Joint Bone Spine. 81 (2): 118–24. doi:10.1016/j.jbspin.2014.01.001. PMID24556284.
^ Portincasa P (2016). “Colchicine, Biologic Agents and More for the Treatment of Familial Mediterranean Fever. The Old, the New, and the Rare”. Current Medicinal Chemistry. 23 (1): 60–86. doi:10.2174/0929867323666151117121706. PMID26572612.
^ Nidorf SM, Fiolet AT, Mosterd A, Eikelboom JW, Schut A, Opstal TS, et al. (August 2020). “Colchicine in Patients with Chronic Coronary Disease”. The New England Journal of Medicine. 383(19): 1838–1847. doi:10.1056/NEJMoa2021372. PMID32865380.
^ Geiger, Ph. L. (1833) “Ueber einige neue giftige organische Alkalien” (On some new poisonous organic alkalis) Annalen der Pharmacie, 7 (3) : 269-280; colchicine is discussed on pages 274-276.
^“About Colcrys”. Colcrys. URL Pharma. Retrieved 11 September 2011.
^ Jump up to:ab Kesselheim AS, Solomon DH (June 2010). “Incentives for drug development–the curious case of colchicine”. The New England Journal of Medicine. 362 (22): 2045–7. doi:10.1056/NEJMp1003126. PMID20393164.
^ Leete E (1963). “The biosynthesis of the alkaloids of Colchicum: The incorporation of phenylalaline-2-C14 into colchicine and demecolcine”. J. Am. Chem. Soc. 85 (22): 3666–3669. doi:10.1021/ja00905a030.
^ Herbert, Richard B. (2001). “The biosynthesis of plant alkaloids and nitrogenous microbial metabolites”. Nat. Prod. Rep. 18 (1): 50–65. doi:10.1039/A809393H. PMID11245400.
^ Dewick PM (2009). Medicinal natural products: A biosynthetic approach. Wiley. pp. 360–362.
To treat schizophrenia in adults and certain aspects of bipolar I disorder in adults
LYBALVI is a combination of olanzapine, an atypical antipsychotic, and samidorphan (as samidorphan L-malate), an opioid antagonist.
Olanzapine is 2-methyl-4-(4-methyl-1-piperazinyl)-10H-thieno[2,3-b][1,5]benzodiazepine. The molecular formula of olanzapine is: C17H20N4S and the molecular weight is 312.44 g/mol. It is a yellow crystalline powder and has pKa values of 7.80 and 5.44. The chemical structure is:
Samidorphan L-malate is morphinan-3-carboxamide, 17-(cyclopropylmethyl)-4, 14-dihydroxy-6-oxo-, (2S)-2-hydroxybutanedioate. The molecular formula of samidorphan L-malate is C21H26N2O4 • C4H6O5 and the molecular weight is 504.54 g/mol. It is a white to off-white crystalline powder and has pKa values of 8.3 (amine) and 10.1 (phenol). The chemical structure is:
LYBALVI is intended for oral administration and is available as film-coated, bilayer tablets in the following strengths: 5 mg/10 mg, 10 mg/10 mg, 15 mg/10 mg, and 20 mg/10 mg of olanzapine and samidorphan (equivalent to 13.6 mg of samidorphan L-malate).
Inactive ingredients include colloidal silicon dioxide, crospovidone, lactose monohydrate, magnesium stearate, and microcrystalline cellulose. The film coating ingredients include hypromellose, titanium dioxide, triacetin, and color additives [iron oxide yellow (5 mg/10 mg); iron oxide yellow and iron oxide red (10 mg/10 mg); FD&C Blue No. 2/ indigo carmine aluminum lake (15 mg/10 mg); iron oxide red (20 mg/10 mg)].
to treat schizophrenia
alone for short-term (acute) or maintenance treatment of manic or mixed episodes that happen with bipolar I disorder
in combination with valproate or lithium to treat manic or mixed episodes that happen with bipolar I disorder
Olanzapine is an effective atypical antipsychotic that, like other antipsychotics, is associated with weight gain, metabolic dysfunction, and increased risk of type II diabetes.5,6 Samidorphan is a novel opioid antagonist structurally related to naltrexone, with a higher affinity for opioid receptors, more potent μ-opioid receptor antagonism, higher oral bioavailability, and a longer half-life, making it an attractive candidate for oral dosing.1,5,11 Although antipsychotic-induced weight gain is incompletely understood, it is thought that the opioid system plays a key role in feeding and metabolism, such that opioid antagonism may be expected to ameliorate these negative effects. Samidorphan has been shown in animal models and clinical trials to ameliorate olanzapine-induced weight gain and metabolic dysfunction.5,6
Samidorphan was first approved as a variety of fixed-dose combination tablets with olanzapine by the FDA on May 28, 2021, and is currently marketed under the trademark LYBALVI by Alkermes Inc.11
Samidorphan (INN, USAN) (developmental code names ALKS-33, RDC-0313), also known as 3-carboxamido-4-hydroxynaltrexone,[2] is an opioid antagonist that preferentially acts as an antagonist of the μ-opioid receptor (MOR). It is under development by Alkermes for the treatment of major depressive disorder and possibly other psychiatric conditions.[3]
However, it has attracted much more attention as part of the combination productALKS-5461 (buprenorphine/samidorphan), where samidorphan is combined with the mixed MOR weak partial agonist and κ-opioid receptor (KOR) antagonist buprenorphine, as an antidepressant. Buprenorphine has shown antidepressant effects in some human studies, thought to be because of its antagonist effects at the KOR, but has not been further developed for this application because of its MOR agonist effects and consequent abuse potential. By combining buprenorphine with samidorphan to block the MOR agonist effects, the combination acts more like a selective KOR antagonist, and produces only antidepressant effects, without typical MOR effects such as euphoria or substance dependence being evident.[6][7]
Samidorphan is also being studied in combination with olanzapine, as ALKS-3831 (olanzapine/samidorphan), for use in schizophrenia.[8] A Phase 3 study found that the addition of samidorphan to olanzapine significantly reduced weight gain compared to olanzapine alone.[9] The combination is now under review for approval by the US Food and Drug Administration.[10]
As such, samidorphan is primarily an antagonist, or extremely weak partial agonist of the MOR.[11][12] In accordance with its in vitro profile, samidorphan has been observed to produce some side effects that are potentially consistent with activation of the KOR such as somnolence, sedation, dizziness, and hallucinations in some patients in clinical trials at the doses tested.[13]
(A) Synthesis of 3-Carboxyamido-naltrexone 2[029] The triflate 11 of naltrexone was prepared according to the method of Wentland et al. (Bioorg. Med. Chem. Lett. 9, 183-187 (2000)), and the carboxamide 2 was prepared by the method described by Wentland et al. [(Bioorg. Med. Chem. Lett. ϋ, 623-626 (2001); and Bioorg. Med. Chem. Lett. 11, 1717-1721 (2001)] involving Pd-catalyzed carbonylation of the triflate 11 in the presence of ammonia and the Pd(O) ligand, DPPF ([l,l’-bis(diphenylρhosphino)ferrocene]) and DMSO.(B) Synthesis of 3-Carboxyamido-4-hydroxy-naltrexone derivative 3[030] Zinc dust (26 mg, 0.40 mmol) was added in portions to a solution of 2 (50 mg, 0.14 mmol) in HCl (37%, 0.2 mL) and AcOH (2 mL) at reflux. After heating at reflux for a further 15 min, the reaction was cooled by the addition of ice/water (10 mL) and basified (pH=9) with NH3/H2O, and the solution was extracted with EtOAc (3×10 mL). The organic extracts were washed with brine, dried, and concentrated. The residue was purified by column chromatography (SiO2, CH2Cl2, CH3OH : NH3/H2O = 15:1:0.01) to give compound 3 as a foam (25 mg, 50%). 1H NMR (CDC13) δl3.28(s, IH, 4-OH), 7.15(d, IH, J=8.1, H-2), 6.47(d, IH, J=8.4, H- 1), 6.10(br, IH, N-H), 4.35(br, IH, N-H), 4.04(dd,lH, J=I.8, 13.5, H-5), 3.11( d, IH, J=6), 2.99( d, IH, J=5.7), 2.94( s, IH), 2.86( d, IH, J= 6), 2.84-2.75(m, 2H), 2.65-2.61(m, 2H), 2.17-2.05(m, IH), 1.89-1.84(m, 2H), 0.85(m, IH), 0.56-0.50(m, 2H), 0.13-0.09(m, 2H). [α]D25= -98.4° (c=0.6, CH2Cl2). MS m/z (ESI) 371(MH+).
Opioid binding affinities were assessed for a series of cyclazocine analogues where the prototypic 8-OH substituent of cyclazocine was replaced by amino and substituted-amino groups. For μ and κ opioid receptors, secondary amine derivatives having the (2R,6R,11R)-configuration had the highest affinity. Most targets were efficiently synthesized from the triflate of cyclazocine or its enantiomers using Pd-catalyzed amination procedures.
In response to the unexpectedly high affinity for opioid receptors observed in a novel series of cyclazocine analogues where the prototypic 8-OH was replaced by a carboxamido group, we have prepared the corresponding 3-CONH2 analogues of morphine and naltrexone. High affinity (Ki=34 and 1.7 nM) for μ opioid receptors was seen, however, the new targets were 39- and 11-fold less potent than morphine and naltrexone, respectively.
Abstract
High-affinity binding to μ opioid receptors has been identified in a series of novel 3-carboxamido analogues of morphine and naltrexone.
References
^ Turncliff R, DiPetrillo L, Silverman B, Ehrich E (February 2015). “Single- and multiple-dose pharmacokinetics of samidorphan, a novel opioid antagonist, in healthy volunteers”. Clinical Therapeutics. 37 (2): 338–48. doi:10.1016/j.clinthera.2014.10.001. PMID25456560.
^ Wentland MP, Lu Q, Lou R, Bu Y, Knapp BI, Bidlack, JM (April 2005). “Synthesis and opioid receptor binding properties of a highly potent 4-hydroxy analogue of naltrexone”. Bioorganic & Medicinal Chemistry Letters. 15 (8): 2107–10. doi:10.1016/j.bmcl.2005.02.032. PMID15808478.
^ Hillemacher T, Heberlein A, Muschler MA, Bleich S, Frieling H (August 2011). “Opioid modulators for alcohol dependence”. Expert Opinion on Investigational Drugs. 20 (8): 1073–86. doi:10.1517/13543784.2011.592139. PMID21651459.
^ Clinical trial number NCT01366001 for “ALK33BUP-101: Safety and Pharmacodynamic Effects of ALKS 33-BUP Administered Alone and When Co-administered With Cocaine” at ClinicalTrials.gov
FDA APPROVED 4/15/2021, To prevent pregnancy Nextstellis
New Drug Application (NDA): 214154 Company: MAYNE PHARMALabel (PDF) Letter (PDF) ReviewLabel (PDF)DrospirenoneCAS Registry Number: 67392-87-4 CAS Name: (2¢S,6R,7R,8R,9S,10R,13S,14S,15S,16S)-1,3¢,4¢,6,7,8,9,10,11,12,13,14,15,16,20,21-Hexadecahydro-10,13-dimethylspiro[17H-dicyclopropa[6,7:15,16]cyclopenta[a]phenanthrene-17,2¢(5¢H)-furan]-3,5¢(2H)-dione Additional Names: 6b,7b,15b,16b-dimethylene-3-oxo-4-androstene-[17(b-1¢)-spiro-5¢]perhydrofuran-2¢-one; 6b,7b,15b,16b-dimethylen-3-oxo-17a-pregn-4-ene-21,17-carbolactone; dihydrospirorenone Manufacturers’ Codes: ZK-30595 Molecular Formula: C24H30O3Molecular Weight: 366.49Percent Composition: C 78.65%, H 8.25%, O 13.10% Literature References: Synthetic progestogen exhibiting antimineralocorticoid and antiandrogenic activity. Prepn: R. Wiechert et al.,DE2652761; eidem,US4129564 (both 1978 to Schering AG); D. Bittler et al.,Angew. Chem.94, 718 (1982). HPLC determn in human plasma: W. Krause, U. Jakobs, J. Chromatogr.230, 37 (1982). Pharmacological profile: P. Muhn et al.,Contraception51, 99 (1995). Review of synthesis: H. Laurent et al.,J. Steroid Biochem.19, 771-776 (1983); of pharmacology and clinical experience: W. Oelkers, Mol. Cell. Endocrinol.217, 255-261 (2004). Properties: mp 201.3°. [a]D22 -182° (c = 0.5 in chloroform). uv (methanol): 265 nm (e 19000). Melting point: mp 201.3° Optical Rotation: [a]D22 -182° (c = 0.5 in chloroform) Derivative Type: Mixture with ethinyl estradiolTrademarks: Angeliq (Schering AG); Yasmin (Schering AG)Literature References: Clinical trial as oral contraceptive: K. S. Parsey, A. Pong, Contraception61, 105 (2000); in treatment of menopausal symptoms: R. Schürmann et al., Climacteric7, 189 (2004). Therap-Cat: Progestogen. In combination with estrogen as oral contaceptive and in treatment of menopausal symptoms.Keywords: Progestogen; Contraceptive (Oral).SYNhttps://www.sciencedirect.com/science/article/abs/pii/S0039128X15002135
Abstract
A general methodology for the synthesis of different steroidal 17-spirolactones is described. This method uses lithium acetylide of ethyl propiolate as the three carbon synthon and the method was successfully applied for the process development of drospirenone.
The synthesis of drospirenone (27.4.12) is believed to have been described for the first time in Wiechert et al [79], with a total yield of approximately 2 to 3% via the pathway presented in Scheme 27.4.
Each compound produced after each reaction step was purified by column chromatography.
Androsta-5,15-diene-3-ol-17-one was methylenated at the 15,16-position (27.4.13) and reacted with organometallic reagent (3,3-dimethoxypropyl)lithium prepared from 3-bromo-1,1-dimethoxypropane (27.4.14) and lithium in THF to produce the tertiary alcohol (27.4.15), which on short-term reflux with toluenesulfonic acid in acetone transformed to cyclic 21,17-hemiacetal (27.4.16). Oppenanuer oxidation with aluminium isopropoxide in excess of cyclohexanone in toluene was brought to mild oxidation of both secondary alcohol groups, and the simultaneous isomerization of the 5,6 double bond to the 4,5 position produced the compound (27.4.17). The last was oxidized with Jones reagent—chromic trioxide in diluted sulfuric acid—producing conjugated diene-one (27.4.18). Corey methylenation of the obtained product with dimethyloxosulfonium methylide in DMSO containing sodium hydride produced the final compound, the desired drospirenone (27.4.12).
The following patents and publications [80-83], which differ slightly from one another, disclose similar processes for preparing drospirenone and are presented in Scheme 27.5.
In Scheme 27.5, drospirenone (27.4.12) is prepared by converting the key starting compound (27.4.19) into the corresponding chloride (27.4.20) via reaction with triphenylphosphine and tetrachloromethane under mild conditions. Reductive dechlorination with Zn in acetic acid in THF tetrahydrofuran produced 5-hydroxy-15β,16β-methylene-3β-pivaloyloxy-5β-androst-6-en-17-one (27.4.21). The pivaloyl protecting group of the last was removed with the mixture of potassium hydroxide and sodium perchlorate in THF/methanol mixture to produce the diol (27.4.22). Simmons–Smith cyclopropanation reaction was applied to this compound. For that purpose, solution of (27.4.22) in dimethyl Cellosolve was stirred at 80°C with zinc-copper couple and methylene iodide, which produced the desired compound (27.4.23). The compound (27.4.23) underwent ethinylation with propargyl alcohol using potassium methylate in THF as a base to produce the 1,4-butindiol derivative (27.4.24). The triple bond of the 1,4-butindiol derivative (27.4.24) was hydrogenated in aTHF/methanol/pyridine mixture in the presence of palladium on carbon to produce the 1,4-butanediol derivative (27.4.25). The obtained compound underwent oxidation–lactonization at 50°C using a solution of CrO3 in water and pyridine to produce the desired drospirenone (27.4.12).
Several other synthetic routes for the production of drospirenone have been proposed [84-96], one of which [96] is presented in Scheme 27.6.
According to Scheme 27.6, a mixture of the key starting ketodiol (27.4.26), synthesis of which was described previously [84], with ethyl propiolate in THF was added to a solution of lithium hexamethyldisilylamide to produce, after quenching with acetic acid and saturated ammonium chloride solution, ethinyl alcohol (27.4.27). This product was hydrogenated on H2-Pd/C catalyst to produce ethyl 4-hydroxybutanoate (27.4.28). The 3-hydroxy group in the obtained product was oxidized to the keto group with (2,2,6,6-tetramethyl-piperidin-1-yl)oxyl, resulting in the compound (27.4.29). Treatment of the last with potassium hydroxide in a methanol–water mixture affects both hydrolysis of the ester group and dehydration of 5-hydroxy substituent. Acidification of the resulting intermediate results in drospirenone (27.4.12).
Drospirenone is a unique synthetic progestogen derived from 17α-spirolactone; it has a pharmacological profile very similar to that of endogenous progesterone. Drospirenone prevents ovulation and is used in contraceptive pills; it is also used as a postmenopausal hormone replacement. Drospirenone provides reliable and well-tolerated contraception and effective treatment of menopause. It has progestational, antialdosterone, and antiandrogenic properties, but is devoid of any estrogenic, androgenic, glucocorticoid, antiglucocorticoid, and mineralocorticoid activities. The affinity of drospirenone for the mineralocorticoid receptor makes it an antagonist of aldosterone, which is not only important in the renin–angiotensin–aldosterone system, but also means it acts directly on the cardiovascular system. It is progestin with antimineralocorticoid property that acts to suppress gonadotropins. It is thus able to prevent excessive sodium loss and regulate blood pressure. Drospirenone slightly decreases body weight and blood pressure and shares many pharmacodynamic properties with progesterone [97-110].
PATENT
https://patents.google.com/patent/US8334375B2/enDrospirenone is a synthetic steroid with progestin, anti-mineral corticoid and anti androgen activity. Drospirenone is currently being used as a synthetic progestin in oral contraceptive formulations. A regioselective synthesis for drospirenone has been described (see e.g., Angew. Chem. 94, 1982, 718) that uses the 17 keto derivative (1) as a key intermediate.
The synthesis of intermediate (1) and the transformation of intermediate (1) into drospirenone has been described in, for example, U.S. Published Patent Application Nos. 2009/0023914; 20080207575; 2008/0200668; 2008/0076915, 20070049747, and 20050192450; U.S. Pat. Nos. 6,933,395; 6,121,465, and 4,129,564, European Patent No. 0 075 189 and PCT Publication No. WO 2006/061309, all of which are incorporated herein by reference. Many of these routes introduce the required C3 side chain in the 17 position of intermediate (1). These conversions are usually carried out with carbanions, such as propargylalcohol, trimethylsulfoxonium iodide, or the use of the anion generated from a suitably protected derivative of 1-bromopropionaldehyde. After oxidation of the 3-hydroxy substituent to a 3-keto group, and the oxidative formation of the 17-spirolactone, the 3-keto-5-hydroxy-17-spirolactone is transformed via acid catalysis into drospireneone. If the oxidation is performed under acidic conditions at elevated temperatures, the oxidation and elimination can be run without isolation of the intermediate products.Most of these procedures rely on the acid-catalyzed elimination of the 5-hydroxy group in the last step of the synthesis. It has been documented that 15,16-methylene-17-spirolactones are prone to undergo rearrangement to generate the inverted 17-spirolactone under mild acidic conditions (see, for example, Tetrahedron Letters, Vol. 27, No 45, 5463-5466) in considerable amounts. This isomer has very similar physical chemical properties, and typically requires chromatographic separation or repeated fractional recrystallizations to purify the product. This isomerization can make these approaches less desirable from an economical point of view.FIG. 1Experimental Example
A solution of compound (1) (5 g; 15.2 mmol) and tert-butyldimethyl (2-propynyloxy)silane (2.83 g, 16.7 mmol) in 75 ml of dry THF was added dropwise through an addition funnel to a precooled slurry of potassium tert-butoxide (8.49 g, 75.7 mmol) at −10 C. A thick white precipitate is formed during the addition and the resulting mixture was stirred for an hour at 0 C. TLC analysis (70% EtOAc/Hexanes) showed completion of the reaction and showed a less polar product. The reaction was quenched by the addition of ice water (100 ml) and neutralized by adding acetic acid (4.3 ml). The THF layer was separated and the aqueous layer was extracted with EtOAc (2×50 ml). The combined organic layers were washed with water (2×100 ml), brine (100 ml) and dried over anhydrous sodium sulfate. The solvent was removed under vacuum to afford compound (2a) (7.5 g, 99.2%) as a solid which was used in the next step without any purification.NMR (CDCl3) δ 0.139 (s, 6H, S1—CH3), 0.385 (m, 1H), 0.628 (m, 1H), 0.857 (s, 18-Me), 0.896 (s, 19-Me), 0.918 (s, 3H, Si—CH3), 0.927 (s, 6H, Si—CH3), 4.05 (s, 1H), 4.428 (s, 2H, —O—CH2) FTIR (ATR): 3311, 3017, 2929, 2858, 2270, 1058 cm−1
Compound (2a) (5 g, 9.98 mmol) was dissolved in 100 ml of ethyl acetate in a Parr hydrogenation bottle and was mixed with 10% palladium on charcoal (1 g, 0.09 mmol). This mixture was hydrogenated on a Parr apparatus at a pressure of 20 psi for 90 minutes. The catalyst was filtered and washed with ethyl acetate. The solvent was removed in vacuo to afford compound (3a) as a colorless foam (5.01 g, 99%).NMR (CDCl3) δ 0.0758 (s, 6H, Si—CH3), 0.283 (m, 1H), 0.628 (m, 1H), 0.856 (s, 18-Me), 0.893 (s, 19-Me), 0.918 (s, 9H, Si—CH3), 3.69 (m, 2H), 4.05 (s, 1H). FTIR (ATR): 3374, 3017, 2929, 2858, 1259, 1091, 1049, 835 cm−1
Chromium trioxide (4.95 g, 49.5 mmol) was added to a solution of pyridine (7.83 g, 99.05 mmol) in anhydrous dichloromethane (100 ml). The resulting mixture was stirred for 15 minutes during which time the color changed to burgundy. A solution of compound (1a) (5 g, 9.90 mmol) in 50 ml of dichloromethane was added and the mixture was stirred at room temperature for 6 h. The excess oxidizing agent was quenched by adding isopropanol. The reaction mixture was diluted with MTBE (50 ml) and was passed through a short pad of Celite. The solid was washed again with 2:1 MTBE-CH2Cl2 (50 ml x2). The solvent was removed in vacuo to give a residue which was dissolved in 100 ml of EtOAc, was washed with water, and dried over anhydrous sodium sulfate. The solvent was removed in vacuo to afford compound (4a) as a pale yellow foam (4.5 g, 90.3%).NMR (CDCl3) δ 0.08 (s, 6H, Si—CH3), 0.31 (m, 1H), 0.914 (s, 9H, Si—CH3), 0.931 (s, 6H, 18-Me, 19-Me) 3.70 (m, 2H). FTIR (ATR): 3399, 3022, 2950, 2929, 2862, 1708, 1649, 1259, 1041 cm−1
A solution of compound (4a) (5 g, 9.94 mmol) in 50 ml of MeOH was refluxed with NaOH (397 mg, 9.94 mmol) for 3 h. When the reaction was over, as shown by TLC, the reaction mixture was cooled to room temperature and added to ice cold water (150 ml). The mixture was extracted with ethyl acetate (3×50 mL). The combined EtOAc layers were washed with water (100 ml) brine (50 ml) and dried over sodium sulfate. The solvent was removed in vacuo to afford compound (5) as a colorless amorphous solid (4.5 g, 92%).NMR (CDCl3) δ 0.05 (s, 6H, Si—CH3), 0.296 (m, 1H), 0.886 (s, 9H, Si—CH3), 0.908 (s, 3H, 18-Me), 1.07 (s, 3H, 19-Me), 3.68 (m, 2-H), 5.95 (s, 1H). FTIR (ATR): 3450, 3009, 2950, 2858, 1653, 1603, 1095 cm−1
A solution of compound (5a) (5 g, 9.94 mmol) in 30 ml of acetone was cooled to −15 C as a 2.7M solution of Jones reagent (3.68 ml, 9.94 mmol) was added drop wise. The reaction mixture was stirred at 0 C for 2 h, during this time TLC showed completion of the reaction. The reaction was quenched by adding isopropanol and diluted with water. The reaction mixture was extracted with EtOAc. The combined EtOAc layers were washed with water, sat. NaHCO3 and brine. The EtOAc layers were dried over sodium sulfate and solvent was removed by vacuum to afford crude drospirenone as a pale yellow foam (3 g, 82%) Recrystallization from acetone-hexane gave 1.5 g of pure drospirenone as white solid.NMR (CDCl3) δ 0.0548 (m, 1H), 0.88 (m, 1H), 1.008 (s, 3H, 18-Me), 1.11 (s, 3H, 18-Me), 6.03 (s, 1H). FTIR (ATR): 3025, 2971, 2942, 1763, 1654, 1590, 1186 cm−1FIG. 2Experimental Example
Lithium hexamethyldisilylamide (LiHMDS) 1.0 M/THF (75.7 mL, 75.7 mmol) was introduced into a 500 mL, 3-neck flask equipped with an addition funnel and a pierced septa for the introduction of a thermocouple probe. The mixture was diluted with THF (25 mL). The solution was stirred (Teflon paddle) and chilled to an internal temperature of −72° C. A THF (75 mL) solution of ketodiol (5 g, 15.13 mmol) containing ethyl propiolate (3.07 mL, 30.26 mmol) was added dropwise over 1 hour while not allowing the temperature to rise above −65° C. Upon completion of the addition the mixture was stirred for 3 hrs while allowing the temperature to warm slowly to −60° C. Finally, the mixture was warned to −40° C. over 1 hour.The mixture was quenched through the addition of acetic acid (4.25 mL)/water (5.0 mL) followed by the addition of saturated ammonium chloride solution (100 mL). The mixture was stirred for 3 min and then transferred to a separatory funnel. The layers were separated and the upper, THF layer was diluted with ethyl acetate (75 mL). The organic phase was washed with water (3×100 mL) and brine (1×100 mL). All the aqueous washes were extracted with ethyl acetate (2×30 mL). The combined organic extract was dried over sodium sulfate, filtered, and evaporated in vacuo (45° C.) to afford a thick oil. Dichloromethane (ca. 35 mL) was added and evaporated in vacuo. The flask was cooled slightly and dichloromethane (33 ml) was added to give a solid mass. The solid was broken up and stirred until a homogeneous slurry was obtained. Hexanes (35 mL) was added slowly to the stirred mixture and the mixture was stored at 2-4 C overnight. The solid was filtered, washed with 30% dichloromethane/hexanes, and dried in vacuo at ambient temperature for 4 hours to give (2b) 5.86 g (90.4%) of a white powder.
Compound (2b) (5.0 g, 11.67 mmol) was dissolved in THF (50 mL) and 5% Pd/C (622 mg, 0.29 mmol Pd) was added and the mixture was shaken at 15 psi H2 for 2 hours. The mixture was diluted with ethyl acetate (25 mL) and filtered through a pad of Celite. The filter pad was washed with ethyl acetate (3×25 mL) and the filtrate was evaporated to dryness to afford 5.0 g (99.1%) of triol (7b) as a stable foam.
Compound (7a) (5.0 g, 11.56 mmol) was dissolved in dichloromethane (50 mL) and the solution was stirred vigorously and chilled to −15° C. (NaCl/ice) and TEMPO (45.16 mg, 0.29 mmol, 2.5 mol %) was added. The mixture was treated dropwise over about 15-20 min. with a mixture of sodium hypochlorite (12.5%) (11.17 mL, 23.12 mmol) in water (8.0 mL) containing potassium bicarbonate (833 mg, 8.32 mmol). The mixture was allowed to warm to 0 C for 1.25 hrs. Analysis of the reaction by TLC (60% EtOAc/hex) shows the appearance of a slightly less polar product (ΔRf=0.8 cm). The mixture was chilled to −5 C and was quenched through the dropwise addition (ca 10-15 min) of a water (15.0 mL) solution of sodium phosphate (1.27 g, 7.75 mmol) and sodium metabisulfite (1.10 g, 5.78 mmol). The layers were separated and the dichloromethane solution was washed with water (2×) and brine. All aqueous washes were extracted with additional dichloromethane (2×15 mL). The combined dichloromethane extract was dried over sodium sulfate, filtered, and evaporated to give 4.88 g (98.08%) of ketone (8b) as a stable foam.
Compound (8b) was added to a methanol (10 mL) solution containing 8.0 M KOH solution (6.3 mL, 50.36 mmol) preheated to 60 C. The solution was heated at reflux for 2.5 hours. The mixture was chilled in an ice bath and treated with acetic acid (36 mL) and water (5.0 mL). The solution was stirred at 50-60 C for 15 hours. The volatiles were evaporated in vacuo and the acetic acid solution was poured into cold water (150 mL) to give a white precipitate. The aqueous mixture was extracted with ethyl acetate (2×100 mL). The ethyl acetate extracts were washed with water (2×), saturated sodium bicarbonate solution, and brine. The combined ethyl acetate extract was dried over sodium sulfate. Evaporation of the solvent gave a yellow foam. Trituration of the foam with acetone/hexane followed by evaporation gave 4.27 g (92.62%) of a light yellow solid. Recrystallization of the solid from acetone/hexanes gave 3.07 g of drospirenone with an HPLC purity of 99.66%. Evaporation of the mother liquor and recrystallization of the residue affords an additional 0.54 g of slightly impure drospirenone.FIG. 3Experimental Example
Lithium hexamethyldisilylamide (LiHMDS) 1.0 M/THF (75.7 mL, 75.7 mmol) was introduced into a 500 mL, 3-neck flask equipped with an addition funnel and a pierced septa for the introduction of a thermocouple probe. The mixture was diluted with THF (25 mL). The solution was stirred (Teflon paddle) and chilled to an internal temperature of −72° C. A THF (75 mL) solution of ketodiol (5 g, 15.13 mmol) containing ethyl propiolate (3.07 mL, 30.26 mmol) was added dropwise over 1 hour while not allowing the temperature to rise above −65° C. Upon completion of the addition the mixture was stirred for 3 hrs while allowing the temperature to warm slowly to −60° C. Finally, the mixture was warmed to −40° C. over 1 hour.The mixture was quenched through the addition of acetic acid (4.25 mL)/water (5.0 mL) followed by the addition of saturated ammonium chloride solution (100 mL). The mixture was stirred for 3 min and then transferred to a separatory funnel. The layers were separated and the upper, THF layer was diluted with ethyl acetate (75 mL). The organic phase was washed with water (3×100 mL) and brine (1×100 mL). All the aqueous washes were extracted with ethyl acetate (2×30 mL). The combined organic extract was dried over sodium sulfate, filtered, and evaporated in vacuo (45° C.) to afford a thick oil. Dichloromethane (ca. 35 mL) was added and evaporated in vacuo. The flask was cooled slightly and dichloromethane (33 ml) was added to give a solid mass. The solid was broken up and stirred until a homogeneous slurry was obtained. Hexanes (35 mL) was added slowly to the stirred mixture and the mixture was stored at 2-4 C overnight. The solid was filtered, washed with 30% dichloromethane/hexanes, and dried in vacuo at ambient temperature for 4 hours to give (2c) 5.86 g (90.4%) of a white powder.
Propiolate adduct (2c) (5.86 g, 13.67 mmol) was suspended in dichloromethane (60 mL). The mixture was stirred vigorously and chilled to −15° C. (NaCl/ice) and TEMPO (54 mg, 0.35 mmol, 2.5 mol %) was added. The mixture was treated dropwise over about 15-20 min. with a mixture of sodium hypochlorite (12.5%) (13.2 mL, 27.34 mmol) in water (8.0 mL) containing potassium bicarbonate (985 mg, 9.84 mmol). During the addition of the hypochlorite solution, a 5-8 C temperature rise was observed and the mixture became yellow. The mixture was allowed to warm to at 0 C for 2 hrs. Analysis of the reaction by TLC (60% EtOAc/hex) shows the appearance of a slightly less polar product (ΔRf=0.8 cm). The mixture was chilled to −5 C and was quenched through the dropwise addition (ca 10-15 min) of a water (150 mL) solution of sodium phosphate (1.50 g, 9.16 mmol) and sodium metabisulfite (1.30 g, 6.84 mmol). Once again, a temperature rise of 5-8 C was observed and the yellow color was quenched. The layers were separated and the dichloromethane solution was washed with water (2×) and brine. All aqueous washes were extracted with additional dichloromethane (2×15 mL). The combined dichloromethane extract was dried over sodium sulfate and the bulk of the solvent was evaporated in vacuo. Upon the observation of solids in the mixture during the evaporation, the evaporation was discontinued and the residue in the flask diluted with MTBE (35 mL). While stirring, the mixture was slowly diluted with hexanes (35 mL). The mixture was then chilled in an ice bath for 30 min. The solid was filtered, washed with 25% MTBE/hexane, and dried to give intermediate (9c) (4.98 g, 85.31%) as a white solid.NMR (CDCl3) δ 0.462 (q, 1H), 0.699 (m, 1H), 0.924 (s, 18-Me), 0.952 (s, 19-Me), 1.338 (t, J=7 Hz, OCH2CH 3), 2.517 (d, 1H), 3.021 (d, 1H), 4.269 (t, OCH 2CH3) ppm. FTIR (ATR): 3493, 3252, 2948, 2226, 1697, 1241 cm−1.
Alkynyl ketone (9c) (5.37 g, 12.59 mmol) was dissolved in THF (27 mL) in a 250 mL shaker bottle. 5% Pd/C (670 mg, 2.5 mol %) was added to the solution and the mixture was shaken under a hydrogen pressure of 15 psi. Over approximately 30 min, there was observed a rapid up take of hydrogen. The pressure was continually adjusted to 15 psi until the uptake of hydrogen ceased and was shaken for a total of 1.5 hrs. The mixture was diluted with a small amount of methanol and filtered through Celite. The filter pad was washed with methanol (ca. 3×25 mL).NMR (CDCl3) δ 0.353 (q, 1H), 0.704 (m, 2H), 0.930 (s, 18-Me), 0.933 (s, 19-Me), 1.279 (t, J=7 Hz, OCH2CH 3), 2.480 (d, 1H), 2.672 (m, 2H), 3.981 (d, 1H), 4.162 (t, OCH 2CH3) ppm. FTIR (ATR): 3465, 2946, 1712, cm−1.
The filtrate containing compound (8c) described above, was added in one portion to a methanol (10 mL) solution containing 8.0 M KOH solution (6.3 mL, 50.36 mmol) preheated to 60 C. The solution was heated at reflux for 2.5 hours. The mixture was chilled in an ice bath and treated with acetic acid (36 mL) and water (5.0 mL). The solution was stirred at 50-60 C for 15 hours. The volatiles were evaporated in vacuo and the acetic acid solution was poured into cold water (150 mL) to give a white precipitate. The aqueous mixture was extracted with ethyl acetate (2×100 mL). The ethyl acetate extracts were washed with water (2×), saturated sodium bicarbonate solution, and brine. The combined ethyl acetate extract was dried over sodium sulfate. Evaporation of the solvent gave a yellow foam. Trituration of the foam with acetone/hexane followed by evaporation gave 4.27 g (92.62%) of a light yellow solid. Recrystallization of the solid from acetone/hexanes gave 3.07 g of drospirenone with an HPLC purity of 99.66%. Evaporation of the mother liquor and recrystallization of the residue affords an additional 0.54 g of slightly impure drospirenone.FIG. 4Experimental Example
Lithium hexamethyldisilylamide (LiHMDS) 1.0 M/THF (75.7 mL, 75.7 mmol) was introduced into a 500 mL, 3-neck flask equipped with an addition funnel and a pierced septa for the introduction of a theiniocouple probe. The mixture was diluted with THF (25 mL). The solution was stirred (Teflon paddle) and chilled to an internal temperature of −72° C. A THF (75 mL) solution of ketodiol (5 g, 15.13 mmol) containing ethyl propiolate (3.07 mL, 30.26 mmol) was added dropwise over 1 hour while not allowing the temperature to rise above −65° C. Upon completion of the addition the mixture was stirred for 3 hrs while allowing the temperature to warm slowly to −60° C. Finally, the mixture was warmed to −40° C. over 1 hour.The mixture was quenched through the addition of acetic acid (4.25 mL)/water (5.0 mL) followed by the addition of saturated ammonium chloride solution (100 mL). The mixture was stirred for 3 min and then transferred to a separatory funnel. The layers were separated and the upper, THF layer was diluted with ethyl acetate (75 mL). The organic phase was washed with water (3×100 mL) and brine (1×100 mL). All the aqueous washes were extracted with ethyl acetate (2×30 mL). The combined organic extract was dried over sodium sulfate, filtered, and evaporated in vacuo (45° C.) to afford a thick oil. Dichloromethane (ca. 35 mL) was added and evaporated in vacuo. The flask was cooled slightly and dichloromethane (33 ml) was added to give a solid mass. The solid was broken up and stirred until a homogeneous slurry was obtained. Hexanes (35 mL) was added slowly to the stirred mixture and the mixture was stored at 2-4 C overnight. The solid was filtered, washed with 30% dichloromethane/hexanes, and dried in vacuo at ambient temperature for 4 hours to give (2d) 5.86 g (90.4%) of a white powder.
Propiolate adduct (2d) (5.86 g, 13.67 mmol) was suspended in dichloromethane (60 mL). The mixture was stirred vigorously and chilled to −15° C. (NaCl/ice) and TEMPO (54 mg, 0.35 mmol, 2.5 mol %) was added. The mixture was treated dropwise over about 15-20 min. with a mixture of sodium hypochlorite (12.5%) (13.2 mL, 27.34 mmol) in water (8.0 mL) containing potassium bicarbonate (985 mg, 9.84 mmol). During the addition of the hypochlorite solution, a 5-8 C temperature rise was observed and the mixture became yellow. The mixture was allowed to warm to at 0 C for 2 hrs. Analysis of the reaction by TLC (60% EtOAc/hex) shows the appearance of a slightly less polar product (ΔRf=0.8 cm). The mixture was chilled to −5 C and was quenched through the dropwise addition (ca 10-15 min) of a water (15.0 mL) solution of sodium phosphate (1.50 g, 9.16 mmol) and sodium metabisulfite (1.30 g, 6.84 mmol). Once again, a temperature rise of 5-8 C was observed and the yellow color was quenched. The layers were separated and the dichloromethane solution was washed with water (2×) and brine. All aqueous washes were extracted with additional dichloromethane (2×15 mL). The combined dichloromethane extract was dried over sodium sulfate and the bulk of the solvent was evaporated in vacuo. Upon the observation of solids in the mixture during the evaporation, the evaporation was discontinued and the residue in the flask diluted with MTBE (35 mL). While stirring, the mixture was slowly diluted with hexanes (35 mL). The mixture was then chilled in an ice bath for 30 min. The solid was filtered, washed with 25% MTBE/hexane, and dried to give intermediate (9d) (4.98 g, 85.31%) as a white solid.NMR (CDCl3) δ 0.462 (q, 1H), 0.699 (m, 1H), 0.924 (s, 18-Me), 0.952 (s, 19-Me), 1.338 (t, J=7 Hz, OCH2CH 3), 2.517 (d, 1H), 3.021 (d, 1H), 4.269 (t, OCH 2CH3) ppm. FTIR (ATR): 3493, 3252, 2948, 2226, 1697, 1241 cm−1.
Compound (9d) (5.0 g) was dissolved in methanol (50 mL) and treated with 1.0 N sulfuric acid (10 mL). The mixture was heated to reflux for 3 hours, cooled, and neutralized through the addition of saturated sodium bicarbonate solution. Most of the methanol was evaporated in vacuo at ambient temperature and diluted with water. The aqueous mixture was extracted with ethyl acetate. The ethyl acetate extract was washed with water and brine, dried over sodium sulfate, filtered, and evaporated to give 4.95 g of unsaturated ketone (10d) as a stable foam.
Compound (10d) (5.0 g, 12.24 mmol) was dissolved in degassed benzene (50 mL) and treated with chlorotris(triphenylphosphine)rhodium (I) (283.1 mg, 0.31 mmol and the resulting mixture was stirred in a hydrogen atmosphere for 10 hours. The solution was evaporated, reconstituted in 50% ethyl acetate/hexanes, and passed through a short column of neutral alumina. Evaporation of the solvent gave 4.95 g of (11d) as a stable foam.
Compound (11d) (4.95 g, 12.01 mmol) was dissolved in 10% aqueous methanol (50 mL) and solid potassium carbonate (4.98 g, 36.04 mmol) was added. The mixture was stirred at room temperature for 30 min and the bicarbonate was neutralized through the addition of acetic acid (2.06 mL, 36.04 mmol). The methanol was evaporated in vacuo at ambient temperature and diluted with water. The aqueous mixture was extracted with ethyl acetate. The ethyl acetate extract was washed with water and brine, dried over sodium sulfate, filtered, and evaporated to give 4.10 g (93%) of a semi solid. The material was dissolved in dichloromethane and evaporated in vacuo to give a stable foam. The foam was dissolved in ethyl acetate (5 mL) and allowed to stand overnight. The resulting solid was filtered, washed with cold ethyl acetate, and dried in vacuo to afford 2.86 g (66%) of pure drospirenone.FIG. 5Experimental Example 1
A dichloromethane (50 mL) solution of ketodiol (1) (5.0 g, 15.13 mmol) was treated with ethyl vinyl ether (7.24 mL, 75.65 mmol), followed by the addition of pyridinium tosylate (380 mg, 1.15 mmol). The solution was stirred at room temperature for 30 min. The dichloromethane solution was washed with water (2×), brine, and dried over sodium sulfate. Following filtration, evaporation of the solvent gave 6.14 g of the 3-protected compound (1e) as a stable foam.
Compound (1e) (6.14 g, 15.13 mmol) was dissolved in DMSO/THF (15 mL/15 mL), treated with trimethylsulfonium iodide (4.63 g, 22.70 mmol) and the mixture was chilled to −15 C. The mixture was treated portion wise with potassium t-butoxide (3.23 g, 28.82 mmol). The mixture was stirred at −15 C for 45 min and then poured into ice/water (200 mL). The aqueous mixture was extracted with ethyl acetate. The ethyl acetate extract was washed with water (2×) and brine, dried over sodium sulfate, and filtered. Evaporation of the solvent gave 6.21 g (98.42%) of oxirane (12e) as a stable foam.
A THF (30 mL) solution of di-isopropyl amine (12.02 mL, 85.03 mmol) was chilled to −40 C and treated with butyl lithium (2.5 M/hexanes, 34.01 mL, 85.03 mmol) and the mixture was stirred for 15 min. A THF (5 mL) solution of acetonitrile (4.7 mL, 90.79 mmol) was added dropwise to the in situ generated lithium di-isopropylamide (LDA) solution to give a slurry of the acetonitrile anion. After stirring for 15 min at −40° C., compound (12e) (6.21 g, 14.91 mmol) as a THF (25 mL) solution was added dropwise over 10 min. The mixture was stirred for 30 min and then quenched through the addition of saturated ammonium chloride solution (210 mL). The mixture was extracted with ethyl acetate. The ethyl acetate extract was washed with water (3×) and brine, dried over sodium sulfate, and filtered. Evaporation of the solvent gave 7.07 g of the addition product (13e) as a tacky foam.
Compound 13e (5.0 g, 10.93 mmol) was dissolved in acetone (25 mL) and chilled to 0 C. The stirred solution was treated dropwise with 2.7M chromic acid (Jones Reagent) (7.0 mL, 18.91 mmol). After 1.5 hrs, the excess Cr (VI) was quenched through the addition of 2-propanol until the green color of Cr (IV) was evident. Water (300 mL) was added and the mixture was stirred until all the chromium salts were dissolved. The aqueous mixture was extracted with ethyl acetate. The ethyl acetate extract was washed with water (2×) and brine, dried over sodium sulfate, and filtered. Evaporation of the solvent gave 3.83 g (96%) of ketone (14e) as a stable foam.
Compound (14e) (3.83 g, 10.48 mmol) was dissolved in methanol (38 mL) and treated with 8.0 M KOH solution (7.0 mL, 56 mmol) and the mixture was heated at reflux for 5 hours. The mixture was cooled to 0 C and treated with acetic acid (15 mL) and water (6 mL) and the mixture was stirred at 50 C for 6 hours. The solvents were evaporated in vacuo and the residue was diluted with water (200 mL). The aqueous mixture was extracted with ethyl acetate. The ethyl acetate extract was washed with water (2×) and brine, dried over sodium sulfate, and filtered. Evaporation of the solvent gave 3.76 g (94%) of crude drospirenone as a stable foam. The crude drospirenone was dissolved in 60% ethyl acetate/hexanes and passed through a short column of neutral alumina (10× w/w) and the column was eluted with the same solvent. Following evaporation of the solvent, 2.58 g (65%) of crystalline drospirenone was obtained. Recrystallization from acetone/hexanes afforded pure drospirenone.FIG. 5Experimental Example 2
Intermediate (1) (5 g, 15.13 mmol) was dissolved in DMSO/THF (50 mL/50 mL), treated with trimethylsulfonium iodide (4.63 g, 22.70 mmol), and the mixture was chilled to −15 C. The mixture was treated portion wise with potassium t-butoxide (5.03 g, 43.88 mmol). The mixture was stirred at −15 C for 45 min and then poured into ice/water (200 mL). The aqueous mixture was extracted with ethyl acetate. The ethyl acetate extract was washed with water (2×) and brine, dried over sodium sulfate, and filtered. Evaporation of the solvent gave 5.11 g (98%) of oxirane (12f) as a stable foam.
A THF (30 mL) solution of di-isopropyl amine (12.02 mL, 85.03 mmol) was chilled to −40 C and treated with butyl lithium (2.5 M/hexanes, 34.01 mL, 85.03 mmol) and the mixture was stirred for 15 min. A THF (5 mL) solution of acetonitrile (4.7 mL, 90.79 mmol) was added dropwise to the above lithium di-isopropylamide (LDA) solution to give a slurry of the acetonitrile anion. After stirring for 15 min at −40 C, compound (12f) (5.11 g, 14.83 mmol) as a THF (75 mL) solution was added dropwise over 10 min. The mixture was stirred for 30 min and then quenched through the addition of saturated ammonium chloride solution (300 mL). The mixture was extracted with ethyl acetate. The ethyl acetate extract was washed with water (3×) and brine, dried over sodium sulfate, and filtered. Evaporation of the solvent gave 5.75 g of addition product (13f) as a tacky foam.
Compound 13f (5.75 g, 14.95 mmol) was dissolved in acetone (25 mL) and chilled to 0 C. The stirred solution was treated dropwise with 2.7M chromic acid (Jones Reagent) until the orange color of Cr (VI) persisted. After 1.5 hrs, the excess Cr (VI) was quenched through the addition of 2-propanol until the green color of Cr (IV) was evident. Water (300 mL) was added and the mixture was stirred until all the chromium salts were dissolved. The aqueous mixture was extracted with ethyl acetate. The ethyl acetate extract was washed with water (2×) and brine, dried over sodium sulfate, and filtered. Evaporation of the solvent gave 5.5 g (96%) of ketone (14f) as a stable foam.
Compound (14f) (5.5 g, 14.38 mmol) was dissolved in methanol (50 mL) and treated with 8.0 M KOH solution (9.35 mL, 74.77 mmol) and the mixture was heated at reflux for 5 hours. The mixture was cooled to 0 C and treated with acetic acid (25 mL) and water (10 mL) and the mixture was stirred at 50 C for 6 hours. The solvents were evaporated in vacuo and the residue was diluted with water (300 mL). The aqueous mixture was extracted with ethyl acetate. The ethyl acetate extract was washed with water (2×) and brine, dried over sodium sulfate, and filtered. Evaporation of the solvent gave 4.95 g (94%) of crude drospirenone as a stable foam. The crude drospirenone was dissolved in 60% ethyl acetate/hexanes and passed through a short column of neutral alumina (10× w/w) and the column was eluted with the same solvent. Following evaporation of the solvent, 3.43 g (65%) of crystalline drospirenone was obtained. Recrystallization from acetone/hexanes afforded pure drospirenone.
Drospirenone was patented in 1976 and introduced for medical use in 2000.[11][12] It is available widely throughout the world.[7] The medication is sometimes referred to as a “fourth-generation” progestin.[13][14] It is available as a generic medication.[15] In 2018, a formulation of drospirenone with ethinylestradiol was the 167th most commonly prescribed medication in the United States, with more than 3 million prescriptions.[16][17]
Birth control pills containing ethinylestradiol and a progestin are associated with an increased risk of venous thromboembolism (VTE), including deep vein thrombosis (DVT) and pulmonary embolism (PE).[48] The incidence is about 4-fold higher on average than in women not taking a birth control pill.[48] The absolute risk of VTE with ethinylestradiol-containing birth control pills is small, in the area of 3 to 10 out of 10,000 women per year, relative to 1 to 5 out of 10,000 women per year not taking a birth control pill.[49][50] The risk of VTE during pregnancy is 5 to 20 in 10,000 women per year and during the postpartum period is 40 to 65 per 10,000 women per year.[50] The higher risk of VTE with combined birth control pills is thought to be due to the ethinylestradiol component, as ethinylestradiol has estrogenic effects on liver synthesis of coagulation factors which result in a procoagulatory state.[8] In contrast to ethinylestradiol-containing birth control pills, neither progestogen-only birth control nor the combination of transdermalestradiol and an oral progestin in menopausal hormone therapy is associated with an increased risk of VTE.[8][51]
Different progestins in ethinylestradiol-containing birth control pills have been associated with different risks of VTE.[8] Birth control pills containing progestins such as desogestrel, gestodene, drospirenone, and cyproterone acetate have been found to have 2- to 3-fold the risk of VTE of birth control pills containing levonorgestrel in retrospective cohort and nested case–controlobservational studies.[8][49] However, this area of research is controversial, and confounding factors may have been present in these studies.[8][49][52] Other observational studies, specifically prospective cohort and case control studies, have found no differences in risk between different progestins, including between birth control pills containing drospirenone and birth control pills containing levonorgestrel.[8][49][52][53] These kinds of observational studies have certain advantages over the aforementioned types of studies, like better ability to control for confounding factors.[53]Systematic reviews and meta-analyses of all of the data in the mid-to-late 2010s found that birth control pills containing cyproterone acetate, desogestrel, drospirenone, or gestodene overall were associated with a risk of VTE of about 1.3- to 2.0-fold compared to that of levonorgestrel-containing birth control pills.[54][55][49]
Androgenic progestins have been found to antagonize to some degree the effects of ethinylestradiol on coagulation.[56][57][58][59] As a result, more androgenic progestins, like levonorgestrel and norethisterone, may oppose the procoagulatory effects of ethinylestradiol and result in a lower increase in risk of VTE.[8][60] Conversely, this would be the case less or not at all with progestins that are less androgenic, like desogestrel and gestodene, as well as progestins that are antiandrogenic, like drospirenone and cyproterone acetate.[8][60]
In the early 2010s, the FDA updated the label for birth control pills containing drospirenone and other progestins to include warnings for stopping use prior to and after surgery, and to warn that such birth control pills may have a higher risk of blood clots.[46]
Data on risk of breast cancer in women with newer progestins like drospirenone are lacking at present.[66] Progestogen-only birth control is not generally associated with a higher risk of breast cancer.[66] Conversely, combined birth control and menopausal hormone therapy with an estrogen and a progestogen are associated with higher risks of breast cancer.[67][66][68]
Overdose
These have been no reports of serious adverse effects with overdose of drospirenone.[2] Symptoms that may occur in the event of an overdose may include nausea, vomiting, and vaginal bleeding.[2] There is no antidote for overdose of drospirenone and treatment of overdose should be based on symptoms.[2] Since drospirenone has antimineralocorticoid activity, levels of potassium and sodium should be measured and signs of metabolic acidosis should be monitored.[2]
Drospirenone is an antagonist of the MR, the biological target of mineralocorticoids like aldosterone, and hence is an antimineralocorticoid.[69] It has about 100 to 500% of the affinity of aldosterone for the MR and about 50 to 230% of the affinity of progesterone for the MR.[1][3][71][62] Drospirenone is about 5.5 to 11 times more potent as an antimineralocorticoid than spironolactone in animals.[69][75][82] Accordingly, 3 to 4 mg drospirenone is said to be equivalent to about 20 to 25 mg spironolactone in terms of antimineralocorticoid activity.[83][2] It has been said that the pharmacological profile of drospirenone more closely resembles that of progesterone than other progestins due to its antimineralocorticoid activity.[69] Drospirenone is the only clinically used progestogen with prominent antimineralocorticoid activity besides progesterone.[1] For comparison to progesterone, a 200 mg dose of oral progesterone is considered to be approximately equivalent in antimineralocorticoid effect to a 25 to 50 mg dose of spironolactone.[84] Both drospirenone and progesterone are actually weak partial agonists of the MR in the absence of mineralocorticoids.[4][3][62]
Drospirenone is an antagonist of the AR, the biological target of androgens like testosterone and dihydrotestosterone (DHT).[1][3] It has about 1 to 65% of the affinity of the synthetic anabolic steroidmetribolone for the AR.[1][3][4][62] The medication is more potent as an antiandrogen than spironolactone, but is less potent than cyproterone acetate, with about 30% of its antiandrogenic activity in animals.[1][88][69][75] Progesterone displays antiandrogenic activity in some assays similarly to drospirenone,[3] although this issue is controversial and many researchers regard progesterone as having no significant antiandrogenic activity.[89][1][4]
Drospirenone shows antiandrogenic effects on the serumlipid profile, including higher HDLcholesterol and triglyceride levels and lower LDL cholesterol levels, at a dose of 3 mg/day in women.[3] The medication does not inhibit the effects of ethinylestradiol on sex hormone-binding globulin (SHBG) and serum lipids, in contrast to androgenic progestins like levonorgestrel but similarly to other antiandrogenic progestins like cyproterone acetate.[3][1][74] SHBG levels are significantly higher with ethinylestradiol and cyproterone acetate than with ethinylestradiol and drospirenone, owing to the more potent antiandrogenic activity of cyproterone acetate relative to drospirenone.[90]Androgenic progestins like levonorgestrel have been found to inhibit the procoagulatory effects of estrogens like ethinylestradiol on hepatic synthesis of coagulation factors, whereas this may occur less or not at all with weakly androgenic progestins like desogestrel and antiandrogenic progestins like drospirenone.[8][60][56][57][58][59]
The oralbioavailability of drospirenone is between 66 and 85%.[1][3][4]Peak levels occur 1 to 6 hours after an oral dose.[1][3][2][82] Levels are about 27 ng/mL after a single 4 mg dose.[2] There is 1.5- to 2-fold accumulation in drospirenone levels with continuous administration, with steady-state levels of drospirenone achieved after 7 to 10 days of administration.[1][2][3] Peak levels of drospirenone at steady state with 4 mg/day drospirenone are about 41 ng/mL.[2] With the combination of 30 μg/day ethinylestradiol and 3 mg/day drospirenone, peak levels of drospirenone after a single dose are 35 ng/mL, and levels at steady state are 60 to 87 ng/mL at peak and 20 to 25 ng/mL at trough.[3][1] The pharmacokinetics of oral drospirenone are linear with a single dose across a dose range of 1 to 10 mg.[2][3] Intake of drospirenone with food does not influence the absorption of drospirenone.[2]
Drospirenone is excreted in urine and feces, with slightly more excreted in feces than in urine.[2] Only trace amounts of unchanged drospirenone can be found in urine and feces.[2] At least 20 different metabolites can be identified in urine and feces.[3] Drospirenone and its metabolites are excreted in urine about 38% as glucuronideconjugates, 47% as sulfate conjugates, and less than 10% in unconjugated form.[3] In feces, excretion is about 17% glucuronide conjugates, 20% sulfate conjugates, and 33% unconjugated.[3]
The elimination half-life of drospirenone is between 25 and 33 hours.[2][3][1] The half-life of drospirenone is unchanged with repeated administration.[2] Elimination of drospirenone is virtually complete 10 days after the last dose.[3][2]
Drospirenone, also known as 1,2-dihydrospirorenone or as 17β-hydroxy-6β,7β:15β,16β-dimethylene-3-oxo-17α-pregn-4-ene-21-carboxylic acid, γ-lactone, is a syntheticsteroidal17α-spirolactone, or more simply a spirolactone.[7][92] It is an analogue of other spirolactones like spironolactone, canrenone, and spirorenone.[7][92] Drospirenone differs structurally from spironolactone only in that the C7α acetylthiosubstitution of spironolactone has been removed and two methylene groups have been substituted in at the C6β–7β and C15β–16β positions.[93]
Spirolactones like drospirenone and spironolactone are derivatives of progesterone, which likewise has progestogenic and antimineralocorticoid activity.[94][95][96] The loss of the C7α acetylthio group of spironolactone, a compound with negligible progestogenic activity,[97][98] appears to be involved in the restoration of progestogenic activity in drospirenone, as SC-5233, the analogue of spironolactone without a C7α substitution, has potent progestogenic activity similarly to drospirenone.[99]
History
Drospirenone was patented in 1976 and introduced for medical use in 2000.[11][12]Schering AG of Germany has been granted several patents on the production of drospirenone, including WIPO and US patents, granted in 1998 and 2000, respectively.[100][101] It was introduced for medical use in combination with ethinylestradiol as a combined birth control pill in 2000.[11] Drospirenone is sometimes described as a “fourth-generation” progestin based on its time of introduction.[13][14] The medication was approved for use in menopausal hormone therapy in combination with estradiol in 2005.[20] Drospirenone was introduced for use as a progestogen-only birth control pill in 2019.[2] A combined birth control pill containing estetrol and drospirenone was approved in 2021.[102]
Society and culture
Generic names
Drospirenone is the generic name of the drug and its INN, USAN, BAN, and JAN, while drospirénone is its DCF.[7] Its name is a shortened form of the name 1,2-dihydrospirorenone or dihydrospirenone.[7][92] Drospirenone is also known by its developmental code names SH-470 and ZK-30595 (alone), BAY 86-5300, BAY 98-7071, and SH-T-00186D (in combination with ethinylestradiol), BAY 86-4891 (in combination with estradiol), and FSN-013 (in combination with estetrol).[7][92][103][104][105][106][102]
Brand names
Drospirenone is marketed in combination with an estrogen under a variety of brand names throughout the world.[7] Among others, it is marketed in combination with ethinylestradiol under the brand names Yasmin and Yaz, in combination with estetrol under the brand name Nextstellis, and in combination with estradiol under the brand name Angeliq.[7][102]
Drospirenone is marketed widely throughout the world.[7]
Generation
Drospirenone has been categorized as a “fourth-generation” progestin.[62]
Litigation
Many lawsuits have been filed against Bayer, the manufacturer of drospirenone, due to the higher risk of venous thromboembolism (VTE) that has been observed with combined birth control pills containing drospirenone and certain other progestins relative to the risk with levonorgestrel-containing combined birth control pills.[52]
In July 2012, Bayer notified its stockholders that there were more than 12,000 such lawsuits against the company involving Yaz, Yasmin, and other birth control pills with drospirenone.[107] They also noted that the company by then had settled 1,977 cases for US$402.6 million, for an average of US$212,000 per case, while setting aside US$610.5 million to settle the others.[107]
As of July 17, 2015, there have been at least 4,000 lawsuits and claims still pending regarding VTE related to drospirenone.[108] This is in addition to around 10,000 claims that Bayer has already settled without admitting liability.[108] These claims of VTE have amounted to US$1.97 billion.[108] Bayer also reached a settlement for arterial thromboembolic events, including stroke and heart attacks, for US$56.9 million.[108]
^ Jump up to:abc Bayer (March 25, 2013). “Summary of Product Characteristics (SPC): Yasmin”. London: electronic Medicines Compendium (eMC), Datapharm. Retrieved April 24, 2014. 4.3. Contraindications: • Severe chronic kidney disease or acute kidney failure. • Presence or history of severe hepatic disease as long as liver function values have not returned to normal.
^ Jump up to:ab Bayer (April 10, 2012). “Yasmin full prescribing information”(PDF). Silver Spring, Md.: Food and Drug Administration (FDA). Retrieved April 14, 2012. 4. Contraindications: • Renal impairment. • Adrenal insufficiency. • Liver disease.
^ Nelson, Anita L.; Cwiak, Carrie (2011). “Combined oral contraceptives (COCs)”. In Hatcher, Robert A.; Trussell, James; Nelson, Anita L.; Cates, Willard Jr.; Kowal, Deborah; Policar, Michael S. (eds.). Contraceptive Technology (20th revised ed.). New York: Ardent Media. pp. 249–341. ISBN978-1-59708-004-0. ISSN0091-9721. OCLC781956734.
^ Oedingen C, Scholz S, Razum O (May 2018). “Systematic review and meta-analysis of the association of combined oral contraceptives on the risk of venous thromboembolism: The role of the progestogen type and estrogen dose”. Thromb. Res. 165: 68–78. doi:10.1016/j.thromres.2018.03.005. PMID29573722.
^ Jump up to:ab Sitruk-Ware R, Nath A (February 2013). “Characteristics and metabolic effects of estrogen and progestins contained in oral contraceptive pills”. Best Pract. Res. Clin. Endocrinol. Metab. 27 (1): 13–24. doi:10.1016/j.beem.2012.09.004. PMID23384742.
^ Fuhrmann, Ulrike; Krattenmacher, Rolf; Slater, Emily P.; Fritzemeier, Karl-Heinrich (1996). “The novel progestin drospirenone and its natural counterpart progesterone: Biochemical profile and antiandrogenic potential”. Contraception. 54 (4): 243–251. doi:10.1016/S0010-7824(96)00195-3. ISSN0010-7824. PMID8922878.
^ Jump up to:ab Hapgood JP, Africander D, Louw R, Ray RM, Rohwer JM (July 2014). “Potency of progestogens used in hormonal therapy: toward understanding differential actions”. J. Steroid Biochem. Mol. Biol. 142: 39–47. doi:10.1016/j.jsbmb.2013.08.001. PMID23954501. S2CID12142015.
^ Bastianelli C, Farris M, Rosato E, Brosens I, Benagiano G (November 2018). “Pharmacodynamics of combined estrogen-progestin oral contraceptives 3. Inhibition of ovulation”. Expert Rev Clin Pharmacol. 11 (11): 1085–1098. doi:10.1080/17512433.2018.1536544. PMID30325245. S2CID53246678.
^ Endrikat J, Gerlinger C, Richard S, Rosenbaum P, Düsterberg B (December 2011). “Ovulation inhibition doses of progestins: a systematic review of the available literature and of marketed preparations worldwide”. Contraception. 84 (6): 549–57. doi:10.1016/j.contraception.2011.04.009. PMID22078182.
^ Nieschlag, Eberhard; Behre, Hermann M.; Nieschlag, Eberhard; Behre, Hermann M.; Nieschlag, Susan (2012). “The essential role of testosterone in hormonal male contraception”. In Nieschlag, Eberhard; Behre, Hermann M; Nieschlag, Susan (eds.). Testosterone. pp. 470–493. doi:10.1017/CBO9781139003353.023. ISBN9781139003353.
^ Jump up to:ab Stanczyk, Frank Z. (2007). “Structure –Function Relationships, Pharmacokinetics, and Potency of Orally and Parenterally Administered Progestogens”. Treatment of the Postmenopausal Woman. pp. 779–798. doi:10.1016/B978-012369443-0/50067-3. ISBN9780123694430.
^ Giordano A, Frontini A, Cinti S (June 2016). “Convertible visceral fat as a therapeutic target to curb obesity”. Nat Rev Drug Discov. 15(6): 405–24. doi:10.1038/nrd.2016.31. PMID26965204. S2CID2632187.
^ Sitruk-Ware R, Husmann F, Thijssen JH, Skouby SO, Fruzzetti F, Hanker J, Huber J, Druckmann R (September 2004). “Role of progestins with partial antiandrogenic effects”. Climacteric. 7 (3): 238–54. doi:10.1080/13697130400001307. PMID15669548. S2CID23112620.
^ Ménard J (2004). “The 45-year story of the development of an anti-aldosterone more specific than spironolactone”. Mol. Cell. Endocrinol. 217 (1–2): 45–52. doi:10.1016/j.mce.2003.10.008. PMID15134800. S2CID19701784. [Spironolactone] was synthesized after the demonstration of the natriuretic effect of progesterone (Landau et al., 1955).
^ J. Larry Jameson; Leslie J. De Groot (18 May 2010). Endocrinology – E-Book: Adult and Pediatric. Elsevier Health Sciences. pp. 2401–. ISBN978-1-4557-1126-0. [Spironolactone] is a potent antimineralocorticoid which was developed as a progestational analog […]
^Aldosterone. Elsevier Science. 23 January 2019. p. 46. ISBN978-0-12-817783-9. In addition to spironolactone, which is a derivative of progesterone […]
^ Hu X, Li S, McMahon EG, Lala DS, Rudolph AE (2005). “Molecular mechanisms of mineralocorticoid receptor antagonism by eplerenone”. Mini Rev Med Chem. 5 (8): 709–18. doi:10.2174/1389557054553811. PMID16101407.
^ Nakajima ST, Brumsted JR, Riddick DH, Gibson M (1989). “Absence of progestational activity of oral spironolactone”. Fertil. Steril. 52 (1): 155–8. doi:10.1016/s0015-0282(16)60807-5. PMID2744183.
^WO patent 9806738, Mohr, Jörg-Thorsten & Klaus Nickisch, “PROCESS FOR PRODUCING DROSPIRENONE (6ss,7ss;15ss,16ss-DIMETHYLENE-3-OXO-17 alpha -PREGN-4-EN-21,17-CARBOLACTONE, DRSP), AS WELL AS 7 alpha -(3-HYDROXY-1-PROPYL)-6ss,7ss;15ss,16ss-DIMETHYLENE-5ss-ANDROSTANE-3ss,5,17ss-TRIOL (ZK 92836) AND 6ss,7ss;15ss,16ss-DIMETHYLENE-5ss HYDROXY-5-OXO-17 alpha -ANDROSTANE-21, 17-CARBOLACTONE”, issued 1998-02-19, assigned to Shering AG
^US patent 6121465, Mohr, Joerg-Thorston & Klaus Nickisch, “Process for production drospirenone and intermediate products of the process”, issued 2000-09-19, assigned to Scheiring AG and Bayer Schering Pharma
^ Nippe S, General S (September 2011). “Parenteral oil-based drospirenone microcrystal suspensions-evaluation of physicochemical stability and influence of stabilising agents”. Int J Pharm. 416 (1): 181–8. doi:10.1016/j.ijpharm.2011.06.036. PMID21729745.
^ Nippe S, General S (November 2012). “Combination of injectable ethinyl estradiol and drospirenone drug-delivery systems and characterization of their in vitro release”. Eur J Pharm Sci. 47 (4): 790–800. doi:10.1016/j.ejps.2012.08.009. PMID22940138.
^ Nippe S, Preuße C, General S (February 2013). “Evaluation of the in vitro release and pharmacokinetics of parenteral injectable formulations for steroids”. Eur J Pharm Biopharm. 83 (2): 253–65. doi:10.1016/j.ejpb.2012.09.006. PMID23116659.
^ Nippe S, General S (April 2015). “Investigation of injectable drospirenone organogels with regard to their rheology and comparison to non-stabilized oil-based drospirenone suspensions”. Drug Dev Ind Pharm. 41 (4): 681–91. doi:10.3109/03639045.2014.895375. PMID24621345. S2CID42932558.
Archer DF (February 2007). “Drospirenone-containing hormone therapy for postmenopausal women. Perspective on current data”. J Reprod Med. 52 (2 Suppl): 159–64. PMID17477110.
Archer DF (2007). “Drospirenone, a progestin with added value for hypertensive postmenopausal women”. Menopause. 14 (3 Pt 1): 352–4. doi:10.1097/gme.0b013e31804d440b. PMID17414576.
Batur P, Casey PM (February 2017). “Drospirenone Litigation: Does the Punishment Fit the Crime?”. J Womens Health (Larchmt). 26 (2): 99–102. doi:10.1089/jwh.2016.6092. PMID27854556.
Bitzer J, Paoletti AM (2009). “Added benefits and user satisfaction with a low-dose oral contraceptive containing drospirenone: results of three multicentre trials”. Clin Drug Investig. 29 (2): 73–8. doi:10.2165/0044011-200929020-00001. PMID19133702. S2CID10356578.
Dickerson V (November 2002). “Quality of life issues. Potential role for an oral contraceptive containing ethinyl estradiol and drospirenone”. J Reprod Med. 47 (11 Suppl): 985–93. PMID12497673.
Fenton C, Wellington K, Moen MD, Robinson DM (2007). “Drospirenone/ethinylestradiol 3mg/20microg (24/4 day regimen): a review of its use in contraception, premenstrual dysphoric disorder and moderate acne vulgaris”. Drugs. 67 (12): 1749–65. doi:10.2165/00003495-200767120-00007. PMID17683173. S2CID46976925.
Idota N, Kobayashi M, Miyamori D, Kakiuchi Y, Ikegaya H (March 2015). “Drospirenone detected in postmortem blood of a young woman with pulmonary thromboembolism: A case report and review of the literature”. Leg Med (Tokyo). 17 (2): 109–15. doi:10.1016/j.legalmed.2014.10.001. PMID25454533.
Krattenmacher R (July 2000). “Drospirenone: pharmacology and pharmacokinetics of a unique progestogen”. Contraception. 62 (1): 29–38. doi:10.1016/S0010-7824(00)00133-5. PMID11024226.
Lete I, Chabbert-Buffet N, Jamin C, Lello S, Lobo P, Nappi RE, Pintiaux A (2015). “Haemostatic and metabolic impact of estradiol pills and drospirenone-containing ethinylestradiol pills vs. levonorgestrel-containing ethinylestradiol pills: A literature review”. Eur J Contracept Reprod Health Care. 20 (5): 329–43. doi:10.3109/13625187.2015.1050091. PMID26007631. S2CID41601833.
Li J, Ren J, Sun W (March 2017). “A comparative systematic review of Yasmin (drospirenone pill) versus standard treatment options for symptoms of polycystic ovary syndrome”. Eur. J. Obstet. Gynecol. Reprod. Biol. 210: 13–21. doi:10.1016/j.ejogrb.2016.11.013. PMID27923166.
Motivala A, Pitt B (2007). “Drospirenone for oral contraception and hormone replacement therapy: are its cardiovascular risks and benefits the same as other progestogens?”. Drugs. 67 (5): 647–55. doi:10.2165/00003495-200767050-00001. PMID17385938. S2CID22985078.
Oelkers W (December 2002). “Antimineralocorticoid activity of a novel oral contraceptive containing drospirenone, a unique progestogen resembling natural progesterone”. Eur J Contracept Reprod Health Care. 7 Suppl 3: 19–26, discussion 42–3. PMID12659403.
Oelkers W (December 2000). “Drospirenone–a new progestogen with antimineralocorticoid activity, resembling natural progesterone”. Eur J Contracept Reprod Health Care. 5 Suppl 3: 17–24. PMID11246598.
Rapkin RB, Creinin MD (October 2011). “The combined oral contraceptive pill containing drospirenone and ethinyl estradiol plus levomefolate calcium”. Expert Opin Pharmacother. 12 (15): 2403–10. doi:10.1517/14656566.2011.610791. PMID21877996. S2CID40231903.
Rübig A (October 2003). “Drospirenone: a new cardiovascular-active progestin with antialdosterone and antiandrogenic properties”. Climacteric. 6 Suppl 3: 49–54. PMID15018248.
Sehovic N, Smith KP (May 2010). “Risk of venous thromboembolism with drospirenone in combined oral contraceptive products”. Ann Pharmacother. 44 (5): 898–903. doi:10.1345/aph.1M649. PMID20371756. S2CID8248469.
Shulman LP (June 2006). “A review of drospirenone for safety and tolerability and effects on endometrial safety and lipid parameters contrasted with medroxyprogesterone acetate, levonorgestrel, and micronized progesterone”. J Womens Health (Larchmt). 15 (5): 584–90. doi:10.1089/jwh.2006.15.584. PMID16796485.
Simoncini T, Genazzani AR (February 2010). “A review of the cardiovascular and breast actions of drospirenone in preclinical studies”. Climacteric. 13 (1): 22–33. doi:10.3109/13697130903437375. PMID19938948. S2CID4306359.
Thorneycroft IH (November 2002). “Evolution of progestins. Focus on the novel progestin drospirenone”. J Reprod Med. 47 (11 Suppl): 975–80. PMID12497671.
Toni I, Neubert A, Botzenhardt S, Gratzki N, Rascher W (September 2013). “Venous thromboembolism in adolescents associated with drospirenone-containing oral contraceptives – two case reports”. Klin Padiatr. 225 (5): 266–7. doi:10.1055/s-0033-1353169. PMID23975850.
Whitehead M (March 2006). “Hormone replacement therapy with estradiol and drospirenone: an overview of the clinical data”. J Br Menopause Soc. 12 Suppl 1: 4–7. doi:10.1258/136218006775992185. PMID16513012. S2CID38095916.
Zhao X, Zhang XF, Zhao Y, Lin X, Li NY, Paudel G, Wang QY, Zhang XW, Li XL, Yu J (September 2016). “Effect of combined drospirenone with estradiol for hypertensive postmenopausal women: a systemic review and meta-analysis”. Gynecol. Endocrinol. 32 (9): 685–689. doi:10.1080/09513590.2016.1183629. PMID27176003. S2CID9116138.
No development reportedGastric cancer; Nasopharyngeal cancer; Post-transplant lymphoproliferative disorder
01 Jun 2021Phase-II clinical trials in Lymphoma (Combination therapy, Second-line therapy or greater) in North America, Europe, Asia (PO)
18 May 2021Ninatinostat is still in phase I trials for Solid tumour in United Kingdom and Netherlands (Viracta Therapeutics pipeline, May 2021)
18 May 2021Virata Therapeutics has patent protection for dose regimen in NAVAL-1 trial in USA
Nanatinostat is under investigation in clinical trial NCT00697879 (Safety Study of the Histone Deacetylase Inhibitor, CHR-3996, in Patients With Advanced Solid Tumours).
Nanatinostat is an orally bioavailable, second-generation hydroxamic acid-based inhibitor of histone deacetylase (HDAC), with potential antineoplastic activity. Nanatinostat targets and inhibits HDAC, resulting in an accumulation of highly acetylated histones, the induction of chromatin remodeling, and the selective transcription of tumor suppressor genes; these events result in the inhibition of tumor cell division and the induction of tumor cell apoptosis. This agent may upregulate HSP70 and downregulate anti-apoptotic Bcl-2 proteins more substantially than some first-generation HDAC inhibitors. HDACs, upregulated in many tumor cell types, are a family of metalloenzymes responsible for the deacetylation of chromatin histone proteins.
Crystalline hydrate form A of N-hydroxy 2-{6-[(6-fluoro-quinolin-2-ylmethyl)-amino]-3-aza-bicyclo[3.1.0]hex-3-yl}pyrimidine-5-carboxamide ( nanatinostat ) .
Compound 1 is also known as nanatinostat, VRx-3996, or CHR-3996. It has been previously described in patents and patent applications, e.g. US patent 7,932,246 and US patent application 15/959,482, each of which is incorporated by reference in their entirety.
Compound 1
PATENT
WO2021071809 , claiming dosages for HDAC treatment with reduced side effects.





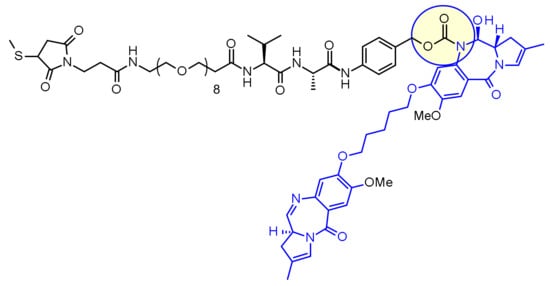


 s own T cells that are harvested and genetically modified ex vivo through transduction with an anti-BCMA02
s own T cells that are harvested and genetically modified ex vivo through transduction with an anti-BCMA02 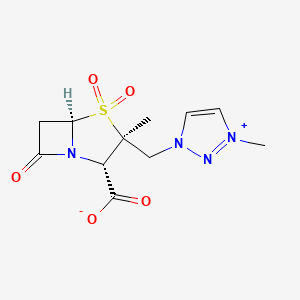








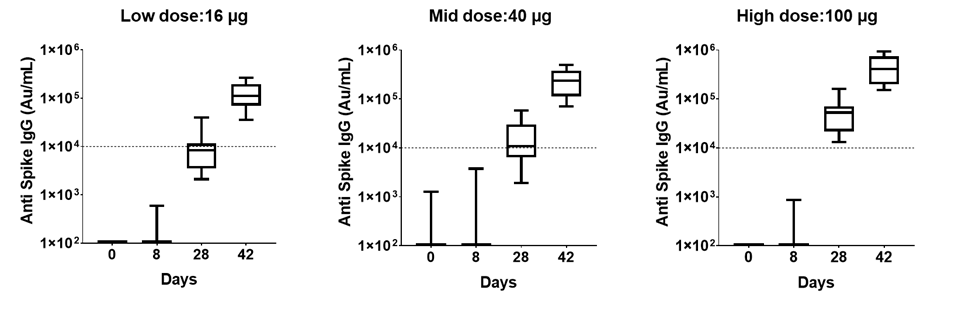
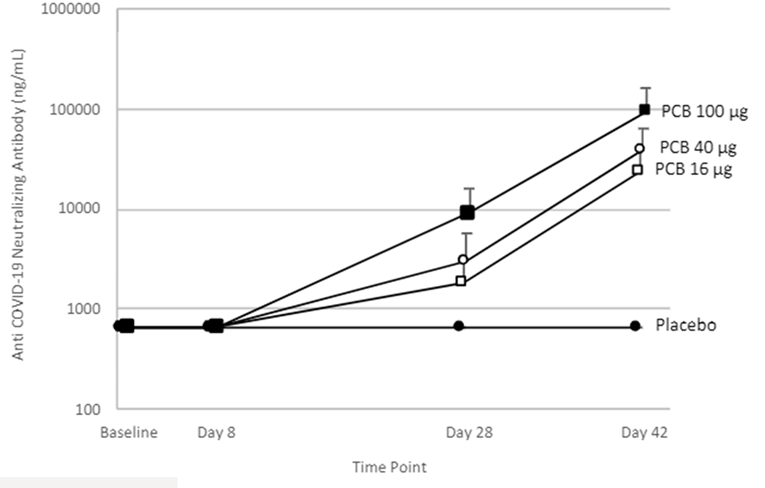












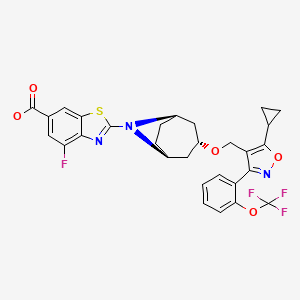

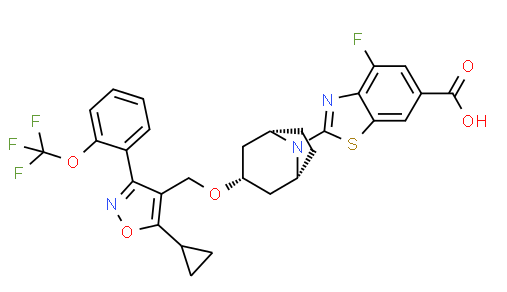



![4-((S)-4-Acryloyl-2-methylpiperazin-1-yl)-6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-1-(2-isopropyl-4-methylpyridin-3-yl)pyrido[2,3-d]pyrimidin-2(1H)-one.png](http://pubchem.ncbi.nlm.nih.gov/image/imgsrv.fcgi?cid=137278711&t=l)
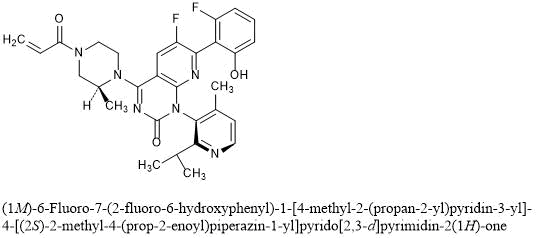









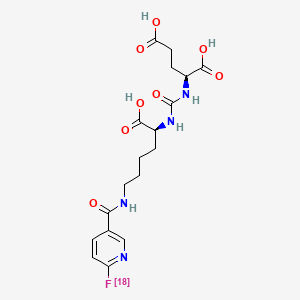
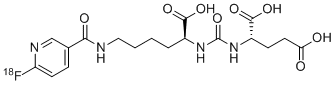


![Radiosynthesis of [ 18 F]DCFPyL](http://www.researchgate.net/publication/301830909/figure/fig4/AS:362696278069251@1463484939571/Radiosynthesis-of-18-FDCFPyL.png)