LIK-066, a new flozin on the horizon
C23 H28 O7 . 2 C6 H11 N O, 642.7795, 1 :2 co-crystal of Example 62 : L-proline. A melting point 176°C…WO2011048112
CAS 1291095-45-8, (1S)-1,5-anhydro-1-C-[3-[(2,3-dihydro-1,4-benzodioxin-6-yl)methyl]-4-ethylphenyl]-D-glucitol (1:1) WITH L-Proline, compd., 1:1 Proline Co-crvstal , 1:1 Proline Co-crvstal …..…WO2011048112
CAS BASE 1291094-73-9, 416.46, C23 H28 O7
(1S)-1,5-Anhydro-1-[3-(2,3-dihydro-1,4-benzodioxin-6-ylmethyl)-4-ethylphenyl]-D-glucitol bis[1-[(2S)-pyrrolidin-2-yl]ethanone]
(2S,3R,4R,5S,6R)-2-[3-(2,3-Dihydro-benzo[1,4]dioxin-6-ylmethyl)-4- ethyl-phenyl]-6-hydroxymethyl-tetrahydro-pyran-3,4,5-triol
Sodium glucose transporter-2 inhibitor
SGLT 1/2 inhibitor
Novartis Ag innovator
Clinical trial……..https://clinicaltrials.gov/ct2/show/NCT01915849
https://clinicaltrials.gov/ct2/show/NCT02470403
- 10 Jun 2015 Novartis initiates enrolment in a phase II trial for Type 2 diabetes mellitus in USA (NCT02470403)
- 02 Apr 2014 Novartis terminates a phase II trial in Type-2 diabetes mellitus in USA, Poland, Argentina, Hungary, Puerto Rico and South Africa (NCT01824264)
- 01 Jan 2014 Novartis completes a phase II trial in Type 2 diabetes mellitus in USA (NCT01915849)
SEE ALSO



LIK-066 is in phase II clinical studies at Novartis for the treatment of type 2 diabetes.
In June 2014, the EMA’s PDCO adopted a positive opinion on a pediatric investigation plan (PIP) for LIK-066 for type 2 diabetes
Diabetes mellitus is a metabolic disorder characterized by recurrent or persistent hyperglycemia (high blood glucose) and other signs, as distinct from a single disease or condition. Glucose level abnormalities can result in serious long-term complications, which include cardiovascular disease, chronic renal failure, retinal damage, nerve damage (of several kinds), microvascular damage and obesity.
Type 1 diabetes, also known as Insulin Dependent Diabetes Mellitus (IDDM), is characterized by loss of the insulin-producing β-cells of the islets of Langerhans of the pancreas leading to a deficiency of insulin. Type-2 diabetes previously known as adult- onset diabetes, maturity-onset diabetes, or Non-Insulin Dependent Diabetes Mellitus (NIDDM) – is due to a combination of increased hepatic glucose output, defective insulin secretion, and insulin resistance or reduced insulin sensitivity (defective responsiveness of tissues to insulin). Chronic hyperglycemia can also lead to onset or progression of glucose toxicity characterized by decrease in insulin secretion from β-cell, insulin sensitivity; as a result diabetes mellitus is self-exacerbated [Diabetes Care, 1990, 13, 610].
Chronic elevation of blood glucose level also leads to damage of blood vessels. In diabetes, the resultant problems are grouped under “microvascular disease” (due to damage of small blood vessels) and “macro vascular disease” (due to damage of the arteries). Examples of microvascular disease include diabetic retinopathy, neuropathy and nephropathy, while examples of macrovascular disease include coronary artery disease, stroke, peripheral vascular disease, and diabetic myonecrosis.
Diabetic retinopathy, characterized by the growth of weakened blood vessels in the retina as well as macular edema (swelling of the macula), can lead to severe vision loss or blindness. Retinal damage (from microangiopathy) makes it the most common cause of blindness among non-elderly adults in the US. Diabetic neuropathy is characterized by compromised nerve function in the lower extremities. When combined with damaged blood vessels, diabetic neuropathy can lead to diabetic foot. Other forms of diabetic neuropathy may present as mononeuritis or autonomic neuropathy. Diabetic nephropathy is characterized by damage to the kidney, which can lead to chronic renal failure, eventually requiring dialysis. Diabetes mellitus is the most common cause of l adult kidney failure worldwide. A high glycemic diet (i.e., a diet that consists of meals that give high postprandial blood sugar) is known to be one of the causative factors contributing to the development of obesity.
Type 2 diabetes is characterized by insulin resistance and/or inadequate insulin secretion in response to elevated glucose level. Therapies for type 2 diabetes are targeted towards increasing insulin sensitivity (such as TZDs), hepatic glucose utilization (such as biguanides), directly modifying insulin levels (such as insulin, insulin analogs, and insulin secretagogues), increasing increttn hormone action (such as exenatide and sitagliptin), or inhibiting glucose absorption from the diet (such as alpha glucosidase inhibitors) [Nature 2001 , 414, 821-827],
Glucose is unable to diffuse across the cell membrane and requires transport proteins. The transport of glucose into epithelial cells is mediated by a secondary active cotransport system, the sodium-D-glucose co-transporter (SGLT), driven by a sodium- gradient generated by the Na+/K+-ATPase. Glucose accumulated in the epithelial cell is further transported into the blood across the membrane by facilitated diffusion through GLUT transporters [Kidney International 2007, 72, S27-S35].
SGLT belongs to the sodium/glucose co-transporter family SLCA5. Two different SGLT isoforms, SGLT1 and SGLT2, have been identified to mediate renal tubular glucose reabsorption in humans [Curr. Opinon in Investigational Drugs (2007): 8(4), 285-292 and references cited herein]. Both of them are characterized by their different substrate affinity. Although both of them show 59% homology in their amino acid sequence, they are functionally different. SGLT1 transports glucose as well as galactose, and is expressed both in the kidney and in the intestine, while SGLT2 is found exclusively in the S1 and S2 segments of the renal proximal tubule.
As a consequence, glucose filtered in the glomerulus is reabsorbed into the renal proximal tubular epithelial cells by SGLT2, a low-affinity/high-capacity system, residing on the surface of epithelial cell lining in S1 and S2 tubular segments. Much smaller amounts of glucose are recovered by SGLT1 , as a high-affinity/low-capacity system, on the more distal segment of the proximal tubule. In healthy human, more than 99% of plasma glucose that is filtered in the kidney glomerulus is reabsorbed, resulting in less than 1 % of the total filtered glucose being excreted in urine. It is estimated that 90% of total renal glucose absorption is facilitated by SGLT2; remaining 10 % is likely mediated by SGLT1 [J. Parenter. Enteral Nutr. 2004, 28, 364-371].
SGLT2 was cloned as a candidate sodium glucose co-transporter, and its tissue distribution, substrate specificity, and affinities are reportedly very similar to those of the low-affinity sodium glucose co-transporter in the renal proximal tubule. A drug with a mode of action of SGLT2 inhibition will be a novel and complementary approach to existing classes of medication for diabetes and its associated diseases to meet the patient’s needs for both blood glucose control, while preserving insulin secretion. In addition, SGLT2 inhibitors which lead to loss of excess glucose (and thereby excess calories) may have additional potential for the treatment of obesity.
Indeed small molecule SGLT2 inhibitors have been discovered and the anti-diabetic therapeutic potential of such molecules has been reported in literature [T-1095 (Diabetes, 1999, 48, 1794-1800, Dapagliflozin (Diabetes, 2008, 57, 1723-1729)].
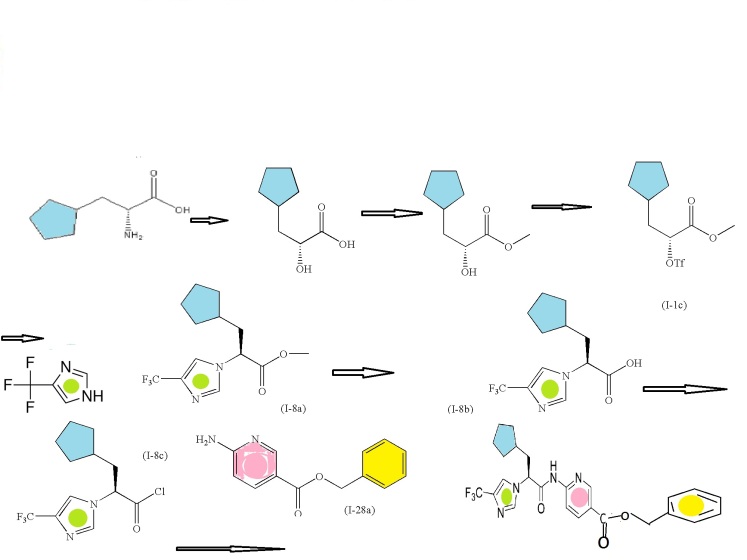
SYNTHESIS


PATENT
WO 2011048112
https://www.google.com/patents/WO2011048112A1?cl=en
Gregory Raymond Bebernitz, Mark G. Bock, Dumbala Srinivas Reddy, Atul Kashinath Hajare, Vinod Vyavahare, Sandeep Bhausaheb Bhosale, Suresh Eknath Kurhade, Videsh Salunkhe, Nadim S. Shaikh, Debnath Bhuniya, P. Venkata Palle, Lili Feng, Jessica Liang,
Example 61-62:

Ex. 61
Example 61 : Acetic acid (2R,3R,4R,5S)-3,4,5-triacetoxy-6-[3-(2,3-dihydro- benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl-phenyl]-tetrahydro-pyran-2-ylmethyl ester
Step I: To a stirred solution of acetic acid (2R,3R,4R,5S)-3,4,5-triacetoxy-6-[4-bromo-3- (2,3-dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-phenyl]-tetrahydro-pyran-2-ylmethyl ester (10.0 g, 15.74 mmol) in toluene (200 mL) was added tricyclohexylphosphine (1.76 g, 6.29 mmol), a solution of potassium phosphate tribasic (13.3 g, 62.9 mmol) in water (15 mL), and ethylboronic acid (3.4 g, 47.2 mmol). The reaction mixture was degassed for 45 min then palladium (II) acetate (529 mg, 2.3 mmol) was added. After refluxing overnight, the reaction mixture was cooled to room temperature, and water was added. The resulting mixture was extracted with ethyl acetate, (2 X 200 mL), washed with water and brine, then dried over sodium sulfate, concentrated and purified by column chromatography to furnish acetic acid (2R,3R,4R,5S)-3,4,5-triacetoxy-6-[3-(2,3-dihydro- benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl-phenyl]-tetrahydro-pyran-2-ylmethyl ester (5.4 g).
Example 62: (2S,3R,4R,5S,6R)-2-[3-(2,3-Dihydro-benzo[1,4]dioxin-6-ylmethyl)-4- ethyl-phenyl]-6-hydroxymethyl-tetrahydro-pyran-3,4,5-triol
Step II: To a stirred solution of acetic acid (2R,3R,4R,5S)-3,4,5-triacetoxy-6-[3-(2,3- dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl-phenyl]-tetrahydro-pyran-2-ylmethyl ester (9.3 g, 15.9 mmol) in methanol:THF:water 3:2:1 (170 mL) was added lithium hydroxide (764 mg, 19.1 mmol). After stirring for 2 h at room temperature, the volatiles were evaporated under reduced pressure. The resulting residue was taken up in ethyl acetate (150 mL) and washed with brine (75 mL), brine containing 5 mL of 5% aqueous KHS04 (75 mL), and brine (20 mL) again, then dried over sodium sulfate and concentrated to furnish (2S,3R,4R,5S,6R)-2-[4-Cyclopropyl-3-(2,3-dihydro- benzo[1 ,4]dioxin-6-ylmethyl)-phenyl]-6-hydroxymethyl-tetrahydro-pyran-3,4,5-triol (6.59)
H NMR (400 MHz, CD3OD): δ 1.07 (t, J = 7.6 Hz, 3H), 2.57 (q, J = 7.6 Hz, 2H), 3.34- 3.50 (m, 4H), 3.68 (dd, J = 12.0, 5.6 Hz, 1 H), 3.85-3.91 (m, 3H), 4.08 (d, J = 9.6 Hz, 1 H), 4.17 (s, 4H), 6.53-6.58 (m, 2H), 6.68 (d, J – 8.4 Hz, 1 H), 7.15-7.25 (m, 3H).
MS (ES) m z 434.2 (M+18).
PICK UP IDEAS FROM HERE
Examples 57-58:

Ex. 57 Ex. 58
Step I: To a stirred solution of 2-bromo-5-iodobenzoic acid (25.0 g, 76.48 mmol) in dichloromethane (200 mL) was added oxalyl chloride (10.3 mL, 114.74 mmol) at 0 °C followed by D F (0.9 mL). After complete addition, the reaction mixture was stirred at room temperature for 3h. Volatiles were evaporated under reduced pressure to furnish 2-bromo-5-iodo-benzoyl chloride (26.4 g). The crude product was used for the next step immediately.
Step II: To a stirred solution of 2-bromo-5-iodo-benzoyl chloride (26.4 g, 76.56 mmol) in dichloromethane (250 mL) was added benzo(1 ,4)-dioxane (10.41 g, 76.26 mmol) at 0 °C. To this reaction mixture, AICI3 (40.78 g, 305.47 mmol) was added in portions. After stirring overnight at room temperature, the reaction mixture was poured into crushed ice. The resulting mixture was extracted with dichloromethane (500 mL X 2). The dichloromethane layers were combined and washed with water (200 mL), saturated aqueous sodium bicarbonate solution (200 mL X 2), and brine (200 mL), then dried over sodium sulfate and concentrated. The solid product was triturated with hexanes, and the triturated product was dried under vacuum to furnish (2-bromo-5-iodo-phenyl)-(2,3- dihydro-benzo[1 ,4]dioxin-6-yl)-methanone (30 g).
1H N R (400 MHz, DMSO-D6): δ 4.29-4.37 (m, 4H), 7.02 (d, J = 8.4 Hz, 1 H), 7.16 (d, J = 2.4 Hz, 1 H), 7.18-7.19 (m, 1 H), 7.53 (d, J = 8.4 Hz, 1 H), 7.77-7.81 (m, 1 H), 7.82 (d, J = 2.0 Hz, 1 H).
Step III: To a stirred solution of (2-bromo-5-iodo-phenyl)-(2,3-dihydro-benzo[1 ,4]dioxin- 6-yl)-methanone (30.0 g, 67.4 mmol) in trifluoroacetic acid (100 mL) was added triethylsilane (86.2 mL, 539.3 mmol) followed by triflic acid (6.0 mL, 67.42 mmol ) at room temperature. After stirring for 25 min at room temperature, volatiles were evaporated under reduced pressure. The resulting residue was taken up in ethyl acetate and washed with saturated aqueous sodium bicarbonate solution (200 mL X 2), water (200 mL), and brine (200 mL), then dried over sodium sulfate, concentrated and purified by silica gel column chromatography to furnish 6-(2-bromo-5-iodo-benzyl)-2,3- dihydro-benzo[1 ,4]dioxine (26.5 g). H NMR (400 MHz, DMSO-D6): δ 3.90 (s, 4H), 4.2 (s, 2H), 6.65 (dd, J = 8.4 Hz, J = 2.0 Hz, H), 6.68 (d, J = 2.0 Hz, 1 H), 6.77 (d, J = 8.4 Hz, H), 7.39 (d, J = 8.4 Hz, 1 H), 7.50 (dd, J = 8.4 Hz, J = 2.4 Hz 1 H), 7.67 (d, J = 2.8 Hz, 1 H).
Step IV: To a stirred solution of 6-(2-bromo-5-iodo-benzyl)-2,3-dihydro- benzo[1 ,4]dioxine (26.5 g, 61.47 mmol) in THF:toluene 2:1 (300 mL) was added 1.6 M solution of n-BuLi in hexanes (42.3 mL, 67.62 mmol) at -78 °C. The reaction mixture was stirred for 1 h, and then transferred to a stirred solution of 2,3,4,6-tetrakis-O- (trimethylsilyl)-D-glucopyranone (28.69 g, 61.47 mmol) in toluene (100 mL) at -78 °C. After stirring for 1 h, 0.6 N methanesulfonic acid in methanol (265 mL) was added dropwise and stirred the reaction mixture for 16 h at room temperature. Reaction was quenched by the addition of aq. NaHC03 solution (~75 mL) and extracted with ethyl acetate (250 mL X 3), dried over sodium sulfate, concentrated and purified by silica gel column chromatography to furnish (3R,4S,5S,6R)-2-[4-Bromo-3-(2,3-dihydro- benzo[1 ,4]dioxin-6-ylmethyl)-phenyl]-6-hydroxymethyl-2-methoxy-tetrahydro-pyran- 3,4,5-triol (28.4 g)
Example 57: [(2R,3R,4R,5S,6S)-3,4,5-triacetoxy-6-[4-bromo-3-(2,3-dihydro-1 ,4- benzodioxin-6-ylmethyl)phenyl]tetrahydropyran-2-yl]methyl acetate
Step V: To a stirred solution of (3R,4S,5S,6R)-2-[4-bromo-3-(2,3-dihydro- benzo[1 ,4]dioxin-6-ylmethyl)-phenyl]-6-hydroxymethyl-2-methoxy-tetrahydro-pyran-3,4,5- triol (28.4 g, 57.1 mmol) in acetonitrile-dichloromethane 1 :1 (250 mL) was added triethylsilane (36.5 mL, 228.4 mmol) and boron trifluoride diethyletharate complex (14.1 mL, 114.2 mmol) at 10 °C. After stirring for 4 h at 10°C, the reaction was quenched with saturated aqueous sodium bicarbonate (~ 100 mL). The organic layer was separated, and the aqueous layer was extracted with ethyl acetate (3 X 150 mL). The organic layers were combined and dried over sodium sulfate, concentrated to furnish (3R,4R,5S,6R)-2- [4-bromo-3-(2,3-dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-phenyl]-6-hydroxymethyl- tetrahydro-pyran-3,4,5-triol (28.4 g). Crude product was used for next reaction without purification. Example 58: [(2R,3R,4R,5S,6S)-3,4,5-triacetoxy-6-[4-bromo-3-(2!3-dihydro-1,4- benzodioxin-6-ylmethyl)phenyl]tetrahydropyran-2-yl]methyl acetate Step V: To a stirred solution of (3R,4R,5S,6R)-2-[4-Bromo-3-(2,3-dihydro- benzo[ 1 ,4]dioxin-6-yl methyl)-phenyl]-6-hydroxymethyl-tetrahyd ro-pyran-3,4 , 5-triol (28.4 g, 60.81 mmol) in dichloromethane (300 mL) was added pyridine (40 mL, 486.5 mmol), acetic anhydride (50 mL, 486.5 mmol) and DMAP (740 mg, 6.08 mmol) at room temperature. After stirring for 2 h, volatiles were evaporated under reduced pressure. The resulting residue was taken up in ethyl acetate (500ml) and washed with 1 N HCI (200 mL X 2) followed by brine (200ml), then dried over sodium sulfate and
concentrated. The resulting crude compound was dissolved in ethanol (320 mL) at 65 °C and allowed to cool to room temperature while stirring. Light yellow solid formed was filtered and washed with cold ethanol (150 mL) followed by hexane (200 mL) to get acetic acid (2R,3R,4R,5S)-3,4,5-triacetoxy-6-[4-bromo-3-(2,3-dihydro-benzo[1 ,4]dioxin- 6-ylmethyl)-phenyl]-tetrahydro-pyran-2-ylmethyl ester powder (22.5 g, purity 98%).
COCRYSTAL
Example 75: 1:1 Proline Co-crvstal with f2S.3R.4R.5S.6R¾-2-r3-f2.3-Dihvdro- benzori.41dioxin-6-ylmethyl)-4-ethyl-phenvn-6-hvdroxymethyl-tetrahydro-pyran- 3.4.5-triol
(2S,3R,4R,5S,6R)-2-[3-(2,3-Dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl- phenyl]-6-hydroxymethyl-tetrahydro-pyran-3,4,5-triol (Example 62) was completely amorphous initially but formed a crystalline complex with proline. This was confirmed by powder X-ray diffraction (PXRD) analysis. The stiochiometry of Example 62 and L- proline in the co-crystal prepared by method 1 was found to be 1 :1 by NMR
spectroscopy & HPLC. Characterization data for co-crystals of Example 62 and proline prepared by method 1 is shown in Table 3. Relative intensities of the most prominent powder x-ray diffraction peaks for co-crystals of Example 62 and proline are shown in Table 3A.
Table 3

Table 3A

3.70 15.78 18.36 25.18
9.68 10.68 18.88 36.33
11.07 21.21 20.42 69.29
14.26 14.81 21.18 27.94
14.80 22.97 22.50 12.25
15.40 4 98 23.78 33.08
16.12 8.45 24.56 6.92
16.59 18.78 25.79 21.69
17.31 100.0 27.46 8.90
17.60 20.35 31.97 7.65
17.98 47.20 32.46 5.98
1:1 Proline Co-crvstal
Example 77: 1:1 Proline Co-crvstal with (2S.3R.4R.5S.6Ri-2-f3-(2.3-Dihvdro- benzoh .41dioxin-6-ylmethvh-4-ethyl-phenvn-6-hvdroxymethyl-tetrahvdro-pyran- 3.4.5-triol
Method 2:
1 :1 Co-Crvstals of Example 62 with L-Proline
(2S,3R,4R,5S,6R)-2-[3-(2,3-Dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl-phenyl]- 6-hydroxymethyl-tetrahydro-pyran-3,4,5-triol (Example 62, 1500mg,3.6mmol), L- proline (415mg, 3.6mmol) and ethanol (23 ml_) were added to a 50 mL 3-neck round bottom flask equipped with nitrogen purging, magnetic stirring bar,
thermometer pocket & calcium chloride guard tube and the mixture was stirred at 25-30°C for 30 min., then heat to reflux. A clear solution was observed which was refluxed for 30 min., then slowly cool to 25-30°C causing percipitation. Di- isopropyl ether (DIPE, 23 mL) was added while maintaining the mixture at 25-30°C and stirring continuously for additional one to two hours at the same temperature. The precipitate was collected by filtration using vacuum (Nitrogen atmosphere), and the filter cake was washed with ethanol-DIPE mixture (1 :1 v/v, 10ml) followed by DIPE (23 mL). The product was vacuum dried at 65-70°C for 5-6 hrs.
1:1 Proline Co-crvstal (ΔΗ 53 J/g) was observed by differential scanning calorimetry (DSC) and is shown in Fig. 1. A powder X-ray diffraction (PXRD) spectrum is shown in Fig. 2.
2:1 Proline Co-crvstal
Example 78: 2:1 Proline Co-crvstal with f2S.3R.4R.5S.6R>-2-r3-f2.3-Pihvdro-benzof1.41dioxin-6-ylmethvH-4-ethyl-phenvn-6-hvdroxymethyl-tetrahvdro-pyran- 3.4.5-triol
Method 3: 1 :2 Co-Crvstals of Example 62 with L-Proline
(2S,3R,4R,5S,6R)-2-[3-(2,3-Dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl-phenyl]-6-hydroxymethyl-tetrahydro-pyran-3,4,5-triol (Example 62, 1 kg) was added to 15 L of ethanol with agitation while maintaining the mixture at 20-25 °C. The mixture was stirred for 10 min at 20-25 °C, then L-proline (537 gm) was added while maintaining the mixture at 20-25 °C. The mixture was stirred at this temperature for 30 min., then heated to reflux and refluxed for 30 min. The mixture was slowly cooled to 25-30°C then stired for 1 hr. DIPE (15 L) was added while maintaining the temperature at 25-30 °C and the mixture was stirred at this temperature for 1 hr. The precipitated product was collected by filtration and the product was washed with DIPE (5 L). The product was air dried at 65-70 °C to yield 1.22 kg
(79%) of a 1 :2 co-crystal of Example 62 : L-proline. A melting point 176°C (ΔΗ 85 J/g) was observed by differential scanning calorimetry (DSC) and is shown in Fig.
3. A powder X-ray diffraction (PXRD) spectrum is shown in Fig. 4. Relative
intensities of the most prominent powder x-ray diffraction peaks for the 1 :2 co- crystals of Example 62 and proline are shown in Table 5.
Table 5

PATENT
WO 2012140597
http://www.google.co.in/patents/WO2012140597A1?cl=en
. TABLE 2:

Intermediate 2: (2S,3R,4R,5S,6R)-2-[3-(2,3-Dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-4-

Intermediate 2
Intermediate 1
Step I: To a stirred solution of acetic acid (2R,3R,4R,5S)-3,4,5-triacetoxy-6-[4-bromo-3- (2,3-dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-phenyl]-tetrahydro-pyran-2-ylmethyl ester (Intermediate 1 , 10.0 g, 15.74 mmol) in toluene (200 mL) was added
tricyclohexylphosphine (1.76 g, 6.29 mmol), a solution of potassium phosphate tribasic (13.3 g, 62.9 mmol) in water (15 mL), and ethylboronic acid (3.4 g, 47.2 mmol). The reaction mixture was degassed for 45 min then palladium (II) acetate (529 mg, 2.3 mmol) was added. After refluxing overnight, the reaction mixture was cooled to room temperature, and water was added. The resulting mixture was extracted with ethyl acetate, (2 X 200 ml_), washed with water and brine, then dried over sodium sulfate, concentrated and purified by column chromatography to furnish acetic acid
(2R,3R,4R,5S)-3,4,5-triacetoxy-6-[3-(2,3-dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl- phenyl]-tetrahydro-pyran-2-ylmethyl ester (5.4 g).
Step II: To a stirred solution of acetic acid (2R,3R,4R,5S)-3,4,5-triacetoxy-6-[3-(2,3- dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl-phenyl]-tetrahydro-pyran-2-ylmethyl ester (9.3 g, 15.9 mmol) in methanol:THF:water 3:2:1 (170 ml.) was added lithium hydroxide (764 mg, 19.1 mmol). After stirring for 2 h at room temperature, the volatiles were evaporated under reduced pressure. The resulting residue was taken up in ethyl acetate (150 ml.) and washed with brine (75 ml_), brine containing 5 ml. of 5% aqueous KHS04 (75 ml_), and brine (20 ml.) again, then dried over sodium sulfate and concentrated to furnish (2S,3R,4R,5S,6R)-2-[4-Cyclopropyl-3-(2,3-dihydro-benzo[1 ,4]dioxin-6-ylmethyl)- phenyl]-6-hydroxymethyl-tetrahydro-pyran-3,4,5-triol (6.5 g)
1H NMR (400 MHz, CD3OD): δ 1.07 (t, J = 7.6 Hz, 3H), 2.57 (q, J = 7.6 Hz, 2H), 3.34- 3.50 (m, 4H), 3.68 (dd, J = 12.0, 5.6 Hz, 1 H), 3.85-3.91 (m, 3H), 4.08 (d, J = 9.6 Hz, 1 H), 4.17 (s, 4H), 6.53-6.58 (m, 2H), 6.68 (d, J = 8.4 Hz, 1 H), 7.15-7.25 (m, 3H).
MS (ES) m/z 434.2 (M+18).
Example 3: Synthesis of phosphoric acid (2R,3S,4R,5R,6S)-6-[3-(2,3-dihydro- benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl-phenyl]-3,4,5-trihydroxy-tetrahydro-pyran-2- ylmethyl ester diethyl ester
To a stirred solution of (2S,3R,4R,5S,6R)-2-[3-(2,3-dihydro-benzo[1 ,4]dioxin-6-ylmethyl)- 4-ethyl-phenyl]-6-hydroxymethyl-tetrahydro-pyran-3,4,5-triol (Intermediate 2, 500 mg, 1.2 mmol) in pyridine (5 ml) was added diethylchlorophosphate (0.27 ml, 1 .9 mmol) at -40°C. After stirring for 1 h at same temperature, reaction was quenched with the addition of 1 N HCI and extracted with ethyl acetate (2 X 10 ml). Combined organic layers were washed with brine (10 ml), dried over sodium sulfate, concentrated and purified by preparative HPLC to give 220 mg of phosphoric acid (2R,3S,4R,5R,6S)-6-[3-(2,3-dihydro- benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl-phenyl]-3,4,5-trihydroxy-tetrahydro-pyran-2-ylmethyl ester diethyl ester as a white solid. 1H NMR (400 MHz, CD3OD): δ 1.07 (t, J = 7.6 Hz, 3H), 1.15 (td J = 7.2, 1.2 Hz, 3H), 1.22 (td, J = 6.8, 0.8 Hz, 3H), 2.57 (q, J = 7.6 Hz, 2H), 3.36-3.46 (m, 3H), 3.53-3.55 (m, 1 H),3.89 (s, 2H), 3.96-4.11 (m, 5H), 4.17 (s, 4H), 4.18-4.22 (m 1 H), 4.30-4.34 (m, 1 H), 6.52 (d, J = 2.0 Hz, 1 H),6.57 (dd, J = 8.4, 2.4 Hz, 1 H), 6.68 (d, J = 8.4 Hz, 1 H), 7.15- 7.22(m, 3H). MS (ES) m/z 553.3 (M+1 ).
Example 4: Synthesis of disodium salt of phosphoric acid mono- {(2R,3S,4R,5R,6S)-6-[3-(2,3-dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl-phenyl]- 3,4,5-trihydroxy-tetrahydro-pyran-2-ylmethyl} ester

To a stirred solution of (2S,3R,4R,5S,6R)-2-[3-(2,3-Dihydro-benzo[1 ,4]dioxin-6- ylmethyl)-4-ethyl-phenyl]-6-hydroxymethyl-tetrahydro-pyran-3,4,5-triol (Intermediate 2, 1.0 g, 2.4 mmol) in THF (15 ml) was added a solution of Diethyl-phosphoramidic acid di- tert-butyl ester (780 mg, 3.12 mmol) in THF (5 ml) at 0°C followed by a solution of tetrazole (435 mg, 6.2 mmol) in DCM (12.5 ml). After stirring for 5 min at same temperature, it was stirred at room temperature for 20 min. Reaction mixture was cooled to -40 °C and added a solution of m-CPBA (830 mg, 4.8 mmol) in DCM (5 ml). The reaction mixture was stirred at same temperature for 5 min and then at room temperature for 2 h. Reaction mixture was cooled to 0°C and quenched by the addition of 10% sodium bisulfite solution (5 ml). This was extracted with ether (3 X 10 ml). Combined organic layer was washed with brine (5 ml), dried over sodium sulfate and concentrated to give 700 mg of phosphoric acid di-tert-butyl ester (2R,3S,4R,5R,6S)-6- [3-(2,3-dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl-phenyl]-3,4,5-trihydroxy-tetrahydro- pyran-2-ylmethyl ester.
To the stirred solution of phosphoric acid di-tert-butyl ester (2R,3S,4R,5R,6S)-6-[3-(2,3- dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl-phenyl]-3,4,5-trihydroxy-tetrahydro-pyran-2- ylmethyl ester (500 mg) in methanol (20 ml) was added amberlyst 15 ion exchange resin (250 mg) and refluxed for overnight. Reaction mixture was cooled to room temperature, filtered through celite bed and filtrate was concentrated to give 300 mg of phosphoric acid mono-{(2R,3S,4R,5R,6S)-6-[3-(2,3-dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl- phenyl]-3,4,5-trihydroxy-tetrahydro-pyran-2-ylmethyl} ester. The crude material was taken up for next reaction.
To a solution of phosphoric acid mono-{(2R,3S,4R,5R,6S)-6-[3-(2,3-dihydro- benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl-phenyl]-3,4,5-trihydroxy-tetrahydro-pyran-2- ylmethyl} ester (300 mg, 0.6 mmol) in methanol (5 ml) was added 1 N sodium bicarbonate solution (80 mg, 0.7 mmol) in water. After stirring at room temperature for 2 h, the volatiles were evaporated under reduced pressure. The resulting solid was triturated with diethyl ether. The resulting residue was purified by preparative HPLC to give 95 mg of disodium salt of phosphoric acid mono-{(2R,3S,4R,5R,6S)-6-[3-(2,3- dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl-phenyl]-3,4,5-trihydroxy-tetrahydro-pyran-2- ylmethyl} ester.
1H NMR (400 MHz, CD3OD): δ 1.06 (t, J = 7.4 Hz, 3H), 2.56 ( q, J = 7.3 Hz, 2H), 3.34- 3.41 (m, 2H), 3.49 (t, J = 8.8 Hz, 1 H), 3.81-3.88 (m, ,3H), 3.92-3.99 (m, 1 H), 4.05 (d, J = 9.3 Hz, 1 H), 4.16 (s, 4H), 4.20-4.25 (m, 1 H), 6.54 (m, 2H), 6.67 (d, J = 7.8 Hz, 1 H), 7.12-7.21 (m, 3H). MS (ES) m/z 497.1 (M+1 ) for phosphoric acid.
![]()
PATENT
SEE INDIAN PATENT
IN 2009DE02173
Glycoside derivatives and uses thereof
REFERENCES
Pediatric investigation plan (PIP) decision: (S)-Pyrrolidine-2-carboxylic acid compound with (2S,3R,4R,5S,6R)-2-(3-((2,3-dihydrobenzo[b][1,4]dioxin-6-yl)methyl)-4-ethylphenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol (2:1) ( LIK066) (EMEA-001527-PIP01-13)
European Medicines Agency (EMA) Web Site 2014, July 24
Safety, tolerability, pharmacokinetics (PK) and pharmacodynamics (PD) assessment of LIK066 in healthy subjects and in patients with type 2 diabetes mellitus (T2DM) (NCT01407003)
ClinicalTrials.gov Web Site 2011, August 07
IN 2009DE02173
| WO2001016147A1 | 24 Aug 2000 | 8 Mar 2001 | Kissei Pharmaceutical | Glucopyranosyloxypyrazole derivatives, medicinal compositions containing the same and intermediates in the production thereof |
| WO2001027128A1 | 2 Oct 2000 | 19 Apr 2001 | Bruce Ellsworth | C-aryl glucoside sglt2 inhibitors |
| WO2001068660A1 | 15 Mar 2001 | 20 Sep 2001 | Hideki Fujikura | Glucopyranosyloxy benzylbenzene derivatives, medicinal compositions containing the same and intermediates for the preparation of the derivatives |
| WO2001074834A1 | 29 Mar 2001 | 11 Oct 2001 | Squibb Bristol Myers Co | O-aryl glucoside sglt2 inhibitors and method |
| WO2003020737A1 | 5 Sep 2002 | 13 Mar 2003 | Squibb Bristol Myers Co | O-pyrazole glucoside sglt2 inhibitors and method of use |
| WO2003043985A1 | 20 Nov 2002 | 30 May 2003 | Andrew Thomas Bach | Heterocyclic compounds and methods of use |
| WO2004018491A1 | 21 Aug 2003 | 4 Mar 2004 | Nobuhiko Fushimi | Pyrazole derivatives, medicinal composition containing the same, medicinal use thereof, and intermediate for production thereof |
| WO2004078163A2 | 26 Feb 2004 | 16 Sep 2004 | Oern Almarsson | Pharmaceutical co-crystal compositions of drugs such as carbamazepine, celecoxib, olanzapine, itraconazole, topiramate, modafinil, 5-fluorouracil, hydrochlorothiazide, acetaminophen, aspirin, flurbiprofen, phenytoin and ibuprofen |
| WO2004080990A1 | 12 Mar 2004 | 23 Sep 2004 | Kazuhiro Ikegai | C-glycoside derivatives and salts thereof |
| WO2004099230A1 | 30 Apr 2004 | 18 Nov 2004 | Eikyu Yoshiteru | Monosaccharide compounds |
| WO2004103995A1 | 19 May 2004 | 2 Dec 2004 | Gary Michael Ksander | N-acyl nitrogen heterocycles as ligands of peroxisome proliferator-activated receptors |
| WO2005011592A2 | 29 Jul 2004 | 10 Feb 2005 | Janssen Pharmaceutica Nv | Substituted indazole-o-glucosides |
| WO2005021566A2 | 20 Aug 2004 | 10 Mar 2005 | Barsoumian Edward Leon | Glucopyranosyloxy- pirazoles, drugs containing said compounds the use and production method thereof |
| WO2005085237A1 | 3 Mar 2005 | 15 Sep 2005 | Kissei Pharmaceutical | Fused heterocycle derivative, medicinal composition containing the same, and medicinal use thereof |
| WO2005085265A1 | 3 Mar 2005 | 15 Sep 2005 | Kissei Pharmaceutical | Fused heterocycle derivative, medicinal composition containing the same, and medicinal use thereof |
| WO2006011502A1 | 27 Jul 2005 | 2 Feb 2006 | Chugai Pharmaceutical Co Ltd | Novel glucitol derivative, prodrug thereof and salt thereof, and therapeutic agent containing the same for diabetes |
| WO2006054629A1 | 17 Nov 2005 | 26 May 2006 | Kissei Pharmaceutical | 1-SUBSTITUTED-3-β-D-GLUCOPYRANOSYLATED NITROGENOUS HETERO- CYCLIC COMPOUNDS AND MEDICINES CONTAINING THE SAME |
| WO2008016132A1 | 3 Aug 2007 | 7 Feb 2008 | Daiichi Sankyo Co Ltd | Benzyl phenyl glucopyranoside derivative |
| WO2011048112A1 * | 19 Oct 2010 | 28 Apr 2011 | Novartis Ag | Glycoside derivatives and uses thereof |
| US20030114390 * | 4 Oct 2002 | 19 Jun 2003 | Washburn William N. | C-aryl glucoside SGLT2 inhibitors and method |
| US20040018998 | 21 Sep 2001 | 29 Jan 2004 | Hideki Fujikura | Glucopyranosyloxybenzylbenzene derivatives and medicinal compositions containing the same |
| US20060009400 | 28 Jun 2005 | 12 Jan 2006 | Boehringer Ingelheim International Gmbh | D-xylopyranosyl-substituted phenyl derivatives, medicaments containing such compounds, their use and process for their manufacture |
| US20060019948 | 15 Jul 2005 | 26 Jan 2006 | Boehringer Ingelheim International Gmbh | Methylidene-D-xylopyranosyl- and oxo-D-xylopyranosyl-substituted phenyl derivatives, medicaments containing such compounds, their use and process for their manufacture |
| US20060025349 | 27 Jul 2005 | 2 Feb 2006 | Boehringer Ingelheim International Gmbh | D-xylopyranosyl-phenyl-substituted cycles, medicaments containing such compounds, their use and process for their manufacture |
| US20060035841 | 9 Aug 2005 | 16 Feb 2006 | Boehringer Ingelheim International Gmbh | D-xylopyranosyl-phenyl-substituted cycles, medicaments containing such compounds, their use and process for their manufacture |
| US20060074031 | 30 Sep 2005 | 6 Apr 2006 | Boehringer Ingelheim International Gmbh | D-pyranosyl-substituted phenyl derivatives, medicaments containing such compounds, their use and process for their manufacture |
| US20060293252 | 14 Aug 2006 | 28 Dec 2006 | Sanofi-Aventis Deutschland Gmbh | Novel Thiophene Glycoside Derivatives, Processes for The Preparation, Medicaments Comprising These Compounds, and The Use Thereof |
| US20080027014 | 26 Jul 2007 | 31 Jan 2008 | Tanabe Seiyaku Co., Ltd. | Novel SGLT inhibitors |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2015032272A1 * | 19 Aug 2014 | 12 Mar 2015 | Jiangsu Hansoh Pharmaceutical Co., Ltd. | C-aryl glucoside derivative, preparation method for same, and medical applications thereof |
| US9034921 | 1 Jun 2012 | 19 May 2015 | Green Cross Corporation | Diphenylmethane derivatives as SGLT2 inhibitors |
INVENTORS OF LIK 066
Gregory Raymond Bebernitz, Mark G. Bock, Dumbala Srinivas Reddy, Atul Kashinath Hajare, Vinod Vyavahare, Sandeep Bhausaheb Bhosale, Suresh Eknath Kurhade, Videsh Salunkhe, Nadim S. Shaikh, Debnath Bhuniya, P. Venkata Palle, Lili Feng, Jessica Liang,
| BEBERNITZ, Gregory, Raymond; (US). BOCK, Mark, G.; (US). REDDY, Dumbala Srinivas; (IN). HAJARE, Atul Kashinath; (IN). VYAVAHARE, Vinod; (IN). BHOSALE, Sandeep Bhausaheb; (IN). KURHADE, Suresh Eknath; (IN). SALUNKHE, Videsh; (IN). SHAIKH, Nadim, S.; (IN). BHUNIYA, Debnath; (IN). PALLE, P., Venkata; (IN). FENG, Lili; (US). LIANG, Jessica; (US) |
 Mark G Bock
Mark G Bock
BEBERNITZ, Gregory, Raymond….PIC NOT AVAILABLE
 Dr. Srinivasa Reddy
Dr. Srinivasa Reddy
 NADEEM SHAIKH
NADEEM SHAIKH
 Venkata Palle
Venkata Palle
ONLY FEW…………………….
//////see……..http://medcheminternational.blogspot.in/2015/11/lik-066-novartis-for-treatment-of-type.html
Filed under: Phase2 drugs, Uncategorized Tagged: flozin, IN 2009DE02173, LIK 066, novartis, of type 2 diabetes, phase 2, Treatment, WO2011048112, WO2012140597








 .
.













































 Originally posted on
Originally posted on .jpg)

















 Known compounds having the general formula (Va) which are used in the above Step D-1 or step D-2, which can be prepared according to step A-C, as otherwise, it is disclosed in Patent Document 5 It can be prepared by method (the following scheme).
Known compounds having the general formula (Va) which are used in the above Step D-1 or step D-2, which can be prepared according to step A-C, as otherwise, it is disclosed in Patent Document 5 It can be prepared by method (the following scheme).

 In the present invention, compounds having formula (IX) prepared via the process F from Step A (and / or its enantiomer) may be very produced as pure compounds. Compounds of formula (IX) which can be obtained by the present invention typically have a quality below.
In the present invention, compounds having formula (IX) prepared via the process F from Step A (and / or its enantiomer) may be very produced as pure compounds. Compounds of formula (IX) which can be obtained by the present invention typically have a quality below.





































































































































































 Food in mumbai
Food in mumbai mumbai skyline
mumbai skyline


































































 .
.