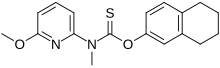
Liranaftate
リラナフタート
88678-31-3
(6-Methoxy-2-pyridinyl)methylcarbamothioic Acid O-(5,6,7,8-Tetrahydro-2-naphthalenyl) Ester
O-(5,6,7,8-Tetrahydronaphthalen-2-yl) (6-methoxypyridin-2-yl)methylcarbamothioate
Zefnart;Piritetrate;M-732
лиранафтат
ليرانافتات
利拉萘酯
| Formula | C18H20N2O2S |
|---|---|
| CAS | 88678-31-3 |
| Mol weight | 328.4286 |
| Efficacy | Antifungal, Ergosterol biosynthesis inhibitor |
|---|---|
| Comment | Thiocarbamate |
Liranaftate (trade name Zefnart) is a topical antifungal drug.[1] It is used as a 2% cream used to treat tinea pedis (athlete’s foot), tinea corporis (ringworm), and tinea cruris (jock itch).[2] It was approved for use in Japan in August 2000.[3][4]
Liranaftate works by inhibiting the fungal enzyme squalene epoxidase that is necessary for the fungus to synthesize sterols which are essential for cell membrane integrity.[5]
SYN
IN 2010MU02699
PAPER
Journal of Chemical and Pharmaceutical Research (2013), 5(11), 219-222,
PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2007010744
Conventionally, 0-aryl N- (6-alkoxy-2-pyridyl) -N-alkylthio-force rubamate has generally been produced by a method using thiophosgen. For example, in Patent Document 1, 0- (5, 6, 7, 8-tetrahydro-2-naphthyl) N- (6-methoxy-2-pyridyl) -N- represented by the following reaction formula 0 or ii) A method for producing methylthiolbamate (4) is disclosed.
(Example 1)
1) Sodium 5, 6, 7, 8-Tetrahydro-2-naphthoside synthesis
[hua 6]
,She
To methanol (10 ml), 0.54 g (10.0 mmol) of sodium methoxide was added, and the mixture was stirred at room temperature. There, 1.50 g (10.0 mmol) of 5,6,7,8-tetrahydro-2-naphthol was added and he stirred for 1 hour at room temperature. The solvent was distilled off under reduced pressure to obtain 3.75 g ( q uant.) Of white powder. I left it overnight in a desiccator.
2) 2- [Ν- (1-imidazolithiocarbol) -Ν’-methyl] amino-6-methoxypyridin compound
[hua 7]
To ethyl acetate (30 ml), 2.07 g (15.0 mmol) of 6-methoxy-2-methylaminoviridin and 2.67 g (15.0 mmol) of 1,1, -thiocarboldiimidazole were added, and the mixture was heated under reflux for 2 hours. After allowing to cool, the solvent was distilled off under reduced pressure to obtain 3.70 g of brown oil. (Yield 99.3%). If necessary, further purification was performed by silica gel column chromatography (hexane: ethyl acetate = 10: 1) to obtain pale yellow crystals.
Melting point: 58.0~60.0°C
NMR(CDC1 3 ) δ ppm:3.86(3H,s), 3.87(3H,s), 6.38 (lH’dd, J=7.5Hz, 0.7Hz), 6.61 (1H
,dd, J=8.3Hz, 0.7Hz), 6.82 (lH,t, J=1.0Hz) , 7.03 (lH,t, J=1.0Hz) , 7.46 (lH’dd, J= 8.3Hz, 7.5Hz), 7.72 (lH,t, J=1.0Hz)
IR(KBr)cm_1: 1604, 1590, 1571, 1465, 1359, 1303, 1120, 1013, 986, 822, 798 MS m/z: 248(M+)
3) Synthesis of 0- (5, 6, 7, 8-tetrahydro-2-naphthyl) -N- (6-methoxy-2-pyridyl) -N-methylthiocarbamate
Dissolve 2- [N- (1-imidazolithiocarbol) -N-methyl] amino-6-methoxypyridin 250 mg (1.0 mmol) in N, N-dimethylformamide (4 ml), and then dissolve. At room temperature, Natrium 5, 6, 7, 8-tetrahydro-2-naphthoside 360 mg (2.0 mmol) was added. -After stirring at room temperature, the reaction solution was extracted with ethyl acetate (10 mlx2), and the insoluble material was filtered off on the way. The organic layer was washed with saturated brine, dried over magnesium sulfate, filtered off magnesium sulfate, and the solvent was distilled off under reduced pressure. Purification by silica gel column chromatography (eco-gel C-200, hexane: ethyl acetate = 10: 1) gave the title compound 266.6 mg (yield 81.3%).
Melting point: 99~100°C
NMR(CDCl 3) δ ppm:1.77(4H,bs), 2.75(4H,bs), 3.75(3H,s), 3.93(3H,s), 6.65(lH,d, J
=8.0Hz), 6.78-7.08(4H,m), 7.64(lH,t,J=8.0Hz)
IR(KBr) cm_1 : 1603, 1460, 1413, 1369, 1325, 1262, 1175, 1035, 808, 785
MS m/z: 328(M+)
(Example 2)
0- (5, 6, 7, 8-tetrahydro-2-naphthyl) N- (6-methoxy-2-pyridyl) -N-methylthio force Rubamate synthesis
[Chemical 9]
1.34 g (33.6 mmol) of 60% sodium hydride was added to N, N-dimethylformamide (20 ml), followed by the addition of 5, 6, 7, 8-tetrahydro-2-naphthol 4.65 g (30.5 mmol). After gas generation is complete, add 2- [N- (1-imidazolthiocarbonyl) -N-methyl] amino-6-methoxypyridin 7.45 g (30.0 mmol) and zinc chloride 2.05 g (15.0 mmol). rice field. After heating and stirring at 60 ° C for 3 hours and allowing to cool, the reaction solution was extracted with ethyl acetate (150 mlx2), and the insoluble material was filtered off on the way. The organic layer is washed with saturated brine, dried over magnesium sulfate, and filtered through magnesium sulfate.
Separately, the solvent was distilled off under reduced pressure. The obtained crystals were purified by one of the following methods.
[0028] A) Purification was performed by silica gel column chromatography (eco-gel C 200, hexane: ethyl silicate = 10: 1) to obtain 9.80 g of the indicated compound (yield 99.5%).
B) Suspended in hexane (10 ml), stirred for 30 minutes, and then the crystals were collected by filtration to obtain 9.65 g of crystals. Further, the mixture was suspended in methanol (10 ml), stirred for 30 minutes, and then the crystals were collected by filtration to obtain 8.62 g (yield 87.5%) of the indicated compound.
The physics and physics data of the obtained compound were consistent with the compounds obtained in the examples.
(Example 3)
1) Synthesis of 2- [N- [1-2 (1H) -pyridonylthiocarbol] -N-methyl] amino-6-methoxypyridine
[Chemical 10]
OMe
Add 6-methoxy-2-methylaminoviridin 690 mg (5.0 mmol) and 1, 1, -thiocarbol-di-2 (1H) -pyridone 1.16 g (5.0 mmol) to ethyl acetate (15 ml). Heated and refluxed for 1 hour. After allowing to cool, the solvent was distilled off under reduced pressure, and purification was performed by silica gel column chromatography (hexane: ethyl acetate = 10: 1)! ヽ, 297.4 mg of brown oil was obtained. (Yield 21.6%).
NMR(CDC1 3 ) δ ppm:3.77(3H,s), 3.93(3H,s), 6.66 (lH’dd, J=8.0Hz, 0.7Hz), 7.07 ( lH,d, J=8.0Hz), 7.14 (lH,d, J=7.5Hz) , 7.25 (lH’dd, J=8.0Hz, 4.0Hz) , 7.62 (lH’dd , J=8.0Hz, 7.5Hz), 7.78 (lH’dd, J=2.0Hz, 0.7Hz) , 8.43 (lH’dd, J=4.0Hz, 0.7Hz)
MS m/z: 275(M+)
[0031] 2) Synthesis of 0- (5, 6, 7, 8-tetrahydro-2-naphthyl) N- (6-methoxy-2-pyridyl) -N-methylthiocarbamate
[Chemical 11]
OMe
N, N-dimethylformamide (2 ml), 2- [N- [1-2 (1H) -pyridonylthiocarbol] –N-methyl] amino-6-methoxypyridin 297 mg (1.08 mmol) and sodium 5 , 6, 7, 8-Tetrahydro-2-naphthoside 390 mg (2.16 mmol) was added and stirred overnight at room temperature. The reaction mixture was extracted with ethyl acetate (50 mlx2), the organic layer was washed with saturated brine, dried over magnesium sulfate, magnesium sulfate was filtered off, and the solvent was distilled off under reduced pressure. The obtained crystals were purified by silica gel column chromatography (eco-gel C-200, hexane: ethyl acetate = 10: 1) to obtain the title compound 288.2 mg (81.4%).
SYN
CN 104725302
| Liranafate is a new-generation antifungal drug, a squalene cyclooxygenase inhibitor and a cell wall synthesis inhibitor, with the chemical name of 6-methoxy-2-N-methyl-pyridylamino-thio Formic acid-(5,6,7,8-tetrahydro)-β-naphthyl ester. A new type of antifungal drug jointly developed by Tosoh Corporation of Japan and Zenyaku Kogyo Corporation was first listed in Japan by Torii Corporation in August 2000. The antifungal drug exerts antifungal activity by inhibiting the squalene epoxidation reaction of fungal cells and inhibiting the synthesis of ergosterol, a component of cell membranes. effect is particularly evident. Today, with the increasing concern of the world about environmental pollution, the development of new green and effective drug synthesis methods is an important task faced by the research of drug synthesis. In recent years, room temperature ionic liquids have been widely used in various organic synthesis reactions as a new type of environmentally friendly reaction media. Compared with traditional organic solvents, ionic liquids have many advantages, such as extremely low vapor pressure, non-flammability, good thermal stability and recyclability. |
| At present, the main synthetic route of liranaftate is as follows: |
| |
| Among the four synthetic routes, the pyridine derivative intermediates of routes C and D need to be prepared through multi-step reactions, the routes are long, the steps are cumbersome, the actual operation is cumbersome, the cost is high, and they are not suitable for industrialized large-scale production. Although route A has simple steps, the yield of pyridine derivatives is low. Each intermediate structure in route B is relatively simple and easy to prepare, but this route uses 6-methoxy-2-methylaminopyridine and 5,6,7,8-tetrahydro-2-naphthoxysulfuryl chloride as raw materials to synthesize the In the process of lanaphthalate, isopropanol-water is used as the reaction medium, and the experiment shows that with the progress of the reaction, the reaction solution becomes viscous, and the reaction is difficult to complete. |
| Example 1 |
| (1) Ionic liquid [bmim]BF 4 Synthesis |
| |
| Add N-methylimidazole (14.8g, 0.18mol) and trichloroethane (80mL) to a dry 250mL three-neck flask, stir to make the mixture uniform, add 20.4mL of freshly distilled n-bromine to the dropping funnel Butane (26.03g, 0.19mol) was added dropwise for about 30min, and the reaction was refluxed for 4-5h (the reflux temperature was about 78±1℃). With the progress of the reaction, the reaction solution changed from colorless and transparent to white turbidity, light yellow turbidity, and the color gradually became darker until brownish red. After the reaction is completed, the liquids are separated into layers, the upper layer is lighter in color, which is the trichloroethane layer, and the lower layer is darker in color (brown red), which is the ionic liquid [bmim]Br layer. The prepared ionic liquid [bmim]Br and trichloroethane were separated, and the ionic liquid [bmim]Br was washed twice with trichloroethane, and then the trichloroethane in the ionic liquid [bmim]Br was washed with a water pump. The alkane was pumped away until the ionic liquid [bmim]Br liquid was no longer turbid, and then dried in a vacuum drying oven at 90 °C for 10-12 h to obtain relatively pure ionic liquid [bmim]Br. |
| |
| Then prepare 0.03mol NaBF 4 of aqueous solution. Add 6.58g (about 0.03mol) ionic liquid [bmim]Br and 5-10mL water to a 100mL round-bottomed single diameter flask, stir, ice-water bath, and dropwise add NaBF 4 The solution (completed dropwise addition in about 5min), continue to stir for 10-20min, the solution is yellow and transparent, pour it into a separatory funnel, extract twice with dichloromethane, combine the dichloromethane layers, and wash the dichloromethane layer 2 with 50 mL of water times, and then the dichloromethane layer was washed with anhydrous MgSO 4 Dry, filter, evaporate the dichloromethane under normal pressure in a water bath (50-52°C), and dry the remaining dark yellow viscous liquid in a vacuum drying oven at 90°C for 10-12h to obtain the ionic liquid [bmim]BF 4 。 |
| |
| (2) Synthesis of 6-methoxy-2-chloropyridine 2 |
| 2,6-dichloropyridine (10g, 0.068mol) and sodium methoxide (24.5g, 0.136mol) were put into the reaction flask, heated under reflux for 4-5h, and the reaction was completed by TLC (ethyl acetate: petroleum ether=1 : 15), concentrated to remove methanol, added 100 mL of water, extracted with ethyl acetate, combined the organic phases, washed with saturated brine, dried, filtered, and the filtrate was concentrated to obtain 9 g of a crude colorless oily product with a yield of 92.5%. used for the next reaction. |
| (3) Synthesis of 6-methoxy-2-methylaminopyridine 3 |
| Take 6-methoxy-2-chloropyridine 2 (9g, 0.127mol), cuprous chloride (1.72g, 0.0017mol) and methylamine aqueous solution (29mL, mass concentration is 25%-30%) and add it to the autoclave , sealed and heated to 120 °C for 7 h, the reaction was stopped, ethyl acetate was added for extraction, the organic phases were combined, washed with saturated brine, dried, and the filtrate was concentrated to obtain 6.18 g of brown oil, the yield was 71.2%, and the HPLC purity was 98% . |
| (4) Synthesis of 5,6,7,8-tetrahydro-2-naphthyloxysulfuryl chloride 4 |
| Mix 50 mL of ethyl acetate, thiophosgene (4.25 mL, 0.056 mol) and 5,6,7,8-tetrahydro-2-naphthol (6.3 g, 0.0425 mol), and cool it in an ice-salt bath to below 0 °C. Add 10 mL of potassium carbonate (3 g, 0.022 mol) solution, continue to stir the reaction after the dropwise addition, and check by TLC (developing solvent: petroleum ether) that the reaction is complete, add 100 mL of water, extract with ethyl acetate, wash the organic phase with saturated brine, Dry, filter, and concentrate the filtrate to obtain 8.7 g of yellow oil with a yield of 90.4%, which can be directly used in the next reaction without purification. |
| (5) Synthesis of Liranaftate 1 |
| The prepared ionic liquid [bmim]BF 4 (100mL), 6-methoxy-2-methylaminopyridine 3 (5.7g, 0.0413mol) and potassium carbonate (5.7g, 0.0413mol) were mixed, cooled with ice water, and slowly added dropwise 5,6,7,8 -Tetrahydro-2-naphthyloxysulfuryl chloride 4 (8.7g, 0.0385mol) was added dropwise for 4h, slowly added 150mL of water under full stirring, continued to stir for 20min, filtered, washed with deionized water to obtain 12.2g of crude product, collected The yield was 96.81%, and acetone was recrystallized to obtain 11 g of white crystalline powder, the yield was 90%, and the HPLC purity was 99.7%. mp: 98.8-99.5°C, IR (2973cm -1 , 2930cm -1 , 2852cm -1 , 1416cm -1 , 1264cm -1 , 1037cm -1 ), 1 HNMR: 1.8 (m, 4H); 6.68(d, 1H) ;6.86(dd,1H);3.78(s,3H);3.98(s,3H);6.68(d,1H);6.86(dd,1H);7.05(d,1H);7.10(d.1H); 7.65 (dd, 1H), MS (m/z: 328, 181, 165, 108). |
| Example 2 |
| Under the same conditions, the ionic liquid 1-n-butyl-3-methylimidazolium tetrafluoroborate ([bmim]BF 4 ), N-ethylpyridine tetrafluoroborate ([EPy]BF 4 ), 1-n-butyl-3-methylimidazolium hexafluorophosphate ([bmim]PF 6 ), 1-hydroxyethyl-2,3-dimethylimidazolium chloride (LOH), 1-cyanopropyl-3-methylimidazolium chloride (LCN), 1-carboxyethyl-3-methylimidazole Chloride salt (LOOH), [Hnmp]HSO 4 The effects of and [bmim]OH on the synthesis of liranaftate are shown in Table 1. The results show that different ionic liquids have little effect on the yield of the synthesis and the yields are relatively high. |
| Table 1 Effects of different ionic liquids on the reaction yield |
| ionic liquidYield/%[bmim] BF 496.81[EPy]BF 496.83[bmim]PF 696.82LOH96.75LCN96.67LOOH96.05[Hnmp]HSO 496.06[bmim]OH95.98 |
| Example 3 |
| Whether the reaction medium used can be recovered and reused is an important content of “green chemistry”. This example specifically examines the reuse of ionic liquid for synthesizing liranaftate. After 5 times of use of ionic liquid, the product yield It just started to decrease, which shows that the ionic liquid can be recovered and reused effectively, and the reuse performance is good. It is a recyclable green solvent. |
SYN
| Comparative Example 1: |
| Put 10 g of 2,6-dichloropyridine, 100 ml of methanol, and 15 g of sodium methoxide into a reaction flask, heat under reflux for about 4 to 5 hours, concentrate to remove methanol, add 150 ml of water, extract with ethyl acetate, and concentrate under reduced pressure to remove ethyl acetate. 6-Methoxy2-chloropyridine was obtained as a colorless oil. |
| 9 g of 6-methoxy 2-chloropyridine, 1.72 g of cuprous chloride, and 29 ml of 30% methylamine aqueous solution were put into the reaction flask, heated and added with a mass fraction of 11.6 g of cuprous chloride, and the temperature was kept at 120 ° C for the reaction 8h, extracted three times with 150 ml of ethyl acetate, washed with saturated brine, concentrated under reduced pressure to remove the ethyl acetate to obtain 6.18 g of 6-methoxy-2-methylaminopyridine as a brown oily product. The two-step yield was 71.2%. |
| 50ml of carbon tetrachloride, 4.25g of thiophosgene, 6.3g of 5,6,7,8-tetrahydro-2-naphthol were added to the reaction flask, the ice-salt bath was lowered to below 0°C, and 10ml of 3g potassium carbonate aqueous solution was added dropwise. , Continue the reaction at 0°C after the dropwise addition, and detect by TLC (developing solvent: petroleum ether) after the reaction is completed, separate the organic phase, wash three times with saturated brine, and concentrate under reduced pressure to obtain red oily products 5, 6, 7 , 8.7g of 8-tetrahydro-2-naphthyloxysulfuryl chloride was directly used in the next reaction. |
| 100ml of acetone, 5.7g of 6-methoxy-2-methylaminopyridine and 5.7g of potassium carbonate were added to the reaction flask, cooled with ice water, and 5,6,7,8-tetrahydro-2-naphthyloxysulfuryl chloride was added dropwise 8.7g, continue to stir and react for 4h after dropping, add 150ml of water, continue to stir for 30min, and filter to obtain the crude product. The crude product was recrystallized with acetone to obtain 11 g of off-white crystalline powder. The weight yield was 174.6% based on 5,6,7,8-tetrahydro-2-naphthol. The maximum single impurity content determined by HPLC was 1.5%, which did not meet the requirements of the Pharmacopoeia. |
SYN
CN 106632018
| Example 1 |
| A preparation method of liranaftate of the present invention comprises the following steps: |
| (1) preparation of Liranaftate crude product: |
| Feeding: 250g of absolute ethanol was added to the reaction flask, 12.5g of 2-methoxy-6-methylaminopyridine, 8.8g of anhydrous sodium carbonate and 31.3g of purified water were added to the reaction flask in turn, stirred for 30 minutes, slowly 18.8 g of 2-(5,6,7,8-tetrahydronaphthyloxy) thioformate chloride was added, and the addition was completed in 2 hours; |
| Reaction: control the temperature at 20°C for 2 hours, add 125.0g of purified water, and stir for 30 minutes; |
| Suction filtration: the reaction solution was suction filtered, and the filter cake was washed three times with purified water, and the consumption of purified water was 25.0 g each time; |
| Drying: put the wet product into a drying box, control the temperature to 45 ℃ and dry for 4 hours, to obtain 24 g of the crude product of lira naphthate; |
| The synthesis yield is 81%; |
| (2) preparation of Liranaftate fine product: |
| Impurity removal: put 23g of Liranaftate crude product and 115g of absolute ethanol into the reaction flask, add 1.38g of medicinal charcoal, decolorize at 55°C under temperature control, remove impurities for 30 minutes, filter, transfer the filtrate to the reaction flask, control the temperature Crystallize at 55°C, centrifuge, dry, pulverize, and pack to obtain 22g of Lira naphthate fine product. |
| The purification yield was 92%. |
| Example 2 |
| A preparation method of liranaftate of the present invention comprises the following steps: |
| (1) preparation of Liranaftate crude product: |
| Feeding: 500g of absolute ethanol was added to the reaction flask, 25g of 2-methoxy-6-methylaminopyridine, 17.6g of anhydrous sodium carbonate and 62.6g of purified water were added to the reaction flask in turn, stirred for 30 minutes, and slowly added 2-(5,6,7,8-tetrahydronaphthyloxy) chlorothioformate 37.6g, added in 2.5 hours; |
| Reaction: control the temperature at 25°C for 2.5 hours, add 250 g of purified water, and stir for 30 minutes; |
| Suction filtration: the reaction solution was suction filtered, and the filter cake was washed three times with purified water, 50 g each time; |
| Drying: put the wet product into a drying box, control the temperature to 55 ℃ and dry for 4 hours to obtain 49 g of the crude product of lira naphthate; |
| The synthesis yield is 82%; |
| (2) preparation of Liranaftate fine product: |
| Impurity removal: put 49g of Liranaftate crude product and 245g of absolute ethanol into the reaction flask, add 2.9g of medicinal charcoal, decolorize at 55~65 ℃ of temperature, remove impurities for 30 minutes, filter, and transfer the filtrate to the reaction flask, The temperature was controlled at 65°C for crystallization, centrifugation, drying, pulverization, and packaging to obtain 45g of fine lanaftate. |
| The purification yield was 92%. |
| Example 3 |
| A preparation method of liranaftate of the present invention comprises the following steps: |
| (1) preparation of Liranaftate crude product: |
| Feeding: 250g of absolute ethanol was added to the reaction flask, 12.5g of 2-methoxy-6-methylaminopyridine, 8.8g of anhydrous sodium carbonate and 31.3g of purified water were added to the reaction flask in turn, stirred for 30 minutes, slowly 18.8 g of 2-(5,6,7,8-tetrahydronaphthyloxy) thioformate chloride was added, and the addition was completed in 2 hours; |
| Reaction: control the temperature at 20°C for 2 hours, add 125.0g of purified water, and stir for 30 minutes; |
| Suction filtration: the reaction solution was suction filtered, and the filter cake was washed three times with purified water, 25.0 g each time; |
| Drying: put the wet product into a drying oven, control the temperature to 45~55 ℃ and dry for 4 hours, to obtain the crude product, 23.3 g of the crude liranaftate; |
| The synthesis yield is 82%; |
| (2) preparation of Liranaftate fine product: |
| Removal of impurities: 140g of absolute ethanol was added to the reaction flask, 23.3g of crude liranaftate was added, the temperature was controlled at 50°C and stirred for 30 minutes, 1.5g of medicinal charcoal was added, the temperature was controlled at 60°C for decolorization for 30 minutes, filtered, and the temperature was controlled Crystallize at 60°C, centrifuge, dry, pulverize, and package to obtain 23g of Lira naphthate fines. |
| The purification yield was 92%. |
SYN
///////////////////////////////////////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
References
- ^ Koga H, Nanjoh Y, Makimura K, Tsuboi R (2009). “In vitro antifungal activities of luliconazole, a new topical imidazole”. Medical Mycology. 47 (6): 640–7. doi:10.1080/13693780802541518. PMID 19115136.
- ^ “Torii Pharmaceutical to Launch Antifungal Agent for External Use, “ZEFNART SOLUTION 2%”, in Japan” (Press release). Torii Pharmaceutical Co. Retrieved June 27, 2021.
- ^ “Liranaftate”. ncats.io. Retrieved June 27, 2021.
- ^ “Liranaftate”. Adis Insight. Retrieved June 27, 2021.
- ^ “Liranaftate”. targetmol.com. Retrieved June 27, 2021.
///////////////////Liranaftate , リラナフタート , Zefnart, Piritetrate, M-732, лиранафтат , ليرانافتات , 利拉萘酯 , ANTIFUNGAL, JAPAN 2000
