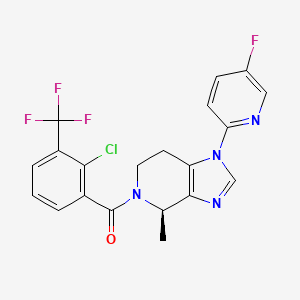

JNJ 54166060
JNJ-54166060; JNJ 54166060; JNJ54166060.
(R)-(2-chloro-3-(trifluoromethyl)phenyl)(1-(5-fluoropyridin-2-yl)-4-methyl-1,4,6,7-tetrahydro-5H-imidazo[4,5-c]pyridin-5-yl)methanone
[2-chloro-3-(trifluoromethyl)phenyl]-[(4R)-1-(5-fluoropyridin-2-yl)-4-methyl-6,7-dihydro-4H-imidazo[4,5-c]pyridin-5-yl]methanone
CAS 1627900-42-8
Chemical Formula: C20H15ClF4N4O
Exact Mass: 438.0871
JNJ-54166060 is a potent P2X7 antagonist. Bioactivity data of JNJ-54166060: rP2X7 IC50=4 nM; rP2X7 IC50=115nM; HLM/RLM = 0.35/0.64, ED50 = 2.3 mg/kg in rats. JNJ-54166060 shows high oral bioavailability and low-moderate clearance in preclinical species, acceptable safety margins in rats, and a predicted human dose of 120 mg of QD. Additionally, JNJ-54166060 possesses a unique CYP profile and was found to be a regioselective inhibitor of midazolam CYP3A metabolism.
The P2X7 receptor is a ligand-gated ion channel and is present on a variety of cell types, largely those known to be involved in the inflammatory and/ or immune process, specifically, macrophages and monocytes in the periphery and predominantly in glial cells (microglia and astrocytes) of the CNS. (Duan and Neary, Glia 2006, 54, 738-746; Skaper et al., FASEB J 2009, 24, 337-345;
Surprenant and North, Annu. Rev. Physiol. 2009, 71, 333-359). Activation of the P2X7 receptor by extracellular nucleotides, in particular adenosine triphosphate, leads to the release of proinflammatory cytokines IL-1 β and IL-18 (Muller, et. Al. Am. J. Respir. Cell Mol. Biol. 201 1 , 44, 456-464), giant cell formation
(macrophages/ microglial cells), degranulation (mast cells) and L-selectin shedding (lymphocytes) (Ferrari et al., J. Immunol. 2006, 176, 3877-3883; Surprenant and North, Annu. Rev. Physiol. 2009, 71, 333-359). P2X7 receptors are also located on antigen-presenting cells (keratinocytes, salivary acinar cells (parotid cells)), hepatocytes, erythrocytes, erythroleukaemic cells, monocytes, fibroblasts, bone marrow cells, neurones, and renal mesangial cells.
The importance of P2X7 in the nervous system arises primarily from experiments using P2X7 knock out mice. These mice demonstrate the role of P2X7 in the development and maintenance of pain as these mice were protected from the development of both adjuvant-induced inflammatory pain and partial nerve ligation induced neuropathic pain (Chessell et al., Pain 2005, 114, 386-396). In addition P2X7 knock out mice also exhibit an anti-depressant phenotype based on reduced immobility in forced swim and tail suspension tests (Basso et al., Behav. Brain Res. 2009, 798, 83-90.). Moreover, the P2X7 pathway is linked to the release of the pro-inflammatory cytokine, IL-1 β, which has been linked to precipitation of mood disorders in humans (Dantzer, Immunol. Allergy Clin. North Am. 2009, 29, 247-264; Capuron and Miller, Pharmacol. Ther. 201 1 , 730, 226-238). In addition, in murine models of Alzheimer’s disease, P2X7 was upregulated around amyloid plaques indicating a role of this target in such pathology as well (Parvathenani et al., J. Biol. Chem. 2003, 278, 13309-13317).
In view of the clinical importance of P2X7, the identification of compounds that modulate P2X7 receptor function represents an attractive avenue into the development of new therapeutic agents. Such compounds are provided herein.
PAPER
Identification of (R)-(2-Chloro-3-(trifluoromethyl)phenyl)(1-(5-fluoropyridin-2-yl)-4-methyl-6,7-dihydro-1H-imidazo[4,5-c]pyridin-5(4H)-yl)methanone (JNJ 54166060), a Small Molecule Antagonist of the P2X7 receptor

The synthesis and SAR of a series of 4,5,6,7-tetrahydro-imidazo[4,5-c]pyridine P2X7 antagonists are described. Addressing P2X7 affinity and liver microsomal stability issues encountered with this template afforded methyl substituted 4,5,6,7-tetrahydro-imidazo[4,5-c]pyridines ultimately leading to the identification of 1 (JNJ 54166060). 1 is a potent P2X7 antagonist with an ED50 = 2.3 mg/kg in rats, high oral bioavailability and low-moderate clearance in preclinical species, acceptable safety margins in rats, and a predicted human dose of 120 mg of QD. Additionally, 1 possesses a unique CYP profile and was found to be a regioselective inhibitor of midazolam CYP3A metabolism.
PATENT
https://www.google.com/patents/WO2014152604A1?cl=en
Example 40. (R* -(2-chloro-3-(trifluoromethyl phenyl (l-(5-fluoropyridin-2-yl -4-methyl- -dihydro-lH-imidazor4,5-c1pyridin-5(‘4H -yl methanone.

The title compound, absolute configuration unknown, was obtained as a single enantiomer by Chiral SFC purification of Example 11 performed using CHIRALCEL OD-H (5μιη, 250x20mm) and a mobile phase of 70% CO2, 30% EtOH. The enantiomeric purity was confirmed by analytical SFC using a CHIRALCEL OD-H (250×4.6mm) and a mobile phase of 70% CO2, 30% EtOH over 7 minutes. (100% single enantiomer, 2.29 min retention time). MS (ESI): mass calculated for C2oH15ClF4N40, 438.1; m/z found, 439.3 [M+H]+.
Example 11. (2-Chloro-3-(trifluoromethyl)phenyl)(l-(5-fluoropyridin-2-yl)-4-methyl-6,7- dihvdro-lH-imidazor4,5-c1pyridin-5(4H)-yl)methanone.

Step A. (2-Chloro-3 -(trifluoromethyl)phenylX 1 -(5 -fluoropyridin-2-yl)-4-methyl- 1 H- imidazor4.5-c1pyridin-5(‘4H)-yl)methanone.
To a solution of Intermediate 1 (0.70 g, 3.27 mmol) in THF (20 mL) was added
Intermediate 12 (0.87 g, 3.60 mmol) dropwise. The reaction was allowed to stir for 1 h then cooled to – 78 °C. To the cooled solution was added 3M MeMgBr in Et20 (1.31 mL, 3.92 mmoL) and the reaction was let come to room temperature. The mixture was then quenched with IN NaOH (50 mL) and extracted with EtOAc (3 x 30 mL). The organic layers were combined, dried (Na2S04), and concentrated. Chromatography of the resulting residue (Si02; MeOH (NH3):DCM) gave the title compound (770 mg, 54%). XH NMR (400 MHz, CDC13) δ 8.43 – 8.34 (m, 1H), 7.92 – 7.73 (m, 2H), 7.70 – 7.33 (m, 4H), 6.08 (dtd, J = 19.7, 11.7, 8.0 Hz, 3H), 1.54 (t, J = 7.0 Hz, 3H). MS (ESI): mass calculated for C2oH13ClF4 40, 436.07; m/z found 437.1 [M+H]+.
Step B. (2-Chloro-3-(trifluoromethyl)phenyl)(l-(5-fluoropyridin-2-yl)-4-methyl-6J- dihydro-lH-imidazo[4.5-clpyridin-5(4H)-yl)methanone.
To a solution of (2-chloro-3-(trifluoromethyl)phenyl)(l-(5-fluoropyridin-2-yl)-4-methyl- lH-imidazo[4,5-c]pyridin-5(4H)-yl)methanone (0.80 g, 1.83 mmol) in degassed EtOH (25 mL) was added 10% palladium on carbon (0.20 g, 0.19 mmol). The reaction was placed under an atmosphere of hydrogen and let stir for 48 h. The reaction was diluted with DCM and filtered through a pad of Celite ©. The solvent was concentrated and chromatography of the resulting residue (Si02; MeOH (NH3):DCM) gave the title compound (500 mg, 62%). ¾ NMR (500 MHz, CDC13) δ 8.45 – 8.30 (m, 1H), 7.94 (dd, J = 18.2, 10.7 Hz, 1H), 7.76 (d, J = 5.7 Hz, 1H), 7.67 – 7.43 (m, 3H), 7.43 – 7.30 (m, 1H), 5.81 (dd, J = 13.3, 6.7 Hz, 1H), 5.07 (d, J = 5.6 Hz, 1H), 4.52 (d, J = 6.7 Hz, 1H), 3.61 – 3.31 (m, 1H), 3.08 – 2.69 (m, 1H), 1.63 – 145 (m, 3H). MS (ESI): mass calculated for C20H15ClF4N4O, 438.08; m/z found 439.1 [M+H]+.
Intermediate 12: 2-Chloro-3-(trifluoromethyl)benzoyl chloride.

To a suspension of 2-chloro-3-(trifluoromethyl)benzoic acid (15 g, 67 mmol) and catalytic DMF (0.06 mL, 0.67 mmol) in DCM (150 mL) was added oxalyl chloride (6.8 mL, 80 mmol) dropwise. The reaction was let stir (vigorous bubbling) for 4 h and concentrated to an oily solid which became solid after overnight drying on high vacuum.
Intermediate 1 : l-(‘5-Fluoropvridin-2-vl)-lH-imidazor4,5-clpvridine.

A solution of 5-azabenzimidazole (1.00 g, 8.40 mmol), 2-bromo-5-fluoropyridine (1.48 g, 8.40 mmol), copper (I) oxide (0.13 g, 0.84 mmol), 8-hydroxyquinoline (0.24 g, 1.68 mmol), and CS2CO3 (5.47 g, 16.8 mmol) in DMSO (4 mL) was irradiated in a microwave apparatus for 1 hour at 140 °C. The reaction was diluted with H2O (100 mL) and extracted with EtOAc (75 mL x 3). The organic layers were combined, dried (Na2S04), and concentrated. Chromatography of the resulting residue (S1O2; MeOH (NH3):DCM) gave the title compound (0.45 g, 25%). MS (ESI): mass calculated for C11H7FN4, 214.07; m/z found 215.1 [M+H]+.
| Patent ID | Date | Patent Title |
|---|---|---|
| US2016039809 | 2016-02-11 | P2X7 MODULATORS |
| US2015322062 | 2015-11-12 | P2X7 MODULATORS |
| US2014275015 | 2014-09-18 | P2X7 MODULATORS |
///////JNJ 54166060, JNJ-54166060, JNJ54166060, 1627900-42-8, P2X7 antagonists
ClC1=C(C(N2CCC(N(C3=CC=C(F)C=N3)C=N4)=C4[C@H]2C)=O)C=CC=C1C(F)(F)F
Filed under: Preclinical drugs Tagged: 1627900-42-8, JNJ 54166060, JNJ54166060, P2X7 antagonists







