Afimetoran is an immunomodulator and an antagonist of toll-like receptors 7 and 8.1,2 It is also is under investigation in clinical trial NCT04269356 (Study to Assess the Way the Body Absorbs, Distributes, Breaks Down and Eliminates Radioactive BMS-986256 in Healthy Male Participants).
The invention further pertains to pharmaceutical compositions containing at least one compound according to the invention that are useful for the treatment of conditions related to TLR modulation, such as inflammatory and autoimmune diseases, and methods of inhibiting the activity of TLRs in a mammal.
Toll/IL-1 receptor family members are important regulators of inflammation and host resistance. The Toll-like receptor family recognizes molecular patterns derived from infectious organisms including bacteria, fungi, parasites, and viruses (reviewed in Kawai, T. et al., Nature Immunol., 11:373-384 (2010)). Ligand binding to the receptor induces dimerization and recruitment of adaptor molecules to a conserved cytoplasmic motif in the receptor termed the Toll/IL-1 receptor (TIR) domain. With the exception of TLR3, all TLRs recruit the adaptor molecule MyD88. The IL-1 receptor family also contains a cytoplasmic TIR motif and recruits MyD88 upon ligand binding (reviewed in Sims, J.E. et al., Nature Rev. Immunol., 10:89-102 (2010)).
Toll-like receptors (TLRs) are a family of evolutionarily conserved, transmembrane innate immune receptors that participate in the first-line defense. As pattern recognition receptors, the TLRs protect against foreign molecules, activated by pathogen associated molecular patterns (PAMPs), or from damaged tissue, activated by danger associated molecular patterns (DAMPs). A total of 13 TLR family members have been identified, 10 in human, that span either the cell surface or the endosomal compartment. TLR7-9 are among the set that are endosomally located and respond to single-stranded RNA (TLR7and TLR8) or unmethylated single-stranded DNA containing cytosine-phosphate-guanine (CpG) motifs (TLR9).
Activation of TLR7/8/9 can initiate a variety of inflammatory responses (cytokine production, B cell activation and IgG production, Type I interferon response). In the case of autoimmune disorders, the aberrant sustained activation of TLR7/8/9 leads to worsening of disease states. Whereas overexpression of TLR7 in mice has been shown to exacerbate autoimmune disease, knockout of TLR7 in mice was found to be protective against disease in lupus-prone MRL/lpr mice. Dual knockout of TLR7 and 9 showed further enhanced protection.
As numerous conditions may benefit by treatment involving modulation of cytokines, IFN production and B cell activity, it is immediately apparent that new compounds capable of modulating TLR7 and/or TLR8 and/or TLR9 and methods of using these compounds could provide substantial therapeutic benefits to a wide variety of patients.
The present invention relates to a new class of [1,2,4]triazolo[1,5-a]pyridinyl substituted indole compounds found to be effective inhibitors of signaling through TLR7/8/9. These compounds are provided to be useful as pharmaceuticals with desirable stability, bioavailability, therapeutic index, and toxicity values that are important to their drugability.
6-(3-isopropyl-5-(piperidin-4-yl)-1H-indol-2-yl)-7,8-dimethyl-[1,2,4]triazolo[1,5-a]pyrid ine, 2 HCl (47.66 g, 104 mmol), DCE (220 mL), DBU (62.4 mL, 414 mmol), and 2-bromoacetamide (17.14 g, 124 mmol). The reaction flask was capped. The reaction mixture was stirred overnight at room temperature. The reaction mixture was concentrated, diluted with water, and stirred for 30 minutes then filtered. The solid was recrystallized using ethanol to afford 2-(4-(2-(7,8-dimethyl-[1,2,4]triazolo[1,5-a] pyridin-6-yl)-3-isopropyl-1H-indol-5-yl)piperidin-1-yl)acetamide (42.3 g, 93 mmol,
Zevotrelvir (Compound 52) is a coronavirus inhibitor with IC50 ranges of <0.1 μM and <0.1mM for 229E hCoV and SARS-CoV-23C-like (3CL) proteases, respectively. Zevotrelvir has the potential to study viral infections.
Coronaviruses are enveloped, positive-sense, single-stranded RNA viruses. The genomic RNA of CoVs has a 5′-cap structure and 3′-poly-A tail and contains at least 6 open reading frames (ORFs). The first ORF (ORF 1a/b) directly translates two polyproteins: pp1a and pp1ab. These polyproteins are processed by a 3C-Like protease (3CLpro), also known as the main protease (Mpro), into 16 non-structural proteins. These non-structural proteins engage in the production of subgenomic RNAs that encode four structural proteins, namely envelope, membrane, spike, and nucleocapsid proteins, among other accessory proteins. As a result, it is understood that 3C-Like protease has a critical role in the coronavirus life cycle.
3CLpro is a cysteine protease involved in most cleavage events within the precursor polyprotein. Active 3CLpro is a homodimer containing two protomers and features a Cys-His dyad located in between domains I and II.3CLpro is conserved among coronaviruses and several common features are shared among the substrates of 3CLpro in different coronaviruses. As there is no human homolog of 3CLpro, it is an ideal antiviral target. Although compounds have been reported to inhibit 3CLpro activity, they have not been approved as coronavirus therapies. (Refer to
WO 2004101742 A2, US 2005/0143320 Al, US 2006/0014821 Al, US 2009/0137818
Al, WO 2013049382 A2, WO 2013166319 A1, WO2018042343, WO2018023054, WO 2022013684, WO 2021252644, WO2022020711, WO 2022020242, US 11,174,231 B1, US 11,124,497 B1, WO 2005113580, and WO2006061714).
There is a need in the art for novel therapeutic agents that treat, ameliorate or prevent SARS-CoV-2 infection. The present invention provides the process of novel compounds which act in inhibiting or preventing SARS-CoV-2 viral replication and thus are used in the treatment of COVID-19 (see PCT/US21/60247).
Synthesis of substituted spirooxindole and its intermediate has been previously published (Refer to PCT/US21/60247, WO2019086142, WO 2020221811, WO2020221826, J. Med. Chem.2012, 55, 9069). However, the scale-up using previous process is very challenging due to the safety concern associated with certain intermediates, instability of certain intermediates as well as lack of purification process other than column chromatograph. Thus, there is a strong need for developing a safe and efficient processes for the large-scale preparation of these novel substituted spirooxindole derivatives.
Example 15. Preparation of Preparation of (3R,5’S)-1′-(N-methyl-N-(4,6,7-trifluoro-1H-indole-2-carbonyl)-L-leucyl)-2-oxospiro[indoline-3,3′-pyrrolidine]-5′-carboxamide (Compound (n))
DMF (760 kg, 8V) was added into the reaction at 0 °C (-5~5 °C) followed by compound (j) (63 kg, 1.05 eq) and N-Methylmorpholine (56 kg, 2 eq), HATU
(106 kg, 1.0 eq) and Compound (m-1) (100 kg, 1.0 eq). The reactor was rinsed with DMF (190 kg, 2V) under and warmed up to 25 °C (20~30 °C) and stirred for 5 h (3~6 h) at 25 °C (20~30 °C). After that, additional HATU (0.1 eq) was added and the reaction mixture was stirred for 16-24 h.25% Ammonium hydroxide (38 kg) was added to the reaction mixture at 25 °C (20~30 °C) and stirred for 2 h (1~3 h) at 25 °C (20~30 °C). The reaction mixture was then added to water (5000 kg, 50V) at 20-30°C over 2 h and the resulting slurry was stirred for 2 h (1~5 h) at 25 °C (20~30 °C). The mixture was filtered and the cake was rinsed with water (500 kg, 5 V). The cake was dissolved in ethyl acetate (1350 kg, 15 V) and washed with 10% sodium chloride solution (500 kg) for three times. The organic layer was separated to 1.5-2.5V at not more than 45℃ under vacuum. The solution was cooled to 25 °C (20~30 °C) and Dichloromethane (660 kg, 5V) was added. The mixture was stirred for 2 h (2~5 h) at 25 °C (20~30 °C) and a slurry was formed. n-Heptane (137 kg, 2V) was added dropwise over 0.5 h (0.5~2 h) at 25 °C (20~30 °C) and stirred for additional 2 h (1~3 h) at 25 °C (20~30 °C). The reaction mixture was filtered and the wet cake was rinsed with DCM/heptane (5/2). The wet cake was dried at 50 °C (45~55 °C) for 20 h (15~25 h) to provide Compound (n) as the white solid in 80-85% yield.
Example 16. Preparation of (3R,5’S)-1′-(N-methyl-N-(4,6,7-trifluoro-1H-indole-2-carbonyl)-L-leucyl)-2-oxospiro[indoline-3,3′-pyrrolidine]-5′-carboxamide (Compound (n))
DMF solution of Compound (m-2) (1 kg, 1.0 eq.) was added to a reactor at around 0-10oC. Compound (l) (600 g, 1.0 eq.), NMM (3.00 eq., 850 g) and HATU (1.00 eq., 1.06 kg) was added to the reactor while maintaining the temperature at 0-10oC; The reaction was warmed to 20±5oC, and stirred for at least 6 hours at 20±5oC. HATU (0.20 eq., 210 g) was added to the reactor at 20±5oC and stirred for at least 6 hours at 20±5oC.25% Ammonium hydroxide (390 g, 1.0 eq) was added to the reaction mixture at 20 °C and stirred for 2 h (1~3 h) at 20 °C. EtOAc (14.0 V) and water (14 V) was added at around 25oC over 20 minutes, and the
solution was stirred for at least 30 min. Aqueous phase was extracted with EtOAc for three times and the organic phase was combined, and washed with 10% aq. NaCl for three times at 20±5oC. The organic phase was concentrated to 6 V then EtOH (7.0 V) was charged. The EtOAc-EtOH solvent swap was repeated for three times and concentrated to 5 V before water (7.0 v) was added at 20±5oC. The mixture was cooled to 0-10oC and stirred for 1 h before being filtered. The filter cake was dissolved in ethyl acetate (15 V) and washed with 10% sodium chloride solution for three times. The organic layer was concentrated to 2-3V at not more than 45℃ under vacuum. The solution was cooled to 25 °C (20~30 °C) and Dichloromethane (5V) was added. The mixture was stirred for 2 h (2~5 h) at 25 °C (20~30 °C) and a slurry was formed. n-Heptane (2V) was added dropwise over 0.5 h (0.5~2 h) at 25 °C (20~30 °C) and stirred for additional 2 h (1~3 h) at 25 °C (20~30 °C). The reaction mixture was filtered and wet cake was rinsed with DCM/heptane (5/2). The wet cake was dried at 50 °C (45~55 °C) for 20 h (15~25 h) to provide Compound (n) as the white solid in about 70-75% yield over two steps.
Example 17. Preparation of (3R,5’S)-1′-(N-methyl-N-(4,6,7-trifluoro-1H-indole-2-carbonyl)-L-leucyl)-2-oxospiro[indoline-3,3′-pyrrolidine]-5′-carboxamide (Compound (n))
DMF (10.0 v) was added to a reactor at 25 °C followed by Compound (l) (4.4 kg, 1.0 eq.), NMM (3.0 eq.) Compound (m-3) (1.0 eq.) and HATU (1.0 eq) at 20-25oC. The reaction mixture was stirred for at least 12 hours at 20-25 °C. Once reaction was complete, aqueous ammonium hydroxide (1.0 eq.) was to the reaction system at 20-25 °C, then stirred for at least 2 hours at 20-25oC. The reaction mixture was then added to water (220 kg, 50V) at 20-30°C over 2 h and the resulting slurry was stirred for 2 h (1~5 h) at 25 °C (20~30 °C). The mixture was filtered and the cake was rinsed with water (22 kg, 5 V). The cake was dissolved in ethyl acetate (135 g, 15 V) and washed with 10% sodium chloride solution (22 kg) for three times. The organic layer was separated to 1.5-2.5V at not more than 45 ℃ under vacuum. The solution was cooled to 25 °C (20~30 °C) and Dichloromethane (5V) was added. The mixture was stirred for 2 h (2~5 h) at 25 °C (20~30 °C) and a slurry was formed. n-Heptane (2V) was added dropwise over 0.5 h (0.5~2 h) at 25 °C (20~30 °C) and stirred for additional 2 h (1~3 h) at 25 °C (20~30 °C). The reaction mixture was filtered and wet cake was rinsed with DCM/heptane (5/2). The wet cake was dried at 50 °C (45~55 °C) for 20 h (15~25 h) to provide Compound (n) as the white solid in 80-85% yield.
Ethyl acetate (630 kg, 10 V) was added into reactor (R1) followed by Compound (n) (70 kg). Make sure the water content was less than 0.20% (w/w). The reaction was cooled to 0 °C (-5 – 5°C) and then triethylamine (89.6 kg) was added followed by trifluoroacetic anhydride (92.4 kg) at 0 °C (-5 – 5°C). The reaction was stirred for 1 h (0.5~2 h) at 0 °C (-5 – 5°C). Once the reaction was complete, the reaction mixture was added slowly to 0.2 N aqueous HCl solution (700 kg) over 1 h at 0 °C (-5~5 °C). The resulting solution was stirred for 30 min at 0 °C (-5~5 °C) and the organic layer was separated.1% aqueous ammonium hydroxide (700 kg) was added to the organic layer and stirred at 20 °C for 30 min (15~25 °C). The organic layer was separated and washed with 10% brine for four times. Then the organic layer was separated and distilled to 2-3 V. Toluene-EtOAc swap was performed until precipitate was observed at 3-4 V. Then Toluene (5-6 V) was added and the slurry was stirred at 50 oC for 2 h. Then the solution was cooled down to 20 oC over 1-2 h and stirred for 10 hr (6~14 hr) at 20 °C (15~25 °C). The reaction mixture was filtered and the wet cake was rinsed with toluene (120 kg, 2V). The wet cake was then dried at 50˚C (45~55 °C) for 48 hr to provide desired compound (o) as a white solid in 80-85% yield.
Seladelpar was approved for medical use in the United States in August 2024.[1][5]
Seladelpar is a peroxisome proliferator-activated receptor (PPAR)-delta (δ) agonist. Seladelpar is a single enantiomer of the R-configuration.5 On August 14, 2024, seladelpar was granted accelerated approval by the FDA for the treatment of primary biliary cholangitis,6 which is a condition associated with aberrant bile acid metabolism. Seladelpar works to block bile acid synthesis.1
Medical uses
Seladelpar is indicated for the treatment of primary biliary cholangitis in combination with ursodeoxycholic acid in adults who have an inadequate response to ursodeoxycholic acid, or as monotherapy in people unable to tolerate ursodeoxycholic acid.[1]
Clinically, Seladelpar reduces pruritus and IL-31 in patients with primary biliary cholangitis.[6]
^ Billin AN (October 2008). “PPAR-beta/delta agonists for Type 2 diabetes and dyslipidemia: an adopted orphan still looking for a home”. Expert Opinion on Investigational Drugs. 17 (10): 1465–1471. doi:10.1517/13543784.17.10.1465. PMID18808307. S2CID86564263.
Preparation of 5-chloro-2-fluoro-4-((4-fluoro-2-(3-(methylamino)pyridin-1-yl)phenyl)amino)-N-(thiazol-4-yl)benzenesulfonamide hydrochloride
Step 1) Preparation of tert-butyl (1-(2-amino-5-fluorophenyl)pyridin-3-yl)(methyl)carbamate
2,4-Difluoro-1-nitrobenzene (2.0 g, 12.6 ng/mol) and tert-butyl methyl (pyridin-3-yl)carbamate (2.5 g, 1.0 eq.) were dissolved in DMF (20 mL), and K2C03 ( 2.6 g , 1.5 eq .) was added. The internal temperature was maintained at 60–70 ° C and the mixture was stirred for 2 hours. The completion of the reaction was confirmed by TLC when the reaction solution turned deep yellow. After cooling to room temperature, ethyl acetate (EA)/H20 was added, stirred, and the layers were separated. MgS04 was added to the separated organic layer, stirred, dried, and filtered. After concentrating the filtrate under reduced pressure, the residue was dissolved in EtOH (10 mL) and distilled water (10 mL), and then Na 2 S 2 0 4 (13.0 g, 6 eq.) was added. After stirring for 2 hours while maintaining the internal temperature at 60 to 70 ° C, the completion of the reaction was confirmed by TLC when the yellow color of the reaction solution lightened and became almost colorless. After cooling to room temperature, distilled water (50 mL) was added and extracted twice with EA (100 mL). MgS0 4 was added to the organic layer, stirred, dried, and filtered. The filtrate was concentrated under reduced pressure, and the obtained residue was separated by column chromatography (n-Hexane/EA = 3/1) to obtain the title compound (2.0 g, 51. ).
Step 2) Preparation of tert-butyl thiazol-4-ylcarbamate
Thiazole-4-carboxylic acid (5.0 g, 38.8 vol) was dissolved in t-Bu0H (100 mL), and then TEA (8.1 mL, 1.5 eq.) and DPPA (7.1 mL, 1.5 eq.) were added. The internal temperature was maintained at 90–100 ° C, and the mixture was stirred for 3 days. The completion of the reaction was confirmed by TLC. The product was concentrated under reduced pressure, distilled water (50 mL) was added, and the solution was washed with EA (100 mL).
It was extracted twice. MgSQ 4 was added to the organic layer, stirred, dried, and filtered.
After concentrating the filtrate under reduced pressure, the residue was added to a small amount of EA, slurried, and the resulting solid was filtered to obtain the white title compound (4.0 g, 51.5%).
Step 3) Preparation of tert-butyl ((4-bromo-5-chloro-2-fluorophenyl)sulfonyl)(thiazol-4-yl)carbamate
Step 2) The tert-Butyl thiazol-4-ylcarbamate (4.0 g, 20.0 ng ol) prepared in the reaction vessel was placed in a reaction vessel and the interior was replaced with nitrogen gas. After dissolving in THF (32 mL), it was cooled to _78 ° C using dry ice— acetone. After cooling, LiHMDS (22.4 mL, 1.5 eq.) was slowly added and the reaction mass was stirred for 30 minutes. 4-Bromo-5-chloro-2-fluorobenzenesulfonyl chloride (6.0 g, 1.0 eq.) was dissolved in THF (10 mL) and slowly added to the reaction mixture. The reaction mass was stirred overnight and the completion of the reaction was confirmed by TLC. Distilled water (50 mL) was added and extracted twice with EA (100 mL). MgS0 4 was added to the organic layer, stirred, dried, and filtered. After concentrating the filtrate under reduced pressure, the residue was crystallized from THF/n-hexane to obtain the title compound (4.4 g, 59.0%).
Step 4) Preparation of tert-butyl (l-(2-((4-(N-(tert-butyloxycarbonyl)-N-(thiazol-4-yl)sulfamoyl)-2-chloro-5-fluorophenyl)amino)-5-fluorophenyl)pyrlidin-3-yl)(methyl)carbamate
Tert-butyl (1-(2-amino-5-fluorophenyl)pyrlidin-3-yl)(methyl)carbamate (0.5 g, 1.1 ng ol) prepared in Step 1) and tert-butyl ((4-bromo-5-chloro-2-fluorophenyl)sulfonyl)(thiazol-4-yl)carbamate (0.9 g, 1.2 eq.) prepared in Step 3) were dissolved in 1,4-dioxane (10 mL). Pd(OAc) 2 (0.03 g, 0.1 eq), rac-BINAP (0.19 g, 0.2 eq.), Cs 2 C0 3 (1.5 g, 3.0 eq.) were added to the reaction solution. After reacting at 120 ° C for 30 minutes using a microwave initiator, the completion of the reaction was confirmed by TLC. Distilled water (50 mL) was added and extracted twice with EA (100 mL).
MgS0 4 was added to the organic layer, stirred, filtered and dried. The filtrate was concentrated under reduced pressure, and the residue was separated by column chromatography (EA/n-Hexane = 1/1). This was repeated twice to obtain the title compound (2.0 g, 88.2%).
Step 5) Preparation of 5-chloro-2-fluoro-4-((4-fluoro-2-(3-(methylamino)pyridin-1-yl)phenyl)amino)-N-(thiazol-4-yl)benzenesulfonamide hydrochloride
Step 4) was prepared by adding 1.25 M HCl in MeOH (15 mL) to tert-butyl (1-(2-((4-(Ν-(tert-butoxycarbonyl)-N-(thiazol-4-yl)sulfamoyl)—2-chloro-5-fluorophenyl)amino)-5-fluorophenyl)pyrlidin-3-yl) (methyl)carbamate (2.0 g, 2.9 µl). The mixture was heated to 40–50 ° C and stirred overnight, and the completion of the reaction was confirmed by TLC. The product was concentrated, and methylene chloride (15 mL) was added to the residue, which was stirred for 1 hour, and the resulting solid was filtered to obtain the title compound (0.9 g, 58.8%).
Palopegteriparatide is a human parathyroid hormone analogue corresponding to amino acid residues 1 – 34 of human parathyroid hormone, to which a methoxy polyethylene glycol (molecular weight: ca. 43,000) is bound via a cleavable linker (pegylation site: S1). Palopegteriparatide is a pegylated synthetic peptide (molecular weight: ca. 48,000) consisting of 34 amino acid residues.
Palopegteriparatide was approved for medical use in the European Union in November 2023,[2] and in the United States in August 2024.[1][5]
Medical uses
Palopegteriparatide is indicated for the treatment of adults with hypoparathyroidism.[1][2]
Adverse effects
The US Food and Drug Administration (FDA) prescription label for palopegteriparatide includes warnings for a potential risk of risk of unintended changes in serum calcium levels related to number of daily injections and total delivered dose, serious hypocalcemia and hypercalcemia (blood calcium levels that are too high), osteosarcoma (a rare bone cancer) based on findings in rats, orthostatic hypotension (dizziness when standing), and a risk of a drug interaction with digoxin (a medicine for certain heart conditions).[5]
History
The effectiveness of palopegteriparatide was evaluated in a 26-week, randomized, double-blind, placebo-controlled trial that enrolled 82 adults with hypoparathyroidism.[5] Prior to randomization, all participants underwent an approximate four-week screening period in which calcium and active vitamin D supplements were adjusted to achieve an albumin-corrected serum calcium concentration between 7.8 and 10.6 mg/dL, a magnesium concentration ≥1.3 mg/dL and below the upper limit of the reference range, and a 25(OH) vitamin D concentration between 20 to 80 ng/mL.[5] During the double-blind period, participants were randomized to either palopegteriparatide (N = 61) or placebo (N= 21), at a starting dose of 18 mcg/day, co-administered with conventional therapy (calcium and active vitamin D).[5] Study drug and conventional therapy were subsequently adjusted according to the albumin-corrected serum calcium levels.[5] At the end of the trial, 69% of the participants in the palopegteriparatide group compared to 5% of the participants in the placebo group were able to maintain their calcium level in the normal range, without needing active vitamin D and high doses of calcium (calcium dose ≤ 600 mg/day).[5]
In September 2023, the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) adopted a positive opinion, recommending the granting of a marketing authorization for the medicinal product Yorvipath, intended for the treatment of chronic hypoparathyroidism in adults.[4][6] The applicant for this medicinal product is Ascendis Pharma Bone Diseases A/S.[4] Palopegteriparatide was approved for medical use in the European Union in November 2023.[2]
^ Jump up to:abcdef“Yorvipath EPAR”. European Medicines Agency. 19 October 2020. Archived from the original on 10 December 2023. Retrieved 11 December 2023. Text was copied from this source which is copyright European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
^“Yorvipath Product information”. Union Register of medicinal products. 20 November 2023. Archived from the original on 26 November 2023. Retrieved 11 December 2023.
^ Jump up to:abc“Yorvipath: Pending EC decision”. European Medicines Agency. 15 September 2023. Archived from the original on 24 September 2023. Retrieved 24 September 2023. Text was copied from this source which is copyright European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
^“EU/3/20/2350”. European Medicines Agency. 15 September 2023. Archived from the original on 24 September 2023. Retrieved 24 September 2023.
^World Health Organization (2021). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 86”. WHO Drug Information. 35 (3). hdl:10665/346562.
^World Health Organization (2023). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 89”. WHO Drug Information. 37 (1). hdl:10665/366661.
Clinical trial number NCT04701203 for “A Trial Investigating the Safety, Tolerability and Efficacy of TransCon PTH Administered Daily in Adults With Hypoparathyroidism (PaTHway)” at ClinicalTrials.gov
To a mixture of (5)-2-(3-morpholino-5-(trifluoromethyl)picolinoyl)-N-2-oxo-5-phenyl-2,3-dihydro-lH-benzo[e] [l,4]diazepin-3-yl)hydrazine-l-carboxamide (1.4 kg, 1 eq.) in DCM (11.2 L) in a flask was charged with 4A-MS (1.4 kg) and stirred at 20±5 °C for 2hrs. Then, it was cooled to 0°C, charged with triethylamine (0.62 Kg, 2.5 eq.) and stirred for 10 min. /^-Toluenesulfonyl chloride (0.7 kg, 1.5 eq.) in DCM (1.4 L) solution was dropwise added to the reaction mixture with maintaining below 5°C and stirred at at 0±5 °C for 5 hrs. The reaction mixture was filtered and washed with DCM (2 X 4.2 L). The filtrate was treated with water (4.2 L) at 0°C and stirred between 0 and 10°C for 5 min. After separation, the organic phase was washed with 5% aqueous NaHCC solution (7 L), water (7 L) and brine (7 L) successively and separated. The DCM layer was concentrated in vacuo at below 30°C to leave ~7L of organic layer. MTBE (7 L) was added to organic layer and concentrated in vacuo to leave ~ 7 L of organic layer (This step was repeated once). The organic layer was charged with water (7 L) and stirred at 20±5 °C for 4 hrs. The solid was filtered and washed with MTBE (3 X 2.1 L) and purified water (2.8 L). The wet cake was stirred with ethyl acetate (7 L) for 12 hrs, charged with n-heptane (14 L) and stirred at 20±5 °C for 5 hrs. The solid was filtered, washed with n-heptane (2 X 2.8 L) and dried under vacuum at ambient temperature to provide the title compound (0.776 kg, 99.6% purity by HPLC, 97.8%
chiral purity by chiral HPLC) as a pale yellowish solid. LC-MS(ESI, m/z): 550.17 [M+H]+;
Example 253 was prepared using a procedure similar to that used to prepare Example 160 where ethyl 3-chloro-5-(trifluoromethyl)picolinate was used in place of methyl 5-bromo-3-fluoropicolinate. ESI-MS m/z: 550.2 [M+H] +.
Example 160 Step c Example 160 was prepared using a procedure similar to that used to prepare Example 152 where methyl 5-cyano-3-morpholinopicolinate was used in place of ethyl 2-morpholino-4-(trifluoromethyl)benzoate. ESI-MS m/z: 507.2 [M+H] +. 1H NMR (400 MHz, DMSO-d 6) δ 3.02-3.04 (m, 4H), 3.71-3.73 (m, 4H), 5.19-5.21 (d, J=8.0 Hz, 1H), 7.26-7.30 (m, 1H), 7.34-7.36 (m, 2H), 7.44-7.55 (m, 5H), 7.65-7.70 (m, 1H), 8.13 (s, 1H), 8.72 (s, 1H), 9.42-9.45 (m, 1H), 10.98 (s, 1H).
Example 1. Preparation of isopropyl ((((R,S)-(2R,3R,4R,5R)-5-(2-amino-6-(methylamino)-9H-purin-9-yl)-4-fluoro-3-hydroxy-4-methyltetrahydrofuran-2-yl)methoxy)-phenoxy-phosphoryl)-L-alaninate
Step 1. Preparation of ((2R,3R,4R,5R)-3-(benzoyloxy)-5-bromo-4-fluoro-4-methyltetrahydrofuran-2-yl)methyl benzoate (2)
To a solution of (2R)-3,5-di-O-benzoyl-2-fluoro-2-C-methyl-D-ribono-γ-lactone (24.8 g, 66.6 mmol) in dry THF (333 mL), under a nitrogen atmosphere and cooled to −30° C., was added lithium tri-tert-butoxyaluminum hydride (1.0 M in THF, 22.6 mL, 22.6 mmol) dropwise. After completion of the addition the reaction mixture was slowly warmed up to −15° C. over 90 min then EtOAc was added (300 mL) and the mixture was quenched with a saturated aq. NH 4Cl solution (200 mL). The resulting solution was filtered on Celite® and the filtrate was extracted twice with EtOAc. The combined organics were dried (Na 2SO 4), filtered and concentrated. The residue was taken up in dry DCM (225 mL) under a nitrogen atmosphere, cooled to −20° C., then PPh 3 (19.1 g, 72.8 mmol) was added. After 10 min of stirring at −20° C., CBr 4 (26.0 g, 78.4 mmol) was added and the reaction mixture was allowed to slowly warm up to 0° C. over 2 h. The resulting mixture was poured onto a silica gel column and eluted with PE/EtOAc (gradient 100:0 to 80:20). The fractions containing the α-bromofuranoside were collected and concentrated to afford the product 2 (18.1 g, 41.3 mmol, 62% over two steps) as a thick colorless oil.
Step 2. Preparation of (2R,3R,4R,5R)-5-(2-amino-6-chloro-9H-purin-9-yl)-2-(benzoyloxymethyl)-4-fluoro-4-methyltetrahydrofuran-3-yl benzoate (3)
2-Amino-6-chloropurine (2.63 g, 15.5 mmol) was suspended in t-BuOH (54 mL) under a nitrogen atmosphere. The reaction mixture was heated to 30° C. then potassium tert-butoxide (1.69 g, 15.1 mmol) was added. After 45 min a solution of bromofuranoside 2 (2.24 g, 5.12 mmol) dissolved in anhydrous MeCN (6 mL) was added, the reaction mixture was heated to 65° C. for 16 h then cooled down to room temperature. A saturated aq. NH 4Cl solution (70 mL) was added and the resulting solution was extracted with EtOAc (3×60 mL). The combined organics were dried (Na 2SO 4), filtered and concentrated. The residue was purified twice by column chromatography (gradient PE/EtOAc 80:20 to 0:100 then 60:40 to 20:80) to afford the product 3 (1.56 g, 2.96 mmol, 57%) as a white solid.
Step 3. Preparation of (2R,3R,4R,5R)-5-(2-amino-6-(methylamino)-9H-purin-9-yl)-4-fluoro-2-(hydroxymethyl)-4-methyltetrahydrofuran-3-ol (4)
To a solution of compound 3 (575 mg, 1.09 mmol) in MeOH (9 mL) was added methylamine (33% in absolute EtOH, 1.7 mL, 1.81 mmol). The reaction mixture was heated to 85° C. in a sealed tube for 16 h, cooled down to room temperature and concentrated. The residue was purified by column chromatography (gradient DCM/MeOH 100:0 to 85:15) then reverse phase column chromatography (gradient H 2O/MeOH 100:0 to 0:100) to afford the product 4 (286 mg, 0.91 mmol, 84%) as a white solid.
Step 4. Preparation of isopropyl ((((R,S)-(2R,3R,4R,5R)-5-(2-amino-6-(methylamino)-9H-purin-9-yl)-4-fluoro-3-hydroxy-4-methyltetrahydrofuran-2-yl)methoxy)-phenoxy-phosphoryl)-L-alaninate (5)
To a solution of compound 4 (114 mg, 365 μmol) in dry THF (4 mL), under a nitrogen atmosphere and cooled to 0° C. was added t-butyl magnesium chloride (1.0 M in THF, 0.66 mL, 660 μmol) dropwise over 10 min. The reaction mixture was stirred 15 min at 0° C. then another 15 min at room temperature. The reaction mixture was cooled down to 0° C. then a solution of isopropyl ((R,S)-(pentafluorophenoxy)-phenoxy-phosphoryl)-L-alaninate, Ross, B. S., Reddy, P. G., Zhang, H. R., Rachakonda, S., and Sofia, M. J., J. Org, Chem., (2011), (253 mg, 558 μmol) dissolved in dry THF (1 mL) was added dropwise over 10 min. The reaction mixture was stirred at 0° C. for 30 min followed by 18 h at room temperature then quenched with a saturated aq. NH 4Cl solution (4 mL) and extracted with EtOAc (3×5 mL). The combined organics were dried, filtered (Na 2SO 4) and concentrated. The residue was purified by column chromatography (gradient DCM/MeOH 100:0 to 90:10) then reverse phase column chromatography (gradient H 2O/MeOH 100:0 to 0:100) to afford product 5 (a mixture of diastereomers, 101 mg, 174 μmol, 48%) as a white solid.
1H NMR (300 MHz, CD 3OD) δ 7.83 (s, 0.55H), 7.82 (s, 0.45H), 7.38-7.16 (m, 5H), 6.15 (d, J=18.5 Hz, 0.45H), (d, J=18.8 Hz, 0.55H), 4.99-4.88 (overlapped with H 2O, m, 1H), 4.65-4.36 (m, 3H), 4.25-4.17 (m, 1H), 3.97-3.85 (m, 1H), 3.05 (br s, 3H), 1.32-1.28 (m, 3H), 1.25-1.15 (m, 9H). 19F NMR (282 MHz, CD 3OD) δ −162.8 (s), −163.3 (s). 31P NMR (121 MHz, CD 3OD) δ 4.10 (s), 3.99 (s). MS (ESI) m/z calcd. for C 24H 34FN 7O 7P [M+H] + 582.2; found 582.2.
Vorasidenib was approved for medical use in the United States in August 2024.[2][3] It is the first approval by the US Food and Drug Administration (FDA) of a systemic therapy for people with grade 2 astrocytoma or oligodendroglioma with a susceptible isocitrate dehydrogenase-1 or isocitrate dehydrogenase-2 mutation.[2]
Medical uses
Vorasidenib is indicated for the treatment of people aged twelve years of age and older with grade 2 astrocytoma or oligodendroglioma with a susceptible isocitrate dehydrogenase-1 or isocitrate dehydrogenase-2 mutation, following surgery including biopsy, sub-total resection, or gross total resection.[2]
Side effects
The most common adverse reactions include fatigue, headache, increased risk of COVID-19 infection, musculoskeletal pain, diarrhea, nausea, and seizures.[2] The most common grade 3 or 4 laboratory abnormalities include increased alanine aminotransferase, increased aspartate aminotransferase, GGT increased, and decreased neutrophils.[2]
History
Efficacy was evaluated in 331 participants with grade 2 astrocytoma or oligodendroglioma with a susceptible isocitrate dehydrogenase-1 or isocitrate dehydrogenase-2 mutation following surgery enrolled in INDIGO (NCT04164901), a randomized, multicenter, double-blind, placebo-controlled trial.[2] Participants were randomized 1:1 to receive vorasidenib 40 mg orally once daily or placebo orally once daily until disease progression or unacceptable toxicity.[2] Isocitrate dehydrogenase-1 or isocitrate dehydrogenase-2 mutation status was prospectively determined by the Life Technologies Corporation Oncomine Dx Target Test.[2] Participants randomized to placebo were allowed to cross over to vorasidenib after documented radiographic disease progression.[2] Participants who received prior anti-cancer treatment, including chemotherapy or radiation therapy, were excluded.[2]
Society and culture
Legal status
Vorasidenib was approved for medical use in the United States in August 2024.[2]
Step 3: Preparation of 6-(6-chloropyridin-2-yl)-N2,N4-bis((R)-1,1,1-trifluoro propan-2-yl)-1,3,5-triazine-2,4-diamine
A mixture of 2,4-dichloro-6-(6-chloro-pyridin-2-yl)-1,3,5-triazine (0.27 g, 1.04 mol), (R)-1,1,1-trifluoropropan-2-amine hydrochloride (0.39 g, 2.6 mol), and potassium carbonate (0.43 g, 3.1 mol) in dry 1,4-dioxane (2.5 mL) was stirred under the atmosphere of N2 at 50° C. for 36 hr then at 100° C. for another 36 hr until the reaction was complete. The resulting mixture was filtered through Celite and the cake was washed with EtOAc. The filtrate was concentrated and the residue was purified by standard methods to give the desired product.
The procedure set forth in Example 10 was used to produce the following compounds using the appropriate starting materials.Compound 6-(6-Chloropyridin-2-yl)-N2,N4-bis((S)-1,1,1-trifluoropropan-2-yl)-1,3,5-triazine-2,4-diamine
Mellinghoff IK, van den Bent MJ, Blumenthal DT, Touat M, Peters KB, Clarke J, et al. (August 2023). “Vorasidenib in IDH1- or IDH2-Mutant Low-Grade Glioma”. The New England Journal of Medicine. 389 (7): 589–601. doi:10.1056/NEJMoa2304194. PMID37272516.
Clinical trial number NCT04164901 for “Study of Vorasidenib (AG-881) in Participants With Residual or Recurrent Grade 2 Glioma With an IDH1 or IDH2 Mutation (INDIGO)” at ClinicalTrials.gov
Clinical trial number NCT02481154 for “Study of Orally Administered AG-881 in Patients With Advanced Solid Tumors, Including Gliomas, With an IDH1 and/or IDH2 Mutation” at ClinicalTrials.gov
Clinical trial number NCT03343197 for “Study of AG-120 and AG-881 in Subjects With Low Grade Glioma” at ClinicalTrials.gov
////////Vorasidenib, Voranigo, FDA 2024, APPROVALS 2024, AG 881, AG-881, AG881
Zelatriazin (NBI-1065846 or TAK-041) is a small-molecule agonist of GPR139. It was developed for schizophrenia and anhedonia in depression but trials were unsuccessful and its development was discontinued in 2023.[1][2][3][4][5][6][7]
Example 2: (S)-2-(4-oxobenzo[d][l,2,3]triazin-3(4H)-yl)-N-(l-(4-(trifluoromethoxy)phenyl)ethyl)acetamide
[0166] To a vial containing 2-(4-oxobenzo[d][l,2,3]triazin-3(4H)-yl)acetic acid (15 mg, 0.073 mmol), HOBT (15 mg, 0.095 mmol) and EDC (21 mg, 0.110 mmol) was added DMF (244 μΕ). After stirring at RT for 5 min, (S)- 1 -(4-(trifluoromethoxy)phenyl)ethanamine (18 mg, 0.088 mmol) and DIPEA (64, 0.366 mmol) were added. The reaction mixture was
allowed to stir at RT for 1 h then water was added (5 mL). The solid was filtered off and washed with water to yield the title compound as a white solid (20 mg, 71 % yield). XH NMR
Example 2(S)-2-(4-oxobenzo[d][1,2,3]triazin-3(4H)-yl)-N-(1-(4-(trifluoromethoxy)phenyl)ethyl)acetamide
To a vial containing 2-(4-oxobenzo[d][1,2,3]triazin-3(4H)-yl)acetic acid (15 mg, 0.073 mmol), HOBT (15 mg, 0.095 mmol) and EDC (21 mg, 0.110 mmol) was added DMF (244 μL). After stirring at RT for 5 min, (S)-1-(4-(trifluoromethoxy)phenyl)ethanamine (18 mg, 0.088 mmol) and DIPEA (64, 0.366 mmol) were added. The reaction mixture was allowed to stir at RT for 1 h then water was added (5 mL). The solid was filtered off and washed with water to yield the title compound as a white solid (20 mg, 71% yield). 1H NMR (500 MHz, DMSO-d6) δ ppm 1.40 (d, J=6.8 Hz, 3H), 4.98 (quin, J=7.1 Hz, 1H), 5.09 (s, 2H), 7.33 (d, J=7.8 Hz, 2H), 7.44-7.49 (m, 2H), 7.93-7.98 (m, 1H), 8.09-8.15 (m, 1H), 8.21-8.29 (m, 2H), 8.85 (d, J=7.8 Hz, 1H); ESI-MS m/z [M+H]+ 393.9.
Deuruxolitinib, sold under the brand name Leqselvi, is a medication used for the treatment of alopecia areata.[1] It is a Janus kinase inhibitor selective for JAK1 and JAK2.[2] Although the relative effectiveness of deuruxolitinib and another Janus kinase inhibitor—baricitinib—for alopecia areata may vary depending on the population studied, both drugs are more effective than alternative treatments.[3]
Deuruxolitinib was approved for medical use in the United States in July 2024.[1][4]
Medical uses
Deuruxolitinib is indicated for the treatment of adults with severe alopecia areata.[1]
Side effects
The FDA prescribing label for deuruxolitinib contains a boxed warning for serious infections; malignancies; cardiovascular death, myocardial infarction, and stroke; and thrombosis.[5]
The synthesis of Compound 10, or a pharmaceutically acceptable salt thereof (such as the phosphate salt) may be readily achieved, e.g., reaction of CTP-543 under conditions suitable to provide hydrolysis of the nitrile functionality of CTP-543. CTP-543 can be prepared, e.g., according to the methods described in U.S. Pat. No. 9,249,149 and US Patent Pub. No. 2019/0160068 (the teachings of which are incorporated herein by reference), to produce CTP-543 and/or its phosphate salt. CTP-543 phosphate salt may be transformed into Compound 10 or its phosphate salt according to Scheme 1 below.
To a round bottom flask, equipped with a magnetic stir bar, was charged sulfuric acid (2 mL) followed by careful addition of CTP-543 Phosphate (4.05 g, 9.8 mmol). To the mixture was added another portion of sulfuric acid (2 mL) and water (0.8 mL). The reaction was stirred at room temperature for 4 hours, then quenched by addition of a potassium carbonate solution (80 g, 30% w/w). The product was extracted using isopropyl alcohol. The organic phase was concentrated under vacuum to dryness. The product was dissolved in isopropyl alcohol (100 mL) and phosphoric acid (1 mL, 85% w/w) was added to crystallize the product as the phosphate salt. The precipitate was filtered and dried in a vacuum oven (5 torr, room temp, slight nitrogen purge) to yield the desired compound as an off-white solid (1.82 g, 4.2 mmol, 43% yield). The product was analyzed by HPLC, HRMS, and NMR.
HPLC method summary: column=Waters XBridge C18, 4.6×150 mm, 3.5 μm column; gradient elution: mobile phase A=10 mM ammonium formate, pH 3.9; mobile phase B=acetonitrile; detection=ultraviolet absorbance at 254 nm. Result: Compound (I)=98.7 area %; retention time=11.1 min.
HRMS: Agilent 6530 Q-TOF LC/MS system with electrospray ionization in positive mode. The measured time-of-flight mass-to-charge ratio (m/z) is 333.22839 (theoretical value=333.22735).
King B, Mesinkovska N, Mirmirani P, Bruce S, Kempers S, Guttman-Yassky E, Roberts JL, McMichael A, Colavincenzo M, Hamilton C, Braman V, Cassella JV: Phase 2 randomized, dose-ranging trial of CTP-543, a selective Janus Kinase inhibitor, in moderate-to-severe alopecia areata. J Am Acad Dermatol. 2022 Aug;87(2):306-313. doi: 10.1016/j.jaad.2022.03.045. Epub 2022 Mar 29. [Article]Yan T, Wang T, Tang M, Liu N: Comparative efficacy and safety of JAK inhibitors in the treatment of moderate-to-severe alopecia areata: a systematic review and network meta-analysis. Front Pharmacol. 2024 Apr 10;15:1372810. doi: 10.3389/fphar.2024.1372810. eCollection 2024. [Article]Barati Sedeh F, Michaelsdottir TE, Henning MAS, Jemec GBE, Ibler KS: Comparative Efficacy and Safety of Janus Kinase Inhibitors Used in Alopecia Areata: A Systematic Review and Meta-analysis. Acta Derm Venereol. 2023 Jan 25;103:adv00855. doi: 10.2340/actadv.v103.4536. [Article]Sardana K, Bathula S, Khurana A: Which is the Ideal JAK Inhibitor for Alopecia Areata – Baricitinib, Tofacitinib, Ritlecitinib or Ifidancitinib – Revisiting the Immunomechanisms of the JAK Pathway. Indian Dermatol Online J. 2023 Jun 28;14(4):465-474. doi: 10.4103/idoj.idoj_452_22. eCollection 2023 Jul-Aug. [Article]FDA Approved Drug Products: LEQSELVI (deuruxolitinib) tablets, for oral use [Link]AJMC: FDA Approves Deuruxolitinib for Alopecia Areata [Link]
^World Health Organization (2021). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 86”. WHO Drug Information. 35 (3). hdl:10665/346562.
^“Deuruxolitinib”. American Medical Association. Retrieved 27 July 2024.
Clinical trial number NCT04518995 for “Study to Evaluate the Efficacy and Safety of CTP-543 in Adults With Moderate to Severe Alopecia Areata (THRIVE-AA1) (THRIVE-AA1)” at ClinicalTrials.gov
Clinical trial number NCT04797650 for “Study to Evaluate the Efficacy and Safety of CTP-543 in Adults With Moderate to Severe Alopecia Areata (THRIVE-AA2) (THRIVE-AA2)” at ClinicalTrials.gov
////Deuruxolitinib, alopecia areata, Leqselvi , approvals 2024, fda 2024, C-21543, CTP 543, CTP-543, CTP543, UNII-0CA0VSF91Y, WHO 11622
Class Anti-inflammatories; Antiulcers; Azetidines; Imidazoles; Methylamines; Pyridines; Small molecules
Mechanism of Action Potassium-competitive acid blockers
Highest Development Phases
Registered Erosive oesophagitis
Phase III Gastric ulcer; Peptic ulcer
19 Jul 2024Onconic Therapeutics completes a phase III trial in Gastric ulcer in South Korea (PO) (NCT05448001)
03 Jun 2024Onconic Therapeutics plans a phase III trial for Peptic ulcer (Prevention) in South Korea (PO, Capsule) (NCT06439563)
29 May 2024Interim efficacy data from a phase III ZERO-1 trial in erosive esophagitis released by Onconic Therapeutics
Zastaprazan (JP-1366) is a proton pump inhibitor (WO2018008929). Zastaprazan can be used for the research of gastrointestinal inflammatory diseases or gastric acid-related diseases.
26 Jun 2024Janssen Research & Development initiates a phase III VENTURA-7 trial for Major depressive disorder (Adjunctive treatment) in USA (PO, Tablet) (NCT06514742) (EudraCT2024-511557-21-00)
01 Oct 2023Janssen Pharmaceuticals is now called Johnson & Johnson Innovative Medicine (Janssen Pharmaceuticals website, October 2023)
19 May 2023Chemical structure information added
Aticaprant, also known by its developmental codes JNJ-67953964, CERC-501, and LY-2456302, is a κ-opioid receptor (KOR) antagonist which is under development for the treatment of major depressive disorder.[2][3][4] A regulatory application for approval of the medication is expected to be submitted by 2025.[2] Aticaprant is taken by mouth.[1]
Aticaprant was originally developed by Eli Lilly, was under development by Cerecor for a time, and is now under development by Janssen Pharmaceuticals.[2] As of July 2022, it is in phase 3clinical trials for major depressive disorder.[2] Like other kappa opioid antagonists currently under clinical investigation for the treatment of major depression, its efficacy may be compromised by the countervailing activation of pro-inflammatory cytokines in microglia within the CNS.[7]
Aticaprant is a potent, selective, short-acting (i.e., non-“inactivating”) antagonist of the KOR (Ki = 0.81 nM vs. 24.0 nM and 155 nM for the μ-opioid receptor (MOR) and δ-opioid receptor (DOR), respectively; approximately 30-fold selectivity for the KOR).[8][9][10] The drug has been found to dose-dependently block fentanyl-induced miosis at 25 mg and 60 mg in humans (with minimal to no blockade at doses of 4 to 10 mg), suggesting that the drug significantly occupies and antagonizes the MOR at a dose of at least 25 mg but not of 10 mg or less.[10] However, a more recent study assessing neuroendocrine effects of the drug in normal volunteers and subjects with a history of cocaine dependence reported observations consistent with modest MOR antagonism at the 10 mg dose.[11] In animal models of depression, aticaprant has been found to have potent synergistic efficacy in combination with other antidepressants such as citalopram and imipramine.[12]
Positron emission tomography imaging revealed that brain KORs were almost completely saturated by the drug 2.5 hours following a single dose of 10 mg, which supported the 4 mg to 25 mg dosages that aticaprant is being explored at in clinical trials.[13][14] Occupancy was 35% for a 0.5 mg dose and 94% for a 10 mg dose.[15][14] At 24 hours post-dose, receptor occupancy was 19% for 0.5 mg and 82% for 25 mg.[15][14] No serious side effects were observed, and all side effects seen were mild to moderate and were not thought to be due to aticaprant.[14]
In February 2015, Cerecor Inc. announced that they had acquired the rights from Eli Lilly to develop and commercialize LY-2456302 (under the new developmental code CERC-501).[19]
In August 2017, it was announced that Cerecor had sold its rights to aticaprant to Janssen Pharmaceuticals.[22][21] Janssen was also experimenting with esketamine for the treatment of depression as of 2017.[21]
N-Methoxy-N-methyl-4-chlorobutyramide (S1). To a mixture of N,O-dimethylhydroxylamine hydrochloride (95.0 mmol, 9.27 g) in CH2Cl2 (150 mL) was added 2 M NaOH (300 mmol, 150 mL) and 4-chlorobutyryl chloride (100 mmol, 11.2 mL) at 0 ˚C. The mixture was stirred for 42 h at room temperature. The organic phase was separated, and the aqueous phase was extracted with CH2Cl2 (2 × 50 mL). The combined organic phase was washed with 2 M NaOH (100 mL), dried over Na2SO4, filtered, and concentrated to afford the title comlund in 75% yield as a colorless liquid. 1H NMR (400 MHz, CDCl3) : 2.08-2.15 (m, 2H), 2.63 (t, J = 7.0 Hz, 2H), 3.19 (s, 3H), 3.64 (t, J = 6.3 Hz, 2H), 3.71 (s, 3H). 13C{ 1H} NMR (100 MHz, CDCl3) : 27.1, 28.6, 32.1, 44.6, 61.1. IR (max/cm-1 ): 2965, 2940, 2821, 1656, 14421, 1417, 1387, 1178, 1107, 997. HRMS (ESI+): calculated for [M+Na]+ : 188.0449, found: 188.0450.
4-Chloro-1-(3,5-dimethylphenyl)butan-1-one (S2). To a mixture of N-methoxy-N-methyl-4-chlorobutyramide (S1, 65.0 mmol, 10.8 g) in anhydrous Et2O (100 mL) was added dropwise 3,5-dimethylphenylmagnesium bromide (ca. 1 M in Et2O, ca. 130 mmol, prepared from 1-bromo-3,5-dimethylbenzene (130 mmol, 17.7 mL) and Mg turnings (169 mmol, 4.11 g) in anhydrous Et2O (130 mL)) over 1 h at -40 ˚C under Ar. The reaction mixture was stirred at room temperature for 20 h. After cooling to 0 ˚C, saturated NH4Cl solution (200 mL) was added. The organic phase was separated, washed with water (100 mL) and brine (100 mL), dried over Na2SO4, and filtered. After concentration, the residue was purified by column chromatography (silica gel, hexane/EtOAc as eluent) to afford the title compound in 91% yield as a greenish yellow liquid. 1H NMR (400 MHz, CDCl3) : 2.18-2.25 (m, 2H), 2.38 (s, 6H), 3.15 (t, J = 7.0 Hz, 2H), 3.67 (t, J = 6.3 Hz, 2H), 7.21 (s, 1H), 7.58 (s, 2H). 13C{ 1H} NMR (100 MHz, CDCl3) : 21.2, 26.8, 35.4, 44.7, 125.8, 134.8, 136.8, 138.3, 199.4. IR (max/cm-1 ): 3047, 3006, 2961, 2920, 2868, 1443, 1411, 1322, 1303, 1181, 1159, 844, 785, 687. HRMS (APCI+): calculated for [M+H]+ : 211.0884, found: 211.0884.
(RS)-N-(4-Chloro-1-(3,5-dimethylphenyl)butylidene)-tertbutanesulfinamide (S3). Ti(OEt)4 (100 mol, 21.0 mL) was added to a mixture of (RS)-tert-butanesulfinamide (1.0 M in THF, 50 mmol, 50 mL) and 4-chloro1-(3,5-dimethylphenyl)butan-1-one (S2, 50.0 mmol, 10.5 g) under N2. The mixture was refluxed for 48 h. After cooling to room temperature, brine (100 mL) was added, and the resulting mixture was filtered over Celite using EtOAc (ca. 300 mL). The organic was separated, dried over Na2SO4, and filtered. After concentration under reduced pressure, the residue was purified by column chromatography (silica gel, hexane/EtOAc as eluent) to afford the title compound in 57% yield as a brown viscous liquid. 1H NMR (400 MHz, CDCl3) : 1.33 (s, 9H), 2.10-2.22 (m, 2H), 2.36 (s, 6H), 3.27 (s, 1H), 3.43 (s, 1H), 3.64 (t, J = 6.5 Hz, 2H), 7.13 (s, 1H), 7.47 (s, 2H). 13C{ 1H} NMR (100 MHz, CDCl3) : 21.3, 22.7, 30.2, 31.6, 44.7, 57.7, 125.2, 133.4, 137.6, 138.2, 178.6. IR (max/cm-1 ): 3046, 2958, 2922, 2866, 1599, 1577, 1455, 1361, 1320, 1308, 1069, 856. HRMS (ESI+): calculated for [M+H] + : 314.1340, found: 314.1344. []D 20 +11.0 (c = 1.01, CH2Cl2).
(RS,S)-1-tert-Butylsulfinyl-2-(3,5-dimethylphenyl)pyrrolidine (S4). To a solution of (RS)-N-(4-chloro-1-(3,5-dimethylphenyl)butylidene)-tert-butanesulfinamide (S3, 25.6 mmol, 8.06 g) in anhydrous THF (100 mL) at -78 °C was added LiBEt3H (28 mmol, 0.5 M in THF, 28.2 mL) under Ar. The reaction was stirred at -78 °C for 1 h, subsequently allowed to warm up to room temperature and stirred for additional 20 h. Saturated NaHCO3 solution (80 mL) was slowly added. The mixture was filtered and extracted with EtOAc (3 × 100 mL). The combined organic phase was dried over Na2SO4 and filtered. After concentration, the residue was purified by column chromatography (silica gel, hexane/EtOAc as eluent) to afford the title compound in 72% yield as pale yellow solid. mp.: 56 ˚C. 1H NMR (400 MHz, CDCl3) : 1.12 (s, 9H), 1.74-1.90 (m, 3H), 1.93-2.02 (m, 1H), 2.18-2.27 (m, 1H), 2.30 (s, 6H), 2.94-3.02 (m, 1H), 3.85-3.91 (m, 1H), 4.55-4.59 (m, 1H), 6.88 (s, 1H), 6.90 (s, 2H). 13C{ 1H} NMR (100 MHz, CDCl3) : 21.3, 23.8, 26.3, 36.0, 42.1, 57.2, 69.2, 125.0, 128.7, 137.7, 143.2. IR (max/cm-1 ): 3023, 2957, 2920, 2866, 1607, 1471, 1360, 1061, 957, 847. HRMS (ESI+): calculated for [M+Na]+ : 302.1549, found: 302.1548. []D 20 -137 (c = 0.49, CH2Cl2)
(S)-2-(3,5-Dimethylphenyl)pyrrolidine hydrochloride (1j•HCl). To a solution of (RS,S)-1-tert-butylsulfinyl-2-(3,5-dimethylphenyl)pyrrolidine (S4, 14.7 mmol, 4.12 g) in dioxane (250 mL) was added dropwise HCl (ca. 150 mmol, 4 M in dioxane, 38 mL). The mixture was stirred for 1 h at room temperature under N2, and then the mixture was concentrated under reduced pressure. Then, Et2O (200 mL) was added to the residue and the mixture was cooled to 0 ˚C. The precipitate was collected by filtration, washed with Et2O (40 mL), and dried under reduced pressure to afford the title compound in 94% yield as white solid. mp.: 198 ˚C. 1H NMR (400 MHz, D2O) : 2.00-2.15 (m, 3H), 2.18 (s, 6H), 2.27-2.35 (m, 1H), 3.27-3.36 (m, 2H), 4.45 (t, J = 8.0 Hz, 1H), 6.97 (s, 2H), 7.01 (s, 1H). 13C{ 1H} NMR (100 MHz, D2O) : 20.9, 24.19, 30.9, 46.0, 63.8, 119.79, 125.6, 131.4, 135.3, 140.1. IR (max/cm-1 ): 3033, 3012, 2970, 2855, 2743, 2571, 2480, 1608, 1590, 1414, 850. HRMS (ESI+): calculated for [M-Cl] + : 176.1434, found: 176.1435. []D 20 +7.1 (c = 1.01, MeOH).
(S)-2-(3,5-Dimethylphenyl)pyrrolidine (1j). To a suspension of (S)-2-(3,5-dimethylphenyl)pyrrolidine hydrochloride (1j•HCl, 13.5 mmol, 2.86 g) in anhydrous Et2O (200 mL) was added a saturated solution of NaHCO3 (200 mL). The resulting mixture was stirred for 20 min at room temperature. The organic was separated and the aqueous phase was extracted with Et2O (2 × 100 mL). The combined organic phase was dried over MgSO4 and filtered. The solvent was removed under reduced pressure to afford the title compound as a pale yellow liquid in 99% yield. 1H NMR (400 MHz, CDCl3) : 1.60-1.71 (m, 1H), 1.78-1.96 (m, 2H), 1.98 (s, 1H), 2.11-2.19 (m, 1H), 2.30 (s, 6H), 2.95-3.02 (m, 1H), 3.17-3.23 (m, 1H), 4.03 (t, J = 7.7 Hz, 1H), 6.87 (s, 1H), 6.97 (s, 2H). 13C{ 1H} NMR (100 MHz, CDCl3) : 21.3, 25.5, 34.2, 46.9, 62.6, 124.2, 128.4, 137.8, 144.7. IR (max/cm-1 ): 3332, 3010, 2960, 2915, 2869, 1605, 1458, 1101, 845. HRMS (ESI+): calculated for [M+H]+ : 176.1434, found: 176.1436. []D 20 -30.5 (c = 1.01, MeOH). Chiral HPLC (ChiralPak ODH, 4.6 mm × L 250 mm, hexane:2-propanol = 90:10, 0.5 mL/min, = 254 nm): tR/min = 18.7 (1%), 19.8 (99%).
3-Fluoro-4-(4-formylphenoxy)benzonitrile2 (S5). A mixture of 3,4- difluorobenzonitrile (35.0 mmol, 4.87 g), 4-hydroxybenzaldehyde (35.0 mmol, 4.27 g), and K2CO3 (70.0 mmol, 9.67 g) in N,N-dimethylacetamide (90 mL) was stirred at 100 ˚C for 2 h under N2. After cooling, the reaction mixture was poured into ice water. White precipitate was collected by filtration, washed with water, and dried under reduced pressure to afford the title compound as pale yellow solid in 82% yield. mp.: 101 ˚C. 1H NMR (400 MHz, CDCl3) : 7.11-7.15 (m, 2H), 7.20 (t, J = 8.2 Hz, 1H), 7.49-7.51 (m, 1H), 7.54 (dd, J = 9.7, 1.9 Hz, 1H), 7.91-7.94 (m, 2H), 9.98 (s, 1H). 13C{ 1H} NMR (100 MHz, CDCl3) : 109.1 (d, 3 JC-F = 8.2 Hz), 117.1 (d, 4 JC-F = 2.5 Hz), 117.9, 121.3 (d, 2 JC-F = 21.3 Hz), 122.5 (d, 4 JC-F = 1.6 Hz), 129.6 (d, 3 JC-F = 4.1 Hz), 132.1, 132.7, 147.0 (d, 2 JC-F = 11.5 Hz), 153.6 (d, 1 JCF = 254.8 Hz), 160.7, 190.4. IR (max/cm-1 ): 3100, 3060, 2846, 2812, 2761, 2232, 1697, 1687, 1585, 1497, 1277, 1216, 1166, 1114, 836. HRMS (APCI+): calculated for [M+H]+ : 242.0612, found: 242.0616.
3-Fluoro-4-(4-formylphenoxy)benzamide2 (2f). To a mixture of 3- fluoro-4-(4-formylphenoxy)benzonitrile (S5, 26.0 mmol, 6.27 g) and K2CO3 (13.0 mmol, 1.80 g) in DMSO (24 mL) was added dropwise 35% H2O2 (ca. 29 mmol, 3.1 mL) at 10 ˚C over 5 min. The reaction mixture was stirred at room temperature for 2 h. The reaction mixture was poured into ice water. White precipitate was collected by filtration, washed with water, and dried under reduced pressure to afford the title compound as white solid in 92% yield. mp. 129 ˚C. 1H NMR (400 MHz, (D3C)2SO) : 9.96 (s, 1H), 8.12 (s, 1H), 7.96 (d, J = 8.2 Hz, 2H), 7.93 (dd, J = 1.9, 10.0 Hz, 1H),
^ Jump up to:ab Browne CA, Wulf H, Lucki I (2022). “Kappa Opioid Receptors in the Pathology and Treatment of Major Depressive Disorder”. In Liu-Chen LY, Inan S (eds.). The Kappa Opioid Receptor. Handbook of Experimental Pharmacology. Vol. 271. pp. 493–524. doi:10.1007/164_2020_432. ISBN978-3-030-89073-5. PMID33580854. S2CID231908782.
^ Jump up to:abc Reed B, Butelman ER, Kreek MJ (2022). “Kappa Opioid Receptor Antagonists as Potential Therapeutics for Mood and Substance Use Disorders”. In Liu-Chen LY, Inan S (eds.). The Kappa Opioid Receptor. Handbook of Experimental Pharmacology. Vol. 271. pp. 473–491. doi:10.1007/164_2020_401. ISBN978-3-030-89073-5. PMID33174064. S2CID226305229.
^ Rorick-Kehn LM, Witkin JM, Statnick MA, Eberle EL, McKinzie JH, Kahl SD, et al. (February 2014). “LY2456302 is a novel, potent, orally-bioavailable small molecule kappa-selective antagonist with activity in animal models predictive of efficacy in mood and addictive disorders”. Neuropharmacology. 77: 131–144. doi:10.1016/j.neuropharm.2013.09.021. PMID24071566. S2CID3230414.
^ Lowe SL, Wong CJ, Witcher J, Gonzales CR, Dickinson GL, Bell RL, et al. (September 2014). “Safety, tolerability, and pharmacokinetic evaluation of single- and multiple-ascending doses of a novel kappa opioid receptor antagonist LY2456302 and drug interaction with ethanol in healthy subjects”. Journal of Clinical Pharmacology. 54 (9): 968–978. doi:10.1002/jcph.286. PMID24619932. S2CID14814449.
^ Jump up to:ab Urbano M, Guerrero M, Rosen H, Roberts E (May 2014). “Antagonists of the kappa opioid receptor”. Bioorganic & Medicinal Chemistry Letters. 24 (9): 2021–2032. doi:10.1016/j.bmcl.2014.03.040. PMID24690494.
^ Mitch CH, Quimby SJ, Diaz N, Pedregal C, de la Torre MG, Jimenez A, et al. (December 2011). “Discovery of aminobenzyloxyarylamides as κ opioid receptor selective antagonists: application to preclinical development of a κ opioid receptor antagonist receptor occupancy tracer”. Journal of Medicinal Chemistry. 54 (23): 8000–8012. doi:10.1021/jm200789r. PMID21958337.
Helal MA, Habib ES, Chittiboyina AG (December 2017). “Selective kappa opioid antagonists for treatment of addiction, are we there yet?”. European Journal of Medicinal Chemistry. 141: 632–647. doi:10.1016/j.ejmech.2017.10.012. PMID29107424.
McHugh KL, Kelly JP (2018). “Modulation of the central opioid system as an antidepressant target in rodent models”. The Opioid System as the Interface between the Brain’s Cognitive and Motivational Systems. Progress in Brain Research. Vol. 239. pp. 49–87. doi:10.1016/bs.pbr.2018.07.003. ISBN9780444641670. PMID30314569.
Banks ML (2020). “The Rise and Fall of Kappa-Opioid Receptors in Drug Abuse Research”. In Nader MA, Hurd YL (eds.). Substance Use Disorders. Handbook of Experimental Pharmacology. Vol. 258. pp. 147–165. doi:10.1007/164_2019_268. ISBN978-3-030-33678-3. PMC7756963. PMID31463605.
Jacobson ML, Browne CA, Lucki I (January 2020). “Kappa Opioid Receptor Antagonists as Potential Therapeutics for Stress-Related Disorders”. Annual Review of Pharmacology and Toxicology. 60: 615–636. doi:10.1146/annurev-pharmtox-010919-023317. PMID31914893. S2CID210121357.
Zamaporvint (RXC004) is an orally active and selective inhibitor of Wnt. Zamaporvint targete membrane-bound o-acyltransferase Porcupine and inhibited Wnt ligand palmitoylation, secretion, and pathway activation. Zamaporvint displays a favorable pharmacokinetic profile and shows potent antiproliferative effects in Wnt ligand-dependent colorectal and pancreatic cell lines. Zamaporvint possesses multiple antitumor mechanisms and can be used in cancer research.
SCHEME
PATENT
Redx Pharma PLC
WO2016055786
SIMILAR BUT NOT SAME
[00281] Example 10: 2-[4-(2-fluoro-4-pyridyl)-5-methyl-imidazol-1-yl]-N-(5-pyrazin-2-yl-2-pyridyl)acetamide
[0312] To a stirred mixture of (1R,4S,6S)-5-(tert-butoxycarbonyl)-5-azaspiro[bicyclo[2.2.1]heptane-2,1′-cyclopropane]-6-carboxylic acid (120 mg, 0.449 mmol, 1.0 eq.) and o-(7-Azabenzotriazol-1-yl)-N,N,N’,N’-tetramethyluronium hexafluorophosphate (204 mg, 0.539 mmol, 1.2 eq.) in DMF (2 mL) was added N-ethyl-N-isopropylpropan-2-amine (348 mg, 2.69 mmol, 6.0 eq.). The mixture was stirred for 10 min at 0 °C, and then (2S)-2-amino-3-[(3S)-2-oxopyrrolidin-3-yl]propanamide hydrochloride (102 mg, 0.494 mmol, 1.1 eq.) was added. The mixture was stirred for 1 h at rt. The crude product was purified by C18 column with CH3CN:Water (0.05% FA). The desired fractions were concentrated under reduced pressure to provide tert-butyl (1R,4S,6S)-6-{[(1S)-1-carbamoyl-2-[(3S)-2-oxopyrrolidin-3-yl]ethyl]carbamoyl}-5-azaspiro[bicyclo[2.2.1]heptane-2,1′-cyclopropane]-5-carboxylate (120 mg, 60 %) as a white solid. LC-MS (ESI, m/z): 421 [M+H]+.
[0313] To a stirred mixture of tert-butyl (1R,4S,6S)-6-{[(1S)-1-carbamoyl-2-[(3S)-2-oxopyrrolidin-3-yl]ethyl]carbamoyl}-5-azaspiro[bicyclo[2.2.1]heptane-2,1′-cyclopropane]-5-carboxylate (140 mg, 0.333 mmol, 1.0 eq.) in DCM (1 mL) was added hydrogen chloride (3 mL, 2M in Et2O). The mixture was stirred for 1 h at rt, and then concentrated under reduced pressure to afford (2S)-2-[(1R,4S,6S)-5-azaspiro[bicyclo[2.2.1]heptane-2,1′-cyclopropan]-6-ylformamido]-3-[(3S)-2-oxopyrrolidin-3-yl]propanamide hydrochloride (110 mg, crude) as a white solid. LC-MS (ESI, m/z): 321 [M+H]+.
[0314] To a stirred mixture of (2S)-3,3-dimethyl-2-(2,2,2-trifluoroacetamido)butanoic acid (70.7 mg, 0.311 mmol, 1.1 eq.) and o-(7-Azabenzotriazol-1-yl)-N,N,N’,N’-tetramethyluronium hexafluorophosphate (129 mg, 0.340 mmol, 1.2 eq.) in DMF (2 mL) were added N-ethyl-N-isopropylpropan-2-amine (219 mg, 1.69 mmol, 6.0 eq.). The mixture was stirred for 10 min at 0 °C, and then (2S)-2-[(1R,4S,6S)-5-azaspiro[bicyclo[2.2.1]heptane-2,1′-cyclopropan]-6-ylformamido]-3-[(3S)-2-oxopyrrolidin-3-yl]propanamide hydrochloride (101 mg, 0.283 mmol, 1.0 eq.) was added. The mixture was stirred for 1 h at rt and purified by C18 column with CH3CN/Water (0.05% FA). The desired fractions were concentrated under reduced pressure to provide (2S)-2-[(1R,4S,6S)-5-[(2S)-3,3-dimethyl-2-(2,2,2-trifluoroacetamido)butanoyl]-5-azaspiro[bicyclo[2.2.1]heptane-2,1′-cyclopropan]-6-ylformamido]-3-[(3S)-2-oxopyrrolidin-3-yl]propanamide (90.0 mg, 57 %) as a white solid. LC-MS (ESI, m/z): 530 [M+H]+.
[0315] To a stirred mixture of (2S)-2-[(1R,4S,6S)-5-[(2S)-3,3-dimethyl-2-(2,2,2- trifluoroacetamido)butanoyl]-5-azaspiro[bicyclo[2.2.1]heptane-2,1′-cyclopropan]-6- ylformamido]-3-[(3S)-2-oxopyrrolidin-3-yl]propanamide (90.0 mg, 0.170 mmol, 1.0 eq.) and pyridine (53.7 mg, 0.680 mmol, 4.0 eq.) in DCM (2 mL) was added trifluoroacetic anhydride (64.2 mg, 0.306 mmol, 1.8 eq.). The mixture was stirred for 1 h at rt. The reaction was quenched with water (10 mL). The mixture was extracted with dichloromethane (3 x 10 mL). The organic layers were combined, washed with brine (2 x 10 mL), dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure to afford the crude product. The crude product was purified by prep-HPLC with the following conditions (Column: Mobile Phase B: ACN; Flow rate: 25 mL/min; Gradient: 38% B to 68% B in 7 min, 68% B; Wave Length: 254 nm; RT1(min): 5.07) to afford (1R,4S,6S)-N-[(1S)-1-cyano-2-[(3S)-2- oxopyrrolidin-3-yl]ethyl]-5-[(2S)-3,3-dimethyl-2-(2,2,2-trifluoroacetamido)butanoyl]-5- azaspiro[bicyclo[2.2.1]heptane-2,1′-cyclopropane]-6-carboxamide (18.2 mg, 20%) as a white solid. 1H NMR (400 MHz, 8.45-9.03 (m, 1H), 7.30- 7.65 (m, 1H), 4.80-4.98 (m, 1H), 4.42-4.76 (m, 2H), 4.02-4.18 (m, 1H), 3.10-3.30 (m, 2H), 2.30-2.44 (m, 1H), 1.97-2.25 (m, 3H), 1.59-1.97 (m, 5H), 1.40-1.58 (m, 1H), 0.90-1.06 (m, 9H), 0.61-0.83 (m, 2H), 0.21-0.54 (m, 2H). LC-MS (ESI, m/z): 512 [M+H]+.
Huntington’s disease (HD) is a progressive, autosomal dominant neurodegenerative disorder of the brain, having symptoms characterized by involuntary movements, cognitive impairment, and mental deterioration. Death, typically caused by pneumonia or coronary artery disease, usually occurs 13 to 15 years after the onset of symptoms. The prevalence of HD is between three and seven individuals per 100,000 in populations of western European descent. In North America, an estimated 30,000 people have HD, while an additional 200,000 people are at risk of inheriting the disease from an affected parent. The disease is caused by an expansion of uninterrupted trinucleotide CAG repeats in the “mutant” huntingtin (Htt) gene, leading to production of HTT (Htt protein) with an expanded poly-glutamine (polyQ) stretch, also known as a “CAG repeat” sequence. There are no current small molecule therapies targeting the underlying cause of the disease, leaving a high unmet need for medications that can be used for treating or ameliorating HD. Consequently, there remains a need to identify and provide small molecule compounds for treating or ameliorating HD.
REFERENCES [1]. Sydorenko, et al. Preparation of heterocyclic and heteroaryl compounds for treating Huntington’s disease. World Intellectual Property Organization, WO2020005873 A1. 2020-01-02.
20240216369THE USE OF A SPLICING MODULATOR FOR A TREATMENT SLOWING PROGRESSION OF HUNTINGTON’S DISEASE
20240132509HETEROCYCLIC AND HETEROARYL COMPOUNDS FOR TREATING HUNTINGTON’S DISEASE
20230405000TABLET FOR USE IN TREATING HUNTINGTON’S DISEASE AND METHOD OF MAKING THE SAME
ClassAnti-inflammatories; Antibacterials; Antidementias; Antineoplastics; Antiparkinsonians; Neuroprotectants; Small molecules
Mechanism of ActionPeptide hydrolase inhibitors
Phase II/IIIAlzheimer’s disease
Phase IIPeriodontal disorders
PreclinicalParkinson’s disease; Squamous cell cancer
27 Jan 2023COR 388 licensed to Lighthouse Pharmaceuticals in the US
01 Aug 2022Atuzaginstat is available for licensing as of 01 Aug 2022. http://www.quincetx.com
01 Aug 2022Cortexyme is now called Quince Therapeutics
You need to be a logged in or subscribed to view this content
This small molecule is an orally available protease inhibitor targeting the lysine proteases of the periodontal pathogen Porphyromonas gingivalis. Known as gingipains, these proteases penetrate gingival tissue and cause inflammation at the site of periodontitis (O’Brien-Simpson et al., 2009). Periodontitis has been linked epidemiologically to cognitive impairment, and P. gingivalis bacterial lipopolysaccharide has been detected in postmortem brain tissue of people with AD (Poole et al., 2013). Oral P. gingivalis has been called a risk factor for Alzheimer’s disease (Kanagasingam et al., 2020).
Cortexyme’s approach is based on the theory that P. gingivalis invades the brain, where gingipains contribute to Alzheimer’s pathology (see Sabbagh and Decourt, 2022). The company reported elevated gingipain in brain tissue from people with AD, and a correlation between levels of gingipain and tau proteins in postmortem middle temporal gyrus from AD and healthy control tissue. P. gingivalis DNA was detected in postmortem cortices from people with AD and healthy controls, and in CSF of AD patients (Jan 2019 news on Dominy et al., 2019). In the same study, they show that in mice, oral P. gingivalis infection led to the appearance of bacterial DNA in the brain, increased brain Aβ42 production, neuroinflammation, and hippocampal degeneration. The first three findings were reported to be reduced by atuzaginstat; results for hippocampal cell death were not reported.
In preclinical work from other labs, infection with P. gingivalis was reported to worsen AD pathology and cognitive impairment in AD transgenic mice, and to cause neuroinflammation, memory impairment, neurodegeneration, micro- and astrogliosis, increased brain Aβ and phospho-tau, and neurofibrillary tangles in wild-type C57Bl6 mice (Ishida et al., 2017; Ilievski et al., 2018; Ding et al., 2018). For a review of the preclinical literature, see Costa et al., 2021.
In human neurons grown in culture, P. gingivalis infection led to tau phosphorylation and degradation, synapse loss, and cell death (Haditsch et al., 2020).
P. gingivalis is associated with cardiovascular disease. In rabbits, oral infection was reported to increase arterial plaque and levels of the inflammatory marker CRP. Both were reversed by treatment with COR388 (2020 AAIC abstract). In aged dogs with periodontal disease, ninety days of COR388 reduced oral bacterial load and gum pathology (Arastu-Kapur et al., 2020). In addition, older dogs had bacterial antigens and ribosomal RNA in their brains, consistent with systemic infection seen in humans.
Findings
Two Phase 1 trials of atuzaginstat were completed by June 2019. In a single-dose study of 5 to 250 mg capsules in 34 healthy adults, the compound was safe and well-tolerated. A multiple-dose study assessed safety and tolerability in 24 healthy older adults (mean age of 60 years) and nine with AD (mean age 72). According to a company press release and a poster presentation at the 2018 CTAD conference, healthy adults received 25, 50, or 100 mg COR388 or placebo every 12 hours for 10 days; AD patients took 50 mg or placebo every 12 hours for 28 days. The pharmacokinetic profiles of COR388 in AD and controls were reported to be similar. All volunteers with AD had P. gingivalis DNA fragments in their CSF at baseline. COR388 caused no serious adverse reactions, and no one withdrew. Gingipains also were reported to degrade ApoE, and 28 days of treatment with COR388 was claimed to reduce CSF ApoE fragments (2020 AAIC abstract).
A Phase 2/3 trial (GAIN) evaluating a 48-week course of COR388 in 643 people with mild to moderate AD began in April 2019. Participants took either 40 mg, 80 mg, or placebo twice daily. The primary endpoint was to be ADAS-Cog11 score, and the ADCS-ADL was added later as a co-primary functional endpoint. Further outcomes included CDR-SB, MMSE, NPI, the Winterlight Speech Assessment, MRI brain scans, and change in periodontal disease status. Investigators assessed CSF Aβ and tau, plus P. gingivalis DNA and gingipains in CSF, blood, and saliva, before and after treatment. A dental substudy of 228 participants is assessing effects of COR388 on periodontal disease. This trial involves 93 sites in the U.S. and Europe. The U.S. sites are offering a 48-week open-label extension.
According to a presentation at the 2020 CTAD, GAIN was fully enrolled. At baseline, more than 80 percent of participants had CSF Aβ and tau levels consistent with amyloid positivity or an AD diagnosis. All had detectable antibodies to P. gingivalis in their blood. In the dental substudy, 90 percent had periodontal disease. In December 2020, an independent data-monitoring committee recommended continuing the trial after a planned futility analysis of 300 patients treated for six months (press release).
In February 2021, the FDA placed a partial clinical hold on GAIN because of liver abnormalities in some participants (press release). Dosing in the open-label extension was stopped, but the placebo-controlled portion of GAIN continued. Cortexyme characterized the liver effects as reversible and showing no risk of long-term effects.
In October 2021, Cortexyme announced top-line results indicating the trial had missed its co-primary endpoints of ADAS-Cog11 and ADCS-ADL (press release). The company reported a statistically significant 57 percent slowing of decline on the ADAS-Cog11 in a subgroup with detectable saliva P. gingivalis DNA at baseline who took the higher dose; a 42 percent slowing on the lower dose did not reach statistical significance. This prespecified subgroup analysis included 242 participants; it found no effect on the ADCS-ADL. Improvements in ADAS-Cog and other cognitive endpoints correlated with reductions in saliva P. gingivalis DNA, according to data presented at CTAD 2021 in November. The most common treatment-related adverse events were gastrointestinal, occurring in 12 to 15 percent of treated participants. The treatment groups had dose-related liver enzyme elevations greater than three times the upper limit of normal, in 7 and 15 percent of participants on low and high doses, respectively, with bilirubin elevation reported in two participants on the high dose. The elevations occurred mainly in the first six weeks of treatment, and all resolved without long-term effects. Discontinuations due to transaminase elevations numbered one on placebo, and five and 17 in the 40 mg and 80 mg groups, respectively. The overall dropout rate was 25 percent in the placebo group, and 40 percent in atuzaginstat groups. There were five deaths in the high dose arm, and one in the low dose. All were deemed unrelated to drug. There was no evidence of ARIA or other imaging abnormalities.
At CTAD, the company announced plans for a confirmatory trial, pending discussions with regulators. The plan was to test atuzaginstat in people with mild to moderate AD and evidence of P. gingivalis infection, at the lower dose of 40 mg twice daily, reached by titration to minimize liver effects. The company was also planning a trial in Parkinson’s disease to begin in 2022. These trials were never registered.
In September 2021, Cortexyme began a Phase 1 trial of a second-generation lysin-gingipain inhibitor, COR588 (press release). This compound is expected to require only once-daily dosing. Results were expected in May 2022.
In January 2022, the company announced that the FDA had placed a full clinical hold on atuzaginstat due to concerns about liver toxicity (press release). The company said it intended to develop its backup compound, COR588, for Alzheimer’s disease, pending Phase 1 results. In July 2022, Cortexyme announced that COR588 had met safety and tolerability endpoints in a single- and multiple-ascending dose study in healthy adults (press release).
In August 2022, Cortexyme discontinued the gingipain inhibitor program, and offered it for external licensing (press release). The company changed its name to Quince, and its focus to bone disease. In January 2023, Quince put out word that it had sold Cortexyme’s legacy small molecule protease inhibitor portfolio to Lighthouse Pharmaceuticals, a company co-founded by a former Cortexyme CEO (press release).
Example 1. Preparation of (S)-N-(7-amino-2-oxo-1-(2,3,6-trifluorophenoxy)heptan-3- yl)cyclopentanecarboxamide(1)hydrochloride
[0224] To a mixture of compound 1.4 (23.0 g, 67.2 mmol, 1.00 eq) in THF (200 mL) was added NMM (6.79 g, 67.2 mmol, 7.38 mL, 1.00 eq), isobutyl carbonochloridate (9.17 g, 67.2 mmol, 8.82 mL, 1.00 eq), and diazomethane (5.65 g, 134 mmol, 2.00 eq) at -40 °C under N2 (15 psi). The mixture was stirred at 0 °C for 30 min. LCMS showed the reaction was completed. FLO (200 mL) was added to the reaction and extracted with two 300-mL portions of ethyl acetate. The combined organic phase was washed with two 200-mL portions of brine (200, dried with anhydrous Na2SO4, filtered and concentrated under vacuum to provide crude compound 1.3 (30.0 g, crude) as a yellow oil.
[0225] To a mixture of compound 1.3 (20.0 g, 54.6 mmol, 1.00 eq) in EtOAc (300 mL) was
added hydrogen bromide(29.8 g, 121.7 mmol, 20.0 mL, 33% purity, 2.23 eq) at -20 °C under
N2 (15 psi). The mixture was stirred at -20 °C for 10 min. TLC (petroleum ether : ethyl
acetate = 0:1) showed the reaction was completed. The reaction was basified by addition of
saturated NaHCO3 until the pH of the mixture reached 8, and the mixture was extracted with
three 500-mL portions of EtOAc. The combined organic phase was washed with two 200-mL portions of brine, dried over anhydrous Na2SO4, filtered and concentrated under vacuum
to afford crude compound 1.2 (15.0 g, crude) as a yellow solid.
[0226] To a mixture of compound 1.2 (4.00 g, 9.54 mmol, 1.00 eq) in DMF (40.0 mL) was
Vorbipiprant (CR6086) is an EP4 receptor antagonist, serving as a targeted immunomodulator. Thus, Vorbipiprant is also a potential immune checkpoint inhibitor, to turn cold tumors into hot tumors. Vorbipiprant also antagonizes PGE2-stimulated cAMP production (IC50=22 nM). Vorbipiprant exhibit striking DMARD effects in rodents, and anti-inflammatory activity to inhibt immune-mediated inflammatory diseases.
SCHEME
PATENT
Rottapharm S.p.A.
World Intellectual Property Organization, WO2013004290
Example 7: 4-(1-(6-(4-(trifluoromethyl)benzyl)-6-azaspiro[2.5]octane-5-carboxamido)cyclopropyl)benzoic acid (single unknown enantiomer) (E7)
Procedure A:
The title compound (E7) (54 mg) was prepared according to the general procedure for esters hydrolysis (Method B) starting from methyl 4-(1 -(6-(4-(trifluoromethyl)benzyl)-6-azaspiro[2.5]octane-5-carboxamido)cyclopropyl)benzoate (D122b) (100mg). (LiOH: 4 eq; Reaction time: 18 hrs; RT)
methyl 4-(1 -(6-(4-(trifluoromethyl)benzyl)-6-azaspiro[2.5]octane-5-carboxamido)cyclopropyl)benzoate (D123)) (17.7 g, 36.38 mmol) was partitioned between dioxane (485 ml) and water (242 ml) prior addition of LiOH H2O (6.1 g,
145.5 mmol). The mixture was stirred at RT for 10 hrs. Water (200 ml) was added followed by addition of acetic acid (5.27 ml). Dioxane was evaporated off and acetic acid was added until the pH of the aqueous solution reached the value of ~ 4. The white solid was filtered from the reaction and dried under vacuum overnight then 24 hrs under vacuum at 40 °C affording the title compound (E7) (16.7g).
A peptide having the amino acid sequence of SEQ ID NO: 1 (LQVVYLH: SEQ ID NO: 1) was produced by Peptron Inc. Specifically, coupling was performed one by one starting from the C-terminus using the Fmoc SPPS (9-Fluorenylmethyloxycarbonyl solid phase peptide synthesis) method using an automatic synthesizer (ASP48S, Peptron Inc).
NH 2 -His(Trt)-2-chloro-Trityl Resin , in which the first amino acid at the C-terminus of the peptide was attached to the resin, was used. All amino acid raw materials used in peptide synthesis have the N-terminus protected by Fmoc, and all residues are trityl (Trt), t-butyloxycarbonyl (Boc), t-butyl (t-Bu), etc., which are removed by acid. The protected one was used. As a coupling reagent, HBTU (2-(1H-Benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate)/HOBt (Hydroxxybenzotriazole)/NMM (N-methylmorpholine) was used. (1) Protected amino acid (8 equivalents) and coupling reagent HBTU (8 equivalents)/HOBt (8 equivalents)/NMM (16 equivalents) were dissolved in DMF (Dimethylformamide) and added, followed by reaction at room temperature for 2 hours. (2) Fmoc removal was performed twice for 5 minutes at room temperature by adding 20% piperidine in DMF. After repeating reactions (1) and (2) to create the basic peptide skeleton, TFA (trifluoroacetic acid)/EDT (1,2-ethanedithiol)/Thioanisole/TIS (triisopropylsilane)/H 2 O=90/ 2.5 / Peptides were separated from the resin using 2.5/2.5/2.5. After purification by reverse phase HPLC using a Vydac Everest C18 column (250 mm × 22 mm, 10 μm), water-acetonitrile linear gradient (10~75% ( v/v) of acetonitrile) method. The molecular weight of the purified peptide was confirmed using LC/MS (Agilent HP1100 series) and lyophilized.
This is a prodrug of homotaurine, a modified amino acid previously developed under the names tramiprosate and Alzhemed. ALZ-801 is converted to homotaurine in vivo, but is more easily absorbed and lasts longer in the blood than tramiprosate.
Tramiprosate was reported to inhibit Aβ42 aggregation into toxic oligomers (Gervais et al., 2007; Kocis et al., 2017). Both ALZ-801 and tramiprosate are metabolized to 3-sulfopranpanoic acid (3-SPA), which is normally found in brain and also inhibits Aβ42 aggregation (Hey et al., 2018). A more recent study found that homotaurine activates GABA receptors, and suggests an alternative mechanism of action for ALZ-801 (Meera et al., 2023).
After tramiprosate failed in Phase 3, its maker, NeuroChem, marketed it as a nutritional supplement. Years later, a subgroup analysis of the trial data indicated a potential positive effect in participants who carried two copies of ApoE4 (Abushakra et al., 2016; Abushakra et al., 2017). Alzheon licensed ALZ-801 from NeuroChem and is developing it for Alzheimer’s disease.
ALZ-801 is a potent and orally available small-molecule β-amyloid (Aβ) anti-oligomer and aggregation inhibitor, valine-conjugated proagent of Tramiprosate with substantially improved PK properties and gastrointestinal tolerability compared with the parent compound. ALZ-801 is an advanced and markedly improved candidate for the treatment of alzheimer’s disease.
General/Typical Procedure: [0311] (i) The solid material was dissolved in water (25 mL). The solution was passed through a Dowex Marathon C ion-exchange column (strongly acidic, 110 g (5 eq), prewashed). The strong acidic fractions were combined and treated with concentrated HCl (10 mL). The mixture was stirred at 50° C. for 30 minutes, and then was concentrated to dryness. The residual material was co-evaporated with EtOH (ethanol) to completely remove water. EtOH (100 mL) was added to the residue. The mixture was stirred at reflux for 1 h, and then cooled to room temperature. The solid material was collected by filtration. The solid material was dissolved in water (10 mL). The solution was added drop wise to EtOH (100 mL). The product slowly crystallized. The suspension was stirred at room temperature for 30 minutes. The solid material was collected by filtration and it was dried in a vacuum oven (60° C.). ID A2. 1H NMR (D2O).δ. 0.87-0.90 (m, 6H), 1.83 (qt, J = 7.2 Hz, 2H), 2.02-2.09 (m, 1H), 2.79 (t, J = 7.8 Hz, 2H), 3.20-3.29 (m, 2H), 3.60 (d, J = 6.3 Hz, 2H); 13C NMR (D2O).δ. 17.20, 17.77, 24.11, 30.00, 38.29, 48.63, 58.96, 169.35; m/z 237 (M-1).
Avenciguat (BI-685509) is a potent and orally active sGC activator. Avenciguat restores cyclic guanosine monophosphate (cGMP) and improves functionality of nitric oxide (NO) pathways. Avenciguat can be used in research of chronic kidney disease (CKD) and diabetic kidney disease (DKD).
Avenciguat is under investigation in clinical trial NCT05282121 (A Study to Test Whether BI 685509 Alone or in Combination With Empagliflozin Helps People With Liver Cirrhosis Caused by Viral Hepatitis or Non-alcoholic Steatohepatitis (NASH) Who Have High Blood Pressure in the Portal Vein (Main Vessel Going to the Liver)).
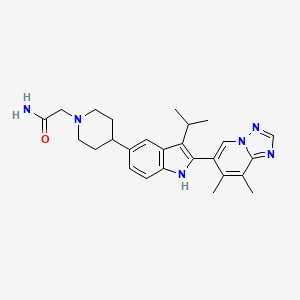
























































































 .
.




