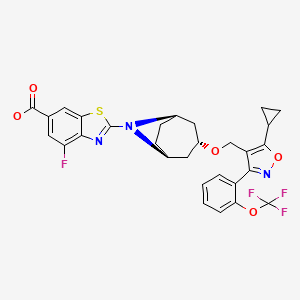

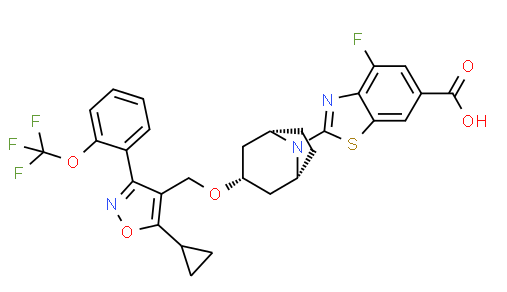
TROPIFEXOR
トロピフェクサー;
PHASE 2, NASH, PBC, liver fibrosis, bile acid diarrhea, non-alcoholic fatty liver disease
| Formula | C29H25F4N3O5S |
|---|---|
| CAS | 1383816-29-2 |
| Mol weight | 603.5845 |
TROPIFEXORLJN 452;LJN-452;LJN452;CS-2712;CPD1549;Tropifexor;Tropifexor (LJN452);LJN452;LJN452,Tropifexor;2-[(1R,3r,5S)-3-({5-cyclopropyl-3-[2-(trifluoromethoxy)phenyl]-1,2-oxazol-4-yl}methoxy)-8-azabicyclo[3.2.1]octan-8-yl]-4-fluoro-1,3-benzothiazole-6-carboxylic acidтропифексор [Russian] [INN]
تروبيفيكسور [Arabic] [INN]
曲匹法索 [Chinese] [INN]2-[(3-endo)-3-({5-Cyclopropyl-3-[2-(trifluormethoxy)phenyl]-1,2-oxazol-4-yl}methoxy)-8-azabicyclo[3.2.1]oct-8-yl]-4-fluor-1,3-benzothiazol-6-carbonsäure [German] [ACD/IUPAC Name]
2-[(3-endo)-3-({5-Cyclopropyl-3-[2-(trifluoromethoxy)phenyl]-1,2-oxazol-4-yl}methoxy)-8-azabicyclo[3.2.1]oct-8-yl]-4-fluoro-1,3-benzothiazole-6-carboxylic acid [ACD/IUPAC Name]
6-Benzothiazolecarboxylic acid, 2-[(3-endo)-3-[[5-cyclopropyl-3-[2-(trifluoromethoxy)phenyl]-4-isoxazolyl]methoxy]-8-azabicyclo[3.2.1]oct-8-yl]-4-fluoro- [ACD/Index Name]
Acide 2-[(3-endo)-3-({5-cyclopropyl-3-[2-(trifluorométhoxy)phényl]-1,2-oxazol-4-yl}méthoxy)-8-azabicyclo[3.2.1]oct-8-yl]-4-fluoro-1,3-benzothiazole-6-carboxylique [French] [ACD/IUPAC Name]
NMZ08KM76Z
Tropifexor fast facts
| CAS Reg. No. | 1383816-29-2 |
| Molar mass | 603.58 g/mol |
| Empirical formula | C29H25F4N3O5S |
| Appearance | White crystals |
| Melting point | 221 ºC |
| Water solubility | 6 mg/L |
| Efficacy | Anti-inflammatory, Farnesoid X receptor (FXR) agonist |
|---|---|
| Comment | Treatment of non-alcoholic steatohepatitis |
Novartis is developing tropifexor, a non-bile acid farnesoid X receptor agonist, and its analog LJP-305, for treating NASH, PBC, liver fibrosis, bile acid diarrhea and non-alcoholic fatty liver disease. In June 2021, this drug was reported to be in phase 2 clinical development.
Nonalcoholic steatohepatitis (NASH) is a liver disease that is becoming more prevalent as worldwide obesity and type 2 diabetes increase. It is characterized by accumulation of fat in the liver, inflammation, hepatocyte ballooning, and fibrosis.
Another liver disease, primary biliary cholangitis (PBC), is a cholestatic condition in which bile flow from the liver to the intestine is reduced or interrupted. It is thought to be autoimmune.
PBC is associated with decreased expression of the farnesoid X receptor (FXR), a ligand-activated nuclear receptor that is highly expressed in the liver and other organs. FXR is a key regulator of bile acid production, conjugation, and transport. FXR activation also suppresses lipogenesis; thus, it has been proposed as a treatment for NASH.
Recently, David C. Tully and colleagues at the Genomics Institute of the Novartis Research Foundation (San Diego) and the Novartis Institutes for Biomedical Research (Emeryville, CA) discovered tropifexor, a highly potent FXR agonist. They began by replacing an indole group in an existing partial FXR agonist with a 2-substituted benzothiazole-6-carboxylic acid, a change that resulted in a dramatic increase in potency. Further changes, including optimization of the benzothiazole substituent, resulted in more potent, orally bioavailable tropifexor.
Tropifexor is an investigational drug which acts as an agonist of the farnesoid X receptor (FXR). It was discovered by researchers from Novartis and Genomics Institute of the Novartis Research Foundation. Its synthesis and pharmacological properties were published in 2017.[1] It was developed for the treatment of cholestatic liver diseases and nonalcoholic steatohepatitis (NASH). In combination with cenicriviroc, a CCR2 and CCR5 receptor inhibitor, it is undergoing a phase II clinical trial for NASH and liver fibrosis.[2]
Rats treated orally with tropifexor (0.03 to 1 mg/kg) showed an upregulation of the FXR target genes, BSEP and SHP, and a down-regulation of CYP8B1. Its EC50 for FXR is between 0.2 and 0.26 nM depending on the biochemical assay.
The patent which covers tropifexor and related compounds was published in 2010.[3]
PATENT
WO-2021104022
Novel, stable crystalline polymorphic form II of tropifexor , useful for treating non-alcoholic steatohepatitis (NASH), fatty liver and primary biliary cholangitis (PBC).Tropifexor was originally developed by Novartis and then licensed to Pfizer for cooperative development. It is a non-steroidal FXR (farnesoid receptor) agonist, currently in clinical phase II of indications for NASH (non-alcoholic steatohepatitis), fatty liver and primary biliary cholangitis.
The structure of Tropifexor is shown in the following formula (1):
Drug polymorphism is a common phenomenon in drug development and an important factor affecting drug quality. Different crystal forms of the same drug may have significant differences in physical and chemical properties such as appearance, fluidity, solubility, storage stability, bioavailability, etc., and there may be great differences, which will affect the storage transfer, application, stability, and efficacy of the drug In order to obtain an effective crystal form that is conducive to production or pharmaceutical preparations, it is necessary to conduct a comprehensive investigation of the crystallization behavior of the drug to obtain a crystal form that meets the production requirements.
At present, there is no literature that discloses the crystal form of Tropifexor, and there is no related literature report.
The present invention obtains a new crystal form of the compound through a large number of experimental studies on the Tropifexor compound. The new crystal form has the advantages of high solubility, good stability, low moisture absorption, simple preparation process and easy operation, etc., and has excellent properties in industrial production. Superiority.Example 1 Preparation method of Tropifexor crystal form II[0049]After mixing 60.3 mg of Tropifexor and p-aminobenzoic acid (13.7 mg), they were added to ethanol (3.0 ml), stirred at 27° C. to obtain a clear solution, and then allowed to stand at room temperature for about 2 days to precipitate a solid product. It was filtered with suction and placed in a drying box at 50°C and vacuum dried to constant weight to obtain 51.3 mg of solid powder. The obtained crystal was detected by XPRD and confirmed to be Tropifexor crystal form II; its X-ray powder diffraction pattern was basically consistent with Fig. 1, its DSC pattern was basically the same as Fig. 2, and its TGA pattern was basically the same as Fig. 3.[0050]Example 2 Preparation method of Tropifexor crystal form II[0051]After mixing 60.3 mg of Tropifexor and p-hydroxybenzoic acid (13.8 mg), they were added to ethanol (3.0 ml), stirred at 27° C. to obtain a clear solution, and then allowed to stand at room temperature for about 2 days to precipitate a solid product. It was filtered with suction and placed in a drying box at 50°C and vacuum dried to constant weight to obtain 48.5 mg of solid powder. The obtained crystal was detected by XPRD and confirmed to be Tropifexor crystal form II; its X-ray powder diffraction pattern was basically consistent with Fig. 1, its DSC pattern was basically the same as Fig. 2, and its TGA pattern was basically the same as Fig. 3.[0052]Example 3 Preparation method of Tropifexor crystal form II[0053]After mixing 60.3 mg of Tropifexor and salicylic acid (13.8 mg), they were added to ethanol (3.0 ml), stirred at 27°C to obtain a clear solution, and then allowed to stand at room temperature for about 2 days to precipitate a solid product. Filter with suction and place in a drying box at 50°C and vacuum dry to constant weight to obtain 50.0 mg of solid powder. The obtained crystal was detected by XPRD and confirmed to be Tropifexor crystal form II; its X-ray powder diffraction pattern was basically consistent with Fig. 1, its DSC pattern was basically the same as Fig. 2, and its TGA pattern was basically the same as Fig. 3.[0054]Example 4 Preparation method of Tropifexor crystal form II[0055]After mixing 60.3 mg of Tropifexor and 2,4-dihydroxybenzoic acid (15.4 mg), they were added to ethanol (3.0 ml), stirred at 27°C to obtain a clear solution, and then allowed to stand at room temperature for about 2 days to precipitate a solid product. It was filtered with suction and placed in a drying box at 50°C and vacuum dried to constant weight to obtain 49.5 mg of solid powder. The obtained crystal was detected by XPRD and confirmed to be Tropifexor crystal form II; its X-ray powder diffraction pattern was basically consistent with Fig. 1, its DSC pattern was basically the same as Fig. 2, and its TGA pattern was basically the same as Fig. 3.
PATENT
WO2021104021 ,
claiming crystalline polymorphic form I of tropifexor,Example 1 Preparation method of Tropifexor crystal form I
50.0 mg of Tropifexor was added to ethanol (1.0 ml), heated to 60° C. and stirred to obtain a clear solution, and then water (3 ml) was added dropwise to the Tropifexor solution. Stir and precipitate solid product. It was filtered with suction and placed in a drying box at 50°C and vacuum dried to constant weight to obtain 38.5 mg of solid powder. The obtained crystal was detected by XPRD and confirmed to be Tropifexor crystal form I; its X-ray powder diffraction pattern was basically consistent with Figure 1, its DSC pattern was basically consistent with Figure 2, and its TGA pattern was basically consistent with Figure 3
PATENT
product pat, WO2012087519 , https://patents.google.com/patent/WO2012087519A1/en
has protection in the EU until November 2031, and expire in US in February 2032 with US154 extension.
PATENT
WO 2016097933
Example 1
2-r(1 R,3r,5S)-3-(f5-cvclopropyl-3-r2-(trifluoromethoxy)phenyll-1 ,2-oxazol-4-yl)methoxy)-8- azabicvcloi3.2.1 loctan-8-yll-4-fluoro-1 ,3-benzothiazole-6-carboxylic acid (1 -1 B) and
-r(1 R,3r,5S)-3-(f5-cvclopropyl-3-r2-(trifluoromethyl)phenyll-1 ,2-oxazol-4-yl)methoxy)-8-
R1a = OCF3 (1 -1A, 1 -1 B)
R a = CF3 (1-2A, 1-2B)
Methyl 2-[(1 R,3r,5S)-3-(i5-cvclopropyl-3-r2-(trifluoromethoxy)phenyll-1 ,2-oxazol-4- yl}methoxy)-8-azabicvcloi3.2.1 loctan-8-yll-4-fluoro-1 ,3-benzothiazole-6-carboxylate (1 -1 A). Into a 25-mL round-bottom flask equipped with a stir bar was added sequentially 4-(((1 R,3r,5S)- 8-azabicyclo[3.2.1 ]octan-3-yloxy)methyl)-5-cyclopropyl-3-(2-(trifluoromethoxy)phenyl)isoxazole (1 .29 mmol), N,N-dimethylacetamide (3.6 mL), cesium carbonate (3.31 mmol), and methyl 2- bromo-4-fluorobenzo[d]thiazole-6-carboxylate (3.87 mmol). After stirring the resulting slurry at room temperature for 10 minutes, the mixture was then warmed to 60 °C and stirred for 1 h. The reaction slurry was allowed to cool to room temperature, and was diluted with 200 mL of ethyl acetate and washed with water (3 χ 30 mL). The organic extracts were concentrated under vacuum and directly purified using normal phase silica gel chromatography (40 g silica column) with a 15 min gradient of 10 % to 60 % ethyl acetate/hexanes. Desired fractions were concentrated in vacuo, and the resulting residue crystallized upon standing to give methyl 2- [(1 R,3r,5S)-3-({5-cyclopropyl-3-[2-(trifluoromethoxy)phenyl]-1 ,2-oxazol-4-yl}methoxy)-8- azabicyclo[3.2.1 ]octan-8-yl]-4-fluoro-1 ,3-benzothiazole-6-carboxylate (1-1 A) as a white crystalline solid. MS (m/z) : 618.2 (M+1 ).
2-r(1 R,3r,5S)-3-(i5-cvclopropyl-3-r2-(trifluoromethoxy)phenyll-1 ,2-oxazol-4-yl}methoxy)- 8-azabicvcloi3.2.1 loctan-8-yll-4-fluoro-1 ,3-benzothiazole-6-carboxylic acid (1 -1 B). To a 25-mL round-bottom flask equipped with a stir bar was added the ester (0.89 mmol), THF (4 mL),
MeOH (2 mL), and 3 N aqueous KOH solution (1 mL, 3 mmol). The resulting homogenous solution was stirred for 1 hour at 70 °C, cooled to room temperature, and then quenched with AcOH (roughly 0.2 mL of glacial acetic, 3 mmol) until pH=6 was achieved (Whatman class pH strip paper). At this time the reaction was diluted with ethyl acetate (40 mL) and washed with water (3 5 mL). The ethyl acetate fraction was concentrated under vacuum to give to an oily residue. To the resulting oil was then added MeOH (6 mL). The oil quickly dissolved, then immediately began to crystallize. Upon standing for 2.5 hrs, the mother liquor was withdrawn and crystals washed (3 x 2 mL of ice cold MeOH). The crystals were dried via vacuum (10 mm Hg pressure at 45 °C overnight) and then recrystallized from acetonitrile, filtered, and dried under vacuum to give 2-[(1 R,3r,5S)-3-({5-cyclopropyl-3-[2-(trifluoromethoxy)phenyl]-1 ,2-oxazol-4-yl}methoxy)-8-azabicyclo[3.2.1 ]octan-8-yl]-4-fluoro-1 ,3-benzothiazole-6-carboxylic acid (1 -1 B). 2-[(1 R,3r,5S)-3-({5-cyclopropyl-3-[2-(trifluoromethyl)phenyl]-1 ,2-oxazol-4-yl}methoxy)-8-azabicyclo[3.2.1 ]octan-8-yl]-4-fluoro-1 ,3-benzothiazole-6-carboxylic acid (1 -2B).
Examples 1 -2A and the corresponding acid 1 -2B can be prepared following the same procedures, from the reaction of intermediate 4-((8-azabicyclo[3.2.1 ]octan-3-yloxy)methyl)-5-cyclopropyl-3-(2-(trifluoromethyl)phenyl)isoxazole.
PAPER
European journal of medicinal chemistry (2021), 209, 112910
https://www.sciencedirect.com/science/article/abs/pii/S0223523420308825

Abstract
Farnesoid X receptor (FXR) agonists are emerging as potential therapeutics for the treatment of various metabolic diseases, as they display multiple effects on bile acid, lipid, and glucose homeostasis. Although the steroidal obeticholic acid, a full FXR agonist, was recently approved, several side effects probably due to insufficient pharmacological selectivity impede its further clinical application. Activating FXR in a partial manner is therefore crucial in the development of novel FXR modulators. Our efforts focusing on isoxazole-type FXR agonists, common nonsteroidal agonists for FXR, led to the discovery a series of novel FXR agonists bearing aryl urea moieties through structural simplification of LJN452 (phase 2). Encouragingly, compound 11k was discovered as a potent FXR agonist which exhibited similar FXR agonism potency but lower maximum efficacy compared to full agonists GW4064 and LJN452 in cell-based FXR transactivation assay. Extensive in vitro evaluation further confirmed partial efficacy of 11k in cellular FXR-dependent gene modulation, and revealed its lipid-reducing activity. More importantly, orally administration of 11k in mice exhibited desirable pharmacokinetic characters resulting in promising in vivo FXR agonistic activity.
References
- ^ Tully DC, Rucker PV, Chianelli D, Williams J, Vidal A, Alper PB, et al. (December 2017). “Discovery of Tropifexor (LJN452), a Highly Potent Non-bile Acid FXR Agonist for the Treatment of Cholestatic Liver Diseases and Nonalcoholic Steatohepatitis (NASH)”. Journal of Medicinal Chemistry. 60 (24): 9960–9973. doi:10.1021/acs.jmedchem.7b00907. PMID 29148806.
- ^ Clinical trial number NCT03517540 for “Safety, Tolerability, and Efficacy of a Combination Treatment of Tropifexor (LJN452) and Cenicriviroc (CVC) in Adult Patients With Nonalcoholic Steatohepatitis (NASH) and Liver Fibrosis. (TANDEM)” at ClinicalTrials.gov
- ^ WO Application Filing 2012087519, Alper PB, Chianelli D, Mutnick D, Vincent P, Tully DC, “Compositions and methods for modulating fxr”, published 2012-06-28, assigned to Genomics Institute of the Novartis Research Foundation. Retrieved 17 May 2019.
| Clinical data | |
|---|---|
| ATC code | None |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 1383816-29-2 |
| PubChem CID | 121418176 |
| UNII | NMZ08KM76Z |
| KEGG | D11548 |
| Chemical and physical data | |
| Formula | C29H25F4N3O5S |
| Molar mass | 603.59 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| show |
///////////TROPIFEXOR, トロピフェクサー, NOVARTIS, PHASE 2, тропифексор , تروبيفيكسور , 曲匹法索 , LJN 452, LJN-452, LJN452, CS-2712, CPD1549, Tropifexor, Tropifexor (LJN452), LJN452, LJN452, PHASE 2, NASH, PBC, liver fibrosis, bile acid diarrhea, non-alcoholic fatty liver disease
1ccc(c(c1)c2c(c(on2)C3CC3)CO[C@H]4C[C@H]5CC[C@@H](C4)N5c6nc7c(cc(cc7s6)C(=O)O)F)OC(F)(F)F
