IVERMECTIN
MK933
22,23-dihydroavermectin B1a + 22,23-dihydroavermectin B1b
70288-86-7  71827-03-7
71827-03-7
| C95H146O28 | |
| Molecular Weight: | 1736.15894 g/mol |
|---|
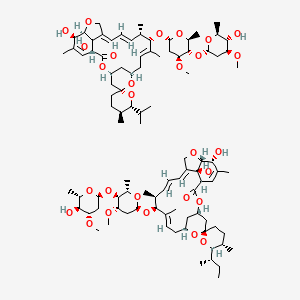
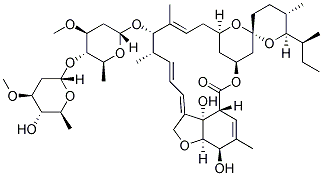 C48H74O14, 875.09
C48H74O14, 875.09
Ivermectin is a macrocyclic lactone derived from Streptomyces avermitilis with antiparasitic activity. Ivermectin exerts its anthelmintic effect via activating glutamate-gated chloridechannels expressed on nematode neurons and pharyngeal muscle cells. Distinct from the channel opening induced by endogenous glutamate transmitter, ivermectin-activated channels open very slowly but essentially irreversibly. As a result, neurons or muscle cells remain at either hyperpolarisation or depolarization state, thereby resulting in paralysis and death of the parasites. Ivermectin does not readily pass the mammal blood-brain barrier to the central nervous system where glutamate-gated chloride channels locate, hence the hosts are relatively resistant to the effects of this agent.
This drug, ivermectin, was developed by William C. Campbell of Drew University and Satoshi Ōmura of Japan’s Kitasato University. They were awarded the Nobel Prize in Physiology or Medicine. Originally, the drug was used to treat parasites in livestock and pets before becoming the mainstay of the global campaigns to combat lymphatic filariasis and onchocerciasis.
A workhorse of a drug that a few weeks ago earned its developers a Nobel prize for its success in treating multiple tropical diseases is showing early promise as a novel and desperately needed tool for interrupting malaria transmission, according to new findings presented today at the American Society of Tropical Medicine and Hygiene (ASTMH) Annual Meeting.
At ASTMH annual meeting, new studies explore advances in using ivermectin in ‘mass drug administration’ campaigns to reduce infections in Africa and slow spread of drug resistance in Asia…http://www.pharmpro.com/news/2015/10/nobel-prize-winning-drug-could-also-fight-malaria?et_cid=4908183&et_rid=577220619&type=cta
This new finding was presented today at the American Society of Tropical Medicine and Hygiene (ASTMH) Annual Meeting by researchers from Colorado State University.
Ivermectin has been used for decades, given once per year as a part of Mass Drug Administration (MDA) programs, to reduce the disabling worm infections onchocerciasis, which causes river blindness, and filariasis, the cause of the hugely swollen legs (elephantiasis). Merck has generously donated the entire supply of drug; other companies have followed suit with different drugs for other neglected tropical diseases.
Ivermectin (22,23-dihydroavermectin B1a + 22,23-dihydroavermectin B1b) is a broad-spectrum antiparasitic drug in theavermectin family. It is sold under brand names Heartgard, Sklice[1] and Stromectol[2] in the United States, Ivomecworldwide by Merial Animal Health, Mectizan in Canada by Merck, Iver-DT[3] in Nepal by Alive Pharmaceutical and Ivextermin Mexico by Valeant Pharmaceuticals International. In Southeast Asian countries, it is marketed by Delta Pharma Ltd. under the trade name Scabo 6. While in development, it was assigned the code MK-933 by Merck.[4]
It is taken internally or used topically, depending on the treated condition.
It is on the World Health Organization’s List of Essential Medicines, a list of the most important medication needed in a basichealth system.[5]
It is a drug for the treatment of Onchocerciasis.
The disease is also known as river blindness. It is sometimes called Robles’ disease, after the Guatemalan doctor Rodolfo Robles, who first linked the blindness with an insect a century ago (1915).
Medical uses
Ivermectin is a broad-spectrum antiparasitic agent, traditionally against parasitic worms. It is mainly used in humans in the treatment of onchocerciasis (river blindness), but is also effective against other worm infestations (such as strongyloidiasis,ascariasis, trichuriasis, filariasis and enterobiasis), and some epidermal parasitic skin diseases, including scabies.
Ivermectin is currently being used to help eliminate river blindness (onchocerciasis) in the Americas, and to stop transmissionof lymphatic filariasis and onchocerciasis around the world in programs sponsored by the Carter Center using ivermectin donated by Merck.[6][7][8] The disease is endemic in 30 African countries, six Latin American countries, and Yemen, according to studies conducted by the World Health Organization.[9] The drug rapidly kills microfilariae, but not the adult worms. A single oral dose of ivermectin, taken annually for the 10- to 15-year lifespan of the adult worms, is all that is needed to protect the individual from onchocerciasis.[10]
An Ivermectin cream called Soolantra has been approved by the FDA for treatment of rosacea.[11][12]
SOOLANTRA (ivermectin) cream, 1% is a white to pale yellow hydrophilic cream. Each gram of SOOLANTRA cream contains 10 mg of ivermectin. It is intended for topical use.
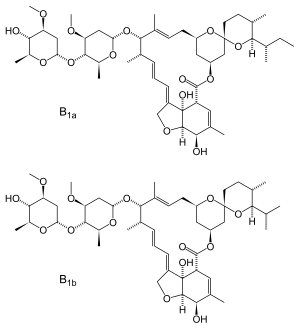
Ivermectin is a semi-synthetic derivative isolated from the fermentation of Streptomyces avermitilis that belongs to the avermectin family of macrocyclic lactones.
Ivermectin is a mixture containing not less than 95.0 % and not more than 102.0 % of 5-O-demethyl-22,23-dihydroavermectin A1a plus 5-O-demethyl-25-de(1-methylpropyl)-25-(1-methylethyl)-22,23-dihydroavermectin A1a, generally referred to as 22,23-dihydroavermectin B1a and B1b or H2B1a and H2B1b, respectively; and the ratio (calculated by area percentage) of component H2B1a/(H2B1a + H2B1b)) is not less than 90.0 %.
The respective empirical formulas of H2B1a and H2B1b are C48H74O14and C47H72O14 with molecular weights of 875.10 and 861.07 respectively.
The structural formulas are:
 |
Component H2B1a: R = C2H5, Component H2B1b: R = CH3.SOOLANTRA cream contains the following inactive ingredients: carbomer copolymer type B, cetyl alcohol, citric acid monohydrate, dimethicone, edetate disodium, glycerin, isopropyl palmitate, methylparaben, oleyl alcohol, phenoxyethanol, polyoxyl 20 cetostearyl ether, propylene glycol, propylparaben, purified water, sodium hydroxide, sorbitan monostearate, and stearyl alcohol.
River blindness?
The disease is also known as river blindness. It is sometimes called Robles’ disease, after the Guatemalan doctor Rodolfo Robles, who first linked the blindness with an insect a century ago (1915).
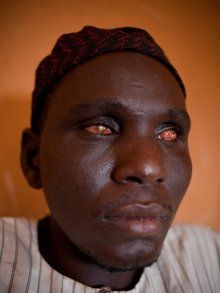
The infection is associated with a nematode worm Onchocerca volvulus, which are transmitted by Simulium blackflies which live and breed near fast-flowing streams and rivers. The worms carry parasitic Wolbachia bacteria. The bite of the flies enables the worm larvae to enter the human’s body; after maturing into adults, followed by breeding, the larvae (microfilariae) formed move towards the skin, and release the bacteria when they die. The bacteria trigger an immune response which leads to lesions on the eye and possible blindness (the “river blindness”).


Left: the Simulium fly (from http://flipper.diff.org/app/items/6730). Right: Simplified life cycle of Onchocerciasis volvulus, modified from the original at: http://emedicine.medscape.com/article/224309-overview#a0104.
Arthropod
More recent evidence supports its use against parasitic arthropods and insects:
- Mites such as scabies:[13][14][15] It is usually limited to cases that prove to be resistant to topical treatments or that present in an advanced state (such as Norwegian scabies).[15]
- Lice:[16][17] Ivermectin lotion (0.5%) is FDA-approved for patients six months of age and older.[18] After a single, 10-minute application of this formulation on dry hair, 78% of subjects were found to be free of lice after two weeks.[19] This level of effectiveness is equivalent to other pediculicide treatments requiring two applications.[20]
- Bed bugs:[21] Early research shows that the drug kills bed bugs when taken by humans at normal doses. The drug enters the human bloodstream and if the bedbugs bite during that time, they will die in a few days.
Contraindications
Ivermectin is contraindicated in children under the age of five, or those who weigh less than 15 kg (33 lb);[22] and those who are breastfeeding, and have a hepatic or renal disease.[23]
Side effects
The main concern is neurotoxicity, which in most mammalian species may manifest as central nervous system depression, and consequent ataxia, as might be expected from potentiation of inhibitory GABA-ergic synapses.
Dogs with defects in the P-glycoprotein gene (MDR1), often collie-like herding dogs, can be severely poisoned by ivermectin.
Since drugs that inhibit CYP3A4 enzymes often also inhibit P-glycoprotein transport, the risk of increased absorption past the blood-brain barrier exists when ivermectin is administered along with other CYP3A4 inhibitors. These drugs include statins, HIV protease inhibitors, many calcium channel blockers, and glucocorticoids such as dexamethasone, lidocaine, and the benzodiazepines.[24]
For dogs, the insecticide spinosad may have the effect of increasing the potency of ivermectin.[25]
Pharmacology
Pharmacodynamics
Ivermectin and other avermectins (insecticides most frequently used in home-use ant baits) are macrocyclic lactones derived from the bacterium Streptomyces avermitilis. Ivermectin kills by interfering with nervous system and muscle function, in particular by enhancing inhibitory neurotransmission.
The drug binds and activates glutamate-gated chloride channels (GluCls).[26] GluCls are invertebrate-specific members of the Cys-loop family of ligand-gated ion channelspresent in neurons and myocytes.
Pharmacokinetics
Ivermectin can be given either by mouth or injection. It does not readily cross the blood–brain barrier of mammals due to the presence of P-glycoprotein,[27] (the MDR1 gene mutation affects function of this protein). Crossing may still become significant if ivermectin is given at high doses (in which case, brain levels peak 2–5 hr after administration). In contrast to mammals, ivermectin can cross the blood–brain barrier in tortoises, often with fatal consequences.
Ecotoxicity
Field studies have demonstrated the dung of animals treated with ivermectin supports a significantly reduced diversity of invertebrates, and the dung persists longer.[28]
History
The discovery of the avermectin family of compounds, from which ivermectin is chemically derived, was made by Satoshi Ōmura of Kitasato University, Tokyo and William C. Campbell of the Merck Institute for Therapeutic research. Ōmura identified avermectin from the bacterium Streptomyces avermitilis. Campbell purified avermectin from cultures obtained from Ōmura and led efforts leading to the discovery of ivermectin, a derivative of greater potency and lower toxicity.[29] Ivermectin was introduced in 1981.[30] Half of the 2015 Nobel Prize in Physiology or Medicine was awarded jointly to Campbell and Ōmura for discovering avermectin, “the derivatives of which have radically lowered the incidence of river blindness and lymphatic filariasis, as well as showing efficacy against an expanding number of other parasitic diseases”.[31]
It started with the avermectins. In 1974, a group of researchers headed by Professor Satoshi Ōmura of the Kitasato Institute, isolated an organism with promising antimicrobial properties in a soil sample (sample OS-3153) picked up near a golf course at Kawana, Ito City, Shizuoka Prefecture, Japan. This was passed on to researchers at the Merck, Sharpe and Dohme (MSD) research laboratories in the USA, who isolated a small family of natural products that became known as avermectins. For many years, scientists have looked in soil samples for the source of potential medicines, like the tetracyclines or streptomycin . There are 8 avermectins, molecules with closely related structures. They are made by fermentation from the bacterium Streptomyces avermitilis.
 Professor Satoshi Ōmura
Professor Satoshi Ōmura

The 8 different avermectins, with the differences between them shown in the table below.
| Name | R1 | R2 | X-Y |
|---|---|---|---|
| Avermectin A1a | Me | Et | CH=CH |
| Avermectin A1b | Me | Me | CH=CH |
| Avermectin A2a | Me | Et | CH2CH(OH) |
| Avermectin A2b | Me | Me | CH2CH(OH) |
| Avermectin B1a | H | Et | CH=CH |
| Avermectin B1b | H | Me | CH=CH |
| Avermectin B2a | H | Et | CH2CH(OH) |
| Avermectin B2b | H | Me | CH2CH(OH) |
The avermectins proved to be have biocidal activity against a wide range of parasites – such as roundworms, lungworms, mites, lice and arachnids; one of these parasites is the tick Rhipicephalus (Boophilus) microplus, one of the most important cattle parasites in tropical regions. Those with the -CH=CH- function are the more active; the most potent was Avermectin B1, occurring as an 80:20 mixture of the similar molecules B1a and B1b, particularly the B1a component. Commercially it is known as Abamectin.
Veterinary use
In veterinary medicine ivermectin is used against many intestinal worms (but not tapeworms), most mites, and some lice. Despite this, it is not effective for eliminating ticks, flies, flukes, or fleas. It is effective against larval heartworms, but not against adult heartworms, though it may shorten their lives. The dose of the medicine must be very accurately measured as it is very toxic in over-dosage. It is sometimes administered in combination with other medications to treat a broad spectrum of animal parasites. Some dog breeds (especially the Rough Collie, the Smooth Collie, the Shetland Sheepdog, and the Australian Shepherd), though, have a high incidence of a certain mutation within the MDR1 gene (coding for P-glycoprotein); affected animals are particularly sensitive to the toxic effects of ivermectin.[32][33] Clinical evidence suggests kittens are susceptible to ivermectin toxicity.[34] A 0.01% ivermectin topical preparation for treating ear mites in cats (Acarexx) is available.
Ivermectin is sometimes used as an acaricide in reptiles, both by injection and as a diluted spray. While this works well in some cases, care must be taken, as several species of reptiles are very sensitive to ivermectin. Use in turtles is particularly contraindicated.
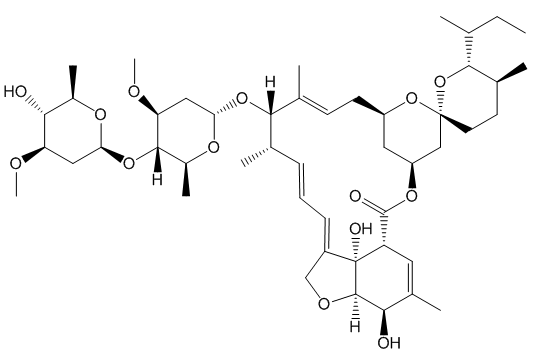 IVERMECTIN
IVERMECTIN
Chlorotris(triphenylphosphine)rhodium(I), [RhCl(PPh3)3]
http://www.chm.bris.ac.uk/motm/wilcat/wilcath.htm
Such selectivity found an important application in the synthesis of Ivermectin (MectizanTM). Avermectin is a naturally-occurring molecule with anthelmintic and insecticidal properties; selectively reducing one double bond using Wilkinson’s catalyst afforded Ivermectin. The resultant small change in molecular shape makes Ivermectin a much more effective drug to combat onchocerciasis (river blindness), a disease which affects many millions of people, mainly in poor African communities.

You need to add just two hydrogen atoms to reduce a C=C bond in avermectin.
Notes and references
- “SKLICE- ivermectin lotion (NDC Code(s): 49281-183-71)”. DailyMed. February 2012. Retrieved 2015-09-09.
- “STROMECTOL- ivermectin tablet (NDC Code(s): 0006-0032-20)”. DailyMed. May 2010. Retrieved 2015-09-09.
- Adhikari, Santosh (2014-05-27). “ALIVE PHARMACEUTICAL (P) LTD.: Iver-DT”. ALIVE PHARMACEUTICAL (P) LTD. Retrieved 2015-10-07.
- Pampiglione S, Majori G, Petrangeli G, Romi R (1985). “Avermectins, MK-933 and MK-936, for mosquito control”. Trans R Soc Trop Med Hyg 79 (6): 797–9. doi:10.1016/0035-9203(85)90121-X. PMID 3832491.
- “WHO Model List of Essential Medicines” (PDF). World Health Organization. October 2013. Retrieved 22 April 2014.
- The Carter Center. “River Blindness (Onchocerciasis) Program”. Retrieved2008-07-17..
- The Carter Center. “Lymphatic Filariasis Elimination Program”. Retrieved 2008-07-17..
- WHO. “African Programme for Onchocerciasis Control”. Retrieved 2009-11-12..
- United Front Against Riverblindness. “Onchocerciasis or Riverblindness”..
- United Front Against Riverblindness. “Control of Riverblindness”..
- Galderma Receives FDA Approval of Soolantra (Ivermectin) Cream for Rosacea“
- “SOOLANTRA- ivermectin cream (NDC Code(s): 0299-3823-30, 0299-3823-45, 0299-3823-60)”. DailyMed. December 2014. Retrieved 2015-09-09.
- Brooks PA, Grace RF (August 2002). “Ivermectin is better than benzyl benzoate for childhood scabies in developing countries”. J Paediatr Child Health 38 (4): 401–4.doi:10.1046/j.1440-1754.2002.00015.x. PMID 12174005.
- Victoria J, Trujillo R (2001). “Topical ivermectin: a new successful treatment for scabies”. Pediatr Dermatol 18 (1): 63–5. doi:10.1046/j.1525-1470.2001.018001063.x.PMID 11207977.
- ^ Jump up to:a b Strong M, Johnstone PW (2007). Strong, Mark, ed. “Interventions for treating scabies”. Cochrane Database of Systematic Reviews (Online) (3): CD000320.doi:10.1002/14651858.CD000320.pub2. PMID 17636630.
- Dourmishev AL, Dourmishev LA, Schwartz RA (December 2005). “Ivermectin: pharmacology and application in dermatology”. International Journal of Dermatology 44(12): 981–8. doi:10.1111/j.1365-4632.2004.02253.x. PMID 16409259.
- Strycharz JP, Yoon KS, Clark JM (January 2008). “A new ivermectin formulation topically kills permethrin-resistant human head lice (Anoplura: Pediculidae)”. Journal of Medical Entomology 45 (1): 75–81. doi:10.1603/0022-2585(2008)45[75:ANIFTK]2.0.CO;2.ISSN 0022-2585. PMID 18283945.
- “Sklice lotion”.
- David M. Pariser, M.D., Terri Lynn Meinking, Ph.D., Margie Bell, M.S., and William G. Ryan, B.V.Sc. (November 1, 2012). “Topical 0.5% Ivermectin Lotion for Treatment of Head Lice”. New England Journal of Medicine 367: 1687–1693.doi:10.1056/NEJMoa1200107.
- Study shows ivermectin ending lice problem in one treatment, Los Angeles Times, Nov 5, 2012
- DONALD G. MCNEIL JR. (2012-12-31). “Pill Could Join Arsenal Against Bedbugs”. The New York Times. Retrieved 2013-04-05.
- Jump up^ Dourmishev AL, Dourmishev LA, Schwartz RA (December 2005). “Ivermectin: pharmacology and application in dermatology”. International Journal of Dermatology 44(12): 981–988. doi:10.1111/j.1365-4632.2004.02253.x. PMID 16409259.
- Huukelbach J, Winter B, Wilcke T, et al. (August 2004). “Tratmient masivo selectivo con ivermectina contra las helmintiasis intestinales y parasitos cutáneas en una población gravemente afectada”. Bull World Health Organ 82 (7): 563–571. doi:10.1590/S0042-96862004000800005.
- Goodman and Gilman’s Pharmacological Basis of Therapeutics, 11th edition, pages 122, 1084-1087.
- Jump up^ “COMFORTIS® and ivermectin interaction Safety Warning Notification”. U.S. Food and Drug Administration (FDA) Center for Veterinary Medicine (CVM).
- Yates DM, Wolstenholme AJ (August 2004). “An ivermectin-sensitive glutamate-gated chloride channel subunit from Dirofilaria immitis”. Int. J. Parasitol. 34 (9): 1075–81.doi:10.1016/j.ijpara.2004.04.010. PMID 15313134.
- Borst P, Schinkel AH (June 1996). “What have we learnt thus far from mice with disrupted P-glycoprotein genes?”. European Journal of Cancer 32 (6): 985–990.doi:10.1016/0959-8049(96)00063-9.
- Iglesias LE, Saumell CA, Fernández AS, et al. (December 2006). “Environmental impact of ivermectin excreted by cattle treated in autumn on dung fauna and degradation of faeces on pasture”. Parasitology Research 100 (1): 93–102. doi:10.1007/s00436-006-0240-x. PMID 16821034.
- Fisher MH, Mrozik H (1992). “The chemistry and pharmacology of avermectins”. Annu. Rev. Pharmacol. Toxicol. 32: 537–53. doi:10.1146/annurev.pa.32.040192.002541.PMID 1605577.
- W. C. CAMPBELL; R. W. BURG, , M. H. FISHER, and , R. A. DYBAS (June 26, 1984).“The Discovery of Ivermectin and Other Avermectins”. American Chemical Society. pp. 5–20. ISBN 9780841210837.
|chapter=ignored (help) - “The Nobel Prize in Physiology or Medicine 2015” (PDF). Nobel Foundation. Retrieved7 October 2015.
- “MDR1 FAQs”, Australian Shepherd Health & Genetics Institute, Inc.
- “Multidrug Sensitivity in Dogs”, Washington State University’s College of Veterinary Medicine
- Frischke H, Hunt L (April 1991). “Suspected ivermectin toxicity”. Canadian Veterinary Journal 32 (4): 245. PMC 1481314. PMID 17423775.
External links
- Stromectol
- The Carter Center River Blindness (Onchocerciasis) Control Program
- Mectizan Donation Program
- American NGDO Treating River Blindness
- MERCK. 25 Years: The MECTIZAN® Donation Program
- Trinity College Dublin. Prof William Campbell – The Story of Ivermectin
- “IVERMECTIN- ivermectin tablet (NDC Code(s): 42799-806-01)”. DailyMed. November 2014. Retrieved 2015-09-09
Bibliography
- Chapman and Hall Combined Chemical Dictionary compound code number CMD99-Y (Ivermectin); CLF13-X (Avermectin).
- The World Health Organisation page on Onchocerciasis
- “Organic Chemists Fighting Blindness” – the Mectizan story from the RSC Organic Division
- A. Crump and K. Otoguro, Trends in Parasitology, 2005, 21, 126-132. (Ōmura’s work)
- Nobel committee’s announcement.
Avermectin
- R. W. Burg, B. M. Miller, E. E. Baker, J. Birnbaum, S. A. Currie, R. Hartman, Y.-L. Kong, R. L. Monaghan, G. Olson, I. Putter, J. B. Tunac, H. Wallick, E. O. Stapley, R. Oiwa, and S. Ōmura, Antimicrob. Agents Chemother., 1979, 15, 361-367 (production of avermectins)
- T. W. Miller, L. Chaiet, D. J. Cole, L. J. Cole, J. E. Flor, R. T. Goegelman, V. P. Gullo, H. Joshua, A. J. Kempf, W. R. Krellwitz, R. L. Monaghan, R. E. Ormond, K. E. Wilson, G. Albers-Schönberg and I. Putter., Antimicrob. Agents Chemother., 1979, 15, 368-371 (isolation of avermectins)
- J. R. Egerton, D. A. Ostlind, L. S. Blair, C. H. Eary, D. Suhayda, S. Cifelli, R. F. Riek and W. C. Campbell, Antimicrob. Agents Chemother., 1979, 15, 372-378 (efficacy of avermectins)
- M. H. Fisher, Pure Appl. Chem., 1990, 62, 1231-1240 (avermectin review)
- Y. J. Yoon, E.-S. Kim, Y.-S. Hwang and C.-Y. Choi, Appl. Microbiol. Biotechnol., 2004, 63, 626–634 (biosynthesis)
Ivermectin
- J. C. Chabala, H. Mrozik, R. L. Tolman, P. Eskola, A. Lusi, L. H. Peterson, M. F. Woods, M. H. Fisher and W. C. Campbell, J. Med. Chem., 1980, 23, 1134-1136 (synth)
- W. C. Campbell, M. H. Fisher, E. O. Stapley, G. Albers-Schönberg and T. A. Jacob, Science, 1983, 221, 823–828 (ivermectin as a new antiparasitic agent)
- S. Ōmura and A. Crump, Nat. Rev. Microbiol., 2004, 2, 984-989. (“The life and times of ivermectin – a success story”).
- K. Collins, Perspect. Biol. Med., 2004, 47, 100-109. (History of the Merck Mectizan donation program)
- A. D. Hopkins, Eye, 2005, 19, 1057–1066 (improvements upon ivermectin treatment)
- T. G. Geary, Trends in Parasitology, 2005, 21, 530–532 (20 years of ivermectin)
- S. Ōmura, Int. J. Antimicrob. Ag., 2008, 31, 91–98 (25 years of Ivermectin)
- A. G. Canga, A. M. S. Prieto, M. J. D. Liébana, N. F. Martínez, M. S.Vega and J. J. G. Vieitez, Vet. J., 2009, 179, 25–37 (pharmacokinetics and metabolism of ivermectin in domestic animal species)
- A. Crump and S. Ōmura, Proc. Jpn. Acad., Ser. B., 2011, 87, 13-28 (ivermectin review)
- I. Farrell, Education in Chemistry, November 2013. (“One in the eye for river blindness”), online.
- The Mectizan donation program
- Colombia eliminates river blindness
Doramectin
- K. Stutzman-Engwall, S. Conlon, R. Fedechko, H. McArthur, K. Pekrun, Y. Chen, S. Jenne, C. La, N. Trinh, S. Kim, Y.-X. Zhang, R. Fox, C. Gustafsson and A. Krebber, Metabolic Engineering, 2005, 7, 27–37 (synth.)
- J.-B. Wang, H.-X. Pan and G.-L. Tang, Bioorg. Med. Chem. Lett., 2011, 21, 3320–3323 (synth.)
|
|
| Systematic (IUPAC) name | |
|---|---|
|
22,23-dihydroavermectin B1a + 22,23-dihydroavermectin B1b
|
|
| Clinical data | |
| Trade names | Stromectol |
| AHFS/Drugs.com | monograph |
| MedlinePlus | a607069 |
| Pregnancy category |
|
| Legal status |
|
| Routes of administration |
Oral, topical |
| Pharmacokinetic data | |
| Protein binding | 93% |
| Metabolism | Liver (CYP450) |
| Biological half-life | 18 hours |
| Excretion | Feces; <1% urine |
| Identifiers | |
| CAS Registry Number | 70288-86-7 71827-03-7 |
| ATC code | D11AX22 P02CF01 QP54AA01QS02QA03 |
| PubChem | CID: 9812710 |
| DrugBank | DB00602 |
| ChemSpider | 7988461 |
| UNII | 8883YP2R6D |
| KEGG | D00804 |
| ChEMBL | CHEMBL341047  |
| PDB ligand ID | IVM (PDBe, RCSB PDB) |
| Chemical data | |
| Formula | C 48H 74O 14 (22,23-dihydroavermectin B1a) C 47H 72O 14 (22,23-dihydroavermectin B1b) |
| Molecular mass | 875.10 g/mol |
SIMILAR
Doramectin is a similar molecule, used to treat parasites in animals, such as cattle, horses, sheep and pigs.

/////////////////ivermectin, MALARIA
Filed under: Malaria Tagged: ivermectin, Malaria

















































 Lucy Shapiro is a Professor in the Department of Developmental Biology at Stanford University School of Medicine where she holds the Virginia and D. K. Ludwig Chair in Cancer Research and is the Director of the Beckman Center for Molecular and Genetic Medicine. She is a member of the Scientific Advisory Board of Ludwig Institute for Cancer Research and is a member of the Board of Directors of Pacific Biosciences, Inc. She founded the anti-infectives discovery company, Anacor Pharmaceuticals, and is a member of the Anacor Board of Directors. Professor Shapiro has been the recipient of multiple honors, including: election to the American Academy of Arts and Sciences, the US National Academy of Sciences, the US Institute of Medicine, the American Academy of Microbiology, and the American Philosophical Society. She was awarded the FASEB Excellence in Science Award, the 2005 Selman Waksman Award from the National Academy of Sciences, the Canadian International 2009 Gairdner Award, the 2009 John Scott Award, the 2010 Abbott Lifetime Achievement Award, the 2012 Horwitz Prize and President Obama awarded her the National Medal of Science in 2012. Her studies of the control of the bacterial cell cycle and the establishment of cell fate has yielded valuable paradigms for understanding the bacterial cell as an integrated system in which the transcriptional circuitry is interwoven with the three-dimensional deployment of key regulatory and morphological proteins, adding a spatial dimension to the systems biology of regulatory networks.
Lucy Shapiro is a Professor in the Department of Developmental Biology at Stanford University School of Medicine where she holds the Virginia and D. K. Ludwig Chair in Cancer Research and is the Director of the Beckman Center for Molecular and Genetic Medicine. She is a member of the Scientific Advisory Board of Ludwig Institute for Cancer Research and is a member of the Board of Directors of Pacific Biosciences, Inc. She founded the anti-infectives discovery company, Anacor Pharmaceuticals, and is a member of the Anacor Board of Directors. Professor Shapiro has been the recipient of multiple honors, including: election to the American Academy of Arts and Sciences, the US National Academy of Sciences, the US Institute of Medicine, the American Academy of Microbiology, and the American Philosophical Society. She was awarded the FASEB Excellence in Science Award, the 2005 Selman Waksman Award from the National Academy of Sciences, the Canadian International 2009 Gairdner Award, the 2009 John Scott Award, the 2010 Abbott Lifetime Achievement Award, the 2012 Horwitz Prize and President Obama awarded her the National Medal of Science in 2012. Her studies of the control of the bacterial cell cycle and the establishment of cell fate has yielded valuable paradigms for understanding the bacterial cell as an integrated system in which the transcriptional circuitry is interwoven with the three-dimensional deployment of key regulatory and morphological proteins, adding a spatial dimension to the systems biology of regulatory networks.





















































 Originally posted on
Originally posted on 
































































 .
.


![[1860-5397-7-5-i1]](http://beilstein-journals.org/bjoc/content/inline/1860-5397-7-5-i1.png?max-width=550&background=FFFFFF)