Clik here to view.

Clik here to view.
![4-((S)-4-Acryloyl-2-methylpiperazin-1-yl)-6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-1-(2-isopropyl-4-methylpyridin-3-yl)pyrido[2,3-d]pyrimidin-2(1H)-one.png](http://pubchem.ncbi.nlm.nih.gov/image/imgsrv.fcgi?cid=137278711&t=l)
Sotorasib
6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-1-(4-methyl-2-propan-2-ylpyridin-3-yl)-4-[(2S)-2-methyl-4-prop-2-enoylpiperazin-1-yl]pyrido[2,3-d]pyrimidin-2-one
AMG 510
AMG-510
AMG510
| Formula | C30H30F2N6O3 |
|---|---|
| CAS | 2296729-00-3 |
| Mol weight | 560.5944 |
FDA APPROVED, 2021/5/28 Lumakras
Antineoplastic, Non-small cell lung cancer (KRAS G12C-mutated)
ソトラシブ (JAN);
Sotorasib
Clik here to view.
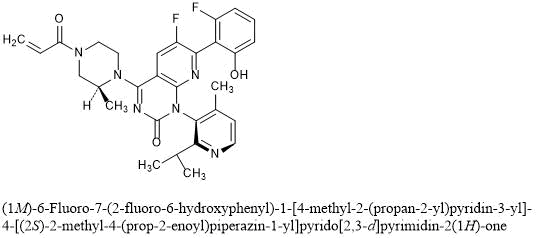
(1M)-6-Fluoro-7-(2-fluoro-6-hydroxyphenyl)-1-[4-methyl-2-(propan-2-yl)pyridin-3-yl]-4-[(2S)-2-methyl-4-(prop-2-enoyl)piperazin-1-yl]pyrido[2,3-d]pyrimidin-2(1H)-one
C30H30F2N6O3 : 560.59
[2296729-00-3]
Sotorasib is an inhibitor of the RAS GTPase family. The molecular formula is C30H30F2N6O3, and the molecular weight is 560.6 g/mol. The chemical name of sotorasib is 6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-(1M)-1-[4-methyl-2-(propan-2-yl)pyridin-3-yl]-4-[(2S)-2-methyl-4-(prop-2enoyl) piperazin-1-yl]pyrido[2,3-d]pyrimidin-2(1H)-one. The chemical structure of sotorasib is shown below:
| Image may be NSFW. Clik here to view.  |
Sotorasib has pKa values of 8.06 and 4.56. The solubility of sotorasib in the aqueous media decreases over the range pH 1.2 to 6.8 from 1.3 mg/mL to 0.03 mg/mL.
LUMAKRAS is supplied as film-coated tablets for oral use containing 120 mg of sotorasib. Inactive ingredients in the tablet core are microcrystalline cellulose, lactose monohydrate, croscarmellose sodium, and magnesium stearate. The film coating material consists of polyvinyl alcohol, titanium dioxide, polyethylene glycol, talc, and iron oxide yellow.
FDA grants accelerated approval to sotorasib for KRAS G12C mutated NSCLC
On May 28, 2021, the Food and Drug Administration granted accelerated approval to sotorasib (LumakrasImage may be NSFW.
Clik here to view. , Amgen, Inc.), a RAS GTPase family inhibitor, for adult patients with KRAS G12C ‑mutated locally advanced or metastatic non-small cell lung cancer (NSCLC), as determined by an FDA ‑approved test, who have received at least one prior systemic therapy.
, Amgen, Inc.), a RAS GTPase family inhibitor, for adult patients with KRAS G12C ‑mutated locally advanced or metastatic non-small cell lung cancer (NSCLC), as determined by an FDA ‑approved test, who have received at least one prior systemic therapy.
FDA also approved the QIAGEN therascreen® KRAS RGQ PCR kit (tissue) and the Guardant360® CDx (plasma) as companion diagnostics for Lumakras. If no mutation is detected in a plasma specimen, the tumor tissue should be tested.
Approval was based on CodeBreaK 100, a multicenter, single-arm, open label clinical trial (NCT03600883) which included patients with locally advanced or metastatic NSCLC with KRAS G12C mutations. Efficacy was evaluated in 124 patients whose disease had progressed on or after at least one prior systemic therapy. Patients received sotorasib 960 mg orally daily until disease progression or unacceptable toxicity.
The main efficacy outcome measures were objective response rate (ORR) according to RECIST 1.1, as evaluated by blinded independent central review and response duration. The ORR was 36% (95% CI: 28%, 45%) with a median response duration of 10 months (range 1.3+, 11.1).
The most common adverse reactions (≥ 20%) were diarrhea, musculoskeletal pain, nausea, fatigue, hepatotoxicity, and cough. The most common laboratory abnormalities (≥ 25%) were decreased lymphocytes, decreased hemoglobin, increased aspartate aminotransferase, increased alanine aminotransferase, decreased calcium, increased alkaline phosphatase, increased urine protein, and decreased sodium.
The recommended sotorasib dose is 960 mg orally once daily with or without food.
The approved 960 mg dose is based on available clinical data, as well as pharmacokinetic and pharmacodynamic modeling that support the approved dose. As part of the evaluation for this accelerated approval, FDA is requiring a postmarketing trial to investigate whether a lower dose will have a similar clinical effect.
View full prescribing information for Lumakras.
This indication is approved under accelerated approval based on overall response rate and duration of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory trial(s).
This review was conducted under Project Orbis, an initiative of the FDA Oncology Center of Excellence. Project Orbis provides a framework for concurrent submission and review of oncology drugs among international partners. For this review, FDA collaborated with the Australian Therapeutic Goods Administration (TGA), the Brazilian Health Regulatory Agency (ANVISA), Health Canada, and the United Kingdom Medicines and Healthcare products Regulatory Agency (MHRA). The application reviews are ongoing at the other regulatory agencies.
This review used the Real-Time Oncology Review (RTOR) pilot program, which streamlined data submission prior to the filing of the entire clinical application, the Assessment Aid, and the Product Quality Assessment Aid (PQAA), voluntary submissions from the applicant to facilitate the FDA’s assessment. The FDA approved this application approximately 10 weeks ahead of the FDA goal date.
This application was granted priority review, fast-track, breakthrough therapy and orphan drug designation. A description of FDA expedited programs is in the Guidance for Industry: Expedited Programs for Serious Conditions-Drugs and Biologics.
Sotorasib, sold under the brand name Lumakras is an anti-cancer medication used to treat non-small-cell lung cancer (NSCLC).[1][2] It targets a specific mutation, G12C, in the protein KRAS which is responsible for various forms of cancer.[3][4]
The most common side effects include diarrhea, musculoskeletal pain, nausea, fatigue, liver damage and cough.[1][2]
Sotorasib is an inhibitor of the RAS GTPase family.[1]
Sotorasib is the first approved targeted therapy for tumors with any KRAS mutation, which accounts for approximately 25% of mutations in non-small cell lung cancers.[2] KRAS G12C mutations represent about 13% of mutations in non-small cell lung cancers.[2] Sotorasib was approved for medical use in the United States in May 2021.[2][5]
Sotorasib is an experimental KRAS inhibitor being investigated for the treatment of KRAS G12C mutant non small cell lung cancer, colorectal cancer, and appendix cancer.
Sotorasib, also known as AMG-510, is an acrylamide derived KRAS inhibitor developed by Amgen.1,3 It is indicated in the treatment of adult patients with KRAS G12C mutant non small cell lung cancer.6 This mutation makes up >50% of all KRAS mutations.2 Mutant KRAS discovered in 1982 but was not considered a druggable target until the mid-2010s.5 It is the first experimental KRAS inhibitor.1
The drug MRTX849 is also currently being developed and has the same target.1
Sotorasib was granted FDA approval on 28 May 2021.6
Medical uses
Sotorasib is indicated for the treatment of adults with KRAS G12C-mutated locally advanced or metastatic non-small cell lung cancer (NSCLC), as determined by an FDA-approved test, who have received at least one prior systemic therapy.[1][2]
Clinical development
Sotorasib is being developed by Amgen. Phase I clinical trials were completed in 2020.[6][7][8] In December 2019, it was approved to begin Phase II clinical trials.[9]
Because the G12C KRAS mutation is relatively common in some cancer types, 14% of non-small-cell lung cancer adenocarcinoma patients and 5% of colorectal cancer patients,[10] and sotorasib is the first drug candidate to target this mutation, there have been high expectations for the drug.[10][11][12] The Food and Drug Administration has granted a fast track designation to sotorasib for the treatment of metastatic non-small-cell lung carcinoma with the G12C KRAS mutation.[13]
Chemistry and pharmacology
Sotorasib can exist in either of two atropisomeric forms and one is more active than the other.[10] It selectively forms an irreversible covalent bond to the sulfur atom in the cysteine residue that is present in the mutated form of KRAS, but not in the normal form.[10]
History
Researchers evaluated the efficacy of sotorasib in a study of 124 participants with locally advanced or metastatic KRAS G12C-mutated non-small cell lung cancer with disease progression after receiving an immune checkpoint inhibitor and/or platinum-based chemotherapy.[2] The major outcomes measured were objective response rate (proportion of participants whose tumor is destroyed or reduced) and duration of response.[2] The objective response rate was 36% and 58% of those participants had a duration of response of six months or longer.[2]
The U.S. Food and Drug Administration (FDA) granted the application for sotorasib orphan drug, fast track, priority review, and breakthrough therapy designations.[2] The FDA collaborated with the Australian Therapeutic Goods Administration (TGA), the Brazilian Health Regulatory Agency (ANVISA), Health Canada and the United Kingdom Medicines and Healthcare products Regulatory Agency (MHRA).[2] The application reviews are ongoing at the other regulatory agencies.[2]
The FDA granted approval of Lumakras to Amgen Inc.[2]
Society and culture
Economics
Sotorasib costs US$17,900 per month.[5]
Names
Sotorasib is the recommended international nonproprietary name (INN).[14]
PAPER
Nature (London, United Kingdom) (2019), 575(7781), 217-223
https://www.nature.com/articles/s41586-019-1694-1
KRAS is the most frequently mutated oncogene in cancer and encodes a key signalling protein in tumours1,2. The KRAS(G12C) mutant has a cysteine residue that has been exploited to design covalent inhibitors that have promising preclinical activity3,4,5. Here we optimized a series of inhibitors, using novel binding interactions to markedly enhance their potency and selectivity. Our efforts have led to the discovery of AMG 510, which is, to our knowledge, the first KRAS(G12C) inhibitor in clinical development. In preclinical analyses, treatment with AMG 510 led to the regression of KRASG12C tumours and improved the anti-tumour efficacy of chemotherapy and targeted agents. In immune-competent mice, treatment with AMG 510 resulted in a pro-inflammatory tumour microenvironment and produced durable cures alone as well as in combination with immune-checkpoint inhibitors. Cured mice rejected the growth of isogenic KRASG12D tumours, which suggests adaptive immunity against shared antigens. Furthermore, in clinical trials, AMG 510 demonstrated anti-tumour activity in the first dosing cohorts and represents a potentially transformative therapy for patients for whom effective treatments are lacking.
Paper
Scientific Reports (2020), 10(1), 11992
PAPER
European journal of medicinal chemistry (2021), 213, 113082.
https://www.sciencedirect.com/science/article/abs/pii/S0223523420310540
Clik here to view.

KRAS is the most commonly altered oncogene of the RAS family, especially the G12C mutant (KRASG12C), which has been a promising drug target for many cancers. On the basis of the bicyclic pyridopyrimidinone framework of the first-in-class clinical KRASG12C inhibitor AMG510, a scaffold hopping strategy was conducted including a F–OH cyclization approach and a pyridinyl N-atom working approach leading to new tetracyclic and bicyclic analogues. Compound 26a was identified possessing binding potency of 1.87 μM against KRASG12C and cell growth inhibition of 0.79 μM in MIA PaCa-2 pancreatic cancer cells. Treatment of 26a with NCI–H358 cells resulted in down-regulation of KRAS-GTP levels and reduction of phosphorylation of downstream ERK and AKT dose-dependently. Molecular docking suggested that the fluorophenol moiety of 26a occupies a hydrophobic pocket region thus forming hydrogen bonding to Arg68. These results will be useful to guide further structural modification.
PAPER
Journal of Medicinal Chemistry (2020), 63(1), 52-65.
https://pubs.acs.org/doi/10.1021/acs.jmedchem.9b01180
Clik here to view.

KRASG12C has emerged as a promising target in the treatment of solid tumors. Covalent inhibitors targeting the mutant cysteine-12 residue have been shown to disrupt signaling by this long-“undruggable” target; however clinically viable inhibitors have yet to be identified. Here, we report efforts to exploit a cryptic pocket (H95/Y96/Q99) we identified in KRASG12C to identify inhibitors suitable for clinical development. Structure-based design efforts leading to the identification of a novel quinazolinone scaffold are described, along with optimization efforts that overcame a configurational stability issue arising from restricted rotation about an axially chiral biaryl bond. Biopharmaceutical optimization of the resulting leads culminated in the identification of AMG 510, a highly potent, selective, and well-tolerated KRASG12C inhibitor currently in phase I clinical trials (NCT03600883).
AMG 510 [(R)-38]. (1R)-6-Fluoro-7-(2-fluoro-6-hydroxyphenyl)-1-[4-methyl-2-(1-methylethyl)-3-pyridinyl]-4-[(2S)-2-methyl-4-(1-oxo-2-propen-1-yl)-1-piperazinyl]-pyrido[2,3-d]pyrimidin-2(1H)-one
………… concentrated in vacuo. Chromatographic purification of the residue (silica gel; 0–100% 3:1 EtOAc–EtOH/heptane) followed by chiral supercritical fluid chromatography (Chiralpak IC, 30 mm × 250 mm, 5 μm, 55% MeOH/CO2, 120 mL/min, 102 bar) provided (1R)-6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-1-[4-methyl-2-(1-methylethyl)-3-pyridinyl]-4-[(2S)-2-methyl-4-(1-oxo-2-propen-1-yl)-1-piperazinyl]pyrido[2,3-d]pyrimidin-2(1H)-one (AMG 510; (R)-38; 2.25 g, 43% yield) as the first-eluting peak. 1H NMR (600 MHz, DMSO-d6) δ ppm 10.20 (s, 1H), 8.39 (d, J = 4.9 Hz, 1H), 8.30 (d, J = 8.9 Hz, 0.5H), 8.27 (d, J = 8.7 Hz, 0.5H), 7.27 (q, J = 8.4 Hz, 1H), 7.18 (d, J = 4.9 Hz, 1H), 6.87 (dd, J = 16.2, 10.8 Hz, 0.5H), 6.84 (dd, J = 16.2, 10.7 Hz, 0.5H), 6.74 (d, J = 8.4 Hz, 1H), 6.68 (t, J = 8.4 Hz, 1H), 6.21 (d, J = 16.2 Hz, 0.5H), 6.20 (d, J = 16.2 Hz, 0.5H), 5.76 (d, J = 10.8 Hz, 0.5H), 5.76 (d, J = 10.7 Hz, 0.5H), 4.91 (m, 1H), 4.41 (d, J = 12.2 Hz, 0.5H), 4.33 (d, J = 12.2 Hz, 1H), 4.28 (d, J = 12.2 Hz, 0.5H), 4.14 (d, J = 12.2 Hz, 0.5H), 4.02 (d, J = 13.6 Hz, 0.5H), 3.69 (m, 1H), 3.65 (d, J = 13.6 Hz, 0.5H), 3.52 (t, J = 12.2 Hz, 0.5H), 3.27 (d, J = 12.2 Hz, 0.5H), 3.15 (t, J = 12.2 Hz, 0.5H), 2.72 (m, 1H), 1.90 (s, 3H), 1.35 (d, J = 6.7 Hz, 3H), 1.08 (d, J = 6.7 Hz, 3H), 0.94 (d, J = 6.7 Hz, 3H).
19F NMR (376 MHz, DMSO-d6) δ −115.6 (d, J = 5.2 Hz, 1 F), −128.6 (br s, 1 F).
13C NMR (151 MHz, DMSO-d6) δ ppm 165.0 (1C), 163.4 (1C), 162.5 (1C), 160.1 (1C), 156.8 (1C), 153.7 (1C), 151.9 (1C), 149.5 (1C), 148.3 (1C), 145.2 (1C), 144.3 (1C), 131.6 (1C), 130.8 (1C), 127.9 (0.5C), 127.9 (0.5C), 127.8 (0.5C), 127.7 (0.5C), 123.2 (1C), 122.8 (1C), 111.7 (1C), 109.7 (1C), 105.7 (1C), 105.3 (1C), 51.4 (0.5C), 51.0 (0.5C), 48.9 (0.5C), 45.4 (0.5C), 44.6 (0.5C), 43.7 (0.5C), 43.5 (0.5C), 41.6 (0.5C), 29.8 (1C), 21.9 (1C), 21.7 (1C), 17.0 (1C), 15.5 (0.5C), 14.8 (0.5C).
FTMS (ESI) m/z: [M + H]+ calcd for C30H30F2N6O3 561.24202. Found 561.24150.
Clik here to view.

d (1R)-6-Fluoro7-(2-fluoro-6-hydroxyphenyl)-1-[4-methyl-2-(1-methylethyl)-3-pyridinyl]-4-[(2S)-2-methyl-4-(1-oxo-2-propen-1-yl)-1- piperazinyl]-pyrido[2,3-d]pyrimidin-2(1H)-one ((R)-38; AMG 510; 2.25 g, 43% yield) as the first-eluting peak.1 H NMR (600 MHz, DMSO-d6) δ ppm 10.20 (s, 1H), 8.39 (d, J = 4.9 Hz, 1H), 8.30 (d, J = 8.9 Hz, 0.5H), 8.27 (d, J = 8.7 Hz, 0.5H), 7.27 (q, J = 8.4 Hz, 1H), 7.18 (d, J = 4.9 Hz, 1H), 6.87 (dd, J = 16.2, 10.8 Hz, 0.5H), 6.84 (dd, J = 16.2, 10.7 Hz, 0.5H), 6.74 (d, J = 8.4 Hz, 1H), 6.68 (t, J = 8.4 Hz, 1H), 6.21 (d, J = 16.2 Hz, 0.5H), 6.20 (d, J = 16.2 Hz, 0.5H), 5.76 (d, J = 10.8 Hz, 0.5H), 5.76 (d, J = 10.7 Hz, 0.5H), 4.91 (m, 1H), 4.41 (d, J = 12.2 Hz, 0.5H), 4.33 (d, J = 12.2 Hz, 1H), 4.28 (d, J = 12.2 Hz, 0.5H), 4.14 (d, J = 12.2 Hz, 0.5H), 4.02 (d, J = 13.6 Hz, 0.5H), 3.69 (m, 1H), 3.65 (d, J = 13.6 Hz, 0.5H), 3.52 (t, J = 12.2 Hz, 0.5H), 3.27 (d, J = 12.2 Hz, 0.5H), 3.15 (t, J = 12.2 Hz, 0.5H), 2.72 (m, 1H), 1.90 (s, 3H), 1.35 (d, J = 6.7 Hz, 3H), 1.08 (d, J = 6.7 Hz, 3H), 0.94 (d, J = 6.7 Hz, 3H).
19F NMR (376 MHz, DMSO-d6) δ –115.6 (d, J = 5.2 Hz, 1 F), –128.6 (br. s., 1 F).
13C NMR (151 MHz, DMSO-d6) δ ppm 165.0 (1C), 163.4 (1C), 162.5 (1C), 160.1 (1C), 156.8 (1C), 153.7 (1C), 151.9 (1C), 149.5 (1C), 148.3 (1C), 145.2 (1C), 144.3 (1C), 131.6 (1C), 130.8 (1C), 127.9 (0.5C), 127.9 (0.5C), 127.8 (0.5C), 127.7 (0.5C), 123.2 (1C), 122.8 (1C), 111.7 (1C), 109.7 (1C), 105.7 (1C), 105.3 (1C), 51.4 (0.5C), 51.0 (0.5C), 48.9 (0.5C), 45.4 (0.5C), 44.6 (0.5C), 43.7 (0.5C), 43.5 (0.5C), 41.6 (0.5C), 29.8 (1C), 21.9 (1C), 21.7 (1C), 17.0 (1C), 15.5 (0.5C), 14.8 (0.5C).
FTMS (ESI) m/z: [M+H]+ Calcd for C30H30F2N6O3 561.24202; Found 561.24150. Atropisomer configuration (R vs. S) assigned crystallographically.The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.9b01180.
PATENT
WO 2021097212
The present disclosure relates to an improved, efficient, scalable process to prepare intermediate compounds, such as compound of Formula 6A, having the structure,
Image may be NSFW.
Clik here to view.
useful for the synthesis of compounds for the treatment of KRAS G12C mutated cancers.
BACKGROUND
[0003] KRAS gene mutations are common in pancreatic cancer, lung adenocarcinoma, colorectal cancer, gall bladder cancer, thyroid cancer, and bile duct cancer. KRAS mutations are also observed in about 25% of patients with NSCLC, and some studies have indicated that KRAS mutations are a negative prognostic factor in patients with NSCLC. Recently, V-Ki-ras2 Kirsten rat sarcoma viral oncogene homolog (KRAS) mutations have been found to confer resistance to epidermal growth factor receptor (EGFR) targeted therapies in colorectal cancer; accordingly, the mutational status of KRAS can provide important information prior to the prescription of TKI therapy. Taken together, there is a need for new medical treatments for patients with pancreatic cancer, lung adenocarcinoma, or colorectal cancer, especially those who have been diagnosed to have such cancers characterized by a KRAS mutation, and including those who have progressed after chemotherapy.
Related Synthetic Processes
[0126] The following intermediate compounds of 6-Fluoro-7-(2-fluoro-6-hydroxyphenyl)-1-(4-methyl-2-(2-propanyl)-3-pyridinyl)-4-((2S)-2-methyl-4-(2-propenoyl)-1-piperazinyl)pyrido[2,3-d]pyrimidin-2(1H)-one are representative examples of the disclosure and are not intended to be construed as limiting the scope of the present invention.
[0127] A synthesis of Compound 9 and the relevant intermediates is described in U.S. Serial No.15/984,855, filed May 21, 2018 (U.S. Publication No.2018/0334454, November 22, 2018) which claims priority to and the benefit claims the benefit of U.S. Provisional Application No.62/509,629, filed on May 22, 2017, both of which are incorporated herein by reference in their entireties for all purposes. 6-Fluoro-7-(2-fluoro-6-hydroxyphenyl)-1-(4-methyl-2-(2-propanyl)-3-pyridinyl)-4-((2S)-2-methyl-4-(2-propenoyl)-1-piperazinyl)pyrido[2,3-d]pyrimidin-2(1H)-one was prepared using the following process, in which the isomers of the final product were isolated via chiral chromatography.
Clik here to view.
[0128] Step 1: 2,6-Dichloro-5-fluoronicotinamide (Intermediate S). To a mixture of 2,6-dichloro-5-fluoro-nicotinic acid (4.0 g, 19.1 mmol, AstaTech Inc., Bristol, PA) in dichloromethane (48 mL) was added oxalyl chloride (2M solution in DCM, 11.9 mL, 23.8 mmol), followed by a catalytic amount of DMF (0.05 mL). The reaction was stirred at room temperature overnight and then was concentrated. The residue was dissolved in 1,4-dioxane (48 mL) and cooled to 0 °C. Ammonium hydroxide solution (28.0-30% NH3 basis, 3.6 mL, 28.6 mmol) was added slowly via syringe. The resulting mixture was stirred at 0 °C for 30 min and then was concentrated. The residue was diluted with a 1:1 mixture of EtOAc/Heptane and agitated for 5 min, then was filtered. The filtered solids were discarded, and the remaining mother liquor was partially concentrated to half volume and filtered. The filtered solids were washed with heptane and dried in a reduced-pressure oven (45 °C) overnight to provide 2,6-dichloro-5-fluoronicotinamide. 1H NMR (400 MHz, DMSO-d6) δ ppm 8.23 (d, J = 7.9 Hz, 1 H) 8.09 (br s, 1 H) 7.93 (br s, 1 H). m/z (ESI, +ve ion): 210.9 (M+H)+.
[0129] Step 2: 2,6-Dichloro-5-fluoro-N-((2-isopropyl-4-methylpyridin-3-yl)carbamoyl)nicotinamide. To an ice-cooled slurry of 2,6-dichloro-5-fluoronicotinamide (Intermediate S, 5.0 g, 23.9 mmol) in THF (20 mL) was added oxalyl chloride (2 M solution in DCM, 14.4 mL, 28.8 mmol) slowly via syringe. The resulting mixture was heated at 75 °C for 1 h, then heating was stopped, and the reaction was concentrated to half volume. After cooling to 0 °C, THF (20 mL) was added, followed by a solution of 2-isopropyl-4-methylpyridin-3-amine (Intermediate R, 3.59 g, 23.92 mmol) in THF (10 mL), dropwise via cannula. The resulting mixture was stirred at 0 °C for 1 h and then was quenched with a 1:1 mixture of brine and saturated aqueous ammonium chloride. The mixture was extracted with EtOAc (3x) and the combined organic layers were dried over anhydrous sodium sulfate and concentrated to provide 2,6-dichloro-5-fluoro-N-((2-isopropyl-4-methylpyridin-3-yl)carbamoyl)nicotinamide. This material was used without further purification in the following step. m/z (ESI, +ve ion): 385.1(M+H)+.
[0130] Step 3: 7-Chloro-6-fluoro-1-(2-isopropyl-4-methylpyridin-3-yl)pyrido[2,3-d]pyrimidine-2,4(1H,3H)-dione. To an ice-cooled solution of 2,6-dichloro-5-fluoro-N-((2-isopropyl-4-methylpyridin-3-yl)carbamoyl)nicotinamide (9.2 g, 24.0 mmol) in THF (40 mL) was added KHMDS (1 M solution in THF, 50.2 mL, 50.2 mmol) slowly via syringe. The ice bath was removed and the resulting mixture was stirred for 40 min at room temperature. The reaction was quenched with saturated aqueous ammonium chloride and extracted with EtOAc (3x). The combined organic layers were dried over anhydrous sodium sulfate and concentrated. The residue was purified by silica gel chromatography (eluent: 0-50% 3:1 EtOAc-EtOH/heptane) to provide 7-chloro-6-fluoro-1-(2-isopropyl-4-methylpyridin-3-yl)pyrido[2,3-d]pyrimidine-2,4(1H,3H)-dione.1H NMR (400 MHz, DMSO-d6) δ ppm 12.27 (br s, 1H), 8.48-8.55 (m, 2 H), 7.29 (d, J = 4.8 Hz, 1 H), 2.87 (quin, J = 6.6 Hz, 1 H), 1.99-2.06 (m, 3 H), 1.09 (d, J = 6.6 Hz, 3 H), 1.01 (d, J = 6.6 Hz, 3 H).19F NMR (376 MHz, DMSO-d6) δ: -126.90 (s, 1 F). m/z (ESI, +ve ion): 349.1 (M+H)+.
[0131] Step 4: 4,7-Dichloro-6-fluoro-1-(2-isopropyl-4-methylpyridin-3-yl)pyrido[2,3-d]pyrimidin-2(1H)-one. To a solution of 7-chloro-6-fluoro-1-(2-isopropyl-4-methylpyridin-3-yl)pyrido[2,3-d]pyrimidine-2,4(1H,3H)-dione (4.7 g, 13.5 mmol) and DIPEA (3.5 mL, 20.2 mmol) in acetonitrile (20 mL) was added phosphorus oxychloride (1.63 mL, 17.5 mmol), dropwise via syringe. The resulting mixture was heated at 80 °C for 1 h, and then was cooled to room temperature and concentrated to provide 4,7-dichloro-6-fluoro-1-(2-isopropyl-4-methylpyridin-3-yl)pyrido[2,3-d]pyrimidin-2(1H)-one. This material was used without further purification in the following step. m/z (ESI, +ve ion): 367.1 (M+H)+.
[0132] Step 5: (S)-tert-Butyl 4-(7-chloro-6-fluoro-1-(2-isopropyl-4-methylpyridin-3-yl)-2-oxo-1,2-dihydropyrido[2,3-d]pyrimidin-4-yl)-3-methylpiperazine-1-carboxylate. To an ice-cooled solution of 4,7-dichloro-6-fluoro-1-(2-isopropyl-4-methylpyridin-3-yl)pyrido[2,3-d]pyrimidin-2(1H)-one (13.5 mmol) in acetonitrile (20 mL) was added DIPEA (7.1 mL, 40.3 mmol), followed by (S)-4-N-Boc-2-methyl piperazine (3.23 g, 16.1 mmol, Combi-Blocks, Inc., San Diego, CA, USA). The resulting mixture was warmed to room temperature and stirred for 1 h, then was diluted with cold saturated aqueous sodium bicarbonate solution (200 mL) and EtOAc (300 mL). The mixture was stirred for an additional 5 min, the layers were separated, and the aqueous layer was extracted with more EtOAc (1x). The combined organic layers were dried over anhydrous sodium sulfate and concentrated. The residue was purified by silica gel chromatography (eluent: 0-50% EtOAc/heptane) to provide (S)-tert-butyl 4-(7-chloro-6-fluoro-1-(2-isopropyl-4-methylpyridin-3-yl)-2-oxo-1,2-dihydropyrido[2,3-d]pyrimidin-4-yl)-3-methylpiperazine-1-carboxylate. m/z (ESI, +ve ion): 531.2 (M+H)+.
[0133] Step 6: (3S)-tert-Butyl 4-(6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-1-(2-isopropyl-4-methylpyridin-3-yl)-2-oxo-1,2-dihydropyrido[2,3-d]pyrimidin-4-yl)-3-methylpiperazine-1-carboxylate. A mixture of (S)-tert-butyl 4-(7-chloro-6-fluoro-1-(2-isopropyl-4-methylpyridin-3-yl)-2-oxo-1,2-dihydropyrido[2,3-d]pyrimidin-4-yl)-3-methylpiperazine-1-carboxylate (4.3 g, 8.1 mmol), potassium trifluoro(2-fluoro-6-hydroxyphenyl)borate (Intermediate Q, 2.9 g, 10.5 mmol), potassium acetate (3.2 g, 32.4 mmol) and [1,1′-bis(diphenylphosphino)ferrocene]dichloropalladium(II), complex with dichloromethane (661 mg, 0.81 mmol) in 1,4-dioxane (80 mL) was degassed with nitrogen for 1 min. De-oxygenated water (14 mL) was added, and the resulting mixture was heated at 90 °C for 1 h. The reaction was allowed to cool to room temperature, quenched with half-saturated aqueous sodium bicarbonate, and extracted with EtOAc (2x) and DCM (1x). The combined organic layers were dried over anhydrous sodium sulfate and concentrated. The residue was purified by silica gel chromatography (eluent: 0-60% 3:1 EtOAc-EtOH/heptane) to provide (3S)-tert-butyl 4-(6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-1-(2-isopropyl-4-methylpyridin-3-yl)-2-oxo-1,2-dihydropyrido[2,3-d]pyrimidin-4-yl)-3-methylpiperazine-1-carboxylate.1H NMR (400 MHz, DMSO-d6) δ ppm 10.19 (br s, 1 H), 8.38 (d, J = 5.0 Hz, 1 H), 8.26 (dd, J = 12.5, 9.2 Hz, 1 H), 7.23-7.28 (m, 1 H), 7.18 (d, J = 5.0 Hz, 1 H), 6.72 (d, J = 8.0 Hz, 1 H), 6.68 (t, J = 8.9 Hz, 1 H), 4.77-4.98 (m, 1 H), 4.24 (br t, J = 14.2 Hz, 1 H), 3.93-4.08 (m, 1 H), 3.84 (br d, J=12.9 Hz, 1 H), 3.52-3.75 (m, 1 H), 3.07-3.28 (m, 1 H), 2.62-2.74 (m, 1 H), 1.86-1.93 (m, 3 H), 1.43-1.48 (m, 9 H), 1.35 (dd, J = 10.8, 6.8 Hz, 3 H), 1.26-1.32 (m, 1 H), 1.07 (dd, J = 6.6, 1.7 Hz, 3 H), 0.93 (dd, J = 6.6, 2.1 Hz, 3 H).19F NMR (376 MHz, DMSO-d6) δ: -115.65 (s, 1 F), -128.62 (s, 1 F). m/z (ESI, +ve ion): 607.3 (M+H)+.
[0134] Step 7: 6-Fluoro-7-(2-fluoro-6-hydroxyphenyl)-1-(4-methyl-2-(2-propanyl)-3-pyridinyl)-4-((2S)-2-methyl-4-(2-propenoyl)-1-piperazinyl)pyrido[2,3-d]pyrimidin-2(1H)-one. Trifluoroacetic acid (25 mL, 324 mmol) was added to a solution of (3S)-tert-butyl 4-(6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-1-(2-isopropyl-4-methylpyridin-3-yl)-2-oxo-1,2-dihydropyrido[2,3-d]pyrimidin-4-yl)-3-methylpiperazine-1-carboxylate (6.3 g, 10.4 mmol) in DCM (30 mL). The resulting mixture was stirred at room temperature for 1 h and then was concentrated. The residue was dissolved in DCM (30 mL), cooled to 0 °C, and sequentially treated with DIPEA (7.3 mL, 41.7 mmol) and a solution of acryloyl chloride (0.849 mL, 10.4 mmol) in DCM (3 mL; added dropwise via syringe). The reaction was stirred at 0 °C for 10 min, then was quenched with half-saturated aqueous sodium bicarbonate and extracted with DCM (2x). The combined organic layers were dried over anhydrous sodium sulfate and concentrated. The residue was purified by silica gel chromatography (eluent: 0-100% 3:1 EtOAc-EtOH/heptane) to provide 6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-1-(4-methyl-2-(2-propanyl)-3-pyridinyl)-4-((2S)-2-methyl-4-(2-propenoyl)-1-piperazinyl)pyrido[2,3-d]pyrimidin-2(1H)-one.1H NMR (400 MHz, DMSO-d6) δ ppm 10.20 (s, 1 H), 8.39 (d, J = 4.8 Hz, 1 H), 8.24-8.34 (m, 1 H), 7.23-7.32 (m, 1 H), 7.19 (d, J = 5.0 Hz, 1 H), 6.87 (td, J = 16.3, 11.0 Hz, 1 H), 6.74 (d, J = 8.6 Hz, 1 H), 6.69 (t, J = 8.6 Hz, 1 H), 6.21 (br d, J = 16.2 Hz, 1 H), 5.74-5.80 (m, 1 H), 4.91 (br s, 1 H), 4.23-4.45 (m, 2 H), 3.97-4.21 (m, 1 H), 3.44-3.79 (m, 2 H), 3.11-3.31 (m, 1 H), 2.67-2.77 (m, 1 H), 1.91 (s, 3 H), 1.35 (d, J = 6.8 Hz, 3 H), 1.08 (d, J = 6.6 Hz, 3 H), 0.94 (d, J = 6.8 Hz, 3 H).19F NMR (376 MHz, DMSO-d6) δ ppm -115.64 (s, 1 F), -128.63 (s, 1 F). m/z (ESI, +ve ion): 561.2 (M+H)+.
[0135] Another synthesis of Compound 9 and the relevant intermediates was described in a U.S. provisional patent application filed November 16, 2018, which is incorporated herein by reference in its entirety for all purposes.
Clik here to view.
Representative Synthetic Processes
[0136] The present disclosure comprises the following steps wherein the synthesis and utilization of the boroxine intermediate is a novel and inventive step in the manufacture of AMG 510 (Compound 9):
Clik here to view.
Raw Materials
Clik here to view.
Clik here to view.
Step la
Clik here to view.
Clik here to view.
[0137] To a solution of 2,6-dichloro-5-fluoro-3-pyridinecarboxylic acid (25kg; 119. lmol) in dichloromethane (167kg) and DMF (592g) was added Oxalyl chloride (18.9kg; 148.9mol) while maintaining an internal temp between 15-20 °C. Additional dichloromethane (33kg) was added as a rinse and the reaction mixture stirred for 2h. The reaction mixture is cooled then quenched with ammonium hydroxide (40.2L; 595.5mol) while maintaining internal temperature 0 ± 10°C. The resulting slurry was stirred for 90min then the product collected by filtration. The filtered solids were washed with DI water (3X 87L) and dried to provide 2,6-dichloro-5-fluoronicotinamide (Compound 1).
Step 1b
Clik here to view.
Clik here to view.
[0138] In reactor A, a solution of 2,6-dichloro-5-fluoronicotinamide (Compound 1) (16.27kg; 77.8mol) in dichloromethane (359.5kg) was added oxalyl chloride (11.9kg;
93.8mol) while maintaining temp ≤ 25°C for 75min. The resulting solution was then headed to 40°C ± 3°C and aged for 3h. Using vacuum, the solution was distilled to remove dichloromethane until the solution was below the agitator. Dichloromethane (300 kg) was then added and the mixture cooled to 0 ± 5°C. To a clean, dry reactor (reactor B) was added,2-isopropyl-4-methylpyridin-3-amine (ANILINE Compound 2A) (12.9kg; 85.9mol) followed by dichloromethane (102.6 kg). The ANILINE solution was azeodried via vacuum distillation while maintaining an internal temperature between 20-25 °), replacing with additional dichloromethane until the solution was dry by KF analysis (limit ≤ 0.05%). The solution volume was adjusted to approx. 23L volume with dichloromethane. The dried ANILINE solution was then added to reactor A while maintaining an internal temperature of 0 ± 5°C throughout the addition. The mixture was then heated to 23 °C and aged for 1h. the solution was polish filtered into a clean reactor to afford 2,6-dichloro-5-fluoro-N-((2- isopropyl-4-methylpyridin-3-yl)carbamoyl)nicotinamide (Compound 3) as a solution in DCM and used directly in the next step.
Step 2
Clik here to view.
Clik here to view.
[0139] A dichloromethane solution of 2,6-dichloro-5-fluoro-N-{[4-methyl-2-(propan-2- yl)pyridin-3-yl]carbamoyl}pyridine-3-carboxamide (UREA (Compound 3)) (15kg contained; 38.9mol) was solvent exchanged into 2-MeTHF using vacuum distillation while maintaining internal temperature of 20-25 °C. The reactor volume was adjusted to 40L and then
additional 2-MeTHF was charged (105.4 kg). Sodium t-butoxide was added (9.4 kg;
97.8mol) while maintaining 5-10 °C. The contents where warmed to 23 °C and stirred for 3h. The contents where then cooled to 0-5C and ammonium chloride added (23.0kg; 430mol) as a solution in 60L of DI water. The mixture was warmed to 20 C and DI water added (15L) and further aged for 30min. Agitation was stopped and the layers separated. The aqueous layer was removed and to the organic layer was added DI water(81.7L). A mixture of conc HCl (1.5kg) and water (9L) was prepared then added to the reactor slowly until pH measured between 4-5. The layers were separated, and the aqueous layer back extracted using 2-MeTHF (42.2kg). The two organic layers combined and washed with a 10% citric acid solution (75kg) followed by a mixture of water (81.7L) and saturated NaCl (19.8 kg). The organic layer was then washed with saturated sodium bicarbonate (75kg) repeating if necessary to achieve a target pH of ≥ 7.0 of the aqueous. The organic layer was washed again with brine (54.7kg) and then dried over magnesium sulfate (5kg). The mixture was filtered to remove magnesium sulfate rinsing the filtered bed with 2-MeTHF (49.2 kg). The combined filtrate and washes where distilled using vacuum to 40L volume. The concentrated solution was heated to 55 °C and heptane (10-12kg) slowly added until cloud point. The solution was cooled to 23 °C over 2h then heptane (27.3 kg) was added over 2h. The product slurry was aged for 3h at 20-25 °C then filtered and washed with a mixture of 2-MeTHF (2.8kg) and heptane (9kg). The product was dried using nitrogen and vacuum to afford solid 7-chloro-6-fluoro-1-(2-isopropyl-4-methylpyridin-3-yl)pyrido[2,3-d]pyrimidine-2,4(1H,3H)-dione (rac-DIONE (Compound 4)).
Step 3
Clik here to view.
Clik here to view.
[0140] To a vessel, an agitated suspension of Compound 4, (1.0 eq.) in 2- methylterahydrofuran (7.0 L/kg) was added (+)-2,3-dibenzoyl-D-tartaric acid (2.0 eq.) under an atmosphere of nitrogen. 2-MeTHF is chiral, but it is used as a racemic mixture. The different enantiomers of 2-MeTHF are incorporated randomly into the co-crystal. The resulting suspension was warmed to 75°C and aged at 75°C until full dissolution was observed (< 30 mins.). The resulting solution was polish filtered at 75°C into a secondary vessel. To the polish filtered solution was charged n-Heptane (2.0 L/kg) at a rate that maintained the internal temperature above 65°C. The solution was then cooled to 60°C, seeded with crystals (0.01 kg/kg) and allowed to age for 30 minutes. The resulting suspension was cooled to 20°C over 4 hours and then sampled for chiral purity analysis by HPLC. To the suspension, n-Heptane (3.0 L/kg) was charged and then aged for 4 hours at 20°C under an atmosphere of nitrogen. The suspension was filtered, and the isolated solids were washed two times with (2:1) n-Heptane:2-methyltetrahydrofuran (3.0 L/kg). The material was dried with nitrogen and vacuum to afford M-Dione:DBTA: Me-THF complex (Compound 4a).
Step 4
Clik here to view.
Clik here to view.
[0141] To vessel A, a suspension of disodium hydrogen phosphate (21.1 kg, 2.0 equiv) in DI water (296.8 L, 6.3 L/kg) was agitated until dissolution was observed (≥ 30 min.). To vessel B, a suspension of the M-Dione:DBTA: Me-THF complex (Composition 4a)[46.9 kg (25.9 kg corrected for M-dione, 1.0 equiv.)] in methyl tert-butyl ether (517.8 L, 11.0 L/kg) was agitated for 15 to 30 minutes. The resulting solution from vessel A was added to vessel B, and then the mixture was agitated for more than 3 hours. The agitation was stopped, and the biphasic mixture was left to separate for more than 30 minutes. The lower aqueous phase was removed and then back extracted with methyl tert-butyl ether (77.7 L, 1.7 L/kg). The organic phases were combined in vessel B and dried with magnesium sulfate (24.8 kg, 0.529 kg/kg). The resulting suspension from vessel B was agitated for more than three hours and then filtered into vessel C. To vessel B, a methyl tert-butyl ether (46.9 L, 1.0 L/kg) rinse was charged and then filtered into vessel C. The contents of vessel C were cooled to 10 °C and then distilled under vacuum while slowly being warmed to 35°C. Distillation was continued until 320-350 kg (6.8-7.5 kg/kg) of methyl tert-butyl ether was collected. After cooling the contents of vessel C to 20°C, n-Heptane (278.7 L, 5.9 L/kg) was charged over one hour and then distilled under vacuum while slowly being warmed to 35°C. Distillation was continued until a 190-200 kg (4.1-4.3 kg/kg) mixture of methyl tert-butyl ether and n-Heptane was collected. After cooling the contents of vessel C to 20°C, n-Heptane (278.7 L, 5.9 L/kg) was charged a second time over one hour and then distilled under vacuum while slowly being warmed to 35°C. Distillation was continued until a 190-200 kg (4.1-4.3 kg/kg) mixture of methyl tert-butyl ether and n-Heptane was collected. After cooling the contents of vessel C to 20°C, n-Heptane (195.9 L, 4.2 L/kg) was charged a third time over one hour and then sampled for solvent composition by GC analysis. The vessel C suspension continued to agitate for more than one hour. The suspension was filtered, and then washed with a n-Heptane (68.6 L, 1.5 L/kg) rinse from vessel C. The isolated solids were dried at 50°C, and a sample was submitted for stock suitability. Afforded 7-chloro-6-fluoro-(1M)-1-[4-methyl-2-(propan-2-yl)pyridin-3-yl]pyrido[2,3-d]pyrimidine-2,4(1H,3H)-dione (M-DIONE) Compound 5M.
[0142] The first-generation process highlighted above has been successfully scaled on 200+ kg of rac-dione starting material (Compound 4). In this process, seeding the crystallization with the thermodynamically-stable rac-dione crystal form (which exhibits low solubility) would cause a batch failure. Based on our subsequent studies, we found that increasing the DBTA equivalents and lowering the seed temperature by adjusting heptane
charge schedule improves robustness of the process. The improved process is resistant to the presence of the thermodynamically-stable rac-dione crystal form and promotes successful separation of atropisomers. Subsequent batches will incorporate the improved process for large scale manufacture.
Step 5
Clik here to view.
Clik here to view.
Note: All L/kg amounts are relative to M-Dione input; All equiv. amounts are relative to M-Dione input after adjusted by potency.
[0143] M-Dione (Compound 5M, 1.0 equiv.) and Toluene-1 (10.0 L/kg) was charged to Vessel A. The resulting solution was dried by azeotropic distillation under vacuum at 45 °C until 5.0 L/kg of solvents has been removed. The contents of Vessel A were then cooled to 20 °C.
[0144] Vessel C was charged with Toluene-3 (4.5 L/kg), Phosphoryl chloride (1.5 equiv.) and N,N-Diisopropylethylamine-1 (2.0 equiv.) while maintaining the internal temperature below 20 ± 5 °C.
Upon finishing charging, Vessel C was warmed to 30 ± 5 °C. The contents of Vessel A were then transferred to Vessel C over 4 hours while maintaining the internal temperature at 30 ± 5°C. Vessel A was rinsed with Toluene-2 (0.5 L/kg) and transferred to Vessel C. The contents of Vessel C were agitated at 30°C for an additional 3 hours. The contents of Vessel C were cooled to 20 ± 5 °C. A solution of (s)-1-boc-3-methylpiperazine (1.2 equiv.), N,N-Diisopropylethylamine-2 (1.2 equiv.) in isopropyl acetate-1 (1.0 L/kg) was prepared in Vessel D. The solution of Vessel D was charged to vessel C while maintaining a batch temperature of 20 ± 5 °C (Note: Exotherm is observed). Upon the end of transfer, Vessel D was rinsed with additional dichloromethane (1.0 L/kg) and transferred to Vessel C. The contents of Vessel C were agitated for an additional 60 minutes at 20 °C. A solution of sodium bicarbonate [water-1 (15.0 L/kg + Sodium bicarbonate (4.5 equiv.)] was then charged into Vessel C over an hour while maintaining an internal temperature at 20 ± 5 °C throughout the addition. The contents of Vessel C were agitated for at least 12 hours at which point the Pipazoline (Compound 6) product was isolated by filtration in an agitated filter dryer. The cake was washed with water-2 and -3 (5.0 L/kg x 2 times, agitating each wash for 15 minutes) and isopropyl acetate-2 and 3 (5.0 L/kg x 2 times, agitating each wash for 15 min). The cake as dried under nitrogen for 12 hours.
Acetone Re-slurry (Optional):
[0145] Pipazoline (Compound 6) and acetone (10.0 L/kg) were charged to Vessel E. The suspension was heated to 50 °C for 2 hours. Water-4 (10.0 L/kg) was charged into Vessel E over 1 hour. Upon completion of water addition, the mixture was cooled to 20 °C over 1 hour. The contents of Vessel E were filtered to isolate the product, washing the cake with 1:1 acetone/water mixture (5.0 L/kg). The cake was dried under nitrogen for 12 hours.
Step 6
Clik here to view.
General Note: All equivalents and volumes are reported in reference to Pipazoline input
Clik here to view.
Note: All L/kg and kg/kg amounts are relative to Pipazoline input
[0146] Reactor A is charged with Pipazoline (Compound 6, 1.0 equiv), degassed 2- MeTHF (9.0 L/kg) and a solution of potassium acetate (2.0 equiv) in degassed water (6.5 L/kg). The resulting mixture is warmed to 75 ± 5 °C and then, charge a slurry of
Pd(dpePhos)Cl2 (0.003 equiv) in 2-MeTHF (0.5 L/kg). Within 2 h of catalyst charge, a solution of freshly prepared Boroxine (Compound 6A, 0.5 equiv) in wet degassed 2-MeTHF (4.0 L/kg, KF > 4.0%) is charged over the course of >1 hour, but < 2 hours, rinsing with an additional portion of wet 2-MeTHF (0.5 L/kg) after addition is complete. After reaction completion ( <0.15 area % Pipazoline remaining, typically <1 h after boroxine addition is complete), 0.2 wt% (0.002 kg/kg) of Biaryl seed is added as a slurry in 0.02 L/kg wet 2- MeTHF, and the resulting seed bed is aged for > 60 min. Heptane (5.0 L/kg) is added over 2 hours at 75 ± 5 °C. The batch is then cooled to 20 ± 5 °C over 2 hours and aged for an additional 2 h. The slurry is then filtered and cake washed with 1 x 5.0L/kg water, 1 x 5.0L/kg 1:1 iPrOH:water followed by 1 x 5.0 L/kg 1:1 iPrOH:heptane (resuspension wash: the cake is resuspended by agitator and allow to set before filtering) . The cake (Biaryl, Compound 7) is then dried under vacuum with a nitrogen sweep.
Note: If the reaction stalls, an additional charge of catalyst and boroxine is required
Step 7 Charcoal Filtration for Pd removal
Image may be NSFW.
Clik here to view.
General Note: All equivalents and volumes are reported in reference to crude Biaryl input
Clik here to view.
Note: All L/kg and kg/kg amounts are relative to crude Biaryl input
[0147] In a clean Vessel A, charge crude Biaryl (1 equiv) and charge DCM (10 L/kg). Agitate content for > 60 minutes at 22 ± 5 °C, observing dissolution. Pass crude Biaryl from Vessel A, through a bag filter and carbon filters at a flux ≤ 3 L2/min/m and collect filtrate in clean Vessel B. Charge DCM rinse (1 L/kg) to Vessel A, and through carbon filters to collect in vessel B.
[0148] From filtrate in Vessel B, pull a solution sample for IPC Pd content. Sample is concentrated to solid and analyzed by ICP-MS. IPC: Pd ≤ 25 ppm with respect to Biaryl. a. If Pd content is greater than 25 ppm with respect to Biaryl on first or second IPC sample, pass solution through carbon filter a second time at ≤ 3 L2/min/m2, rinsing with 1 L/kg DCM; sample filtrate for IPC.
b. If Pd content remains greater than 25 ppm after third IPC, install and condition fresh carbon discs. Pass Biaryl filtrate through refreshed carbon filter, washing with 1 L/kg DCM. Sample for IPC.
[0149] Distill and refill to appropriate concentration. Prepare for distillation of recovered filtrate by concentrating to ≤ 4 L/kg DCM, and recharge to reach 5.25 ± 0.25 L/kg DCM prior to moving into Step 7 Boc-deprotection reaction.
Step 7
Image may be NSFW.
Clik here to view. General Note: All equivalents and volumes are reported in reference to crude Biaryl input
Clik here to view.
Note: All L/kg and kg/kg amounts are relative to Biaryl input
[0150] To Reactor A was added: tert-butyl (3S)-4-{6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-(1M)-1-[4-methyl-2-(propan-2-yl)pyridin-3-yl]-2-oxo-1,2-dihydropyrido[2,3-d]pyrimidin-4-yl}-3-methylpiperazine-1-carboxylate (Biaryl) (1.0 equiv), dichloromethane (5.0 L/kg), and the TFA (15.0 equiv, 1.9 L/kg) is charged slowly to maintain the internal temperature at 20 ± 5 °C. The reaction was stirred for 4 h at 20 ± 5 °C.
[0151] To Reactor B was added: potassium carbonate (18.0 equiv), water (20.0 L/kg), and NMP (1.0) to form a homogenous solution. While agitating at the maximum acceptable rate for the equipment, the reaction mixture in A was transferred into the potassium carbonate solution in B over 30 minutes (~ 0.24 L/kg/min rate). The mixture was stirred at 20 ± 5 °C for an additional 12 h.
[0152] The resulting slurry was filtered and rinsed with water (2 x 10 L/kg). The wet cake was dried for 24 h to give 6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-4-[(2S)-2-methylpiperazin- 1-yl]-(1M)-1-[4-methyl-2-(propan-2-yl)pyridin-3-yl]pyrido[2,3-d]pyrimidin-2(1H)-one (Des- Boc, Compound 8).
Step 8
Clik here to view.
Clik here to view.
Note: All L/kg and kg/kg amounts are relative to Des-Boc input
[0153] Des-Boc (Compound 8, 1.0 equiv) and NMP (4.2 L/kg) are charged to Vessel A under nitrogen, charge the TFA (1.0 equiv.) slowly to maintain the Tr <25 °C. The mixture is aged at 25 °C until full dissolution is observed (about 0.5 hour). The solution is then polish filtered through a 0.45 micron filter into Vessel B, washing with a NMP (0.8 L/kg). The filtrate and wash are combined, and then cooled to 0 °C. To the resulting solution, Acryloyl Chloride (1.3 equiv.) is added while maintaining temperature < 10 C. The reaction mixture is then aged at 5 ±5°C until completed by IPC (ca.1.5 hrs).
Preparation of Aqueous Disodium Phosphate Quench:
[0154] Disodium Phosphate (3.0 equiv) and Water (15.0 L/kg) are charged to Vessel C. The mixture is aged at 25 °C until full dissolution is observed. The solution is warmed to 45 ±5°C. A seed slurry of AMG 510 (0.005 equiv.) in Water (0.4 L/kg) is prepared and added to Vessel C while maintaining temperature at 45 ±5°C.
[0155] The reaction mixture in Vessel B is transferred to Vessel C (quench solution) while maintaining temperature at 45 ±5°C (ca.1 hrs). Vessel B is washed with a portion of NMP (0.5 L/kg). The product slurry is aged for 2 hrs at 45 ±5°C, cooled to 20 °C over 3 hrs, aged at 20 °C for a minimum of 12 hrs, filtered and washed with Water (2 x 10.0 L/kg). The product is dried using nitrogen and vacuum to afford Crude AMG 510 (Compound 9A).
Step 9
Image may be NSFW.
Clik here to view. General Note: All equivalents and volumes are reported in reference to crude AMG 510 input
Clik here to view.
Note: All L/kg and kg/kg amounts are relative to Crude AMG 510 input
[0156] Reactor A was charged with 6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-(1M)-1-[4- methyl-2-(propan-2-yl)pyridin-3-yl]-4-[(2S)-2-methyl-4-(prop-2-enoyl)piperazin-1- yl]pyrido[2,3-d]pyrimidin-2(1H)-one (Crude AMG 510) (1.0 equiv), ethanol (7.5 L/kg), and water (1.9 L/kg). The mixture heated to 75 °C and polish filtered into a clean Reactor B. The solution was cool to 45 °C and seeded with authentic milled AMG 510 seed (0.015 േ 0.005
1 Seed performs best when reduced in particle size via milling or with other type of mechanical grinding if mill is not available (mortar/ pestle). Actual seed utilized will be based on seed availability. 1.0- 2.0% is seed is target amount.
kg/kg); the resulting slurry was aged for 30 min. Water (15.0 L/kg) was added over 5h while maintaining an internal temperature > 40 °C; the mixture was aged for an additional 2h.
[0157] The mixture was cooled to 20 °C over 3 hours and aged for 8h, after which the solid was collected by filtration and washed using a mixture of ethanol (2.5 L/kg) and water (5.0 L/kg). The solid was dried using vacuum and nitrogen to obtain 6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-(1M)-1-[4-methyl-2-(propan-2-yl)pyridin-3-yl]-4-[(2S)-2-methyl-4-(prop-2-enoyl)piperazin-1-yl]pyrido[2,3-d]pyrimidin-2(1H)-one (AMG 510, Compound 9).
Compound 6A Boroxine Synthesis:
Lithiation/borylation
Clik here to view.
Clik here to view.
Clik here to view.
[0158] Reactor A was charged with THF (6 vol), a secondary amine base, Diisopropylamine (1.4 equiv), and a catalyst, such as triethylamine hydrochloride (0.01 equiv.). The resulting solution was cooled to -70 °C and a first base, n-BuLi (2.5 M in hexane, 1.5 equiv) was slowly added. After addition is complete, a solution of 3-fluoroanisole (1.0 equiv) in THF (6 vol) was added slowly and kept at -70 °C for 5 min. Concurrently or subsequently, a reagent, B(EtO)3 (2.0 equiv), was added slowly and kept at -70 °C for 10 min. The reaction mixture was quenched with an acid, 2N HCl. The quenched reaction mixture was extracted with MTBE (3 x 4 vol). The combined organic phases were concentrated to 1.5-3 total volumes. Heptane (7-9 vol) was added drop-wise and the mixture was cooled to 0-10 °C and stirred for 3 h. The mixture was filtrated and rinsed with heptane (1.5 vol). The solid was dried under nitrogen at < 30 °C to afford (2-fluoro-6-methoxyphenyl)boronic acid.
Demethylation:
Clik here to view.
Clik here to view.
Note: All L/kg and kg/kg amounts are relative to (2-fluoro-6-methoxyphenyl)boronic acid input
[0159] To a reactor, charge dichloromethane (solvent, 4.0 L/kg) and an acid, BBr3 (1.2 equiv), and cool to -20 °C. To this solution, a suspension of (2-fluoro-6-methoxyphenyl)boronic acid (1.0 equiv) in dichloromethane (4.0 L/kg) was added into the BBr3/DCM mixture while keeping temperature -15 to -25 °C. The reaction was allowed to proceed for approximately 2 hours while monitored by HPLC [≤1% (2-fluoro-6-methoxyphenyl)boronic acid] before reverse quenching into water (3.0 L/kg). The precipitated solid was then isolated by filtration and slurried with water (3.0 L/kg) on the filter prior to deliquoring. The filtrates were adjusted to pH 4-6 by the addition of sodium bicarbonate. The bottom organic phase was separated and the resulting aqueous layer was washed with dichloromethane (solvent, 5.0 Vol) and adjusted to pH = 1 by addition of concentrated hydrochloric acid. The resulting solids were isolated by filtration, washing the cake with water (2 x 5.0 L/kg)
Purification via Reslurry (required)
[0160] The combined crude solids were charged into a reactor and slurried with 5% EtOH/water (5.0 L/kg) at 20 °C for >1 h. The purified product was then isolated by filtration and rinsed with water (2 x 3 L/kg) before drying on the filter at < 30 °C to with nitrogen/vacuum to afford 2,2′,2”-(1,3,5,2,4,6-trioxatriborinane-2,4,6-triyl)tris(3-fluorophenol) (Boroxine, Compound 6A).
PATENT
WO 2020102730
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2020102730
PATENT
US 20180334454
References
- ^ Jump up to:a b c d e “Lumakras- sotorasib tablet, coated”. DailyMed. Retrieved 6 June 2021.
- ^ Jump up to:a b c d e f g h i j k l m n “FDA Approves First Targeted Therapy for Lung Cancer Mutation Previously Considered Resistant to Drug Therapy”. U.S. Food and Drug Administration (FDA). 28 May 2021. Retrieved 28 May 2021. Image may be NSFW.
Clik here to view.![]() This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ “KRAS mutant-targeting AMG 510”. NCI Drug Dictionary. National Cancer Institute. 2 February 2011. Retrieved 16 November2019.
- ^ Canon J, Rex K, Saiki AY, Mohr C, Cooke K, Bagal D, et al. (November 2019). “The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity”. Nature. 575 (7781): 217–23. Bibcode:2019Natur.575..217C. doi:10.1038/s41586-019-1694-1. PMID 31666701.
- ^ Jump up to:a b “FDA approves Amgen drug for lung cancer with specific mutation”. CNBC. 28 May 2021. Retrieved 28 May 2021.
- ^ Hong DS, Fakih MG, Strickler JH, Desai J, Durm GA, Shapiro GI, et al. (2020). “KRASG12C inhibition with sotorasib in advanced solid tumors”. N Engl J Med. doi:10.1056/NEJMoa1917239. PMC 7571518.
- ^ Clinical trial number NCT03600883 for “A Phase 1/2, Study Evaluating the Safety, Tolerability, PK, and Efficacy of AMG 510 in Subjects With Solid Tumors With a Specific KRAS Mutation ” at ClinicalTrials.gov
- ^ “The Discovery Of Amgen’s Novel Investigational KRAS(G12C) Inhibitor AMG 510 Published In Nature” (Press release). Amgen. 30 October 2019. Retrieved 16 November 2019.
- ^ Irving M (24 December 2019). “Drug targeting common cancer cause enters phase 2 clinical trials”. New Atlas. Retrieved 24 December 2019.
- ^ Jump up to:a b c d Halford B (3 April 2019). “Amgen unveils its KRas inhibitor in human clinical trials: AMG 510 shuts down a mutant version of the cancer target via covalent interaction”. Chemical & Engineering News. 97 (4). Retrieved 16 November 2019.
- ^ Al Idrus A (9 September 2019). “Amgen’s KRAS drug continues to deliver but faces ‘curse’ of high expectations”. fiercebiotech.com. Retrieved 16 November 2019.
- ^ Kaiser J (30 October 2019). “Two new drugs finally hit ‘undruggable’ cancer target, providing hope for treatments”. Science Magazine. AAAS. Retrieved 16 November 2019.
- ^ Astor L (9 September 2019). “FDA Grants AMG 510 Fast Track Designation for KRAS G12C+ NSCLC”. targetedonc.com. Retrieved 16 November 2019.
- ^ World Health Organization (2021). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 85” (PDF). WHO Drug Information. 35 (1).
Further reading
- Hong DS, Fakih MG, Strickler JH, Desai J, Durm GA, Shapiro GI, et al. (September 2020). “KRASG12C Inhibition with Sotorasib in Advanced Solid Tumors”. N Engl J Med. 383 (13): 1207–17. doi:10.1056/NEJMoa1917239. PMC 7571518. PMID 32955176.
- Lanman BA, Allen JR, Allen JG, Amegadzie AK, Ashton KS, Booker SK, et al. (January 2020). “Discovery of a Covalent Inhibitor of KRASG12C (AMG 510) for the Treatment of Solid Tumors”. J Med Chem. 63 (1): 52–65. doi:10.1021/acs.jmedchem.9b01180. PMID 31820981.
External links
- “Sotorasib”. Drug Information Portal. U.S. National Library of Medicine.
- Clinical trial number NCT03600883 for “A Phase 1/2, Study Evaluating the Safety, Tolerability, PK, and Efficacy of AMG 510 in Subjects With Solid Tumors With a Specific KRAS Mutation (CodeBreaK 100)” at ClinicalTrials.gov
| Clinical data | |
|---|---|
| Trade names | Lumakras |
| Other names | AMG 510 |
| License data | US DailyMed: Sotorasib |
| Routes of administration | By mouth |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only [1][2] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 2252403-56-6 |
| PubChem CID | 137278711 |
| DrugBank | DB15569 |
| ChemSpider | 72380148 |
| UNII | 2B2VM6UC8G |
| KEGG | D12055 |
| Chemical and physical data | |
| Formula | C30H30F2N6O3 |
| Molar mass | 560.606 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI |
////////Sotorasib, ソトラシブ , FDA 2021, APPROVALS 2021, Lumakras, CANCER, ANTINEOPLASTIC, AMG 510, AMG-510, AMG510, AMGEN, priority review, fast-track, breakthrough therapy, orphan drug
CC1CN(CCN1C2=NC(=O)N(C3=NC(=C(C=C32)F)C4=C(C=CC=C4F)O)C5=C(C=CN=C5C(C)C)C)C(=O)C=C
Clik here to view.
Clik here to view.
Sotorasib
6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-1-(4-methyl-2-propan-2-ylpyridin-3-yl)-4-[(2S)-2-methyl-4-prop-2-enoylpiperazin-1-yl]pyrido[2,3-d]pyrimidin-2-one
AMG 510
AMG-510
AMG510
| Formula | C30H30F2N6O3 |
|---|---|
| CAS | 2296729-00-3 |
| Mol weight | 560.5944 |
FDA APPROVED, 2021/5/28 Lumakras
Antineoplastic, Non-small cell lung cancer (KRAS G12C-mutated)
ソトラシブ (JAN);
Sotorasib
Clik here to view.
(1M)-6-Fluoro-7-(2-fluoro-6-hydroxyphenyl)-1-[4-methyl-2-(propan-2-yl)pyridin-3-yl]-4-[(2S)-2-methyl-4-(prop-2-enoyl)piperazin-1-yl]pyrido[2,3-d]pyrimidin-2(1H)-one
C30H30F2N6O3 : 560.59
[2296729-00-3]
Sotorasib is an inhibitor of the RAS GTPase family. The molecular formula is C30H30F2N6O3, and the molecular weight is 560.6 g/mol. The chemical name of sotorasib is 6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-(1M)-1-[4-methyl-2-(propan-2-yl)pyridin-3-yl]-4-[(2S)-2-methyl-4-(prop-2enoyl) piperazin-1-yl]pyrido[2,3-d]pyrimidin-2(1H)-one. The chemical structure of sotorasib is shown below:
| Image may be NSFW. Clik here to view. |
Sotorasib has pKa values of 8.06 and 4.56. The solubility of sotorasib in the aqueous media decreases over the range pH 1.2 to 6.8 from 1.3 mg/mL to 0.03 mg/mL.
LUMAKRAS is supplied as film-coated tablets for oral use containing 120 mg of sotorasib. Inactive ingredients in the tablet core are microcrystalline cellulose, lactose monohydrate, croscarmellose sodium, and magnesium stearate. The film coating material consists of polyvinyl alcohol, titanium dioxide, polyethylene glycol, talc, and iron oxide yellow.
FDA grants accelerated approval to sotorasib for KRAS G12C mutated NSCLC
On May 28, 2021, the Food and Drug Administration granted accelerated approval to sotorasib (LumakrasImage may be NSFW.
Clik here to view., Amgen, Inc.), a RAS GTPase family inhibitor, for adult patients with KRAS G12C ‑mutated locally advanced or metastatic non-small cell lung cancer (NSCLC), as determined by an FDA ‑approved test, who have received at least one prior systemic therapy.
FDA also approved the QIAGEN therascreen® KRAS RGQ PCR kit (tissue) and the Guardant360® CDx (plasma) as companion diagnostics for Lumakras. If no mutation is detected in a plasma specimen, the tumor tissue should be tested.
Approval was based on CodeBreaK 100, a multicenter, single-arm, open label clinical trial (NCT03600883) which included patients with locally advanced or metastatic NSCLC with KRAS G12C mutations. Efficacy was evaluated in 124 patients whose disease had progressed on or after at least one prior systemic therapy. Patients received sotorasib 960 mg orally daily until disease progression or unacceptable toxicity.
The main efficacy outcome measures were objective response rate (ORR) according to RECIST 1.1, as evaluated by blinded independent central review and response duration. The ORR was 36% (95% CI: 28%, 45%) with a median response duration of 10 months (range 1.3+, 11.1).
The most common adverse reactions (≥ 20%) were diarrhea, musculoskeletal pain, nausea, fatigue, hepatotoxicity, and cough. The most common laboratory abnormalities (≥ 25%) were decreased lymphocytes, decreased hemoglobin, increased aspartate aminotransferase, increased alanine aminotransferase, decreased calcium, increased alkaline phosphatase, increased urine protein, and decreased sodium.
The recommended sotorasib dose is 960 mg orally once daily with or without food.
The approved 960 mg dose is based on available clinical data, as well as pharmacokinetic and pharmacodynamic modeling that support the approved dose. As part of the evaluation for this accelerated approval, FDA is requiring a postmarketing trial to investigate whether a lower dose will have a similar clinical effect.
View full prescribing information for Lumakras.
This indication is approved under accelerated approval based on overall response rate and duration of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory trial(s).
This review was conducted under Project Orbis, an initiative of the FDA Oncology Center of Excellence. Project Orbis provides a framework for concurrent submission and review of oncology drugs among international partners. For this review, FDA collaborated with the Australian Therapeutic Goods Administration (TGA), the Brazilian Health Regulatory Agency (ANVISA), Health Canada, and the United Kingdom Medicines and Healthcare products Regulatory Agency (MHRA). The application reviews are ongoing at the other regulatory agencies.
This review used the Real-Time Oncology Review (RTOR) pilot program, which streamlined data submission prior to the filing of the entire clinical application, the Assessment Aid, and the Product Quality Assessment Aid (PQAA), voluntary submissions from the applicant to facilitate the FDA’s assessment. The FDA approved this application approximately 10 weeks ahead of the FDA goal date.
This application was granted priority review, fast-track, breakthrough therapy and orphan drug designation. A description of FDA expedited programs is in the Guidance for Industry: Expedited Programs for Serious Conditions-Drugs and Biologics.
Sotorasib, sold under the brand name Lumakras is an anti-cancer medication used to treat non-small-cell lung cancer (NSCLC).[1][2] It targets a specific mutation, G12C, in the protein KRAS which is responsible for various forms of cancer.[3][4]
The most common side effects include diarrhea, musculoskeletal pain, nausea, fatigue, liver damage and cough.[1][2]
Sotorasib is an inhibitor of the RAS GTPase family.[1]
Sotorasib is the first approved targeted therapy for tumors with any KRAS mutation, which accounts for approximately 25% of mutations in non-small cell lung cancers.[2] KRAS G12C mutations represent about 13% of mutations in non-small cell lung cancers.[2] Sotorasib was approved for medical use in the United States in May 2021.[2][5]
Sotorasib is an experimental KRAS inhibitor being investigated for the treatment of KRAS G12C mutant non small cell lung cancer, colorectal cancer, and appendix cancer.
Sotorasib, also known as AMG-510, is an acrylamide derived KRAS inhibitor developed by Amgen.1,3 It is indicated in the treatment of adult patients with KRAS G12C mutant non small cell lung cancer.6 This mutation makes up >50% of all KRAS mutations.2 Mutant KRAS discovered in 1982 but was not considered a druggable target until the mid-2010s.5 It is the first experimental KRAS inhibitor.1
The drug MRTX849 is also currently being developed and has the same target.1
Sotorasib was granted FDA approval on 28 May 2021.6
Medical uses
Sotorasib is indicated for the treatment of adults with KRAS G12C-mutated locally advanced or metastatic non-small cell lung cancer (NSCLC), as determined by an FDA-approved test, who have received at least one prior systemic therapy.[1][2]
Clinical development
Sotorasib is being developed by Amgen. Phase I clinical trials were completed in 2020.[6][7][8] In December 2019, it was approved to begin Phase II clinical trials.[9]
Because the G12C KRAS mutation is relatively common in some cancer types, 14% of non-small-cell lung cancer adenocarcinoma patients and 5% of colorectal cancer patients,[10] and sotorasib is the first drug candidate to target this mutation, there have been high expectations for the drug.[10][11][12] The Food and Drug Administration has granted a fast track designation to sotorasib for the treatment of metastatic non-small-cell lung carcinoma with the G12C KRAS mutation.[13]
Chemistry and pharmacology
Sotorasib can exist in either of two atropisomeric forms and one is more active than the other.[10] It selectively forms an irreversible covalent bond to the sulfur atom in the cysteine residue that is present in the mutated form of KRAS, but not in the normal form.[10]
History
Researchers evaluated the efficacy of sotorasib in a study of 124 participants with locally advanced or metastatic KRAS G12C-mutated non-small cell lung cancer with disease progression after receiving an immune checkpoint inhibitor and/or platinum-based chemotherapy.[2] The major outcomes measured were objective response rate (proportion of participants whose tumor is destroyed or reduced) and duration of response.[2] The objective response rate was 36% and 58% of those participants had a duration of response of six months or longer.[2]
The U.S. Food and Drug Administration (FDA) granted the application for sotorasib orphan drug, fast track, priority review, and breakthrough therapy designations.[2] The FDA collaborated with the Australian Therapeutic Goods Administration (TGA), the Brazilian Health Regulatory Agency (ANVISA), Health Canada and the United Kingdom Medicines and Healthcare products Regulatory Agency (MHRA).[2] The application reviews are ongoing at the other regulatory agencies.[2]
The FDA granted approval of Lumakras to Amgen Inc.[2]
Society and culture
Economics
Sotorasib costs US$17,900 per month.[5]
Names
Sotorasib is the recommended international nonproprietary name (INN).[14]
PAPER
Nature (London, United Kingdom) (2019), 575(7781), 217-223
https://www.nature.com/articles/s41586-019-1694-1
KRAS is the most frequently mutated oncogene in cancer and encodes a key signalling protein in tumours1,2. The KRAS(G12C) mutant has a cysteine residue that has been exploited to design covalent inhibitors that have promising preclinical activity3,4,5. Here we optimized a series of inhibitors, using novel binding interactions to markedly enhance their potency and selectivity. Our efforts have led to the discovery of AMG 510, which is, to our knowledge, the first KRAS(G12C) inhibitor in clinical development. In preclinical analyses, treatment with AMG 510 led to the regression of KRASG12C tumours and improved the anti-tumour efficacy of chemotherapy and targeted agents. In immune-competent mice, treatment with AMG 510 resulted in a pro-inflammatory tumour microenvironment and produced durable cures alone as well as in combination with immune-checkpoint inhibitors. Cured mice rejected the growth of isogenic KRASG12D tumours, which suggests adaptive immunity against shared antigens. Furthermore, in clinical trials, AMG 510 demonstrated anti-tumour activity in the first dosing cohorts and represents a potentially transformative therapy for patients for whom effective treatments are lacking.
Paper
Scientific Reports (2020), 10(1), 11992
PAPER
European journal of medicinal chemistry (2021), 213, 113082.
https://www.sciencedirect.com/science/article/abs/pii/S0223523420310540
Clik here to view.
KRAS is the most commonly altered oncogene of the RAS family, especially the G12C mutant (KRASG12C), which has been a promising drug target for many cancers. On the basis of the bicyclic pyridopyrimidinone framework of the first-in-class clinical KRASG12C inhibitor AMG510, a scaffold hopping strategy was conducted including a F–OH cyclization approach and a pyridinyl N-atom working approach leading to new tetracyclic and bicyclic analogues. Compound 26a was identified possessing binding potency of 1.87 μM against KRASG12C and cell growth inhibition of 0.79 μM in MIA PaCa-2 pancreatic cancer cells. Treatment of 26a with NCI–H358 cells resulted in down-regulation of KRAS-GTP levels and reduction of phosphorylation of downstream ERK and AKT dose-dependently. Molecular docking suggested that the fluorophenol moiety of 26a occupies a hydrophobic pocket region thus forming hydrogen bonding to Arg68. These results will be useful to guide further structural modification.
PAPER
Journal of Medicinal Chemistry (2020), 63(1), 52-65.
https://pubs.acs.org/doi/10.1021/acs.jmedchem.9b01180
Clik here to view.
KRASG12C has emerged as a promising target in the treatment of solid tumors. Covalent inhibitors targeting the mutant cysteine-12 residue have been shown to disrupt signaling by this long-“undruggable” target; however clinically viable inhibitors have yet to be identified. Here, we report efforts to exploit a cryptic pocket (H95/Y96/Q99) we identified in KRASG12C to identify inhibitors suitable for clinical development. Structure-based design efforts leading to the identification of a novel quinazolinone scaffold are described, along with optimization efforts that overcame a configurational stability issue arising from restricted rotation about an axially chiral biaryl bond. Biopharmaceutical optimization of the resulting leads culminated in the identification of AMG 510, a highly potent, selective, and well-tolerated KRASG12C inhibitor currently in phase I clinical trials (NCT03600883).
AMG 510 [(R)-38]. (1R)-6-Fluoro-7-(2-fluoro-6-hydroxyphenyl)-1-[4-methyl-2-(1-methylethyl)-3-pyridinyl]-4-[(2S)-2-methyl-4-(1-oxo-2-propen-1-yl)-1-piperazinyl]-pyrido[2,3-d]pyrimidin-2(1H)-one
………… concentrated in vacuo. Chromatographic purification of the residue (silica gel; 0–100% 3:1 EtOAc–EtOH/heptane) followed by chiral supercritical fluid chromatography (Chiralpak IC, 30 mm × 250 mm, 5 μm, 55% MeOH/CO2, 120 mL/min, 102 bar) provided (1R)-6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-1-[4-methyl-2-(1-methylethyl)-3-pyridinyl]-4-[(2S)-2-methyl-4-(1-oxo-2-propen-1-yl)-1-piperazinyl]pyrido[2,3-d]pyrimidin-2(1H)-one (AMG 510; (R)-38; 2.25 g, 43% yield) as the first-eluting peak. 1H NMR (600 MHz, DMSO-d6) δ ppm 10.20 (s, 1H), 8.39 (d, J = 4.9 Hz, 1H), 8.30 (d, J = 8.9 Hz, 0.5H), 8.27 (d, J = 8.7 Hz, 0.5H), 7.27 (q, J = 8.4 Hz, 1H), 7.18 (d, J = 4.9 Hz, 1H), 6.87 (dd, J = 16.2, 10.8 Hz, 0.5H), 6.84 (dd, J = 16.2, 10.7 Hz, 0.5H), 6.74 (d, J = 8.4 Hz, 1H), 6.68 (t, J = 8.4 Hz, 1H), 6.21 (d, J = 16.2 Hz, 0.5H), 6.20 (d, J = 16.2 Hz, 0.5H), 5.76 (d, J = 10.8 Hz, 0.5H), 5.76 (d, J = 10.7 Hz, 0.5H), 4.91 (m, 1H), 4.41 (d, J = 12.2 Hz, 0.5H), 4.33 (d, J = 12.2 Hz, 1H), 4.28 (d, J = 12.2 Hz, 0.5H), 4.14 (d, J = 12.2 Hz, 0.5H), 4.02 (d, J = 13.6 Hz, 0.5H), 3.69 (m, 1H), 3.65 (d, J = 13.6 Hz, 0.5H), 3.52 (t, J = 12.2 Hz, 0.5H), 3.27 (d, J = 12.2 Hz, 0.5H), 3.15 (t, J = 12.2 Hz, 0.5H), 2.72 (m, 1H), 1.90 (s, 3H), 1.35 (d, J = 6.7 Hz, 3H), 1.08 (d, J = 6.7 Hz, 3H), 0.94 (d, J = 6.7 Hz, 3H).
19F NMR (376 MHz, DMSO-d6) δ −115.6 (d, J = 5.2 Hz, 1 F), −128.6 (br s, 1 F).
13C NMR (151 MHz, DMSO-d6) δ ppm 165.0 (1C), 163.4 (1C), 162.5 (1C), 160.1 (1C), 156.8 (1C), 153.7 (1C), 151.9 (1C), 149.5 (1C), 148.3 (1C), 145.2 (1C), 144.3 (1C), 131.6 (1C), 130.8 (1C), 127.9 (0.5C), 127.9 (0.5C), 127.8 (0.5C), 127.7 (0.5C), 123.2 (1C), 122.8 (1C), 111.7 (1C), 109.7 (1C), 105.7 (1C), 105.3 (1C), 51.4 (0.5C), 51.0 (0.5C), 48.9 (0.5C), 45.4 (0.5C), 44.6 (0.5C), 43.7 (0.5C), 43.5 (0.5C), 41.6 (0.5C), 29.8 (1C), 21.9 (1C), 21.7 (1C), 17.0 (1C), 15.5 (0.5C), 14.8 (0.5C).
FTMS (ESI) m/z: [M + H]+ calcd for C30H30F2N6O3 561.24202. Found 561.24150.
Clik here to view.
d (1R)-6-Fluoro7-(2-fluoro-6-hydroxyphenyl)-1-[4-methyl-2-(1-methylethyl)-3-pyridinyl]-4-[(2S)-2-methyl-4-(1-oxo-2-propen-1-yl)-1- piperazinyl]-pyrido[2,3-d]pyrimidin-2(1H)-one ((R)-38; AMG 510; 2.25 g, 43% yield) as the first-eluting peak.1 H NMR (600 MHz, DMSO-d6) δ ppm 10.20 (s, 1H), 8.39 (d, J = 4.9 Hz, 1H), 8.30 (d, J = 8.9 Hz, 0.5H), 8.27 (d, J = 8.7 Hz, 0.5H), 7.27 (q, J = 8.4 Hz, 1H), 7.18 (d, J = 4.9 Hz, 1H), 6.87 (dd, J = 16.2, 10.8 Hz, 0.5H), 6.84 (dd, J = 16.2, 10.7 Hz, 0.5H), 6.74 (d, J = 8.4 Hz, 1H), 6.68 (t, J = 8.4 Hz, 1H), 6.21 (d, J = 16.2 Hz, 0.5H), 6.20 (d, J = 16.2 Hz, 0.5H), 5.76 (d, J = 10.8 Hz, 0.5H), 5.76 (d, J = 10.7 Hz, 0.5H), 4.91 (m, 1H), 4.41 (d, J = 12.2 Hz, 0.5H), 4.33 (d, J = 12.2 Hz, 1H), 4.28 (d, J = 12.2 Hz, 0.5H), 4.14 (d, J = 12.2 Hz, 0.5H), 4.02 (d, J = 13.6 Hz, 0.5H), 3.69 (m, 1H), 3.65 (d, J = 13.6 Hz, 0.5H), 3.52 (t, J = 12.2 Hz, 0.5H), 3.27 (d, J = 12.2 Hz, 0.5H), 3.15 (t, J = 12.2 Hz, 0.5H), 2.72 (m, 1H), 1.90 (s, 3H), 1.35 (d, J = 6.7 Hz, 3H), 1.08 (d, J = 6.7 Hz, 3H), 0.94 (d, J = 6.7 Hz, 3H).
19F NMR (376 MHz, DMSO-d6) δ –115.6 (d, J = 5.2 Hz, 1 F), –128.6 (br. s., 1 F).
13C NMR (151 MHz, DMSO-d6) δ ppm 165.0 (1C), 163.4 (1C), 162.5 (1C), 160.1 (1C), 156.8 (1C), 153.7 (1C), 151.9 (1C), 149.5 (1C), 148.3 (1C), 145.2 (1C), 144.3 (1C), 131.6 (1C), 130.8 (1C), 127.9 (0.5C), 127.9 (0.5C), 127.8 (0.5C), 127.7 (0.5C), 123.2 (1C), 122.8 (1C), 111.7 (1C), 109.7 (1C), 105.7 (1C), 105.3 (1C), 51.4 (0.5C), 51.0 (0.5C), 48.9 (0.5C), 45.4 (0.5C), 44.6 (0.5C), 43.7 (0.5C), 43.5 (0.5C), 41.6 (0.5C), 29.8 (1C), 21.9 (1C), 21.7 (1C), 17.0 (1C), 15.5 (0.5C), 14.8 (0.5C).
FTMS (ESI) m/z: [M+H]+ Calcd for C30H30F2N6O3 561.24202; Found 561.24150. Atropisomer configuration (R vs. S) assigned crystallographically.The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.9b01180.
PATENT
WO 2021097212
The present disclosure relates to an improved, efficient, scalable process to prepare intermediate compounds, such as compound of Formula 6A, having the structure,
Image may be NSFW.
Clik here to view.
useful for the synthesis of compounds for the treatment of KRAS G12C mutated cancers.
BACKGROUND
[0003] KRAS gene mutations are common in pancreatic cancer, lung adenocarcinoma, colorectal cancer, gall bladder cancer, thyroid cancer, and bile duct cancer. KRAS mutations are also observed in about 25% of patients with NSCLC, and some studies have indicated that KRAS mutations are a negative prognostic factor in patients with NSCLC. Recently, V-Ki-ras2 Kirsten rat sarcoma viral oncogene homolog (KRAS) mutations have been found to confer resistance to epidermal growth factor receptor (EGFR) targeted therapies in colorectal cancer; accordingly, the mutational status of KRAS can provide important information prior to the prescription of TKI therapy. Taken together, there is a need for new medical treatments for patients with pancreatic cancer, lung adenocarcinoma, or colorectal cancer, especially those who have been diagnosed to have such cancers characterized by a KRAS mutation, and including those who have progressed after chemotherapy.
Related Synthetic Processes
[0126] The following intermediate compounds of 6-Fluoro-7-(2-fluoro-6-hydroxyphenyl)-1-(4-methyl-2-(2-propanyl)-3-pyridinyl)-4-((2S)-2-methyl-4-(2-propenoyl)-1-piperazinyl)pyrido[2,3-d]pyrimidin-2(1H)-one are representative examples of the disclosure and are not intended to be construed as limiting the scope of the present invention.
[0127] A synthesis of Compound 9 and the relevant intermediates is described in U.S. Serial No.15/984,855, filed May 21, 2018 (U.S. Publication No.2018/0334454, November 22, 2018) which claims priority to and the benefit claims the benefit of U.S. Provisional Application No.62/509,629, filed on May 22, 2017, both of which are incorporated herein by reference in their entireties for all purposes. 6-Fluoro-7-(2-fluoro-6-hydroxyphenyl)-1-(4-methyl-2-(2-propanyl)-3-pyridinyl)-4-((2S)-2-methyl-4-(2-propenoyl)-1-piperazinyl)pyrido[2,3-d]pyrimidin-2(1H)-one was prepared using the following process, in which the isomers of the final product were isolated via chiral chromatography.
Clik here to view.
[0128] Step 1: 2,6-Dichloro-5-fluoronicotinamide (Intermediate S). To a mixture of 2,6-dichloro-5-fluoro-nicotinic acid (4.0 g, 19.1 mmol, AstaTech Inc., Bristol, PA) in dichloromethane (48 mL) was added oxalyl chloride (2M solution in DCM, 11.9 mL, 23.8 mmol), followed by a catalytic amount of DMF (0.05 mL). The reaction was stirred at room temperature overnight and then was concentrated. The residue was dissolved in 1,4-dioxane (48 mL) and cooled to 0 °C. Ammonium hydroxide solution (28.0-30% NH3 basis, 3.6 mL, 28.6 mmol) was added slowly via syringe. The resulting mixture was stirred at 0 °C for 30 min and then was concentrated. The residue was diluted with a 1:1 mixture of EtOAc/Heptane and agitated for 5 min, then was filtered. The filtered solids were discarded, and the remaining mother liquor was partially concentrated to half volume and filtered. The filtered solids were washed with heptane and dried in a reduced-pressure oven (45 °C) overnight to provide 2,6-dichloro-5-fluoronicotinamide. 1H NMR (400 MHz, DMSO-d6) δ ppm 8.23 (d, J = 7.9 Hz, 1 H) 8.09 (br s, 1 H) 7.93 (br s, 1 H). m/z (ESI, +ve ion): 210.9 (M+H)+.
[0129] Step 2: 2,6-Dichloro-5-fluoro-N-((2-isopropyl-4-methylpyridin-3-yl)carbamoyl)nicotinamide. To an ice-cooled slurry of 2,6-dichloro-5-fluoronicotinamide (Intermediate S, 5.0 g, 23.9 mmol) in THF (20 mL) was added oxalyl chloride (2 M solution in DCM, 14.4 mL, 28.8 mmol) slowly via syringe. The resulting mixture was heated at 75 °C for 1 h, then heating was stopped, and the reaction was concentrated to half volume. After cooling to 0 °C, THF (20 mL) was added, followed by a solution of 2-isopropyl-4-methylpyridin-3-amine (Intermediate R, 3.59 g, 23.92 mmol) in THF (10 mL), dropwise via cannula. The resulting mixture was stirred at 0 °C for 1 h and then was quenched with a 1:1 mixture of brine and saturated aqueous ammonium chloride. The mixture was extracted with EtOAc (3x) and the combined organic layers were dried over anhydrous sodium sulfate and concentrated to provide 2,6-dichloro-5-fluoro-N-((2-isopropyl-4-methylpyridin-3-yl)carbamoyl)nicotinamide. This material was used without further purification in the following step. m/z (ESI, +ve ion): 385.1(M+H)+.
[0130] Step 3: 7-Chloro-6-fluoro-1-(2-isopropyl-4-methylpyridin-3-yl)pyrido[2,3-d]pyrimidine-2,4(1H,3H)-dione. To an ice-cooled solution of 2,6-dichloro-5-fluoro-N-((2-isopropyl-4-methylpyridin-3-yl)carbamoyl)nicotinamide (9.2 g, 24.0 mmol) in THF (40 mL) was added KHMDS (1 M solution in THF, 50.2 mL, 50.2 mmol) slowly via syringe. The ice bath was removed and the resulting mixture was stirred for 40 min at room temperature. The reaction was quenched with saturated aqueous ammonium chloride and extracted with EtOAc (3x). The combined organic layers were dried over anhydrous sodium sulfate and concentrated. The residue was purified by silica gel chromatography (eluent: 0-50% 3:1 EtOAc-EtOH/heptane) to provide 7-chloro-6-fluoro-1-(2-isopropyl-4-methylpyridin-3-yl)pyrido[2,3-d]pyrimidine-2,4(1H,3H)-dione.1H NMR (400 MHz, DMSO-d6) δ ppm 12.27 (br s, 1H), 8.48-8.55 (m, 2 H), 7.29 (d, J = 4.8 Hz, 1 H), 2.87 (quin, J = 6.6 Hz, 1 H), 1.99-2.06 (m, 3 H), 1.09 (d, J = 6.6 Hz, 3 H), 1.01 (d, J = 6.6 Hz, 3 H).19F NMR (376 MHz, DMSO-d6) δ: -126.90 (s, 1 F). m/z (ESI, +ve ion): 349.1 (M+H)+.
[0131] Step 4: 4,7-Dichloro-6-fluoro-1-(2-isopropyl-4-methylpyridin-3-yl)pyrido[2,3-d]pyrimidin-2(1H)-one. To a solution of 7-chloro-6-fluoro-1-(2-isopropyl-4-methylpyridin-3-yl)pyrido[2,3-d]pyrimidine-2,4(1H,3H)-dione (4.7 g, 13.5 mmol) and DIPEA (3.5 mL, 20.2 mmol) in acetonitrile (20 mL) was added phosphorus oxychloride (1.63 mL, 17.5 mmol), dropwise via syringe. The resulting mixture was heated at 80 °C for 1 h, and then was cooled to room temperature and concentrated to provide 4,7-dichloro-6-fluoro-1-(2-isopropyl-4-methylpyridin-3-yl)pyrido[2,3-d]pyrimidin-2(1H)-one. This material was used without further purification in the following step. m/z (ESI, +ve ion): 367.1 (M+H)+.
[0132] Step 5: (S)-tert-Butyl 4-(7-chloro-6-fluoro-1-(2-isopropyl-4-methylpyridin-3-yl)-2-oxo-1,2-dihydropyrido[2,3-d]pyrimidin-4-yl)-3-methylpiperazine-1-carboxylate. To an ice-cooled solution of 4,7-dichloro-6-fluoro-1-(2-isopropyl-4-methylpyridin-3-yl)pyrido[2,3-d]pyrimidin-2(1H)-one (13.5 mmol) in acetonitrile (20 mL) was added DIPEA (7.1 mL, 40.3 mmol), followed by (S)-4-N-Boc-2-methyl piperazine (3.23 g, 16.1 mmol, Combi-Blocks, Inc., San Diego, CA, USA). The resulting mixture was warmed to room temperature and stirred for 1 h, then was diluted with cold saturated aqueous sodium bicarbonate solution (200 mL) and EtOAc (300 mL). The mixture was stirred for an additional 5 min, the layers were separated, and the aqueous layer was extracted with more EtOAc (1x). The combined organic layers were dried over anhydrous sodium sulfate and concentrated. The residue was purified by silica gel chromatography (eluent: 0-50% EtOAc/heptane) to provide (S)-tert-butyl 4-(7-chloro-6-fluoro-1-(2-isopropyl-4-methylpyridin-3-yl)-2-oxo-1,2-dihydropyrido[2,3-d]pyrimidin-4-yl)-3-methylpiperazine-1-carboxylate. m/z (ESI, +ve ion): 531.2 (M+H)+.
[0133] Step 6: (3S)-tert-Butyl 4-(6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-1-(2-isopropyl-4-methylpyridin-3-yl)-2-oxo-1,2-dihydropyrido[2,3-d]pyrimidin-4-yl)-3-methylpiperazine-1-carboxylate. A mixture of (S)-tert-butyl 4-(7-chloro-6-fluoro-1-(2-isopropyl-4-methylpyridin-3-yl)-2-oxo-1,2-dihydropyrido[2,3-d]pyrimidin-4-yl)-3-methylpiperazine-1-carboxylate (4.3 g, 8.1 mmol), potassium trifluoro(2-fluoro-6-hydroxyphenyl)borate (Intermediate Q, 2.9 g, 10.5 mmol), potassium acetate (3.2 g, 32.4 mmol) and [1,1′-bis(diphenylphosphino)ferrocene]dichloropalladium(II), complex with dichloromethane (661 mg, 0.81 mmol) in 1,4-dioxane (80 mL) was degassed with nitrogen for 1 min. De-oxygenated water (14 mL) was added, and the resulting mixture was heated at 90 °C for 1 h. The reaction was allowed to cool to room temperature, quenched with half-saturated aqueous sodium bicarbonate, and extracted with EtOAc (2x) and DCM (1x). The combined organic layers were dried over anhydrous sodium sulfate and concentrated. The residue was purified by silica gel chromatography (eluent: 0-60% 3:1 EtOAc-EtOH/heptane) to provide (3S)-tert-butyl 4-(6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-1-(2-isopropyl-4-methylpyridin-3-yl)-2-oxo-1,2-dihydropyrido[2,3-d]pyrimidin-4-yl)-3-methylpiperazine-1-carboxylate.1H NMR (400 MHz, DMSO-d6) δ ppm 10.19 (br s, 1 H), 8.38 (d, J = 5.0 Hz, 1 H), 8.26 (dd, J = 12.5, 9.2 Hz, 1 H), 7.23-7.28 (m, 1 H), 7.18 (d, J = 5.0 Hz, 1 H), 6.72 (d, J = 8.0 Hz, 1 H), 6.68 (t, J = 8.9 Hz, 1 H), 4.77-4.98 (m, 1 H), 4.24 (br t, J = 14.2 Hz, 1 H), 3.93-4.08 (m, 1 H), 3.84 (br d, J=12.9 Hz, 1 H), 3.52-3.75 (m, 1 H), 3.07-3.28 (m, 1 H), 2.62-2.74 (m, 1 H), 1.86-1.93 (m, 3 H), 1.43-1.48 (m, 9 H), 1.35 (dd, J = 10.8, 6.8 Hz, 3 H), 1.26-1.32 (m, 1 H), 1.07 (dd, J = 6.6, 1.7 Hz, 3 H), 0.93 (dd, J = 6.6, 2.1 Hz, 3 H).19F NMR (376 MHz, DMSO-d6) δ: -115.65 (s, 1 F), -128.62 (s, 1 F). m/z (ESI, +ve ion): 607.3 (M+H)+.
[0134] Step 7: 6-Fluoro-7-(2-fluoro-6-hydroxyphenyl)-1-(4-methyl-2-(2-propanyl)-3-pyridinyl)-4-((2S)-2-methyl-4-(2-propenoyl)-1-piperazinyl)pyrido[2,3-d]pyrimidin-2(1H)-one. Trifluoroacetic acid (25 mL, 324 mmol) was added to a solution of (3S)-tert-butyl 4-(6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-1-(2-isopropyl-4-methylpyridin-3-yl)-2-oxo-1,2-dihydropyrido[2,3-d]pyrimidin-4-yl)-3-methylpiperazine-1-carboxylate (6.3 g, 10.4 mmol) in DCM (30 mL). The resulting mixture was stirred at room temperature for 1 h and then was concentrated. The residue was dissolved in DCM (30 mL), cooled to 0 °C, and sequentially treated with DIPEA (7.3 mL, 41.7 mmol) and a solution of acryloyl chloride (0.849 mL, 10.4 mmol) in DCM (3 mL; added dropwise via syringe). The reaction was stirred at 0 °C for 10 min, then was quenched with half-saturated aqueous sodium bicarbonate and extracted with DCM (2x). The combined organic layers were dried over anhydrous sodium sulfate and concentrated. The residue was purified by silica gel chromatography (eluent: 0-100% 3:1 EtOAc-EtOH/heptane) to provide 6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-1-(4-methyl-2-(2-propanyl)-3-pyridinyl)-4-((2S)-2-methyl-4-(2-propenoyl)-1-piperazinyl)pyrido[2,3-d]pyrimidin-2(1H)-one.1H NMR (400 MHz, DMSO-d6) δ ppm 10.20 (s, 1 H), 8.39 (d, J = 4.8 Hz, 1 H), 8.24-8.34 (m, 1 H), 7.23-7.32 (m, 1 H), 7.19 (d, J = 5.0 Hz, 1 H), 6.87 (td, J = 16.3, 11.0 Hz, 1 H), 6.74 (d, J = 8.6 Hz, 1 H), 6.69 (t, J = 8.6 Hz, 1 H), 6.21 (br d, J = 16.2 Hz, 1 H), 5.74-5.80 (m, 1 H), 4.91 (br s, 1 H), 4.23-4.45 (m, 2 H), 3.97-4.21 (m, 1 H), 3.44-3.79 (m, 2 H), 3.11-3.31 (m, 1 H), 2.67-2.77 (m, 1 H), 1.91 (s, 3 H), 1.35 (d, J = 6.8 Hz, 3 H), 1.08 (d, J = 6.6 Hz, 3 H), 0.94 (d, J = 6.8 Hz, 3 H).19F NMR (376 MHz, DMSO-d6) δ ppm -115.64 (s, 1 F), -128.63 (s, 1 F). m/z (ESI, +ve ion): 561.2 (M+H)+.
[0135] Another synthesis of Compound 9 and the relevant intermediates was described in a U.S. provisional patent application filed November 16, 2018, which is incorporated herein by reference in its entirety for all purposes.
Clik here to view.
Representative Synthetic Processes
[0136] The present disclosure comprises the following steps wherein the synthesis and utilization of the boroxine intermediate is a novel and inventive step in the manufacture of AMG 510 (Compound 9):
Clik here to view.
Raw Materials
Clik here to view.
Clik here to view.
Step la
Clik here to view.
Clik here to view.
[0137] To a solution of 2,6-dichloro-5-fluoro-3-pyridinecarboxylic acid (25kg; 119. lmol) in dichloromethane (167kg) and DMF (592g) was added Oxalyl chloride (18.9kg; 148.9mol) while maintaining an internal temp between 15-20 °C. Additional dichloromethane (33kg) was added as a rinse and the reaction mixture stirred for 2h. The reaction mixture is cooled then quenched with ammonium hydroxide (40.2L; 595.5mol) while maintaining internal temperature 0 ± 10°C. The resulting slurry was stirred for 90min then the product collected by filtration. The filtered solids were washed with DI water (3X 87L) and dried to provide 2,6-dichloro-5-fluoronicotinamide (Compound 1).
Step 1b
Clik here to view.
Clik here to view.
[0138] In reactor A, a solution of 2,6-dichloro-5-fluoronicotinamide (Compound 1) (16.27kg; 77.8mol) in dichloromethane (359.5kg) was added oxalyl chloride (11.9kg;
93.8mol) while maintaining temp ≤ 25°C for 75min. The resulting solution was then headed to 40°C ± 3°C and aged for 3h. Using vacuum, the solution was distilled to remove dichloromethane until the solution was below the agitator. Dichloromethane (300 kg) was then added and the mixture cooled to 0 ± 5°C. To a clean, dry reactor (reactor B) was added,2-isopropyl-4-methylpyridin-3-amine (ANILINE Compound 2A) (12.9kg; 85.9mol) followed by dichloromethane (102.6 kg). The ANILINE solution was azeodried via vacuum distillation while maintaining an internal temperature between 20-25 °), replacing with additional dichloromethane until the solution was dry by KF analysis (limit ≤ 0.05%). The solution volume was adjusted to approx. 23L volume with dichloromethane. The dried ANILINE solution was then added to reactor A while maintaining an internal temperature of 0 ± 5°C throughout the addition. The mixture was then heated to 23 °C and aged for 1h. the solution was polish filtered into a clean reactor to afford 2,6-dichloro-5-fluoro-N-((2- isopropyl-4-methylpyridin-3-yl)carbamoyl)nicotinamide (Compound 3) as a solution in DCM and used directly in the next step.
Step 2
Clik here to view.
Clik here to view.
[0139] A dichloromethane solution of 2,6-dichloro-5-fluoro-N-{[4-methyl-2-(propan-2- yl)pyridin-3-yl]carbamoyl}pyridine-3-carboxamide (UREA (Compound 3)) (15kg contained; 38.9mol) was solvent exchanged into 2-MeTHF using vacuum distillation while maintaining internal temperature of 20-25 °C. The reactor volume was adjusted to 40L and then
additional 2-MeTHF was charged (105.4 kg). Sodium t-butoxide was added (9.4 kg;
97.8mol) while maintaining 5-10 °C. The contents where warmed to 23 °C and stirred for 3h. The contents where then cooled to 0-5C and ammonium chloride added (23.0kg; 430mol) as a solution in 60L of DI water. The mixture was warmed to 20 C and DI water added (15L) and further aged for 30min. Agitation was stopped and the layers separated. The aqueous layer was removed and to the organic layer was added DI water(81.7L). A mixture of conc HCl (1.5kg) and water (9L) was prepared then added to the reactor slowly until pH measured between 4-5. The layers were separated, and the aqueous layer back extracted using 2-MeTHF (42.2kg). The two organic layers combined and washed with a 10% citric acid solution (75kg) followed by a mixture of water (81.7L) and saturated NaCl (19.8 kg). The organic layer was then washed with saturated sodium bicarbonate (75kg) repeating if necessary to achieve a target pH of ≥ 7.0 of the aqueous. The organic layer was washed again with brine (54.7kg) and then dried over magnesium sulfate (5kg). The mixture was filtered to remove magnesium sulfate rinsing the filtered bed with 2-MeTHF (49.2 kg). The combined filtrate and washes where distilled using vacuum to 40L volume. The concentrated solution was heated to 55 °C and heptane (10-12kg) slowly added until cloud point. The solution was cooled to 23 °C over 2h then heptane (27.3 kg) was added over 2h. The product slurry was aged for 3h at 20-25 °C then filtered and washed with a mixture of 2-MeTHF (2.8kg) and heptane (9kg). The product was dried using nitrogen and vacuum to afford solid 7-chloro-6-fluoro-1-(2-isopropyl-4-methylpyridin-3-yl)pyrido[2,3-d]pyrimidine-2,4(1H,3H)-dione (rac-DIONE (Compound 4)).
Step 3
Clik here to view.
Clik here to view.
[0140] To a vessel, an agitated suspension of Compound 4, (1.0 eq.) in 2- methylterahydrofuran (7.0 L/kg) was added (+)-2,3-dibenzoyl-D-tartaric acid (2.0 eq.) under an atmosphere of nitrogen. 2-MeTHF is chiral, but it is used as a racemic mixture. The different enantiomers of 2-MeTHF are incorporated randomly into the co-crystal. The resulting suspension was warmed to 75°C and aged at 75°C until full dissolution was observed (< 30 mins.). The resulting solution was polish filtered at 75°C into a secondary vessel. To the polish filtered solution was charged n-Heptane (2.0 L/kg) at a rate that maintained the internal temperature above 65°C. The solution was then cooled to 60°C, seeded with crystals (0.01 kg/kg) and allowed to age for 30 minutes. The resulting suspension was cooled to 20°C over 4 hours and then sampled for chiral purity analysis by HPLC. To the suspension, n-Heptane (3.0 L/kg) was charged and then aged for 4 hours at 20°C under an atmosphere of nitrogen. The suspension was filtered, and the isolated solids were washed two times with (2:1) n-Heptane:2-methyltetrahydrofuran (3.0 L/kg). The material was dried with nitrogen and vacuum to afford M-Dione:DBTA: Me-THF complex (Compound 4a).
Step 4
Clik here to view.
Clik here to view.
[0141] To vessel A, a suspension of disodium hydrogen phosphate (21.1 kg, 2.0 equiv) in DI water (296.8 L, 6.3 L/kg) was agitated until dissolution was observed (≥ 30 min.). To vessel B, a suspension of the M-Dione:DBTA: Me-THF complex (Composition 4a)[46.9 kg (25.9 kg corrected for M-dione, 1.0 equiv.)] in methyl tert-butyl ether (517.8 L, 11.0 L/kg) was agitated for 15 to 30 minutes. The resulting solution from vessel A was added to vessel B, and then the mixture was agitated for more than 3 hours. The agitation was stopped, and the biphasic mixture was left to separate for more than 30 minutes. The lower aqueous phase was removed and then back extracted with methyl tert-butyl ether (77.7 L, 1.7 L/kg). The organic phases were combined in vessel B and dried with magnesium sulfate (24.8 kg, 0.529 kg/kg). The resulting suspension from vessel B was agitated for more than three hours and then filtered into vessel C. To vessel B, a methyl tert-butyl ether (46.9 L, 1.0 L/kg) rinse was charged and then filtered into vessel C. The contents of vessel C were cooled to 10 °C and then distilled under vacuum while slowly being warmed to 35°C. Distillation was continued until 320-350 kg (6.8-7.5 kg/kg) of methyl tert-butyl ether was collected. After cooling the contents of vessel C to 20°C, n-Heptane (278.7 L, 5.9 L/kg) was charged over one hour and then distilled under vacuum while slowly being warmed to 35°C. Distillation was continued until a 190-200 kg (4.1-4.3 kg/kg) mixture of methyl tert-butyl ether and n-Heptane was collected. After cooling the contents of vessel C to 20°C, n-Heptane (278.7 L, 5.9 L/kg) was charged a second time over one hour and then distilled under vacuum while slowly being warmed to 35°C. Distillation was continued until a 190-200 kg (4.1-4.3 kg/kg) mixture of methyl tert-butyl ether and n-Heptane was collected. After cooling the contents of vessel C to 20°C, n-Heptane (195.9 L, 4.2 L/kg) was charged a third time over one hour and then sampled for solvent composition by GC analysis. The vessel C suspension continued to agitate for more than one hour. The suspension was filtered, and then washed with a n-Heptane (68.6 L, 1.5 L/kg) rinse from vessel C. The isolated solids were dried at 50°C, and a sample was submitted for stock suitability. Afforded 7-chloro-6-fluoro-(1M)-1-[4-methyl-2-(propan-2-yl)pyridin-3-yl]pyrido[2,3-d]pyrimidine-2,4(1H,3H)-dione (M-DIONE) Compound 5M.
[0142] The first-generation process highlighted above has been successfully scaled on 200+ kg of rac-dione starting material (Compound 4). In this process, seeding the crystallization with the thermodynamically-stable rac-dione crystal form (which exhibits low solubility) would cause a batch failure. Based on our subsequent studies, we found that increasing the DBTA equivalents and lowering the seed temperature by adjusting heptane
charge schedule improves robustness of the process. The improved process is resistant to the presence of the thermodynamically-stable rac-dione crystal form and promotes successful separation of atropisomers. Subsequent batches will incorporate the improved process for large scale manufacture.
Step 5
Clik here to view.
Clik here to view.
Note: All L/kg amounts are relative to M-Dione input; All equiv. amounts are relative to M-Dione input after adjusted by potency.
[0143] M-Dione (Compound 5M, 1.0 equiv.) and Toluene-1 (10.0 L/kg) was charged to Vessel A. The resulting solution was dried by azeotropic distillation under vacuum at 45 °C until 5.0 L/kg of solvents has been removed. The contents of Vessel A were then cooled to 20 °C.
[0144] Vessel C was charged with Toluene-3 (4.5 L/kg), Phosphoryl chloride (1.5 equiv.) and N,N-Diisopropylethylamine-1 (2.0 equiv.) while maintaining the internal temperature below 20 ± 5 °C.
Upon finishing charging, Vessel C was warmed to 30 ± 5 °C. The contents of Vessel A were then transferred to Vessel C over 4 hours while maintaining the internal temperature at 30 ± 5°C. Vessel A was rinsed with Toluene-2 (0.5 L/kg) and transferred to Vessel C. The contents of Vessel C were agitated at 30°C for an additional 3 hours. The contents of Vessel C were cooled to 20 ± 5 °C. A solution of (s)-1-boc-3-methylpiperazine (1.2 equiv.), N,N-Diisopropylethylamine-2 (1.2 equiv.) in isopropyl acetate-1 (1.0 L/kg) was prepared in Vessel D. The solution of Vessel D was charged to vessel C while maintaining a batch temperature of 20 ± 5 °C (Note: Exotherm is observed). Upon the end of transfer, Vessel D was rinsed with additional dichloromethane (1.0 L/kg) and transferred to Vessel C. The contents of Vessel C were agitated for an additional 60 minutes at 20 °C. A solution of sodium bicarbonate [water-1 (15.0 L/kg + Sodium bicarbonate (4.5 equiv.)] was then charged into Vessel C over an hour while maintaining an internal temperature at 20 ± 5 °C throughout the addition. The contents of Vessel C were agitated for at least 12 hours at which point the Pipazoline (Compound 6) product was isolated by filtration in an agitated filter dryer. The cake was washed with water-2 and -3 (5.0 L/kg x 2 times, agitating each wash for 15 minutes) and isopropyl acetate-2 and 3 (5.0 L/kg x 2 times, agitating each wash for 15 min). The cake as dried under nitrogen for 12 hours.
Acetone Re-slurry (Optional):
[0145] Pipazoline (Compound 6) and acetone (10.0 L/kg) were charged to Vessel E. The suspension was heated to 50 °C for 2 hours. Water-4 (10.0 L/kg) was charged into Vessel E over 1 hour. Upon completion of water addition, the mixture was cooled to 20 °C over 1 hour. The contents of Vessel E were filtered to isolate the product, washing the cake with 1:1 acetone/water mixture (5.0 L/kg). The cake was dried under nitrogen for 12 hours.
Step 6
Clik here to view.
General Note: All equivalents and volumes are reported in reference to Pipazoline input
Clik here to view.
Note: All L/kg and kg/kg amounts are relative to Pipazoline input
[0146] Reactor A is charged with Pipazoline (Compound 6, 1.0 equiv), degassed 2- MeTHF (9.0 L/kg) and a solution of potassium acetate (2.0 equiv) in degassed water (6.5 L/kg). The resulting mixture is warmed to 75 ± 5 °C and then, charge a slurry of
Pd(dpePhos)Cl2 (0.003 equiv) in 2-MeTHF (0.5 L/kg). Within 2 h of catalyst charge, a solution of freshly prepared Boroxine (Compound 6A, 0.5 equiv) in wet degassed 2-MeTHF (4.0 L/kg, KF > 4.0%) is charged over the course of >1 hour, but < 2 hours, rinsing with an additional portion of wet 2-MeTHF (0.5 L/kg) after addition is complete. After reaction completion ( <0.15 area % Pipazoline remaining, typically <1 h after boroxine addition is complete), 0.2 wt% (0.002 kg/kg) of Biaryl seed is added as a slurry in 0.02 L/kg wet 2- MeTHF, and the resulting seed bed is aged for > 60 min. Heptane (5.0 L/kg) is added over 2 hours at 75 ± 5 °C. The batch is then cooled to 20 ± 5 °C over 2 hours and aged for an additional 2 h. The slurry is then filtered and cake washed with 1 x 5.0L/kg water, 1 x 5.0L/kg 1:1 iPrOH:water followed by 1 x 5.0 L/kg 1:1 iPrOH:heptane (resuspension wash: the cake is resuspended by agitator and allow to set before filtering) . The cake (Biaryl, Compound 7) is then dried under vacuum with a nitrogen sweep.
Note: If the reaction stalls, an additional charge of catalyst and boroxine is required
Step 7 Charcoal Filtration for Pd removal
Image may be NSFW.
Clik here to view.
General Note: All equivalents and volumes are reported in reference to crude Biaryl input
Clik here to view.
Note: All L/kg and kg/kg amounts are relative to crude Biaryl input
[0147] In a clean Vessel A, charge crude Biaryl (1 equiv) and charge DCM (10 L/kg). Agitate content for > 60 minutes at 22 ± 5 °C, observing dissolution. Pass crude Biaryl from Vessel A, through a bag filter and carbon filters at a flux ≤ 3 L2/min/m and collect filtrate in clean Vessel B. Charge DCM rinse (1 L/kg) to Vessel A, and through carbon filters to collect in vessel B.
[0148] From filtrate in Vessel B, pull a solution sample for IPC Pd content. Sample is concentrated to solid and analyzed by ICP-MS. IPC: Pd ≤ 25 ppm with respect to Biaryl. a. If Pd content is greater than 25 ppm with respect to Biaryl on first or second IPC sample, pass solution through carbon filter a second time at ≤ 3 L2/min/m2, rinsing with 1 L/kg DCM; sample filtrate for IPC.
b. If Pd content remains greater than 25 ppm after third IPC, install and condition fresh carbon discs. Pass Biaryl filtrate through refreshed carbon filter, washing with 1 L/kg DCM. Sample for IPC.
[0149] Distill and refill to appropriate concentration. Prepare for distillation of recovered filtrate by concentrating to ≤ 4 L/kg DCM, and recharge to reach 5.25 ± 0.25 L/kg DCM prior to moving into Step 7 Boc-deprotection reaction.
Step 7
Image may be NSFW.
Clik here to view. General Note: All equivalents and volumes are reported in reference to crude Biaryl input
Clik here to view.
Note: All L/kg and kg/kg amounts are relative to Biaryl input
[0150] To Reactor A was added: tert-butyl (3S)-4-{6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-(1M)-1-[4-methyl-2-(propan-2-yl)pyridin-3-yl]-2-oxo-1,2-dihydropyrido[2,3-d]pyrimidin-4-yl}-3-methylpiperazine-1-carboxylate (Biaryl) (1.0 equiv), dichloromethane (5.0 L/kg), and the TFA (15.0 equiv, 1.9 L/kg) is charged slowly to maintain the internal temperature at 20 ± 5 °C. The reaction was stirred for 4 h at 20 ± 5 °C.
[0151] To Reactor B was added: potassium carbonate (18.0 equiv), water (20.0 L/kg), and NMP (1.0) to form a homogenous solution. While agitating at the maximum acceptable rate for the equipment, the reaction mixture in A was transferred into the potassium carbonate solution in B over 30 minutes (~ 0.24 L/kg/min rate). The mixture was stirred at 20 ± 5 °C for an additional 12 h.
[0152] The resulting slurry was filtered and rinsed with water (2 x 10 L/kg). The wet cake was dried for 24 h to give 6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-4-[(2S)-2-methylpiperazin- 1-yl]-(1M)-1-[4-methyl-2-(propan-2-yl)pyridin-3-yl]pyrido[2,3-d]pyrimidin-2(1H)-one (Des- Boc, Compound 8).
Step 8
Clik here to view.
Clik here to view.
Note: All L/kg and kg/kg amounts are relative to Des-Boc input
[0153] Des-Boc (Compound 8, 1.0 equiv) and NMP (4.2 L/kg) are charged to Vessel A under nitrogen, charge the TFA (1.0 equiv.) slowly to maintain the Tr <25 °C. The mixture is aged at 25 °C until full dissolution is observed (about 0.5 hour). The solution is then polish filtered through a 0.45 micron filter into Vessel B, washing with a NMP (0.8 L/kg). The filtrate and wash are combined, and then cooled to 0 °C. To the resulting solution, Acryloyl Chloride (1.3 equiv.) is added while maintaining temperature < 10 C. The reaction mixture is then aged at 5 ±5°C until completed by IPC (ca.1.5 hrs).
Preparation of Aqueous Disodium Phosphate Quench:
[0154] Disodium Phosphate (3.0 equiv) and Water (15.0 L/kg) are charged to Vessel C. The mixture is aged at 25 °C until full dissolution is observed. The solution is warmed to 45 ±5°C. A seed slurry of AMG 510 (0.005 equiv.) in Water (0.4 L/kg) is prepared and added to Vessel C while maintaining temperature at 45 ±5°C.
[0155] The reaction mixture in Vessel B is transferred to Vessel C (quench solution) while maintaining temperature at 45 ±5°C (ca.1 hrs). Vessel B is washed with a portion of NMP (0.5 L/kg). The product slurry is aged for 2 hrs at 45 ±5°C, cooled to 20 °C over 3 hrs, aged at 20 °C for a minimum of 12 hrs, filtered and washed with Water (2 x 10.0 L/kg). The product is dried using nitrogen and vacuum to afford Crude AMG 510 (Compound 9A).
Step 9
Image may be NSFW.
Clik here to view. General Note: All equivalents and volumes are reported in reference to crude AMG 510 input
Clik here to view.
Note: All L/kg and kg/kg amounts are relative to Crude AMG 510 input
[0156] Reactor A was charged with 6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-(1M)-1-[4- methyl-2-(propan-2-yl)pyridin-3-yl]-4-[(2S)-2-methyl-4-(prop-2-enoyl)piperazin-1- yl]pyrido[2,3-d]pyrimidin-2(1H)-one (Crude AMG 510) (1.0 equiv), ethanol (7.5 L/kg), and water (1.9 L/kg). The mixture heated to 75 °C and polish filtered into a clean Reactor B. The solution was cool to 45 °C and seeded with authentic milled AMG 510 seed (0.015 േ 0.005
1 Seed performs best when reduced in particle size via milling or with other type of mechanical grinding if mill is not available (mortar/ pestle). Actual seed utilized will be based on seed availability. 1.0- 2.0% is seed is target amount.
kg/kg); the resulting slurry was aged for 30 min. Water (15.0 L/kg) was added over 5h while maintaining an internal temperature > 40 °C; the mixture was aged for an additional 2h.
[0157] The mixture was cooled to 20 °C over 3 hours and aged for 8h, after which the solid was collected by filtration and washed using a mixture of ethanol (2.5 L/kg) and water (5.0 L/kg). The solid was dried using vacuum and nitrogen to obtain 6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-(1M)-1-[4-methyl-2-(propan-2-yl)pyridin-3-yl]-4-[(2S)-2-methyl-4-(prop-2-enoyl)piperazin-1-yl]pyrido[2,3-d]pyrimidin-2(1H)-one (AMG 510, Compound 9).
Compound 6A Boroxine Synthesis:
Lithiation/borylation
Clik here to view.
Clik here to view.
Clik here to view.
[0158] Reactor A was charged with THF (6 vol), a secondary amine base, Diisopropylamine (1.4 equiv), and a catalyst, such as triethylamine hydrochloride (0.01 equiv.). The resulting solution was cooled to -70 °C and a first base, n-BuLi (2.5 M in hexane, 1.5 equiv) was slowly added. After addition is complete, a solution of 3-fluoroanisole (1.0 equiv) in THF (6 vol) was added slowly and kept at -70 °C for 5 min. Concurrently or subsequently, a reagent, B(EtO)3 (2.0 equiv), was added slowly and kept at -70 °C for 10 min. The reaction mixture was quenched with an acid, 2N HCl. The quenched reaction mixture was extracted with MTBE (3 x 4 vol). The combined organic phases were concentrated to 1.5-3 total volumes. Heptane (7-9 vol) was added drop-wise and the mixture was cooled to 0-10 °C and stirred for 3 h. The mixture was filtrated and rinsed with heptane (1.5 vol). The solid was dried under nitrogen at < 30 °C to afford (2-fluoro-6-methoxyphenyl)boronic acid.
Demethylation:
Clik here to view.
Clik here to view.
Note: All L/kg and kg/kg amounts are relative to (2-fluoro-6-methoxyphenyl)boronic acid input
[0159] To a reactor, charge dichloromethane (solvent, 4.0 L/kg) and an acid, BBr3 (1.2 equiv), and cool to -20 °C. To this solution, a suspension of (2-fluoro-6-methoxyphenyl)boronic acid (1.0 equiv) in dichloromethane (4.0 L/kg) was added into the BBr3/DCM mixture while keeping temperature -15 to -25 °C. The reaction was allowed to proceed for approximately 2 hours while monitored by HPLC [≤1% (2-fluoro-6-methoxyphenyl)boronic acid] before reverse quenching into water (3.0 L/kg). The precipitated solid was then isolated by filtration and slurried with water (3.0 L/kg) on the filter prior to deliquoring. The filtrates were adjusted to pH 4-6 by the addition of sodium bicarbonate. The bottom organic phase was separated and the resulting aqueous layer was washed with dichloromethane (solvent, 5.0 Vol) and adjusted to pH = 1 by addition of concentrated hydrochloric acid. The resulting solids were isolated by filtration, washing the cake with water (2 x 5.0 L/kg)
Purification via Reslurry (required)
[0160] The combined crude solids were charged into a reactor and slurried with 5% EtOH/water (5.0 L/kg) at 20 °C for >1 h. The purified product was then isolated by filtration and rinsed with water (2 x 3 L/kg) before drying on the filter at < 30 °C to with nitrogen/vacuum to afford 2,2′,2”-(1,3,5,2,4,6-trioxatriborinane-2,4,6-triyl)tris(3-fluorophenol) (Boroxine, Compound 6A).
PATENT
WO 2020102730
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2020102730
PATENT
US 20180334454
References
- ^ Jump up to:a b c d e “Lumakras- sotorasib tablet, coated”. DailyMed. Retrieved 6 June 2021.
- ^ Jump up to:a b c d e f g h i j k l m n “FDA Approves First Targeted Therapy for Lung Cancer Mutation Previously Considered Resistant to Drug Therapy”. U.S. Food and Drug Administration (FDA). 28 May 2021. Retrieved 28 May 2021. Image may be NSFW.
Clik here to view.![]() This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ “KRAS mutant-targeting AMG 510”. NCI Drug Dictionary. National Cancer Institute. 2 February 2011. Retrieved 16 November2019.
- ^ Canon J, Rex K, Saiki AY, Mohr C, Cooke K, Bagal D, et al. (November 2019). “The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity”. Nature. 575 (7781): 217–23. Bibcode:2019Natur.575..217C. doi:10.1038/s41586-019-1694-1. PMID 31666701.
- ^ Jump up to:a b “FDA approves Amgen drug for lung cancer with specific mutation”. CNBC. 28 May 2021. Retrieved 28 May 2021.
- ^ Hong DS, Fakih MG, Strickler JH, Desai J, Durm GA, Shapiro GI, et al. (2020). “KRASG12C inhibition with sotorasib in advanced solid tumors”. N Engl J Med. doi:10.1056/NEJMoa1917239. PMC 7571518.
- ^ Clinical trial number NCT03600883 for “A Phase 1/2, Study Evaluating the Safety, Tolerability, PK, and Efficacy of AMG 510 in Subjects With Solid Tumors With a Specific KRAS Mutation ” at ClinicalTrials.gov
- ^ “The Discovery Of Amgen’s Novel Investigational KRAS(G12C) Inhibitor AMG 510 Published In Nature” (Press release). Amgen. 30 October 2019. Retrieved 16 November 2019.
- ^ Irving M (24 December 2019). “Drug targeting common cancer cause enters phase 2 clinical trials”. New Atlas. Retrieved 24 December 2019.
- ^ Jump up to:a b c d Halford B (3 April 2019). “Amgen unveils its KRas inhibitor in human clinical trials: AMG 510 shuts down a mutant version of the cancer target via covalent interaction”. Chemical & Engineering News. 97 (4). Retrieved 16 November 2019.
- ^ Al Idrus A (9 September 2019). “Amgen’s KRAS drug continues to deliver but faces ‘curse’ of high expectations”. fiercebiotech.com. Retrieved 16 November 2019.
- ^ Kaiser J (30 October 2019). “Two new drugs finally hit ‘undruggable’ cancer target, providing hope for treatments”. Science Magazine. AAAS. Retrieved 16 November 2019.
- ^ Astor L (9 September 2019). “FDA Grants AMG 510 Fast Track Designation for KRAS G12C+ NSCLC”. targetedonc.com. Retrieved 16 November 2019.
- ^ World Health Organization (2021). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 85” (PDF). WHO Drug Information. 35 (1).
Further reading
- Hong DS, Fakih MG, Strickler JH, Desai J, Durm GA, Shapiro GI, et al. (September 2020). “KRASG12C Inhibition with Sotorasib in Advanced Solid Tumors”. N Engl J Med. 383 (13): 1207–17. doi:10.1056/NEJMoa1917239. PMC 7571518. PMID 32955176.
- Lanman BA, Allen JR, Allen JG, Amegadzie AK, Ashton KS, Booker SK, et al. (January 2020). “Discovery of a Covalent Inhibitor of KRASG12C (AMG 510) for the Treatment of Solid Tumors”. J Med Chem. 63 (1): 52–65. doi:10.1021/acs.jmedchem.9b01180. PMID 31820981.
External links
- “Sotorasib”. Drug Information Portal. U.S. National Library of Medicine.
- Clinical trial number NCT03600883 for “A Phase 1/2, Study Evaluating the Safety, Tolerability, PK, and Efficacy of AMG 510 in Subjects With Solid Tumors With a Specific KRAS Mutation (CodeBreaK 100)” at ClinicalTrials.gov
| Clinical data | |
|---|---|
| Trade names | Lumakras |
| Other names | AMG 510 |
| License data | US DailyMed: Sotorasib |
| Routes of administration | By mouth |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only [1][2] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 2252403-56-6 |
| PubChem CID | 137278711 |
| DrugBank | DB15569 |
| ChemSpider | 72380148 |
| UNII | 2B2VM6UC8G |
| KEGG | D12055 |
| Chemical and physical data | |
| Formula | C30H30F2N6O3 |
| Molar mass | 560.606 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI |
////////Sotorasib, ソトラシブ , FDA 2021, APPROVALS 2021, Lumakras, CANCER, ANTINEOPLASTIC, AMG 510, AMG-510, AMG510, AMGEN, priority review, fast-track, breakthrough therapy, orphan drug
CC1CN(CCN1C2=NC(=O)N(C3=NC(=C(C=C32)F)C4=C(C=CC=C4F)O)C5=C(C=CN=C5C(C)C)C)C(=O)C=C
Clik here to view.
