Estrogenic substances are commonly used in methods of Hormone Replacement Therapy (HRT) and methods of female contraception. These estrogenic substances can be divided in natural estrogens and synthetic estrogens. Examples of natural estrogens that have found pharmaceutical application include estradiol, estrone, estriol and conjugated equine estrogens. Examples of synthetic estrogens, which offer the advantage of high oral bioavailability include ethinyl estradiol and mestranol.Recently, estetrol has been found effective as an estrogenic substance for use in HRT, disclosure of which is given in the Applicant’s co-pending application WO 02/094276 . Estetrol is a biogenic estrogen that is endogeneously produced by the fetal liver during human pregnancy. Other important applications of estetrol are in the fields of contraception, therapy of auto-immune diseases, prevention and therapy of breast and colon tumors, enhancement of libido, skin care, and wound healing as described in the Applicant’s co-pending applications WO 02/094276 , WO 02/094279 , WO 02/094278 , WO 02/094275 , EP 1511496 A1 , EP 1511498 A1 , WO 03/041718 , WO 03/018026 , EP 1526856 A1 and WO 04/0278032 .[0004]The synthesis of estetrol and derivatives thereof on a laboratory scale basis is known in the art: Fishman J., Guzik H., J. Org. Chem. 33, 3133 – 3135 (1968); Nambara T. et al., Steroids 27, 111 – 121 (1976); or Suzuki E. et al., Steroids 60, 277 – 284(1995).[0005]
Fishman J., Guzik H., J. Org. Chem. 33, 3133 – 3135 (1968) discloses a successful synthesis of estetrol from an estrone derivative (compound (III); cf. for a synthesis of compound (III) Cantrall, E.W., Littell, R., Bernstein, S. J. Org. Chem 29, 214 – 217 (1964)). In a first step, the carbonyl group at C17 of compound (III) was reduced with LiAlH4 to estra-1,3,5(10),15-tetraene-3,17-diol (compound VIa) that was isolated as the diacetate (compound VIb). Compound VIb was subjected to cis-hydroxylation of the double bond of ring D by using OsO4 which resulted into the formation of estra-1,3,5(10)-triene-3,15α,16α,17β-tetraol-3,17-diacetate (compound Ib) that under heating with K2CO3 in methanol produces estetrol (Scheme 1).
[0006]
The overall yield of this three step process is, starting from estrone derivative III, only about 7%. It is worth noting that the protected derivative 17,17-ethylenedioxyestra-1,3,5(10),15-tetraene-3-ol-3-acetate (compound IV) could be cis-hydroxylated to its 15α,16α-diol derivative (compound Va), but that thereafter the dioxolane group could not be removed (p-toluene sulfonic acid in acetone at room temperature) or that the hydrolysis (aqueous sulfuric acid in warm dioxane) of the dioxolane group resulted in a mixture containing a multitude of products (Scheme 2).
[0007]Nambara T. et al., Steroids 27, 111 – 121 (1976) discloses another synthesis of estetrol wherein estrone is the starting material. The carbonyl group of estrone is first protected by treatment with ethylene glycol and pyridine hydrochloride followed by acetylation of the hydroxy group at C3. The next sequence of steps involved a bromination/base catalyzed dehydrobromination resulting into the formation of 17,17-ethylenedioxyestra-1,3,5(10),15-tetraene-3-ol (compound IVa). This compound IVa was subsequently acetylated which produced 17,17-ethylenedioxyestra-1,3,5(10),15-tetraene-3-ol-3-acetate (compound IVb). In a next step, the dioxolane group of compound IVb was hydrolysed by using p-toluene sulfonic acid to compound Vb, followed subsequently by reduction of the carbonyl group at C17 (compound Vc) and oxidation of the double bond of ring D thereby forming estra-1,3,5(10)-triene-3,15α,16α,17β-tetraol-3,17-diacetate (compound VIb). See Scheme 3.[0008]
Suzuki E. et al., Steroids 60, 277 – 284 (1995) also discloses the synthesis of estetrol by using compound Vb of Nambara T. et al. as starting material. The carbonyl group at C17 of this compound was first reduced followed by acetylation yielding estra-1,3,5(10),15-tetraene-3,17-diol-3,17-diacetate (compound 2b). The latter was subjected to oxidation with OsO4 which provided estra-1,3,5(10)-triene-3,15α,16α,17β-tetraol-3,17-diacetate (compound 3b) in 46% yield.
[0009]According to the Nambara T. et al. and Suzuki E. et al., the synthesis of estetrol can be performed with a yield of approximately 8%, starting from estrone.0010]
Poirier D., et al., Tetrahedron 47, 7751 – 7766 (1991) discloses the following compounds which were prepared according to methods that have been used to prepare similar compounds:
[0011]Dionne, P. et al., Steriods 62, 674 – 681 (1997) discloses the compound shown above wherein R is either methyl or t-butyldimethylsilyl.[0012]Magnus, P. et al., J. Am. Chem. Soc. 120, 12486 -12499 (1998) discloses that the main methods for the synthesis of α,β-unsaturated ketones from saturated ketones are (a) halogenation followed by dehydrohalogenation, (b) utilising sulphur or selenium derivatives, (c) DDQ and (d) utilizing palladium(II) complexes.[0013]Furthermore, it has also been found that by following the prior art methods mentioned above, estetrol of high purity was obtained only in low yield when using an acetyl group as a protecting group for the 3-hydroxy group of estra-1,3,5(10),15-tetraen-3-ol-17-one, in particular because its sensitivity to hydrolysis and solvolysis. In particular, the lability of the acetyl group lead not only to an increased formation of byproducts during the reactions, but also during chromatography and crystallisation for purification of intermediate products when protic solvents such as methanol were used. Therefore, it is difficult to isolate purified estetrol and intermediates thereof in good yield.
Example 7 3-Benzyloxy-estra-1,3,5 (10),15-tetraen-17-ol (compound 5; A = benzyl)
[0088]To a solution of 3-benzyl-dehydroestrone (compound 6; A = benzyl; 58 g, 162 mmol) in a mixture of MeOH (900 mL) and THF (200 mL) at room temperature was added CeCl3 heptahydrate (66.4 g, 178 mmol). After stirring for 1 h the mixture was cooled to 0-5°C using an ice/water bath. Then NaBH4 (12.2 g, 324 mmol) was added in small portions maintaining a temperature below 8°C. After stirring for 2 h at 0-5°C (TLC showed the reaction to be complete) 1 N NaOH (300 mL) and DCM (1 L) were added and the mixture was stirred for ½ h at room temperature. The layers were separated and the aqueous layer was extracted with DCM (200 mL). The organic layers were combined, dried (Na2SO4) and concentrated in vacuo to give an off-white solid (55.0 g, 152.8 mmol, 94%) TLC: Rf = 0.25 (heptanes/ethyl acetate = 4:1); HPLC-MS: 93% β-isomer, 2% α-isomer; DSC: Mp. 149.7°C, purity 96.6%; 1H-NMR (200 MHz, CDCl3) δ 7.48 (m, 5H), 7.27 (d, 1H, J = 8.4 Hz), 6.85 (dd, 1H, J1 = 2.8 Hz, J2 = 8.6 Hz), 6.81 (d, 1H, J = 2.4 Hz), 6.10 (d, 1H, J = 5.8 Hz), 5.79 (dd, 1H, J1 = 1.8 Hz, J2 = 3.4 Hz), 5.11 (s, 2H), 4.48 (d, 1H, J = 7.6), 2.96 (m, 2H), 2.46 – 1.64 (m, 9H), 0.93 (s, 3H) ppm.
Example 8 17-Acetyloxy-3-benzyloxy-estra-1,3,5 (10),15-tetraene (compound 4; A = benzyl, C = acetyl)
[0089]A solution of 3-Benzyloxy-estra-1,3,5 (10),15-tetraen-17-ol (compound 5; A = benzyl; 55.0 g, max. 153 mmol) in pyridine (400 mL) was treated with Ac2O (50 mL, 0.53 mol) and 4-dimethylaminopyridine (1.5 g, 12.3 mmol). The mixture was stirred for 2 h at room temperature (TLC showed the reaction to be complete). It was concentrated in vacuo. The residue was dissolved in EtOAc (400 mL), washed with water (200 mL) and brine (150 mL), dried (Na2SO4) and concentrated in vacuo to yield a yellow solid (54.0 g, 49.8 mmol, 88%). The product was purified by recrystallization from heptanes/ EtOAc/ EtOH (1:0.5:1) to afford a white solid (45.0 g, 112 mmol, 73%) TLC: Rf = 0.6 (heptanes/ethyl acetate = 4/1); HPLC-MS: 98% β-isomer, 1% α-isomer, 1.3% ß-estradiol; DSC: Mp. 122.8°C, purity 99.8%; 1H-NMR (200 MHz, CDCl3) δ 7.44 (m, 5H), 7.27 (d, 1H, J = 8.4 Hz), 6.86 (dd, 1H, J1 = 2.6 Hz, J2 = 8.4 Hz), 6.80 (d, 1H, J = 2.6 Hz), 6.17 (d, 1H, J = 5.8 Hz), 5.78 (dd, 1H, J1 = 1.4 Hz, J2 = 3.2 Hz), 5.45 (m, 1H), 5.11 (s, 2H), 2.96 (m, 2H), 2.40 – 1.54 (m, 10H), 2.18 (s, 3H), 0.93 (s, 3H) ppm.
Example 9 17-Acetyl-3-Benzyl estetrol (compound 3; A = benzyl, C = acetyl)
[0090]OsO4 on PVP (9 g, ~5% w/w OsO4 on PVP, prepared according to Cainelli et al. Synthesis, 45 – 47 (1989) was added to a solution of 17-Acetyloxy-3-benzyloxy-estra-1,3,5 (10),15-tetraene (compound 4; A = benzyl, C = acetyl; 45 g, 112 mmol) in THF (450 mL) and the mixture was heated to 50°C. Trimethylamine-N-oxide dihydrate (24.9 g, 224 mmol) was added portion-wise over 2 h. After stirring for 36 h at 50°C (TLC showed the reaction to be complete) the reaction mixture was cooled to room temperature. The solids were filtered off, washed with THF (100 mL) and the filtrate was concentrated. The residue was taken up in EtOAc (250 mL) and water (250 mL) was added. The aqueous layer was acidified with 1 N HCl (ca. 10 mL). The layers were separated and the aqueous layer was extracted with EtOAc (150 mL). The organic layers were combined, dried (Na2SO4) and concentrated in vacuo. The residue was triturated with heptanes/EtOAc (1:1, 100 mL), stirred for 2 h and the resulting white precipitate was filtered off to give the product as a white solid (41 g, 94 mmol, 84%). The product was purified by recrystallization from heptanes/ ethyl acetate/ EtOH (2:1:1) three times to afford a white solid (21 g, 48.2 mmol, 43%). HPLC-MS: 99.5% βαα-isomer; DSC: Mp. 159.3°C, purity 98.7%; 1H-NMR (200 MHz, CDCl3) δ 7.49 (m, 5H), 7.27 (d, 1H, J = 8.4 Hz), 6.84 (dd, 1H, J1 = 2.6 Hz, J2 = 8.4 Hz), 6.81 (d, 1H, J = 2.4 Hz), 5.11 (s, 2H), 4.45 (d, 1H, J = 4.4), 4.11 (m, 3H), 3.12 (m, 1H) 2.95 (m, 2H), 2.46 -1.64 (m, 10H), 2.24 (s, 3H), 0.93 (s, 3H) ppm.
Example 10 17-Acetyl estetrol (compound 2; C = acetyl)
[0091]To a solution of 17-acetyl-3-benzyl estetrol (compound 3; A = benzyl, C = acetyl; 21 g, 48.2 mmol) in MeOH (600 mL, HPLC-grade) was added a preformed suspension of 10% Palladium on activated carbon (2 g) in methanol (50 mL). The mixture was placed under an atmosphere of H2 at 1 atm and stirred for 24 h (TLC showed the reaction to be completed) at room temperature. It was filtered over Celite® and the filter cake was washed with MeOH (200 mL). The filtrate was concentrated in vacuo to give 17-acetyl estetrol as a white solid (15 g, 43.4 mmol, 90%). TLC: Rf = 0.2 (heptanes/ethyl acetate = 1/1); HPLC-MS: 99.2%, DSC: Mp. 212.2°C, purity 98.9%; 1H-NMR (200 MHz, CD3OD) δ 7.14 (d, 1H, J = 8.0 Hz), 6.60 (dd, 1H, J1 = 2.6 Hz, J2 = 8.8 Hz), 6.56 (d, 1H, J = 2.4 Hz), 4.81 (dd, 1H, J1 = 3.4 Hz, J2 = 6.4 Hz), 4.07 (m, 3H), 3.12 (m, 1H), 2.85 (m, 2H), 2.37 – 1.37 (m, 10H), 2.18 (s, 3H), 0.91 (s, 3H) ppm.
Example 11 Estetrol
[0092]17-Acetyl-estetrol (compound 2; C = acetyl; 15 g, 43.4 mmol) and K2CO3 (6 g, 43.4 mmol) were suspended in MeOH (500 mL, HPLC-grade) and stirred for 4 h at room temperature (TLC showed the reaction to be complete). The solvents were evaporated in vacuo. Water (200 mL) and CHCl3 (70 mL) were added and the mixture was stirred and neutralized with 0.1 N HCl (50 mL). The product was collected by filtration, washed with water (100 mL) and CHCl3 (100 mL) to give estetrol as a white solid (12.2 g, 40.1 mmol, 92.5%, overall yield from estrone 10.8%) after drying at 40°C in an air-ventilated oven. TLC: Rf = 0.05 (heptanes/ethyl acetate = 1/1); HPLC-MS: 99.1%, DSC: Mp. 243.7°C, purity 99.5%; 1H-NMR (200 MHz, CD3OD) δ 7.14 (d, 1H, J = 8.6 Hz), 6.61 (dd, 1H, J1 = 2.6 Hz, J2 = 8.4 Hz), 6.56 (d, 1H, J = 2.4 Hz), 4.83 (m, 1H), 3.93 (m, 3H), 3.50 (d, 1H, J = 5.2), 3.38 (m, 2H), 2.84 (m, 2H), 2.32 (m, 3H), 1.97 (m, 1H), 1.68 – 1.24 (m, 5H), 0.86 (s, 3H) ppm.
Estetrol (E4), or oestetrol, is a weak estrogensteroid hormone, which is found in detectable levels only during pregnancy in humans.[1][2] It is produced exclusively by the fetalliver.[1] Estetrol is closely related to estriol (E3), which is also a weak estrogen that is found in high quantities only during pregnancy.[1][2] Along with estradiol (E2), estrone (E1), and E3, estetrol (E4) is a major estrogen in the body, although only during pregnancy.[1]
In addition to its role as a natural hormone, estetrol is under clinical development for use as a medication, for instance in hormonal contraception (in combination with drospirenone) and as menopausal hormone therapy; for information on estetrol as a medication, see the estetrol (medication) article.
Biological function
Estetrol is an estrogen and has estrogenic effects in various tissues.[1] Estetrol interacts with nuclear Estrogen Receptor (ERα) in a manner identical to that of the other estrogens and distinct from that observed with Selective Estrogen Receptor Modulators (SERMs).[3][4] So far the physiological function of estetrol is unknown. The possible use of estetrol as a marker for fetal well-being has been studied quite extensively. However, due to the large intra- and inter-individual variation of maternal estetrol plasma levels during pregnancy this appeared not to be feasible.[5][6][7][8][9]
Biological activity
Estetrol is an agonist of the estrogen receptors (ERs), and hence is an estrogen.[10][11] It has moderate affinity for ERα and ERβ, with Ki values of 4.9 nM and 19 nM, respectively.[10][12] As such, estetrol has 4- to 5-fold preference for the ERα over the ERβ.[10][12] The estrogen has low affinity for the ERs relative to estradiol, and both estetrol and the related estrogen estriol require substantially higher concentrations than estradiol to produce similar effects to estradiol.[10] The affinity of estetrol for the ERs is about 0.3% (rat) to 6.25% (human) of that of estradiol, and its in vivopotency in animals is about 2 to 3% of that of estradiol.[10] Estetrol shows high selectivity for the ERs.[10][12]
Biochemistry
Biosynthesis
Estetrol is synthesized during pregnancy only in the fetalliver from estradiol (E2) and estriol (E3) by the two enzymes 15α- and 16α-hydroxylase.[13][14][15] Alternatively, estetrol is synthesized with 15α-hydroxylation of 16α-hydroxy-DHEA sulfate as an intermediate step.[16] It appears in maternal urine at around week 9 of pregnancy.[2] After birth the neonatal liver rapidly loses its capacity to synthesize estetrol because these two enzymes are no longer expressed.
Estetrol reaches the maternal circulation through the placenta and was already detected at nine weeks of pregnancy in maternal urine.[17][18] During the second trimester of pregnancy high levels were found in maternal plasma, with steadily rising concentrations of unconjugated estetrol to about 1 ng/mL (>3 nM) towards the end of pregnancy.[1]
vteStructures of major endogenous estrogensEstrone (E1)Estradiol (E2)Estriol (E3)Estetrol (E4)Note the hydroxyl (–OH) groups: estrone (E1) has one, estradiol (E2) has two, estriol (E3) has three, and estetrol (E4) has four.
Estetrol was discovered in 1965 by Egon Diczfalusy and coworkers at the Karolinska Institute in Stockholm, Sweden, via isolation from the urine of pregnant women.[10][23]
^ Jump up to:abc Reproductive Endocrinology: Physiology, Pathophysiology, and Clinical Management, 3rd ed., SSC Yen and RB Jaffe (eds.), pp. 936–981, Copyright Elsevier/Saunders 1991
^ J. Heikkilä, T. Luukkainen, Urinary excretion of estriol and 15a-hydroxyestriol in complicated pregnancies, Am. J. Obstet. Gynecol. 110 (1971) 509-521.
^ D. Tulchinsky, F.D. Frigoletto, K.J. Ryan, J. Fishman, Plasma estetrol as an index of fetal well-being, J. Clin. Endocrinol. Metab. 40 (1975) 560-567
^ A.D. Notation, G.E. Tagatz, Unconjugated estriol and 15a-hydroxyestriol in complicated pregnancies, Am. J. Obstet. Gynecol. 128 (1977) 747-756.
^ N. Kundu, M. Grant, Radioimmunoassay of 15a-hydroxyestriol (estetrol) in pregnancy serum, Steroids 27 (1976) 785-796.
^ N. Kundu, M. Wachs, G.B. Iverson, L.P. Petersen, Comparison of serum unconjugated estriol and estetrol in normal and complicated pregnancies, Obstet. Gynecol. 58 (1981) 276-281.
^ Jump up to:abc Visser M, Foidart JM, Coelingh Bennink HJ (2008). “In vitro effects of estetrol on receptor binding, drug targets and human liver cell metabolism”. Climacteric. 11 Suppl 1: 64–8. doi:10.1080/13697130802050340. PMID18464025.
^ J. Schwers, G. Eriksson, N. Wiqvist, E. Diczfalusy, 15a-hydroxylation: A new pathway of estrogen metabolism in the human fetus and newborn, Biochim. Biophys. Acta. 100 (1965) 313-316
^ J. Schwers, M. Govaerts-Videtsky, N. Wiqvist, E. Diczfalusy, Metabolism of oestrone sulphate by the previable human foetus, Acta Endocrinol. 50 (1965) 597-610.
^ S. Mancuso, G. Benagiano, S. Dell’Acqua, M. Shapiro, N. Wiqvist, E. Diczfalusy, Studies on the metabolism of C-19 steroids in the human foeto-placental unit, Acta Endocrinol. 57 (1968) 208-227.
^ J. Heikkilä, H. Adlercreutz, A method for the determination of urinary 15α-hydroxyestriol and estriol, J. Steroid Biochem. 1 (1970) 243-253
^ J. Heikkilä, Excretion of 15α-hydroxyestriol and estriol in maternal urine during normal pregnancy, J. Steroid Biochem. 2 (1971) 83-93.
^ Visser M, Holinka CF, Coelingh Bennink HJ (2008). “First human exposure to exogenous single-dose oral estetrol in early postmenopausal women”. Climacteric. 11 Suppl 1: 31–40. doi:10.1080/13697130802056511. PMID18464021.
^ Hammond GL, Hogeveen KN, Visser M, Coelingh Bennink HJ (2008). “Estetrol does not bind sex hormone binding globulin or increase its production by human HepG2 cells”. Climacteric. 11 Suppl 1: 41–6. doi:10.1080/13697130701851814. PMID18464022.
^ Warmerdam EG, Visser M, Coelingh Bennink HJ, Groen M (2008). “A new route of synthesis of estetrol”. Climacteric. 11 Suppl 1: 59–63. doi:10.1080/13697130802054078. PMID18464024.
^ Hagen AA, Barr M, Diczfalusy E (June 1965). “Metabolism of 17-beta-oestradiol-4-14-C in early infancy”. Acta Endocrinol. 49: 207–20. doi:10.1530/acta.0.0490207. PMID14303250.
A COVID-19 vaccine comprising a dimeric form of SARS-CoV-2 receptor-binding domain (RBD) produced in China hamster ovary (CHO) cells and adjuvanted with aluminum hydroxide (Anhui Zhifei Longcom/Institute of Microbiol. China Academy of Sciences)
Recombinant vaccine
Anhui Zhifei Longcom Biopharmaceutical, Institute of Microbiology of the Chinese Academy of Sciences
China, Uzbekistan
CHO Cells Recombinant Vaccine
ZF-2001
ZF-UZ-VAC2001
Chinese Academy of Sciences (Originator)
Zhifei Longcom (Originator)
Human SARS-CoV-2 (Covid-19 coronavirus) vaccine consisting of recombinant dimer comprising two RBD domains (R319-K527) of the spike glycoprotein of SARS-CoV-2 fused via a disulfide link; expressed in CHO cells
ZF-2001 is a recombinant coronavirus vaccine jointly developed by the Institute of Microbiology of the Chinese Academy of Sciences and Zhifei Longcom. The vaccine became available in 2021 in Uzbekistan under an emergency use authorization for the prevention of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection (COVID-19). The vaccine is currently evaluated in phase III clinical trials.
This vaccine candidate, developed in China, uses SARS-CoV-2 protein subunits that are entirely engineered, created, and secreted by Chinese Hamster Ovary (CHO) cells1. The vaccine candidate is sponsored by Anhui Zhifei Longcom Biologic Pharmacy Co., Ltd. and is undergoing phase I clinical trials to evaluate safety and tolerability.
ZF2001 employs technology similar to other protein-based vaccines in Phase III trials from Novavax, Vector Institute, and Medicago.[5] It is administered in 3 doses over a period of 2 months.[6]
ZF2001 was first approved for use in Uzbekistan and later China.[7][8] Production capacity is expected to be one billion doses a year.[6] Phase II results published in The Lancet on the three dose administration showed seroconversion rates of neutralizing antibodies of between 92% to 97%.[9]
Anhui Zhifei Longcom Biopharmaceuticals began a phase 3 clinical trial for its recombinant protein vaccine candidate in December, according to the WHO. State-run China Global Television Network in November reported that a one-year trial would take place in Uzbekistan and aim to recruit 5,000 volunteers. Anhui Zhifei is a unit of private firm Chongqing Zhifei Biological Products. It is co-developing the vaccine with the Chinese Academy of Sciences, a government institution.
Emergency Use Authorization received in UZ by Zhifei Longcom for the prevention of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection (COVID-19)
Description
As described in Cell, the CoV spike receptor-binding domain (RBD) is an attractive vaccine target for coronaviruses but is constrained by limited immunogenicity, however a dimeric form of MERS-CoV RBD offers greater protection. The RBD-dimer significantly increases neutralizing antibodies compared to a conventional monomeric form and protected mice against MERS-CoV infection. CoV RBD-dimer have been produced at high yields in pilot scale production.[10]
Rather than injecting a whole virus, subunit vaccines contains virus particles specially selected to stimulate an immune response. Because the fragments are incapable of causing disease, subunit vaccines are considered very safe.[11] Subunit vaccines in widespread use include the Hepatitis B vaccine and Pertussis vaccine. However, as only a few viral components are included in the vaccine which does not display the full complexity of the virus, their efficacy may be limited.[12] Subunit vaccines are delivered alongside adjuvants and booster doses may be required.[11]
According to industry experts, production for this kind of vaccine is stable and reliable, and easier to achieve large-scale industrial production at home and overseas. However it was noted it can be very inconvenient for people to come back for a second and third dose.[6]
ZF2001 (Anhui Zhifei Longcom Biopharmaceutical/Chinese Academy of Medical Sciences)
The latest subunit vaccine candidate to enter Phase 3 clinical studies is the adjuvanted RBD-dimeric antigen designed by Anhui Zhifei Longcom Biopharmaceutical and the Institute of Microbiology of the Chinese Academy of Medical Sciences. Phase 3 clinical study was launched on December104 and will be initially carried out in China and Uzbekistan while Indonesia, Pakistan and Ecuador will follow as study sites (Clinical Trial Identifier: NCT04646590 and Registration Number: ChiCTR2000040153). The design of the study involves recruitment of 22,000 volunteers from China and 7000 subjects outside China for a total of 29,000 volunteers. There are still no published results on this candidate, however data from its Phase 2 placebo-controlled clinical trial (Clinical Trial Identifier: NCT04466085) conducted on a total of 900 participants ranging from 18 to 59 years old suggest that a 2 or 3 dose regimen is evaluated. Each immunization will be separated by the next by 4 weeks.
Development
Phase I and II trials and results
In June, Longcom began a double-blind, randomized, placebo parallel controlled Phase I trial with 50 participants aged 18–59 in Chongqing divided into low-dose, high-dose, and placebo groups.[13]
In July, Longcom began a randomized, double-blind, placebo-controlled Phase II trial with 900 participants aged 18–59 in Changsha, Hunan divided into low-dose, high-dose, and placebo groups.[14] In August, an additional Phase II trial was launched with 50 participants aged 60 and above.[15][1]
In Phase II results published in The Lancet, on the two-dose schedule, seroconversion rates of neutralizing antibodies after the second dose were 76% (114 of 150 participants) in a 25 μg group and 72% (108 of 150) in a 50 μg group. On the three-dose schedule, seroconversion rate of neutralizing antibodies after the third dose were 97% (143 of 148 participants) in the 25 μg group and 93% (138 of 148) in the 50 μg group. 7 to 14 days after the administration of the third dose, the GMTs of neutralizing antibodies reached levels that were significantly higher than observed in human convalescent serum of recovering COVID-19 patients, especially in the 25 μg group.[9]
Phase III trials
In December, Longcom began enrollment of a Phase III randomized, double-blind, placebo-controlled clinical trial for 29,000 participants, including 750 participants between 18-59 and 250 participants 60 and older in China and 21,000 participants between 18-59 and 7,000 participants 60 and older outside China.[16][17]
In December, Malaysia‘s MyEG announced it would conduct Phase III trials. If the trials were successful, MyEG would be the sole distributor of ZF2001 in Malaysia for 3 years.[4]
In December, Uzbekistan began a year-long Phase III trial of ZF2001 with 5,000 volunteers between 18 and 59.[18][19]
In December, Ecuador‘s Minister of Health, Juan Carlos Zevallos announced Phase III trials would involve between 5,000 and 8,000 volunteers.[20]
In February, Pakistan‘s Drug Regulatory Authority (DRAP) approved Phase III trials with approximately 10,000 participants to be conducted at UHS Lahore, National Defense Hospital, and Agha Khan Hospital.[21]
Discussions to begin Phase III trials are also underway in Indonesia.[17][22]
COVID-19 Variants
In February, lab studies of twelve serum samples taken from recipients of BBIBP-CorV and ZF2001 retained neutralizing activity against the Beta variant although with weaker activity than against the original virus.[23] For ZF-2001, geometric mean titers declined by 1.6-fold, from 106.1 to 66.6, which was less than antisera from mRNA vaccine recipients with a 6-folds decrease.[24] Preliminary clinical data from Novavax and Johnson & Johnson also showed they were less effective in preventing COVID-19 in South Africa, where the new variant is widespread.[23]
Manufacturing
The company’s vaccine manufacturing facility was put into use in September.[17] In February 2021, Pu Jiang, General Manager of Zhifei Longcom, said the company had an annual production capacity of 1 billion doses.[6]
On March 1, Uzbekistan granted approval for ZF2001 (under tradename ZF-UZ-VAC 2001) after having taken part in the Phase III trials.[8] In March, Uzbekistan received 1 million doses and started vaccinations in April.[25] By May, a total of 3 million doses had been delivered.[26]
On March 15, China approve of ZF2001 for emergency use after being approved by Uzbekistan earlier in the month.[7]
^ Clinical trial number NCT04445194 for “Phase I Clinical Study of Recombinant Novel Coronavirus Vaccine” at ClinicalTrials.gov
^ Clinical trial number NCT04466085 for “A Randomized, Blinded, Placebo-controlled Trial to Evaluate the Immunogenicity and Safety of a Recombinant New Coronavirus Vaccine (CHO Cell) With Different Doses and Different Immunization Procedures in Healthy People Aged 18 to 59 Years” at ClinicalTrials.gov
^ Clinical trial number NCT04550351 for “A Randomized, Double-blind, Placebo-controlled Phase I Clinical Trial to Evaluate the Safety and Tolerability of Recombinant New Coronavirus Vaccines (CHO Cells) in Healthy People Aged 60 Years and Above” at ClinicalTrials.gov
^ Clinical trial number NCT04646590 for “A Phase III Randomized, Double-blind, Placebo-controlled Clinical Trial in 18 Years of Age and Above to Determine the Safety and Efficacy of ZF2001, a Recombinant Novel Coronavirus Vaccine (CHO Cell) for Prevention of COVID-19” at ClinicalTrials.gov
MKIEEGKLVIWINGDKGYNGLAEVGKKFEKDTGIKVTVEHPDKLEEKFPQVAATGDGPDIIFWAHDRFGGYAQSGLLAEITPDKAFQDKLYPFTWDAVRYNGKLIAYPIAVEALSLIYNKDLLPNPPKTWEEIPALDKELKAKGKSALMFNLQEPYFTWPLIAADGGYAFKYENGKYDIKDVGVDNAGAKAGLTFLVDLIKNKHMNADTDYSIAEAAFNKGETAMTINGPWAWSNIDTSKVNYGVTVLPTFKGQPSKPFVGVLSAGINAASPNKELAKEFLENYLLTDEGLEAVNKDKPLGAVALKSYEEELAKDPRIAATMENAQKGEIMPNIPQMSAFWYAVRTAVINAASGRQTVDEALKDAQTNSSSNNNNNNNNNNLGDNGPQNQRNAPRITFGGPSDSTGSNQNGERSGARSKQRRPQGLPNNTASWFTALTQHGKEDLKFPRGQGVPINTNSSPDDQIGYYRRATRRIRGGDGKMKDLSPRWYFYYLGTGPEAGLPYGANKDGIIWVATEGALNTPKDHIGTRNPANNAAIVLQLPQGTTLPKGFYAEGSRGGSQASSRSSSRSRNSSRNSTPGSSRGTSPARMAGNGGDAALALLLLDRLNQLESKMSGKGQQQQGQTVTKKSAAEASKKPRQKRTATKAYNVTQAFGRRGPEQTQGNFGDQELIRQGTDYKHWPQIAQFAPSASAFFGMSRIGMEVTPSGTWLTYTGAIKLDDKDPNFKDQVILLNKHIDAYKTFPPTEPKKDKKKKADETQALPQRQKKQQTVTLLPAADLDDLSKQLQQSMSSADSTQA. “Carrier protein sequence”.
EpiVacCorona
Federal Budgetary Research Institution State Research Center of Virology and Biotechnology
EpiVacCorona (Russian: ЭпиВакКорона, tr.EpiVakKorona) is a peptide-based vaccine againstCOVID-19 developed by the VECTOR center of Virology.[1][2][3] It consists of three chemically synthesized peptides (short fragments of a viral spike protein) that are conjugated to a large carrier protein. This protein is a fusion product of a viral nucleocapsid protein and a bacterial MBP protein.The third phase of a clinical trial, which should show whether the vaccine is able to protect people from COVID-19 or not, was launched in November 2020 with more than three thousand participants.[2] It is assumed it will be completed in August 2021.[2] According to the vaccine developers, the peptides and the viral part of the chimeric protein should immunize people who received this vaccine against SARS-CoV-2 and trigger the production of protective antibodies. However, some experts in the field have expressed concerns about the selection of peptides for use as vaccine antigens.[3][4] In addition, there are also serious concerns about the vaccine immunogenicity data, which have fueled independent civic research efforts[5][6][7] and criticism by some experts.[3][8][4][9][10] Meanwhile, the EpiVacCorona has received vaccine emergency authorization in a form of government registration and is available for vaccination outside the clinical trials.[11] The vaccine delivered via intramuscular route and aluminum hydroxide serves as an immunological adjuvant.
The vaccine includes three chemically synthesized short fragments of the viral spike protein – peptides, which, according to the developers of EpiVacCorona represent the protein regions containing B-cell epitopes that should be recognized by the human immune system.
These peptides are represented by following amino acid sequences:
In the vaccine all peptides are conjugated to a carrier protein, which is an expression product of the chimeric gene. This chimeric gene was created by fusion of two genes originating from different organisms, namely a gene encoding a viral nucleocapsid protein and a gene encoding a bacterial maltose-binding protein (MBP). The fusion chimeric gene expressed in Escherichia coli. The sequence of the chimeric protein is available from the patent.[4] The genetic construct of the chimeric gene also includes a short genetic fragment encoding a polyhistidine-tag, which is used to purify the chimeric protein from E. coli lysate. After the purification, the protein is conjugated with three peptides in a way that only one variant of the peptide molecule is attached to each protein molecule. As a result, three types of conjugated molecules are created: chimeric protein with attached peptide number 1, the same protein with peptide number 2, and finally the same protein with peptide number 3. All three types of conjugated molecules are included in the vaccine.[citation needed]
EpiVacCorona: antigens origin and composition
Vaccine antigens and antibodies
According to the developers’ publications,[14][5][6] vaccine antigens are three peptides of the spike protein and a chimeric protein consisting of two parts (viral nucleocapsid protein and bacterial maltose-binding protein). In addition, the polyhistidine-tag – a short peptide that is introduced into a vaccine composition to purify a chimeric protein from a bacterial lysate – is also a vaccine antigen against which antibodies can form in those who have received the vaccine. A person vaccinated with EpiVacCorona can develop antibodies not only to the peptides of the spike protein, but also to other antigens present in the vaccine. According to Anna Popova who is a head of the Federal Service for Supervision of Consumer Rights Protection and Human Welfare, it takes 42 days for those vaccinated with EpiVacCorona to develop immunity.[15]
Development
Immunogenic peptide screening in rabbits for EpiVacCorona design
Preclinical studies
The primary screening of peptides for the search for the most immunogenic ones was carried out in animals. The level of antibodies that was triggered by each tested peptide after administration to rabbits was measured. In the test, hemocyanin protein was used as a carrier protein for the studied peptides. Further, on six species of animals (mice, rats, rabbits, African green monkeys, rhesus monkeys, guinea pigs), the vaccine was shown to be harmless in terms of such parameters as general toxicity, allergic properties, and mutagenic activity. In four species of animals (hamsters, ferrets, African green monkeys, rhesus monkeys), specific activity was shown: immunogenicity and protective properties against SARS-CoV-2. The main results of preclinical studies are published in the “Bulletin of the Russian Academy of Medical Sciences”.[12][13]
Clinical studies
The studies development timeline was reported in Russian media in January 2021.[16] There are currently two clinical trials of EpiVacCorona registered in the ClinicalTrials.gov database.[17][18][2]
Phase I-II
The trial “Study of the Safety, Reactogenicity and Immunogenicity of “EpiVacCorona” Vaccine for the Prevention of COVID-19 (EpiVacCorona)”[18] was registered in clinical trial database with ClinicalTrials.gov identifier: NCT04780035. Another trial with the same title was registered with ClinicalTrials.gov Identifier: NCT04527575. Results of the trial that included data on 86 participants were published in Russian Journal of Infection and Immunity, indicating preliminary evidence of safety and an immune response.[1] The publication reports preliminary results of the first two phases of clinical trials of the vaccine in volunteers, of which 14 people aged 18-30 years participated in the first phase, and 86 volunteers aged 18-60 years in the second phase. It is claimed that antibodies were formed in 100% of the volunteers, and the vaccine is also claimed to be safe.[1]
EpiVacCorona Vaccine Development Timeline
Phase III
The third phase of a clinical trial, which should show whether the vaccine is able to protect people from COVID-19 or not, was launched in November 2020 with more than three thousand participants planned. It is expected to be completed in September 2021.[2] In the clinical trials database the phase III trial etitled “Study of the Tolerability, Safety, Immunogenicity and Preventive Efficacy of the EpiVacCorona Vaccine for the Prevention of COVID-19[2]” was registered only in March 2021 with ClinicalTrials.gov Identifier: NCT04780035. Phase 3-4 trial was registered in Russia at 18.11.2020 with 4991 participants planned.[19]
Intellectual property
The following patents of the Russian Federation for invention have been published, which protect the EpiVacCorona vaccine:
In all of these patents, the carrier protein is referred to as a chimeric fusion protein with an amino acid sequence derived from two parts, a bacterial maltose binding protein and a viral nucleocapsid protein.[20]
In Russia phase III clinical study is called post-registration study. Therefore, government registration of the vaccine means permission to perform phase III clinical research and public vaccination outside of clinical trials as well.[21] Since December 2020, the vaccine has been released for public vaccination in Russia.[22]
As of March 2021, Turkmenistan is the only foreign state to register EpiVacCorona with full authorization.[23][24]
Russia’s Chief Health Officer Anna Popova said: “In December 2020 the EpiVacCorona documents were presented to the World Health Organization, and we are expecting a decision from WHO.”[25] However, Deutsche Welle reports “As of March 1, the WHO had yet to receive an Expression of Interest (EOI) from EpiVacCorona’s developers, “VECTOR,” to enable WHO experts to evaluate their vaccine.”[26]
Export
The Deputy Director-General of the World Health Organization (WHO) Dr. Soumya Swaminathan during news conference in Geneva that took place in October 2020, told: “We will only be able to have a position on a vaccine when we see results of the phase III clinical trials.”[27] According to the center’s director Rinat Maksyutov, many government and non-government organizations want to test or be involved in the production of the vaccine.[28] As of March 30, Venezuela obtained 1000 doses of the Russian EpiVacCorona vaccine for a trial.[29] Venezuela also has reached a deal to purchase doses of the vaccine, as well as manufacture it locally, Vice President Delcy Rodriguez provided this information on June 4, 2021.[30] Turkmenistan expects to receive EpiVacCorona, as the vaccine has already been approved for use in that country.[31]
Controversy
Independent study of clinical trial participants
Ministry of Health’s response to a request from trial participants to perform independent antibody screening tests
English translation of Ministry of Health’s response to a request from trial participants to perform independent antibody screening tests.
At the start of the Phase III, trial participants and those vaccinated outside the trial began to form a community through the Telegram messenger network. On January 18, 2021, the members of the community turned to the Ministry of Health of the Russian Federation with an open letter, in which they stated that the production of antibodies after vaccination among them is much lower than declared by vaccine developers. Study participants claimed that antibodies were not found in more than 50% of those who documented their participation in the study, although only 25% of the participants should have had a placebo according to the study design. The trial participants also claimed that negative results were obtained using the a special ELISA test developed and recommended by VECTOR for EpiVacCorona detection.[5][6][4] More questions about the quality and protectiveness of antibodies induced by EpiVacCorona appeared along with the first results of a special antibody VECTOR’s test, when, with a positive special test, negative results of all other commercially available tests were otained: LIAISON SARS-CoV-2 S1 / S2 IgG – DiaSorin, IgM / IgG – Mindray, SARS-CoV-2 IgG – Abbott Architect, Anti-SARS-CoV-2 ELISA (IgG) – Euroimmun, Access SARS-CoV-2 IgG (RBD) – Beckman Coulter, “SARS-CoV-2-IgG-ELISA -BEST “-” Vector-Best “,” Anti-RBD IgG “- Gamaleya Research Center.[5][6][4][8] Clinical trial participants conducted their own antibody mini-study that was performed in independent Russian laboratory. The study participants asked Dr. Alexander Chepurnov, the former head of the infectious diseases department at VECTOR, who now works at another medical institute, to check neutralizing antibodies presence in their serum samples.[3] They also sent to Dr. Chepurnov control serum samples from former COVID-19 patients or people vaccinated with another Russian vaccine, Sputnik V, which is known to trigger the production of neutralizing antibodies.[32] All serum samples were blinded before antibody tests. On 23 March 2021, the participants reported the results of their mini-study in an open letter to the Ministry of Health of the Russian Federation.[6][7] According to the letter, even with the help of the VECTOR antibody detection system, antibodies were detected only in 70-75% of those vaccinated with EpiVacCorona. However, the level of antibodies was very low. Moreover, according to the letter, virus-neutralizing antibodies were not detected in the independent research Dr. Alexander Chepurnov laboratory at all.[3][6][7] The trial participants asked Ministry of Health in their open letter to perform independent study for the verification of their findings.[3][6][7] In addition, the letter reports 18 cases of COVID-19 cases as of March 22, 2021 among those who received the vaccine and became ill (sometimes severe) three weeks or later after the second dose of EpiVacCorona.[33][6][7] April 20, 2021 the study participants got a reply, with refusal of performing any additional verification antibody tests or investigation of sever COVID-19 cases among vaccinated individuals. The reply include the following text: “Considering that the listed immunobiological preparations (vaccines) for the prevention of COVID-19 are registered in the prescribed manner, their effectiveness and safety have been confirmed.”
Vaccine criticism by independent experts
Some independent experts criticized the vaccine design[3][4] and clinical data presentation in the publication.[8][9][10] The experts are saying that peptide selection is “crucial” for the innovative peptide approach, which VECTOR uses for EpiVacCorona design. However, some researchers are not convinced that the viral spike protein peptides selected for the vaccine are actually “visible” by human immune system.[3][4][34] They stated that these peptides do not overlap[35] with peptides that have been shown in several publications to contain human linear B cell epitopes in spike protein of SARS-CoV-2.[36][37][38][39][40] Moreover, the study was criticized for the lack of positive control of convalescent plasma samples in reports related to neutralizing antibody titers in vaccinated individuals.[1][10] The same study was also criticized for presence of detectable antibodies in negative controls samples that were not discussed by authors.[1][10] In addition, vaccine developers have been criticized for aggressively advertising their vaccine efficacy prior to the completion of phase III clinical trial. The most substantial criticism came from Dr. Konstantin Chumakov, who currently serves as the Associate Director for Research at the FDA Office of Vaccines Research and Review. Dr. Chumakov said: “I would not be in a hurry to call this peptide formulation a vaccine yet, because its effectiveness has not yet been proven…For the introduction of such a vaccine, the level of evidence must be much higher, and therefore the developers of EpiVacCorona, before launching their vaccine on the market, had to conduct clinical trials and prove that their vaccine actually protects against the disease. However, such tests were not carried out, which is absolutely unacceptable.”[41]
The title page of the “EpiVacCorona” patent with Anna’s Popova name among inventors
Conflict of interest
The vaccine design was protected by several already issued patents (see section above). In each patent one of its co-authors is a namesake of Anna Popova who is a head of the Federal Service for Supervision of Consumer Rights Protection and Human Welfare. This patent authorship represents an issue as far as Anna Popova is a head of the Russian agency that is charged with overseeing vaccine safety and efficacy. As a co-author of these patents, she might have an interest in promoting the vaccine despite its shortcomings.
^MKIEEGKLVIWINGDKGYNGLAEVGKKFEKDTGIKVTVEHPDKLEEKFPQVAATGDGPDIIFWAHDRFGGYAQSGLLAEITPDKAFQDKLYPFTWDAVRYNGKLIAYPIAVEALSLIYNKDLLPNPPKTWEEIPALDKELKAKGKSALMFNLQEPYFTWPLIAADGGYAFKYENGKYDIKDVGVDNAGAKAGLTFLVDLIKNKHMNADTDYSIAEAAFNKGETAMTINGPWAWSNIDTSKVNYGVTVLPTFKGQPSKPFVGVLSAGINAASPNKELAKEFLENYLLTDEGLEAVNKDKPLGAVALKSYEEELAKDPRIAATMENAQKGEIMPNIPQMSAFWYAVRTAVINAASGRQTVDEALKDAQTNSSSNNNNNNNNNNLGDNGPQNQRNAPRITFGGPSDSTGSNQNGERSGARSKQRRPQGLPNNTASWFTALTQHGKEDLKFPRGQGVPINTNSSPDDQIGYYRRATRRIRGGDGKMKDLSPRWYFYYLGTGPEAGLPYGANKDGIIWVATEGALNTPKDHIGTRNPANNAAIVLQLPQGTTLPKGFYAEGSRGGSQASSRSSSRSRNSSRNSTPGSSRGTSPARMAGNGGDAALALLLLDRLNQLESKMSGKGQQQQGQTVTKKSAAEASKKPRQKRTATKAYNVTQAFGRRGPEQTQGNFGDQELIRQGTDYKHWPQIAQFAPSASAFFGMSRIGMEVTPSGTWLTYTGAIKLDDKDPNFKDQVILLNKHIDAYKTFPPTEPKKDKKKKADETQALPQRQKKQQTVTLLPAADLDDLSKQLQQSMSSADSTQA. “Carrier protein sequence”.
EpiVacCorona Vaccine, developed by the Vektor State Research Center of Virology and Biotechnology in Russia, is based on peptide-antigens that facilitate immunity to the SARS-CoV-2 virus1. It is currently being tested in Phase I/II clinical trials for safety and immunogenicity (NCT04527575)1,2.
Precision Vaccinations: VACCINE INFO EpiVacCorona Vaccine [Link]
The Pharma Letter: Russia’s EpiVacCorona vaccine post-registration trials started [Link]
In February 2021, global data from Phase III trials and 101 COVID cases showed that the vaccine had a 65.7% efficacy in preventing moderate symptoms of COVID-19, and 91% efficacy in preventing severe disease.[8] It has similar efficacy to Johnson & Johnson’s Ad26.COV2.S, another one-shot adenovirus vector vaccine with 66% efficacy in a global trial.[9][1] Convidecia is similar to other viral vector vaccines like AZD1222, Gam-COVID-Vac, and Ad26.COV2.S.[10] Its single-dose regimen and normal refrigerator storage requirement (2°to 8 °C) could make it a favorable vaccine option for many countries.[9]
Convidecia is approved for use by some countries in Asia,[11][12][13] Europe,[14][15] and Latin America.[16][17][18] Production capacity for Ad5-NCov should reach 500 million doses in 2021. Manufacturing will take place in China,[19] Malaysia,[13] Mexico,[20] and Pakistan.[21]
Ad5-nCoV is a recombinant adenovirus type-5 vector (Ad5) vaccine currently being investigated for prophylaxis against SARS-CoV-2.1,2 It is being developed by CanSino Biologics Inc., in partnership with the Beijing Institute of Biotechnology, who in March 2020 announced the approval of a phase I clinical trial (ChiCTR2000030906)1 with an expected completion in December 2020. The study will evaluate antibody response in healthy patients between the ages of 18 and 60 who will receive one of three study doses, with follow-up taking place at weeks 2 and 4 and months 3 and 6 post-vaccination.2
Chinese Clinical Trial Register: A phase I clinical trial for recombinant novel coronavirus (2019-COV) vaccine (adenoviral vector) [Link]
In February 2021, data released from an interim analysis of Phase III trials with 30,000 participants and 101 COVID cases showed that globally, the vaccine had an efficacy of 65.7% at preventing moderate cases of COVID-19 and 90.98% efficacy at preventing severe cases. In the Pakistan trial subset, the vaccine had an efficacy of 74.8% at preventing symptomatic cases 100% for preventing severe disease.[8]
While the efficacy rates were lower than the Pfizer–BioNTech and Moderna vaccines, its single-dose regimen and normal refrigerator storage requirement (2 to 8 °C) could make it a favorable option for many countries. It has similar efficacy to Johnson & Johnson’s Ad26.COV2.S, another one-shot adenovirus vaccine found to be 66% effective in a global trial.[9][1]
Clinical trials
Phase I-II
In early 2020, Chen Wei led a joint team of the Institute of Biotechnology, the Academy of Military Medical Sciences and CanSino Biologics to develop AD5-nCOV. According to the Chinese state media, the team registered an experimental COVID-19 vaccine for Phase Itrial in China on 17 March 2020 to test its safety. The trial was conducted on 108 healthy adults aged 18 to 60 in two medical facilities in Wuhan, Hubei province.[23]
In April, Ad5-nCoV became the first COVID-19 vaccine candidate in the world to begin Phase II trials.[24] The Phase II trial results were published in the peer-reviewed journal The Lancet in August 2020, and noted neutralizing antibody and T cell responses based on statistical analyses of data involving 508 eligible participants.[25] In September, Zeng Guang, chief scientist of the Chinese Center for Disease Control and Prevention said the amount of COVID-19 antibodies in subjects from the Phase I trials remained high six months after the first shot. Zeng said the high levels of antibodies suggested the shots may provide immunity for an extended period of time, although Phase III results were still required.[26] On September 24, CanSino began Phase IIb trials on 481 participants to evaluate the safety and immunogenicity of Ad5-nCoV for children ages 6–17 and elderly individuals ages 56 and above.[27]
On 16 May 2020, Canadian Prime Minister Justin Trudeau announced Health Canada had approved Phase II trials to be conducted by the Canadian Center for Vaccinology (CCfV) on the COVID-19 vaccine produced by CanSino. Scott Halperin, director of the CCfV said the vaccine would not be the only one going into clinical trials in Canada, and any potential vaccine would not be publicly available until after Phase 3 is complete.[29][30] If the vaccine trials were successful, then the National Research Council would work with CanSino to produce and distribute the vaccine in Canada.[30] In August 2020, the National Research Council disclosed the vaccine had not been approved by Chinese customs to ship to Canada, after which the collaboration between CanSino and the Canadian Center for Vaccinology was abandoned.[31]
Nasal spray trials
In September, CanSino began a Phase I trial in China with 144 adults to determine the safety and immunogenicity of the vaccine to be administered as a nasal spray, in contrast with most COVID-19 vaccine candidates which require intramuscular injection.[32] On June 3, 2021, Chen Wei announced the expansion of clinical trials was approved by the NMPA, in the meantime, they are applying for Emergency Use Listing for the nasal spray.[33]
Phase III
In August, Saudi Arabia confirmed it would begin Phase III trials on 5,000 people for Ad5-nCoV in the cities of Riyadh, Dammam, and Mecca.[7]
In October, Mexico began Phase III trials on 15,000 volunteers.[34][4]
In September, Russia began Phase III trials on 500 volunteers,[35] which Petrovax later received approval from the government to expand to 8,000 more volunteers.[36][6]
In September, Pakistan began Phase III trials on 40,000 volunteers as part of a global multi-center study.[5] As of December, about 13,000 volunteers have participated in trials of Ad5-nCoV.[22]
In April 2021, a new trial was registered in Jiangsu involving one dose of Convidecia followed by a dose of ZF2001 28 or 56 days later using different technologies as a way to further boost efficacy.[38]
Manufacturing
In February, Chen Wei who lead the development of the vaccine, said annual production capacity for Ad5-NCov could reach 500 million doses in 2021.[19]
In February, Mexico received the first batch of active ingredients for Convidecia, which is being packaged in Querétaro by Drugmex.[20]
In Malaysia, final filling and packaging of the vaccine for distribution would be completed by Solution Biologics.[13]
In May, Pakistan began filling and finishing 3 million doses a month at the National Institute of Health, which would be branded as PakVac for domestic distribution.[39]
If the vaccine is approved in Russia, Petrovax said it would produce 10 million doses per month in 2021.[40]
Marketing and deployment
Full authorization Emergency authorization Eligible COVAX recipient (ongoing assessment)[41]
On 25 June 2020, China approved the vaccine for limited use by the military.[42] In February 2021, China approved the vaccine for general use.[11]
In February, Malaysia‘s Solution Biologics agreed to supply 3.5 million doses to the government.[43] The doses would be delivered starting in April with 500,000 complete doses, with the rest in bulk to be finished by Solution Biologics.[13]
In October, Indonesia reached an agreement with CanSino to deliver 100,000 doses in November 2020, with the expectation that an additional 15 to 20 million doses would be delivered in 2021.[44]
In February, Pakistan approved the vaccine for emergency use.[45] The country purchased 20 million doses of the vaccine[12] of which the first 3 million doses are to arrive in May.[12]
Europe
In March, Hungary granted emergency use approval for the vaccine.[14]
In March, Moldova authorized use of the vaccine.[46]
North America
In December 2020, Mexico‘s Foreign Minister Marcelo Ebrard signed an agreement for 35 million doses.[47] In February, Mexico approved the vaccine for emergency use.[48] Mexico received active ingredients for 2 million doses with a total of 6 million doses expected to arrive in February.[16]
South America
In June, Argentina approved emergency use of the vaccine and ordered 5.4 million doses.[17]
In June, Brazil announced plans to purchase 60 million doses.[49] In May, Brazil began reviewing the vaccine for emergency use.[50]
In March, Chile signed a deal for 1.8 million doses for delivery between May and June,[51] for which emergency use approval was granted in April.[18]
In June, Ecuador approved emergency use and ordered 6 million doses for delivery between June and August 2021.[52]
An emergency license was issued for the use of the Iranian-made “CovIran Barakat” vaccine against the coronavirus yesterday on June 13, the Iranian Minister of Health and Medical Education Saeed Namaki said, Trend reports citing IRNA.
He made the remark in an event dedicated to the launch of a number of health and medical facilities in Iran’s Markazi Province today on June 14.
Namaki said that moreover, a license for the using of the Iranian-made “Pastor” vaccine against the coronavirus will be issued next week.
“Also, the licenses for the using of Iranian-made “Razi” and “Fakhra” vaccines will be issued in the near future,” he added.
According to the minister, the Iranian population will be vaccinated fully by the end of autumn with the opportunities created in connection with the production of vaccines in Iran.
Reportedly, about 10 million people in Iran are planned to be vaccinated with the “CovIran Barakat’ vaccine next week. The production of “CovIran Barakat” vaccine in Iran is expected to reach 50 million doses per month by the end of the summer.
On June 14, 26 health and medical facilities were launched in Iran’s Markazi Province. A total of 1.45 trillion rials (about $34.5 million) has been spent on these facilities.
Iran continues to monitor the coronavirus situation in the country. According to recent reports from Iranian officials, over 3.03 million people have been infected, and 82,217 people have already died.
Meanwhile, over 2.66 million people have reportedly recovered from the disease.
The country continues to apply strict measures to contain further spread. Reportedly, the disease was brought to Iran by a businessman from Iran’s Qom city, who went on a business trip to China, despite official warnings. The man died later from the disease.
The Islamic Republic only announced its first infections and deaths from the coronavirus on Feb. 19.
The outbreak in the Chinese city of Wuhan – which is an international transport hub – began at a fish market in late December 2019.
The World Health Organization (WHO) on March 11 declared COVID-19 a pandemic. Some sources claim the coronavirus outbreak started as early as November 2019.
A total of 5.2 million people have been vaccinated in Iran so far. About 4.35 million people were vaccinated on the first stage, and 851,000 people were vaccinated on the second stage.
COVIran Barakat is a COVID-19 vaccine developed by Iranian state-owned Shifa Pharmed Industrial Group. It has successfully been tested on animals and has been approved by the Iran Food and Drug Administration for testing on humans.[1][2][3]Phase 2/3 (II/III) clinical trial began on 13 March 2021,[4] and the first participants were inoculated on March 29.[5] Finally, the vaccine consumption license was issued on June 13, 2021.[6] Around 650 people worked in 3 shifts around the clock to develop the vaccine.[7]
Dr. Minoo Mohraz has been selected as the lead of the “Corona vaccine project in Iran”.[8] Dr. Mohraz is an Iranian physician, scientist, and AIDS specialist. She is a Full Professor (Emeritus) of Infectious Diseases at Tehran University of Medical Sciences and head of the Iranian Centre for HIV/AIDS.[9] Dr. Mohraz has also served as within the World Health Organization as an expert on HIV/AIDS in Iran and the Eastern Mediterranean.[10]
This vaccine has been authorised for emergency use by the Iranian authorities. This makes it the first locally developed to be approved for emergency use in the Middle East.[11]
Technology
On 29 December 2020, human trials of Iran’s first domestic COVID-19 vaccine candidate were started. The mechanism of production of this vaccine is based on the inactivated vaccine. In other words, “it is made of a coronavirus that has been weakened or killed by chemicals, similar to how polio immunizations are made.”[12]
Development
Iran’s first domestic COVID-19 vaccine candidate was started
Tayyebeh Mokhber, the first volunteer who receives a shot of COVIran Barakat was the daughter of Mohammad Mokhber director of setad. Minister of Health Saeed Namaki and Vice President for Science and Technology Sorena Sattari participated at the ceremony of vaccine injection. According to reports, there are more than 65,000 Iranians volunteered to test the vaccine and 56 selected people took part in the first phase of human trials which last 45 to 60 days.[13] The initial phase of human-testing for this vaccine started with the injection of 56 volunteers who were at the age of 18-50.[14][15][16]
The second/third group of volunteers were also injected with the vaccine.[17][18] According to the head of the vaccine production team at the Setad, the results show that this vaccine also neutralizes the British mutated COVID-19 virus.[19][20][21]
In March 2021, the Executive Office of Imam Khomeini’s Order began a Phase II–III clinical trial of COVIran Barakat with 280 participants in cities including Tehran, Mashhad, Karaj, Esfahan, Shiraz. According to the allowance of medical equipment department, the second phase coincided with third phase.[22][23] The vaccine has reached its third phase of human-testing;[24] and the first injection(s) of the 3rd phase began 25 April 2021.[25]
As official in charge of manufacturing Iran Barakat vaccines, Mohammad Reza Salehi said, “some neighboring countries tend to enter the third phase of the clinical trial of the Iranian “COVIran Barakat””. They are reviewing recommendations to let them participate.[14]
Production
According to Setad (the Executive Headquarters of Imam’s Directive), under the direct control of the Supreme Leader of Iran, “production of the vaccine developed by one of its companies, Shifa Pharmed, could reach 12 million doses per month, six months after a successful trial ends”.[26] On 15 March 2021, he stated that EIKO has already a capacity of three million doses per month and that by end of June the capacity will be 15-20 million doses per month.[27][28]
On 29 March 2021, the Tehran Times reported that a capacity of three million doses per month was achieved;[citation needed] and the production line of 25 million doses per month of Iran Koo vaccine was discharged on 26 April 2021.[29]
On 10 May 2021, the first product of mass production of the Iranian corona vaccine called “COVIran Barakat” was unveiled in phase one of the vaccine production factory associated with Execution of Imam Khomeini’s Order (EIKO). Therefore, 2 industrial lines have been set up. The first production line is prepared and the second line is being prepared. By the end of September (taking into account the capacity of three million doses of the first line), 20 million doses of Iran Barakat vaccine will be available in the month.[30]
QazCovid-inVaccinePhase I/II/IIIThe QazCovid-in vaccine is an inactivated vaccine. Inactive viral vaccines are created by propagating viruses in cell culture (such as in Vero cells) and/or by inactivation using a chemical reagent (such as beta-propiolactone or formaldehyde). Upon vaccination, this allows the body to generate a diverse immune response against numerous viral antigens while having no threat of actually being infected because the virus is inactive.NEWS FEED December 31, 2020The Republic of Khazakstan’s QazCovid-in COVID19 vaccine enters phase 3 with an expected 3000 participants. August 28, 2020QazCovid-in, an inactive viral vaccine manufactured by Research Institute for Biological Safety Problems Republic of Kazakhstan enters Phase 1/2 clinical trials.ORGANIZATIONSResearch Institute for Biological Safety Problems, National Scientific Center for Phthisiopulmonology of the Republic of Kazakhstan, City polyclinic No. 4 of the UZO of Almaty, Clinic of the International Institute of Postgraduate Education, City Multidisciplinary Hospital of the Health Department of the Akimat of Zhambyl RegionCOUNTRIES INVOLVED TRIAL PARTICIPANTS
A new vaccine on the scene: Kazakhstan begins rollout of homegrown QazVac
The world’s approved COVID-19 vaccines have all come from large economies such as the U.S., China, the U.K., Russia, and India. Until today.
On Monday, Kazakhstan started rolling out its homegrown vaccine, now known as QazVac. Before a rebranding at the end of last month, it was called QazCovid-in, but the central Asian country’s government decided that name might be a turnoff for the public.
The vaccine was developed by Kazakhstan’s Research Institute for Biological Safety Problems, which claimed 96% efficacy in the second stage of clinical trials. The final phase is still ongoing, with a conclusion expected in July, but Kazakh health authorities decided it was fine to begin the rollout as long as the 3,000-participant Phase III trial was at least halfway finished.
This isn’t an adenovirus vector vaccine like those from Johnson & Johnson and AstraZeneca—though it does share their relatively mild refrigeration requirements—nor is it an mRNA-based jab like the BioNTech/Pfizer and Moderna vaccines. Instead, it uses an inactivated form of the SARS-CoV-2 virus itself, much like China’s CoronaVac and India’s Covaxin, which are both in use, and Valneva’s vaccine, which isn’t there yet. The QazVac regimen comprises two doses, to be administered three weeks apart.
‘Turn the tide’
Health Minister Alexei Tsoi was one of the first QazVac recipients on Monday morning. Tsoi was at the start of this month on the receiving end of a public dressing-down by President Kassym-Jomart Tokayev, who was furious about the sluggish start to the country’s inoculation campaign amid rising case numbers.
“You must turn the tide, otherwise a personnel decision that is going to be very disappointing for you will follow,” Tokayev told Tsoi. The vaccination campaign, which had previously focused on frontline workers, then reportedly sprang to life for others too in the oil-rich country.
Thus far, Kazakhstan’s vaccination drive has been powered by Russia’s Sputnik V, which has been produced locally for the past couple of months (Tokayev opted for the Russian shot, rather than waiting for QazVac). By late last week, just over 800,000 people had received their first dose. Kazakhstan has a population of 18.8 million people; the government plans to inoculate 2 million each month.
Tokayev tweeted Friday that domestic production would provide vaccine availability to all citizens. If so, that would be a remarkable turnaround—Almaty health officials said five weeks ago that the largest Kazakh city had run out of vaccines, and mass vaccination would not be realistic in the near future.
QazVac may have given Tokayev the opportunity to praise Kazakhstan’s scientific prowess, but production remains a bottleneck. The first batch to be distributed runs to only 50,000 doses, and the next tranche, to be produced in May, will be of the same volume.
Tsoi said Monday that the Kazakh government was talking to Turkish manufacturers about increasing production capacity.
QazCovid-in, commercially known as QazVac,[1][2] is a COVID-19 vaccine developed by the Research Institute for Biological Safety Problems in Kazakhstan.[3][4][5] QazCoVac-P is a second COVID-19 vaccine developed by the Kazakh Biosafety Research Institute and in clinical trials.[6]
Clinical research
QazVac is currently in Phase 3 (III) of the Clinical Trial, which is expected to be fully completed by 9 July 2021.[7][8] It is unclear when the first preliminary results will be published.[9][10]
The administration of the vaccine for the general population began at the end of April 2021.[11] The Research Institute Kunsulu Zakarya’s Director General’s justification is that the trial is almost 50% completed and “people who have received [the] vaccine feel well; there have been no side-effects and the effectiveness of the vaccine is high”.[12]
Production
The vaccine was first manufactured by Kazakhstan’s Research Institute of Biological Safety Problems. Production capacity has been capped at 50,000 doses per month.
Beginning in June 2021, the vaccine is slated[13] to be packaged in large bulk to be bottled in Turkey by a major Turkish company.[14][15] This will allow for a production capacity of 500,000-600,000 doses per month.[16] The contract is still being negotiated,[17] despite earlier claims that suggesting the deal had already been finalized.[18][19]
Vaccine innoculation
The first batch of 50,000 doses was delivered on 26 April 2021, and vaccination began shortly after.[20] In June 2021, the capacity will increase to 100,000 doses per month, regardless of the contract for bottling in Turkey.[21]
The vaccine can be stored at standard refrigeration temperatures (2°C-8°C) and is a two-dose régime with the doses administered twenty-one days apart.[22]
The QazCovid-in vaccine, an inactivated vaccine, was developed and tested in the Kazakh Research Institute for Biological Safety Problems1. It demonstrated high efficacy, safety, and immunogenicity at 96% in initial Phase I and II trials (NCT04530357), and will now be undergoing upcoming Phase III trials2,3.
The Astana Times: Kazakhstan Begins Vaccinating 3,000 Volunteers With Self-Made QazCovid-in [Link]
The Lancet: COVID-19 response in central Asia [Link]
Cuba says Abdala vaccine 92.28% effective against coronavirus
The announcement came just days after the government said another homegrown vaccine, Soberana 2, has proved to be 62% effective with just two of its three doses.
The announcement came just days after the government said another homegrown vaccine, Soberana 2, has proved to be 62% effective with just two of its three doses.
“Hit by the pandemic, our scientists at the Finlay Institute and Center for Genetic Engineering and Biotechnology have risen above all the obstacles and given us two very effective vaccines,” President Miguel Diaz-Canel tweeted.
The announcement came from state-run biopharmaceutical corporationBioCubaFarma, which oversees Finlay, the maker of Soberana 2, and the Center for Genetic Engineering and Biotechnology, the producer of Abdala.
Both vaccines are expected to be granted emergency authority by local regulators shortly.
Cuba, whose biotech sector has exported vaccines for decades, has five coronavirus vaccine candidates.
The Caribbean’s largest island is facing its worst Covid-19 outbreak since the start of the pandemic following the arrival of more contagious variants, setting new records for daily coronavirus cases.
The Communist-run country has opted not to import foreign vaccines but to rely on its own. Some experts said it was a risky bet but it appears to have paid off, putting Cuba in position to burnish its scientific reputation, generate much-needed hard currency through exports and strengthen the vaccination drive worldwide.
Several countries from Argentina and Jamaica to Mexico, Vietnam and Venezuela have expressed an interest in buying Cuba’s vaccines. Iran started producing Soberana 2 earlier this year as part of late-phase clinical trials.
Cuba’s authorities have already started administering the experimental vaccines en masse as part of “intervention studies” they hope will slow the spread of the virus.
About a million of the country’s 11.2 million residents have been fully vaccinated to date.
Daily cases have halved in the capital, Havana, since the start of the vaccination campaign a month ago, using Abdala, according to official data.
Cuba has reported a total of 169,365 Covid-19 cases and 1,170 deaths.
ABDALA, technical name CIGB-66, is a COVID-19 vaccine candidate developed by the Center for Genetic Engineering and Biotechnology in Cuba.[1][2] This vaccine candidate, named after a patriotic drama by Cuban independence hero José Martí, is a protein subunit vaccine containing COVID-derived proteins that trigger an immune response.[3] However, none of the clinical trial full results have been published. This candidate followed a previous one called CIGB-669 (MAMBISA).[4]
In July 2020, CIGB-66 commenced phase I/II clinical trials.[8]
Phase III
The Phase III trial compares 3 doses of the vaccine administered at 0, 14 and 28 days against a placebo, with the primary outcome measuring the proportion of cases reported for each group 14 days after the third dose.
The trial was registered on 18 March 2021. The first dose was administered on 22 March and by April 4, the 48,000 participants had received their first dose,[9][10] and second doses started being administered from April 5.[11][12] Third doses have started being administered on 19 April[13][14][15] and on May 1, 97% of the original participants had received their 3 doses, the others 3% were lost in the process.
Intervention study
124,000 people aged 19 to 80 received 3 doses of the vaccine as part of an intervention study, with the primary outcome measuring the proportion of cases and deaths for the vaccinated compared to the unvaccinated population.[16]
A wider intervention study with the 1.7 million inhabitants of Havana is expected to start in May with the ABDALA and Soberana 2 vaccine.[17]
Efficacy
From May 3, the efficacy of the vaccine will start being evaluated.[18][19][20]
The “first evaluation of efficacy” can begin when there is 50 cases, then there is a second evaluation at 100 cases and a definitive efficacy can “finally be demonstrated” at 150 cases, Cuban Center for Genetic Engineering and Biotechnology director said.[21]
Production outside Cuba
Venezuela has claimed that it will manufacture the vaccine[22] but this claim has not yet materialised.[23] State-owned EspromedBIO will manufacture the vaccine but some “arrangements” are needed to start production.[24] In April, Nicolás Maduro said that a capacity of 2 Million doses per month is hoped to be reach by “August, September approximately”.[25
In June 2021, Vietnam’s Ministry of Health announced that negotiations were ongoing between Cuba and Vietnam for Abdala vaccine production. The Institute of Vaccines and Medical Biologicals (IVAC) was named as the focal point for receiving technology transfer.[26]
Hadassah Medical Center; Sheba Medical Center Hospital
The SARS-CoV-2 virus is responsible for the COVID-19 pandemic. The pandemic emerged from Wuhan Province in China in December 2019 and was declared by the WHO Director-General a Public Health Emergency of International Concern on 30 January 2020.
In this study, a vaccine developed by IIBR for SARS-CoV-2 virus will be assessed for its safety and potential efficacy in volunteers. The study is comprised of two phases, a dose-escalation phase (phase I) during which subjects (18-55 years old) will be randomly allocated to receive a single administration of IIBR-100 100 at low, mid or high dose or saline or two administrations of IIBR-100 at low dose, or saline, 28 days apart.
Based on results obtained during phase I, and cumulative phase I data review, the expansion phase (phase II) has begun, during which larger cohorts as well as elderly age subjects will be randomly allocated to receive a single administration of IIBR-100 at low, mid or high dose or saline, or two administrations of IIBR-100 at low, mid or high dose (prime-boost) or saline, 28 days apart. Additional top-dose (prime-boost) may be implemented when immunogenicity of any prime-boost arm is considered insufficient.
Based on immunogenicity preliminary data and DSMB recommendations, the two administrations of mid, high and top dose (prime-boost) or saline will continue.
The subjects will be followed for a period of up to 12 months post last vaccine administration to assess the safety and efficacy of the vaccine.
^ Clinical trial number NCT04608305 for “Phase I/II Randomized, Multi-Center, Placebo-Controlled, Dose-Escalation Study to Evaluate the Safety, Immunogenicity and Potential Efficacy of an rVSV-SARS-CoV-2-S Vaccine (IIBR-100) in Adults” at ClinicalTrials.gov
Israeli institute’s COVID vaccine candidate said very effective in animal trials
Secretive Israeli research center’s shot shows near 100% efficacy in non-human trials, is on par with US company Moderna’s candidate, TV report says
Israeli researchers at a top secret research center have made progress on a coronavirus vaccine that shows a high level of effectiveness in animals, according to a Friday TV report.
However, there is no guarantee that the vaccine under development will be effective in humans, or will be available soon.
The Israel Institute for Biological Research (IIBR), a secretive unit that works under the Prime Minister’s Office, developed a vaccine that shows close to 100 percent protection against the virus in lab animals, the Channel 12 report said, citing “a security source.”
The vaccine under development is on par in effectiveness with a vaccine being developed by US biotechnology company Moderna, the report said.
Unlike vaccines developed abroad, the domestic vaccine will first be delivered to Israeli citizens, it added. If successful, it was expected to provide protection against the disease with a single dose.
The institute has not started human trials but was preparing to manufacture 10 to 15 million doses, report said.
Hebrew media have reported on potential breakthroughs at the shadowy institute several times before, starting in mid-March, with the Defense Ministry pushing back on some of the claims to tamper expectations.
Magen David Adom medical workers test Israelis for the coronavirus at a drive-through site in Lod, on July 10, 2020. (Yossi Aloni/Flash90)
IIBR said last month that it had completed successful coronavirus vaccine trials on rodents, paving the way for further testing on other animals and then possibly human trials.
In a paper published on the website of bioRxiv, an online repository for papers that haven’t yet been peer-reviewed, the institute, which is based in Ness Ziona, said it hopes to have a finished vaccine in a year, or possibly even earlier.
In the abstract of the report, the researchers say their vaccine, which they tested on hamsters, “results in rapid and potent induction of neutralizing antibodies against SARS-CoV-2,” the virus that causes COVID-19.
Earlier this month a vaccine adviser to the government cautioned that there was no guarantee that the shots being developed will prove widely effective.
In May, the institute confirmed that it had isolated an antibody it believed could be used to develop treatments against the virus. The development would not be useful in the creation of a vaccine, but would rather be a move toward a drug treatment for those who have already contracted the disease.
Tal Zaks, Moderna’s Israeli chief medical officer, described to Channel 12 on Friday the company’s push into Phase 3 testing of its vaccine candidate, which was developed with the National Institutes of Health, and began its first injections Monday.
The trial, the world’s largest vaccine study, plans to test the vaccine on 30,000 volunteers.
There’s still no guarantee that the experimental vaccine, developed by the National Institutes of Health and Moderna Inc., will really offer protection.
“The first time we saw the first model, that the vaccine, even if it’s just in mice, successfully stimulated the immune system to identify the virus and neutralize it, I knew that we hadn’t missed anything, that we had the correct vaccine,” he said.
“And of course the second ‘ah-ha’ moment was when we saw the first clinical results, when it was clear that in humans we weren’t just getting to antibody levels we were seeing in sick people, which is what we aspired to, but we were getting to even higher levels,” Zaks said.
A Nurse gives a volunteer an injection, as the world’s biggest study of a possible COVID-19 vaccine, developed by the US National Institutes of Health and Moderna Inc., gets underway on July 27, 2020, in Binghamton, NY. (AP Photo/Hans Pennink)
Last month Israel signed a deal with Moderna for the potential purchase of its coronavirus vaccine if it ends up proving effective.
Moderna said the vaccination was administered in Savannah, Georgia, the first site to get underway among more than seven dozen trial sites scattered around the country.
Several other vaccines made by China and by Britain’s Oxford University earlier this month began smaller final-stage tests in Brazil and other hard-hit countries.
The massive studies aren’t just to test if the shots work — they’re needed to check each potential vaccine’s safety. And following the same study rules will let scientists eventually compare all the shots.
It normally takes years to create a new vaccine from scratch, but scientists are setting speed records this time around, spurred by knowledge that vaccination is the world’s best hope against the pandemic.
If everything goes right with the final studies, it still will take months for the first data to trickle in from the Moderna test, followed by the Oxford one.
Governments around the world are trying to stockpile millions of doses of those leading candidates so if and when regulators approve one or more vaccines, immunizations can begin immediately. But the first available doses will be rationed, presumably reserved for people at highest risk from the virus.
Coronavirus cases in Israel rose by 1,791 in 24 hours on Friday and the national death toll hit 512, according to the latest Health Ministry figures.
The total case count stood at 70,970, with 320 patients in serious condition, including 98 on ventilators. The number of recovered patients reached 43,850.
Israel has the fifth-highest number of new coronavirus infections per capita in the world, overtaking the United States, according to data compiled by a scientific publication based at Oxford University.
And while Israel has seen the number of new coronavirus cases rocket to more than 2,000 a day in recent weeks, a new Hebrew University report published on Thursday asserted that Israel has managed to gain control of the second wave of the coronavirus, thanks to a recent stabilization in the number of seriously and moderately ill patients.
The curve for seriously and moderately ill patients began to spike in late June before stabilizing in recent days, the researchers reported. They credited the restrictions imposed by the government in recent weeks to limit crowding for helping to flatten the curve.
According to the report, the death toll will climb by roughly 200 in the coming three weeks as a result of the high infection rate over the past month.
Experts have blamed a too-speedy reopening and the lack of an effective contact-tracing program as main factors in the virus resurgence, which has come as new daily coronavirus cases around the world have also reached record highs.
launched 2012, as forxiga in EU, FDA 2014, JAPAN PMDA 2014
Dapagliflozin propanediol monohydrate was first approved by European Medicine Agency (EMA) on November 12, 2012, then approved by the U.S. Food and Drug Administration (FDA) on January 8, 2014, and approved by Pharmaceuticals and Medical Devices Agency of Japan (PMDA) on March 24, 2014. It was co-developed and co-marketed as Forxiga® by Bristol-Myers Squibb and AstraZeneca in EU.
Dapagliflozin propanediol monohydrate is a sodium-glucose co-transporter 2 (SGLT2) inhibitor indicated as an adjunct to diet and exercise to improve glycemic control in adults with type 2 diabetes mellitus.
Forxiga® is available as tablet for oral use, containing 5 mg or 10 mg of free Dapagliflozin. The recommended starting dose is 5 mg once daily in the morning.
Dapagliflozin propanediol is a solvate containing 1:1:1 ratio of the dapagliflozin, (S)-(+)-1,2-propanediol, and water.
US——-In 2011, the product was not recommended for approval by the FDA’s Endocrinologic and Metabolic Drugs Advisory Committee. In 2011, the FDA assigned a complete response letter to the application. A new application was resubmitted in 2013 by Bristol-Myers Squibb and AstraZeneca in the U.S
AstraZeneca (NYSE:AZN) and Bristol-Myers Squibb Company (NYSE:BMY) today announced the U.S. Food and Drug Administration’s (FDA) Endocrinologic and Metabolic Drugs Advisory Committee (EMDAC) voted 13-1 that the benefits of dapagliflozin use outweigh identified risks and support marketing of dapagliflozin as an adjunct to diet and exercise to improve glycemic control in adults with type 2 diabetes mellitus. The Advisory Committee also voted 10-4 that the data provided sufficient evidence that dapagliflozin, relative to comparators, has an acceptable cardiovascular risk profile.
The FDA is not bound by the Advisory Committee’s recommendation but takes its advice into consideration when reviewing the application for an investigational agent. The Prescription Drug User Fee Act (PDUFA) goal date for dapagliflozin is Jan. 11, 2014.
Dapagliflozin is being reviewed by the FDA for use as monotherapy, and in combination with other antidiabetic agents, as an adjunct to diet and exercise to improve glycemic control in adults with type 2 diabetes. It is a selective and reversible inhibitor of sodium-glucose cotransporter 2 (SGLT2) that works independently of insulin to help remove excess glucose from the body. Dapagliflozin, an investigational compound in the U.S., was the first SGLT2 inhibitor to be approved anywhere in the world. Dapagliflozin is currently approved under the trade name [Forxiga](TM) for the treatment of adults with type 2 diabetes, along with diet and exercise, in 38 countries, including the European Union and Australia.
https://patents.google.com/patent/WO2017206808A1/enDaggliflozin (English name: Dapagliflozin) is a new Sodium glucose co-transporters 2 (SGLT-2) inhibitor developed by Bristol-Myers Squibb and AstraZeneca. Approved by the European Commission on November 14, 2012, and marketed in the United States on January 8, 2014, to improve glycemic control in adult patients with type 2 diabetes by combining diet and exercise; the trade name is Farxiga, currently offering 5 mg and 10 mg tablets. At the same time, a combination of dapagliflozin and metformin hydrochloride has also been marketed.The chemical name of dapagliflozin is (2S,3R,4R,5S,6R)-2-(3-(4-ethoxybenzyl)-4-chlorophenyl)-6-hydroxymethyltetrahydro-2H – pyran-3,4,5-triol, the chemical formula is C 21 H 25 ClO 6 , CAS No. 461432-26-8, the structural formula is shown as 2, clinically used as a pharmaceutical for dapagliflozin (S) -1,2-propanediol monohydrate, the structural formula is as shown in 1.
The synthesis of β-type C-aryl glycosidic bonds is a key point in the synthetic route during the preparation of dapagliflozin. At present, there are four synthetic methods for the synthesis of dapagliflozin reported in the literature and patents.Route 1: The synthetic route of dapagliflozin reported in patent WO03099836A1 is as follows:
The route uses 2-chloro-5-bromobenzoic acid (12) as raw material to react with phenethyl ether to form intermediate 11 and then triethylsilane to obtain intermediate 10; intermediate 10 and n-butyl The lithium is reacted at -78 ° C, and then subjected to a nucleophilic addition reaction with the intermediate 9, and then methoxylated to obtain the intermediate 8; the intermediate 8 is subjected to acylation reduction and deprotection to obtain the intermediate 2. The disadvantage of this method is that the β-type C-aryl glycosidic bond synthesis of the compound is carried out at a low temperature of -78 ° C, which is obviously difficult to meet the needs of industrial production; and, through nucleophilic addition, methoxylation, The five-step reaction of acetylation, reduction and hydrolysis can synthesize the β-type C-aryl glycosidic bond. The procedure is relatively long, and the purity of the intermediate 2 is only 94%.Route 2: The synthetic route of dapagliflozin reported in the literature OrgLett.2012, 14, 1480 is as follows:
The intermediate 14 of the route is reacted with di-n-butyl-n-hexylmagnesium for 48 hours at 0 ° C, and then reacted with zinc bromide to prepare an organozinc reagent by Br/Mg/Zn exchange reaction, and then with intermediate 4 Intermediate 3 was prepared by nucleophilic substitution reaction; finally, intermediate 2 was obtained by deprotection with sodium methoxide. The synthesis method is relatively novel, and the synthesis step is short. However, the research experiment is conducted only as a synthesis method, and the post treatment of the intermediate 3 is performed by column chromatography. The purity of the intermediate 2 produced was not reported. Moreover, the di-n-butyl-n-hexylmagnesium reagent used in the route is not a commonly used reagent, and is not commercially available in China. It can only be prepared by reacting dibutylmagnesium with n-hexyllithium reagent before the test, and the operation is cumbersome and difficult to mass. use.Route 3: The synthetic route of dapagliflozin reported in patent WO2013068850A2 is as follows:
The route uses 1,6-anhydroglucose (20) as a raw material, protects the 2,4-hydroxyl group by tert-butyldiphenylchlorosilane, and then protects the 3-position hydroxyl group with phenylmagnesium bromide. Intermediate 18. The intermediate 14 is subjected to an Br/Mg/Al exchange reaction to prepare an organoaluminum reagent 16, which is reacted with an intermediate 18 to form an intermediate 15, and finally, deprotected to obtain an intermediate 2. The synthesis method is very novel and is also used as a synthetic methodological study. The purification of the intermediates is carried out by column chromatography. The 1,6-anhydroglucose (20) used in the route is very expensive; and the multi-step reaction in the route uses a format reagent, a preparation format reagent or an organoaluminum reagent, which is cumbersome and cumbersome to perform, and is difficult to scale synthesis. The purity of the intermediate 2 produced was not reported.Route 4: The synthetic route of dapagliflozin reported in patent WO2013152476A1 is as follows:
The route uses 2-chloro-5-iodobenzoic acid (24) as raw material to form intermediate 22 by Friedel acylation and reduction reaction, and exchange with I-Mg at -5 ° C with isopropyl magnesium chloride lithium chloride. The intermediate 8 is obtained by nucleophilic addition and methoxylation with the intermediate 9, and then the intermediate 2 is obtained by reduction with triethylsilane, and the intermediate 2 is further purified by co-crystallizing with L-valine. Finally, The pure intermediate 2 was obtained by removing L-valine. This route is a modified route of Route 1, which replaces n-butyllithium with isopropylmagnesium chloride chloride to raise the reaction temperature of the reaction from -78 °C to -5 °C. However, the problem of a long step of synthesizing a β-type C-aryl glycosidic bond still exists. The obtained intermediate 2 is not optically pure, and needs to be purified by co-crystallizing with L-valine, and the work amount of post-treatment is increased, and finally the purity of the intermediate 2 is 99.3%.Among the four synthetic routes described above for dapagliflozin, route one and route four are commonly used synthetic methods for β-type C-aryl glycosidic bonds, and the route is long, and the optical purity of the obtained product is not high, and further purification is required. Post processing is cumbersome. Moreover, the reaction required at -78 °C in Route 1 requires high equipment and high energy consumption, which undoubtedly increases the cost. Although both Route 2 and Route 3 are new methods, most of the purification of intermediates used is column chromatography. Such a process is not suitable for scale production in factories; and some of the synthetic routes are used. Reagents are not commercially available or expensive, and there is no advantage in such route costs. Therefore, there is an urgent need to find a new method for the synthesis of dapagliflozin, and to enable industrial production, and the route has a cost advantage.Repeating the procedure reported in the literature in Equation 2, the yield of Intermediate 3 was only 46%. The organic zinc reagent is prepared by Br/Mg/Zn exchange reaction, and the exchange reaction yield is 78%; and the raw material is prepared by X/Li/Zn exchange reaction to prepare an organic zinc reagent, and the exchange reaction yield is 98.5%, which is also the two Different reaction pathways lead to the essential reason for the different yields of intermediate 3. Moreover, the price of commercially available 1.0 mol/L di-n-butyl magnesium n-heptane solution 500 mL is 1380 yuan, and the price of 1.6 mol/L n-hexyl lithium n-hexane solution 500 mL is 950 yuan, and 2.5 mol/L n-butyl lithium. The price of 500 mL of n-hexane solution is only 145 yuan. Therefore, the method for preparing dapagliflozin by preparing an organozinc reagent by X/Li/Zn and then synthesizing the β-type C-aryl glycosidic bond designed by the invention has the advantages of cost, ease of operation and industrialization. Very obvious advantage.In order to solve this problem, the original compound company uses a eutectic method in the production of dapagliflozin to make dapagliflozin together with a solvent or an amino acid compound, since the compound 2 sugar ring structure contains four hydroxyl groups and is easy to absorb moisture and deteriorate. The crystal is made into a relatively stable solid, easy to store, stable and controllable in quality, and easy to prepare. Among them, the marketed dapagliflozin forms a stable eutectic with (S)-1,2-propanediol and water (1). The original crystal form patent (CN101479287B, CN103145773B) reported that all 11 crystal forms are dapagliflozin solvate or dapagliflozin. Crystal. Among them, there are two preparation methods for the da forme (S)-1,2-propanediol monohydrate (1) having a crystal structure of type Ia:Method 1: The preparation method is as follows:
Compound 7 is deprotected with sodium hydroxide to obtain compound 2, then compound 2 is extracted with isopropyl acetate, (S)-1,2-propanediol ((S)-PG) is added, and seed crystal of compound 1 is added. Then, cyclohexane was added to crystallize and separated to obtain a eutectic of the compound (1) of the type Ia.Method 2: The preparation method is as follows:
Compound 8 is subjected to reduction of methoxy group by triethylsilane and boron trifluoride diethyl ether complex, and then the reaction solution is extracted with methyl tert-butyl ether (MTBE), and (S)-1,2-propanediol ( (S)-PG), a seed crystal of the compound 1 is added, and then cyclohexane is added to crystallize, and the mixture is separated and dried to obtain a eutectic of the compound (1) of the type Ia.The above two methods for preparing the eutectic are all used in the cyclohexane solvent, which is listed in the appendix of the 2015 edition of the Pharmacopoeia (four parts) as the second type of solvent that should be restricted, with a residual limit of 0.388%. The solvent residue of the final product obtained must reach the specified limit, and the post-treatment process is complicated, time-consuming and labor-intensive, and the production cost is correspondingly increased. The invention finds a suitable solvent on the basis of the synthetic route to prepare a medicinal crystal form, and has obvious advantages in both the method and the process operation steps.The synthetic route is as follows:
Comparative Example 1, (1S)-2,3,4,6-tetra-O-pivaloyl-1,5-anhydro-1-[3-(4-ethoxyphenylmethyl)-4- Preparation of chlorophenyl]glucosamine (Compound 3)Under nitrogen protection, 1.0 mol/L di-n-butylmagnesium-n-heptane solution (16 mL) was cooled to 0 ° C, and 1.6 mol/L n-hexane lithium n-hexane solution (10 mL) was slowly added dropwise. After the addition was completed, 0 ° C After stirring for 15 h, dry n-butyl ether (2.5 mL) was added to prepare a solution of di-n-butyl-n-hexylmagnesium lithium solution, which was calibrated with iodine and stored for use.Zinc bromide (2.7 g) and lithium bromide (1.04 g) were added with n-butyl ether (20 mL), heated to 50 ° C for 4 h, and cooled for use. 4-(2-Chloro-5-bromo-benzyl) phenyl ether (6.513 g) was added with toluene (8 mL) and n-butyl ether (5 mL) under nitrogen, cooled to 0 ° C, and 0.61 mol/L was added dropwise. n-Butyl-n-hexylmagnesium lithium solution (13.1 mL), after the addition is completed, the reaction was kept at 0 ° C for 48 h, and the above-mentioned alternate zinc bromide and lithium bromide n-butyl ether solution were added, and the reaction was kept at 0 ° C for 1 h, and added 2 , 3,4,6-tetra-O-pivaloyl-α-D-bromoglucopyranose (14.49 g) in toluene (25 mL), heated to 100 ° C to stir the reaction, after TLC detection reaction, add 1 mol / L diluted hydrochloric acid (60 mL), taken after stirring extraction, the organic phase was washed with water (40 mL), then washed with saturated brine (40 mL), dried over anhydrous Na 2 SO 4, concentrated under reduced pressure, column chromatography (petroleum ether / Ethyl acetate = 20:1) 10.38 g of Compound 3 as a pale yellow oil. Yield: 46%. Purity: 99.02%. The organozinc reagent prepared by the method has an iodine calibration yield of 78%.The calibration method of the concentration of the prepared organic zinc reagent: accurately weighed iodine (1 mmol), placed in a three-necked flask, replaced nitrogen, and added anhydrous 0.5 mol/L LiCl tetrahydrofuran solution (5 mL), stirred and dissolved, and cooled to 0 ° C. The prepared organozinc reagent was slowly added dropwise until the color of the brownish yellow solution disappeared.Example 2 (1S)-2,3,4,6-tetra-O-pivaloyl-1,5-anhydro-1-[3-(4-ethoxyphenylmethyl)-4-chloro Preparation of phenyl]glucitol (compound 3)Zinc bromide (2.25 g) and lithium bromide (0.87 g) were added with n-butyl ether (30 mL), heated to 50 ° C for 2 h, and cooled for use. 4-(2-Chloro-5-iodo-benzyl) phenyl ether (7.45 g) was added with toluene (10 mL) and n-butyl ether (10 mL) under nitrogen, cooled to -20 ° C, and slowly added dropwise 1.6 mol / L-n-hexyl lithium n-hexane solution (14mL), control the internal temperature does not exceed -10 ° C, after the completion of the addition, the temperature is incubated at -20 ° C for 0.5 h, adding the above-mentioned spare zinc bromide and lithium bromide n-butyl ether solution, The reaction was stirred at 20 ° C for 3 h. Add 2,3,4,6-tetra-O-pivaloyl-α-D-bromoglucopyranose (11.59g) toluene (50mL) solution, heat to 120 ° C and stir the reaction for 4h, after TLC detection reaction, was added 1mol / L diluted hydrochloric acid (40 mL), water (20 mL), and extracted, the organic phase was washed with water (40 mL), dried over anhydrous Na 2 SO 4, concentrated with n-heptane (15mL) and methanol (60 mL) and recrystallized 10.8 g of Compound 3 as a white solid was obtained in a yield: 72.42%. Purity: 99.47%. Melting point: 99.5 to 101.6 °C. (The organic zinc reagent prepared by this method was iodine-calibrated in a yield of 98.5%.) ESI-MS (m/z): 767.30 [M+Na] + . 1 H-NMR (400 MHz, CDCl 3 ): δ 7.33 (1H, d), 7.14-7.17 (2H, m), 7.05 (2H, d), 6.79-6.81 (2H, dd), 5.39 (1H, t ), 5.21-5.31 (2H, m), 4.33 (1H, d), 4.17-4.20 (1H, dd), 3.94-4.11 (5H, m), 3.79-3.83 (1H, m), 1.39 (3H, t ), 1.20 (9H, s), 1.16 (9H, s), 1.11 (9H, s), 0.86 (9H, s).Example 3, (1S)-2,3,4,6-tetra-O-pivaloyl-1,5-anhydro-1-[3-(4-ethoxyphenylmethyl)-4-chloro Preparation of phenyl]glucitol (compound 3) PrepareZinc bromide (3.38 g) and lithium bromide (1.3 g) were added with n-butyl ether (40 mL), heated to 50 ° C for 2 h, and cooled for use. 4-(2-Chloro-5-iodo-benzyl) phenyl ether (7.45 g) was added with toluene (20 mL) and n-butyl ether (5 mL) under nitrogen, cooled to -50 ° C, and slowly added dropwise 2.5 mol / L-butyllithium hexane solution (8mL), control the internal temperature does not exceed -30 ° C, after the addition is completed, the reaction is kept at -50 ° C for 10 h, adding the above-mentioned alternate zinc bromide and lithium bromide n-butyl ether solution, The reaction was stirred at -20 ° C for 10 h. Add 2,3,4,6-tetra-O-pivaloyl-α-D-bromoglucopyranose (34.77g) toluene (80mL) solution, heat to 100 ° C and stir the reaction for 24h, after TLC detection reaction, was added 1mol / L diluted hydrochloric acid (60 mL), water (50 mL), and extracted, the organic phase was washed with water (40 mL), dried over anhydrous Na 2 SO 4, concentrated with n-heptane (15mL) and methanol (60 mL) and recrystallized 10.854 g of Compound 3 as a white solid. Yield: 72.81%. Purity: 99.53%.Example 4, (1S)-2,3,4,6-tetra-O-pivaloyl-1,5-anhydro-1-[3-(4-ethoxyphenylmethyl)-4-chloro Preparation of phenyl]glucitol (compound 3)N-butyl ether (50 mL) was added to zinc iodide (3.19 g) and lithium iodide (1.34 g), and the mixture was heated to 50 ° C for 1.5 h, and cooled for use. 4-(2-Chloro-5-iodo-benzyl) phenyl ether (7.45 g) was added with toluene (15 mL) and n-butyl ether (5 mL) under nitrogen, cooled to -60 ° C, and slowly added dropwise 1.6 mol / L-n-hexyl lithium n-hexane solution (13.8mL), control the internal temperature does not exceed -20 ° C, after the addition is completed, the reaction is kept at -60 ° C for 5 h, and the above-mentioned alternate zinc iodide and lithium iodide n-butyl ether solution is added. The reaction was stirred at 25 ° C for 1 h. Add 2,3,4,6-tetra-O-pivaloyl-α-D-bromoglucopyranose (23.2g) toluene (50mL) solution, heat to 140 ° C reflux reaction for 0.5h, after TLC detection reaction was added 1mol / L diluted hydrochloric acid (50 mL), water (50 mL), and extracted, the organic phase was washed with water (40 mL), dried over anhydrous SO 4 Na 2, concentrated by weight of n-heptane (15mL) and methanol (60 mL) Crystallization gave 10.51 g of Compound 3 as a white solid, yield 70.5%. Purity: 99.41%.Example 5, (1S)-2,3,4,6-tetra-O-pivaloyl-1,5-anhydro-1-[3-(4-ethoxyphenylmethyl)-4-chloro Preparation of phenyl]glucitol (compound 3)To the zinc bromide (2.25 g) and lithium bromide (0.87 g), cyclopentyl methyl ether (30 mL) was added, and the mixture was heated to 50 ° C for 3 hours, and cooled for use. 4-(2-Chloro-5-iodo-benzyl) phenyl ether (7.45 g) was added with toluene (10 mL) and cyclopentyl methyl ether (10 mL) under nitrogen, cooled to -5 ° C, and slowly added dropwise. Mol / L n-hexyl lithium n-hexane solution (12.5mL), control the internal temperature does not exceed 0 ° C, after the addition is completed, the reaction is kept at -5 ° C for 3 h, adding the above-mentioned spare zinc bromide and lithium bromide cyclopentyl methyl ether The solution was incubated at -5 ° C for 4 h, and a solution of 2,3,4,6-tetra-O-pivaloyl-α-D-bromoglucopyranose (17.39 g) in toluene (40 mL) was added and heated to 80 ℃ reaction was stirred 6h, after completion of the reaction by TLC, was added 1mol / L diluted hydrochloric acid (50 mL), water (50 mL), and extracted, the organic phase was washed with water (40 mL), dried over anhydrous 2 SO 4 Na, and concentrated under reduced pressure, Recrystallization of n-heptane (15 mL) and methanol (60 mL) gave 8.15 g of Compound 3 as a white solid. Purity: 99.39%.Example 6, (1S)-2,3,4,6-tetra-O-pivaloyl-1,5-anhydro-1-[3-(4-ethoxyphenylmethyl)-4-chloro Preparation of phenyl]glucitol (compound 3)Zinc bromide (4.5 g) and lithium bromide (1.74 g) were added with n-butyl ether (60 mL), heated to 50 ° C for 3 h, and cooled for use. 4-(2-Chloro-5-bromo-benzyl) phenyl ether (6.513 g) was added with toluene (15 mL) and n-butyl ether (5 mL) under nitrogen, cooled to -30 ° C, and slowly added dropwise 2.5 mol / L-butyllithium n-hexane solution (8.4mL), control the internal temperature does not exceed -20 ° C, after the addition is completed, the reaction is kept at -30 ° C for 3 h, and the above-mentioned alternate zinc bromide and lithium bromide n-butyl ether solution is added. The reaction was incubated at -5 ° C for 4 h, and a solution of 2,3,4,6-tetra-O-pivaloyl-α-D-bromoglucopyranose (14.49 g) in toluene (50 mL) was added and heated to 120 ° C for stirring. the reaction 4h, after completion of the reaction by TLC, was added 1mol / L diluted hydrochloric acid (50 mL), water (40 mL), and extracted, the organic phase was washed with water (40 mL), dried over anhydrous Na 2 SO 4, and concentrated under reduced pressure, n-heptyl Recrystallization of the alkane (15 mL) and methanol (60 mL) gave 10.38 g of Compound 3 as a white solid. Purity: 99.54%.Example 7, (1S)-2,3,4,6-tetra-O-pivaloyl-1,5-anhydro-1-[3-(4-ethoxyphenylmethyl)-4-chloro Preparation of phenyl]glucitol (compound 3)Methyl bromide (40 mL) was added to zinc bromide (2.25 g) and lithium bromide (0.87 g), and the mixture was heated to 50 ° C for 3 h, and cooled for use. 4-(2-Chloro-5-iodo-benzyl) phenyl ether (7.45 g) was added with toluene (15 mL), methyl tert-butyl ether (15 mL), cooled to -40 ° C, and slowly added dropwise. 1.6mol/L n-hexyl lithium n-hexane solution (13.8mL), control the internal temperature does not exceed -30 ° C, after the addition is completed, the reaction is kept at -40 ° C for 4 h, and the above-mentioned alternate zinc bromide and lithium bromide are added. The butyl ether solution was incubated at 5 ° C for 7 h, and a solution of 2,3,4,6-tetra-O-pivaloyl-α-D-bromoglucopyranose (17.39 g) in toluene (50 mL) was added and heated. to 90 deg.] C the reaction was stirred 8h, after completion of the reaction by TLC, was added 1mol / L diluted hydrochloric acid (40 mL), water (40 mL), and extracted, the organic phase was washed with water (40 mL), dried over anhydrous Na 2 SO 4, and concentrated under reduced pressure Recrystallization from n-heptane (15 mL) and methanol (60 mL) gave 9.41 g of Compound 3 as a white solid. Purity: 99.42%. Example 8. Preparation of dapagliflozin (S)-1,2-propanediol monohydrate eutectic (Compound 1)To the compound 3 (37.27 g), methanol (190 mL) was added, and sodium methoxide (10.8 g) was added thereto, and the mixture was heated under reflux for 3 hours. After the TLC reaction was completed, methanol was concentrated, and isopropyl acetate (100 mL) was added to the residue, and water was added. (60 mL), extracted with stirring and the organic phase washed with water (50 mL). (S)-1,2-propanediol (3.8g) and water (0.9g) were added to the organic phase, stirred until it was dissolved, and n-heptane (200 mL) was added, and the mixture was stirred for 2 hours under ice-cooling, suction filtration, filter cake Washing with n-heptane and drying at 30 ° C gave 23.89 g of Compound 1 as a white solid. Yield: 95%. Purity: 99.79%. Melting point: 69.1 to 75.6 °C. The product obtained was subjected to KF = 3.74% (theoretical value: 3.58%). ESI-MS (m/z): 431.22 [M+Na] + . 1 H-NMR (400 MHz, CD 3 OD): δ 7.33 – 7.37 (2H, m), 7.28-7.30 (1H, dd), 7.11 (2H, d), 6.80-6.83 (2H, dd), 4.1 ( 1H, d), 3.98-4.05 (4H, m), 3.88-3.91 (1H, dd), 3.74-3.82 (1H, m), 3.68-3.73 (1H, m), 3.37-3.49 (5H, m), 3.28-3.34 (1H, m), 1.37 (1H, t), 1.15 (3H, d).The crystal form of the obtained product was subjected to thermogravimetric analysis (TGA) by a Universal V4.7A TA instrument, and the TGA curve (Fig. 1) showed a weight loss of about 18.52% from about room temperature to about 240 ° C. The original form Ia crystal form The TGA plot shows a value of 18.7%.The crystal form of the obtained product was subjected to differential scanning calorimetry (DSC) by a Universal V4.7A TA instrument, and the DSC curve (Fig. 2) showed endotherm in the range of about 60 ° C to 85 ° C. The DSC plot shows a range of approximately 50 ° C to 78 ° C.
The crystal form of the obtained product was examined by a Bruker D8advance instrument for powder X-ray diffraction (PXRD), and the 2X value of the PXRD pattern (Fig. 3) (CuKα).
There are characteristic peaks at 3.749°, 7.52°, 7.995°, 8.664°, 15.134°, 15.708°, 17.069°, 18.946°, 20.049°, which are completely consistent with the characteristic peaks of the PXRD pattern of the Ia crystal form in the original patent.In combination with the nuclear magnetic data and melting point of the prepared crystal form, the crystal form of the product (Compound 1) obtained by the present invention is consistent with the pharmaceutically acceptable crystalline form Ia reported in the original patent.
Patent Citations
Publication numberPriority datePublication dateAssigneeTitleCN101479287A *2006-06-282009-07-08布里斯托尔-迈尔斯斯奎布公司Crystalline solvates and complexes of (is) -1, 5-anhydro-l-c- (3- ( (phenyl) methyl) phenyl) -d-glucitol derivatives with amino acids as sglt2 inhibitors for the treatment of diabetesCN104496952A *2014-11-282015-04-08深圳翰宇药业股份有限公司Synthesis method of dapagliflozinCN105153137A *2015-09-172015-12-16上海应用技术学院Preparation method of empagliflozinFamily To Family CitationsCN104829572B *2014-02-102019-01-04江苏豪森药业集团有限公司Dapagliflozin novel crystal forms and preparation method thereofCN105399735A *2015-12-292016-03-16上海应用技术学院Empagliflozin intermediate, and preparation method and application thereof* Cited by examiner, † Cited by third party
Non-Patent Citations
TitleCHEN DEJIN ET AL., CHINA MASTER’S THESES FULL-TEXT DATABASE, ENGINEERING TECHNOLOGY I, vol. B016-731, no. 3, 15 March 2016 (2016-03-15) *LEMAIRE S. ET AL.: “Stereoselective C-glycosylation of furanosyl halides with arylzinc reagents”, PURE APPL. CHEM., vol. 86, no. 3, 4 March 2014 (2014-03-04), pages 329 – 333 *LEMAIRE S. ET AL.: “Stereoselective C-Glycosylation Reactions with Arylzinc Reagents”, ORGANIC LETTERS, vol. 14, no. 6, 2 March 2012 (2012-03-02), pages 1480 – 1483, XP055069093 ** Cited by examiner, † Cited by third partyCLIP
Chemical Synthesis
Dapagliflozin propanediol hydrate, an orally active sodium glucose cotransporter type 2 (SGLT-2) inhibitor, was developed by Bristol-Myers Squibb (BMS) and AstraZeneca for the once-daily treatment of type 2 diabetes. As opposed to competitor SGLT-2 inhibitors, dapagliflozin was not associated with renal toxicity or long-term deterioration of renal function in phase III clinical trials. The drug exhibits excellent SGLT2 potency with more than 1200 fold selectivity over the SGLT1 enzyme.
The IC50 for SGLT2 is less than one thousandth of the IC50 for SGLT1 (1.1 versus 1390 nmol/l), so that the drug does not interfere with the intestinal glucose absorption.[7
dapagliflozin being an inhibitor of sodiumdependent glucose transporters found in the intestine and kidney (SGLT2) and to a method for treating diabetes, especially type II diabetes, as well as hyperglycemia, hyperinsulinemia, obesity, hypertriglyceridemia, Syndrome X, diabetic
complications, atherosclerosis and related diseases, employing such C-aryl glucosides alone or in combination with one, two or more other type antidiabetic agent and/or one, two or more other type therapeutic agents such as hypolipidemic agents.
Approximately 100 million people worldwide suffer from type II diabetes (NIDDM – non-insulin-dependent diabetes mellitus), which is characterized by hyperglycemia due to excessive hepatic glucose production and peripheral insulin resistance, the root causes for which are as yet unknown. Hyperglycemia is considered to be the major risk factor for the development of diabetic complications, and is likely to contribute directly to the impairment of insulin secretion seen in advanced NIDDM. Normalization of plasma glucose in NIDDM patients would be predicted to improve insulin action, and to offset the development of diabetic complications. An inhibitor of the sodium-dependent glucose transporter SGLT2 in the kidney would be expected to aid in the normalization of plasma glucose levels, and perhaps body weight, by enhancing glucose excretion.
Dapagliflozin can be prepared using similar procedures as described in U.S. Pat. No. 6,515,117 or international published applications no. WO 03/099836 and WO 2008/116179
WO 03/099836 A1 refers to dapagliflozin having the structure according to formula 1 .
formula 1
WO 03/099836 A1 discloses a route of synthesis on pages 8-10, whereby one major step is the purification of a compound of formula 2
formula 2
The compound of formula 2 provides a means of purification for providing a compound of formula 1 since it crystallizes. Subsequently the crystalline form of the compound of formula 2 can be deprotected and converted to dapagliflozin. Using this process, dapagliflozin is obtained as an amorphous glassy off-white solid containing 0.1 1 mol% of EtOAc. Crystallization of a pharmaceutical drug is usually advantageous as it provides means for purification also suitable for industrial scale preparation. However, for providing an active pharmaceutical drug a very high purity is required. In particular, organic impurities such as EtOAc either need to be avoided or further purification steps are needed to provide the drug in a
pharmaceutically acceptable form, i.e. substantially free of organic solvents. Thus, there is the need in the art to obtain pure and crystalline dapagliflozinwhich is substantially free of organic solvents.
WO 2008/002824 A1 discloses several alternative solid forms of dapagliflozin, such as e.g. solvates containing organic alcohols or co-crystals with amino acids such as proline and phenylalanine. For instance, the document discloses crystalline
dapagliflozin solvates which additionally contain water molecules (see e.g.
Examples 3-6), but is silent about solid forms of dapagliflozin which do not contain impurities such as organic alcohols. As described above, it is desirable to provide the pharmaceutical active drug in a substantially pure form, otherwise triggering further expensive and time-consuming purification steps. In contrast, the document relates to dapagliflozin solvates where an alcohol and water are both incorporated into the crystal lattice. Hence, there is the need in the art to obtain pure and crystalline dapagliflozin suitable for pharmaceutical production.
WO 2008/1 16179 A1 refers to an immediate release pharmaceutical composition comprising dapagliflozin and propylene glycol. Propylene glycol is a chiral
substance and (S)-propylene glycol used is very expensive. Consequently, also the immediate release pharmaceutical composition is more expensive.
Crystalline forms (in comparision to the amorphous form) often show desired different physical and/or biological characteristics which may assist in the manufacture or formulation of the active compound, to the purity levels and uniformity required for regulatory approval. As described above, it is desirable to provide the pharmaceutical active drug in a substantially pure form, otherwise triggering further expensive and time-consuming purification steps.
PATENT
WO 2008/ 1 16179 Al seems to disclose an immediate release formulation comprising dapagliflozin and propylene glycol hydrate. WO 2008/ 116195 A2 refers to the use of an SLGT2 inhibitor in the treatment of obesity
Example 2 Dapagliflozin (S) PGS—(2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-ethoxybenzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol (S)-propane-1,2-diol hydrate (1:1:1)
Dapagliflozin (S) propylene glycol hydrate (1:1:1) can be prepared using similar procedures as described in published applications WO 08/002824 and WO 2008/116179, the disclosures of which are herein incorporated by reference in their entirety for any purpose. SGLT2 EC50=1.1 nM.
Example 3 Dapagliflozin (R) PGS—(2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-ethoxybenzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol (R)-propane-1,2-diol hydrate (1:1:1)
Dapagliflozin (R) propylene glycol hydrate (1:1:1) can be prepared using similar procedures as described in WO 08/002824 and WO 2008/116179, the disclosures of which are herein incorporated by reference in their entirety for any purpose. SGLT2 EC50=1.1 nM.
WO 2008/002824 A1 discloses several alternative solid forms of dapagliflozin, such as e.g. solvates containing organic alcohols or co-crystals with amino acids such as proline and phenylalanine. For instance, the document discloses crystalline
dapagliflozin solvates which additionally contain water molecules (see e.g.
Examples 3-6), but is silent about solid forms of dapagliflozin which do not contain impurities such as organic alcohols. As described above, it is desirable to provide the pharmaceutical active drug in a substantially pure form, otherwise triggering further expensive and time-consuming purification steps. In contrast, the document relates to dapagliflozin solvates where an alcohol and water are both incorporated into the crystal lattice. Hence, there is the need in the art to obtain pure and crystalline dapagliflozin suitable for pharmaceutical production.
WO 2008/1 16179 A1 refers to an immediate release pharmaceutical composition comprising dapagliflozin and propylene glycol. Propylene glycol is a chiral
substance and (S)-propylene glycol used is very expensive. Consequently, also the immediate release pharmaceutical composition is more expensive.
Surprisingly, amorphous dapagliflozin can be purified with the process of the present invention. For instance amorphous dapagliflozin having a purity of 99,0% can be converted to crystalline dapagliflozin hydrate having a purity of 100% (see examples of the present application). Moreover, said crystalline dapagliflozin hydrate does not contain any additional solvent which is desirable. Thus, the process of purifying dapagliflozin according to the present invention is superior compared with the process of WO 03/099836 A1 .
Additionally, the dapagliflozin hydrate obtained is crystalline which is advantageous with respect to the formulation of a pharmaceutical composition. The use of expensive diols such as (S)-propanediol for obtaining an immediate release pharmaceutical composition as disclosed in WO 2008/1 16179 A1 can be avoided
HRMS calcd for C21H25ClNaO6 (M + Na)+ 431.1237, found 431.1234. Anal. Calcd for C21H25ClO6: C, 61.68; H, 6.16. Found: C, 61.16; H, 6.58.
HPLC
HPLC measurements were performed with an Agilent 1100 series instrument equipped with a UV-vis detector set to 240 nm according to the following method: Column: Ascentis Express RP-Amide 4.6 x 150 mm, 2.7 mm; Column temperature: 25 °C – Eluent A: 0.1 % formic acid in water – Eluent B: 0.1 % formic acid in acetonitrile – Injection volume: 3 mL – Flow: 0.7 mL/min – Gradient:Time [min][%] B0.02525.06526.07029.07029.52535.025……………………..
EXAMPLE 24 – Synthesis of 2,4-di-6>-ieri-butyldiphenylsilyl-l-C-(4-chloro-3-(4- ethoxybenzyl)phenyl)- -D-glucopyranoside 2,4-di-6>-TBDPS-dapagliflozin; (IVj”))
[0229] l-(5-Bromo-2-chlorobenzyl)-4-ethoxybenzene (1.5 g, 4.6 mmol) and magnesium powder (0.54 g, 22.2 mmol) were placed in a suitable reactor, followed by THF (12 mL) and 1,2- dibromoethane (0.16 mL). The mixture was heated to reflux. After the reaction had initiated, a solution of l-(5-bromo-2-chlorobenzyl)-4-ethoxybenzene (4.5 g, 13.8 mmol) in THF (28 mL) was added dropwise. The mixture was allowed to stir for another hour under reflux, and was then cooled to ambient temperature, and then titrated to determine the concentration. The above prepared 4-chloro-3-[(4-ethoxyphenyl)methyl]phenyl magnesium bromide (31 mL, 10 mmol, 0.32 M in THF) and A1C13 (0.5 M in THF, 8.0 mL, 4.0 mmol) were mixed at ambient temperature to give a black solution, which was stirred at ambient temperature for 1 hour. To a solution of
I, 6-anhydro-2,4-di-6>-ieri-butyldiphenylsilyl- -D-glucopyranose (0.64 g, 1.0 mmol) in PhOMe (3.0 mL) at ambient temperature was added phenylmagnesium bromide (0.38 mL, 1.0 mmol, 2.6 M solution in Et20). After stirring for about 5 min the solution was then added into the above prepared aluminum mixture via syringe, followed by additional PhOMe (1.0 mL) to rinse the flask. The mixture was concentrated under reduced pressure (50 torr) at 60 °C (external bath temperature) to remove low-boiling point ethereal solvents and then PhOMe (6mL) was added. The reaction mixture was heated at 130 °C (external bath temperature) for 8 hours at which time HPLC assay analysis indicated a 51% yield of 2,4-di-6>-ieri-butyldiphenylsilyl-l-C-(4-chloro-3- (4-ethoxybenzyl)phenyl)- -D-glucopyranoside. After cooling to ambient temperature, the reaction was treated with 10% aqueous NaOH (1 mL), THF (10 mL) and diatomaceous earth at ambient temperature, then the mixture was filtered and the filter cake was washed with THF. The combined filtrates were concentrated and the crude product was purified by silica gel column chromatography (eluting with 1:30 EtOAc/77-heptane) affording the product 2,4-di-6>- ieri-butyldiphenylsilyl- 1 – -(4-chloro-3 -(4-ethoxybenzyl)phenyl)- β-D-glucopyranoside (0.30 g, 34%) as a white powder.
[0230] A solution of the 2,4-di-6>-ieri-butyldiphenylsilyl-l-C-(4-chloro-3-(4- ethoxybenzyl)phenyl)- -D-glucopyranoside (60 mg, 0.068 mmol) in THF (3.0 mL) and TBAF (3.0 mL, 3.0 mmol, 1.0 M in THF) was stirred at ambient temperature for 15 hours. CaC03 (0.62 g), Dowex^ 50WX8-400 ion exchange resin (1.86 g) and MeOH (5mL) were added to the product mixture and the suspension was stirred at ambient temperature for 1 hour and then the mixture was filtrated through a pad of diatomaceous earth. The filter cake was rinsed with MeOH and the combined filtrates was evaporated under vacuum and the resulting residue was purified by column chromatography (eluting with 1 : 10 MeOH/DCM) affording dapagliflozin (30 mg).
Example 1 – Synthesis of l,6-anhydro-2,4-di-6>-ieri-butyldiphenylsilyl- -D-glucopyranose (II”)
III II”
[0206] To a suspension solution of l,6-anhydro- -D-glucopyranose (1.83 g, 11.3 mmol) and imidazole (3.07 g, 45.2 mmol) in THF (10 mL) at 0 °C was added dropwise a solution of TBDPSC1 (11.6 mL, 45.2 mmol) in THF (10 mL). After the l,6-anhydro-P-D-gJucopyranose was consumed, water (10 mL) was added and the mixture was extracted twice with EtOAc (20 mL each), washed with brine (10 mL), dried (Na2S04) and concentrated. Column
WO 2016147197, DAPAGLIFLOZIN, NEW PATENT, HARMAN FINOCHEM LIMITED
LINK>>> (WO2016147197) A NOVEL PROCESS FOR PREPARING (2S,3R,4R,5S,6R)-2-[4-CHLORO-3-(4-ETHOXYBENZYL)PHENY 1] -6-(HY DROXY METHYL)TETRAHYDRO-2H-PY RAN-3,4,5-TRIOL AND ITS AMORPHOUS FORM
PATENT
PATENT
WO2016018024, CRYSTALLINE COMPOSITE COMPRISING DAPAGLIFLOZIN AND METHOD FOR PREPARING SAME
HANMI FINE CHEMICAL CO., LTD. [KR/KR]; 59, Gyeongje-ro, Siheung-si, Gyeonggi-do 429-848 (KR)
Dapagliflozin, sold under the brand name Farxiga among others, is a medication used to treat type 2 diabetes and, with certain restrictions, type 1 diabetes.[2] It is also used to treat adults with certain kinds of heart failure.[3][4][5]
It was developed by Bristol-Myers Squibb in partnership with AstraZeneca. In 2018, it was the 227th most commonly prescribed medication in the United States, with more than 2 million prescriptions.[10][11]
Medical uses
Dapagliflozin is used along with diet and exercise to improve glycemic control in adults with type 2 diabetes and to reduce the risk of hospitalization for heart failure among adults with type 2 diabetes and known cardiovascular disease or other risk factors.[12][3] It appears more useful than empagliflozin.[13][verification needed]
In addition, dapagliflozin is indicated for the treatment of adults with heart failure with reduced ejection fraction to reduce the risk of cardiovascular death and hospitalization for heart failure.[3][4][5] It is also indicated to reduce the risk of kidney function decline, kidney failure, cardiovascular death and hospitalization for heart failure in adults with chronic kidney disease who are at risk of disease progression.[14]
In the European Union it is indicated in adults:
for the treatment of insufficiently controlled type 2 diabetes mellitus as an adjunct to diet and exercise:
as monotherapy when metformin is considered inappropriate due to intolerance;
in addition to other medicinal products for the treatment of type 2 diabetes;
for the treatment of insufficiently controlled type 1 diabetes mellitus as an adjunct to insulin in patients with BMI ≥ 27 kg/m2, when insulin alone does not provide adequate glycaemic control despite optimal insulin therapy; and
for the treatment of heart failure with reduced ejection fraction.[5]
Adverse effects
Since dapagliflozin leads to heavy glycosuria (sometimes up to about 70 grams per day) it can lead to rapid weight loss and tiredness. The glucose acts as an osmotic diuretic (this effect is the cause of polyuria in diabetes) which can lead to dehydration. The increased amount of glucose in the urine can also worsen the infections already associated with diabetes, particularly urinary tract infections and thrush (candidiasis). Rarely, use of an SGLT2 drug, including dapagliflozin, is associated with necrotizing fasciitis of the perineum, also called Fournier gangrene.[15]
Dapagliflozin can cause dehydration, serious urinary tract infections and genital yeast infections.[3] Elderly people, people with kidney problems, those with low blood pressure, and people on diuretics should be assessed for their volume status and kidney function.[3] People with signs and symptoms of metabolic acidosis or ketoacidosis (acid buildup in the blood) should also be assessed.[3] Dapagliflozin can cause serious cases of necrotizing fasciitis of the perineum (Fournier gangrene) in people with diabetes and low blood sugar when combined with insulin.[3]
To lessen the risk of developing ketoacidosis (a serious condition in which the body produces high levels of blood acids called ketones) after surgery, the FDA has approved changes to the prescribing information for SGLT2 inhibitor diabetes medicines to recommend they be stopped temporarily before scheduled surgery. Canagliflozin, dapagliflozin, and empagliflozin should each be stopped at least three days before, and ertugliflozin should be stopped at least four days before scheduled surgery.[17]
Symptoms of ketoacidosis include nausea, vomiting, abdominal pain, tiredness, and trouble breathing.[17]
Use is not recommended in patients with eGFR < 45ml/min/1.73m2, though data from 2021 shows the reduction in the kidney failure risks in people with chronic kidney disease using dapagliflozin.[18]
Mechanism of action
Dapagliflozin inhibits subtype 2 of the sodium-glucose transport proteins (SGLT2) which are responsible for at least 90% of the glucose reabsorption in the kidney. Blocking this transporter mechanism causes blood glucose to be eliminated through the urine.[19] In clinical trials, dapagliflozin lowered HbA1c by 0.6 versus placebo percentage points when added to metformin.[20]
Regarding its protective effects in heart failure, this is attributed primarily to haemodynamic effects, where SGLT2 inhibitors potently reduce intravascular volume through osmotic diuresis and natriuresis. This consequently may lead to a reduction in preload and afterload, thereby alleviating cardiac workload and improving left ventricular function.[21]
Selectivity
The IC50 for SGLT2 is less than one thousandth of the IC50 for SGLT1 (1.1 versus 1390 nmol/L), so that the drug does not interfere with intestinal glucose absorption.[22]
In July 2016, the fixed-dose combination of saxagliptin and dapagliflozin was approved for medical use in the European Union and is sold under the brand name Qtern.[28] The combination drug was approved for medical use in the United States in February 2017, where it is sold under the brand name Qtern.[29][30]
In May 2019, the fixed-dose combination of dapagliflozin, saxagliptin, and metformin hydrochloride as extended-release tablets was approved in the United States to improve glycemic control in adults with type 2 diabetes when used in combination with diet and exercise. The FDA granted the approval of Qternmet XR to AstraZeneca.[31] The combination drug was approved for use in the European Union in November 2019, and is sold under the brand name Qtrilmet.[32]
Dapagliflozin was found effective in several studies in participants with type 2 and type 1 diabetes.[5] The main measure of effectiveness was the level of glycosylated haemoglobin (HbA1c), which gives an indication of how well blood glucose is controlled.[5]
In two studies involving 840 participants with type 2 diabetes, dapagliflozin when used alone decreased HbA1c levels by 0.66 percentage points more than placebo (a dummy treatment) after 24 weeks.[5] In four other studies involving 2,370 participants, adding dapagliflozin to other diabetes medicines decreased HbA1c levels by 0.54-0.68 percentage points more than adding placebo after 24 weeks.[5]
In a study involving 814 participants with type 2 diabetes, dapagliflozin used in combination with metformin was at least as effective as a sulphonylurea (another type of diabetes medicines) used with metformin.[5] Both combinations reduced HbA1c levels by 0.52 percentage points after 52 weeks.[5]
A long-term study, involving over 17,000 participants with type 2 diabetes, looked at the effects of dapagliflozin on cardiovascular (heart and circulation) disease.[5] The study indicated that dapagliflozin’s effects were in line with those of other diabetes medicines that also work by blocking SGLT2.[5]
In two studies involving 1,648 participants with type 1 diabetes whose blood sugar was not controlled well enough on insulin alone, adding dapagliflozin 5 mg decreased HbA1c levels after 24 hours by 0.37% and by 0.42% more than adding placebo.[5]
Dapagliflozin was approved for medical use in the European Union in November 2012.[5] It is marketed in a number of European countries.[33]
Dapagliflozin was approved for medical use in the United States in January 2014.[34][14]
In 2020, the U.S. Food and Drug Administration (FDA) expanded the indications for dapagliflozin to include treatment for adults with heart failure with reduced ejection fraction to reduce the risk of cardiovascular death and hospitalization for heart failure.[3] It is the first in this particular drug class, sodium-glucose co-transporter 2 (SGLT2) inhibitors, to be approved to treat adults with New York Heart Association’s functional class II-IV heart failure with reduced ejection fraction.[3]
Dapagliflozin was shown in a clinical trial to improve survival and reduce the need for hospitalization in adults with heart failure with reduced ejection fraction.[3] The safety and effectiveness of dapagliflozin were evaluated in a randomized, double-blind, placebo-controlled study of 4,744 participants.[3] The average age of participants was 66 years and more participants were male (77%) than female.[3] To determine the drug’s effectiveness, investigators examined the occurrence of cardiovascular death, hospitalization for heart failure, and urgent heart failure visits.[3] Participants were randomly assigned to receive a once-daily dose of either 10 milligrams of dapagliflozin or a placebo (inactive treatment).[3] After about 18 months, people who received dapagliflozin had fewer cardiovascular deaths, hospitalizations for heart failure, and urgent heart failure visits than those receiving the placebo.[3]
In July 2020, the FDA granted AstraZeneca a Fast Track Designation in the US for the development of dapagliflozin to reduce the risk of hospitalisation for heart failure or cardiovascular death in adults following a heart attack.[35]
In August 2020, it was reported that detailed results from the Phase III DAPA-CKD trial showed that AstraZeneca’s FARXIGA® (dapagliflozin) on top of standard of care reduced the composite measure of worsening of renal function or risk of cardiovascular (CV) or renal death by 39% compared to placebo (p<0.0001) in patients with chronic kidney disease (CKD) Stages 2-4 and elevated urinary albumin excretion. The results were consistent in patients both with and without type 2 diabetes (T2D)[36]
In April 2021, the FDA expanded the indications for dapagliflozin (Farxiga) to include reducing the risk of kidney function decline, kidney failure, cardiovascular death and hospitalization for heart failure in adults with chronic kidney disease who are at risk of disease progression.[14] The efficacy of dapagliflozin to improve kidney outcomes and reduce cardiovascular death in people with chronic kidney disease was evaluated in a multicenter, double-blind study of 4,304 participants.[14]
Research
One study found that it had no benefit on heart disease risk or overall risk of death in people with diabetes.[37] Another study found that in heart failure with a reduced ejection fraction, dapagliflozin reduced the risk of worsening of heart failure or progression to death from cardiovascular causes, irrespective of diabetic status.[38]
^ Ptaszynska, Agata; Johnsson, Kristina M.; Parikh, Shamik J.; De Bruin, Tjerk W. A.; Apanovitch, Anne Marie; List, James F. (2014). “Safety Profile of Dapagliflozin for Type 2 Diabetes: Pooled Analysis of Clinical Studies for Overall Safety and Rare Events”. Drug Safety. 37 (10): 815–829. doi:10.1007/s40264-014-0213-4. PMID25096959. S2CID24064402.
^ Zelniker TA, Wiviott SD, Raz I, et al. (January 2019). “SGLT2 inhibitors for primary and secondary prevention of cardiovascular and renal outcomes in type 2 diabetes: a systematic review and meta-analysis of cardiovascular outcome trials”. Lancet. 393(10166): 31–9. doi:10.1016/S0140-6736(18)32590-X. PMID30424892. S2CID53277899. However, in patients with atherosclerotic cardiovascular disease, the effect of empagliflozin on cardiovascular death was more pro-nounced than that of canagliflozin or dapagliflozin
Clinical trial number NCT00528372 for “A Phase III Study of BMS-512148 (Dapagliflozin) in Patients With Type 2 Diabetes Who Are Not Well Controlled With Diet and Exercise” at ClinicalTrials.gov
Clinical trial number NCT00643851 for “An Efficacy & Safety Study of BMS-512148 in Combination With Metformin Extended Release Tablets” at ClinicalTrials.gov
Clinical trial number NCT00859898 for “Study of Dapagliflozin in Combination With Metformin XR to Initiate the Treatment of Type 2 Diabetes” at ClinicalTrials.gov
Clinical trial number NCT00528879 for “A Phase III Study of BMS-512148 (Dapagliflozin) in Patients With Type 2 Diabetes Who Are Not Well Controlled on Metformin Alone” at ClinicalTrials.gov
Clinical trial number NCT00660907 for “Efficacy and Safety of Dapagliflozin in Combination With Metformin in Type 2 Diabetes Patients” at ClinicalTrials.gov
Clinical trial number NCT00680745 for “Efficacy and Safety of Dapagliflozin in Combination With Glimepiride (a Sulphonylurea) in Type 2 Diabetes Patients” at ClinicalTrials.gov
Clinical trial number NCT01392677 for “Evaluation of Safety and Efficacy of Dapagliflozin in Subjects With Type 2 Diabetes Who Have Inadequate Glycaemic Control on Background Combination of Metformin and Sulfonylurea” at ClinicalTrials.gov
Clinical trial number NCT00673231 for “Efficacy and Safety of Dapagliflozin, Added to Therapy of Patients With Type 2 Diabetes With Inadequate Glycemic Control on Insulin” at ClinicalTrials.gov
Clinical trial number NCT02229396 for “Phase 3 28-Week Study With 24-Week and 52-week Extension Phases to Evaluate Efficacy and Safety of Exenatide Once Weekly and Dapagliflozin Versus Exenatide and Dapagliflozin Matching Placebo” at ClinicalTrials.gov
Clinical trial number NCT02413398 for “A Study to Evaluate the Effect of Dapagliflozin on Blood Glucose Level and Renal Safety in Patients With Type 2 Diabetes (DERIVE)” at ClinicalTrials.gov
Clinical trial number NCT01730534 for “Multicenter Trial to Evaluate the Effect of Dapagliflozin on the Incidence of Cardiovascular Events (DECLARE-TIMI58)” at ClinicalTrials.gov
Clinical trial number NCT03036124 for “Study to Evaluate the Effect of Dapagliflozin on the Incidence of Worsening Heart Failure or Cardiovascular Death in Patients With Chronic Heart Failure (DAPA-HF)” at ClinicalTrials.gov
Schubert-Zsilavecz, M, Wurglics, M, Neue Arzneimittel 2008/2009
more1) Pal, Manojit et al; Improved Process for the preparation of SGLT2 inhibitor dapagliflozin via glycosylation of 5-bromo-2-Chloro-4′-ethoxydiphenylmethane with Gluconolactone ;. Indian Pat Appl,. 2010CH03942 , 19 Oct 20122) Lemaire, Sebastien et al; Stereoselective C-Glycosylation Reactions with Arylzinc Reagents ;
Organic Letters , 2012, 14 (6), 1480-1483;3) Zhuo, Biqin and Xing, Xijuan; Process for preparation of Dapagliflozin amino acid cocrystals ;
Faming Zhuanli Shenqing , 102 167 715, 31 Aug 20114) Shao, Hua et al; Total synthesis of SGLT2 inhibitor Dapagliflozin ;
Hecheng Huaxue , 18 (3), 389-392; 20105) Liou, Jason et al; Processes for the preparation of C-Aryl glycoside amino acid complexes as potential SGLT2 Inhibitors ;. PCT Int Appl,.
WO20100223136) Seed, Brian et al; Preparation of Deuterated benzyl-benzene glycosides having an inhibitory Effect on sodium-dependent glucose co-transporter; . PCT Int Appl,.
WO20100092437) Song, Yanli et al; Preparation of benzylbenzene glycoside Derivatives as antidiabetic Agents ;. PCT Int Appl,.
WO20090265378) Meng, Wei et al; D iscovery of Dapagliflozin: A Potent, Selective Renal Sodium-Dependent Glucose cotransporter 2 (SGLT2) Inhibitor for the Treatment of Type 2 Diabetes ;
Journal of Medicinal chemistr y, 2008, 51 (5), 1145 -1149;9) Gougoutas, Jack Z. et al; Solvates Crystalline complexes of amino acid with (1S)-1 ,5-anhydro-LC (3 – ((phenyl) methyl) phenyl)-D-glucitol were prepared as for SGLT2 Inhibitors the treatment of Diabetes ;. PCT Int Appl,.
WO200800282410) Deshpande, Prashant P. et al; Methods of producing C-Aryl glucoside SGLT2 Inhibitors ;..
A lipid-enabled and UnlockedNucleomonomer Agent modified RNA (LUNAR) of self-replicating RNA for vaccination against spike protein of SARS-CoV-2 (Arcturus)
Self-replicating RNA vaccine
Arcturus Therapeutics and Duke-NUS Medical School, Singapore
ARCT-021: Currently undergoing phase 1/2 clinical trials, it combines two technologies, i.e., saRNA STARR and LUNAR® lipid-mediated delivery method. It was designed to enhance and extend antigen expression, enabling vaccination at lower doses [87]. In addition, LUNAR® lipids are pH-sensitive and biodegradable, causing minimal lipid accumulation in cells after multiple dosing [87]The Arcturus COVID-19 vaccine, commonly known as ARCT-021 and LUNAR-COV19, is a COVID-19 vaccine candidate developed by Arcturus Therapeutics.
54. de Alwis R, Gan ES, Chen S, Leong YS, Tan HC, Zhang SL. et al. A Single Dose of Self-Transcribing and Replicating RNA Based SARS-CoV-2 Vaccine Produces Protective Adaptive Immunity In Mice. bioRxiv. 2020. 2020 09.03.280446 Development
Arcturus Therapeutics partnered with Singapore’s Duke–NUS Medical School to develop a COVID-19 vaccine.[1] The company also partnered with Catalent, a contract development and manufacturing organization, to manufacture multiple batches of Arcturus’ COVID-19 mRNA vaccine candidate.[2]
Clinical research
Phase I-II
LUNAR-COV19 clinical trials in humans began in July 2020.[3] On 4 January 2021, Arcturus Therapeutics started phase-2 clinical trials.[4]
Phase 2 study to be conducted in the U.S. and Singapore, and will evaluate both single dose and two dose priming regimens of ARCT-021 in up to 600 participants
Anticipate interim Phase 2 data in early 2021; targeting global Phase 3 study start in Q2 2021 which could allow application for emergency use authorization/conditional approval in H2 2021January 04, 2021 07:01 AM Eastern Standard Time
SAN DIEGO–(BUSINESS WIRE)–Arcturus Therapeutics Holdings Inc. (the “Company”, “Arcturus”, Nasdaq: ARCT), a leading clinical-stage messenger RNA medicines company focused on the development of infectious disease vaccines and significant opportunities within liver and respiratory rare diseases, today announced that the Company has received allowance of the Investigational New Drug (IND) application from the U.S. Food and Drug Administration (FDA) for the Phase 2 clinical study of its vaccine candidate ARCT-021 following review of data from the Phase 1/2 study.
Arcturus Therapeutics Receives FDA Allowance to Proceed with Phase 2 Study of ARCT-021 (LUNAR-COV19) Vaccine Candidate in the United States
Arcturus previously announced that the ARCT-021 Phase 2 study had been approved to proceed by the Singapore Health Sciences Authority (HSA), who reviewed the same data as reviewed by the FDA. These Phase 1/2 study results demonstrated favorable tolerability and both humoral and cellular immunogenicity following administration of ARCT-021.
The Phase 2 study will enroll 600 participants, with 450 receiving ARCT-021 and 150 receiving placebo. Both older and younger adult participants will be included. Early interim analyses of safety and immunogenicity will be performed to inform dose selection for a Phase 3 study, which is targeted to start in Q2 2021, if the Phase 2 study is successful.
“Allowance of the IND for our ARCT-021 Phase 2 clinical study represents an important milestone for the program and we look forward to starting to screen study participants at U.S. and Singapore clinical sites very soon,” said Steve Hughes, M.D., Chief Medical Officer of Arcturus. “We have advanced ARCT-021 to Phase 2 based on promising interim results from our Phase 1/2 study and extensive preclinical data. Our prior clinical results show that ARCT-021 administration results in humoral and cellular immunogenicity, and we are encouraged by an increasing body of evidence highlighting the potential importance of T cells in providing protection against SARS-CoV-2 infection and COVID-19. We believe that ARCT-021 holds promise to be a highly effective vaccine with a differentiated clinical profile, including the potential to only require a single dose for protection.”
About Arcturus Therapeutics
Founded in 2013 and based in San Diego, California, Arcturus Therapeutics Holdings Inc. (Nasdaq: ARCT) is a clinical-stage mRNA medicines and vaccines company with enabling technologies: (i) LUNAR® lipid-mediated delivery, (ii) STARR mRNA Technology and (iii) mRNA drug substance along with drug product manufacturing expertise. Arcturus’ diverse pipeline of RNA therapeutic and vaccine candidates includes self-replicating mRNA vaccine programs for SARS-CoV-2 (COVID-19) and Influenza, and other programs to potentially treat Ornithine Transcarbamylase (OTC) Deficiency, Cystic Fibrosis, and Cardiovascular Disease along with partnered programs including Glycogen Storage Disease Type 3, Hepatitis B Virus, and non-alcoholic steatohepatitis (NASH). Arcturus’ versatile RNA therapeutics platforms can be applied toward multiple types of nucleic acid medicines including messenger RNA, small interfering RNA, replicon RNA, antisense RNA, microRNA, DNA, and gene editing therapeutics. Arcturus’ technologies are covered by its extensive patent portfolio (205 patents and patent applications, issued in the U.S., Europe, Japan, China and other countries). Arcturus’ commitment to the development of novel RNA therapeutics has led to collaborations with Janssen Pharmaceuticals, Inc., part of the Janssen Pharmaceutical Companies of Johnson & Johnson, Ultragenyx Pharmaceutical, Inc., Takeda Pharmaceutical Company Limited, CureVac AG, Synthetic Genomics Inc., Duke-NUS Medical School, and the Cystic Fibrosis Foundation. For more information visit www.ArcturusRx.com. In addition, please connect with us on Twitter and LinkedIn.
^ Clinical trial number NCT04480957 for “Phase 1/2 Ascending Dose Study of Investigational SARS-CoV-2 Vaccine ARCT-021 in Healthy Adult Subjects” at ClinicalTrials.gov
Uprifosbuvir is under investigation in clinical trial NCT02332707 (Efficacy and Safety of Grazoprevir (MK-5172) and Uprifosbuvir (MK-3682) With Elbasvir (MK-8742) or Ruzasvir (MK-8408) for Chronic Hepatitis C Genotype (GT)1 and GT2 Infection (MK-3682-011)).Hepatitis C viruss (HCV) have the newly-increased patients of 3-4 million every year, and World Health Organization (WHO) is estimated in global sense More than 200,000,000, in China more than 10,000,000 patients, HCV belongs to flaviviridae hepatovirus virus to dye person.Long-term hepatitis C virus Gently to inflammation, weight is to liver cirrhosis, hepatocarcinoma for poison infection.And during hepatitis C cirrhosis patients in decompensation, can there are various complication, such as abdomen Water abdominal cavity infection, upper gastrointestinal hemorrhage, hepatic encephalopathy, hepatorenal syndrome, liver failure etc. are showed.The side of HCV infection is treated initially Method is interferon and interferon and ribavirin combination therapy, and only 50% therapist has reaction, and interferon to the method With obvious side effect, such as flu-like symptoms, body weight lower and fatigue and weak, and interferon and ribavirin Conjoint therapy then produces sizable side effect, including haemolysis, anemia and tired etc..U.S. FDA have approved multiple HCV medicines, including the polymerization of protease inhibitor, ucleosides and non-nucleoside in recent years Enzyme inhibitor and NS5A inhibitor etc..The protease inhibitor class medicine of FDA approvals has three:VX‐950 (Telaprevir), SCH-503034 (Boceprevir) and TMC435 (Simeprevir), the shortcoming of protease inhibitor is It is also easy to produce that mutation, toxicity is big, poor bioavailability, it is effective to individual other gene type.Eggs of the Telaprevir as the first generation White enzyme inhibitor has logged out market.The second filial generation and third generation protease inhibitor of high activity and wide spectrum is mainly used as and other One of component of drug combination of hepatitis C medicine.NS5A inhibitor is the highly active anti-HCV medicament of a class.The most representative Daclatasive for having BMS, The Ombitasvir of the Ledipasvir and AbbVie of Gilead, as this kind of medicine independent medication is easy to produce drug resistance, They treat one of drug component of HCV primarily as drug combination.The AG14361 of hepatitis C is generally divided into two kinds of ucleosides and non-nucleoside.At present, clinically only Suo Feibu One ucleosides hepatitis C medicine of Wei is listed by FDA approvals, and other are still in the anti-hepatitis C virus medicine of ucleosides of clinical experimental stage Thing also has the MK-3682 (IDX21437) of Mo Shadong, the AL-335 of the ACH-3422 and Alios of Achillion drugmakers.Third Hepatitis virus have the features such as Multi-genotype and fast variation, and single medicine treatment hepatitis C has generation drug resistance fast, to part Genotype cure rate is low and the various defects such as course for the treatment of length.In order to overcome these defects, the treatment of drug combination is primarily now taken Scheme, in order to overcome these defects, primarily now takes the therapeutic scheme of drug combination, the Sovaldi conducts of FDA approval listings The key component of drug combination, for the patient of 4 type of 1 type of gene and gene be Suo Feibuwei, profit Ba Wei woodss and Polyethylene Glycol-α- The drug combination of interferon three, the course for the treatment of are 12 weeks;For 1 type of gene and the patient of 3 types, the big woods joints of Suo Feibuwei and Li Ba Medication, the course for the treatment of are respectively 12 weeks and 24 weeks.- 2016 years 2013, FDA ratified Suo Feibuwei and NS3 protein inhibitors again in succession Simeprevir shares the patient of 1 type of therapeutic gene;The NS5A inhibitor Daclatavir therapeutic genes 1 of Suo Feibuwei and BMS With the patient of 3 types.Harvoni is the patient that Suo Feibuweijia NS5A inhibitor Ledipasvir is used for 1 type of gene.Even if using Same nucleoside, the NS5A inhibitor and/or NS3 protease inhibitor for sharing varying strength can effectively extend composition of medicine Clinical application range and Shorten the Treatment Process.In June, 2016, FDA have approved Suo Feibuwei and more potent secondary NS5A inhibitor Velpatasvir shares the hepatitis C patient suitable for all gene types, it is not necessary to carry out genetic test.Just in three phases clinic Suo Feibuwei, NS5A inhibitor Velpatasvir and NS3 protease inhibitor Voxilaprevir goes for all of disease People, is try to the course for the treatment of and shortened to 8 weeks from 12 weeks.Suo Feibuwei just in clinical trial target spots different with hepatitis C virus are directed to Drug regimen (such as Suo Feibuweijia new type NS 5A inhibitor Velpatasvir and/or protease inhibitor GS5816), its knot Fruit show than single drug more wide spectrum, effectively, and can be with Shorten the Treatment Process.MSD Corp. is by MK-3682 and NS5A inhibitor Grazoprevir and/or protease inhibitor Elbasvir is used as new drug regimen, effective for all genotype of HCV, And further shorten to the course for the treatment of of 8 weeks.New deuterated nucleoside phosphoric acid ester compound disclosed in patent of the present invention, especially The double deuterated compound such as VI-1b2 in 5 ‘-position, shows than the more preferable bioavailability of former compound MK-3682 and longer partly declines Phase.In addition, this kind of novel nucleoside phosphoramidate is significantly superior to the Suo Feibuwei of clinical practice in terms of anti-hepatitis C activity, On sugared ring, chlorine atom replaces fluorine atom, and cytotoxicity is significantly reduced in surveyed cell line.By to base, sugared ring With the transformation and optimization of prodrug moiety system, the anti-hepatitis C activity of partial synthesis compound is higher than Suo Feibuwei 2-10 times, meanwhile, In the optimization of metabolism key position, synthesis compound shows that in blood plasma the higher metabolic stabilities of peso Fei Buwei and chemistry are steady It is qualitative.Therefore this kind of new deuterated nucleotide phosphate and NS5A inhibitor and/or egg as shown in formula a, a1, a2, b, b1, b2 The newtype drug combination constituted by white enzyme inhibitor is with extremely wide application prospect.Deuterium is the naturally occurring hydrogen isotope of nature, the deuterated isotopic body in common drug all containing trace.Deuterium without It is malicious, “dead”, it is safe to human body, C-D keys are more stable (6-9 times) than c h bond, hydrogen is replaced with after deuterium, can extend medicine Half-life, while pharmacologically active (shape difference of H and D is little, J Med Chem.2011,54,2529-2591) is not affected, in addition Deuterated medicine usually shows more preferable bioavailability and less toxicity, and the active ribonucleoside triphosphote of its metabolism is more stable, So deuterated nucleoside phosphoramidate will be better than corresponding nucleoside medicine in the curative effect of clinical practice.For example, 2013 It is exactly a deuterated compound that the nucleoside anti hepatitis C virus drug ACH-3422 of clinical trial is in the approval of year FDA, with non-deuterium (WO2014169278, WO are 2014169280) than having higher bioavailability and longer half-life for the former compound phase in generation. Based on above-mentioned present Research, we design and are prepared for the new deuterated nucleoside that compound VI-1b2 is representative Phosphoramidate.Below we will be described in the architectural feature of deuterated nucleoside phosphoramidate of our inventions, preparation method, Antiviral activity experimental result and it as anti-hepatitis c virus drug combination key component and NS5A inhibitor and/ Or the drug regimen of protease inhibitor is in the application of anti-virus aspect.
The EPA awarded the greener reaction conditions to the pharmaceutical company Merck & Co. for building a prodrug synthesis that eliminated the use of toxic reagents. Prodrugs are molecules that get metabolized by our bodies into an active pharmaceutical. Some hepatitis C and HIV medications are prodrugs and get synthesized through a method call pronucleotide (ProTide) synthesis. The method uses toxic and corrosive thionyl chloride, plus an excess of expensive pentafluorophenol that generates a lot of waste. Merck’s new method creates their target compounds in 90 to 92% yields without these reagents and eliminates the need for halogenated solvents entirely through strategic catalyst loading and the use of different starting materials from the traditional route.
The design of greener chemicals award went to the development of more environmentally friendly versions of chemicals called thermoset binders, which can serve as carpet adhesives and are involved in the manufacture of mineral and fiberglass products. Generally, these chemicals are based on formaldehyde or polycarboxylic acids, and they can give off toxic formaldehyde and often use small amounts of sulfuric and hypophosphorous acid as catalysts to activate them. The insulation and commercial roofing company Johns Manville created a new binder based on the reaction between renewable dextrose, fructose, and other simple sugars, bound together by the α-carbon-containing cross-linking agent glyoxal. The reaction also uses a biodegradable acid in water as a catalyst. The binder can be made in just one step instead of the traditional multistep synthesis. Also, the synthesis can be done directly at the manufacturing site, instead of beforehand like with the traditional approach, meaning this new binder creates fewer of the health and environmental hazards that come from storage and transportation.
A novel application of the synthesis of pronucleotide (ProTide) 5′-phosphoramidate monoesters promoted by aluminum-based Lewis acids is described. In the multikilogram synthesis of uprifosbuvir (MK-3682, 1), a clinical candidate for the treatment of hepatitis C, this methodology provided >100:1 diastereoselectivity at the phosphorus stereocenter and >100:1 selectivity for the 5′-mono phosphorylation over undesired bisphosphorylation side products. The high diastereoselectivity and mono/bis ratio achieved enabled elimination of the tedious workup associated with the tert-butyl magnesium chloride protocol commonly used to install this functionality in similar nucleotide prodrugs, achieving a near doubling of the isolated yield from 45% to 81%. The process development and purity control strategy of MK-3682, as well as handling of the pyrophoric reagent on scale, will also be discussed.
PAPER
Science (Washington, DC, United States) (2020), 369(6504), 725-730.
Science (Washington, DC, United States) (2017), 356(6336), 426-430.
[00273] A 5 L flange flask was fitted with a thermometer, nitrogen inlet, pressure equalizing dropping funnel, bubbler, and a suba»seal. Methyl lithium solution (1.06 L, 1.6 M in diethylether, 1.7 equiv.) was added, and the solution was cooled to about -25 °C.Diisopropyl amine (238 ml, 1.7 equiv.) was added using the dropping funnel over about 40 minutes. The reaction was left stirring, allowing to warm to ambient temperature overnight. C02(s)/acetone cooling was applied to the LDA solution, cooling to about -70 °C.[00274] i?-Glyceraldehyde dimethylacetal solution (50% in DCM) was evaporated down to -100 mbar at a bath temp of 35 °C, to remove the DCM, then azeotroped with anhydrous hexane (200 ml), under the same Buchi conditions. 1H NMR was used to confirm that all but a trace of DCM remained.[00275] The fresh aldehyde (130 g, 1 mol) and ethyl 2-chloropropionionate (191 ml, 1.5 equiv.) were placed in a 1 L round bottom flask, which was filled with toluene (800 ml). This solution was cooled in a C02(s)/acetone bath, and added via cannula to the LDA solution over about 50 minutes, keeping the internal temperature of the reaction mixture cooler than -60 °C. The mixture was stirred with cooling (internal temp, slowly fell to ~ -72 °C) for 90 min, then warmed to room temperature over 30 minutes using a water bath. This solution was added to a sodium dihydrogen phosphate solution equivalent to 360 g of NaH2P04 in 1.5 L of ice/water, over about 10 minutes, with ice-bath cooling. The mixture was stirred for 20 minutes, then transferred to a sep. funnel, and partitioned. The aqueous layer was further extracted with EtOAc (2 x 1 L), and the combined organic extracts were dried over sodium sulfate. The volatiles were removed in vacuo (down to 20 mbar). The resultant oil was hydrolyzed crude.
H O CI[00276] The crude oil A2 was taken up in acetic acid (1.5 L, 66% in water) and heated to 90 °C over one hour, then at held at that temperature for one hour. Once the mixture had cooled to room temperature, the volatiles were removed in vacuo, and azeotroped with toluene (500 ml). The resultant oil was combined with some mixed material from an earlier synthesis and columned in two portions (each -1.25 L of silica, 38→ 75% EtOAc in DCM). The lower of the two main spots is the desired material; fractions containing this material as the major component were combined and the solvent removed in vacuo to give 82 g of orange solid whose 1 H NMR showed the material to be of about 57% purity (of the remainder 29% was the indicated epimer). This material was recrystallized fromtoluene/butanone (600 ml / -185 ml), the butanone being the ‘good’ solvent. The resultant solid was filtered washing with toluene and hexane, and dried in vacuo to give product of about 92% purity (30 g).(2R,3R,4R)-2-[(benzoyIoxy)methyI]-4-chIoro-4-methyI-5-oxooxoIan-3-yI benzoate(A5):
[00277] A 2 L 3 -neck round bottom flask was fitted with an overhead stirrer, thermometer and pressure equalizing dropping funnel (→N2). The intermediate A4 (160 mmol) in acetonitrile (1 L) was added, followed by 4-dimethylaminopyridine (3.2 mmol) and benzoyl chloride (352 mmol). Finally triethylamine (384 mmol) was added over 10 minutes using the dropping funnel. The addition of the triethylamine is accompanied by a mild exotherm, which obviated the addition of a cold water bath to keep the internal temperature below 25 °C. The reaction was stirred at ambient temperature for 2.5 hours. The reaction mixture was transferred to a sep. funnel with EtOAc (2 L) and half saturated brine (2 L), and partitioned. The aqueous layer was re-extracted with EtOAc (1 L). The combined organic layers were washed with 50%> sodium bicarbonate/25%) brine (1.5 L) and dried over sodium sulfate, to give 62 g of solid. This was recrystallized from 1.8 L of 1 : 1 toluene/trimethylpentane (95 °C), to give 52.4 g of product.[00278] 1H NMR (CDCls, 400 MHz): δ (ppm) 1.91 (s, 3H), 4.57 (dd, J= 5.12Hz and J = 12.57Hz, 1H), 4.77 (dd, J= 3.29Hz and J= 12.68Hz, 1H), 4.92-4.96 (m, 1H), 5.60 (d, J = 8.36Hz, 1H), 7.38-7.66 (m, 6H), 7.97-7.99 (m, 2H), 8.08-8.10 (m, 2H); MS (ESI) m/z= 411.1(MNa ).
[00279] To a solution of A5 (14.48 mmol) in anhydrous tetrahydrofurane (70 ml) was added under inert atmosphere at -35°C, LiAlH(OtBu)3 (1M in tetrahydrofurane, 21.7 mmol) over a 30 min period. The reaction mixture was stirred for 1 hour at -20 °C and quenched by addition of a saturated NH4C1 solution, keeping the temperature bellow 0 °C. Ethyl acetate was added and the white suspension was filtered through a pad of celite and washed with ethyl acetate. The filtrate was extracted with ethyl acetate twice. The combined organic layers were dried over anhydrous sodium sulfate, filtered and evaporated under reduced pressure. The residue was purified by chromatography on silica gel (eluent: petroleum ether/ethyl acetate 0 to 20%). The product was dried in vacuum (50 °C) overnight to afford expected intermediate as a colorless oil in 96% yield (mixture α/β: 45/55).[00280] 1H NMR (CDC13, 400 MHz): δ (ppm) 1.74 (s, 1.75HP), 1.76 (s, 1.25Ha), 4.42-4.69 (m, 3H), 5.30 (d, J= 12.8Hz, 0.55HP), 5.43-5.47 (m, 0.45Ha), 5.60 (d, J= 7.0Hz, 0.55HP), 5.78 (d, J= 7.0Hz , 0.45Ha), 7.35-7.41 (m, 2H), 7.45-7.56 (m, 3H), 7.59-7.65 (m, 1H), 7.96- 8.04 (m, 2H), 8.06-8.14 (m, 2H); MS (ESI) m/z= 413 (MNa+).3,5-Di-0-benzoyl-2-C-chloro-2-C-methyl-D-arabinofuranosyl bromide (A7):
[00281] To a solution of A6 (12.80 mmol) in anhydrous dichloromethane (80 ml) was added under inert atmosphere at -20 °C, triphenylphosphine (18.0 mmol). The reaction mixture was stirred for 15 minutes at -20 °C and CBr4 (19.20 mmol) was added. The reaction mixture was then stirred for 1 hour at -20 °C. The crude was partially concentrated under reduced pressure (bath temperature bellow 30 °C) and directly purified by chromatography on silica gel (eluent: petroleum ether/ethyl acetate 0 to 30%) to afford a mixture of β sugar A7a (1.67 g) and a sugar A7b (2.15 g) as a colorless gum in 66%> global yield.[00282] 1H NMR (CDC13, 400 MHz): β sugar δ (ppm) 1.93 (s, 3H), 4.60-4.88 (m, 3H), 6.08 (d, J= 7.9 Hz, 1H), 6.62 (s, 1H), 7.31-7.38 (m, 2H), 7.41-7.55 (m, 3H), 7.59-7.65 (m, 1H), 8.00-8.05 (m, 2H), 8.06-8.12 (m, 2H); a sugar δ (ppm) 1.88 (s, 3H), 4.66-4.89 (m, 3H), 5.37 (d, J= 4.88Hz, 1H), 6.44 (s, 1H), 7.41-7.55 (m, 4H), 7.54-7.65 (m, 2H), 8.00-8.05 (m, 2H), 8.14-8.20 (m, 2H); MS (ESI) m/z= 476/478 (MNa+).3 ,5′-Di-0-benzoyl-2′-C-chloro-2′-C-methyl-4-benzoyl-cytidine (A8):
[00283] To a suspension of N-benzoyl cytosine (9.48 mmol), and a catalytic amount of ammonium sulfate in 4-chlorobenzene (24 ml) was added HMDS (28.44 mmol). The reaction mixture was heated during 2 hours at 140 °C. The solvent was removed under inert atmosphere and the residue was taken in 4-chlorobenzene (15 ml). Then, A7b (4.74 mmol) in chlorobenzene (10 ml) was added dropwise to the reaction mixture followed by SnCl4 (14.22 mmol) dropwise. The reaction mixture was stirred at 70 °C overnight, cooled to room temperature and diluted with dichloromethane and a saturated NaHC03 solution. The white suspension was filtered through a pad of celite and washed with dichloromethane. The filtrate was extracted with dichloromethane twice. The combined organic layers were dried over anhydrous Na2S04, filtered and evaporated under reduced pressure to afford expected intermediate as a white solid in 89% yield.[00284] 1H NMR (DMSO, 400 MHz): δ (ppm) 1.58 (s, 3H), 4.68-4.81 (m, 3H), 5.68 (brs, 1H), 6.55 (brs, 1H), 7.36 (d, J= 7.84 Hz, 1H), 7.39-7.76 (m, 9H), 7.88-8.07 (m, 6H), 8.30 (d, J= 7.84 Hz, 1H); MS (ESI) m/z= 588 (MH+).3′,5′-Di-0-benzoyl-2,-C-chloro-2,-C-methyluridine (A9):
[00285] A suspension of A8 (4.19 mmol) in an acetic acid/water mixture (67 ml/17 ml, v/v), was heated at 110 °C for 3 hours. The reaction mixture was evaporated to dryness and co-evaporated with toluene (three times) to afford expected intermediate in quantitative yield as an oil which was directly used for the next step; MS (ESI) m/z= 485 (MH+). 2 -C-Chloro-2 -C-methyluridine (301):
H O CI[00286] Intermediate A9 (4.19 mmol) in 7 N methanolic ammonia (80 ml) was stirred at room temperature for 24 hours. The mixture was evaporated to dryness, diluted with water and transferred into a separatory funnel. The aqueous layer was extracted withdichloromethane and water was removed under reduced pressure. The residue was purified by flash RP18 gel chromatography (eluent: water/acetonitrile 0 to 40%) to afford pure expected compound as a white foam in 79% yield.[00287] 1H NMR (DMSO, 400 MHz): δ (ppm) 1.44 (s, 3H), 3.60-3.68 (m, 1H), 3.80-3.94 (m, 3H), 5.39 (t, J= 4.45 Hz, 1H), 5.63 (d, J= 8.26 Hz, 1H), 5.93 (d, J= 5.72 Hz, 1H), 6.21 (s, 1H), 8.16 (d, J= 8.90 Hz, 1H), 11.44 (m, 1H); MS (ESI) m/z= 277 (MH+).2′-C-Chloro-2′-C-methyl-3-benzyloxymethyluridine (Al 1):
H O CI[00288] To a solution of 301 (0.361 mmol) in anhydrous DMF (4 ml) was added at -5 °C, DBU (0.723 mmol) followed by benzyloxymethylchloride (0.542 mmol). The reaction mixture was stirred for 45 minutes between -5 °C and 5 °C. The solvent was evaporated under reduced pressure and the residue was purified by chromatography on silica gel (eluent: dichloromethane/methanol 0 to 10%) to afford pure expected intermediate as a white solid in 80% yield.[00289] 1H NMR (DMSO, 400 MHz): δ (ppm) 1.41 (s, 3H), 3.61-3.69 (m, 1H), 3.82-3.95 (m, 3H), 4.57 (s, 2H), 5.32 (s, 2H), 5.43 (t, J= 4.46Hz, 1H), 5.80 (d, J= 8.08Hz, 1H), 5.96 (d, J= 4.46 Hz, 1H), 6.23 (s, 1H), 7.22-7.36 (m, 5H), 8.25 (d, J= 8.22Hz, 1H); MS (ESI) m/z= 397 (MH+). Isopropyl (2S)-2-[[chloro(phenoxy)phosphoryl]amino]propanoate (A12a):
[00290] To a solution of aminoester, HC1 salt (0.434 mmol) in anhydrous dichloromethane (or acetonitrile) (4 ml) (3 times vacuo/nitrogen) under nitrogen was added at -30°C phenyldichlorophosphate (0.434 mmol) followed by N-methylimidazole (2.90 mmol)(or only 1.45 mmol for A12b). The reaction mixture was stirred at -30°C during 1 hour. The reaction was monitored by LC/MS (the sample was quenched by methanol or water) to check the complete formation of expected intermediate A12a [MS (ESI) m/z= 302 (MH+)(-OMe compounder A12b [MS (ESI) m/z= 314 (MH~)].Compound (A13a), (A13b) or (83ii):[00291] To the previous reaction mixture containing A12 was added All (or 302) (0.29 mmol) at -25°C under nitrogen. The reaction mixture was allowed to warm up slowly to room temperature overnight, and then diluted with dichloromethane and water (or with NaHCC”3 and EtOAc). The organic layer was extracted, dried, filtered and evaporated under reduced pressure. The crude residue was purified by chromatography on silica gel (eluent: dichloromethane/methanol 0 to 10%) (followed by preparative HPLC for A29).Compound (A13a):
[00292] Mixture of diastereoisomers; MS (ESI) m/z= 666 (MH+). Compound (A13b):
[00293] Mixture of diastereoisomers; MS (ESI) m/z= 692.3 (MH ).Compound (83ii):
[0251] A 3-neck 100 mL jacketed round bottom flask with nitrogen inlet and mechanical stirrer was charged with compound 4 (3.0 g, 10.8 mmol), compound 13 (0.484 g, 2.17 mmol, 0.20 equiv), 2-butanone (21 mL), and 2,6-lutidine (2.53 mL, 21.7 mmol, 2.0 equiv). The resulting slurry was cooled to −15° C., then a solution of compound 12 (7.96 g, 13.0 mmol) in 2-butanone (3 mL) was added over 14 hours. The reaction mixture was allowed to stir at −15° C. for an additional 25 hours and then warmed to 20° C. n-Heptane (16 mL) was added with stirring over a 1 hour period then the mixture was allowed to stir at 25° C. for 3 hours, then filtered through a fitted funnel. The filter cake was slurry-washed with a 3:2 mixture of 2-butanone and n-heptane (10 mL and then 15 mL), then dried by pulling nitrogen stream through the fritted funnel. The filter cake was slurried in a 10:1 mixture of water and 2-butanone (21 mL) and then filtered. This slurrying and filtration sequence was repeated two more times. The resulting filter cake was dried with nitrogen stream through the fritted funnel to provide compound 6.
Example 21Alternate Preparation of Compound A
[0252]
[0253] Compound 6 (0.072 mmol, 1 equiv), K2HPO4 (63.0 mg, 0.361 mmol) and compound 14 (5.45 mg, 0.018 mmol) were added to a 1 dram vial with 4 A mol sieves (40 mg). To the resulting mixture was added DCM (800 μl), then the resulting reaction was allowed to stir for 5 minutes. To the reaction mixture was then added compound 14 (28.7 mg, 0.094 mmol, 1.3 equiv) and the resulting reaction was allowed to stir for about 15 hours at room temperature to provide Compound A.
[0256]
[0257] A 100 mL reactor with nitrogen inlet and mechanical stirrer was charged with compound 4 (7.00 g, 25.3 mmol), compound 15 (0.225 g, 0.506 mmol, 0.020 equiv), 1,3-dioxolane (42 mL), and 2,6-lutidine (4.42 mL, 38.0 mmol, 1.5 equiv). The mixture was cooled to −10° C. and a 33 wt % solution of compound 12 in isopropyl acetate (29 mL, 30 mmol) was added over 1 hour. The reaction mixture was allowed to stir at −10° C. for additional 40 hours, then isopropyl acetate (28 mL) was added, and the resulting mixture was warmed to 0° C. A 10 wt % aqueous NaHSO4 solution was added (14 mL), and the mixture was allowed to stir at 30° C. for 30 minutes, then the layers were separated. To the organic layer was added an aqueous solution containing 5 wt % NaHCO3 and 5 wt % Na2SO4 (21 mL). The mixture was allowed to stir at 50° C. for 6 h. The layers were separated. To the organic layer was added 10 wt % aqueous NaCl solution (21 mL). The mixture was allowed to stir at 50° C. for 30 min. The organic layer was separated, combined with isopropyl acetate (5 mL) and concentrated in vacuo to half volume at 20000 pa in a 50° C. bath. The resulting solution was solvent-switched with isopropanol (4×35 mL) to 60 g weight. The mixture was seeded with 100 mg of compound A at 60° C. The resulting slurry was allowed to stir at 55° C. for 30 minutes, then n-Heptane (35 mL) was added over 1 hour at 55° C. The resulting slurry was allowed to stir for an additional 1 hour at 55° C., then cooled to room temperature and filtered. The filter cake was washed with a 1:1 mixture of isopropanol and n-heptane (3×14 mL), followed by n-heptane (14 mL), then dried under nitrogen to provide Compound A.
Uprifosbuvir is an antiviral agent developed for treatment of chronic hepatitis C infections. Its original synthesis route requires twelve steps with an overall yield of only 1 %. Such a difficult and time-consuming synthesis approach is acceptable for the early trial phase of a new drug, but impractical for broad application as hepatitis C treatment or for repurposing against novel viral diseases.
Artis Klapars, John Y. L. Chung, and colleagues, Merck & Co., Inc., Rahway, NJ, USA, and WuXi STA, Shanghai, China, have developed a synthesis route for uprifosbuvir requiring only five steps and starting from readily available uridine. Initially, uridine is selectively oxidized after OH-acylation with pivaloyl chloride in an acyl migration/oxidation process driven by complexation with the Lewis acid BF3*OEt2 in toluene. In the second step, methylation is achieved by MeMgBr/MgCl2 in a toluene/anisole mixture where a more reactive methyl-manganese species is formed in-situ from the Grignard reagent, providing high yield and a good diastereomeric ratio (dr). Subsequently, the tertiary chloride group is introduced. Due to the high functional-group density, a cyclodehydration step is required before chlorination to avoid side reactions. The chlorination is carried out using dichlorodimethylsilane with FeCl3*6H2O and tetramethyldisiloxane as additives which avoids the hazardous use of HCl gas under pressure required in the initial synthesis. In the final step, the regioselective phosphoramidation is achieved using a chlorophosphoramidate precursor and a dimeric chiral imidazole carbamate catalyst which led to a dr of 97:3 starting from a 1:1 diastereomeric mixture of the chlorophosphoramidate reagent.
Uprifosbuvir was synthesized with an overall yield of 50 %, a vast improvement compared to the 1 % of the original synthesis route. Additionally, the newly developed synthesis steps have the potential to provide easier access to other nucleoside-based antiviral agents.
Efficient Synthesis of Antiviral Agent Uprifosbuvir Enabled by New Synthetic Methods, Artis Klapars, John Chung, John Limanto, Ralph Calabria, Louis-Charles Campeau, Kevin Campos, Wenyong Chen, Stephen M Dalby, Tyler A Davis, Daniel DiRocco, Alan Hyde, Amude M Kassim, Mona Utne Larsen, Guiquan Liu, Peter Maligres, Aaron Moment, Feng Peng, Rebecca Ruck, Michael Shevlin, Bryon L Simmons, Zhiguo Jake Song, Lushi Tan, Timothy J Wright, Susan Zultanski, Chemical Science2021. https://doi.org/10.1039/D1SC01978C
Efficient synthesis of antiviral agent uprifosbuvir enabled by new synthetic methods†
All publication charges for this article have been paid for by the Royal Society of Chemistry
Abstract
An efficient route to the HCV antiviral agent uprifosbuvir was developed in 5 steps from readily available uridine in 50% overall yield. This concise synthesis was achieved by development of several synthetic methods: (1) complexation-driven selective acyl migration/oxidation; (2) BSA-mediated cyclization to anhydrouridine; (3) hydrochlorination using FeCl3/TMDSO; (4) dynamic stereoselective phosphoramidation using a chiral nucleophilic catalyst. The new route improves the yield of uprifosbuvir 50-fold over the previous manufacturing process and expands the tool set available for synthesis of antiviral nucleotides.
Scheme 1 Synthetic approaches to uprifosbuvir 1 with the two main challenges highlighted. (a) Me2NH, AcOH, EtOH/MeOH, 80 °C, 1.5 h; (b) Ca(OH)2, water, 70 °C, 24 h, 19% over 2 steps.9
Scheme 3 Complexation-driven selective acyl migration/oxidation to access 12. (a) PivCl, pyridine, 0 °C, 16 h; (b) BF3·OEt2, PhMe, 40 °C, 10 h; (c) TEMPO, Bu4NBr, AcOOH, dioctyl sulphide, PhMe, −10 °C to 20 °C, 24 h, 83% from 5.
Scheme 6 Completion of uprifosbuvir synthesis. (a) TMS-Cl, iPrOH, 70 °C, 12 h; (b) NEt3, iPrOAc, wiped film evaporation, 80%; (c) PhOP(O)Cl2, NEt3, iPrOAc, −20 °C, 2 h, 90%; (d) C6F5OH, NEt3, iPrOAc, −5 °C to 10 °C, 18 h, 76%;26 (e) 4, 3 mol% 24, 2,6-lutidine, 1,3-dioxolane, −10 °C, 24 h, 88%; (f) 4, tBuMgCl, THF, −5 °C to 5 °C, 15 h, 50%;27 (g) 4, Me2AlCl, 2,6-lutidine, THF, 35 °C, 16 h, 81%.27
Scheme 7 Summary of uprifosbuvir synthesis. AY = assay yield; IY = isolated yield.
^ Soriano V, Fernandez-Montero JV, de Mendoza C, Benitez-Gutierrez L, Peña JM, Arias A, Barreiro P (August 2017). “Treatment of hepatitis C with new fixed dose combinations”. Expert Opinion on Pharmacotherapy. 18 (12): 1235–1242. doi:10.1080/14656566.2017.1346609. PMID28644739. S2CID205819421.
^ Borgia G, Maraolo AE, Nappa S, Gentile I, Buonomo AR (March 2018). “NS5B polymerase inhibitors in phase II clinical trials for HCV infection”. Expert Opinion on Investigational Drugs. 27 (3): 243–250. doi:10.1080/13543784.2018.1420780. PMID29271672. S2CID3672885.
^ Lawitz E, Gane E, Feld JJ, Buti M, Foster GR, Rabinovitz M, et al. (September 2019). “Efficacy and safety of a two-drug direct-acting antiviral agent regimen ruzasvir 180 mg and uprifosbuvir 450 mg for 12 weeks in adults with chronic hepatitis C virus genotype 1, 2, 3, 4, 5 or 6”. Journal of Viral Hepatitis. 26 (9): 1127–1138. doi:10.1111/jvh.13132. PMID31108015. S2CID160014275.
^ Soriano V, Fernandez-Montero JV, de Mendoza C, Benitez-Gutierrez L, Peña JM, Arias A, Barreiro P (August 2017). “Treatment of hepatitis C with new fixed dose combinations”. Expert Opinion on Pharmacotherapy. 18 (12): 1235–1242. doi:10.1080/14656566.2017.1346609. PMID28644739. S2CID205819421.
^ Borgia G, Maraolo AE, Nappa S, Gentile I, Buonomo AR (March 2018). “NS5B polymerase inhibitors in phase II clinical trials for HCV infection”. Expert Opinion on Investigational Drugs. 27 (3): 243–250. doi:10.1080/13543784.2018.1420780. PMID29271672. S2CID3672885.
^ Lawitz E, Gane E, Feld JJ, Buti M, Foster GR, Rabinovitz M, et al. (September 2019). “Efficacy and safety of a two-drug direct-acting antiviral agent regimen ruzasvir 180 mg and uprifosbuvir 450 mg for 12 weeks in adults with chronic hepatitis C virus genotype 1, 2, 3, 4, 5 or 6”. Journal of Viral Hepatitis. 26 (9): 1127–1138. doi:10.1111/jvh.13132. PMID31108015. S2CID160014275.
A new multivalent COVID-19 vaccine developed by Australian company Vaxine to tackle the new virus variants could be game-changer in the fight against COVID-19
The world desperately needs a vaccine that blocks virus transmission and protects against all the variants. Covax-19 vaccine may soon change history”— Sharen Pringle, Vaxine Business Mananager
ADELAIDE, SA, AUSTRALIA, May 16, 2021 /EINPresswire.com/ — Professor Petrovsky, who is the Chairman and Research Director of Australian-based Vaxine Pty Ltd, explains that the two biggest challenges to tackling the COVID-19 pandemic are to develop a vaccine that completely prevents virus transmission something other COVID-19 have not been completely successful in achieving, and the second being to find a vaccine that protects equally against all the evolving immune-escape variants.
Professor Petrovsky has been researching coronavirus vaccines for the last 17 years, having previously published scientific papers on vaccines against both the SARS and MERS coronaviruses, which were highly protective in relevant animal models. He also recently published data from a collaboration with the US Army on development of a promising Ebola vaccine that protected mice against this most lethal disease after just a single vaccine dose. He has now successfully taken the same approach to design a protein-based vaccine against COVID-19.
Studies in a broad range of animal models including mice, hamsters, ferrets and monkeys, have recently revealed the high potential of this vaccine that is currently known as Covax-19(TM), but which likely will be soon rebranded as in its latest iteration it moves into late stage human trials in a number of countries.
Recent breakthrough data generated by Vaxine’s partner, Professor Kaissar Tabynov who leads the International Center for Vaccinology at the Kazakh National Agrarian University has shown that Vaxine’s unique spike protein antigen which is produced using insect cells in culture, was unique in that it not only totally protected hamsters from infection themselves but also prevented them from transmitting the virus to unvaccinated animals that were placed in the same cage two days after the vaccinated animals had been challenged with virus. Protection against transmission was not seen in hamsters given other vaccines making this finding unique to Vaxine’s spike protein antigen.
This hamster data reinforced findings in hamster, ferret and monkey challenge study performed by collaborating US Universities, who showed that two doses of Vaxine’s Covax-19 vaccine provided complete clearance of recoverable virus from the lungs and nose of animals when sampled just days after an infectious challenge.
“COVAX-19 vaccine has now been shown to be highly protective against the original Wuhan strain of the virus in hamster, ferret and monkey infection models performed by independent academic institutions in multiple countries, attesting to the strength of our protein-based vaccine approach”, says Prof. Petrovsky.
“A key element in the success of Covax-19 vaccine is the inclusion of Vaxine’s Advax adjuvant technology which acts as a turbocharger to drive an optimal immune response against the virus” explains Prof. Petrovsky who has been working on this promising vaccine adjuvant technology for the last 20 years with funding support from the US National Institutes of Health.
“We have now shown that our COVAX-19 vaccine can provide effective immunity including an ability to block nasal virus replication and this in turn successfully prevents transmission of the virus to vaccine-naïve animals,” he explains.
Follow on studies to confirm and expand upon these initial findings are currently underway at several US universities as well as Kazakh National Agrarian University, with a manuscript describing some of the initial animal data currently under review at a leading vaccine journal.
In another major breakthrough the team has now developed the vaccine into a multivariant format designed to protect against all the recently described variant strains of COVID-19, with work also underway on the most recently described Indian strains.
While the data is still preliminary says Prof. Petrovsky, the immune responses to the multivalent vaccine in mice are generating equally strong antibody binding activity against all the major virus variants. “This is extremely exciting as the world desperately needs vaccines able to protect against all the new strains of the virus including the UK, South African and Brazilian strains. By contrast , the currently available vaccines are clearly not as strong against some of these variants as they are against the original Wuhan strain” he explains.
Already there have been multiple confirmed cases of vaccine breakthrough where otherwise healthy individuals who have received mRNA, adenovirus or inactivated whole virus vaccines have become infected generally with either the South African or Brazilian variants.
This problem of immune-escape will only get worse over time as more complex variants emerge which is why Vaxine has been putting all its energy into finding a robust solution to this issue before proceeding with Phase 3 clinical trials of its Covax-19 vaccine.
Dr. Petrovsky went on to conclude “Now we have a multivalent formulation of Covax-19 vaccine that is showing high promise in animal studies, we plan to work as fast as we can to advance this new vaccine formulation in human trials, while expanding manufacturing capacity to ensure we are able to produce enough vaccine to meet the enormous global demand that will be attracted by such a successful vaccine.”
“To help us in this task Vaxine is looking to assemble a global network of partner organisations in countries around the world to assist Vaxine with vaccine development, clinical trials, manufacturing, distribution and sales. This is going to be a mammoth effort as we go to war against this insidious virus that continues to wreak havoc around the globe, with WHO recently predicting that the second year of the pandemic is likely to be much worse even than the first, an ominous warning for many countries that still remain poorly prepared and lacking in local vaccine manufacturing capability.
Vaxine wishes to help developing countries to establish their own local state-of-the-art vaccine manufacturing facilities, providing advice on appropriate facility design and undertaking technology transfer of its state of the art protein production technology to such facilities.
Countries in the developing world can no longer afford to sit and wait for outside organisations like COVAX to solve their vaccine supply problems, instead Vaxine proposes to help such countries find their own local solutions to the vaccine supply bottleneck for this.
Currently, the Australian influenza vaccine and adjuvant specialist and the Polish protein drug maker have just inked a memorandum of understanding, so the terms of a future contract remain to be defined. However, the technology behind is interesting.
The partners intent to utilize an insect cell-based recombinant spike protein of SARS-CoV–2 in combination with Vaxine’s proprietary Advax adjuvant and have already started Phase I testing in Australia with first result expected later this month. The company announced it will use artificial intelligence to evalutate clinical data in real time and announced the ambition to complete Phase II and III trials at the end of this year. “Supported by Microsoft technology, we aim to collect and analyse the COVAX-19 trial data in real time, rather than waiting until the end of the trial before seeing if the vaccine is working, which is the traditional process,” said Vaxine’s Research Director Professor Nikolai Petrovsky from Flinders University in Adelaide.
Preclinically, Vaxine Pty Ltd’s syntetic spike protein with the company’s non-inflammatory Advax adjuvant, induced antibody and T-cell immune responses against the co-administered antigen. In various animal models, Covax-19 vaccination provided robust protection against an infection with the novel coronavirus.
CAS Registry Number: 4759-48-2 CAS Name: 13-cis-Retinoic acid
Additional Names: 2-cis-vitamin A acid; neovitamin A acid
Manufacturers’ Codes: Ro-4-3780Trademarks: Accutane (Roche); Isotrex (Stiefel); Oratane (Douglas); Roaccutane (Roche) Molecular Formula: C20H28O2Molecular Weight: 300.44Percent Composition: C 79.95%, H 9.39%, O 10.65% Literature References: Naturally occurring metabolite of vitamin A, q.v.; inhibits sebum production. Prepn: C. D. Robeson et al.,J. Am. Chem. Soc.77, 4111 (1955). Stereoselective process: R. Lucci, EP111325; idem,US4556518 (1984, 1985 both to Hoffmann-La Roche). Toxicology and teratogenicity study: J. J. Kamm, J. Am. Acad. Dermatol.6, 652 (1982). Identification as endogenous metabolite of all-trans-retinoic acid: M. E. Cullum, M. H. Zile, J. Biol. Chem.260, 10590 (1985). HPLC determn in serum: G. Tang, R. M. Russell, J. Lipid Res.31, 175 (1990). Review of pharmacology and clinical efficacy in acne: A. R. Shalita et al.,Cutis42, Suppl. 6A, 1-19 (1988). Symposium on clinical experience: Dermatology195, Suppl. 1, 1-37 (1997). Properties: Reddish-orange plates from isopropyl alcohol, mp 174-175°. uv max: 354 nm (e 39800). LD50 (20 day) in mice, rats (mg/kg): 904, 901 i.p.; 3389, >4000 orally (Kamm).
Isotretinoin, also known as 13-cis-retinoic acid and sold under the brand name Accutane among others, is a medication primarily used to treat severe acne. It is also used to prevent certain skin cancers (squamous-cell carcinoma), and in the treatment of other cancers. It is used to treat harlequin-type ichthyosis, a usually lethal skin disease, and lamellar ichthyosis. It is a retinoid, meaning it is related to vitamin A, and is found in small quantities naturally in the body. Its isomer, tretinoin, is also an acne drug.
The most common adverse effects are a transient worsening of acne (lasting 1–4 months), dry lips (cheilitis), dry and fragile skin, and an increased susceptibility to sunburn. Uncommon and rare side effects include muscle aches and pains (myalgias), and headaches. Isotretinoin is known to cause birth defects due to in-utero exposure because of the molecule’s close resemblance to retinoic acid, a natural vitamin A derivative which controls normal embryonic development. It is also associated with psychiatric side effects, most commonly depression but also, more rarely, psychosis and unusual behaviours. Other rare side effects include hyperostosis, and premature epiphyseal closure, have been reported to be persistent.
In the United States, a special procedure is required to obtain the pharmaceutical. In most other countries, a consent form is required which explains these risks. In other countries, such as Israel, it is prescribed like any other medicine from a dermatologist (after proper blood tests).
Women taking isotretinoin must not get pregnant during and for one month after the discontinuation of isotretinoin therapy. Sexual abstinence or effective contraception is mandatory during this period. Barrier methods by themselves (e.g., condoms) are not considered adequate due to the unacceptable failure rates of approximately 3%. Women who become pregnant while taking isotretinoin therapy are generally counseled to have an abortion.
It was patented in 1969 and approved for medical use in 1982.[2] It sold well, but in 2009, Roche decided to discontinue manufacturing due to diminishing market share due to the availability of the many generic versions and the settling of multiple lawsuits over side effects. It continues to be manufactured as of 2019 by Absorica, Amnesteem, Claravis, Myorisan, Sotret, and Zenatane.[3]
Medical uses
Isotretinoin is used primarily for severe cystic acne and acne that has not responded to other treatments.[4][5][6][7] Many dermatologists also support its use for treatment of lesser degrees of acne that prove resistant to other treatments, or that produce physical or psychological scarring.[8] Isotretinoin is not indicated for treatment of prepubertal acne and is not recommended in children less than 12 years of age.[9]
Isotretinoin therapy has furthermore proven effective against genital warts in experimental use, but is rarely used for this indication as there are more effective treatments. Isotretinoin may represent an efficacious and safe alternative systemic form of therapy for recalcitrant condylomata acuminata (RCA) of the cervix. In most countries this therapy is currently unapproved and only used if other therapies failed.[11][12]
Prescribing restrictions
Isotretinoin is a teratogen; there is about a 20–35% risk for congenital defects in infants exposed to the drug in utero, and about 30–60% of children exposed to isotretinoin prenatally have been reported to show neurocognitive impairment.[13] Because of this, there are strict controls on prescribing isotretinoin to women who may become pregnant and women who become pregnant while taking isotretinoin are strongly advised to terminate their pregnancies.[13]
In most countries, isotretinoin can only be prescribed by dermatologists or specialist physicians; some countries also allow limited prescription by general practitioners and family doctors. In the United Kingdom[14] and Australia,[15][16] isotretinoin may be prescribed only by or under the supervision of a consultant dermatologist. Because severe cystic acne has the potential to cause permanent scarring over a short period, restrictions on its more immediate availability have proved contentious.[17] In New Zealand, isotretinoin can be prescribed by any doctor but subsidised only when prescribed by a vocationally-registered general practitioner, dermatologist or nurse practitioner.[18]
In the United States, since March 2006 the dispensing of isotretinoin is run through a website called iPLEDGE. The FDA required the companies marketing the drug in the US, which at the time that iPLEDGE was launched were Roche, Mylan, Barr, and Ranbaxy, to put this website in place as a risk evaluation and mitigation strategy. These companies formed a group called the Isotretinoin Products Manufacturing Group, and it hired Covance to run the website.[19][20] Prescribers, pharmacists, and all people to whom the drug is prescribed need to register on the site and log information into it. Women with child-bearing potential must commit to using two forms of effective contraception simultaneously for the duration of isotretinoin therapy and for a month immediately preceding and a month immediately following therapy. Additionally they must have two negative pregnancy tests 30 days apart and have negative pregnancy tests before each prescription is written.[21][22]
The compound 13-cis retinoic acid was first studied in the 1960s at Roche Laboratories in Switzerland by Werner Bollag as a treatment for skin cancer. Experiments completed in 1971 showed that the compound was likely to be ineffective for cancer and, surprisingly, that it could be useful to treat acne. However, they also showed that the compound was likely to cause birth defects, so in light of the events around thalidomide, Roche abandoned the product. In 1975, Gary Peck and Frank Yoder independently rediscovered the drug’s use as a treatment of cystic acne while studying it as a treatment for lamellar ichthyosis, and published that work. Roche resumed work on the drug. In clinical trials, subjects were carefully screened to avoid including women who were or might become pregnant. Roche’s New Drug Application for isotretinoin for the treatment of acne included data showing that the drug caused birth defects in rabbits. The FDA approved the application in 1982.
Scientists involved in the clinical trials published articles warning of birth defects at the same time the drug was launched in the US, but nonetheless isotretinoin was taken up quickly and widely, both among dermatologists and general practitioners. Cases of birth defects showed up in the first year, leading the FDA to begin publishing case reports and to Roche sending warning letters to doctors and placing warning stickers on drug bottles, and including stronger warnings on the label. Lawsuits against Roche started to be filed. In 1983 the FDA’s advisory committee was convened and recommended stronger measures, which the FDA took and were that time unprecedented: warning blood banks not to accept blood from people taking the drug, and adding a warning to the label advising women to start taking contraceptives a month before starting the drug. However use of the drug continued to grow, as did the number of babies born with birth defects. In 1985 the label was updated to include a boxed warning. In early 1988 the FDA called for another advisory committee, and FDA employees prepared an internal memo estimating that around 1,000 babies had been born with birth defects due to isotretinoin, that up to around 1,000 miscarriages had been caused, and that between 5,000 and 7,000 women had had abortions due to isotretinoin. The memo was leaked to the New York Times[77] a few days before the meeting, leading to a storm of media attention. In the committee meeting, dermatologists and Roche each argued to keep the drug on the market but to increase education efforts; pediatricians and the CDC argued to withdraw the drug from the market. The committee recommended to restrict physicians who could prescribe the drug and to require a second opinion before it could be prescribed. The FDA, believing it did not have authority under the law to restrict who had the right to prescribe the drug, kept the drug on the market but took further unprecedented measures: it required to Roche to make warnings yet more visible and graphic, provide doctors with informed consent forms to be used when prescribing the drug, and to conduct follow up studies to test whether the measures were reducing exposure of pregnant women to the drug. Roche implemented those measures, and offered to pay for contraception counseling and pregnancy testing for women prescribed the drug; the program was called the “Pregnancy Prevention Program”.
A CDC report published in 2000[78] showed problems with the Pregnancy Prevention Program and showed that the increase in prescriptions was from off-label use, and prompted Roche to revamp its program, renaming it the “Targeted Pregnancy Prevention Program” and adding label changes like requirements for two pregnancy tests, two kinds of contraception, and for doctors to provide pharmacists with prescriptions directly; providing additional educational materials, and providing free pregnancy tests. The FDA had another advisory meeting in late 2000 that again debated how to prevent pregnant women from being exposed to the drug; dermatologists testified about the remarkable efficacy of the drug, the psychological impact of acne, and demanded autonomy to prescribe the drug; others argued that the drug be withdrawn or much stricter measures be taken. In 2001 the FDA announced a new regulatory scheme called SMART (the System to Manage Accutane Related Teratogenicity) that required Roche to provide defined training materials to doctors, and for doctors to sign and return a letter to Roche acknowledging that they had reviewed the training materials, for Roche to then send stickers to doctors, which doctors would have to place on prescriptions they give people after they have confirmed a negative pregnancy test; prescriptions could only be written for 30 days and could not be renewed, thus requiring a new pregnancy test for each prescription.[citation needed]
In February 2002, Roche’s patents for isotretinoin expired, and there are now many other companies selling cheaper generic versions of the drug. On June 29, 2009, Roche Pharmaceuticals, the original creator and distributor of isotretinoin, officially discontinued both the manufacture and distribution of their Accutane brand in the United States due to what the company described as business reasons related to low market share (below 5%), coupled with the high cost of defending personal-injury lawsuits brought by some people who took the drug.[79] Generic isotretinoin will remain available in the United States through various manufacturers. Roche USA continues to defend Accutane and claims to have treated over 13 million people since its introduction in 1982. F. Hoffmann-La Roche Ltd. apparently will continue to manufacture and distribute Roaccutane outside of the United States.[80]
Among others, actor James Marshall sued Roche over allegedly Accutane-related disease that resulted in removal of his colon.[81] The jury, however, decided that James Marshall had a pre-existing bowel disease.[82]
Several trials over inflammatory bowel disease claims have been held in the United States thus far, with many of them resulting in multimillion-dollar judgments against the makers of isotretinoin.[83]
As of 2017 it was marketed as a topical combination drug with erythromycin under the brand names Isotrex Eritromicina, Isotrexin, and Munderm.[1]
Research
While excessive bone growth has been raised a possible side effect, a 2006 review found little evidence for this.[84]
syn
C. D. Robeson et al., J. Am. Chem. Soc. 77, 4111 (1955). Stereoselective process: R. Lucci, EP 111325; idem, US 4556518 (1984, 1985 both to Hoffmann-La Roche). doi:10.1021/jo00349a001.
syn
J Chem Soc 1968,(16),1982-83
The reaction of vinyl-beta-ionol (I) with triphenylphosphonium bromide (II) in ethanol gives the corresponding phosphonium salt (III), which is condensed through a Wittig reaction with cis-beta-formylcrotonic acid (IV) by means of sodium ethoxide in ethanol to afford a mixture of cis-2-cis-4-vitamin A acid (V) and the desired product. Finally, compound (V) is isomerized bv irradiation with diffuse light in ether in the presence of iodine.
syn
Tetrahedron 2000,56(37),7211
The formylation of the beta-ionone (I) with methyl formate and NaOMe gives the enol (II), which by reaction with methanol and H2SO4 yields the dimethylacetal (III). The reaction of (III) with methylenetriphenylphosphorane (IV) affords the methylene compound (V), which is treated with formic acid to provide the aldehyde (VI). The condensation of (VI) with isopropylidenemalonic acid dimethyl ester (VII) by means of NaOH gives the polyenic malonic acid (VIII) as a mixture of isomers that is separated by crystallization in ethyl ether to yield the desired all-trans-isomer (IX). Finally, this malonic acid is selectively monodecarboxylated by means of refluxing 2,6-dimethylpyridine to afford the target (E,E,E,Z)-isomer.
^ Strauss JS, Krowchuk DP, Leyden JJ, Lucky AW, Shalita AR, Siegfried EC, Thiboutot DM, Van Voorhees AS, Beutner KA, Sieck CK, Bhushan R (April 2007). “Guidelines of care for acne vulgaris management”. Journal of the American Academy of Dermatology. 56 (4): 651–63. doi:10.1016/j.jaad.2006.08.048. PMID17276540.
^ Sehgal VN, Srivastava G, Sardana K (June 2006). “Isotretinoin–unapproved indications/uses and dosage: a physician’s reference”. International Journal of Dermatology. 45 (6): 772–7. doi:10.1111/j.1365-4632.2006.02830.x. PMID16796650.
^ Specifically, doctors who are fellows of the Australasian College of Dermatologists (FACD); cf. Pharmaceutical Services Branch, Guide to poisons and therapeutic goods legislation for medical practitioners and dentists, Sydney: NSW Department of Health; 2006.[page needed]
^ DiGiovanna JJ (November 2001). “Isotretinoin effects on bone”. Journal of the American Academy of Dermatology. 45 (5): S176-82. doi:10.1067/mjd.2001.113721. PMID11606950.
^ Ellis CN, Madison KC, Pennes DR, Martel W, Voorhees JJ (1984). “Isotretinoin therapy is associated with early skeletal radiographic changes”. Journal of the American Academy of Dermatology. 10 (6): 1024–9. doi:10.1016/S0190-9622(84)80329-1. PMID6588057.
^ Scheinfeld N, Bangalore S (May 2006). “Facial edema induced by isotretinoin use: a case and a review of the side effects of isotretinoin”. Journal of Drugs in Dermatology. 5 (5): 467–8. PMID16703787.
^ Jump up to:abcd Borovaya A, Olisova O, Ruzicka T, Sárdy M (September 2013). “Does isotretinoin therapy of acne cure or cause depression?”. International Journal of Dermatology. 52 (9): 1040–52. doi:10.1111/ijd.12169. PMID23962262.
^ Jump up to:ab Rowe C, Spelman L, Oziemski M, Ryan A, Manoharan S, Wilson P, Daubney M, Scott J (May 2014). “Isotretinoin and mental health in adolescents: Australian consensus”. The Australasian Journal of Dermatology (Review). 55 (2): 162–7. doi:10.1111/ajd.12117. PMID24283385. S2CID29178483.
^ Goodman AB (May 1996). “Congenital anomalies in relatives of schizophrenic probands may indicate a retinoid pathology”. Schizophrenia Research. 19 (2–3): 163–70. doi:10.1016/0920-9964(96)88523-9. PMID8789914. S2CID12089905.
^ Lowenstein EB, Lowenstein EJ (2011). “Isotretinoin systemic therapy and the shadow cast upon dermatology’s downtrodden hero”. Clinics in Dermatology. 29 (6): 652–61. doi:10.1016/j.clindermatol.2011.08.026. PMID22014987.
^ Kremer I, Gaton DD, David M, Gaton E, Shapiro A (1994). “Toxic effects of systemic retinoids on meibomian glands”. Ophthalmic Research. 26 (2): 124–8. doi:10.1159/000267402. PMID8196934.
^ Griffin JN, Pinali D, Olds K, Lu N, Appleby L, Doan L, Lane MA (November 2010). “13-Cis-retinoic acid decreases hypothalamic cell number in vitro”. Neuroscience Research. 68 (3): 185–90. doi:10.1016/j.neures.2010.08.003. PMID20708044. S2CID207152111.
^ Sakai Y, Crandall JE, Brodsky J, McCaffery P (June 2004). “13-cis Retinoic acid (accutane) suppresses hippocampal cell survival in mice”. Annals of the New York Academy of Sciences. 1021 (1): 436–40. Bibcode:2004NYASA1021..436S. doi:10.1196/annals.1308.059. PMID15251924.
^ Jump up to:ab Peck GL, Olsen TG, Yoder FW, Strauss JS, Downing DT, Pandya M, Butkus D, Arnaud-Battandier J (February 1979). “Prolonged remissions of cystic and conglobate acne with 13-cis-retinoic acid”. The New England Journal of Medicine. 300 (7): 329–33. doi:10.1056/NEJM197902153000701. PMID153472.
^ Shalita A (2001). “The integral role of topical and oral retinoids in the early treatment of acne”. Journal of the European Academy of Dermatology and Venereology. 15: 43–9. doi:10.1046/j.0926-9959.2001.00012.x. PMID11843233.
^[unreliable medical source?]Farrell LN, Strauss JS, Stranieri AM (December 1980). “The treatment of severe cystic acne with 13-cis-retinoic acid. Evaluation of sebum production and the clinical response in a multiple-dose trial”. Journal of the American Academy of Dermatology. 3 (6): 602–11. doi:10.1016/S0190-9622(80)80074-0. PMID6451637.
^ Toyoda M, Nakamura M, Makino T, Kagoura M, Morohashi M (June 2002). “Sebaceous glands in acne patients express high levels of neutral endopeptidase”. Experimental Dermatology. 11 (3): 241–7. doi:10.1034/j.1600-0625.2002.110307.x. PMID12102663. S2CID23468315.
^ Wysowski DK, Swartz L (May 2005). “Relationship between headache and depression in users of isotretinoin”. Archives of Dermatology. 141 (5): 640–1. doi:10.1001/archderm.141.5.640. PMID15897395.
^ Ng CH, Schweitzer I (February 2003). “The association between depression and isotretinoin use in acne”. The Australian and New Zealand Journal of Psychiatry. 37 (1): 78–84. doi:10.1046/j.1440-1614.2003.01111.x. PMID12534661. S2CID8475675.
^ Halverstam CP, Zeichner J, Lebwohl M (2006). “Lack of significant skeletal changes after long-term, low-dose retinoid therapy: case report and review of the literature”. Journal of Cutaneous Medicine and Surgery. 10 (6): 291–9. doi:10.2310/7750.2006.00065. PMID17241599. S2CID36785828.
Chidamide is being researched as a treatment for pancreatic cancer.[4][5][6] However, it is not US FDA approved for the treatment of pancreatic cancer.
Chidamide (Epidaza®), a class I HDAC inhibitor, was discovered and developed by ChipScreen and approved by the CFDA in December 2014 for the treatment of recurrent of refractory peripheral T-cell lymphoma. Chidamide, also known as CS055 and HBI- 8000, is an orally bioavailable benzamide type inhibitor of HDAC isoenzymes class I 1–3, as well as class IIb 10, with potential antineoplastic activity. It selectively binds to and inhibits HDAC, leading to an increase in acetylation levels of histone protein H3.74 This agent also inhibits the expression of signaling kinases in the PI3K/ Akt and MAPK/Ras pathways and may result in cell cycle arrest and the induction of tumor cell apoptosis. Currently, phases I and II clinical trials are underway for the treatment of non-small cell lung cancer and for the treatment of breast cancer, respectively.
Chemical Synthesis
The scalable synthetic approach to chidamide very closely follows the discovery route. The sequence began with the condensation of commercial nicotinaldehyde (52) and malonic acid (53) in a mixture of pyridine and piperidine. Next, activation of acid 54 with N,N0-carbonyldiimidazole (CDI) and subsequent reaction with 4-aminomethyl benzoic acid (55) under basic conditions afforded amide 56 in 82% yield. Finally, activation of 56 with CDI prior to treatment with 4-fluorobenzene- 1,2-diamine (57) and subsequent treatment with TFA and THF yielded chidamide (VIII) in 38% overall yield from 52. However, no publication reported that mono-N-Boc-protected bis-aniline was used to approach Chidamide.
^ Qiao Z, Ren S, Li W, Wang X, He M, Guo Y, et al. (April 2013). “Chidamide, a novel histone deacetylase inhibitor, synergistically enhances gemcitabine cytotoxicity in pancreatic cancer cells”. Biochemical and Biophysical Research Communications. 434 (1): 95–101. doi:10.1016/j.bbrc.2013.03.059. PMID23541946.
^ Guha M (April 2015). “HDAC inhibitors still need a home run, despite recent approval”. Nature Reviews. Drug Discovery. 14 (4): 225–6. doi:10.1038/nrd4583. PMID25829268. S2CID36758974.
The CVnCov Vaccine (or CV07050101) is in development by CureVac AG. The vaccine uses mRNA technology to create a protein associated with SARS-CoV2, and upon administration and replication, to initiate subsequent immune responses in the body. As of June 2020, the company received regulatory approval from German and Belgian Authorities to commence Phase 1 clinical trials of this vaccine (NCT04449276).
Efficacy
On 16 June 2021,[4] CureVac said its vaccine showed 47% efficacy from its Phase III trial. This was based on interim analysis of 134 COVID cases in its Phase III study conducted in Europe and Latin America. The final analysis for the trials requires a minimum of 80 additional cases.[2]
Manufacturing of mRNA vaccines can be performed rapidly in high volume,[10] including use of portable, automated printers (“RNA microfactories”) for which CureVac has a joint development partnership with Tesla.[11]
mRNA vaccines require stringent cold chain refrigeration throughout manufacturing, distribution and storage.[12][13] The CureVac technology for CVnCoV uses a non-modified, more natural mRNA less affected by hydrolysis, enabling storage at 5 °C (41 °F) and relatively simplified cold chain requirements that facilitate up to three months of storage and distribution to world regions that do not have specialized ultracold equipment.[6][10]
CureVac has a European-based network to accelerate manufacturing of CVnCoV, if proven safe and effective, for production of up to 300 million doses in 2021 and 600 million doses in 2022.[10][14] An estimated 405 million doses will be provided to EU states.[14]
Clinical trials
In November 2020, CureVac reported results of a Phase I-II clinical trial that CVnCoV (active ingredient zorecimeran) was well-tolerated, safe, and produced a robust immune response.[15][16]
In December 2020, CureVac began a Phase III clinical trial of CVnCoV with 36,500 participants.[17][18]Bayer will provide clinical trial support and international logistics for the Phase III trial, and may be involved in eventual manufacturing should the vaccine prove to be safe and effective.[19][20] In February 2021, the EU’s CHMP started a rolling review of CVnCoV.[21][22] In April 2021, the same procedure began in Switzerland.[23]
Imeglimin is an experimental drug being developed as an oral anti-diabetic.[1][2] It is an oxidative phosphorylation blocker that acts to inhibit hepatic gluconeogenesis, increase muscle glucose uptake, and restore normal insulin secretion. It will be the first of a new class of anti-diabetic if it is approved.
PATENT
https://patents.google.com/patent/WO2012072663A1/enEXAMPLESExample 1 : Synthesis and isolation of (+)-2-amino-3,6-dihydro-4-dimethylamino-6- methyl-l,3,5-triazine hydrochloride by the process according to the invention
Preliminary step: Synthesis of racemic 2-amino-3,6-dihydro-4-dimethylamino- 6-methyl-l,3,5-triazine hydrochloride:
Metformin hydrochloride is suspended in 4 volumes of isobutanol. Acetaldehyde diethylacetal (1.2 eq.) and para-toluenesulfonic acid (PTSA) (0.05 eq) are added and the resulting suspension is heated to reflux until a clear solution is obtained. Then 2 volumes of the solvent are removed via distillation and the resulting suspension is cooled to 20°C. The formed crystals are isolated on a filter dryer and washed with isobutanol (0.55 volumes). Drying is not necessary and the wet product can be directly used for the next step.Acetaldehyde diethylacetal can be replaced with 2,4,6-trimethyl-l,3,5-trioxane (paraldehyde).- Steps 1 and 2: formation of the diastereoisomeric salt and isolation of the desired diastereoisomer
Racemic 2-amino-3,6-dihydro-4-dimethylamino-6-methyl-l,3,5-triazine hydrochloride wet with isobutanol (obtained as crude product from preliminary step without drying) and L-(+)-Tartaric acid (1 eq.) are dissolved in 2.2 volumes of methanol at 20-40°C. The obtained clear solution is filtered and then 1 equivalent of triethylamine (TEA) is added while keeping the temperature below 30°C. The suspension is heated to reflux, stirred at that temperature for 10 minutes and then cooled down to 55°C. The temperature is maintained at 55°C for 2 hours and the suspension is then cooled to 5- 10°C. After additional stirring for 2 hours at 5-10°C the white crystals are isolated on a filter dryer, washed with methanol (2 x 0.5 Vol) and dried under vacuum at 50°C. The yield after drying is typically in the range of 40-45%
– Steps 3 and 4: transformation of the isolated diastereoisomer of the tartrate salt into the hydrochloride salt and recovery of the salt
γ ethanol HN^NH(+) 2-amino-3,6-dihydro-4-dimethylamino-6-methyl-l,3,5-triazine tartrate salt is suspended in 2 volumes of ethanol and 1.02 equivalents of HCl-gas are added under vacuum (-500 mbar). The suspension is heated to reflux under atmospheric pressure (N2) and 5% of the solvent is removed via distillation. Subsequent filtration of the clear colourless solution into a second reactor is followed by a cooling crystallization, the temperature is lowered to 2°C. The obtained suspension is stirred at 2°C for 3 hours and afterwards the white crystals are isolated with a horizontal centrifuge. The crystal cake is washed with ethanol and dried under vacuum at 40°C. The typical yield is 50-55% and the mother liquors can be used for the recovery of about 25-30%) of (+)-2-amino- 3,6-dihydro-4-dimethylamino-6-methyl-l,3,5-triazine tartrate.Example 2: Modification of the solvent of steps 3 and 4
– Steps 3 and 4: transformation of the isolated diastereoisomer of the tartrate salt into the hydrochloride salt and recovery of the salt
HN^NH acetone HN^NH(+) 2-amino-3,6-dihydro-4-dimethylamino-6-methyl-l,3,5-triazine tartrate salt synthesized according to steps 1 and 2 of example 1 is suspended in 1 volume (based on total amount of (+) 2-amino-3,6-dihydro-4-dimethylamino-6-methyl-l,3,5-triazine tartrate salt) of acetone at 20°C. To this suspension 1.01 equivalents of 37% Hydrochloric acid are added. The suspension is heated to reflux under atmospheric pressure (N2) and water is added until a clear solution is obtained. 1.5 vol of acetone are added at reflux temperature. The compound starts crystallising and the obtained suspension is kept at reflux for 2 hours followed by a cooling crystallization to 0°C. The obtained suspension is stirred at 0°C for 2 hours and the white crystals are isolated by centrifugation. The crystal cake is washed with isopropanol and dried under vacuum at 40°C in a continuous drying oven.
Announcement of Marketing Authorization Approval in Japan and Co-promotion Agreement of UPASITA® IV Injection Syringe for the Treatment of Secondary Hyperparathyroidism in Dialysis Patients
SANWA KAGAKU KENKYUSHO Co., Ltd. (Head Office: Nagoya, President and CEO : Shusaku Isono, Suzuken Group, ; “SANWA KAGAKU”) has received Marketing Authorization approval today for UPASITA® IV Injection Syringes (generic name: Upacicalcet Sodium Hydrate; “UPASITA®”) for the treatment of secondary hyperparathyroidism in patients on hemodialysis.
UPASITA® was created by Ajinomoto Pharmaceuticals Co., Ltd. (currently EA Phama Co., Ltd.) and developed by SANWA KAGAKU for the treatment of secondary hyperparathyroidism under a licensing agreement with EA Pharma. UPASITA® acts on calcium sensing receptor in the parathyroid and suppresses excessive secretions of parathyroid hormones (PTH). UPASITA® is administered by intravenous injection to dialysis patients through dialysis circuit by physicians or medical staffs upon completion of dialysis and such administration is expected to reduce the burden of patients with many oral medications whose drinking water volume is severely restricted.
Regarding provision of medical and drug information, SANWA KAGAKU entered into a co-promotion agreement in Japan with Kissei Pharmaceutical Co., Ltd. (Head Office: Matsumoto, Nagano; Chairman and CEO: Mutsuo Kanzawa ; “Kissei”). SANWA KAGAKU will handle the production, marketing, and distribution of the Product while SANWA KAGAKU and Kissei collaboratively promote it to medical institutions in the field in accordance with the agreement. Through the co-promotion activity in the field, SANWA KAGAKU and Kissei will contribute to the treatment of dialysis patients suffering from secondary hyperparathyroidism.
《Reference》
About secondary hyperparathyroidism (SHPT) SHTP is one of complications that occur as chronic kidney disease (chronic kidney failure) progresses and is a pathological condition where excessive PTH is secreted by the parathyroid gland. It has been reported that excessive secretion of parathyroid hormone promotes efflux of phosphorus and calcium from the bone into the blood, thereby increasing the risk of developing bone fractures and arteriosclerosis due to calcification of the cardiovascular system and affecting the vital prognosis.
Product Summary of UPASITA® IV Injection Syringe for Dialysis Brand name: UPASITA® IV Injection Syringe for Dialysis 25μg UPASITA® IV Injection Syringe for Dialysis 50μg UPASITA® IV Injection Syringe for Dialysis 100μg UPASITA® IV Injection Syringe for Dialysis 150μg UPASITA® IV Injection Syringe for Dialysis 200μg UPASITA® IV Injection Syringe for Dialysis 250μg UPASITA® IV Injection Syringe for Dialysis 300μg
Generic Name (JAN): Upacicalcet Sodium Hydrate
Date of Marketing Approval: June 23, 2021
Indications: Secondary hyperparathyroidism in patients on hemodialysis
Dosage and Administration: In adults, UPASITA® is usually administered into venous line of the dialysis circuit at the end of dialysis session during rinse back at a dose of 25 μg sodium upacicalcet 3 times a week as a starting dose. The starting dose can be 50 μg depending on the concentration of serum calcium. Thereafter, the dose may be adjusted in a range from 25 to 300 μg while parathyroid hormone (PTH) and serum calcium level should be carefully monitored in patients.
SYN
WO 2020204117
PATENT
WO 2011108724
WO 2011108690
JP 2013063971
WO 2016194881
JP 6510136
PATENT
WO 2016194881
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2016194881&tab=FULLTEXT(Example 1) Synthesis of (2S) -2-amino-3-{[(5-chloro-2-hydroxy-3-sulfophenyl) carbamoyl] amino} propanoic acid (Compound 1 ) [Chemical formula 14] CDI 150. 2 g (926.6Mmol, 1.1 eq. vs Boc-DAP-O t Bu) to and stirred at 5 ° C. acetone was added 750mL (3.0L / kg). 250 g (842.6 mmol) of Boc-DAP-OtBu was added in two portions, and the mixture was washed with 125 mL (0.5 L / kg) of acetone. After stirring for 30 minutes, completion of the IC (imidazolylcarbonylation) reaction was confirmed by HPLC. 282.6 g (1263.8 mmol, 1.5 eq.) Of ACHB was added in 3 portions, and the mixture was washed with 125 mL (0.5 L / kg) of acetone. After raising the temperature to 30 ° C. and stirring for 18 hours, the completion of the urea conversion reaction was confirmed by HPLC. After cooling to 5 ° C., 124.5 mL (1432.4 mmol, 1.7 eq.) Of concentrated hydrochloric acid was added, and the mixture was stirred for 1 hour. The precipitated unwanted material was filtered and washed with 1000 mL (4.0 L / kg) of acetone. The filtrate was concentrated to 1018 g (4.1 kg / kg), the temperature was raised to 50 ° C., and 625.0 mL (7187 mmol, 8.5 eq.) Of concentrated hydrochloric acid was added dropwise. After stirring for 30 minutes and confirming the completion of deprotection by HPLC, 750 mL of water was added (3.0 L / kg). This liquid was concentrated under reduced pressure to 1730 g (6.9 kg / kg) to precipitate a solid. After stirring at 20 ° C. for 14 hours, vacuum filtration was performed. The filtered solid was washed with 500 mL (2.0 L / kg) of acetone and then dried under reduced pressure at 60 ° C. for 6 hours to obtain 201.4 g of the target product (64.5%). 1H-NMR (400MHz, DMSO-d6): δ 8.3 (s, 1H), 8.2 (bs, 3H), 8.1 (d, 1H, J = 2.6Hz), 7.3 (t, 1H, J = 6.0Hz), 7.0 (d, 1H, J = 2.6Hz), 4.0-4.1 (m, 1H), 3.6-3.7 (m, 1H), 3.4-3.5 (m, 1H)[0026](Example 2) Synthesis of (2S) -2-amino-3-{[(3-sulfophenyl) carbamoyl] amino} propanoic acid (Compound 2 ) [Chemical formula 15] CDI 120.2 g (741.2 mmol, 1. 600 mL (3.0 L / kg) of acetone was added to 1 eq. Vs Boc-DAP-OtBu), and the mixture was stirred at 5 ° C. 200 g (673.9 mmol) of Boc-DAP-OtBu was added in two portions, and the mixture was washed with 100 mL (0.5 L / kg) of acetone. After stirring for 30 minutes, the completion of the IC reaction was confirmed by HPLC. 175.0 g (1010.8 mmol, 1.5 eq.) Of ABS was added in 3 portions and washed with 100 mL (0.5 L / kg) of acetone. After raising the temperature to 30 ° C. and stirring for 18 hours, the completion of the urea conversion reaction was confirmed by HPLC. After cooling to 5 ° C., 99.6 mL (1145.4 mmol, 1.7 eq.) Of concentrated hydrochloric acid was added, and the mixture was stirred for 1 hour. The precipitated unwanted material was filtered and washed with 1400 mL (7.0 L / kg) of acetone. The filtrate was concentrated to 800.1 g (4.0 kg / kg), heated to 50 ° C., and then 500.0 mL (5750.0 mmol, 8.5 eq.) Of concentrated hydrochloric acid was added dropwise. After stirring for 30 minutes and confirming the completion of deprotection by HPLC, 600 mL of water was added (3.0 L / kg). This liquid was concentrated under reduced pressure to 1653.7 g to precipitate a solid. After aging at 20 ° C. for 15 hours, vacuum filtration was performed. The filtered solid was washed with 400 mL (2.0 L / kg) of acetone and then dried under reduced pressure at room temperature for 6 hours to obtain 140.3 g of the desired product (net 132.2 g, 64.7%). 1H-NMR (400MHz, DMSO-d6): δ 8.8 (s, 1H), 8.2 (bs, 3H), 7.7 (s, 1H), 7.3-7.4 (m, 1H), 7.1-7.2 (m, 2H) , 6.3-6.4 (bs, 1H), 4.0-4.1 (bs, 1H), 3.6-3.7 (bs, 1H), 3.5-3.6 (bs, 1H)[0027](Example 3) Synthesis of (2S) -2-amino-3-{[(3-chloro-2-methyl-5-sulfophenyl) carbamoyl] amino} propanoic acid (Compound 3 ) [Chemical formula 16] CDI 14. To 4 g (88.8 mmol, 1.05 eq. Vs Boc-DAP-OtBu), 75 mL (3.0 L / kg vs DAP-OtBu) of acetone was added and stirred at 5 ° C. After adding 25 g (84.3 mmol) of Boc-DAP-OtBu in two portions and stirring for 30 minutes, the completion of the IC reaction was confirmed by HPLC. 26.1 g (118.0 mmol, 1.4 eq.) Of ACTS was added in 3 portions and washed with 25 mL (1.0 L / kg) of acetone. After the temperature was raised to 30 ° C., the mixture was stirred overnight, and the completion of the urea conversion reaction was confirmed by HPLC. After concentrating under reduced pressure at 10 kPa and 40 ° C. until the solvent was completely removed, 37.5 mL (1.5 L / kg) of water and 22.8 mL (257.6 mmol) of concentrated hydrochloric acid were added to perform deprotection for 2 hours. After confirming the completion of the reaction by HPLC, the mixture was cooled to 5 ° C., 60 mL (2.4 L / kg) of MeCN was added, and the mixture was stirred overnight. Further, when 120 mL (4.8 L / kg) of MeCN was added, stratification occurred, so 10 mL (0.4 L / kg) of water and 2.5 mL (0.1 L / kg) of MeCN were added. The precipitated solid was filtered under reduced pressure, washed with 60 mL of MeCN / water (1/2), and then dried under reduced pressure at 60 ° C. for 14 hours to obtain 20.1 g of the desired product as a white solid (net18.3 g, yield 61). 0.8%). 1H-NMR (400MHz, DMSO-d6): δ 14.70-13.30 (bs, 1H), 8.27 (bs, 3H), 8.15 (s, 1H), 7.98 (d, 1H, J = 1.6Hz), 7.27 (d , 1H, J = 1.6Hz), 6.82 (t, 1H, J = 6.0Hz), 4.04 (bs, 1H), 3.70-3.60 (m, 1H), 3.60-3.50 (m, 1H), 2.22 (s, 3H)[0028](Example 4) Synthesis of compound 3 using phenylchloroformate as a carbonyl group-introducing reagent (Step 1) [Chemical formula 17] MeCN 375 mL (7.5 L / kg vs ACTS), Py for 50 g (225.6 mmol) of ACTS. 38.1 mL (473.7 mmol, 2.1 eq.) Was added and stirred at 25 ° C. 29.9 mL (236.8 mmol, 1.05 eq.) Of ClCO 2 Ph (phenyl chloroformate) was added dropwise, and after stirring for 30 minutes, completion of the CM (carbamate) reaction was confirmed by HPLC. 68.9 g (232.4 mmol) of Boc-DAP-OtBu was added, 97.5 mL (699.3 mmol, 3.1 eq.) Of TEA was added dropwise, and the mixture was stirred at 25 ° C. for 3 hours. The completion of the urea conversion reaction was confirmed by HPLC. Here, 103.5 g of the total amount of 517.43 g was used to move to the next step (down to ACTS 10 g scale). 30 mL of water was added and concentrated to 77.0 g at 40 ° C. and 5 kPa. After 100 mL (10 L / kg) of AcOEt was added and the liquid separation operation was performed, 30 mL of water was added to the organic layer and the liquid separation operation was performed again. The organic layer was concentrated to 47.6 g at 40 ° C. and 10 kPa, and then 15 mL (1.5 L / kg) of AcOEt and 100 mL (10 L / kg) of THF were added. Again, it was concentrated to 50.7 g and THF was added up to 146 g. When it was concentrated again to 35.5 g and added to AcOEt 30 mL (3 L / kg) and THF 100 mL (10 L / kg), a solid was precipitated. It was cooled to 5 ° C. and aged overnight. The precipitated solid was filtered under reduced pressure, washed with 20 mL (2.0 L / kg) of THF, and then dried under reduced pressure at 40 ° C. for 3 hours overnight at 30 ° C. to obtain 24.9 g of the desired product as a white solid (net). 23.0 g, 83.6%). 1 H-NMR (400MHz, DMSO-d6): δ 8.86 (bs, 1H), 8.09 (s, 1H), 7.88 (s, 1H), 7.25 (d, 1H, J = 1.6Hz), 7.14 (d, 1H, J = 7.6Hz), 6.60 (t, 1H, J = 5.6Hz), 4.00-3.90 (m, 1H), 3.60-3.50 (m, 1H), 3.30-3.20 (m, 1H), 3.15-3.05 (m, 6H), 2.19 (s, 3H), 1.50-1.30 (m, 18H), 1.20-1.10 (m, 9H)
(Step 2) [Chemical
formula 18] Compound 4 21.64 g (net. 20.0 g, 68 mL of water (3.4 L / kg vs. compound 4) vs. 32.8 mmol) ) Was added, the mixture was stirred at 50 ° C., and 12 mL (135.6 mmol, 4.1 eq.) Of concentrated hydrochloric acid was added dropwise. After stirring for 1 hour, the temperature was raised to 70 ° C. to dissolve the precipitated solid. After confirming the completion of the reaction by HPLC, the mixture was cooled to 50 ° C. and aged for 1 hour, and then cooled to 5 ° C. over 4 hours. The precipitated solid was filtered under reduced pressure, washed with 40 mL (2.0 L / kg) of MeCN / water (2/1), and then dried under reduced pressure at 60 ° C. for 3 hours to obtain 11.2 g of the desired product as a white solid (11.2 g). net 10.5 g, 91.1%).[0029](Example 5) [Chemical formula 19] MeCN 10.0 mL (10.0 L / kg vs ACSS), Py 0.75 mL (9.25 mmol, 2.05 eq.) For 1.00 g (4.51 mmol) of ACTS. , And stirred at 8 ° C. After dropping 0.59 mL (4.74 mmol, 1.05 eq.) Of ClCO 2 Ph, raising the temperature to room temperature and stirring for 1 hour, completion of the CM conversion reaction was confirmed by HPLC. 1.33 g (4.51 mmol, 1.0 eq.) Of Boc-DAP-OtBu was added, 1.92 mL (13.76 mmol, 3.05 eq.) Of TEA was added dropwise, and the mixture was stirred at 40 ° C. for 1 hour. After confirming the completion of the urea conversion reaction by HPLC, the mixture was concentrated until the solvent was completely removed. 1.0 mL of water and 2.0 mL of concentrated hydrochloric acid (22.6 mmol, 5.0 eq.) Were added, and the mixture was stirred at 50 ° C. for 4 hours. After confirming the completion of deprotection by HPLC, MeCN 7.5 mL (7.5 L / kg), 1 M HCl aq. After adding 4.5 mL, the mixture was stirred at 5 ° C. overnight. The precipitated solid was filtered under reduced pressure, washed with 3.0 mL (3.0 L / kg) of MeCN, and then dried at 60 ° C. overnight to obtain 1.28 g of the desired product as a white solid (net 1.18 g, 77). .0%).[0030](Example 6) (Step 1) 3-({[(2S) -2-amino-3-methoxy-3-oxopropyl] carbamoyl} amino) -5-chloro-4-methylbenzene-1-sulfonic acid ( Synthesis of Compound 5 ) [Chemical formula 20] To 5 g (22.56 mmol) of ACTS, 37.5 mL (7.5 L / kg vs ACTS) of MeCN and 3.81 mL (47.38 mmol, 2.1 eq.) Of Py were added. The mixture was stirred at 25 ° C. 2.99 mL (23.68 mmol, 1.05 eq.) Of ClCO 2 Ph was added dropwise, and after stirring for 30 minutes, the completion of the CM reaction was confirmed by HPLC. 5.92 g (23.23 mmol, 1.03 eq.) Of Boc-DAP-OMe was added, 9.75 mL (69.93 mmol, 3.1 eq.) Of TEA was added dropwise, and the mixture was stirred at 25 ° C. for 3 hours. 0.4 g (1.58 mmol, 0.07 eq.) Of Boc-DAP-OMe and 0.22 mL (1.58 mmol, 0.07 eq.) Of TEA were added, and the completion of the ureaization reaction was confirmed by HPLC. 7.32 mL (112.8 mmol, 5.0 eq.) Of MsOH was added, the temperature was raised to 50 ° C., and the mixture was stirred for 4 hours. After confirming the completion of deprotection by HPLC, the mixture was cooled to 25 ° C. and 37.5 mL (7.5 L / kg) of MeCN and 7.5 mL (1.5 L / kg) of water were added to precipitate a solid. It was cooled to 5 ° C. and aged for 16 hours. The precipitated solid was filtered under reduced pressure, washed with 20 mL (4.0 L / kg) of water / MeCN (1/2), and then dried under reduced pressure at 40 ° C. for 5 hours to obtain 7.72 g of the target product as a white solid (772 g of the target product). net 7.20 g, 87.3%). 1H-NMR (400MHz, DMSO-d6): δ 8.39 (bs, 3H), 8.16 (d, 1H, J = 1.2Hz), 7.90 (d, 1H, J = 1.6Hz), 7.28 (d, 1H, J = 1.6Hz), 6.78 (t, 1H, J = 5.6Hz), 4.20-4.10 (m, 1H), 3.77 (s, 3H), 3.70-3.60 (m, 1H), 3.55-3.45 (m, 1H) , 2.21 (s, 3H) HRMS (FAB – ): calcd for m / z 364.0369 (MH), found The m / z 364.0395 (MH)
(step 2) [Formula 21]
compound 5 10.64 g (net Non 10.0 g, To 27.34 mmol), 18 mL of water (1.8 L / kg vs. compound 5 ) was added and stirred at 8 ° C. 3.42 mL (57.41 mmol, 2.1 eq.) Of a 48% aqueous sodium hydroxide solution was added dropwise, and the mixture was washed with 1.0 mL (1.0 L / kg) of water and then stirred at 8 ° C. for 15 minutes. After confirming the completion of hydrolysis by HPLC, the temperature was raised to 25 ° C. and 48% HBr aq. The pH was adjusted to 5.8 by adding about 3.55 mL. After confirming the precipitation of the target product by dropping 65 mL (6.5 L / kg) of IPA, the mixture was aged for 1 hour. 81 mL (8.1 L / kg) of IPA was added dropwise and aged at 8 ° C. overnight. The precipitated solid was filtered under reduced pressure, washed with 20 mL (2.0 L / kg) of IPA, and then dried under reduced pressure at 40 ° C. for 4 hours to obtain 10.7 g of the desired product as a white solid (net 9.46 g, 92. 6%). 1 H-NMR (400MHz, DMSO-d6): δ8.76 (s, 1H), 7.91 (d, 1H, J = 1.6Hz), 8.00-7.50 (bs, 2H), 7.24 (d, 1H, J = 1.6Hz), 7.20 (t, 1H, J = 5.6Hz), 3.58-3.54 (m, 1H), 3.47-3.43 (m, 1H), 3.42-3.37 (m, 1H), 2.23 (s, 3H)[0031](Example 7) (Step 1) [Chemical formula 22] For 10.0 g (45.1 mmol) of ACTS, 50 mL (5.0 L / kg vs ACTS) of MeCN, 7.46 mL (92.5 mmol, 2.05 eq. ) Was added, and the mixture was stirred at 8 ° C. 5.98 mL (47.4 mmol, 1.05 eq.) Of ClCO 2 Ph was added dropwise, the temperature was raised to 25 ° C., and the mixture was stirred for 1 hour, and then the completion of the CM reaction was confirmed by HPLC. 100 ml of acetone (10.0 L / kg vs ACTS) was added, the mixture was cooled to 8 ° C., and aged for 1 hour. The precipitated solid was filtered under reduced pressure, washed with 30 mL of acetone (3.0 L / kg vs ACTS), and then dried under reduced pressure at 60 ° C. for 2 hours to obtain 17.8 g of the target product (net 14.4 g as a free form). Quant). 1 H-NMR (400MHz, DMSO-d6): δ 9.76 (bs, 1H), 8.93-8.90 (m, 2H), 8.60-8.50 (m, 1H), 8.10-8.00 (m, 2H), 7.60 (s , 1H), 7.50-7.40 (m, 3H), 7.30-7.20 (m, 3H), 2.30 (s, 3H)
(Step 2) [Chemical 23]
Compound 6 To 5.0 g (11.9 mmol), 50 ml of acetonitrile and 3.53 g (11.9 mmol) of Boc-DAP-OtBu were added, and the mixture was stirred at 8 ° C. 3.5 ml (25 mmol) of triethylamine was added dropwise, and the mixture was stirred overnight at room temperature. The solvent was distilled off under reduced pressure, and 25 ml of ethyl acetate and 5 ml of water were added for extraction. The organic layer was washed with 5 ml of water, the solvent was distilled off, 50 ml of tetrahydrofuran was added, the mixture was cooled to 8 ° C., and aged for 1 hour. The precipitated solid was filtered under reduced pressure, washed with 10 ml of tetrahydrofuran, and dried under reduced pressure at 60 ° C. overnight to obtain 6.3 g of the desired product as a white solid.[0032](Example 8) [Chemical formula 24] For 1.08 g (4.89 mmol) of ACTS, 8.1 mL (7.5 L / kg vs ACTS) of MeCN and 827 μL (10.27 mmol, 2.1 eq.) Of Py were added. In addition, it was stirred at room temperature. ClCO 2 Ph 649 μL (5.14 mmol, 1.05 eq.) Was added dropwise, and the mixture was stirred for 30 minutes, and then the completion of the CM conversion reaction was confirmed by HPLC. 1.48 g (5.04 mmol, 1.03 eq.) Of Cbz-DAP-OMe HCl was added, 2.1 mL (15.17 mmol, 3.1 eq.) Of TEA was added dropwise, and the mixture was stirred at room temperature for about 5 hours. After confirming the completion of the urea conversion reaction by HPLC, the mixture was concentrated until the solvent was completely removed. 15.0 mL of 30% HBr / AcOH was added, and the mixture was stirred at room temperature for 70 minutes, and the completion of deprotection was confirmed by HPLC. After concentration to dryness, 10 mL of water and 4 mL of AcOEt were added to carry out an extraction operation, and then the aqueous layer was stirred at room temperature overnight. The precipitated solid was filtered under reduced pressure, washed with 15 mL of water and 10 mL of AcOEt, and then dried at 40 ° C. for 3 hours to obtain 1.45 g of the desired product as a white solid (58.8%).[0033](Example 9) Synthesis of compound 7 ( methyl ester of compound 1 ) using phenyl chloroformate as a carbonyl group introduction reagent [Chemical formula 25] MeCN 73 mL (14.6 L) with respect to 5.00 g (22.4 mmol) of ACHB. / Kg vs ACHB), Py 3.8 mL (47 mmol, 2.1 eq.), Was added and stirred at 40 ° C. After adding 3.0 mL (24 mmol, 1.05 eq.) Of ClCO 2 Ph and stirring for 30 minutes, the completion of the CM conversion reaction was confirmed by HPLC. 5.87 g (23 mmol, 1.0 eq.) Of Boc-DAP-OMe was added, washed with a small amount of MeCN, 9.7 mL (70 mmol, 3.1 eq.) Of TEA was added dropwise, and the mixture was stirred at 40 ° C. for 3 hours. After confirming the completion of the urea conversion reaction by HPLC, the mixture was cooled to room temperature. 7.3 mL (112 mmol, 5.0 eq.) Of MsOH was added, the temperature was raised to 50 ° C., and the mixture was stirred for 7 hours. Further, 1.5 mL (23 mmol, 1.0 eq.) Of MsOH was added, and the reaction was carried out at 50 ° C. overnight. After confirming the completion of deprotection by HPLC, 90 mL of acetone was added to the reaction solution, and the mixture was cooled to room temperature. The precipitated solid was obtained and dried under reduced pressure at 60 ° C. to obtain the desired product. 1 H-NMR (400MHz, DMSO-d6): δ 7.22 (m, 1H), 7.14 (m, 1H), 4.36 (m, 1H), 3.80 (s, 3H), 3.20-3.40 (m, 2H).[0034](Example 10) Synthesis of compound 5 using 4-chlorophenylchloroformate as a carbonyl group-introducing reagent [Chemical formula 26] For 5.00 g (22.6 mmol) of ACTS, 73 mL (14.6 L / kg vs ACTS) of MeCN, 3.8 mL (47 mmol, 2.1 eq.) Of Py was added and stirred at 40 ° C. After adding 3.25 mL (23.7 mmol, 1.05 eq.) Of 4-chloroformic acid 4-chlorophenylate and stirring at 40 ° C. for 1.5 hours, completion of the CM conversion reaction was confirmed by HPLC. Add 5.92 g (23.2 mol, 1.0 eq.) Of Boc-DAP-OMe, wash with a small amount of MeCN, add 9.7 mL (70 mmol, 3.1 eq.) Of TEA, and stir at 40 ° C. for 2 hours. did. After confirming the completion of the urea conversion reaction by HPLC, the mixture was cooled to room temperature. 7.3 mL (113 mmol, 5.0 eq.) Of MsOH was added, the temperature was raised to 50 ° C., and the mixture was stirred for 3.5 hours. After confirming the completion of deprotection by HPLC, the reaction solution was cooled to room temperature, 7.5 mL of water was added, the mixture was cooled to 8 ° C., and the mixture was stirred overnight. The precipitated solid was filtered, washed with a small amount of MeCN water, and dried at 60 ° C. overnight to obtain 6.94 g of the desired product as a white solid (84.1%).[0035](Example 11) Synthesis of compound 5 using 4-nitrophenyl chloroformate as a carbonyl group-introducing reagent [Chemical formula 27] 73 mL (14.6 L / kg vs. ACTS) of MeCN with respect to 5.00 g (22.6 mmol) of ACTS. , Py 3.8 mL (47 mmol, 2.1 eq.), And stirred at 40 ° C. 4.77 mL (23.7 mmol, 1.05 eq.) Of 4-nitrophenyl chloroformate was added dropwise, and the mixture was stirred at 40 ° C. for 3.5 hours, and then the completion of the CM reaction was confirmed by HPLC. Add 5.92 g (23.2 mmol, 1.0 eq.) Of Boc-DAP-OMe, wash with a small amount of MeCN, add 9.7 mL (70 mmol, 3.1 eq.) Of TEA, and stir at 40 ° C. for 2 hours. did. After confirming the completion of the urea conversion reaction by HPLC, the mixture was cooled to room temperature. 7.3 mL (113 mmol, 5.0 eq.) Of MsOH was added, the temperature was raised to 50 ° C., and the mixture was stirred for 3.5 hours. After confirming the completion of deprotection by HPLC, the reaction solution was cooled to room temperature, 7.5 mL of water was added, the mixture was cooled to 8 ° C., and the mixture was stirred overnight. The precipitated solid was filtered, washed with a small amount of MeCN water, and dried at 60 ° C. overnight to obtain 5.96 g of the desired product as a white solid (72.2%).[0036](Example 12) Synthesis of compound 3 using Boc-DAP-OH [Chemical 28] MeCN 73 mL (14.6 L / kg vs ACTS), Py 3.8 mL, relative to 5.00 g (22.6 mmol) of ACTS. (47 mmol, 2.1 eq.) Was added and stirred at 40 ° C. After adding 3.00 mL (23.8 mmol, 1.05 eq.) Of phenylchloroformate and stirring at 40 ° C. for 0.5 hours, the completion of the CM conversion reaction was confirmed by HPLC (CM conversion reaction product: 4.37 minutes). , ACTS: N.D.). Add 4.75 g (23.2 mmol, 1.0 eq.) Of Boc-DAP-OH, wash with a small amount of MeCN, add 9.7 mL (70 mmol, 3.1 eq.) Of TEA, and stir at 40 ° C. for 2 hours. did. After confirming the completion of the urea-forming reaction by HPLC (urea-forming reaction product: 3.81 minutes, CM-forming reaction product: 0.02 area% vs. urea-forming reaction product), the mixture was cooled to room temperature. By adding 7.3 mL (113 mmol, 5.0 eq.) Of MsOH, raising the temperature to 50 ° C., stirring for 4.5 hours, and further adding 1.5 mL (23 mmol, 1.0 eq.) Of MsOH, stirring for 1 hour. , The formation of the target product was confirmed by HPLC (Compound 3: 2.49 minutes, urea conversion reaction product: 0.50 area vs. compound 3, area of compound 3 with respect to the total area excluding pyridine: 71.0 area).
PATENT
JP 6510136
PATENT
WO 2020204117
Reference Example 1 Synthesis of 3-{[(2S) -2-amino-2-carboxyethyl] carbamoylamino} -5-chloro-4-methylbenzenesulfonate sodium (Compound A1) (Step 1) Synthesis of 3 -({[(2S) -2-amino-3-methoxy-3-oxopropyl] carbamoyl} amino) -5-chloro-4-methylbenzene-1-sulfonic acid 3-amino- 37.5 mL (7.5 L / kg vs ACTS) of acetonitrile and 3.81 mL (47.38 mmol, 2.1 eq.) Of pyridine against 5 g (22.56 mmol) of 5-chloro-4-methylbenzenesulfonic acid (ACTS). Was added and stirred at 25 ° C. 2.99 mL (23.68 mmol, 1.05 eq.) Of ClCO 2 Ph was added dropwise, and after stirring for 30 minutes, the completion of the carbamate reaction was confirmed by HPLC. Add 5.92 g (23.23 mmol, 1.03 eq.) Of 3-amino-N- (tert-butoxycarbonyl) -L-alanine methyl ester hydrochloride and 9.75 mL (69.93 mmol, 3.1 eq.) Triethylamine. Was added dropwise, and the mixture was stirred at 25 ° C. for 3 hours. Add 0.4 g (1.58 mmol, 0.07 eq.) Of 3-amino-N- (tert-butoxycarbonyl) -L-alanine methyl ester hydrochloride and 0.22 mL (1.58 mmol, 0.07 eq.) Of triethylamine. Then, the completion of the urea conversion reaction was confirmed by HPLC. 7.32 mL (112.8 mmol, 5.0 eq.) Of methanesulfonic acid was added, the temperature was raised to 50 ° C., and the mixture was stirred for 4 hours. After confirming the completion of deprotection by HPLC, the mixture was cooled to 25 ° C. and 37.5 mL (7.5 L / kg) of acetonitrile and 7.5 mL (1.5 L / kg) of water were added to precipitate a solid. It was cooled to 5 ° C. and aged for 16 hours. The precipitated solid was filtered under reduced pressure, washed with 20 mL (4.0 L / kg) of water / acetonitrile (1/2), and then dried under reduced pressure at 40 ° C. for 5 hours to obtain 7.72 g of the desired product as a white solid (. net 7.20 g, 87.3%).
1 H-NMR (400MHz, DMSO-d6): δ 8.39 (bs, 3H), 8.16 (d, 1H, J = 1.2Hz), 7.90 (d, 1H, J = 1.6Hz), 7.28 (d, 1H, J = 1.6Hz), 6.78 (t, 1H, J = 5.6Hz), 4.20-4.10 (m, 1H), 3.77 (s, 3H), 3.70-3.60 (m, 1H), 3.55-3.45 (m, 1H) ), 2.21 (S, 3H)HRMS (FAB – ): Calcd For M / Z 364.0369 (MH & lt;), Found M / Z 364.0395 (MH & lt;) (Step 2) (2) Compound obtained in step 1 of synthesis of 3-{[(2S) -2-amino-2-carboxyethyl] carbamoylamino} -5-chloro-4-methylbenzenesulfonate . To 64 g (net 10.0 g, 27.34 mmol), 18 mL of water (1.8 L / kg vs. the compound of Step 1) was added, and the mixture was stirred at 8 ° C. 3.42 mL (57.41 mmol, 2.1 eq.) Of a 48% aqueous sodium hydroxide solution was added dropwise, and the mixture was washed with 1.0 mL (1.0 L / kg) of water and then stirred at 8 ° C. for 15 minutes. After confirming the completion of hydrolysis by HPLC, the temperature was raised to 25 ° C. and 48% HBr aq. About 3.55 mL was added to adjust the pH to 5.8. After confirming the precipitation of the desired product by dropping 65 mL (6.5 L / kg) of isopropyl alcohol, the mixture was aged for 1 hour. 81 mL (8.1 L / kg) of isopropyl alcohol was added dropwise and the mixture was aged at 8 ° C. overnight. The precipitated solid was filtered under reduced pressure, washed with 20 mL (2.0 L / kg) of isopropyl alcohol, and then dried under reduced pressure at 40 ° C. for 4 hours to obtain 10.7 g of the desired product as a white solid (net 9.46 g, 92). .6%). 1 H-NMR (400MHz, DMSO-d6): δ8.76 (s, 1H), 7.91 (d, 1H, J = 1.6Hz), 8.00-7.50 (bs, 2H), 7.24 (d, 1H, J = 1.6Hz), 7.20 (t, 1H, J = 5.6Hz), 3.58-3.54 (m, 1H), 3.47-3.43 (m, 1H), 3.42-3.37 (m, 1H), 2.23 (s, 3H)
Tralokinumab is a human monoclonal antibody which targets the cytokine interleukin 13,[1] and is designed for the treatment of asthma and other inflammatory diseases.[2] Tralokinumab was discovered by Cambridge Antibody Technology scientists, using Ribosome Display, as CAT-354[3] and taken through pre-clinical and early clinical development.[4] After 2007 it has been developed by MedImmune, a member of the AstraZeneca group, where it is currently in Ph3 testing for asthma and Ph2b testing for atopic dermatitis.[5][6] This makes it one of the few fully internally discovered and developed drug candidates in AstraZeneca’s late stage development pipeline.
Discovery and development
Tralokinumab (CAT-354) was discovered by Cambridge Antibody Technology scientists[7] using protein optimization based on Ribosome Display.[8] They used the extensive data sets from ribosome display to patent protect CAT-354 in a world-first of sequence-activity-relationship claims.[7] In 2004, clinical development of CAT-354 was initiated with this first study completing in 2005.[9] On 21 July 2011, MedImmune LLC initiated a Ph2b, randomized, double-blind study to evaluate the efficacy of tralokinumab in adults with asthma.[10]
In 2016, MedImmune and AstraZeneca were developing tralokinumab for asthma (Ph3) and atopic dermatitis (Ph2b) while clinical development for moderate-to-severe ulcerative colitis and idiopathic pulmonary fibrosis (IPF) have been discontinued.[9] In July of that year AstraZeneca licensed Tralokinumab to LEO Pharma for skin diseases.[11]
A phase IIb study of Tralokinumab found that treatment was associated with early and sustained improvements in atopic dermatitis symptoms and tralokinumab had an acceptable safety and tolerability profile, thereby providing evidence for targeting IL-13 in patients with atopic dermatitis.[12]
On 15 June 2017, Leo Pharma announced that they were starting phase III clinical trials with tralokinumab in atopic dermatitis.[13]
Society and culture
Legal status
On 22 April 2021, the Committee for Medicinal Products for Human Use (CHMP) adopted a positive opinion, recommending the granting of a marketing authorization for the medicinal product Adtralza, intended for the treatment of moderate‑to‑severe atopic dermatitis.[14]
The applicant for this medicinal product is LEO Pharma A/S.
References
^ Kopf M, Bachmann MF, Marsland BJ (September 2010). “Averting inflammation by targeting the cytokine environment”. Nature Reviews. Drug Discovery. 9 (9): 703–18. doi:10.1038/nrd2805. PMID20811382. S2CID23769909.
^ Clinical trial number NCT01402986 for “A Phase 2b, Randomized, Double-blind Study to Evaluate the Efficacy of Tralokinumab in Adults With Asthma” at ClinicalTrials.gov
N-(2-(2-(Dimethylamino)ethoxy)-4-methoxy-5-((4-(1-methyl-1H-indol-3-yl)-2-pyrimidinyl)amino)phenyl)-2-propenamideThe epidermal growth factor receptor (EGFR, Herl, ErbB l) is a principal member of the ErbB family of four structurally-related cell surface receptors with the other members being Her2 (Neu, ErbB2), Her3 (ErbB3) and Her4 (ErbB4). EGFR exerts its primary cellular functions though its intrinsic catalytic tyrosine protein kinase activity. The receptor is activated by binding with growth factor ligands, such as epidermal growth factor (EGF) and transforming growth factor-alpha (TGF-a), which transform the catalytically inactive EGFR monomer into catalytically active homo- and hetero- dimers. These catalytically active dimers then initiate intracellular tyrosine kinase activity, which leads to the autophosphorylation of specific EGFR tyrosine residues and elicits the downstream activation of signaling proteins. Subsequently, the signaling proteins initiate multiple signal transduction cascades (MAPK, Akt and JNK), which ultimately mediate the essential biological processes of cell growth, proliferation, motility and survival.EGFR is found at abnormally high levels on the surface of many types of cancer cells and increased levels of EGFR have been associated with advanced disease, cancer spread and poor clinical prognosis. Mutations in EGFR can lead to receptor overexpression, perpetual activation or sustained hyperactivity and result in uncontrolled cell growth, i.e. cancer. Consequently, EGFR mutations have been identified in several types of malignant tumors, including metastatic lung, head and neck, colorectal and pancreatic cancers. In lung cancer, mutations mainly occur in exons 18 to 21, which encode the adenosine triphosphate (ATP)-binding pocket of the kinase domain. The most clinically relevant drug- sensitive EGFR mutations are deletions in exon 19 that eliminate a common amino acid motif (LREA) and point mutations in exon 21, which lead to a substitution of arginine for leucine at position 858 (L858R). Together, these two mutations account for nearly 85% of the EGFR mutations observed in lung cancer. Both mutations have perpetual tyrosine kinase activity and as a result they are oncogenic. Biochemical studies have demonstrated that these mutated EGFRs bind preferentially to tyrosine kinase inhibitor drugs such as erlotinib and gefitinib over adenosine triphosphate (ATP).Erlotinib and gefitinib are oral EGFR tyrosine kinase inhibitors that are first line monotherapies for non-small cell lung cancer (NSCLC) patients having activating mutations in EGFR. Around 70% of these patients respond initially, but unfortunately they develop resistance with a median time to progression of 10-16 months. In at least 50% of these initially responsive patients, disease progression is associated with the development of a secondary mutation, T790M in exon 20 of EGFR (referred to as the gatekeeper mutation). The additional T790M mutation increases the affinity of the EGFR kinase domain for ATP, thereby reducing the inhibitory activity of ATP- competitive inhibitors like gefitinib and erlotinib.Recently, irreversible EGFR tyrosine kinase inhibitors have been developed that effectively inhibit the kinase domain of the T790M double mutant and therefore overcome the resistance observed with reversible inhibitors in the clinic. These inhibitors possess reactive electrophilic functional groups that react with the nucleophilic thiol of an active-site cysteine. Highly selective irreversible inhibitors can be achieved by exploiting the inherent non-covalent selectivity of a given scaffold along with the location of a particular cysteine residue within the ATP binding site. The acrylamide moieties of these inhibitors both undergo a Michael reaction with Cys797 in the ATP binding site of EGFRT790M to form a covalent bond. This covalent mechanism is thought to overcome the increase in ATP affinity of the T790M EGRF double mutant and give rise to effective inhibition. However, these inhibitors may cause various undesired toxicities. Therefore, development of new inhibitors for treatment of various EGFR-related cancers is still in high demand. PatentCN201580067776) N-(2-(2-(dimethylamino)ethoxy)-4-methoxy-5-((4-(1-methyl-1H- Indol-3-yl)pyrimidin-2-yl)amino)phenyl)acrylamide (compound of formula I) can be prepared by the following synthetic route:
N-(4-(2-(Dimethylamino)ethoxy)-2-methoxy-5-nitrophenyl)-4-(l-methyl-lH- indol-3-yl)pyrimidin-2-amine (Scheme 1, Intermediate B). To a slurry of NaH (30 mmol, 60% oil dispersion prewashed with hexanes) and 50 mL of 1,4-dioxane was added 2-dimethylaminoethanol (27 mmol, 2.7 mL) dropwise with stirring under N2. After stirring for 1 h, a slurry of A (5.4 mmol) in 50 mL of 1,4-dioxane was added portion-wise over 15 min under a stream of N2. The resulting mixture was stirred overnight, then poured into water and the solid was collected, rinsed with water, and dried under vacuum to yield 2.6 g of product as a yellow solid. A purified sample was obtained from chromatography (silica gel; CH2C12-CH30H gradient). 1H NMR (300 MHz, DMSO) δ 2.26 (s, 6H), 2.70 (t, 2H, J = 6 Hz), 3.87 (s, 3H), 4.01 (s, 3H), 4.32 (t, 2H, J = 6 Hz), 7.00-7.53 (m, 5H), 8.18-8.78 (m, 5H); C24H26N604 m/z MH+ 463.4-(2-(Dimethylamino)ethoxy)-6-methoxy-Nl-(4-(l-methyl-lH-indol-3- yl)pyrimidin-2-yl)benzene-l,3-diamine (Scheme 1, Intermediate C). A suspension of 2.6 g of Intermediate B, 1.6 g of Fe°, 30 mL of ethanol, 15 mL of water, and 20 mL of cone. HC1 was heated to 78 °C for 3 h. The solution was cooled to room temperature, adjusted to pH 10 with 10% NaOH (aq) and diluted with CH2C12. The mixture was filtered through Dicalite, and the filtrate layers were separated. The aqueous phase was extracted with CH2C12 twice, and the combined organic extracts were dried over Na2S04 and concentrated. Column chromatography (silica gel, CH2Cl2-MeOH gradient) afforded 1.2 g of Intermediate C as a solid. C24H28N602 m/z MH+ 433.N-(2-(2-(Dimethylamino)ethoxy)-4-methoxy-5-((4-(l-methyl-lH-indol-3- yl)pyrimidin-2-yl)amino)phenyl)acrylamide (1). To a solution of Intermediate C (2.8 mmol) in 50 mL of THF and 10 mL of water was added 3-chloropropionychloride (2.8 mmol) dropwise with stirring. After 5 h of stirring, NaOH (28 mmol) was added and the mixture was heated at 65°C for 18 h. After cooling to room temperature, THF was partially removed under reduced pressure, and the mixture was extracted with CH2C12, dried over Na2S04, and concentrated. Chromatography of the crude product (silica gel, CH2Cl2-MeOH) afforded 0.583 g of Example 1 as a beige solid. 1H NMR (300 MHz, DMSO) δ 2.28 (s, 6H), 2.50-2.60 (m, 2H), 3.86 (s, 3H), 3.90 (s, 3H), 4.19 (t, 2H, = 5.5 Hz), 5.73-5.77 (m, IH), 6.21-6.27 (m, IH), 6.44-6.50 (m, IH), 6.95 (s, IH), 7.11-7.53 (overlapping m, 3H), 7.90 (s, IH), 8.27-8.30 (overlapping m, 3H), 8.55 (s, IH), 8.84 (s, IH), 9.84 (s, IH) ppm; C27H30N6O3 m/z MH+ 487
PATENT WO2021115425
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2021115425&tab=FULLTEXT&_cid=P20-KQN9F3-73566-1Epidermal growth factor receptors (EGFR, Her1, ErbB1) are the main members of the ErbB family of four structurally related cell surface receptors, and the other members are Her2 (Neu, ErbB2), Her3 (ErbB3) and Her4 (ErbB4). EGFR exerts its main cellular functions through its inherent catalytic tyrosine protein kinase activity. The receptor is activated by binding to growth factor ligands, such as epidermal growth factor (EGF) and transforming growth factor-α (TGF-α). The catalytically inactive EGFR monomer is transformed into a catalytically active homopolymer and Heterodimer. These catalytically active dimers then initiate intracellular tyrosine kinase activity, which leads to autophosphorylation of specific EGFR tyrosine residues and elicits downstream activation of signaling proteins. Subsequently, the signal protein initiates multiple signal transduction cascades (MAPK, Akt, and JNK), which ultimately regulate the basic biological processes of cell growth, proliferation, motility, and survival.
EGFR has been found to have abnormally high levels on the surface of many types of cancer cells, and elevated EGFR levels have been associated with advanced disease, cancer spread, and poor clinical prognosis. Mutations in EGFR can lead to overexpression of the receptor, permanent activation or continuous hyperactivity, leading to uncontrolled cell growth, which is cancer. Therefore, EGFR mutations have been identified in several types of malignant tumors, including metastatic lung cancer, head and neck cancer, colorectal cancer, and pancreatic cancer. In brain cancer, mutations mainly occur in exons 18-21, which encode the adenosine triphosphate (ATP)-binding pocket of the kinase domain. The most clinically relevant drug-sensitive EGFR mutations are deletions in exon 19 and point mutations in exon 21. The former eliminates a common amino acid motif (LREA), and the latter results in position 858 (L858R). The arginine is replaced by leucine. Together, these two mutations account for nearly 85% of the EGFR mutations observed in lung cancer. Both mutations have permanent tyrosine kinase activity, so they are carcinogenic. In at least 50% of patients who initially responded to current therapies, the progression of the disease is related to the development of a secondary mutation, T790M (also known as the goalkeeper mutation) in exon 20 of EGFR. BPI-7711 is a third-generation EGFR-TKI compound developed by Beida Pharmaceuticals and disclosed in International Patent No. WO2017/218892. It is the N-(2-(2-(dimethylamino) )Ethoxy)-4-methoxy-5-((4-(1-methyl-1H-indol-3-yl)pyrimidin-2-yl)amino)phenyl)acrylamide methanesulfonic acid salt:
Need to develop improved properties containing N-(2-(2-(dimethylamino)ethoxy)-4-methoxy-5-((4-(1-methyl-1H-indole-3 -Yl)pyrimidin-2-yl)amino)phenyl)acrylamide pharmaceutically acceptable salt, in particular the pharmaceutical composition of BPI-7711 and its use, and the preparation of said pharmaceutical composition suitable for large-scale production method.
PATENT
WO2021061695 , for another filing, assigned to Beta Pharma, claiming a combination of an EGFR inhibitor (eg BPI-7711) and a CDK4/6 inhibitor, useful for treating cancer.
Novel crystalline polymorphic form A of rezivertinib – presumed to be BPI-7711 – useful for treating diseases mediated by EGFR mutations eg lung cancer, preferably non-small cell lung cancer (NSCLC).Epidermal growth factor receptor (EGFR) is a type of transmembrane receptor tyrosine kinase in the human body. The activation (ie phosphorylation) of this kinase is of great significance to the inhibition of tumor cell proliferation, angiogenesis, tumor invasion, metastasis and apoptosis. EGFR kinase is involved in the disease process of most cancers, and these receptors are overexpressed in many major human tumors. Overexpression, mutations, or high expression of ligands associated with these family members can lead to some tumor diseases, such as non-small cell lung cancer, colorectal cancer, breast cancer, head and neck cancer, cervical cancer, bladder cancer, and thyroid. Cancer, stomach cancer, kidney cancer, etc. In recent years, epidermal growth factor receptor tyrosine kinase has become one of the most attractive targets in current anti-tumor drug research. In 2003, the US FDA approved the first epidermal growth receptor tyrosine kinase inhibitor (EGFR-TKI) drug (gefitinib) for the treatment of advanced non-small cell lung cancer (NSCLC). Development of a generation of EGFR inhibitors. Numerous clinical trials have confirmed that for patients with EGFR-positive non-small cell lung cancer, the therapeutic effect of molecular targeted drugs is significantly better than traditional chemotherapy. Although the first-generation EGFR-inhibiting targeted drugs responded well to the initial treatment of many non-small cell lung cancer (NSCLC) patients, most patients will eventually develop disease progression due to drug resistance (such as EGFR secondary T790M mutation). The emergence of drug resistance is caused by various mechanisms based on the mutations in the original EGFR pathway activity. In the drug resistance research on the first generation of EGFR inhibitors, the research frontier is the irreversible third generation EFGR inhibitor. But so far, the third-generation EGFR inhibitors worldwide, in addition to AstraZeneca O’Higgins imatinib developed, there is no other effective against T790M resistance mutations in patients with drug approved for clinical use; Several drug candidates for the T790M mutation are in clinical development. The chemical structure of this third-generation EGFR inhibitor is completely different from that of the first-generation. The main difference from the first-generation EGFR inhibitors is that they both use a highly selective core structure to replace the low-selective aminoquinoline core structure of the first and second-generation EGFR-TKIs. Compared with wild-type EGFR, these third-generation compounds are highly specific and selective for the T790M mutation after EGFR positive resistance. Chinese Patent Application No. CN201580067776.8 discloses a compound of the following formula I, which also belongs to the third-generation EGFR-TKI class of small molecule targeted drugs. The compound has a high inhibitory effect on non-small cell lung cancer (NSCLC) cells with single-activity mutation and T790M double-mutant EGFR, and its effective inhibitory concentration is significantly lower than the concentration required to inhibit the activity of wild-type EGFR tyrosine kinase. It has good properties, low side effects and good safety.
Chinese Patent Application No. CN201780050034.3 also discloses various salts and corresponding crystal forms of the compound of the above formula I. Example 2 discloses two crystal forms of the methanesulfonate of the compound of formula I, 2A and 2B, respectively.In the following examples, the “room temperature” can be 15-25°C.[0041](1) N-(2-(2-(Dimethylamino)ethoxy)-4-methoxy-5-((4-(1-methyl-1H-indol-3-yl)pyrimidine -2-yl)amino)phenyl)acrylamide (compound of formula I)[0042]
[0043]Known (for example, see CN201580067776.8) N-(2-(2-(dimethylamino)ethoxy)-4-methoxy-5-((4-(1-methyl-1H- Indol-3-yl)pyrimidin-2-yl)amino)phenyl)acrylamide (compound of formula I) can be prepared by the following synthetic route:[0044]
[0045]Step 1-Preparation of Intermediate J:[0046]
[0047]Preparation: In a 10L reaction flask, add 6L of anhydrous tetrahydrofuran solvent, protected by nitrogen, and cool to 0°C. While stirring, slowly add 101 g of sodium hydride (101 g, 2.52 mol), and the internal temperature does not exceed 10° C., and add 234 g of dimethylaminoethanol (234 g, 2.62 mol). After the addition, the temperature is adjusted to room temperature to prepare a sodium alkoxide solution.[0048]In a 30L reaction flask, add N-(4-fluoro-2-methoxy-5-nitrophenyl)-4-(1-methyl-1H-indol-3-yl)-2-pyrimidinamine ( Starting material B) (430g, 1.10mol), then add 9L of tetrahydrofuran, start stirring, dissolve it, control the temperature at 10±10°C, slowly add the prepared sodium alkoxide solution dropwise. Control the temperature at 10±10℃ and keep it for 5.0h. When the raw material content is ≤0.5%, the reaction ends. Control the temperature at 10±10°C, slowly add 3% hydrochloric acid solution dropwise, adjust the pH of the solution to 6-7, stir for 1.5h and then stand for stratification, separate the organic phase, and concentrate to 15-20L. After cooling to 20±5°C, 4.3 kg of water was slowly added dropwise, filtered, and dried to obtain 497 g of yellow powder intermediate J with a yield of 98.0% and an HPLC purity of 99.3%. MS m/z: 463.2 [M+1].[0049]Nuclear magnetic data: 1 HNMR (d 6 -DMSO): δ ppm: 8.78 (s, 1H); 8.42-8.28 (m, 3H); 8.16 (s, 1H); 7.53 (d, 1H, J = 8.28); 7.29- 7.20 (m, 2H); 7.13-7.07 (m, 1H); 7.01 (s, 1H); 4.33 (t, 2H, J = 5.65); 4.02 (s, 3H); 3.88 (s, 3H); 2.71 ( t, 2H, J = 5.77); 2.27 (s, 6H).[0050]Step 2-Preparation of Intermediate K:[0051]
[0052]Preparation: Add 5L of tetrahydrofuran and Intermediate J (350g, 108mmol) to a 10L hydrogenation reactor, add 17.5g of wet palladium charcoal, replace the hydrogenation reactor with hydrogen, adjust the pressure value to 0.2MPa, control the temperature at 25°C, and keep the temperature for reaction. At 9h, HPLC monitors the progress of the reaction, and stops the reaction when the substrate is ≤0.5%. Filter, concentrate the filtrate under reduced pressure until the solvent volume is about 2L, adjust the internal temperature to room temperature, slowly add 4L n-heptane dropwise within 4-7 hours, filter and dry the solid under reduced pressure to obtain 285g of white powder intermediate K The yield was 86%, and the HPLC purity was 99.60%. MS m/z: 433.3 [M+1].
Add 250 mL of anhydrous tetrahydrofuran solvent and Intermediate K (14 g, 32 mmol) to the reaction flask and stir, cool to 0-5° C., add 10% hydrochloric acid (12 ml), and stir for 20 minutes. At 0-5°C, slowly drop 3-chloropropionyl chloride (5.6 g, 45 mmol) into the reaction flask. Stir for 3 hours, after sampling test (K/(U+K)≤0.5%) is qualified, add 36% potassium hydroxide aqueous solution (75ml, 480mmol), heat to 23-25°C, and stir for 12 hours. Raise the temperature to 50-60°C and stir for 4 hours. After the sampling test (U/(U+L)≤0.1%) is qualified, stand still for liquid separation. Separate the organic phase, wash with 10% brine three times, dry, filter, and concentrate the organic phase to 150 ml. The temperature was raised to 40° C., 150 ml of n-heptane was slowly added dropwise, and the temperature was lowered to room temperature to precipitate crystals. Filtered and dried to obtain 10.71 g of light brown solid (compound of formula I), yield 68%, HPLC purity: 99.8% (all single impurities do not exceed 0.15%). MS m/z: 487.3 [M+1].[0057]Nuclear magnetic data (Figure 1): 1 HNMR (d 6 -DMSO): δppm: 9.84 (s, 1H), 8.90 ~ 8.82 (m, 1H), 8.32-8.25 (m, 2H), 7.89 (s, 1H) ,7.51(d,1H,J=8.25), 7.27~7.10(m,1H), 6.94(s,1H), 6.49(dd,1H,J=16.88,10.13), 6.25(dd,1H,J=16.95 ,1.81),5.80~5.75(m,1H),4.19(t,2H,J=5.57),3.88(d,6H,J=14.63,6H),3.34(s,3H),2.58(d,2H, J=5.5), 2.28 (s, 6H).
(2) N-(2-(2-(Dimethylamino)ethoxy)-4-methoxy-5-((4-(1-methyl-1H-indol-3-yl)pyrimidine -2-yl)amino)phenyl)acrylamide methanesulfonate (Form A) preparation Example 1
The compound of formula I (3 g, 6.1 mmol) was dissolved in 24 ml of dimethyl sulfoxide DMSO solvent, the temperature was raised to 65° C., and the mixture was stirred and dissolved. Add an equivalent amount of methanesulfonic acid (0.59 g, 6.1 mmol) to the system. The temperature was lowered to 50°C, and 12ml of isopropyl acetate IPAc was slowly added. Stir at 50°C for 1 hour, then lower the temperature to 15°C. 21ml IPAc was added in 4 hours. The solution was stirred and crystallized at 15°C, filtered under reduced pressure, the filter cake was washed with isopropyl acetate, and washed with acetone to reduce the residual DMSO solvent. Blow drying at 50°C (or vacuum drying at 50°C) to obtain 3.16 g of a pale yellow solid (crystal form A). HPLC purity is 100%, yield is 88%, DMSO: <100ppm; IPAc: <100ppm. MS m/z: 487.2 [M+1-MsOH]. Melting point: 242-244°C. Nuclear magnetic data (figure 2): 1 HNMR(d 6 -DMSO): δppm: 9.57(brs,1H), 9.40(s,1H), 8.71(s,1H), 8.48(s,1H), 8.32(d ,1H,J=7.9),8.29(d,1H,J=5.3),7.96(s,1H),7.51(d,1H,J=8.2),7.23(ddd,1H,J=7.9,7.1,0.8 ), 7.19 (d, 1H, J = 5.4), 7.15 (ddd, 1H, J = 7.8, 7.3, 0.5), 6.94 (s, 1H), 6.67 (dd, 1H, J = 16.9, 10.2), 6.27 ( dd, 1H, J = 16.9, 1.8), 5.57 (dd, 1H, J = 16.9, 1.7), 4.44 (t, 2H, J = 4.6), 3.89 (s, 3H), 3.88 (s, 3H), 3.58 (t, 2H, J=4.6), 2.93 (s, 6H), 2.39 (s, 3H). After testing, the powder X-ray diffraction pattern of crystal form A obtained in this example has diffraction angle 2θ values of 11.06±0.2°, 12.57±0.2°, 13.74±0.2°, 14.65±0.2°, 15.48±0.2°, 16.58±0.2°, 17.83±0.2°, 19.20±0.2°, 19.79±0.2°, 20.88±0.2°, 22.05±0.2°, 23.06±0.2°, 24.23±0.2°, 25.10±0.2°, 25.71±0.2°, 26.15±0.2°, 27.37±0.2°, 27.42±0.2° has a characteristic peak; its XRPD spectrum is shown in Figure 3 and the attached table, DSC diagram is shown in Figure 4, TGA diagram is shown in Figure 5, and infrared spectrum IR diagram is shown in Figure 6. Show. Example 2
[0066]The compound of formula I (28.25 g, 58.1 mmol) was dissolved in 224 ml of dimethyl sulfoxide DMSO solvent, the temperature was raised to 15-35° C., and the mixture was stirred to clear. 0.97 equivalents of methanesulfonic acid (5.4 g, 0.97 mmol) were added to the system in batches. Slowly add 448 ml of methyl isobutyl ketone (MIBK). Stir for 1 hour, then lower the temperature to 10-15°C. The solution was reacted with salt formation at 10-15°C, sampled, and HPLC detected the residue of the compound of formula I in the mother liquor (≤0.4%). After the reaction was completed, vacuum filtration was performed to obtain 32 g of the crude methanesulfonate of the compound of formula I.Add 3g of the crude methanesulfonate of the compound of formula I into 24ml of dimethyl sulfoxide DMSO solvent, stir to clear at 65°C, cool down, slowly add 48ml of methyl isobutyl ketone (MIBK) dropwise, stir and crystallize 6-8 After hours, vacuum filtration, drying at 60° C. (or 60° C. vacuum drying) to obtain the target crystal form A. Melting point: 242-244°C. The XRPD pattern of the crystal form is consistent with Figure 3 (Figure 7), and all characteristic peaks are within the error range.
A SARS-CoV-2 vaccine comprising a conjugate of the spike protein RBD domain with tetanus toxoid (Finlay Vaccine Institute of Cuba) Soberana 02, is a conjugate vaccine developed by Instituto Finlay de Vacunas.[517]
Soberana 02, technical name FINLAY-FR-2, is a COVID-19 vaccine produced by the Finlay Institute, a Cuban epidemiological research institute. It is a conjugate vaccine. This candidate followed a previous one called SOBERANA-01 (FINLAY-FR-1).[2] Professor Ihosvany Castellanos Santos said that the antigen is safe because it contains parts instead of the whole live virus, and therefore it does not require extra refrigeration, like other candidates in the world.[3] According to the WHO candidate landscape vaccine document, this vaccine requires two doses, the second one being administered 28 days after the first shot.[4]
The name of the vaccine, Soberana, is a Spanish word that means “sovereign”.[5]
Efficacy
It has shown an efficacy of 62% after only two doses, according to BioCubaFarma, though a pre-print or details of the study have not been released.[6][7][8]
The Cuban government says it is planning to produce 100 million doses of its vaccine to respond to its own demand and that of other countries.[12][13] Cuba has also suggested that, once it’s approved, it will offer the vaccine to tourists visiting the country.[14][15][16]
The production of the first batch of about 100,000 doses will start in April.[17] José Moya, representative of the World Health Organization and the Pan American Health Organization (PAHO) in Cuba, suggested that after the vaccine passes all clinical stages, it could be included as part of PAHO’s Revolving Fund.[18]
The roll-out began with an “Interventional Trial”[19] that consisted of inoculating 150,000 at-risk participants which seems to be defined as health-care workers.[20][21] On April 11, 2021, the Ministry of Public Health of Cuba announced that 75,000 health-care workers were inoculated with their first dose of either of the two Cuba’s Phase III vaccines (the other being Abdala).[22][23]
Outside Cuba
Vietnam, Iran, Venezuela, Argentina,[24][25][26] Pakistan, India, the African Union, Jamaica and Suriname[27] have expressed interest in purchasing the vaccine, although they are waiting on Phase 3 results.[28][29]
Iran has signed an agreement to manufacture the vaccine[30] and Argentina is negotiating one.[24][25][26] Additionally, the Cuban government offered a “transfer of technology” to Ghana and will also supply “active materials” needed to make the vaccine.[31][32][33]
While the price is currently unknown, the commercialization strategy of the vaccine will be a combination of the “impact on health” and the capability of Cuba’s system to financially support “the production of vaccines and drugs for the country”, per the director of the Finlay Institute, Vicente Vérez.[34]
Clinical trials
Phase I
FINLAY-FR-2, which started being developed in October 2020, had 40 volunteers for its Phase I, according to the Cuban Public Registry of Clinical Trials, with an open, sequential and adaptive study to assess safety, reactogenicity and explore immunogenicity of the vaccine.[35]
Phase II
Phase IIa involved 100 Cubans, and phase IIb of the vaccine will have 900 volunteers between 19 and 80 years.[36][37] Vicente Vérez, director general of the Finlay Vaccine Institute, said that the vaccine has shown to give an immune response after 14 days.[38] The second phase has been supervised by Iranian officials from the Pasteur Institute.[5]
Phase III
Phase III commenced at the beginning of March as originally scheduled,[39][15] and “ready to publish” results are expected by June.[40][41][42] The trial volunteers are divided into three groups: some will receive two doses of the vaccine 28 days apart, another group will get two doses plus a third immune booster (Soberana Plus[43][44][45]), and the third a placebo.[39]
Although the trials involve thousands of adult volunteers recruited in Havana,[46] Cuba’s public health officials have said that they will also need to conduct phase III trials abroad because the island doesn’t have an outbreak of sufficient scale to produce meaningful statistics on vaccine protection.[5][14]
On March 13, 2021, the Cuban Biotechnology and Pharmaceutical Industries Business Group (BioCubaFarma) announced on social media that it had sent 100,000 doses of its Soberana 02 coronavirus vaccine candidate to the Pasteur Institute of Iran for clinical testing, “as part of the collaboration with other countries in the development of COVID-19 vaccines.” [47]
On April 26, 2021, it was reported that a Phase III conducted by the Pasteur Institute of Iran was approved to be started in Iran[48][49][50] It was previously reported that the Institute will host Phase 3 but the pre-requisites were “technology transfer and joint production”.[51][5]
The “Interventional Study” is set both in Havana,[53] Cuba’s capital and Santiago de Cuba, Cuba’s second most populous city [54][55] and in other provinces.[56] On May 6, 2021, the Finlay Institute of Vaccines announced on social media that the following adverse events have been observed: injection site pain (20%), inflammation at the injection site (5%), and general discomfort (5%).[57][58]
^“Coronavirus: Vacuna cubana Soberana 02 alista fase 3 y ensayos”. Deutsche Welle (in Spanish). 5 February 2021. Las expectativas sobre Soberana 02 son tales que el titular del organismo estatal que desarrolló la vacuna, Vicente Vérez, confirmó que mientras se aguarden los resultados de la Fase 3 solo en La Habana, en abril se dará inicio a la producción del primer lote, de alrededor de 100 mil dosis.
^“Cuba anuncia fase 3 de la vacuna Soberana 02”. La Jornada(in Spanish). 7 February 2021. Una vez que superen las etapas clínicas, la OMS podría contar con el fármaco cubano, afirmó Moya, y “pasar a ser parte del grupo de vacunas que se oferten a través del Fondo Rotatorio”, un mecanismo que desde hace cuatro décadas permite gestionar antígenos e insumos a los países de las Américas.

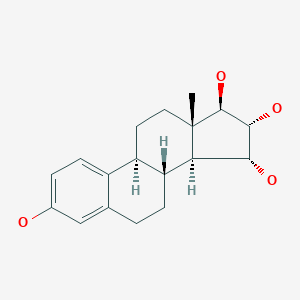




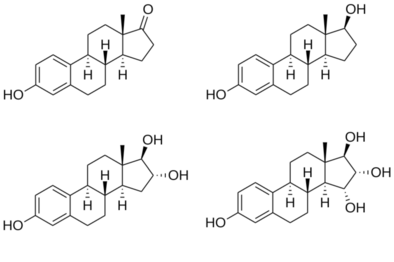 Estrone (E1)Estradiol (E2)Estriol (E3)Estetrol (E4)
Estrone (E1)Estradiol (E2)Estriol (E3)Estetrol (E4)
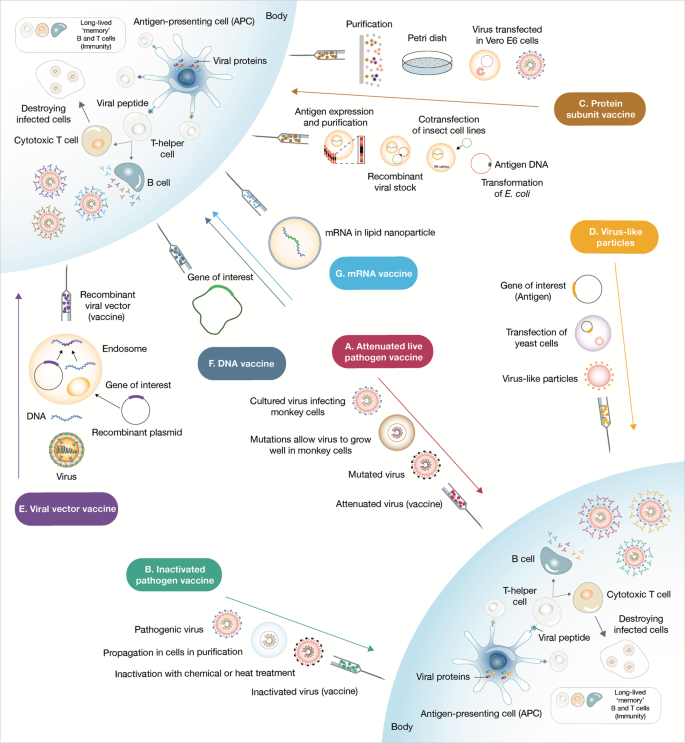




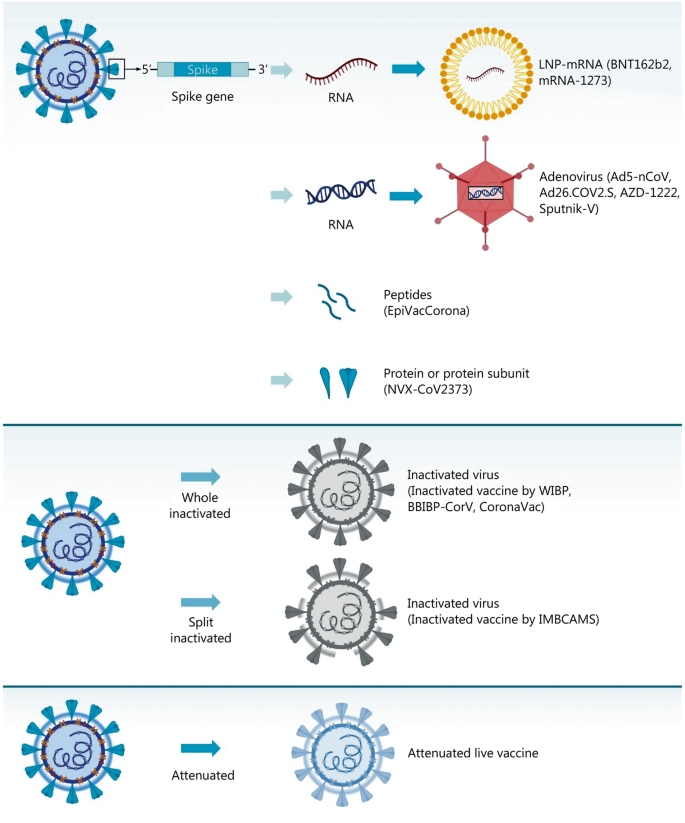










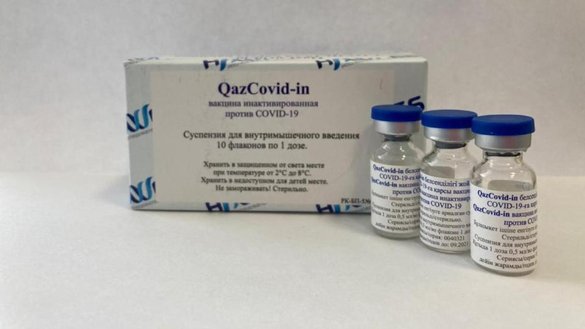
 QazCovid-in
QazCovid-in Vaccine
Vaccine Phase I/II/IIIThe QazCovid-in vaccine is an inactivated vaccine. Inactive viral vaccines are created by propagating viruses in cell culture (such as in Vero cells) and/or by inactivation using a chemical reagent (such as beta-propiolactone or formaldehyde). Upon vaccination, this allows the body to generate a diverse immune response against numerous viral antigens while having no threat of actually being infected because the virus is inactive.NEWS FEED December 31, 2020The Republic of Khazakstan’s QazCovid-in COVID19 vaccine enters phase 3 with an expected 3000 participants. August 28, 2020QazCovid-in, an inactive viral vaccine manufactured by Research Institute for Biological Safety Problems Republic of Kazakhstan enters Phase 1/2 clinical trials.ORGANIZATIONSResearch Institute for Biological Safety Problems, National Scientific Center for Phthisiopulmonology of the Republic of Kazakhstan, City polyclinic No. 4 of the UZO of Almaty, Clinic of the International Institute of Postgraduate Education, City Multidisciplinary Hospital of the Health Department of the Akimat of Zhambyl RegionCOUNTRIES INVOLVED TRIAL PARTICIPANTS
Phase I/II/IIIThe QazCovid-in vaccine is an inactivated vaccine. Inactive viral vaccines are created by propagating viruses in cell culture (such as in Vero cells) and/or by inactivation using a chemical reagent (such as beta-propiolactone or formaldehyde). Upon vaccination, this allows the body to generate a diverse immune response against numerous viral antigens while having no threat of actually being infected because the virus is inactive.NEWS FEED December 31, 2020The Republic of Khazakstan’s QazCovid-in COVID19 vaccine enters phase 3 with an expected 3000 participants. August 28, 2020QazCovid-in, an inactive viral vaccine manufactured by Research Institute for Biological Safety Problems Republic of Kazakhstan enters Phase 1/2 clinical trials.ORGANIZATIONSResearch Institute for Biological Safety Problems, National Scientific Center for Phthisiopulmonology of the Republic of Kazakhstan, City polyclinic No. 4 of the UZO of Almaty, Clinic of the International Institute of Postgraduate Education, City Multidisciplinary Hospital of the Health Department of the Akimat of Zhambyl RegionCOUNTRIES INVOLVED TRIAL PARTICIPANTS





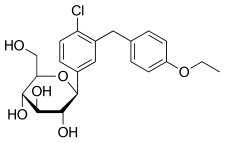

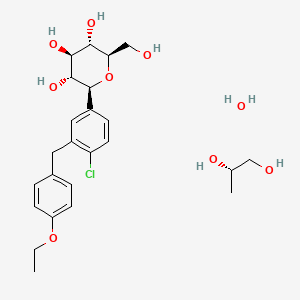



























 and LUNAR® lipid-mediated delivery method. It was designed to enhance and extend antigen expression, enabling vaccination at lower doses [
and LUNAR® lipid-mediated delivery method. It was designed to enhance and extend antigen expression, enabling vaccination at lower doses [


























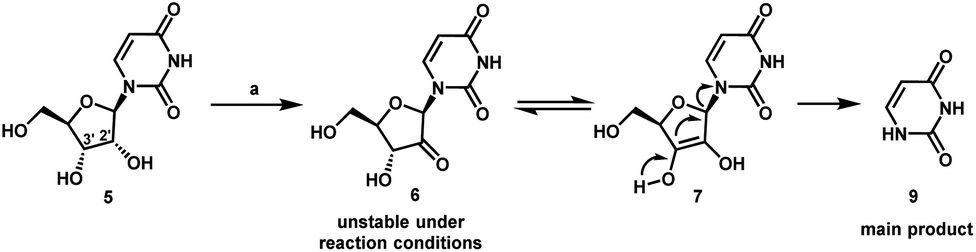










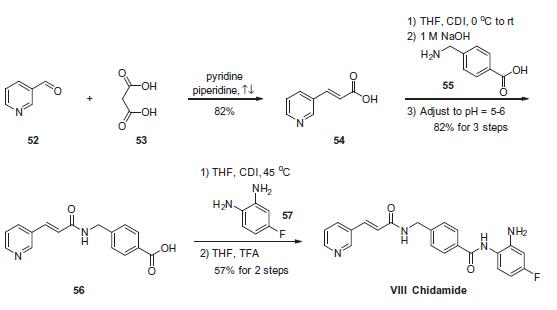








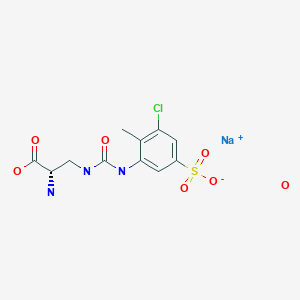
 xH2O
xH2O







 Pain at the injection site (20%).
Pain at the injection site (20%). 