Carebastine is the active metabolite of Ebastine. Carebastine is a histamine H1 receptor antagonist. Carebastine inhibits VEGF-induced HUVEC and HPAEC proliferation, migration and angiogenesis in a dose-dependent manner. Carebastine suppresses the expression of macrophage migration inhibitory factor.
Carebastine is the active metabolite of Ebastine. Carebastine is a histamine H1 receptor antagonist. Carebastine inhibits VEGF-induced HUVEC and HPAEC proliferation, migration and angiogenesis in a dose-dependent manner[1]. Carebastine suppresses the expression of macrophage migration inhibitory factor[2].
Literature References: Nonsedating type histamine H1-receptor antagonist. Prepn: J. M. P. Soto et al., EP 134124; eidem, US 4550116 (both 1985 to Fordonal). Metabolized in vivo to carebastine, its active carboxylic acid metabolite.
These schemes also illustrate the interrelatedness of the processes and intermediates.
EXAMPLE 1
One gram of 9 was dissolved in 20 mL of DMF and 18 mg of P(tBu)3, 41 mg of Pd(dba)2, 230 mg of ZnF2 and 1.2 g of 5 were added. A mixture was stirred at 80° for 18 hours, cooled to room temperature, diluted with ether and washed with water. The organic layer was dried over sodium sulfate, filtered and stripped in vacuo. The resulting product was flash chromatographed on silica gel using 4:1 hexane ethyl acetate to yield 1.0 g (91%) of 10. A repeat of the reaction on larger scale using 15 g of 9 provided 15.2 g (93%) of 10.
EXAMPLE 2
Five grams of 9 was dissolved in 50 mL of methylene chloride and cooled to 0° C. To the solution was added 5.78 g of trimethylsilyl iodide. The mixture was stirred for 30 minutes and excess sodium bisulfite solution was added with vigorous stirring at room temperature. The layers were separated and the aqueous layer extracted twice with methylene chloride. Combined organic layers were dried, filtered and stripped in vacuo to provide 7.7 g (98%) of 1. The reaction was repeated on a larger scale using 15 g of 9 to produce 22.5 g of 1 (96%) yield.
EXAMPLE 3
Six grams of potassium carbonate, 5.8 g of piperidine 2 and 7.6 g of 1 are combined in 100 mL of DMF. The suspension is stirred at room temperature until TLC in 4:1 hexane-ethyl acetate indicates a complete reaction. The reaction mixture is poured into 400 mL of water and extracted three times with methylene chloride. The combined organic extracts are dried, filtered and reduced in vacuo. The resulting product is flash chromatographed on silica gel using ethyl acetate containing 10% triethylamine to yield 3.
EXAMPLE 4
Seven grams of 3 is dissolved in 100 mL of methanol, cooled to 0° C. and 1.1 g of sodium borohydride is added. The mixture is stirred 1 hour, concentrated and partitioned between ethyl acetate and saturated aqueous sodium bicarbonate. The bicarbonate layer is extracted twice with ethyl acetate, the combined organic layers are dried over sodium sulfate and the solution is reduced in vacuo to provide 4.
EXAMPLE 5
Two grams of 4 is dissolved in 30 mL of DMF. To this are added 16.2 mg of P(tBu)3, 36.6 mg of Pd(dba)2, 209 mg of ZnF2 and 1.056 g of 5. The mixture is heated at 80° C., cooled, diluted with ether and worked up as in example 1. The resulting product is flash chromatographed on silica gel using 9:1 ethyl acetate-triethylamine to provide 7.
EXAMPLE 6
One hundred fifty milligrams of 6 is slurried in 5 mL of water and 10 mL of methanol. To the slurry is added 175 mg of sodium hydroxide. The slurry is refluxed for one hour, cooled to room temperature and the methanol removed in vacuo. The resulting aqueous solution is distributed between water and chloroform, the chloroform layer is discarded, the aqueous layer is adjusted to pH 2.3 and extracted with chloroform. The organic layer is dried, filtered and reduced in vacuo to provide carebastine.
EXAMPLE 7
Five grams of 1 was combined with 2.64 g of 2 and 2.0 g of potassium carbonate and 80 mL of DMF. The mixture was stirred at room temperature for two hours, poured into 400 mL of water and extracted three times into methylene chloride. The combined organic layers were dried, filtered and reduced in vacuo. The resulting product was flash chromatographed on silica gel using 9:1 ethyl acetate-triethylamine to provide 2.0 g (54%) of 3.
EXAMPLE 8
One and seven-tenths grams of 3, 90 mg of P(tBu)3, 300 mg of Pd(dba)2, 250 mg of ZnF2 and 1.1 g of 5 were dissolved in 330 mL of DMF under argon. The mixture was heated to 80° for two hours, cooled to room temperature, diluted with ether and worked up as described in example 1. The resulting product was filtered through silica to provide 1.2 g (67.8%) of 6.
EXAMPLE 9
Two grams of 20, 170 mg of P(tBu)3, 560 mg of Pd(acac)2, 474 mg of ZnF2 and 2.0 g of 5 were combined in 50 mL of DMF under argon. The mixture was heated to 80° C. and monitored by HPLC. When reaction was complete, the mixture was cooled to room temperature and 250 mL of water was added. The mixture was extracted three times with ether, dried, filtered and reduced in vacuo. The resulting product was flash chromatographed in 4:1 hexane-ethyl acetate to provide 1.89 g (85%) of 8.
EXAMPLE 10
Two grams of the triflate analog of 20 were reacted as in the foregoing example with 134 mg P(tBu)3, 433 mg of Pd(acac)2, 375 mg of ZnF2 and 1.58 g of 5 to provide 1.56 g (90% yield) of 8.
Example 11
Piperidinol 25 is reacted with chlorodiphenylmethane as described in Fujii et al. Arzneim.-Forsch. 44, 527-538 (1994) to provide 6.
Example 1: Potassium 2-(4-(4-(4-(diphenylmethoxy)piperidin-1-yl)butyryl)phenyl)-2-methylpropionate (carristin potassium salt ) preparation
[0060]
[0061]
Step 1: Preparation of methyl 2-(4-(4-(4-(diphenylmethoxy)piperidin-1-yl)butyryl)phenyl)-2-methylpropionate
[0062]
[0063]
Add 4-(diphenylmethoxy)piperidine hydrochloride (473mg, 1.77mmol), DMAC (4.5ml), K 3 PO 4 (1.13g, 5.3mmol), KI (29mg, 0.177mmol) to a 25ml single-neck bottle. , stir and heat to 100°C. Weigh 2-[4-(4-chloro-1-butyryl)phenyl]-2-methylpropionate methyl ester (600mg, 2.12mmol) and dissolve it in 1ml of DMAC. Add the reaction solution slowly and dropwise, and keep the reaction for 4~ 6h, TLC detects that the raw material reaction is complete. Cool to room temperature, add isopropyl acetate and water, and stir to separate layers. The aqueous phase was then extracted with isopropyl acetate, the organic phases were combined, washed twice with water, dried over anhydrous sodium sulfate, filtered, concentrated, and passed through a silica gel column to obtain 500 mg of the title product, yield 45%, purity: 97.3%.
Step 2: Preparation of 2-(4-(4-(4-(Diphenylmethoxy)piperidin-1-yl)butyryl)phenyl)-2-methylpropionic acid (carristin)
[0067]
[0068]
Add (5-methyl-2-oxo-1,3-dioxo-4-yl)methyl-2-(4-(4-(4-(diphenylmethoxy))piperidine-1 to a 25ml three-necked flask) -Methyl)-butyryl)phenyl)-2-methylpropionate (320 mg, 0.62 mmol), 1.5 ml of methanol, 2 ml of 10% NaOH, heated to 60°C for 2 hours, and the TLC raw material reaction was completed. After the reaction is completed, cool to room temperature, concentrate to dryness, add EA, add hydrochloric acid to adjust the pH to 2~3, layer the layers, wash once with water, dry the organic phase, and concentrate to dryness to obtain 300 mg of the title product. Yield: 95%, purity 95.0%.
Add 2-(4-(4-(4-(diphenylmethoxy)piperidin-1-yl)butyryl)phenyl)-2-methylpropionic acid (499mg, 1mmol) and acetonitrile 3.5 to a 25ml three-necked flask. ml, heated to 60°C, added potassium hydroxide (56 mg, 1 mmol), stirred, cooled down, a white solid precipitated, filtered, and dried to obtain 500 mg of carristine potassium salt, with a yield of 90% and a purity of 98.67%.
Example 2: Sodium 2-(4-(4-(4-(diphenylmethoxy)piperidin-1-yl)butyryl)phenyl)-2-methylpropionate (carristine sodium salt ) preparation
[0077]
[0078]
In this example, the preparation method of 2-(4-(4-(4-(diphenylmethoxy)piperidin-1-yl)butyryl)phenyl)-2-methylpropionic acid is the same as in Example 1.
[0079]
Add 2-(4-(4-(4-(diphenylmethoxy)piperidin-1-yl)butyryl)phenyl)-2-methylpropionic acid (499mg, 1mmol) and acetonitrile 3.5 to a 25ml three-necked flask. ml, heated to 60°C, added sodium hydroxide (40 mg, 1 mmol) and stirred for 1 hour, concentrated to dryness, added methyl tert-butyl ether and stirred, filtered, and dried to obtain 458 mg of carristin sodium salt, yield 85%, purity 96.98 %.
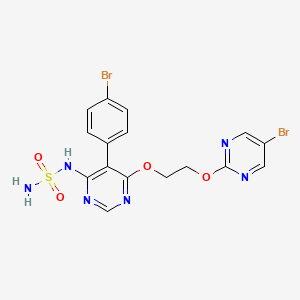
Fda approved, To treat paroxysmal nocturnal hemoglobinuria, 12/5/2023, Fabhalta ‘
Iptacopan is a small-molecule factor B inhibitor previously investigated as a potential treatment for the rare blood disease paroxysmal nocturnal hemoglobinuria (PNH) by inhibiting the complement factor B.1 Factor B is a positive regulator of the alternative complement pathway, where it activates C3 convertase and subsequently C5 convertase.2 This is of particular importance to PNH, where one of the disease hallmarks is the mutation of the PIGA gene. Due to this mutation, all progeny erythrocytes will lack the glycosyl phosphatidylinositol–anchored proteins that normally anchor 2 membrane proteins, CD55 and CD59, that protect blood cells against the alternative complement pathway.3 Additionally, iptacopan has the benefit of targeting factor B, which only affect the alternative complement pathway, leaving the classic and lectin pathway untouched for the body to still mount adequate immune responses against pathogens.2
On December 6th, 2023, Iptacopan under the brand name Fabhalta was approved by the FDA for the treatment of adults with PNH. This approval was based on favorable results obtained from the phase III APPL-PNH and APPOINT-PNH studies, where 82.3% and 77.5% of patients experienced a sustained hemoglobin improvement without transfusions respectively.5
Iptacopan was approved by the US Food and Drug Administration (FDA) for the treatment of adults with paroxysmal nocturnal hemoglobinuria in December 2023.[2][3]
Medical uses
Iptacopan is indicated for the treatment of adults with paroxysmal nocturnal hemoglobinuria.[1][4]
In a clinical study with twelve participants, iptacopan as a single drug led to the normalization of hemolytic markers in most patients, and no serious adverse events occurred during the 12-week study.[5][6]
Iptacopan is also investigated as a drug in other complement-mediated diseases, like age-related macular degeneration and some types of glomerulopathies.[7]
To a 3 L three-necked flask were successively added tetrahydrofuran (150 mL) and 4-bromoxynil (50 g). Isopropylmagnesium chloride lithium chloride coordination complex (1.3 M, 210 mL) was slowly added to the reaction system under nitrogen atmosphere. After the reaction was carried out at room temperature for 2 h, the reaction system was diluted with anhydrous tetrahydrofuran (500 mL) for dilution. The reaction system was cooled to −5° C., and 4-methoxypyridine (25 mL) was added, followed by slowly dropwise addition of benzyl chloroformate (35 mL) (the system temperature was maintained below 0° C.). After the dropwise addition was completed, the reaction system was successively reacted at 0° C. for 2 h, and then warmed to room temperature and reacted at that temperature for 16 h. After the reaction was completed, hydrochloric acid solution (6 M, 150 mL) was added. The mixture was stirred at room temperature for half an hour, added with water (1000 mL) for dilution, and extracted twice with ethyl acetate (500 mL). The extract phase was washed with saturated brine (50 mL), then dried over anhydrous sodium sulfate and filtered. The filtrate was concentrated, and the resulting crude product was separated and purified by a silica gel column (petroleum ether:ethyl acetate=3:1 to 1:1) to give intermediate 1 (23 g, yield: 23%). MS m/z (ESI): 333.0 [M+H].
To a 500 mL single-neck flask were successively added intermediate 1 (28 g), zinc powder (55 g) and acetic acid (200 mL). The reaction mixture was heated to 100° C. and reacted at that temperature for 16 h. After the reaction was completed, the reaction mixture was filtered. The filtrate was added with water (500 mL) for dilution and extracted with ethyl acetate (500 mL). The extract phase was washed twice with saturated aqueous sodium bicarbonate solution (500 mL), washed once with saturated brine (100 mL), dried over anhydrous sodium sulfate and filtered. The filtrate was concentrated under reduced pressure to give intermediate 2 (26 g, yield: 73%). MS m/z (ESI): 334.8 [M+H].
To a 1 L single-neck flask were successively added tetrahydrofuran (100 mL), ethanol (100 mL) and intermediate 2 (26 g), and sodium borohydride (2 g) was added in batches. The mixture was reacted at room temperature for 2 h. After the reaction was completed, the system was cooled to 0° C., and saturated aqueous ammonium chloride solution (100 mL) was added until the temperature did not increase any more. Water (300 mL) was added for dilution, followed by extraction with ethyl acetate (200 mL×2). The extract phase was washed with saturated brine (500 mL), dried over anhydrous sodium sulfate and filtered. The filtrate was concentrated under reduced pressure to give intermediate 3 (25 g, yield: 76%). MS m/z (ESI): 336.9 [M+H].
Dichloromethane (200 mL) was added to a 500 mL single-neck flask, and then intermediate 3 (25 g), imidazole (6.6 g) and tert-butyldiphenylchlorosilane (25 g) were successively added. The mixture was reacted at room temperature for 2 h. After the reaction was completed, water (500 mL) was added for dilution, followed by the extraction with dichloromethane (200 mL). The extract phase was washed with water (50 mL), dried over anhydrous sodium sulfate and filtered. The filtrate was concentrated under reduced pressure. The residue was separated and purified by a silica gel column (petroleum ether:ethyl acetate=10:1) to give intermediate 4 (5.7 g, yield: 13%, R f=0.55; isomer R f=0.50). MS m/z (ESI): 597.0 [M+23].
To a 250 mL single-neck flask were successively added a solution of tetrabutylammonium fluoride in tetrahydrofuran (1 M, 30 mL) and intermediate 4 (5 g). The mixture was reacted at room temperature for 2 h. After the reaction was completed, water (100 mL) was added for dilution, followed by the extraction with ethyl acetate (50 mL×3). The extract phase was washed with saturated brine (100 mL), dried over anhydrous sodium sulfate and filtered. The filtrate was concentrated under reduced pressure. The residue was separated and purified by a silica gel column (petroleum ether:ethyl acetate=3:1 to 0:1) to give a racemic intermediate. The intermediate was subjected to SFC chiral resolution (apparatus: SFC Thar prep 80; column: CHIRALPAK AD-H, 250 mm×20 mm, 5 m; modifier: 35% methanol (0.2% aqueous ammonia); column temperature: 40° C.; column pressure: 60 bar; wavelength: 214/254 nm; flow rate: 40 g/min; Rt=4.78 min) to give intermediate 5 (1.2 g, yield: 41%). MS m/z (ESI): 358.8 [M+23].
To a 100 mL single-neck flask were successively added N,N-dimethylformamide (15 mL) as a solvent, intermediate 5 (1.2 g) and iodoethane (1.1 g). After the reaction system was cooled to 0° C., sodium hydrogen (60%, 243 mg) was added. Then the system was warmed to room temperature and reacted at that temperature for 2 h. After the reaction was completed, water (30 mL) was added for dilution, followed by the extraction with ethyl acetate (50 mL). The extract phase was washed with saturated brine (10 mL), dried over anhydrous sodium sulfate and filtered. The filtrate was concentrated under reduced pressure to give intermediate 6 (1.2 g, yield: 83%). MS m/z (ESI): 386.9 [M+23].
To a 100 mL single-neck flask were successively added methanol (10 mL), water (10 mL), concentrated sulfuric acid (10 mL) and intermediate 6 (1.2 g). The mixture was heated to 80° C. and reacted at that temperature for 48 h. After the reaction was completed, the reaction mixture was concentrated to remove methanol. The residue was made neutral with saturated aqueous sodium hydroxide solution and extracted three times with ethyl acetate (10 mL). The extract phase was washed with saturated brine (5 mL), dried over anhydrous sodium sulfate and filtered. The filtrate was concentrated under reduced pressure to give intermediate 7 (850 mg, yield: 81%). MS m/z (ESI): 264.1 [M+H]. 1H NMR (400 MHz, CDCl 3) δ 8.01 (d, J=8.3 Hz, 2H), 7.49 (d, J=8.3 Hz, 2H), 4.13 (dd, J=11.7, 2.4 Hz, 1H), 3.92 (s, 3H), 3.82-3.70 (m, 1H), 3.62-3.47 (m, 2H), 3.27-3.10 (m, 1H), 3.02-2.88 (m, 1H), 2.07-1.97 (m, 1H), 1.95-1.85 (m, 1H), 1.82-1.62 (m, 2H), 1.27 (t, J=7.0 Hz, 3H).
To a 250 mL single-neck flask were successively added dichloromethane (50 mL), 5-methoxy-7-methyl-1H-indole (3 g), BOC anhydride (5.68 g), 4-dimethylaminopyridine (227 mg) and triethylamine (2.26 g). The mixture was reacted at room temperature for 16 h. After the reaction was completed, the reaction mixture was quenched by adding saturated ammonium chloride solution (5 mL) and extracted three times with dichloromethane (20 mL). The combined organic phases were washed with water (5 mL), dried over anhydrous sodium sulfate and filtered. The filtrate was concentrated. The residue was purified by column chromatography on silica gel (petroleum ether:ethyl acetate=10:1) to give intermediate 8 (4.6 g, yield: 94%). MS m/z (ESI): 262.0 [M+H].
To a 250 mL single-neck flask were successively added dichloromethane (80 mL), N-methylformanilide (3.8 g) and oxalyl chloride (3.6 g). The mixture was stirred at room temperature for 3 h. Then the reaction temperature was lowered to −14° C., and intermediate 8 (2.5 g) was added. The reaction system was naturally warmed to room temperature and stirred for 1 h. After the reaction was completed, the reaction liquid was poured into ice water and extracted three times with dichloromethane (100 mL). The combined extract phases were washed twice with water (10 mL), dried over anhydrous sodium sulfate and filtered. The filtrate was concentrated. The residue was separated and purified by a silica gel column (petroleum ether:ethyl acetate=20:1) to give intermediate 9 (1.3 g, yield: 47%). MS m/z (ESI): 290.0 [M+H]. 1H NMR (400 MHz, CDCl 3) δ 10.65 (s, 1H), 7.65 (d, J=3.4 Hz, 1H), 7.49 (d, J=3.4 Hz, 1H), 6.76 (s, 1H), 3.98 (s, 3H), 2.70 (s, 3H), 1.65 (s, 9H).
To a 50 mL three-necked flask were successively added 1,2-dichloroethane (5 mL), intermediate 7 (127 mg) and intermediate 9 (130 mg). The mixture was reacted at room temperature for 18 h. Then sodium triacetoxyborohydride (438.72 mg) was added, and the system was successively reacted at room temperature for 18 h. After the reaction was completed, dichloromethane (10 mL) was added for dilution, followed by a wash with 10 mL of water. The organic phase was dried over anhydrous sodium sulfate and filtered. The filtrate was concentrated. The residue was separated and purified by a silica gel column (methanol:dichloromethane=1:10) to give intermediate 10 (50 mg, yield: 14.58%). MS m/z (ESI): 437.3 [M+H], RT=1.142 min.
To a 50 mL three-necked flask were successively added tetrahydrofuran (0.5 mL), methanol (0.5 mL), water (0.5 mL), sodium hydroxide (44 mg) and intermediate 10 (50 mg). The mixture was reacted at room temperature for 18 h. After the reaction was completed, the reaction liquid was directly concentrated under reduced pressure and lyophilized to give intermediate 11 (50 mg, yield: 92%). MS m/z (ESI): 423.1 [M+H].
The alternative pathway (AP) of the complement system is a key contributor to the pathogenesis of several human diseases including age-related macular degeneration, paroxysmal nocturnal hemoglobinuria (PNH), atypical hemolytic uremic syndrome (aHUS), and various glomerular diseases. The serine protease factor B (FB) is a key node in the AP and is integral to the formation of C3 and C5 convertase. Despite the prominent role of FB in the AP, selective orally bioavailable inhibitors, beyond our own efforts, have not been reported previously. Herein we describe in more detail our efforts to identify FB inhibitors by high-throughput screening (HTS) and leveraging insights from several X-ray cocrystal structures during optimization efforts. This work culminated in the discovery of LNP023 (41), which is currently being evaluated clinically in several diverse AP mediated indications.
a Reagents and conditions: (a) i PrMgCl·LiCl, Cbz-Cl, THF; (b) Zn, AcOH; (c) LiBH4, THF; (d) TBDPS-Cl, imidazole, DMF; (e) separation of diastereomers by flash chromatography; (f) TBAF, THF; (g) NaH, EtI, DMF; (h) Ba(OH)2, i PrOH, H2O; (i) K2CO3, MeI, DMF; (j) H2, Pd/C, MeOH; (k) (±)-50, DIPEA, DMA; (l) K2CO3, MeOH; then TMS-diazomethane, toluene, MeOH; (m) chiral SFC; (n) LiOH, H2O, MeOH, THF; (o) (2S,4S)-50, NaBH(OAc)3, DCE.
4-((2S,4S)-(4-Ethoxy-1-((5-methoxy-7-methyl-1H-indol-4- yl)methyl)piperidin-2-yl))benzoic Acid (41, LNP023). Step 1: tert-Butyl 4-(((2S,4S)-4-Ethoxy-2-(4-(methoxycarbonyl)phenyl)- piperidin-1-yl)methyl)-5-methoxy-7-methyl-1H-indole-1-carboxylate (58). To a solution of tert-butyl 4-formyl-5-methoxy-7-methyl1H-indole-1-carboxylate (57) (1.5 g, 5.18 mmol) and methyl 4- ((2S,4S)-4-ethoxypiperidin-2-yl)benzoate ((2S,4S)-50) (1.185 g, 4.50 mmol) in DCE (20 mL) was added NaBH(OAc)3 (3 g, 14.1 mmol), and this was stirred at rt for 21.5h. Additional tert-butyl 4-formyl-5- methoxy-7-methyl-1H-indole-1-carboxylate (57) (500 mg, 1.90 mmol) was added, and this was stirred for 20 h. The reaction was diluted with EtOAc, washed successively with 5% aqueous NaHCO3, H2O, and brine, dried over Na2SO4, filtered, and concentrated to provide the title compound (2.415 g, quant) which was used without further purification. MS (ESI+) m/z 537.4 (M + H). The absolutestereochemistry was ultimately determined via cocrystallization of 41 with the catalytic domain of FB. Step 2: 4-((2S,4S)-(4-Ethoxy-1-((5-methoxy-7-methyl-1H-indol-4- yl)methyl)piperidin-2-yl))benzoic Acid (41, LNP023). To a solution of tert-butyl 4-(((2S,4S)-4-ethoxy-2-(4-(methoxycarbonyl)phenyl)- piperidin-1-yl)methyl)-5-methoxy-7-methyl-1H-indole-1-carboxylate (58) (2.415 g, 4.50 mmol) in THF (10 mL) and MeOH (20 mL) was added 1 M LiOH in H2O (15 mL, 15 mmol), and this was stirred at 70 °C for 8 h. The reaction was cooled to rt, diluted with H2O, half saturated aqueous KHSO4 and citric acid, saturated with sodium chloride, then extracted with 9:1 DCM/TFE, dried with Na2SO4, filtered, and concentrated. RP-HPLC-B purification provided the title compound (730 mg, 38% for 2 steps). 1 H NMR (400 MHz, D2O) δ 7.96 (d, J = 8.0 Hz, 2H), 7.58 (d, J = 8.1 Hz, 2H), 7.30 (d, J = 3.2 Hz, 1H), 6.66 (s, 1H), 6.20 (s, 1H), 4.62−4.47 (m, 1H), 4.06 (d, J = 13.2 Hz, 1H), 3.97−3.76 (m, 2H), 3.66−3.48 (m, 5H), 3.43−3.29 (m, 1H), 3.26−3.15 (m, 1H), 2.35 (s, 3H), 2.31−2.11 (m, 2H), 2.00 (d, J = 15.4 Hz, 1H), 1.93−1.74 (m, 1H), 1.25−1.07 (m, 3H). HRMS calcd for C25H31N2O4 (M + H)+ 423.2284, found 423.2263. 4-((2S,4S)-(4-Ethoxy-1-((5-methoxy-7-methyl-1H-indol-4- yl)methyl)piperidin-2-yl))benzoic Acid Hydrochloride (41· HCl). To a solution of 41 (620 mg, 1.47 mmol) in H2O (10 mL) and acetonitrile (3 mL) was added 5 M aqueous HCl (0.5 mL, 2.5 mmol). The mixture was then lyophilized, and the resulting solid was suspended in i PrOH and heated to 70 °C. The mixture turned into a solution after 1.5 h and was then cooled to rt with stirring. After about 5 h, the mixture turned into a suspension and the solid was collected by filtration and dried under high vacuum at 50 °C to provide the title compound as the hydrochloride salt (450 mg, 65%). 1 H NMR (400 MHz, methanol-d4) δ 10.73 (s, 1H), 8.23 (d, J = 8.2 Hz, 2H), 7.74 (d, J = 8.3 Hz, 2H), 7.36−7.31 (m, 1H), 6.77 (s, 1H), 6.42−6.31 (m, 1H), 4.40−4.19 (m, 2H), 3.87−3.80 (m, 1H), 3.76 (s, 3H), 3.68− 3.50 (m, 4H), 3.45−3.38 (m, 1H), 2.51 (s, 3H), 2.30−2.18 (m, 2H), 2.13−1.89 (m, 2H), 1.31 (t, J = 7.0 Hz, 3H). MS (ESI+) m/z 423.3 (M + H).
//////////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
Clinical trial number NCT04558918 for “Study of Efficacy and Safety of Twice Daily Oral LNP023 in Adult PNH Patients With Residual Anemia Despite Anti-C5 Antibody Treatment (APPLY-PNH)” at ClinicalTrials.gov
Clinical trial number NCT04820530 for “Study of Efficacy and Safety of Twice Daily Oral Iptacopan (LNP023) in Adult PNH Patients Who Are Naive to Complement Inhibitor Therapy (APPOINT-PNH)” at ClinicalTrials.gov
Eplontersen, FDA APP, 12/21/2023, To treat polyneuropathy of hereditary transthyretin-mediated amyloidosis, Wainua
AKCEA-TTR-LRx is under investigation in clinical trial NCT04136184 (Neuro-ttransform: A Study to Evaluate the Efficacy and Safety of Akcea-ttr-lrx in Participants With Hereditary Transthyretin-mediated Amyloid Polyneuropathy).
^ Coelho, Teresa; Marques, Wilson; Dasgupta, Noel R.; Chao, Chi-Chao; Parman, Yeşim; França, Marcondes Cavalcante; et al. (October 2023). “Eplontersen for Hereditary Transthyretin Amyloidosis With Polyneuropathy”. The Journal of the American Medical Association. 330 (15): 1448–1458. doi:10.1001/jama.2023.18688. PMC 10540057. PMID37768671.
^ Diep, John K.; Yu, Rosie Z.; Viney, Nicholas J.; Schneider, Eugene; Guo, Shuling; Henry, Scott; et al. (December 2022). “Population pharmacokinetic/pharmacodynamic modelling of eplontersen, an antisense oligonucleotide in development for transthyretin amyloidosis”. British Journal of Clinical Pharmacology. 88 (12): 5389–5398. doi:10.1111/bcp.15468. PMID35869634. S2CID250989659.
^World Health Organization (2021). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 85”. WHO Drug Information. 35 (1). hdl:10665/340684.
External links
Clinical trial number NCT04136184 for “NEURO-TTRansform: A Study to Evaluate the Efficacy and Safety of Eplontersen (Formerly Known as ION-682884, IONIS-TTR-LRx and AKCEA-TTR-LRx) in Participants With Hereditary Transthyretin-Mediated Amyloid Polyneuropathy” at ClinicalTrials.gov
Clinical trial number NCT01737398 for “Efficacy and Safety of Inotersen in Familial Amyloid Polyneuropathy” at ClinicalTrials.gov
Capivasertib is a novel pyrrolopyrimidine derivative, and an orally available inhibitor of the serine/threonine protein kinase AKT (protein kinase B) with potential antineoplastic activity. Capivasertib binds to and inhibits all AKT isoforms. Inhibition of AKT prevents the phosphorylation of AKT substrates that mediate cellular processes, such as cell division, apoptosis, and glucose and fatty acid metabolism. A wide range of solid and hematological malignancies show dysregulated PI3K/AKT/mTOR signaling due to mutations in multiple signaling components. By targeting AKT, the key node in the PIK3/AKT signaling network, this agent may be used as monotherapy or combination therapy for a variety of human cancers.
Medical uses
Capivasertib, used in combination with fulvestrant (Faslodex), is indicated for adults with hormone receptor-positive, human epidermal growth factor receptor 2-negative locally advanced or metastatic breast cancer with one or more PIK3CA/AKT1/PTEN-alterations, as detected by an FDA-approved test, following progression on at least one endocrine-based regimen in the metastatic setting or recurrence on or within twelve months of completing adjuvant therapy.[1][3]
History
Efficacy was evaluated in CAPItello-291 (NCT04305496), a randomized, double-blind, placebo-controlled, multicenter trial in 708 participants with locally advanced or metastatic HR-positive, HER2-negative breast cancer, of which 289 participants had tumors with PIK3CA/AKT1/PTEN-alterations.[3] All participants were required to have progression on aromatase inhibitor-based treatment.[3] Participants could have received up to two prior lines of endocrine therapy and up to one line of chemotherapy for locally advanced or metastatic disease.[3]
HCl (4M in Dioxane) (3.00 mL, 12.00 mmol) was added to (S)-tert-butyl 4-(1-(4-chlorophenyl)-3-hydroxypropylcarbamoyl)-1-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)piperidin-4-ylcarbamate (Intermediate 22) (1.27 g, 2.40 mmol) in dichloromethane (20 mL). The resulting suspension was stirred at 20° C. for 16 hours. The reaction mixture was filtered through a PTFE filter cup and the crude solid was purified by preparative HPLC (Waters XTerra C18 column, 5 μm silica, 19 mm diameter, 100 mm length), using decreasingly polar mixtures of water (containing 1% TFA) and MeCN as eluents. Fractions containing the desired compound were purified by ion exchange chromatography, using an SCX column. The desired product was eluted from the column using 7M NH 3/MeOH and pure fractions were evaporated to dryness to afford (S)-4-amino-N-(1-(4-chlorophenyl)-3-hydroxypropyl)-1-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)piperidine-4-carboxamide (0.200 g, 19.4%) as a white solid. 1H NMR (399.9 MHz, DMSO-d6) δ 1.45 (2H, d), 1.86 (1H, d), 1.90-1.93 (1H, m), 2.19 (2H, s), 3.38 (2H, q), 3.51-3.58 (2H, m), 4.35-4.38 (2H, m), 4.53 (1H, t), 4.88 (1H, d), 6.58 (1H, t), 7.16 (1H, t), 7.32-7.38 (4H, m), 8.12 (1H, s), 8.43 (1H, d), 11.63 (1H, s), m/z (ESI+) (M+H)+=429; HPLC tR=1.46 min.
EXAMPLE 9 ALTERNATIVE ROUTE 1: (S)-4-AMINO-N-(1-(4-CHLOROPHENYL)-3-HYDROXYPROPYL)-1-(7H-PYRROLO[2,3-D]PYRIMIDIN-4-YL)PIPERIDINE-4-CARBOXAMIDE
N-Ethyldiisopropylamine (1.676 ml, 9.62 mmol) was added to (S)-4-amino-N-(1-(4-chlorophenyl)-3-hydroxypropyl)piperidine-4-carboxamide (Intermediate 49) (1 g, 3.21 mmol) and 4-chloro-7H-pyrrolo[2,3-d]pyrimidine (0.493 g, 3.21 mmol) in butan-1-ol (15 ml). The resulting solution was stirred at 60° C. for 18 hours. The reaction mixture was diluted with EtOAc (50 mL), and washed sequentially with water (25 mL) and saturated brine (25 mL). The organic layer was dried over MgSO 4, filtered and evaporated to afford crude product. The crude product was purified by flash silica chromatography, elution gradient 0 to 6% MeOH with ammonia in DCM. Pure fractions were evaporated to dryness to afford (S)-4-amino-N-(1-(4-chlorophenyl)-3-hydroxypropyl)-1-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)piperidine-4-carboxamide (842 mg) as a white foam. (S)-4-amino-N-(1-(4-chlorophenyl)-3-hydroxypropyl)-1-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)piperidine-4-carboxamide was stirred in ethyl acetate (7 mL) for 18 hours. The solid was collected by filtration, washed with a small amount of ethyl acetate and vacuum oven dried at 55° C. for 18 hours to afford (S)-4-amino-N-(1-(4-chlorophenyl)-3-hydroxypropyl)-1-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)piperidine-4-carboxamide (0.585 g, 42.5%) as a white solid.
m/z (ES+) (M+H)+=429; HPLC tR=1.60 min.
1H NMR (400.13 MHz, DMSO-d 6) δ 1.39-1.47 (2H, m), 1.80-2.02 (4H, m), 2.17 (2H, s), 3.35-3.40 (2H, m), 3.50-3.59 (2H, m), 4.34-4.41 (2H, m), 4.53 (1H, t), 4.88 (1H, d), 6.57 (1H, m), 7.14-7.16 (1H, m), 7.31-7.37 (4H, m), 8.12 (1H, s), 8.42 (1H, d), 11.62 (1H, s)
EXAMPLE 9 ALTERNATIVE ROUTE 2: (S)-4-AMINO-N-(1-(4-CHLOROPHENYL)-3-HYDROXYPROPYL)-1-(7H-PYRROLO[2,3-D]PYRIMIDIN-4-YL)PIPERIDINE-4-CARBOXAMIDE
(S)-3-Amino-3-(4-chlorophenyl)propan-1-ol (Intermediate 47) (2.055 g, 11.07 mmol) was added in one portion to 4-(tert-butoxycarbonylamino)-1-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)piperidine-4-carboxylic acid (Intermediate 1) (4 g, 11.07 mmol) and DIPEA (5.80 ml, 33.20 mmol) in DMA (40 ml). HATU (4.63 g, 12.18 mmol) was added and the resulting solution was stirred at 20° C. for 24 hours. The reaction mixture was evaporated to dryness then diluted with EtOAc (300 mL), and washed sequentially with water (50 mL) and saturated brine (50 mL). The organic layer was dried over MgSO 4, filtered and evaporated to afford crude product. The crude product was purified by flash silica chromatography, elution gradient 2 to 6% MeOH with ammonia in DCM. Pure fractions were evaporated to dryness and triturated with dioxane (40 ml) to afford (S)-tert-butyl 4-(1-(4-chlorophenyl)-3-hydroxypropylcarbamoyl)-1-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)piperidin-4-ylcarbamate (Intermediate 22) (4.82 g, 82%) as a white solid. (S)-tert-butyl 4-(1-(4-chlorophenyl)-3-hydroxypropylcarbamoyl)-1-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)piperidin-4-ylcarbamate (Intermediate 22) (4.82 g, 82%) was suspended in dioxane (40.0 ml) and 4M hydrogen chloride in dioxane (7.69 ml, 221.36 mmol) added. The reaction was stirred at ambient temperature for 2 hours. The crude product was purified by ion exchange chromatography, using an SCX column. The desired product was eluted from the column using 3.5M NH 3/MeOH and pure fractions were evaporated to dryness. The crude product was purified by preparative HPLC (Waters XBridge Prep C18 OBD column, 5 μm silica, 19 mm diameter, 100 mm length), using decreasingly polar mixtures of water (containing 1% NH 3) and MeCN as eluents. Fractions containing the desired compound were evaporated to dryness to afford (S)-4-amino-N-(1-(4-chlorophenyl)-3-hydroxypropyl)-1-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)piperidine-4-carboxamide (1.200 g, 25.3%) as a white solid.
m/z (ES+) (M+H)+=429; HPLC tR=1.67 min.
1H NMR matches previous.
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
Clinical trial number NCT04305496 for “Capivasertib+Fulvestrant vs Placebo+Fulvestrant as Treatment for Locally Advanced (Inoperable) or Metastatic HR+/HER2- Breast Cancer (CAPItello-291)” at ClinicalTrials.gov
///////Capivasertib, Truqap, FDA 2023, APPROVALS 2023, AZD 5363
Tacaciclib is a CDK inhibitor, antineoplastic effect.
The present invention is directed to methods of preparation of compound of formula (I) that is useful for inhibiting Cyclin-dependent kinase 7 (CDK7) and for treating diseases or disorders mediated thereby.
CDK7, which complexes with cyclin H and RING-finger protein MAT1, phosphorylates the cell cycle CDKs in the activation of T-loop, to promote their activities (Fisher et al., Cell., Aug 26;78(4):713- 24, 1994). As such, it has been proposed that inhibiting CDK7 would provide a potent means of inhibiting cell cycle progression, which may be especially relevant given that there is compelling evidence from gene knockout studies in mice for lack of an absolute requirement for CDK2, CDK4 and CDK6 for the cell cycle at least in most cell types (M alumbres et al., Nature Cell Biology, 11, 1275 – 1276, 2009), whilst different tumors appear to require some, but they are independent of other interphase CDKs (CDK2, CDK4 , CDK6). Recent genetic and biochemical studies have confirmed the importance of CDK7 for cell cycle progression (Larochelle. et al., Mol Cell., Mar 23;25(6):839-50. 2007; Ganuza et al., EM BO J., May 30; 31(11): 2498-510, 2012).
Cyclin-dependent kinase 7 (CDK7) activates cell cycle CDKs and is a member of the general Transcription factor II Human (TFIIH). CDK7 also plays a role in transcription and possibly in DNA repair. The trimeric Cak complex CDK7/CyclinH/MATl is also a component of TFIIH, the general transcription/DNA repair factor IIH (Morgan, DO., Annu.Rev. Cell Dev. Biol. 13, 261-91, 1997). As a TFIIH subunit, CDK7 phosphorylates the CTD (Carboxy-Terminal-Domain) of the largest subunit of RNA polymerase II (pol II). The CTD of mammalian pol (II) consists of 52 heptad repeats with the consensus sequence 1 YSPTSPS 7 and the phosphorylation status of the Ser residues at positions 2 and 5 has been shown to be
important in the activation of RNAP-II indicating that it is likely to have a crucial role in the function of the CTD. CDK7, which primarily phosphorylates Ser-5 (PSS) of RNAP-II at the promoter as part of transcriptional initiation (Gomes et ah, Genes Dev. 2006 Mar 1; 20(5):601-12, 2006), in contrast with CDK9, which phosphorylates both Ser-2 and Ser-5 of the CTD heptad (Pinhero et al., Eur. J. Biochem., 271, pp. 1004-1014, 2004).
In addition to CDK7, other CDKs have been reported to phosphorylate and regulate RNA pol (II) CTD. The other CDKs include, Cdk9/ Cyclin T1 or T2 that constitute the active form of the positive transcription elongation factor (P-TEFb) (Peterlin and Price, Mol Cell., Aug 4; 23(3): 297-305,2006) and Cdkl2/Cyclin K and Cdkl3/Cyclin K as the latest members of RNAPII CTD kinases (Bartkowiak et al., Genes Dev., Oct 1 5;24(20):2303-16, 2010; Blazek et al., Genes Dev .Oct 15;25(20):2158-72, 2011).
Disruption of RNAP II CTD phosphorylation has been shown to preferentially effect proteins with short half-lives, including those of the anti-apoptotic BCL-2 family. (Konig et al., Blood, 1, 4307-4312, 1997; The transcriptional non-selective cyclin-dependent kinase inhibitor flavopiridol induces apoptosis in multiple myeloma cells through transcriptional repression and down-regulation of Mcl-1; (Gojoet al., Clin. Cancer Res. 8, 3527-3538, 2002).
This suggests that the CDK7 enzyme complexes are involved in multiple functions in the cell: cell cycle control, transcription regulation and DNA repair. It is surprising to find one kinase involved in such diverse cellular processes, some of which are even mutually exclusive. It also is puzzling that multiple attempts to find cell cycle dependent changes in CDK7 kinase activity remained unsuccessful. This is unexpected since activity and phosphorylation state of its substrate, CDC2, fluctuate during the cell cycle. In fact, it is shown that cdk7 activity is required for the activation of both Cdc2/Cyclin A and Cdc2/Cyclin B complexes, and for cell division. (Larochelle, S. et al. Genes Dev 12,370-81, 1998). Indeed, flavopiridol, a non-selective pan-CDK inhibitor that targets CTD kinases, has demonstrated efficacy for the treatment of chronic lymphocytic leukemia (CLL), but suffers from a poor toxicity profile (Lin et al.,). 27, 6012-6018, 2009; Christian et al., Clin. Lymphoma Myeloma, 9, Suppl.
3, S179-S185, 2009).
International publication WO2016193939, which is incorporated herein by reference for all purposes describes CDK7 inhibitors and processes for the preparation thereof. Inhibitors of CDK7 are currently being developed for the treatment of cancer. For drug development, it is typically advantageous to employ individual stereoisomers as they exhibit marked differences in pharmacodynamic, pharmacokinetic, and toxicological properties.
Example- 1: Preparation of compound of formula (I)
Scheme-1: Preparation of KRM-A
Step-4
KRM-A 4
Step-1: Preparation of 2-(3-bromophenyl)-3-methylbutanoic acid (1)
2M LDA (698 mL, 1.38mol) was added to a solution of 2-(3-bromophenyl) acetic acid (XA, 150 g, 0.69 mol) in THF (700mL) at -78 °C over a period of 30 min. The reaction mixture was stirred for 2h at -78 °C followed by a drop wise addition of isopropyl bromide (X B , 255 g, 2.07 mol) over a period of 30 min. The reaction mixture was stirred at room temperature overnight. Then, the reaction mixture was quenched with IN HC1 (pH 2) and the obtained product was extracted to ethyl acetate (500 mL x 3). The combined organic layer was washed with water followed by brine solution. The organic layer was dried over anhydrous sodium sulfate and concentrated under reduced pressure to afford the crude compound which was purified by silica column by eluting with 0-10% ethyl acetate-hexane system to afford the title compound (150 g, 83% yield) , HPLC purity-96%. The compound of formula (1) can also be prepared by the procedure described in CN 110590747.
Step-2: Preparation of Compound 3
2-(3-bromophenyl)-3-methylbutanoic acid (1, 510 g, 1.98 mol) was dissolved in 30% of IP A in water (10.2 L; 3.06 L of IPA-7.14 L of water) and ( 1L\ 2i ?)-cyclohexane-1,2-diamine (2, 113 g, 0.9 mol) was added. The reaction mixture was stirred at room temperature for 10 min until the precipitation was observed, then was heated to 100 °C until the solution became clear and stirred at the same temperature for another 30 min. The reaction mixture was allowed to slowly reach room temperature for 8-12h. The obtained solid was filtered and washed with 500 mL of 30% IPA-water mixture and dried under vacuum to afford the compound 3 (620 g, wet).
Work up (for Chiral purity): Small portion (100 mg) of compound 3 was taken in DCM (2-3 mL) and was added IN HC1 (pH 2) at 0 °C until the clear solution was observed. The compound was extracted into DCM, dried over NaiSCL and the solvent was evaporated to afford the title compound as white solid (20 mg). Chiral HPLC was recorded for this sample and 20.6% of undesired isomer was observed in chiral HPLC.
In order to improve the chiral purity of the title compound, the recrystallization method was performed as described below.
Step-3: Recrystallization
The compound 3 (619.90 g) was taken in 30% of IP A in water (12.4 L), then the mixture was heated to 100 °C until the solution became clear and was stirred at the same temperature for another 30min. The reaction mixture was allowed to reach room temperature slowly for 8-12h.
The obtained solid was filtered and washed with 500mL 30% IPA-water and dried under vacuum to afford a desired compound (360g, wet).
Work up for analysis (for Chiral purity): Small portion (100 mg) from above compound was taken in DCM (2-3mL), was added IN HC1 (pH 2) at 0 °C until the clear solution was observed and the compound was extracted to DCM, dried over NaiSCL and the solvent was evaporated to afford title compound as white solid (35 mg). Chiral HPLC was recorded for this sample and 10.3% of undesired isomer was observed in chiral HPLC.
The recrystallization method was repeated for three more times by using 30% of IPA in water as per the aforesaid procedure to obtain the purity of greater than 98.50% ee along with 0.27% other isomer to afford 286 g of compound 4.
Step-4: Preparation of (S)-2-(3-bromophenyl)-3-methylbutanoic acid (KRM-A)
The compound 4 (286 g) was taken in DCM (1.3 L), then was added IN HC1 at 0 °C until the clear solution was observed, and the compound was extracted to DCM (500 mL x 2). The organic layer was separated, washed with brine solution (500 mL) and dried over NaiSCL. The solvent was evaporated from the reaction mixture to afford title compound as white solid (148 g, 60% yield). Chiral HPLC: 98.50%
Step-1: Synthesis of (S)-2-(3-bromophenyl)-N-(5-cyclopropyl-1H-pyrazol-3-yl)-3-methylbutanamide
Step-la: Preparation ofKRM-D
To a stirred solution of KRM-A (lOOg, O.388mol) in dry DCM (600 mL, 6 vol), a catalytic amount of DMF (10 mL) was added followed by oxalyl chloride (45 mL, 0.525 mol) dropwise at 0°C over a period of 30 min. After completion of addition, the reaction mixture was stirred for 15 min at the same temperature. The reaction mixture was allowed to reach room temperature and stirred for 2 to 4h. After completion of the reaction (reaction was monitored by TLC, acid chloride formation was checked by quenching an aliquot of reaction mixture with MeOH), the reaction mixture was concentrated under vacuum at 40°C-45°C to afford crude (S)- 2-(3-bromophenyl)-3-methylbutanoyl chloride (KRM-D). The crude KRM-D was dissolved in toluene (500mL) and used for next step.
Step-lb: Preparation of compound of formula (II)
(5)-2-(3-bromophcnyl)-3-mcthylbutanoyl chloride in toluene was added slowly to a pre-cooled solution (0 to 5 °C) of ieri-butyl 3-amino-5-cyclopropyl-1H-pyrazole- l-carboxylate (KRM-B, 95.5g, 0.427 mol) and N, N-diisopropylethyl amine (100 mL, 0.583 mol) in toluene (1.2 L) at 0 °C for the period of l-2h. The reaction mixture was allowed to reach RT and stirred overnight. The reaction mixture was then cooled to 0-5°C and washed with ice-cold 1.5N HC1 (3 x 500 mL). The organic layer was washed with sodium bicarbonate solution (500 mL), brine solution (500 mL), dried over anhydrous NaiSCL , filtered and concentrated under vacuum at 45-50°C to afford crude tert-butyl (S)-5-( 2-(3-bromophenyl)-3-methylbutanamido)-3-
cyclopropyl- lH-pyrazole- 1-carboxylate (compound of formula (IG)) as light brown oil (~180g, LCMS: m/z= 461.9 (M+H) + , HPLC: 80.80%, retention time:15.89 min) . The crude product was taken as such for next step without further purification.
Step-1 c: Preparation of compound of formula (I)
To a suspension of tert-butyl (S)-5-(2-(3-bromophenyl)-3-methylbutanamido)-3-cyclopropyl-1H-pyrazole-1-carboxylate (180 g, 1,731 mol) in dioxane (360 mL ) was added 2N aqueous HC1 (360 mL) at 0 °C. The reaction mixture was stirred overnight at room temperature. After completion of the reaction, dioxane was concentrated, and the reaction mixture was diluted with water (500 mL) and basified with solid sodium bicarbonate (until pH-8). The obtained compound was extracted with DCM (700 mL x 3). The combined organic layers were washed with water (300 mL), brine solution (300 mL), and dried over anhydrous NaiSCL . The organic layer was concentrated to obtain a crude (S)-2-(3-bromophenyl)-N-(5-cyclopropyl-lH-pyrazol-3-yl)-3-methylbutanamide (Compound of formula (G)) as a semi-solid. The crude was dissolved in toluene (500 mL) and the solution was stirred for 18 h. The obtained solid was filtered and washed with toluene (100 mL) and n-heptane (200 mL). The solid was further dried under vacuum at 45-50°C for 6 h to afford a title compound (1 lOg, Yield: 78% over two steps). LCMS: m/z= 362 (M+H) + , HPLC: 97.66%, retention time: 24.10 min
Step-2: Preparation of (S, E)-N-(5-(3-(l-((5-cyclopropyl-lH-pyrazol-3-yl) amino)-3-methyl-l-oxobutan-2- yl) phenyl) pyridin-2-yl)-4-morpholinobut-2-enamide (Compound of formula (I))
To a degassed solution of (5)-2-(3-bromophcnyl)-N-(5-cyclopropyl-1 H-pyrazol-3-yl)-3-methylbutanamide (50 g, 0.138 mol) and (E)-4 -morpholino-N-(5-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)pyridin-2-yl)but-2-enamide (KRM-C, 56.6 g, 0.151 mol, 1.1 eq) (prepared according to the procedure described in W02020202001) in 1,4-dioxane (500 mL, 10 vol) and water (100 mL, 2 vol) was added K3PO4 tribasic (73.2 g, 0.345 mol, 2.5 eq) at room temperature The reaction mass was stirred for 20 min with argon purging (degassing). Pd(dppf)Ch.DCM (3.38 g, 0.0042 mol, and 0.03eq) was added to the reaction mixture and the reaction mixture was heated to 90°C for 1-2 h (The reaction was monitored by TLC using 10% methanol in DCM as solvent system).
After completion of the reaction, the reaction mass was cooled to room temperature and filtered through Celite ® bed. The bed was washed with 1, 4-dioxane (200 mL) and the filtrate was concentrated to get crude compound. The crude compound was dissolved in 5% methanol in DCM (400 mL) and washed with water (200 mL x 2). The aqueous layer was separated and
extracted with DCM (100 mL x 2). The combined organic layer was washed with brine solution, filtered and dried over sodium sulfate. The organic layer was concentrated under vacuum at 35-40°C to obtain crude title compound (~80g).
The crude compound of formula (I), (80 g) was dissolved in 700 mL of ethyl acetate. The reaction mixture was cooled to 15°C and 2N HC1 was slowly added (until pH ~1). The reaction mixture was then stirred at room temperature for 20 min and the layers were separated. The aqueous layer (containing the product) was washed with ethyl acetate (300 mL x 3). The aqueous layer was cooled to 0°C and adjusted the pH to ~8 using 20% aqueous NaiCCL solution. The product was extracted with 10% methanol in DCM (300 mL x 3). The combined organic layer was washed with water (300 mL), dried over sodium sulfate and filtered. The filtrate was treated with activated charcoal (16 g, 20% w/w with respect to crude input of 80 g), then the reaction mixture was stirred overnight at room temperature and filtered through Celite ® bed. The bed was washed with 5% methanol in DCM (~ 20 vol, until absence of product by TLC). The filtrate was concentrated under vacuum at 35°C – 40°C to afford compound of formula (I) (70g, HPLC purity: 92.70%, retention time: 15.65 min).
Work-up for improved chiral purity: The above compound of formula (I) was dissolved in ethylacetate (~30 vol, 2L) and washed with aqueous citric acid (2 times, 400 mL x 1 and 200mL x 1), aqueous NaHCCL solution (2%, 500 mL x 1) and aqueous NaCl solution (10%, 500 mL x 1). The combined organic layer was dried over sodium sulfate and filtered. The filtrate was concentrated under vacuum at 35°C – 40°C to afford compound of formula (I) (~60g).
In some embodiments, the compound of formula (I) is (E)-N-(5-(3-(l-((5-cyclopropyl-lH-pyrazol-3-yl)amino)-3-methyl-l-oxobutan-2-yl)phenyl)pyridin-2-yl)-4-morpholinobut-2-enamide or a pharmaceutically acceptable salt or a stereoisomer thereof (Compound 44).
Compound 44 is disclosed in WO 2016/193939 Al, published December 8, 2016, entitled “Substituted heterocyclyl derivatives as cdk inhibitors,” the entire contents of which are incorporated herein by reference. Compound 44A can be in the form of a fumaric acid salt or cocrystal as described in WO 2022/130304 Al, published June 23, 2022, entitled “Cocrystal of a cdk inhibitor,” the entire contents of which are incorporated herein by reference.
Example 3: Synthesis of Compounds 44A & 44B via Chiral Separation
Scheme-1
Step-1: Synthesis of 2-(3-bromophenyl)-3-methylbutanoic acid
[0352] 2M LDA (698 mL, 1.38mol) was added to a solution of 2-(3 -bromophenyl) acetic acid (reagent-1, 150g, 0.69mol) in THF (700mL) at -78 °C over a period of 30 min. The reaction mass was stirred for 2h at -78 °C followed by the drop wise addition of Isopropyl bromide (255 g, 2.07mol) over a period of 30 min at -78 °C. The reaction mass was stirred at room temperature for overnight. The reaction mass was quenched with IN HC1 (pH 2) and product extracted to ethyl acetate (500mL x 3). The combined organic layer washed with water followed by brine, dried and concentrated under reduced pressure to afford the title crude compound which was purified by silica column by eluting with 0-10% ethyl acetate -hexane system to afford the title compound 2 (150g, 83% yield). LCMS: m/z = 254.80 (M-2H)’
Step-2: Synthesis of tert-butyl 3-(2-(3-bromophenyl)-3-methylbutanamido)-5-cyclopropyl-lH-pyrazole-1 -carboxylate
[0353] 2-(3-bromophenyl)-3-methylbutanoic acid (intermediate-2, 70g, 0.0.27mol) was dissolved in dry DCM (500 mL) and added oxalyl chloride (68 mL, 0.78mol) dropwise at 0 °C followed by addition of catalytic amount of DMF (0.8mL) and maintained reaction mass at same temperature for 30min. The reaction mass was allowed to room temperature and stirred for 4h, distilled off the solvent and excess oxalyl chloride under vacuum. Re-dissolved the residue in DCM (250 mL) and added slowly to the cooled solution of tert-butyl 3 -amino-5 -cyclopropyl- 1H-pyrazole-1 -carboxylate (intermediate-3, 49g, 0.218mol) and TEA (55 mL, 0.546mol) in THF (250 mL) at 0 °C for 30min, The reaction was stirred at room temperature for 12h then the reaction mass was concentrated under reduced pressure and the residue was dissolved in DCM, washed with saturated NaHCO3 solution and brine. The organic layer was dried over anhydrous sodium sulphate and concentrated under reduced pressure, the crude was purified by silica gel column chromatography by eluting with 15% ethyl acetate-hexane to afford the title compound 4 (90g, 71% ) LCMS: m/z = 363.80 (M-Boc+2).
Step-3: Synthesis of tert-butyl 5-cyclopropyl-3-(3-methyl-2-(3-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)phenyl)butanamido)-l H -pyr azole- 1 -carboxylate
[0354] To a degassed solution of tert-butyl 3-(2-(3-bromophenyl)-3-methylbutanamido)-5-cyclopropyl-lH-pyrazole-1 -carboxylate (intermediate-4, 90g, 0.193mol) and 4, 4, 4′, 4′, 5, 5, 5′, 5′-octamethyl-2,2′-bi(l,3,2-dioxaborolane) (62g, 0.25 Imol) in 1,4-Dioxane (500 mL) was added potassium acetate (37.80g, 0.386mol). The reaction mass was allowed to stir for 10 min with degassing at RT and added PdC12(dppf).DCM complex (12.5g, 0.015mol). The reaction mass was heated for 3-4 h at 100 °C. Reaction mixture cooled to RT and filtered on celite bed, filtrate evaporated to get dark brown liquid. The crude material was purified by silica column chromatography by eluting with 20% ethyl acetate in hexane to afford the compound 5 (90g, 86%). LCMS: m/z = 410 (M-Boc+1)+.
Step-4: Synthesis of (E)-N-(5-(3-(l-((5-cyclopropyl-lH-pyrazol-3-yl)amino)-3-methyl-l-oxobutan-2-yl)phenyl)pyridin-2-yl)-4-morpholinobut-2-enamide
[0355] To a degassed solution of tert-butyl 5-cyclopropyl-3-(3-methyl-2-(3-(4, 4,5,5-tetramethyl- 1 ,3 ,2-dioxaborolan-2-yl)phenyl)butanamido)- 1 H-pyrazole- 1 -carboxylate, 5 (10g, 0.019mol) and (E)-N-(5-bromopyridin-2-yl)-4-morpholinobut-2-enamide (7.7g, 0.023mol) in
1,4-Dioxane (lOOmL) and water (40mL) followed by Cs2CO3 (14.5g, 0.045mol) were added. The reaction mass was allowed to stir for 10 min with degassing and added Pd(PPh3)4 (1.1g, 0.00095mol), heated the reaction mass for 4 h at 100 °C in a sealed tube. The reaction mass was cooled and diluted with brine solution. The aqueous layer was separated and re-extracted with ethyl acetate. The combined organic layer was evaporated to dryness and crude material was purified by silica column chromatography by eluting with 10%-l 5 % methanol in DCM to get desired pure compound 44 (4.5g, 44%). LCMS: m/z = 529.15 (M+H)+; HPLC: 95.17%, rt: 6.34 min.
[0356] Racemic (E)-N-(5 -(3 -( 1 -((5 -cyclopropyl- 1 H-pyrazol-3 -yl)amino)-3 -methyl- 1 -oxobutan-2-yl)phenyl)pyridin-2-yl)-4-morpholinobut-2-enamide was separated by using chiral preparative HPLC column (Method: Column: Chiral Pak IA (20mm X 250 mm, 5 micron), Elution: isocratic (50:50), A=ACN, B= MeOH, Flow: 20mL/min ) to afford the pure Isomer- 1 and Isomer-2.
Example 4: Preparation of Compound 44-A via Chiral Synthesis
Preparation of KRM-A (chemical precursor to Compound 44-A)
Step-4
KRM-A
Step-1: Preparation of 2-(3-bromophenyl)-3-methylbutanoic acid (1)
[0359] 2M LDA (698 mL, 1.38mol) was added to a solution of 2-(3 -bromophenyl) acetic acid (150 g, 0.69 mol) in THF (700mL) at -78 °C over a period of 30 min. The reaction mixture was stirred for 2h at -78 °C followed by drop wise addition of isopropyl bromide (XB, 255 g, 2.07 mol) over a period of 30 min at -78 °C. The reaction mass was stirred at room temperature overnight. The reaction mass was quenched with IN HC1 (pH 2) and the obtained product was extracted to ethyl acetate (500 mL x 3). The combined organic layer was washed with water followed by brine, dried over anhydrous sodium sulfate and concentrated under reduced pressure to afford the title crude compound which was purified by silica column by eluting with 0-10% ethyl acetate -hexane system to afford the title compound (150 g, 83% yield), HPLC purity-96%. The compound of formula (1) can also be prepared by the procedure described in CN110590747.
Step-2: Preparation of Compound 3
[0360] 2-(3-bromophenyl)-3-methylbutanoic acid (1, 510 g, 1.98 mol) was dissolved in 30% of IPA in water (10.2 L; 3.06 L of IPA-7.14 L of water) and (1R, 27?)-cyclohexane-l,2-diamine (2, 113 g, 0.9 mol) was added. The reaction mixture was stirred at room temperature for 10 min until the precipitation was observed, then heated to 100 °C till the solution becomes clear and was stirred at same temperature for another 30 min. The reaction mixture was allowed to attain room temperature slowly for 8-12h. The obtained solid was filtered and washed with 500 mL of 30% IPA-water mixture and dried under vacuum to afford the compound 3 (620 g, wet).
[0361] Work up for analysis (for Chiral purity): Small portion (100 mg) of compound 3 was taken in DCM (2-3 mL) and was added IN HC1 (pH 2) at 0 °C till the clear solution was observed. The compound was extracted into DCM, dried over Na2SC>4 and the solvent was evaporated to afford the title compound as white solid (20 mg). Chiral HPLC was recorded for this sample and 20.6% of undesired isomer was observed in chiral HPLC.
[0362] In order to improve the chiral purity of the title compound, the recrystallization method was performed as described below.
Step-3: Recrystallization
[0363] The compound 3 (619.90 g) was taken in 30% of IPA in water (12.4 L), then the mixture was heated to 100 °C till the solution becomes clear and stirred at same temperature for another 30min. The reaction mixture was allowed to attain room temperature slowly for 8-12h. The obtained solid was filtered and washed with 500mL 30% IP A- water and dried under vacuum to afford a desired compound (360g, wet).
[0364] Work up for analysis (for Chiral purity): Small portion (100 mg) from above compound was taken in DCM (2-3mL), was added IN HC1 (pH 2) at 0 °C till the clear solution was observed and the compound was extracted to DCM, dried over Na2SCL and the solvent was evaporated to afford title compound as white solid (35 mg). Chiral HPLC was recorded for this sample and 10.3% of undesired isomer was observed in chiral HPLC.
[0365] The recrystallization method was repeated for three more times by using 30% of IPA in water as described above to get the purity >98.50% ee along with 0.27% other isomer to afford 286 g of compound 4.
Step-4: Preparation of (S)-2-(3-bromophenyl)-3-methylbutanoic acid (KRM-A)
[0366] The compound 4 (286 g) was taken in DCM (1.3 L), then was added IN HC1 at 0 °C until the clear solution was observed, and the compound was extracted to DCM (500 mL x 2). The organic layer was separated and washed brine solution (500 mL) and dried over Na2SO4, the solvent was evaporated to afford title compound as white solid (148 g, 60% yield). Chiral HPLC: 98.50%
Step-1: Synthesis of (S)-2-(3-bromophenyl)-N-(5-cyclopropyl-lH-pyrazol-3-yl)-3-methylhutanamide
Step- la: Preparation of KRM-D
[0368] To a stirred solution of KRM-A (100g, 0.388mol) in dry DCM (600 mL, 6 vol), a catalytic amount of DMF (10 mL) was added followed by oxalyl chloride (45 mL, 0.525 mol) dropwise at 0 °C over a period of 30 min. After completion of addition, the reaction mixture was stirred for 15 min at the same temperature. The reaction mixture was allowed to reach room temperature and stirred for 2 to 4h. After completion of the reaction (reaction was monitored by TLC, acid chloride formation was checked by quenching an aliquot of reaction mixture with MeOH), the reaction mixture was concentrated under vacuum at 40°C-45°C to afford crude 2-(3-bromophenyl)-3 -methylbutanoyl chloride (KRM-D). The crude KRM-D was dissolved in toluene (500mL) and used for next step.
Step- lb: Preparation of compound of formula Z
[0369] (S)-2-(3-bromophenyl)-3 -methylbutanoyl chloride in toluene was added slowly to a pre-cooled solution (0 to 5 °C) of te/7-butyl 3 -amino-5 -cyclopropyl- IH-pyrazole-l -carboxylate (KRM-B, 95.5g, 0.427 mol) and N, N-diisopropylethyl amine (100 mL, 0.583 mol) in toluene (1.2 L) at 0 °C for the period of l-2h. The reaction mixture was allowed to attain RT and stirred for overnight. The reaction mixture was then cooled to 0-5°C and washed with ice-cold 1.5N HCI (3 x 500 mL). The organic layer was washed with sodium bicarbonate solution (500 mL),
brine solution (500 mL), dried over anhydrous Na2SO4, filtered and concentrated under vacuum at 45-50°C to afford crude tert-butyl (5)-5-(2-(3-bromophenyl)-3-methylbutanamido)-3-cyclopropyl-lH-pyrazole-1 -carboxylate (compound of formula Z) as light brown oil (~180g, LCMS: m/z= 461.9 (M+H)+, HPLC: 80.80%, retention time: 15.89 min). The crude product was taken as such for next step without further purification.
Step-lc: Preparation of compound of formula Y
[0370] To a suspension of tert-butyl (S)-5-(2-(3-bromophenyl)-3-methylbutanamido)-3-cyclopropyl-lH-pyrazole-1 -carboxylate (180 g, 1.731 mol) in dioxane (360 mL) was added 2N aqueous HC1 (360 mL) at 0 °C. The reaction mixture was stirred overnight at room temperature.
[0371] After completion of the reaction, dioxane was concentrated, and the reaction mixture was diluted with water (500 mL) and basified with solid sodium bicarbonate (until pH-8). The resulted compound was extracted with DCM (700 mL x 3). The combined organic layers were washed with water (300 mL) and brine solution (300 mL), and dried over anhydrous Na2SO4. The organic layer was concentrated to get a crude (<S)-2-(3-bromophenyl)-N-(5-cyclopropyl-lH-pyrazol-3-yl)-3-methylbutanamide (Compound of formula Y) as a semi solid. The crude was dissolved in toluene (500 mL) and the solution was stirred for 18 h. The solid formed was filtered and washed with toluene (100 mL) and n-heptane (200 mL). The solid was further dried under vacuum at 45-50°C for 6 h to afford a title compound (110g, Yield: 78% over two steps). LCMS: m/z= 362 (M+H)+, HPLC: 97.66%, retention time: 24.10 min
[0373] To a degassed solution of (<S)-2-(3-bromophenyl)-N-(5-cyclopropyl-lH-pyrazol-3-yl)-3-methylbutanamide (50 g, 0.138 mol) and (£)-4-morpholino-N-(5-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)pyridin-2-yl)but-2-enamide (KRM-C, 56.6 g, 0.151 mol, 1.1 eq) (prepared according to the procedure described in W02020202001) in 1,4-dioxane (500 mL, 10 vol) and water (100 mL, 2 vol) was added K3PO4 tribasic (73.2 g, 0.345 mol, 2.5 eq) at room temperature The reaction mass was stirred for 20 min with argon purging (degassing). Pd(dppf)C12.DCM [l,l’-Bis(diphenylphosphino)ferrocene]dichloropalladium(II) complex with dichloromethane] (3.38 g, 0.0042 mol, and 0.03eq) was added and the reaction mixture was heated to 90°C for 1-2 h (The reaction was monitored by TLC using 10% methanol in DCM as solvent system).
[0374] After completion of the reaction, the reaction mass was cooled to room temperature and filtered through Celite® bed. The bed was washed with 1, 4-dioxane (200 mL) and the filtrate was concentrated to get crude compound. The crude compound was dissolved in 5% methanol in DCM (400 mL) and washed with water (200 mL x 2). The aqueous layer was separated and extracted with DCM (100 mL x 2). The combined organic layer was washed with brine solution, filtered and dried over sodium sulphate. The organic layer was concentrated under vacuum at 35-40°C to get crude title compound (~80g).
[0375] The crude compound 44A, (80 g) was dissolved in 700 mL of ethyl acetate. The reaction mixture was cooled to 15°C and 2N HC1 was slowly added (until pH ~1). The reaction mixture was then stirred at room temperature for 20 min and the layers were separated. The aqueous layer (containing the product) was washed with ethyl acetate (300 mL x 3). The aqueous layer was cooled to 0°C and adjusted the pH to ~8 using 20 % aqueous Na2COs solution. The product was extracted with 10% methanol in DCM (300 mL x 3). The combined organic layer was washed with water (300 mL), dried over sodium sulphate and filtered. The filtrate was treated with activated charcoal (16 g, 20% w/w with respect to crude input of 80 g), stirred overnight at room temperature and filtered through Celite® bed. The bed was washed with 5% methanol in DCM (~ 20 vol, till absence of product by TLC). The filtrate was concentrated under vacuum at 35°C – 40°C to afford compound 44A (70g, HPLC purity: 92.70%, retention time: 15.65 min).
Sotuletinib, also known as BLZ945, is a potent and selective CSF-1R kinase inhibitor. BLZ945 showed effects of CSF1R inhibition on other tumor-infiltrating immune cells. BLZ945 attenuates the turnover rate of TAMs while increasing the number of CD8+ T cells that infiltrate cervical and breast carcinomas. BLZ945 decreases the growth of malignant cells in the mouse mammary tumor virus-driven polyomavirus middle T antigen (MMTV-PyMT) model of mammary carcinogenesis. BLZ945 prevents tumor progression in the keratin 14-expressing human papillomavirus type 16 (K14-HPV-16) transgenic model of cervical carcinogenesis.
Sotuletinib (BLZ945) is an experimental drug in development for the treatment of amyotrophic lateral sclerosis (ALS). It works as a colony-stimulating factor 1 (CSF1) receptor inhibitor.[1][2][3]
OriginatorCelgene Corporation; Novartis
ClassAmides; Amines; Antineoplastics; Benzothiazoles; Cyclohexanols; Ethers; Pyridines; Small molecules
Mechanism of ActionMacrophage colony stimulating factor receptor antagonists
Phase IIAmyotrophic lateral sclerosis
Phase I/IISolid tumours
05 Dec 2022Novartis Pharmaceuticals terminates a phase I/II trials in Solid tumours (Combination therapy, Late-stage disease, Metastatic disease) in Taiwan, Japan, Israel (PO) in US, Israel, Italy, Japan, Singapore, Spain, Taiwan and Switzerland (EudraCT2015-005806-12) (NCT02829723)
14 Feb 2022Adverse events and pharmacodynamics data from preclinical macaque model study in brain disorders presented at the 29th Conference on Retroviruses and Opportunistic Infections
03 Dec 2020Chemical structure information added
An orally bioavailable inhibitor of colony stimulating factor 1 receptor (CSF-1R; CSF1R), with potential antineoplastic activity. CSF1R inhibitor BLZ945 selectively binds to CSF1R expressed on tumor-associated macrophages (TAMs), blocks the activity of CSF1R, and inhibits CSF1R-mediated signal transduction pathways. This inhibits the activity and proliferation of TAMs, and reprograms the immunosuppressive nature of existing TAMs. Altogether, this reduces TAM-mediated immune suppression in the tumor microenvironment, re-activates the immune system, and improves anti-tumor cell responses mediated by T-cells. CSF1R, also known as macrophage colony-stimulating factor receptor (M-CSFR) and CD115 (cluster of differentiation 115), is a cell-surface receptor for its ligand, colony stimulating factor 1 (CSF1); this receptor is overexpressed by TAMs in the tumor microenvironment, and plays a major role in both immune suppression and the induction of tumor cell proliferation.
PATENT
The free base and salts of the compound of formula (I) may be prepared for example, according to the procedures given in International Patent Application No. PCT/US2007/066898 filed on Apr. 18, 2007 and published as WO2007/121484 on Oct. 25, 2007. The compound of formula (I) has the chemical name: 4-(2-((1R,2R)-2-hydroxycyclohexylamino)benzothiazol-6-yloxy)-N-methylpicolinamide and is also known as BLZ945.
Step 1. Preparation of 4-(2-((lR,2R)-2-aminocyclohexylamino)benzo[d]thiazol-6-yloxy)-N-methylpicolinamide To the solution of N-methyl-4-(2-(methylsulfinyl)benzo[d]thiazol-6-yloxy)picolinamide (15 mg, 43 μmole) in 400 μL of NMP was added (lR,2R)-cyclohexane-1,2-diamine (17 mg, 150 μmole). The reaction solution was stirred at 105°c for 24 hours. The crude reaction solution was purified on prep HPLC and evaporated in vaccuo to give 4-(2-((lR,2R)-2-aminocyclohexylamino)benzo[d]thiazol-6-yloxy)-N-methylpicolinamide (12 mg, 30 μmole) as white powder. ES/MS m/z 398.1(MH+).
^ Martinez-Gonzalez, Loreto; Martinez, Ana (1 February 2023). “Emerging clinical investigational drugs for the treatment of amyotrophic lateral sclerosis”. Expert Opinion on Investigational Drugs. 32 (2): 141–160. doi:10.1080/13543784.2023.2178416.
useful in the treatment of a steroid receptor, in particular androgen receptor (AR), dependent conditions and diseases, and to pharmaceutical compositions containing such compounds.
Prostate cancer is worldwide the most common cancer in men. Even though the 5-year survival rate of patients with localized prostate cancer is high, the prognosis for those patients, who develop castration-resistant prostate cancer (CRPC) within that 5-year follow-up period, is poor.
The androgen receptor (AR) signalling axis is critical in all stages of prostate cancer. In the CPRC stage, disease is characterized by high AR expression, AR amplification and persistent activation of the AR signalling axis by residual tissue/tumor androgens and by other steroid hormones and intermediates of steroid biosynthesis. Thus, treatment of advanced prostate cancer involves androgen deprivation therapy (ADT) such as hormonal manipulation using gonadotropin-releasing hormone (GnRH) agonists/antagonists or surgical castration, AR antagonists or CYP17A1 inhibitors (such as abiraterone acetate in combination with prednisone).
Although therapies can initially lead to disease regression, eventually majority of the patients develop a disease that is refractory to currently available therapies. Increased progesterone levels in patients treated with abiraterone acetate has been hypothesized to be one of the resistance mechanisms. Several nonclinical and clinical studies have indicated upregulation of enzymes that catalyse steroid biosynthesis at the late stage of CRPC. Very recently it has been published that 11β-OH androstenedione can be
metabolized into 11-ketotestosterone (11-K-T) and 11-ketodehydrotestosterone (11-K-DHT) which can bind and activate AR as efficiently as testosterone and dihydrotestosterone. It has been shown that these steroids are found in high levels in plasma and tissue in prostate cancer patients, suggesting their role as AR agonists in CRPC. Furthermore, it has been addressed that prostate cancer resistance to CYP17A1 inhibition may still remain steroid dependent and responsive to therapies that can further suppress de novo intratumoral steroid synthesis upstream of CYP17A1, such as by CYP11A1 inhibition therapy (Cai, C. et al, Cancer Res., 71(20), 6503-6513, 2011).
Cytochrome P450 monooxygenase 11A1 (CYP11A1), also called cholesterol side chain cleavage enzyme, is a mitochondrial monooxygenase which catalyses the conversion of cholesterol to pregnenolone, the precursor of all steroid hormones. By inhibiting CYP11A1, the key enzyme of steroid biosynthesis upstream of CYP17A1, the total block of the whole steroid biosynthesis can be achieved. CYP11A1 inhibitors may therefore have a great potential for treating steroid hormone dependent cancers, such as prostate cancer, even in advanced stages of the disease, and especially in those patients who appear to be hormone refractory. It has been recently shown that a compound having CYP11A1 inhibitory effect significantly inhibited tumor growth in vivo in a murine CRPC xenograft model (Oksala, R. et al, Annals of Oncology, (2017) 28 (suppl.
To a solution of 5-hydroxy-2-(isoindolin-2-ylmethyl)-4H-pyran-4-one (0.10 g, 0.41 mmol) in DMF (2 ml) were added (4-(methylsulfonamidomethyl)cyclohexyl)methyl methanesulfonate (0.14 g, 0.45 mmol) and K2CO3 (0.12 g, 0.8 mmol). The reaction mixture was heated at 80 °C for 2 h. The mixture was cooled to RT, water (10 ml) was added and the product was extracted with EtOAc. The combined extracts were washed with water, dried with Na2SO4, filtered and evaporated. The crude product was purified by column chromatography to afford the title compound (0.06 g). 1H NMR (400 MHz, Chloroform-d) δ ppm 0.92 – 1.11 (m, 4 H) 1.40 – 1.63 (m, 2 H) 1.78 – 2.00 (m, 4 H) 2.91 – 2.99 (m, 5 H) 3.65 (d, J=6.46 Hz, 2 H) 3.77 (s, 2 H) 4.03 (s, 4 H) 5.04 (br t, J=6.31 Hz, 1 H) 6.49 (s, 1 H) 7.20 (s, 4 H) 7.59 (s, 1 H).
To a stirred solution of 2-(chloromethyl)-5-hydroxy-4H-pyran-4-one (2.0 g, 12.5 mmol) in acetonitrile (50 mL) were added DIPEA (3.22 mL, 25.0 mmol) and isoindoline (1.78 g, 25.0 mmol) at RT. When the reaction was complete, the precipitated solid was filtered and washed with EtOAc. The title compound was collected as pale brown solid (1.1 g). LC-MS: m/z 244.1 (M+H)+.
///////////////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
The rising global epidemic of obesity and its comorbidities, e.g., type 2 diabetes mellitus and hyperlipidemia, is placing an enormous burden both on public health (mortality and morbidity) and on the available public health resources required to treat these conditions.
Current drugs that treat hyperlipidemia (e.g., statins, omega-3 fatty acids, fibrates) have mostly neutral effects on glycemic control, whilst drugs targeting glycemic control e.g., insulin, thiazolidinediones (TZDs), have adverse effects upon bodyweight and (for TZDs) other unwanted side-effects restricting their use.
In addition to hyperlipidemia and type 2 diabetes, a marked increase in the prevalence of non-alcoholic fatty liver disease (NAFLD) has occurred. NAFLD has become the most common chronic liver condition in Western populations in relation to the obesity and type 2 diabetes epidemics. The prevalence of non-alcoholic steatohepatitis (NASH), a form of NAFLD that is associated with hepatic inflammation and ballooning of hepatocytes, is expected to increase by 63% between 2015 and 2030 in the United States (Estes, Hepatology, 2018; 67(1): 123-133), where NASH is expected to become the leading cause of liver transplantation by 2020. As liver fibrosis, but not inflammation, is associated with mortality and morbidity in NASH patients, drugs which prevent progression/induce regression of fibrosis are also a focus of biomedical research.
The development of novel compounds that simultaneously target both hyperlipidemia and glycemic control, without the adverse side-effects (e.g., weight gain) typically associated with insulin sensitising drugs is thus a desirable goal. Such compounds would be even more attractive if they could additionally prevent the progression/reverse hepatic fibrosis and reduce hepatic steatosis. The present invention addresses these needs for new treatment methods, compounds, and pharmaceutical compositions.
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
GS-443902 is a bioactive ATP analogue with broad-spectrum antiviral activity [5] and is the same compound formed by remdesivir, though by a different enzymatic pathway. Unlike remdesivir, which is metabolized by enzymes that are highly expressed in the liver, GS-441524 released by obeldesivir is metabolized by enzymes that are evenly expressed throughout the body. Due to their different metabolic pathways, obeldesivir can be administered orally, whereas remdesivir must be administered intravenously for COVID-19 treatment.[citation needed]
The pharmacokinetic properties of obeldesivir and improved was first published by Chinese researchers in May 2022. The Chinese group pursued investigation of obeldesivir independently from Gilead Sciences. Compared to IV administered GS-441524 in rats at 5 mg/kg, orally administered obeldesivir at 25 mg/kg (referred to as “ATV006”) yielded approximately 22% bioavailability.[6] Treatment with obdeldesivir reduced viral load and prevents lung pathology in KI-hACE2 and Ad5-hACE2 mouse models of SARS-CoV-2. A patent filed by Gilead Sciences with a priority date of August 27, 2020,[7] found the bioavailability of GS-441524 after oral administration of obdeldesivir (compound 15) in mice, rats, ferrets, dogs, and cynomolgus macaques to be 41%, 63.9%, 154%, 94%, and 38%, respectively. Across all species evaluated, obeldesivir showed improved oral bioavailability compared to oral administration of the parent nucleoside, GS-441524
Remdesivir 1 is an phosphoramidate prodrug that releases the monophosphate of nucleoside GS-441524 (2) into lung cells, thereby forming the bioactive triphosphate 2-NTP. 2-NTP, an analog of ATP, inhibits the SARS-CoV-2 RNA-dependent RNA polymerase replication and transcription of viral RNA. Strong clinical results for 1 have prompted interest in oral approaches to generate 2-NTP. Here, we describe the discovery of a 5′-isobutyryl ester prodrug of 2 (GS-5245, Obeldesivir, 3) that has low cellular cytotoxicity and 3–7-fold improved oral delivery of 2 in monkeys. Prodrug 3 is cleaved presystemically to provide high systemic exposures of 2 that overcome its less efficient metabolism to 2-NTP, leading to strong SARS-CoV-2 antiviral efficacy in an African green monkey infection model. Exposure-based SARS-CoV-2 efficacy relationships resulted in an estimated clinical dose of 350–400 mg twice daily. Importantly, all SARS-CoV-2 variants remain susceptible to 2, which supports development of 3 as a promising COVID-19 treatment.
Synthesis of ((2R,3S,4R,5R)-5-(4-Aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl Isobutyrate (3)
To a solution of (3aR,4R,6R,6aR)-4-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-6-(hydroxymethyl)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxole-4-carbonitrile 4 (2000 mg, 6.0 mmol) (30) and isobutyric acid (638 mg, 7.2 mmol) in DMF (5 mL), N,N′-diisopropylcarbodiimide (914 mg, 7.2 mmol) was slowly added followed by 4-(dimethylamino)pyridine (737 mg, 6.0 mmol). The reaction mixture was stirred for 4 h and then diluted with ethyl acetate, washed with water and brine, dried over sodium sulfate, and concentrated under reduced pressure. The residue was subjected to silica gel chromatography, eluting with 20% MeOH in CH2Cl2 to provide the intermediate ((3aR,4R,6R,6aR)-6-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-6-cyano-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl)methyl isobutyrate. LCMS m/z = 402.2 (M + 1). To a solution of the intermediate (1500 mg) in THF (10 mL), conc. HCl (2 mL) was added, and the mixture was stirred at rt for 4 h. The reaction mixture was diluted with CH2Cl2, washed with water, saturated aqueous bicarbonate, and brine, dried over sodium sulfate, concentrated, and subjected to silica gel chromatography, eluting with 30% MeOH in CH2Cl2 to afford the title compound (660 mg, 50%). 1H NMR (400 MHz, MeOH-d4) δ 7.88 (s, 1H), 6.96–6.85 (m, 2H), 4.50–4.27 (m, 4H), 4.16 (dd, J = 6.2, 5.3 Hz, 1H), 2.56 (p, J = 7.0 Hz, 1H), 1.14 (dd, J = 7.0, 3.8 Hz, 6H). LCMS m/z: 362.1 (M + 1). 13C NMR (400 MHz, CHCl3–d3) δ 175.9, 155.6, 147.9, 123.5, 110.2, 100.8, 116.9, 116.6, 81.3, 79.0, 74.0, 70.2, 62.9, 33.2, 18.7, 18.6. HRMS m/z: 362.14615, C16H20N5O5 362.14644.
Example 1. (3aR, 4R, 6R, 6aR)-4-(4-aminopyrrole[2,1-f][1,2,4]triazin-7-yl)-6-(hydroxymethyl- Synthesis of 2,2-dimethyltetrahydrofuran[3,4-d][1,3]dioxolyl-4-carbonitrile (compound 5)
Dissolve 5.62g of compound 69-0 in 30mL of acetone, then add 11.50mL of 2,2-dimethoxypropane and 1.34mL of 98% sulfuric acid, stir at 45°C for half an hour, cool to room temperature, and remove by rotary evaporation Organic solvents. Extract with 100 mL of ethyl acetate and 100 mL of saturated sodium bicarbonate solution. Repeat the extraction three times. Combine the ethyl acetate layers, add anhydrous sodium sulfate to dry, and filter to remove sodium sulfate. The organic solvent was removed by rotary evaporation, and 6.20 g of compound 5 (white solid, yield 97%) was obtained through column chromatography (eluent: petroleum ether/ethyl acetate (v/v) = 1/2). The hydrogen spectrum of the obtained compound 5 was detected, and the results are as follows:
Example 2. ((3aR,4R,6R,6aR)-6-(4-aminopyrrole[2,1-f][1,2,4]triazin-7-yl)-6-cyano-2 ,Synthesis of 2-dimethyltetrahydrofuran[3,4-d][1,3]dioxol-4-yl)methylisobutyrate (compound INT-1)
Dissolve 1.50g of compound 5 in 15ml of dichloromethane, then add 0.42mL of isobutyric acid and 55.40mg of 4-dimethylaminopyridine, stir for 10 minutes, add 1.02g of dicyclohexylcarbodiimide, and stir at room temperature. Stir for 24h. After column chromatography separation (eluent: petroleum ether/ethyl acetate (v/v) = 1/1), 1.71 g of compound INT-1 (white solid, yield 94%) was obtained. The hydrogen spectrum and carbon spectrum of the obtained compound INT-1 were detected, and the results are as follows:
Hydrogen spectrum: 1 H NMR (400MHz, CDCl 3 ,ZQF-RD01-2)δ(ppm):7.99(s,1H),6.99(d,J=4.6Hz,1H),6.62(d,J=4.6Hz,1H),5.72(br,2H),5.49(d,J=6.8Hz,1H),4.93-4.90(dd,J=6.8Hz,4.3Hz,1H),4.61-4.58(q,J=4.4Hz,1H),4.44-4.26(m,2H),2.61-2.50(m,1H),1.77(s,3H),1.42(s,3H),1.17-1.14(q,J=3.8Hz,6H)。
Carbon spectrum: 13 C NMR (100MHz, CDCl 3 ,ZQF-RD01-2)δ(ppm):176.7,155.2,147.3,123.5,117.2,116.7,115.6,112.6,100.0,83.8,83.0,82.0,81.4,63.1,33.8,26.4,25.6,18.9。
To a solution of (3aR,4R,6R,6aR)-4-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-6-(hydroxymethyl)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxole-4-carbonitrile, Intermediate 1 (2000 mg, 6.0 mmol) (Siegel et. al. J. Med. Chem.2017, 60, 1648-1661) in tetrahydrofuran (THF) was added N,N-dimethylaminopyridine (DMAP) (0.03 eq). To the reaction mixture, isobutyric anhydride (1.1 eq) was added slowly. After the completion of the staring material, the reaction mixture was concentrated and purified by flash chromatography using 20% methanol in DCM as an eluant to give ((3aR,4R,6R,6aR)-6-(4- aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-6-cyano-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4- yl)methyl isobutyrate, Intermediate 1a. LCMS: MS m/z: 402.2 (M+1).
Concentrated HCl (5 eq, 1mL) was added to a solution of Intermediate 1a (1000 mg) in acetonitrile (10 mL), which was then stirred at room temperature for 2 hours. LCMS showed the product formation. The reaction was stopped after 4 hours. The reaction mixture was diluted with ethyl acetate and quenched with saturated bicarbonate. The organic layer was separated, washed with brine, dried over sodium sulphate, and concentrated. The residue was purified by flash chromatography using 30% methanol DCM as an eluant. The collected fractions were concentrated to give Intermediate 2.1H NMR (400 MHz, Methanol-d4) δ 7.88 (s, 1H), 6.96 – 6.85 (m, 2H), 4.50 – 4.27 (m, 4H), 4.16 (dd, J = 6.2, 5.3 Hz, 1H), 2.56 (p, J = 7.0 Hz, 1H), 1.14 (dd, J = 7.0, 3.8 Hz, 6H); LCMS: MS m/z: 362.1 (M+1).
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
The compound has been disclosed in patent WO2020020374A1. It is an enhancer of Zeste homolog 2 (EZH2) inhibitor, and can be used for preventing or treating EZH2-mediated diseases, including brain cancer, thyroid cancer, cardiac sarcoma, lung cancer, oral cancer, stomach cancer and various other cancers.
Example 4: Preparation of the Compound N-((4,6-dimethyl-2-oxo-1,2-dihydropyridin-3-yl)methyl)-3-(ethyl(tetrahydro-2H-pyran-4-yl)amino)-2-methyl-5-(trans-3-morpholinylcyclobutoxy)benzamide (4)
The title compound was prepared by referring to Example 1 with cyclobutanol replaced by cis-3-morpholinylcyclobutyl-1-ol. The cis-3-morpholinylcyclobutyl-1-ol was prepared as follows:
3-benzyloxy-cyclobutyl-1-one (3 g, 17 mmol, 1 eq) and morpholine (2.97 g, 34 mmol, 2 eq) were dissolved in DCE (30 mL). After stirring at room temperature for 0.5 h, the solution was added with NaBH(OAc)3 (7.23 g, 34 mmol, 2 eq) and stirred at room temperature for 2 h. The reaction mixture was concentrated under reduced pressure to remove the solvent, adjusted to basicity with aqueous NaHCO3 solution, and extracted with ethyl acetate. The organic phase was washed with brine, dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure to give a crude product, which was purified by a phase preparative column to give 4-(3-(benzyloxy)cyclobutyl)morpholine (2.9 g). M/z (ES+), [M+H] +=248.
4-(3-(benzyloxy)cyclobutyl)morpholine (2.9 g, 117.4 mmol) and methane sulfonic acid (20 mL) was added to dichloromethane (40 mL). The mixture was stirred overnight at room temperature and concentrated under reduced pressure to remove the solvent to give a crude product, which was purified by a preparative column to give cis-3-morpholinylcyclobutyl-1-ol; m/z (ES+), [M+H] +=158. The trans-3-morpholinylcyclobutyl-1-ol can also be separated and obtained by this method.
Golcadomide is a modulator of the E3 ubiquitin ligase complex containing cereblon (CRL4-CRBN E3 ubiquitin ligase), with potential immunomodulating and antineoplastic activities. Upon administration, golcadomide specifically binds to cereblon (CRBN), thereby affecting the ubiquitin E3 ligase activity, and targeting certain substrate proteins for ubiquitination. This induces proteasome-mediated degradation of certain transcription factors, some of which are transcriptional repressors in T-cells. This leads to modulation of the immune system, including activation of T-lymphocytes, and downregulation of the activity of other proteins, some of which play key roles in the proliferation of certain cancer cell types. CRBN, the substrate recognition component of the CRL4-CRBN E3 ubiquitin ligase complex, plays a key role in the ubiquitination of certain proteins.
Example 1: Synthesis of (S)-2-(2,6-Dioxopiperidin-3-yl)-4-((2-fluoro-4-((3-morpholinoazetidin-1-yl)methyl)benzyl)amino)isoindoline-1,3-dione (Compound 1)
(S)-2-(2,6-Dioxopiperidin-3-yl)-4-((2-fluoro-4-(hydroxymethyl)benzyl)amino)isoindoline-1,3-dione: A suspension of (S)-4-amino-2-(2,6-dioxopiperidin-3-yl)isoindoline-1,3-dione (5.00 g, 18.3 mmol) and 2-fluoro-4-(hydroxymethyl)benzaldehyde (2.82 g, 18.30 mmol) in 2:1 dioxane-MeOH (75 mL) was cooled to 0° C. and B 10H 14 (4.92 g, 40.3 mmol) was added in small portions over 5 minutes. The reaction flask was fitted with a septum and needle vent (pressure) and vigorously stirred for 10 minutes. The mixture was allowed to reach ambient temperature and stirred for 3 hours. The mixture was concentrated and the residue purified by silica gel chromatography (0-10% MeOH-DCM) to provide (S)-2-(2,6-dioxopiperidin-3-yl)-4-((2-fluoro-4-(hydroxymethyl)benzyl)amino)isoindoline-1,3-dione as a yellow solid (4.23 g, 56%). LCMS (ESI) m/z 411.8 [M+H] +.
(S)-4-((4-(Chloromethyl)-2-fluorobenzyl)amino)-2-(2,6-dioxopiperidin-3-yl)isoindoline-1,3-dione: A solution of (S)-2-(2,6-dioxopiperidin-3-yl)-4-((2-fluoro-4-(hydroxymethyl)benzyl)amino)isoindoline-1,3-dione (0.727 g, 1.77 mmol) in dry NMP (6 mL) was cooled to 0° C. and methane sulfonyl chloride (0.275 mL, 3.35 mmol) and DIEA (0.617 mL, 3.53 mmol) were added sequentially. The reaction mixture was allowed to reach ambient temperature and was stirred for 18 hours. The reaction mixture was slowly added to H 2O (60 mL) cooled to 0° C. with vigorous mixing. The resulting suspension was filtered and the collected solid was washed with H 2O and Et 2O. The solid was dissolved in EtOAc and the solution dried with MgSO 4, filtered and concentrated to provide (S)-4-((4-(chloromethyl)-2-fluorobenzyl)amino)-2-(2,6-dioxopiperidin-3-yl)isoindoline-1,3-dione as a yellow solid (0.600 g, 79%). LCMS (ESI) m/z 430.0 [M+H] +.
(S)-2-(2,6-Dioxopiperidin-3-yl)-4-((2-fluoro-4-((3-morpholinoazetidin-1-yl)methyl)benzyl)amino)isoindoline-1,3-dione: To a solution of (S)-4-((4-(chloromethyl)-2-fluorobenzyl)amino)-2-(2,6-dioxopiperidin-3-yl)isoindoline-1,3-dione (300 mg, 0.698 mmol) in dry DMSO (1.0 mL) was added 4-(azetidin-3-yl)morpholine hydrochloride (125 mg, 0.698 mmol) and DIEA (0.122 mL, 0.698 mmol). The reaction mixture was stirred at ambient temperature for 18 hours and was diluted with DMSO (1 mL). The solution was purified by chiral reverse-phase chromatography to give (S)-2-(2,6-dioxopiperidin-3-yl)-4-((2-fluoro-4-((3-morpholinoazetidin-1-yl)methyl)benzyl)amino)isoindoline-1,3-dione (89 mg, 24%, 97% ee). LCMS (ESI) m/z 536.2 [M+H] +.
Example 1: Synthesis of (S)-2-(2,6-dioxopiperidin-3-yl)-4-((2-fluoro-4-((3- morpholinoazetidin-1-yl)methyl)benzyl)amino)isoindoline -1,3-dione
[00242] Synthesis of Ethyl 4-nitro-1,3-dioxo-isoindoline-2-carboxylate (Compound 10): To a solution of 4-nitroisoindoline-1,3-dione (Compound 11, 440 g, 2.29 mol) and TEA (262 g, 2.59 mol, 359 mL) in dry DMF (2.2 L) was cooled to 0 °C and ethyl chloroformate (313 g, 2.89 mol, 275 mL) was added dropwise over 5 minutes. The reaction mixture was stirred at 22 °C for 10 hours. The mixture was slowly added to chilled water (10 L) and the resulting suspension stirred for 5 minutes. The suspension was filtered and the filter cake was washed with water (1 L). The solid was dissolved with ethyl acetate (5 L) and the organic phase was washed with aqueous HCl (1 M, 1 L), water (2 L) and brine (2 L). The organic phase was dried over sodium sulfate , filtered and concentrated to give Compound 10 (360 g, 59%) as a white solid. 1 H NMR (400 MHz CDCl 3 ) δ ppm 8.24 (d, J = 7.6 Hz, 1H), 8.19 (d, J = 8.4 Hz, 1H), 8.06-8.02 (m, 1H), 4.49 (q, J = 7.2 Hz, 2H), 1.44 (t, J = 6.8 Hz, 3H).
[00243] Synthesis of tert-Butyl (4S)-5-amino-4-(4-nitro-1,3-dioxo-isoindolin-2-yl)-5-oxo-pentanoate (Compound 6): To a solution of Compound 10 (165 g, 625 mmol) and DIEA (113 g, 874 mmol, 153 mL) in dry THF (1700 mL) was added tert-butyl (4S)-4,5-diamino-5-oxo-pentanoate hydrochloride ( 149 g, 625 mmol) and heated at reflux for 10 hours. The reaction mixture was concentrated under reduced pressure. The resulting residue was diluted with methyl tert-butyl ether (5 L) and stirred at 20 °C for 1 hour. The suspension was filtered and the filter cake was dissolved with DCM (4 L). The organic phase was washed with water (1.5 L x 3), brine (1.5 L) and dried over sodium sulfate . The organic phase was filtered and concentrated under reduced pressure to give a light yellow oil. The oil was diluted with hexane / ethyl acetate (10/1, 2 L) and stirred until a light yellow suspension formed. The suspension was filtered and the filter cake was triturated and concentrated in vacuum to give Compound 6 (175 g, 74%) as a light yellow solid. 1 H NMR (400 MHz CDCl3) δ ppm 8.12 (d, J = 8.0 Hz, 2H), 7.94 (t, J = 8.0 Hz, 1H), 6.48 (s, 1H), 5.99 (s, 1H), 4.84- 4.80 (m, 1H), 2.49-2.44 (m, 2H), 2.32-2.27 (m, 2H), 1.38 (s, 9H).
[00244] Synthesis of tert-Butyl (S)-5-amino-4-(4-amino-1,3-dioxoisoindolin-2-yl)-5-oxopentanoate (Compound 5): To a suspension of Compound 6 (170.0 g, 450.5 mmol, 1.00 eq) in DMA (1.00 L) was added palladium on carbon (50.0 g, 10% purity) under nitrogen. The suspension was degassed under vacuum and purged with hydrogen gas several times. The mixture was stirred under hydrogen gas (50 psi) at 25 °C for 16 hours. The mixture was filtered and the filtrate was poured into cooled water (3.0 L). The mixture was stirred at 10 °C for 1 hour and filtered. The filter cake was washed with water (700 mL) and dissolved in DCM (1.00 L). The organic phase was dried over sodium sulfate , filtered and concentrated under reduced pressure to give Compound 5 (107 g, 68%) as a green solid. 1 H NMR (400 MHz DMSO-d 6 ) δ ppm 7.52 (s, 1H), 7.43 (dd, J = 8.4, 7.2 Hz, 1H), 7.13 (s, 1H), 6.95-6.99 (m, 2H), 6.42 (s, 2H), 5.75 (s, 1H), 4.47-4.51 (m, 1H), 2.32-2.33 (m, 1H), 2.14-2.20 (m, 3H), 1.32 (s, 9H); HPLC purity, 100.0%; SFC purity, 100.0% ee.
[00245] Synthesis of 2-fluoro-4-(hydroxymethyl)benzaldehyde (Compound 8): To a solution of 4-(((tert-butyldimethylsilyl)oxy)methyl)-2-fluorobenzaldehyde (370.0 g, 1.38 mol, 1.00 eq ) in THF (1.85 L) was added a solution of p-toluenesulfonic acid monohydrate (78.7 g, 413.6 mmol, 0.30 eq) in water (1.85 L) drop-wise at 10 °C. The mixture was stirred at 27 °C for 16 hours. TEA (80 mL) was added drop-wise and stirred for 10 minutes. The organic phase was separated and the aqueous phase was extracted with ethyl acetate (600 mL × 4). The combined
organic phase was washed with brine (1.50 L), dried over anhydrous sodium sulfate , filtered and concentrated under reduced pressure. The residue was purified by silica gel column chromatography to give Compound 8 (137.5 g, 76%) as a yellow oil. 1 H NMR (400 MHz CDCl 3 ) δ ppm 10.34 (s, 1H), 7.86 (dd, J = 8.0, 7.2 Hz, 1H), 7.25 (s, 1H), 7.22 (d, J = 4.4 Hz, 1H) , 4.79 (d, J = 6.0 Hz, 2H), 1.91 (t, J = 6.0 Hz, 1H).
[00246] Synthesis of tert-Butyl (S)-5-amino-4-(4-((2-fluoro-4-(hydroxymethyl)benzyl)amino)-1,3-dioxoisoindolin-2-yl)-5- oxopentanoate (Compound 2-b): To a solution of Compound 5 (100.0 g, 287.9 mmol, 1.00 eq) and Compound 8 (57.7 g, 374.3 mmol, 1.30 eq) in dry DCM (1.00 L) was added TFA (164.1 g , 1.44 mol, 5.00 eq) at 0 °C. The reaction mixture was stirred at 28 °C for 2 hours. To the solution was added sodium cyanoborohydride (27.1 g, 431.8 mmol, 1.50 eq) at 0 °C. The mixture was stirred at 28 °C for 30 minutes. The reaction mixture was quenched by addition of MeOH (600 mL) and concentrated under reduced pressure. The residue was purified by silica gel column chromatography to give Compound 2-b (110.0 g, 74.0%) as a yellow solid. 1 H NMR (400 MHz, DMSO-d6) δ ppm 7.56 (s, 1H), 7.50 (dd, J = 8.4, 7.2 Hz, 1H), 7.34 (t, J = 8.0 Hz, 1H), 7.02-7.18 ( m, 4H), 6.94-7.01 (m, 2H), 4.57 (d, J = 6.0 Hz, 2H), 4.47-4.53 (m, 3H), 2.31-2.35 (m, 1H), 2.15-2.22 (m, 3H), 1.31 (s, 9H); HPLC purity, 94.0%; SFC purity, 100.0% ee.
[00247] Synthesis of tert-butyl (S)-5-amino-4-(4-((4-(chloromethyl)-2-fluorobenzyl)amino)-1,3-dioxoisoindolin-2-yl)-5-oxopentanoate (Compound 2-a): To a solution of Compound 2-b (100.0 g, 206.0 mmol, 1.00 eq) in NMP (430.0 mL) was added DIEA (79.9 g, 617.9 mmol, 3.00 eq) and MsCl (47.2 g, 411.9 mmol, 2.00 eq) at 0 °C. The ice bath was removed, and the reaction was stirred at 28°C for 10 hours. The reaction was poured into cooled water (<10°C, 2.0 L) and stirred for 10 minutes. The mixture was extracted with methyl tert-butyl ether (750 mL x 3). The combined organic layer was washed with brine (1.25 L), dried over sodium sulfate , filtered and concentrated under reduced pressure. The residue was purified by silica gel column chromatography to give Compound 2-a (86.0 g, 81.2%) as a yellow solid.
[00248] Synthesis of tert-butyl (S)-5-amino-4-(4-((2-fluoro-4-((3-morpholinoazetidin-1-yl)methyl)benzyl)amino)-1,3- dioxoisoindolin-2-yl)-5-oxopentanoate (Compound 2): To a solution of 4-(azetidin-3-yl)morpholine hydrochloride (Compound 7 HCl, 30.5 g, 170.7 mmol, 1.00 eq) and DIEA (66.2 g, 512.0 mmol, 3.00 eq) in DMSO (350.0 mL) was added to a solution of Compound 2-a (86 g, 170.65 mmol, 1.00 eq) in DMSO (350.0 mL) drop-wise at 15 °C. The reaction mixture was stirred at 28 °C for 16 hours. The reaction mixture was poured into cold half saturated brine (<10°C, 2.5 L) and extracted with ethyl acetate (1.50 L, 1.00 L, 800.0 mL). The combined organic phase was washed with saturated brine (1.50 L), dried over sodium sulfate , filtered, and concentrated under reduced pressure. The residue was purified by silica gel column chromatography to give Compound 2 (68.3 g, 65.7%) as a yellow solid. 1 H NMR (400 MHz DMSO-d 6 ) δ ppm 7.55 (s, 1H), 7.50 (dd, J = 8.4, 7.2 Hz, 1H), 7.31 (t, J = 8.0 Hz, 1H), 7.16 (s, 1H), 6.94-7.10 (m, 5H), 4.56 (d, J = 6.4 Hz, 2H), 4.49-4.52 (m, 1H), 3.54-3.55 (m, 6H) 3.31-3.32 (m, 3H), 2.81-2.88 (m, 3H), 2.29-2.38 (m, 1H), 2.15-2.25 (m, 7H), 1.30 (s, 9H); HPLC purity, 100.0%; SFC purity, 100.0% ee.
[00249] Synthesis of (S)-2-(2,6-dioxopiperidin-3-yl)-4-((2-fluoro-4-((3-morpholinoazetidin-1-yl)methyl)benzyl)amino)isoindoline -1,3-dione (Compound 1): A solution of Compound 2 (30.0 g, 49.2 mmol, 1.00 eq) and benzenesulfonic acid (31.1 g, 196.8 mmol, 4.00 eq) in acetonitrile (480.0 mL) was stirred at reflux for 3 hours. The reaction was cooled to 20 °C, poured into cold brine:saturated sodium bicarbonate solution (1:1, <10 °C, 2.0 L) and extracted with ethyl acetate (1.0 L). The organic phase was washed with cold brine:saturated sodium bicarbonate solution (1:1, <10°C, 1.00 L) once more. The combined aqueous phase was extracted with ethyl acetate (500.0 mL x 2). The combined organic phase was washed with cold brine (<10°C, 1.0 L), dried over sodium sulfate , filtered and concentrated under reduced pressure. The crude product was purified by silica gel column chromatography to give Compound 1 (17.5 g, 66.0%) as a yellow solid. 1 H NMR (400 MHz, DMSO-d 6 ) δ ppm 11.10 (s, 1H), 7.54 (t, J = 8.0 Hz, 1H), 7.30 (t, J = 8.0 Hz, 1H), 7.04-7.10 (m , 4H), 7.00 (d, J = 8.4 Hz, 1H), 5.07 (dd, J = 12.8, 5.2 Hz, 1H), 4.58 (d, J = 6.4 Hz, 2H), 3.53-3.55 (m, 6H) , 3.30-3.32 (m, 2H), 2.81-2.89 (m, 4H), 2.54-2.61 (m, 2H), 2.20 (m, 4H) 2.03-2.06 (m, 1H); HPLC purity, 100.0%; SFC purity, 97.2% ee; LCMS (ESI) m/z 536.1 [M+H] + .
Example 2: Synthesis of (S)-2-(2,6-dioxopiperidin-3-yl)-4-((2-fluoro-4-((3- morpholinoazetidin-1-yl)methyl)benzyl)amino)isoindoline -1,3-dione
[00250] Synthesis of tert-butyl (S)-5-amino-4-(4-nitro-1,3-dioxoisoindolin-2-yl)-5-oxopentanoate (Compound 6): Ethyl acetate (245 mL, 5 V ), 3-nitrophthalic anhydride (49.1 g, 0.25 mol, 1 eq), and tert-butyl (S)-4,5-diamino-5-oxopentanoate hydrochloride (59.2 g, 0.25 mol,
1 eq) were charged into a reactor and cooled to 15-20°C. A premade solution of CDI (66.7 g,
0.41 mol, 1.5 eq) in DMF (245 mL, 5 V) was charged and the mixture was stirred at 20-25°C for
1 hour. The reaction was quenched with 15% (wt/wt) aqueous citric acid solution (10 V).
EtOAc (5 V) was added, the mixture was agitated and the phases split and separated. Tea
aqueous layer was extracted with EtOAc (5 V) and the combined organic layers were washed
twice with a 5% (wt/wt) aqueous citric acid solution (5 V each wash). The organic layer was
distilled at reduced pressure to 5 V and further continuously distilled at reduced pressure with the addition of iPrOH (10 V), maintaining a constant volume at 5 V. The final distillate was diluted to 13 V with iPrOH and used in the next step without further handling. 91% solution yield. [00251] Synthesis of tert-butyl (S)-5-amino-4-(4-amino-1,3-dioxoisoindolin-2-yl)-5-oxopentanoate (Compound 5): The solution of Compound 6 in iPrOH was charged to a hydrogenation reactor. 10% palladium on carbon (50% wet, 4.65g 5 wt%) was charged. The reaction mixture was stirred under 50-60 psi H2 at 40-50 o C for 16 hrs. The reaction mixture was filtered and the filter cake was washed three times with iPrOH (1 V each wash). The solution was distilled at reduced pressure to 5 V, cooled to ambient temperature and seeded (1 wt%). Water (20 V) was charged at 20-25 o C. The resulting slurry was cooled to 3-8 o C for 4-8 hrs. The solids were collected by filtration and washed three times with cold water (1.5 V each wash). The solids were dried at 35-45 o C under reduced pressure to give Compound 5 in 87% yield. 1 H NMR (500 MHz DMSO-d 6 ) δ (ppm): 7.52 (s, 1H), 7.43 (dd, J = 8.4, 7.0 Hz, 1H), 7.13 (s, 1H), 6.97 (ddd, J = 10.9, 7.7, 0.61 Hz, 2H), 6.43 (s, 2H), 4.49 (m, 1H), 2.33 (m, 1H), 2.17 (m, 3H), 1.32 (s, 9H); HPLC purity, 99.2%; Chiral purity, 99.9% ee; LCMS (ESI) m/z 348.2, [M+H] + , 292.2 [Mt-Bu+H] + . Residual IPA: 0.7 mol% by 1 H NMR.
[00252] Synthesis of 4-(1-(4-bromo-3-fluorobenzyl)azetidin-3-yl)morpholine (Compound 4): A mixture of 4-bromo-3-flurobenzaldehyde (Compound 14, 82 g, 396 mmol ) and 4-(azetidin-3-yl)-morpholine hydrochloride (Compound 7 HCl, 72 g, 396 mmol) in acetonitrile (820 ml) was agitated at 25±5°C for at least 3 hours. The mixture was cooled to 10±5°C and sodium triacetoxyborohydride (130 g, 594 mmol) was added in four portions while maintaining the temperature of the mixture below 30°C. The temperature of the mixture was adjusted to 25±5°C and stirred for at least 30 min until reaction completion. The mixture was transferred to a precooled (10-15°C) solution of aqueous citric acid (152 g in 400 ml water, 792 mmol) while maintaining the temperature below 30°C. Once the quenching process was complete, the mixture was concentrated to ~ 560 ml (7 volumes) while keeping the temperature at or below 45°C. The mixture was then washed with toluene (320 ml). To the aqueous phase was added THF and the pH was adjusted to above 12 with aqueous NaOH solution (320 ml, 10 N). The phases were separated, and the aqueous phase was removed. The organic phase was washed with brine and subsequently concentrated with addition of THF (~ 3L) until KF ≤ 0.10%. The mixture was filtered to remove any inorganics and the product Compound 4 was isolated as a solution in THF with 95% yield.
[00253] Synthesis of sodium (2-fluoro-4-((3-morpholinoazetidin-1-yl)methyl)phenyl) (hydroxy)methanesulfonate (Compound 13): A solution of Compound 4 (520 g, 1.58 mol) in
THF (380 ml) was cooled to −15 ± 5 °C. A solution of iPrMgCl . LiCl (1.3 M, 1823 ml, 2.37 mol) in THF was added over the course of at least 1 hour while maintaining the temperature below −10 °C. After addition was complete, the temperature of the reaction mixture was adjusted to 0 ± 5 °C and stirred for at least 1 hour. Once magnesiation was complete, the mixture was cooled to −15 ± 5 °C (target −15 °C to −20 °C) and a solution of DMF (245 ml g, 3.16 mol) in THF (260 ml) was added slowly over the course of at least 1 hour while maintaining the temperature below −10 °C. The temperature of the mixture was then adjusted to −15 ± 5 °C and agitated for at least 4 hours.
[00254] Upon reaction completion, the reaction mixture was charged into an aqueous 3 N HCl solution (2600 ml) over the course of at least 1 hour while maintaining the temperature below −5 °C. The temperature of the mixture was then adjusted to 5 ± 5 °C and agitation was stopped, letting the mixture settle for at least 15 minutes. The layers were separated. The lower aqueous layer containing the product was washed with 2-MeTHF (2600 ml). The aqueous layer was then charged with 2-MeTHF (2600 ml) and the temperature of the batch was adjusted to −10 ± 5 °C. To the cooled mixture, an aqueous 5 N NaOH (728 ml, 3.64 mol) solution was added while maintaining the temperature below −5 °C until the pH of the mixture was between 10 and 11. The temperature of the mixture was adjusted to 5 ± 5 °C and agitated for at least 15 minutes. The agitation of the mixture was stopped and the mixture allowed to settle for at least 15 minutes. The layers were separated, and the lower aqueous layer was back extracted two times with 2-MeTHF (2600 ml). The combined organic layer was washed with water (1040 mL) and the organic solution was evaporated to dryness, affording 372 g of crude Compound 3 freebase as an oil (yield 85%). 1 H NMR (DMSO-d 6 ) δ (ppm): 10.18 (s, 1H), 7.78 (t, J =7.7 Hz, 1H), 7.23-7.35 (m, 2H), 3.66 (s, 2H), 3.51 -3.60 (m, 4H), 3.26-3.47 (m, 2H), 2.72-2.97 (m, 3H), 2.12-2.32 (m, 4H).
[00255] The crude Compound 3 freebase (4.3 kg) was adsorbed onto silica gel (8.6 kg) with 100% DCM, loaded onto a 60 L column containing 12.9 kg silica gel (packed with 100% DCM), and eluted with DCM ( 86 L), followed successively by 1% MeOH/DCM (40 L), 3% MeOH/DCM (80 L) and 10% MeOH/DCM (40 L). The fractions were collected and concentrated at or below 38˚C to give Compound 3 as a purified oil (3.345 kg, yield 66%).
[00256] A portion of Compound 3 (1.0 kg, 3.59 mol) was dissolved in ethanol (16.0 L, 16
vol) at 20±5 °C and the mixture heated to 40 °C. A solution of Na2S2O5 (622.0 g, 3.27 mol; 0.91 eq) in water (2 L, 2 vol) was prepared at 20±5 °C and added to the freebase solution at 40 °C to obtain an off-white suspension. The batch was agitated and maintained at 40 °C for 2 hrs, then cooled to 20±5 °C and agitated for 1 to 2 hrs. The batch was filtered and washed with ethanol (2×2.0 L, 2×2 vol) to obtain an off-white solid. The wet cake was dried under vacuum at 40 °C for 18 hrs to afford about 1.88 kg of Compound 13.
[00257] Synthesis of 2-fluoro-4-((3-morpholinoazetidin-1-yl)methyl)benzaldehyde (Compound 3): Compound 13 (1.88 kg) was dissolved in ethyl acetate (15.0 L) at 20±5 °C . A 2 M Na 2 CO 3 solution (total 15.0 L used) was added to adjust the pH to 10.0. The batch was agitated for 1 to 1.5 hrs at 20±5 °C. After the reaction was complete, the phases were separated and the organic layer was washed with brine (2.0 L). The organic layer was concentrated to dryness at 35-38 °C to afford 852.0 g of Compound 3 as a colorless oil (yield 81%).
[00258] Synthesis of 2-fluoro-4-((3-morpholinoazetidin-1-yl)methyl)benzaldehyde bis-oxalic acid salt (Compound 3 bis-oxalic acid salt): A portion of the Compound 3 oil (187 g, 0.67 mol) was dissolved in isopropanol (1125 ml) and water (375 ml). A first portion (~30%) of this freebase mixture (480 ml) was slowly added over the course of at least 30 minutes to a solution of oxalic acid (125 g, 1.38 mol) in IPA (1125 ml)/water (375 ml) at 60 ± 5 °C. A second portion (~20%) of the freebase mixture (320 ml) was slowly added over the course of at least 30 minutes to the reaction mixture at 60 ± 5 °C. The reaction mixture was agitated at 60 ± 5 °C for at least 90 minutes. A third portion (~25%) of the freebase mixture (~ 400 ml) was slowly added over the course of at least 30 minutes to the reaction mixture at 60 ± 5 °C and the reaction mixture was agitated at 60 ± 5 °C for at least 90 minutes. The remaining freebase solution (400 ml) was slowly added over the course of at least 30 minutes to the reaction mixture at 60 ± 5 °C and the reaction mixture was agitated at 60 ± 5 °C for at least 90 minutes. The temperature of the mixture was adjusted to 20 ± 5 °C (target 20 °C) over the course of at least 1 hour and the mixture was agitated for at least 16 hours at 20 ± 5 °C and then filtered. The cake was washed three times with IPA (2 x 375 ml) and dried in the drying oven at ≤ 40 °C with a slow bleed of nitrogen to afford 261 g of Compound 3 bis-oxalic acid salt (yield 85%). 1 H NMR (DMSO-d 6 ) δ (ppm): 10.21 (s, 1H), 7.87 (t, J = 7.6 Hz, 1H), 7.42-7.56 (m, 2H), 4.31 (s, 2H), 3.89 -4.03 (m, 2H), 3.75-3.89 (m, 2H), 3.60 (br t, J = 4.3 Hz, 4H), 3.26 (br t, J = 6.9 Hz, 1H), 2.37 (br s, 4H) .
[00259] Synthesis of tert-butyl (S)-5-amino-4-(4-((2-fluoro-4-((3-morpholinoazetidin-1-yl)methyl)benzyl)amino)-1,3- dioxoisoindolin-2-yl)-5-oxopentanoate (Compound 2):
Acetonitrile (6.8 L, 8.0 X Vol) was added to a 30 L jacketed cylindrical reactor. Compound 5 (0.845 kg, 1.00 X Wt) and Compound 3 bis-oxalic acid salt (1.35 kg, 1.60 The contents of the reactor were equilibrated with agitation at 20 ± 5 °C. Trifluoroacetic acid (0.19 L, 0.22 X Vol) was added dropwise, maintaining the batch temperature at 20 ± 5 °C. The reaction mixture was stirred at 20 ± 5 °C for no less than 5 minutes and then sodium triacetoxyborohydride (0.13 kg, 015 X Wt) was added as a solid, maintaining the batch temperature at 20 ± 5 °C. The process of adding trifluoroacetic acid and then sodium triacetoxyborohydride was repeated an additional 5 times. After the last addition, the reaction mixture was sampled to determine the reaction progress. The reaction was held at 20 ± 5 °C overnight. The reaction mixture was then quenched with water (3.4 L, 4.0 X Vol), maintaining the batch temperature at 20 ± 5 °C. The mixture was then stirred at 20 ± 5 °C for no less than 30 minutes and the resulting slurry filtered through a 3 L sintered glass filter, directing the filtrates to clean containers. The reactor was rinsed with acetonitrile (0.4 L, 0.5 X Vol) and the rinse passed through the contents of the 3 L sintered glass filter, directing the filtrate to the containers containing the main batch. The contents of the containers were concentrated to ~5 X Vol under reduced pressure at a bath temperature of no more than 30 °C. The residue was transferred to a clean reactor, was rinsing with 2-MeTHF (2.5 L, 3.0 X Vol) to complete the transfer. Additional 2-MeTHF (10.1 L, 12.0 X Vol) was added to the reactor, followed by water (3.4 L, 4.0 X Vol). The mixture was agitated for no less than 15 minutes at 20 ± 5 °C, then allowed to settle for no less than 10 minutes at 20 ± 5 °C before transferring the bottom aqueous layer to new containers. An aqueous sodium bicarbonate solution (5.3 L, 6.3 X Vol, 9% wt/wt) was added to the reactor with stirring over 30 minutes, maintaining batch temperature no more than 25 °C. The mixture was agitated for no more than 15 minutes at 20 ± 5 °C, then allowed to settle for no less than 10 minutes at 20 ± 5 °C before the bottom aqueous layer was transferred to new containers. The aqueous sodium bicarbonate wash was repeated an additional 2 times to reach a pH of about 6.6 for the spent aqueous layer. A saturated aqueous solution of NaCl(0.85 L, 1.0 X Vol) was then added to reactor with agitation. The mixture was agitated for no less than 15 minutes at 20 ± 5 °C, then allowed to settle for no less than 10 minutes before the bottom aqueous layer was transferred to new containers. The remaining organics were concentrated under reduced pressure to a batch volume of ~5 X Vol at a bath temperature of about 40 °C. Acetonitrile (5.1 L, 6.0 X Vol) was added to the residual volume and the resulting solution concentrated to a batch volume of ~ 5 X Vol under reduced pressure at bath temperature of about 40 °C. The process of adding acetonitrile and concentrating under vacuum was repeated two more times to reach the distillation endpoint with a water content of about 1%. The acetonitrile solution was transferred to a clean container along with two 1.7 L (2.0 X Vol) rinses and held at 5 °C overnight. The acetonitrile solution was then filtered through a 3 L sintered glass filter, followed by a 1.7 L (2.0 X Vol) acetonitrile rinse, directing the filtrates to a clean container. The filtrate was transferred to a clean reactor and the container rinsed twice with 1.7 L (2.0 X Vol) of acetonitrile to complete the transfer. Enough acetonitrile (roughly 0.6 L) was added to adjust the total volume in the reactor to about 14 L. A solution assay of the contents of the reactor was obtained to calculate the amount of Compound 2 present for use in the next step (result = 1.3 kg = 1.00 X Wt for remainder of process).
[00260] Synthesis of (S)-2-(2,6-dioxopiperidin-3-yl)-4-((2-fluoro-4-((3-morpholinoazetidin-1-yl)methyl)benzyl)amino)isoindoline -1,3-dione bis-besylate salt (Compound 1 bis-besylate salt): The solution of Compound 2 in acetonitrile from the previous step was diluted with acetonitrile (roughly 2 L) such that the total volume in the reactor was about 16 L. The solution was cooled with stirring to 10 ± 5 °C and held within that range for 96 hours. Benzenesulfonic acid (1.86 kg, 1.43 X Wt) was added while sparging the reaction mixture with nitrogen gas and maintaining the batch temperature at 10 ± 10 °C. The temperature of the reactor was then adjusted to 20 ± 5 °C and the mixture stirred at that temperature for 60 minutes. The total volume of reaction mixture was adjusted back to 16 L to account for solvent lost during sparging by the addition of acetonitrile (roughly 0.4 L). The reaction mixture was then heated to 55 ± 5 °C over the course of about 30 minutes and held in that range for 15 to 16 hours for reaction completion. The mixture was then cooled to 50 ± 5 °C and MTBE (3.9 L, 3.0 X Vol) was added, maintaining the batch temperature at 50 ± 5 °C. The mixture was allowed to stir at 50 ± 5 °C for about 1.5 hours to establish a self-seeded slurry. Additional MTBE (3.9 L, 3.0 X Vol) was added to the reactor over the course of about 1.75 hours at 50 ± 5 °C. The slurry was cooled to 20 ± 5 °C over the course of about 1.75 hours and held in that temperature range overnight. The slurry was filtered using a Buchner funnel. The reactor was rinsed twice with
MTBE (3.9 L each, 3.0 X Vol) and the rinse was used to wash the solids in the Buchner funnel. The solids were dried on drying trays for about 23 hours at 40 °C under reduced pressure (15-150 mbar), yielding 1.62 kg (77.9%) of Compound 1 bis-besylate salt.
[00261] Synthesis of (S)-2-(2,6-dioxopiperidin-3-yl)-4-((2-fluoro-4-((3-morpholinoazetidin-1-yl)methyl)benzyl)amino)isoindoline -1,3-dione hydrochloride salt (Compound 1 HCl): A suspension of Compound 1 bis-besylate salt (120 g, 1 equiv.) in 2-MeTHF (25 L/kg) was added to a reactor and agitated at 10 °C. A solution of KHCO 3 (32.5 g, 2.4 equiv) in water (1.8 L, 6 L/kg) was added to the slurry over the course of 40 minutes. The mixture was stirred for an additional 30 minutes. The batch was then allowed to settle, at which point the aqueous (bottom) layer was separated and discarded. An aqueous solution of NaCl (5%, 5 L/kg, 575 ml) was added to the organic layer and the mixture was agitated for 10 minutes, after which point the temperature was raised to 20 °C. The batch was allowed to settle, at which point the aqueous (bottom) layer was discarded. The brine was repeated a second time.
Additional 2-MeTHF (500 ml) was added to dilute the organic layer, resulting in a concentration of about 20 mg product per ml. A solution of HCl (total 0.98 eq.) in 2-MeTHF was prepared and a portion (20% of total, corresponding to ~0.2 eq.) then added to the reaction mixture over the course of about 10 min. Seeds of Compound 1 hydrochloride (~5% wt) were added, but did not dissolve. The batch was held under vigorous agitation for one hour. To the slurry, the remaining portion of the HCl solution (~0.78 eq.) was added over the course of 3 hours at a constant rate. Vigorous agitation was maintained. After addition was complete, the batch was held for one hour, after which the batch was filtered, washed three times with 3 L/kg of 2-MeTHF . The filter cake was placed in a vacuum oven at 22 °C for 12 hours, at which point the temperature was raised to 40 °C. Dry cake of Compound 1 hydrochloride (58g, 75% yield) was obtained and packaged. Achiral HPLC purity: 98.91%; chiral HPLC purity: 99.68%.
Example 4: Synthesis of (S)-2-(2,6-dioxopiperidin-3-yl)-4-((2-fluoro-4-((3- morpholinoazetidin-1-yl)methyl)benzyl)amino)isoindoline -1,3-dione
[00264] Synthesis of tert-butyl (S)-5-amino-4-(4-nitro-1,3-dioxoisoindolin-2-yl)-5-oxopentanoate (Compound 6): To a solution of 3-nitrophthalic anhydride (Compound 12,
mL) and 2-MeTHF (110 mL) at 25°C. 2,6-Lutidine (23.4 mL, 201 mmol, 1.14 eq) was added
slowly to maintain the temperature at or below 25°C. The mixture was aged at 25°C for 1 hour before being cooled to 5°C. CDI (4.17 g, 25.7 mmol, 0.146 eq) was added and stirred until the temperature returned to 5°C. Another portion of CDI (4.62 g, 28.5 mmol, 0.161 eq) was added and stirred until temperature returned to 5°C. CDI (8.87 g, 54.7 mmol, 0.310 eq) was added and stirred until the temperature returned to 5°C. CDI (8.91 g, 54.9 mmol, 0.311 eq) was added and stirred until the temperature returned to 5°C. The mixture was warmed to 20°C and CDI (16.4 g, 101.1 mmol, 0.573 eq) was added, and the mixture was aged at 20°C for 16 hours. The mixture was cooled to 5°C and a solution of 30 wt% citric acid and 5 wt% NaCl (350 mL) was added slowly while maintaining the temperature. The mixture was warmed to 20°C and aged for 30 minutes. The phases were split and separated. The organic phase was diluted with EtOAc (175 mL) and washed with a solution of 5 wt% citric acid (175 mL), and concentrated by distillation (75 torr, 50°C) to a volume of 175 mL EtOAc. The solvent was changed to iPrOH by constant volume distillation (75 torr, 50°C) with 350 mL iPrOH to a final volume of 175 mL. The distillate was diluted with 200 mL iPrOH to afford Compound 6 as a solution for use in the next step. 1 H NMR (500 MHz, CDCl 3 ) δ (ppm): 8.18 – 8.13 (m, 2H), 7.96 (t, J = 7.8 Hz, 1H), 6.34 (s, 1H), 5.59 (s, 1H), 4.90 (dd, J = 10.1, 4.6 Hz, 1H), 2.61 (ddt, J = 14.6, 10.1, 6.1 Hz, 1H), 2.49 (ddt, J = 14.2, 8.7, 5.2 Hz, 1H), 2.44 – 2.29 ( m, 2H), 1.44 (s, 9H).
[00265] Synthesis of tert-butyl (S)-5-amino-4-(4-amino-1,3-dioxoisoindolin-2-yl)-5-oxopentanoate (Compound 5): To a solution of Compound 6 in iPrOH (375 mL) was added 5% palladium on carbon (1.23 g, 3.5 wt%, wet). The mixture was purged with nitrogen five times and with hydrogen three times. The mixture was pressurized with hydrogen (50 psi) and aged at 50°C for 16 hours. The mixture was cooled to room temperature and purged with nitrogen three times, filtered to remove catalyst, and the filter cake was washed with iPrOH (20mL) three times. The filtrate was concentrated to 200 mL, seeded (0.454 g, 1.3 wt%) at 22 °C, and aged for 45 minutes. Water (1325 mL) was added over 3 hours at 22°C. After the addition of water, the mixture was cooled to 8°C over 2 hours and aged for 1 hour at 8°C. The slurry was filtered, and the cake was rinsed with cold water (200 mL) three times and dried under vacuum at 50°C to yield Compound 5 as a yellow solid (47.97 g, 80.6% yield, 99.62% LC purity, 103% 1 H NMR potency). 1 H NMR (500 MHz, CDCl 3 ) δ (ppm): 7.46 (dd, J = 8.3, 7.0 Hz, 1H), 7.19 (d, J = 7.2 Hz, 1H), 6.89 (d, J = 8.3 Hz, 1H), 6.28 (s, 1H), 5.41 (s, 1H), 5.28 (s, 2H), 4.83 (dd, J = 9.3, 6.0 Hz, 1H), 2.52 (p, J = 7.0 Hz, 2H), 2.36 – 2.29 (m, 2H), 1.44 (s, 9H). 13 C NMR (126 MHz, CDCl 3 ) δ (ppm): 171.80, 171.12, 169.64, 168.27, 145.70, 135.50, 132.20, 121.43, 112.98, 80.99, 53.04, 32.23, 28.02 , 24.36. LCMS (ESI): m/z 291.9 [M+H – tBu]
[00266] Synthesis of 4-(1-(4-bromo-3-fluorobenzyl)azetidin-3-yl)morpholine bis-methanesulfonic acid salt (Compound 4 bis-methanesulfonic acid salt): A mixture of 4-bromo-3- fluorobenzaldehyde (Compound 14, 102 g, 493 mmol) and 4-(azetidin-3-yl)morpholine hydrochloride (Compound 7 HCl, 90 g, 493 mmol) in acetonitrile (1000 ml) was agitated at a temperature of about 20 to 25 °C for 2 to 3 hours. The slurry was cooled to temperature of about 10 to 15 °C and sodium triacetoxyborohydride (STAB, 162 g, 739 mmol) was added in 4 portions over the course of about 45 minutes while maintaining the batch temperature at no more than 30 °C. The slurry was stirred at a temperature of about 20 to 25 °C for at least 30 minutes and then quenched by an aqueous citric acid solution (191 g, 986 mmol in 500 ml of water) at a temperature of about 40 to 45 °C over the course of 2 hours. Upon completion of the quenching process, the batch volume was reduced by vacuum distillation to about 700 ml at a temperature of no more than 45 °C. Cyclopentylmethylether (CPME, 400 ml) was added to the aqueous solution to afford a final volume of about 1100 ml. The pH was adjusted to about 8 to 9 by addition of an aqueous solution of 10 N NaOH (added volume about 430 ml). The phases were separated, and the aqueous phase discarded. The organic phase was washed with brine (100 ml) twice such that the pH was no more than 8 and the volume was adjusted to about 1000 ml with addition of extra CPME. The batch was distilled at constant volume under reduced pressure with addition of CPME until KF was no more than 0.15%. CPME was added (if needed) to adjust the batch to a volume of 1000 ml at the end of distillation. The dry CPME solution was seeded (500 to 750 mg) at ambient temperature. The seeded, dry CPME slurry was heated to a temperature of 50 to 60 °C and then charged with methanesulfonic acid in 200 ml of CPME over the course of 4 to 5 hours. The slurry was then cooled to 20 °C over the course of 4 to 5 hours and kept at 20 °C for 3 to 4 hours, filtered, rinsed with CPME and dried in a vacuum oven at 35 to 40 °C over 16 hours to give Compound 4 bis-methanesulfonic acid salt as a white solid. 1 H NMR (500 MHz DMSO-d 6 ) δ (ppm): 10.62 (br s, 1-2H), 7.85 (t, J = 7.8 Hz, 1H), 7.58 (dd, J = 9.5 Hz, 1.9 Hz, 1H), 7.34 (dd, J = 8.2 Hz, 1.8 Hz, 1H), 4.55-4.24 (m, 7H), 3.84 (br s, 4H), 3.14 (m, 4H); HPLC purity, 99.8%, LCMS (ESI) m/z 329.1 /331.1[M/M+2] + . XRPD pattern of the product is shown in FIG.10. DSC thermogram of the product is shown in FIG.11.
[00267] Preparation of 4-(1-(4-bromo-3-fluorobenzyl)azetidin-3-yl)morpholine (Compound 4): A slurry of Compound 4 bis-methanesulfonic acid salt (70 g, 134 mmol) in t -butyl methyl ether was cooled to 10±5°C. An aqueous solution of NaOH (2 N, 201 ml, 403 mmol) was added over the course of at least 30 minutes while maintaining the batch temperature at about 15 °C. After the addition of NaOH , the batch temperature was raised to 20±5°C and agitated over the course of about 20 minutes. The organic layer was separated and washed with water (210 ml) three times. The organic layer was subsequently concentrated with addition of THF (~ 1.05L) until KF ≤ 0.10%. The product Compound 4 was isolated as a solution in THF
with 95% solution yield.
[00268] Preparation of 2-fluoro-4-((3-morpholinoazetidin-1-yl)methyl)benzaldehyde dihydrochloride (Compound 3 di-HCl): A solution of Compound 4 (44 g, 134 mmol) in THF (total volume ~ 350 ml) was then cooled to −20 ± 5 °C. A solution of iPrMgCl . LiCl (1.3 M, 176 ml, 228 mmol) in THF was added over the course of half an hour while maintaining the temperature below −10 °C. After the addition was complete, the batch was stirred at −20 ± 5 °C for 16 to 22 hours. DMF (21 ml, 268 mmol) was then added slowly over the course of 30 minutes while maintaining the batch temperature no more than -15 °C. The batch was stirred at −20 ± 5 °C for 6 to 24 hours. 2-MeTHF (350 ml) was then added to the batch over the course of 30 minutes, followed by slow addition of 3 N HCl (235 ml, 704 mmol) while keeping the batch temperature no more than -10 °C. After the addition of aqueous HCl, the batch was warmed to 0 ± 5 °C and 2 N aqueous NaOH (154 ml, 309 mmol) was added slowly to adjust the solution pH to about 8 to 9. The batch was stirred for about 30 minutes and then warmed to 20 ± 5 °C. The organic layer was separated and washed with 15% aqueous NaCl (3 x 140 ml). The organic layer was subsequently concentrated with addition of 2-MeTHF until KF ≤ 0.10%.
[00269] A portion of the free base of 2-fluoro-4-((3-morpholinoazetidin-1-yl)methyl)benzaldehyde (37.4 g, 134 mmol) so obtained was dissolved in 2-MeTHF (total ~ 420 ml) , to which isopropanol (420 ml) and water (21 ml) were added at 20 ± 5 °C. The batch was then heated to 50 ± 5 °C and a solution of HCl in IPA (5 to 6 N, 28 ml, half of total HCl volume) was added over the course of 1 hour. The batch was seeded with 2-fluoro-4-((3-morpholinoazetidin-1-yl)methyl)benzaldehyde dihydrochloride (700 mg) and aged for 1 hour. The remaining HCl (28 ml) was then added over the course of 1 hour. The batch was agitated at 50 ± 5 °C for 4 hours and then cooled to 20 ± 5 °C for 8 hours. The slurry was filtered, washed with IPA (210 ml), and the filter cake dried under vacuum at 50 ± 5 °C to afford Compound 3 dihydrochloride salt (36 g, yield 75%). 1 H NMR (DMSO-d 6 ) δ (ppm): 12.32-12.55 (m, 1H), 10.23 (s, 1H), 7.93 (t, J =7.6 Hz, 1H), 7.66 (d, J = 10.5 Hz , 1H), 7.58 (d, J = 7.9 Hz, 1H), 4.80 (br s, 2H), 4.48-4.70 (m, 2H), 4.30 (br s, 4H), 3.78-4.00 (m, 5H), 2.93-3.15 (m, 2H). Two polymorphic forms were obtained. XRPD pattern and DSC thermogram of Form A (anhydrous) are shown in FIG.5 and FIG.6, respectively. XRPD pattern, TGA thermogram and DSC thermogram of Form B (hydrate) are shown in FIG.7, FIG.8, and FIG.9, respectively.
[00270] Synthesis of tert-butyl (S)-5-amino-4-(4-((2-fluoro-4-((3-morpholinoazetidin-1-yl)methyl)benzyl)amino)-1,3- dioxoisoindolin-2-yl)-5-oxopentanoate (Compound 2): A mixture of Compound 5 (12 g, 34.5 mmol, 1.0 eq) and Compound 3 dihydrochloride (14.56 g, 41.5 mmol, 1.2 eq) in MeCN (96 ml) was cooled to 0-5 °C. Trifluoroacetic acid (TFA, 2.0 ml, 26 mmol, 0.75 eq) was added, followed by sodium triacetoxyborohydride (STAB, 2.75 g, 12.95 mmol, 0.375 eq) while maintaining the internal temperature below 10 °C. The addition of TFA and STAB was repeated three additional times. After a total of four additions of TFA and STAB, the reaction was aged at 0-5°C for 1 hour. A 10% brine solution (108 ml) was then added to the reaction mixture over the course of 1 hour and partitioned with IPAc (96 ml). The mixture was warmed to 20-25 °C and aged for 30 minutes. The layers were then separated and the organic layer was washed with 2.0 M K3PO4 (114 ml). The pH of the spent aqueous layer should have a pH of about 8.5 – 9.0. The layers were separated again and the organic phase was washed with 8.5% NaHCO 3 (2 x 60 ml), with 30 minutes between each wash, followed by 24% brine (60 ml). The organic fraction was distilled to 72 ml at an internal temperature near 50°C. Toluene (72 ml) was added to bring the volume to 144 ml and distillation continued at constant volume at 50°C with feed and bleed until water content < 0.1. The mixture was heated to 50°C and acetonitrile (48 ml) was added, followed by slow addition of heptane (144 ml) while maintaining the internal temperature above 45 °C. The reaction was held at 50 °C for 2 hours. Once complete, the reaction was slowly cooled to 20-25°C over the course of 4 hours and held at 20-25°C overnight (16 hour). The yellow slurry was then filtered and the yellow cake displacement washed with 1:3:3 mixture of acetonitrile/heptane/toluene (3 x 48 ml). The final cake was then dried under reduced pressure at 50 °C under nitrogen to provide Compound 2 (87.7% isolated molar yield) with >99.0% LCAP. HPLC purity, 99.85%; Chiral purity, >99.9% ee. 1 H NMR (DMSO-d6, 500 MHz) δ (ppm) 7.55 (s, 1H), 7.51 (dd, J = 7.2, 8.4 Hz, 1H), 7.32 (t, J = 7.9 Hz, 1H), 7.16 ( s, 1H), 7.0-7.1 (m, 5H), 4.57 (d, J = 6.3 Hz, 2H), 4.5-4.5 (m, 1H), 3.5-3.6 (m, 6H), 3.3-3.4 (m, 3H), 2.8-2.9 (m, 3H), 2.3-2.4 (m, 1H), 2.1-2.3 (m, 7H), 1.31 (s, 9H); LCMS m/z 610.3 [M+H] + .
[00271] Synthesis of (S)-2-(2,6-dioxopiperidin-3-yl)-4-((2-fluoro-4-((3-morpholinoazetidin-1-yl)methyl)benzyl)amino)isoindoline -1,3-dione bis-besylate salt (Compound 1 bis-besylate): To a suspension of Compound 2 (130 g, 1.0 equiv.) in MeCN (1.56 L, 12 L/kg) agitated at 55 °C was added a solution of benzene sulfonic acid (185 g, 5.5
equiv.) in MeCN (0.39 L, 3 L/kg) and water (0.01 L, 2.0 equiv.). The mixture was stirred at 55 °C for 16 hours. After the reaction age, crystalline seeds (1.3 g, 1 wt%) of bis-besylate salt of Compound 1 were charged into the batch, resulting in formation of a yellow slurry. The slurry was then cooled to 20 °C over the course of 90 minutes. 2-MeTHF (1.3 L, 10 L/kg) was added to the batch slowly over 2 hours at 20 °C. The batch was agitated for an additional 4 hours at 20°C. The yellow slurry was then filtered and the yellow cake re-slurried with MeTHF (1.3 L, 10 L/kg) followed by a displacement MeTHF (0.65 L, 5 L/kg) wash. The final cake was then dried under reduced pressure at 50 °C under nitrogen to give Compound 1 bis-besylate salt (160 g, 88.4% yield). HPLC purity: 98.39%; chiral HPLC purity: 100%. XRPD pattern, TGA thermogram and DSC thermogram of the product are shown in FIG.1, FIG.2, and FIG.3, respectively.
[00272] Synthesis of (S)-2-(2,6-dioxopiperidin-3-yl)-4-((2-fluoro-4-((3-morpholinoazetidin-1-yl)methyl)benzyl)amino)isoindoline -1,3-dione hydrochloride salt (Compound 1 HCl): A suspension of Compound 1 bis-besylate salt (300 g, 1 equiv.) in EtOAc (4.68 L, 15.6 L/kg) and 2-propanol (0.12 L, 0.4 L/kg) was agitated at 15 °C. To the suspension was added a solution of KHCO 3 (82.4 g, 2.5 equiv) in water (1.8 L, 6 L/kg) over 30 minutes. The mixture was heated to 20 °C over 30-60 minutes and then agitated for 30 minutes. The batch was allowed to settle for 30 minutes, at which point the aqueous (bottom) layer was discarded. To the rich organic layer was added water (1.2 L, 4 L/kg) and the reactor contents were agitated for 30 minutes. The batch was allowed to settle for 30 minutes, at which point the aqueous (bottom) layer was discarded. To the rich organic stream was added 2-propanol (2.375 L, 7.9 L/kg) and the stream was then filtered. Water was added to the filtrate to adjust the water content to 8≤KF≤8.2. To the above agitated solution at 20 °C was added 0.2 N HCl (38 mL, 0.025 equiv prepared in EtOAC/IPA 2:1, v/v with 8wt% water) over 10 minutes. To the mixture was added crystalline seeds of Compound 1 hydrochloride salt (1.6 g, 0.5wt%) and the contents of the reactor were agitated at 20 °C for 30 minutes. To the suspension was added 0.2 N HCl (1.44 L, 0.945 equiv. prepared in EtOAC/IPA 2:1, v/v with 8 wt% water) over 4.5 hours. The slurry was agitated for 14 hours, then filtered and washed with EtOAC/IPA (750 mL, 2.5 L/kg, 2:1 v/v with 8 wt% water) followed by IPA (750 mL, 2.5 L/kg). The solids were dried under vacuum at 40 °C to afford Compound 1 hydrochloride salt (170 g, 90% yield). Achiral HPLC purity: 99.91%; chiral HPLC purity: 99.58%. XRPD analysis (FIG.4) confirmed the product (a)
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
Inaxaplin (VX-147) is a small-molecule apolipoprotein L1 inhibitor developed by Vertex Pharmaceuticals for APOL1-mediated kidney disease. In preliminary studies the drug has shown promise in treating people with kidney disease and multiple gain of function mutations on the APOL1 gene.[1][2]
FSGS is a disease of the podocyte (glomerular visceral epithelial cells) responsible for proteinuria and progressive decline in kidney function. NDKD is a disease characterized by hypertension and progressive decline in kidney function. Human genetics support a causal role for the G1 and G2 APOL1 variants in inducing kidney disease. Individuals with 2 APOL1 risk alleles are at increased risk of developing primary (idiopathic) FSGS, human immunodeficiency virus (HIV)-associated FSGS, and NDKD. Currently, FSGS and NDKD are managed with symptomatic treatment (including blood pressure control using blockers of the renin angiotensin system), and patients with FSGS and heavy proteinuria may be offered high dose steroids. Corticosteroids induce remission in a minority of patients and are associated with numerous side effects. These patients, in particular individuals of recent sub-Saharan African ancestry with 2 APOL1 risk alleles, experience rapid disease progression leading to end-stage renal disease (ESRD). Thus, there is an unmet medical need for treatment for FSGS and NDKD.
Synthesis of 3-[5,7-difluoro-2-(4-fluorophenyl)-1H-indol-3-yl]-N-[(3S,4R)-4-hydroxy-2-oxo-pyrrolidin-3-yl]propanamide (2)
Method G: Amide Coupling with CDMT. A 2 L 3-neck RB flask with magnetic stirrer, temperature probe and nitrogen inlet was charged with 3-[5,7-difluoro-2-(4-fluorophenyl)-1H-indol-3-yl]propanoic acid S12 (90.5 g, 283.5 mmol) and (3S,4R)-3-amino-4-hydroxy-pyrrolidin-2-one S2 (39.9 g, 343.6 mmol) in DMF (1.65 L), and stirred for 15 minutes. CDMT (61.1 g, 348 mmol) was added. The mixture was then cooled to −2° C. on an ice bath. N-methylmorpholine was added (131 mL, 1.2 mol) dropwise over 20 minutes and the mixture was heated at 30° C. overnight. The reaction mixture was added into approx. 4.5 L of ice water, and extracted with EtOAc (1.2 L×4). The combined organic layers, were washed with 1.2 L of 1 M HCl (×3) and then water (1.2 L) and brine (1.2 L). The combined organic layers were dried over Na 2SO 4, filtered and concentrated. The mixture was washed through a silica gel plug (1.8 L of silica gel), first eluting with 25% EtOAc in dichloromethane (8 L) to remove impurities, followed by hot EtOAc (8 L), to elute the product. The EtOAc filtrate was concentrated in vacuo. TBME was then added (400 mL), and the mixture allowed to stirred for overnight. Filtration of the resulting solid afforded the product as a white solid. 62 g, 52%) 1H NMR (300 MHz, CD 3OD) δ 7.70-7.58 (m, 2H), 7.29-7.13 (m, 3H), 6.73 (ddd, J=11.1, 9.6, 2.2 Hz, 1H), 4.34 (td, J=7.6, 6.8 Hz, 1H), 4.21 (d, J=7.8 Hz, 1H), 3.56 (dd, J=9.9, 7.6 Hz, 1H), 3.20-3.04 (m, 3H), 2.65-2.53 (m, 2H). LCMS m/z 418.2 [M+H] +. Optical rotation: [α] D20.7=−14.01 (c=1.0, 10 mg in 1 mL of MeOH).
Alternative Procedure for Synthesis of Compound (2)
Step 1. Synthesis of Methyl (E)-3-[5,7-difluoro-2-(4-fluorophenyl)-1H-indol-3-yl]prop-2-enoate (C51)
A solution of 5,7-difluoro-2-(4-fluorophenyl)-1H-indole C50 (100 g, 1.0 equiv) in dichloromethane (850 mL, 8.5 vol) was agitated at 22° C. Methyl 3,3-dimethoxypropionate (63 mL, 1.1 equiv) was charged followed by trifluoroacetic acid (96 mL, 3.1 equiv), which was rinsed forward with dichloromethane (25 mL, 0.25 vol). The batch was heated to 38° C. and stirred at that temperature. After 4 h, the batch was cooled to 22° C. and charged with n-heptane (200 mL, 2 vol). The mixture was stirred for no less than 1 h at 22° C. The slurry was filtered, and the reactor and the filter cake were washed with n-heptane (1×2 vol (200 mL) and 1×3 vol (300 mL)). The resulting solid was dried under vacuum with nitrogen bleed at 45° C. to afford the product C51 (127.7 g, 95% yield).
Step 2. Synthesis of Methyl 3-[5,7-difluoro-2-(4-fluorophenyl)-1H-indol-3-yl]propanoate (C52)
To a hydrogenator was charged methyl (E)-3-[5,7-difluoro-2-(4-fluorophenyl)-1H-indol-3-yl]prop-2-enoate C51 (100.4 g, 1.0 equiv) followed by Pd(OH) 2/C (0.014 equiv). The vessel was sealed and three vacuum/purge cycles with N2 were performed. 2-MeTHF (2000 mL, 20 vol) was charged using residual vacuum and the resulting mixture was stirred at 22° C. The vessel was sealed and three vacuum/purge cycles with N2 were performed followed by one vacuum purge cycle with hydrogen (H 2). The temperature was adjusted to 22° C., and the vessel pressurized with 20 psi H 2. The mixture was agitated at 22° C. for 4 h. Three vacuum/purge cycles with nitrogen N2 were performed. The batch was filtered through a pad of Hyflo® and the filter cake was rinsed with 2-MeTHF (2×300 mL, 2×3 vol). The combined filtrates were placed under vacuum and distilled at ≤45.0° C. to 2.0 to 3.0 total volumes. The batch temperature was adjusted to 22° C. and the vessel was charge with n-heptane (1000 mL, 10 vol) over at least 1 h. A vacuum was applied and the filtrate distilled at 45.0° C. to 3.5 to 4.5 total volumes. The slurry was cooled to 22° C. and allowed to stir for no less than 1 h. The slurry was filtered and the filter cake was washed with n-heptane (1×1 vol (100 mL) and 1×0.5 vol (50 mL)). The solids were dried under vacuum with nitrogen bleed at 45° C. to afford the product C52 (91.9 g, 91% yield).
Step 3. Synthesis of 3-[5,7-difluoro-2-(4-fluorophenyl)-1H-indol-3-yl]propanoic Acid (S12)
A mixture of methyl 3-[5,7-difluoro-2-(4-fluorophenyl)-1H-indol-3-yl]propanoate C52 (80.0 g, 1.0 equiv) and 2-MeTHF (480 mL, 6 vol) was agitated at 22° C. and treated with methanol (72 mL, 0.9 vol). A solution of KOH (27.1 g, 2.0 equiv) in water (107 mL, 1.3 vol) was charged over approximately 20 min. The resulting mixture was heated to an internal temperature of 35° C. and stirred for 3 h. The temperature was adjusted to 22° C. A vacuum was applied and the mixture was distilled at ≤45° C. to 3.0 total volumes. The internal temperature was adjusted to 12° C. The mixture was then charged with water (64 mL, 0.8 vol) and 2-MeTHF (304 mL, 3.8 vol). 6N HCl (75 mL, 0.9 vol) was slowly charged into the mixture with vigorous agitation until the batch attained a pH<3. The internal temperature was adjusted to 22° C., and the biphasic mixture was stirred for no less than 0.5 h. The stirring was stopped and the phases were allowed to separate for no less than 0.5 h. The lower aqueous phase was removed. Water (160 mL, 2 vol) was charged to the reactor at 22° C., and the biphasic mixture stirred for no less than 0.5 h. The stirring was stopped, and the phases allowed separated over no less than 0.5 h. The lower aqueous phase was removed and the batch was filtered through a pad of Hyflo®. The reactor and filter cake were rinsed with 2-MeTHF (160 mL, 2 vol). A vacuum was applied and the combined filtrates distilled at ≤40.0° C. to 2-3 total volumes. The vessel was charged with n-heptane (160 mL, 2 vol), a vacuum was applied and the filtrate distilled at 40.0° C. to 2 total volumes (this step was repeated one additional time). The mixture was then charged with additional n-heptane (144 mL, 1.8 vol). The internal temperature was adjusted to 40° C. and stirred for no less than 2 h. The internal temperature was adjusted to 22° C. over a minimum of 5 h and stirred for no less than 16 hours. The slurry was filtered. The filter cake was washed with n-heptane (3×40 mL, 3×0.5 vol). The solids were dried under vacuum with nitrogen bleed at 45° C. to afford product S12 (72.6 g, 95% yield).
Step 4. Synthesis of 3-[5,7-difluoro-2-(4-fluorophenyl)-1H-indol-3-yl]-N-[(3S,4R)-4-hydroxy-2-oxo-pyrrolidin-3-yl]propanamide (2)
A mixture of S12 (50.0 g, 1.0 equiv), (3S,4R)-3-amino-4-hydroxypyrrolidin-2-one hydrochloride S2 (25.1 g, 1.05 equiv), and CDMT (30.3 g, 1.1 equiv) in DMF (250 mL, 5 vol) was agitated and cooled to 0° C. The reactor was charged with NMM (60 mL, 3.5 equiv) over no less than 1 h, while maintaining the internal temperature at ≤5° C. The batch was stirred at −5° C. for no less than 1 h. The batch was warmed to 22° C. over at least 1 h and stirred at 22° C. for 16 h. The batch was cooled to 0° C. Water (250 mL, 5 vol) was charged, while keeping the internal temperature <20° C. The mixture was charged with a 90/10 mixture of EtOAc/IPA (1000 mL, 20 vol). 6N HCl (40 mL, 0.8 vol) was then charged, while maintaining an internal temperature <10° C., until a pH˜1-3 was achieved. The internal temperature was adjusted to 22° C. and the biphasic mixture stirred for no less than 0.5 h. Stirring was stopped and the phases allowed to separate for no less than 0.5 h. The lower aqueous phase was removed. The aqueous layer was back extracted with a 90/10 mixture of EtOAc/IPA (2×250 mL, 2×5 vol) at 22° C. The combined organic phases from extractions were washed with water (5×500 mL, 5×10 vol) at 22° C., by mixing for no less than 0.5 h and settling for no less than 0.5 h for each wash. The batch was polish filtered. A vacuum was applied and the organic phase distilled at <50° C. to 9.5-10.5 total volumes. The mixture was charged with EtOAc (500 mL, 10 vol), vacuum was applied and the organic phase distilled at <50° C. to 9.5-10.5 total volumes (this step was repeated one more time). The mixture was charged with EtOAc (300 mL, 6 vol) and n-heptane (200 mL, 4 vol). The resulting slurry was heated to 50° C. and stirred for no less than 17 h. The mixture was then cooled to 22° C. over 2 h, and stirred for no less than 1 h. The slurry was filtered. The filter cake was washed with 1:1 EtOAc/n-heptane (2×150 mL, 2×3 vol). The solids were dried under vacuum with nitrogen bleed at ≤45° C. to afford Compound 2 (52.6 g, 80% yield).
Re-Crystallization of Compound 2
Compound 2 (37.6 g, 1.0 equiv) was charged to a reactor followed by a 3:1 mixture of IPA/water (240 mL, 6.4 vol). The slurry was heated to an internal temperature of 75° C. The batch was cooled to an internal temperature of 55° C. and stirred at that temperature for at least 0.5 h. The batch was seeded with 0.5 wt % of a previously generated batch of Compound 2, as a suspension in a mixture of 3:1 IPA/water (4 mL, 0.1 vol). The mixture was stirred at 55° C. for no less than 1.5 h. Water (218 mL, 5.8 vol) was added over minimum period of 5 h while maintaining the temperature at 55° C. The slurry was cooled to 22° C. over no less than 5 h and stirred for no less than 2 h. The slurry was filtered. The filter cake was washed with 2:3 IPA/water (2×114 mL, 2×3 vol). The solids were dried under vacuum with nitrogen bleed at ≤45° C. to afford Compound 2 (34.5 g, 92% yield).
Form A of Compound 2
12.3 kg of Compound 2 was charged to the reactor follow by a 3:1 mixture of 2-propanol/water. Agitation was initiated and the mixture was heated to 75° C. to achieve complete dissolution. The mixture was cooled to 55° C. over 1 hour and agitated at that temperature for 30 minutes. Agitation was continued for 1.5 hours. Water (5.8 vol) was charged over 5 h at 55° C., after which the mixture was cooled to 22° C. over 6 hours. The mixture was agitated at 22° C. for 2 hours then filtered under vacuum. The resulting wet cake was washed with a 3:1 mixture of 2-propanol/water (2.74 vol×2) and pulled dry under vacuum. The wet cake was further dried under vacuum with nitrogen bleed at 45° C. to yield 11.2 kg of Form A.
Hydrate Form A of Compound 2
200 mg of Compound 2 was charged with 10 mL of water. The slurry was cooled to 5° C. and allowed to stir. Hydrate A was observed after 3 days of stirring.
Danicopan, sold under the brand name Voydeya, is a medication used for the treatment of paroxysmal nocturnal hemoglobinuria.[2] It is a complement inhibitor which reversibly binds to factor D to prevent alternative pathway-mediated hemolysis and deposition of complement C3 proteins on red blood cells.[2]
Danicopan was approved for medical use in Japan in January 2024, and in the United States in March 2024.[3][4]
Paroxysmal nocturnal hemoglobinuria (PNH) is a rare acquired hematologic disease characterized by hemolysis, thrombophilia, and bone marrow dysfunction.1,7 Both hemolysis and thrombophilia are mediated primarily by the complement system.1 Standard therapy for PNH involves the use of complement C5 inhibitors (e.g. eculizumab, ravulizumab) which are effective in mitigating complement-mediated intravascular hemolysis and thromboembolism.1 Unfortunately, complement C5 inhibition does not address C3-mediated extravascular hemolysis, which occurs earlier in the complement cascade within the alternative pathway.1,5
Danicopan is a small molecule complement factor D inhibitor that selectively blocks the alternative pathway, thereby working to address extravascular hemolysis when used in conjunction with C5 inhibitors.3 It was first approved in January 2024 in Japan for patients with PNH,2,6 shortly after which the EMA adopted a positive opinion and recommended granting it marketing authorization.2 It was subsequently approved by the FDA in March 2024.4
Step 1: Synthesis of tert-Butyl (2S,4R)-2-((6-bromopyridin-2-yl)carbamoyl) fluoropyrrolidine-1-carboxylate (3): N-Boc-trans-4-Fluoro-L-proline (50.8 kg) was added to DCM (1000 L) in a glass-lined reactor under an atmosphere of nitrogen. The reaction mixture was cooled to 0±5° C. and N-methylimidazole (44.7 kg) was added while maintaining the temperature at 0±5° C. Methanesulfonyl chloride (29.97 kg) was slowly added to the reaction mixture followed by the addition of 2-amino-6-bromopyridine (2). The reaction temperature was warmed to room temperature and stirred for 12 h. The reaction was monitored by HPLC. After completion of the reaction water (2,000 kg) was added, the reaction was stirred and the DCM layer separated. The aqueous layer was once more extracted with DCM (1000 L). The combined DCM layer was washed in succession with dilute HCl, aqueous NaHCO3 and brine. The DCM extract was evaporated to dryness and tert-butyl (2S,4R)-2-((6-bromopyridin-2-yl)carbamoyl)-4-fluoropyrrolidine-1-carboxylate (3) was isolated using DCM heptane mixture and dried. Yield, 71.76 Kg (84.86%))
[0404] Step 2: Synthesis of (2S,4R)-N-(6-Bromopyridin-2-yl)-4-fluoropyrrolidine-2-carboxamide (4): To a solution 4M HCl/Dioxane (168 kg) was added intermediate 3 (40 kg) at 25±5° C. under an atmosphere of nitrogen and the reaction was stirred for 1 h. The reaction was monitored by HPLC and after completion, the reaction was diluted with DCM (800 L) and washed with aqueous NaHCO3. The DCM layer was separated and concentrated. The product, 2S,4R)-N-(6-bromopyridin-2-yl)-4-fluoropyrrolidine-2-carboxamide, (4), was isolated using DCM/heptane and dried. Yield, 25.81 kg, 87%.
[0405] Step 3: Synthesis of tert-Butyl 2-(3-acetyl-5-bromo-1H-indazol-1-yl)acetate (6): 1-(5-Bromo-1H-indazol-yl)ethan-1-one (5, 30 kg) was added to a reactor containing DMF (210 L) under an atmosphere of nitrogen followed by potassium carbonate (4.05 kg). Tert-butyl bromoacetate (3.42 kg) was added to the reaction mixture with stirring and maintaining the temperature at 30±10° C. After addition was complete, the reaction mixture was heated at 50±5° C. for 1 h. After the reaction was complete the reaction mixture was cooled to 25±5° C. and diluted with water (630 L). The precipitated solid was filtered, washed with water (90 L) and dried. Yield, 43.13 kg, 97.13%.
[0406] Step 4: Synthesis of tert-Butyl 2-(3-acetyl-5-(2-methylpyrimidin-5-yl)-1H-indazol-1-yl)acetate (9): Bispinnacolato diboron (14.67 kg) was added to a solution of 4-bromo methylpyrimidine (7, 10 kg) in dioxane (206 kg) under an atmosphere of nitrogen followed by the addition of potassium acetate (17 kg). The reaction mixture was degassed using nitrogen. Pd(dppf)Cl2 (0.94 kg) was added and the reaction mixture heated to 90±5° C. until the pyrimidine was consumed. The reaction mixture was cooled to 25±5° C. and intermediate 6 (16.33 kg) was added followed by potassium carbonate (20.7 kg) and water (16.33 kg) and the reaction was degassed using nitrogen. The reaction was again heated to 90±5° C. until completion. The reaction mixture was cooled to 25±5° C. and diluted with ethyl acetate (269 kg) and water (150 kg) maintaining the temp at 10±5° C. Activated charcoal (1 kg) was added to the mixture with stirring and then filtered through a bed of celite. The ethyl acetate layer was separated, washed with 5% aqueous sodium chloride followed by 5% L-Cysteine solution to remove palladium related impurities. The ethyl acetate layer was evaporated to dryness. The product (9) was isolated from MTBE/heptane. Yield, 11.8 kg, 56%.
[0407] Step 5: Synthesis of 2-(3-Acetyl-5-(2-methylpyrimidin-5-yl)-1H-indazol-1-yl)acetic acid (10): To a stirred solution of intermediate 9 (50 kg) in DCM (465 kg) at 15±5° C. was added TFA (374.5 kg) while maintaining the said temperature. The reaction was warmed to 35±5° C. and stirring continued until completion of the reaction. DCM and TFA were distilled off under reduced pressure. The residue was dissolved in DCM (kg) and stirred with aqueous sodium bicarbonate. The biphasic mixture was acidified with concentrated HCl and the pH was adjusted to 2-3. The precipitated solid was filtered, washed with water and dried. Yield, 42.4 kg, quantitative.
[0408] Step 6: Synthesis of Compound 1: To a solution of intermediate 9 (42 kg) in DMF (277 kg) was added intermediate 4 (38.7 kg) and the reaction was cooled to 10±5° C. Coupling agent TBTU (56.7 kg) was added to the reaction mixture followed by the addition of DIPEA (86.5 kg) while maintaining the reaction temperature at 10±5° C. The reaction was warmed to 25±+5° C. and stirred until complete. The reaction mixture was diluted with ethyl acetate (1344 kg) and washed with water twice. (The reaction may be washed with aq. K2CO3 if fluorine related impurities are present.) Anhydrous sodium sulfate was added to silica gel and added to the ethyl acetate layer and filtered. The ethyl acetate layer was passed over a column of silica gel (40 kg) and the pure fractions were collected. The fractions were treated with activated charcoal and then filtered over celite. The palladium content was checked, and if above 10 ppm, the ethyl acetate layer was treated with palladium scavenging resin (SilabondThiol®). The ethyl acetate was evaporated to dryness under vacuum and the residue was crystallized from IPA (crystalline seed may be added) and heptane to afford Compound 1 Form II. Yield, 60 kg, 78%.
Society and culture
Legal status
In February 2024, the Committee for Medicinal Products for Human Use of the EMA adopted a positive opinion, recommending the granting of a marketing authorization for the medicinal product Voydeya, intended as add-on therapy to ravulizumab or eculizumab for the treatment of residual hemolytic anemia in adults with paroxysmal nocturnal hemoglobinuria (PNH).[2][5] The applicant for this medicinal product is Alexion Europe.[2]
^ Jump up to:abcd“Voydeya EPAR”. European Medicines Agency. 22 February 2024. Archived from the original on 23 February 2024. Retrieved 24 February 2024. Text was copied from this source which is copyright European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
^World Health Organization (2019). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 81”. WHO Drug Information. 33 (1). hdl:10665/330896.
Further reading
Lee JW, Griffin M, Kim JS, Lee Lee LW, Piatek C, Nishimura JI, et al. (December 2023). “Addition of danicopan to ravulizumab or eculizumab in patients with paroxysmal nocturnal haemoglobinuria and clinically significant extravascular haemolysis (ALPHA): a double-blind, randomised, phase 3 trial”. The Lancet. Haematology. 10 (12): e955–e965. doi:10.1016/S2352-3026(23)00315-0. PMID38030318.
fda approved 4/3/2024, To treat certain bloodstream infections, bacterial skin and associated tissue infections, and community-acquired bacterial pneumonia Press Release zevtera
Ceftobiprole, sold under the brand name Zevtera among others, is a fifth-generation[5]cephalosporin antibacterial used for the treatment of hospital-acquired pneumonia (excluding ventilator-associated pneumonia) and community-acquired pneumonia. It is marketed by Basilea Pharmaceutica under the brand names Zevtera and Mabelio.[6][7][8][9][10][11] Like other cephalosporins, ceftobiprole exerts its antibacterial activity by binding to important penicillin-binding proteins and inhibiting their transpeptidase activity which is essential for the synthesis of bacterial cell walls. Ceftobiprole has high affinity for penicillin-binding protein 2a of methicillin-resistant Staphylococcus aureus strains and retains its activity against strains that express divergent mecA gene homologues (mecC or mecALGA251). Ceftobiprole also binds to penicillin-binding protein 2b in Streptococcus pneumoniae (penicillin-intermediate), to penicillin-binding protein 2x in Streptococcus pneumoniae (penicillin-resistant), and to penicillin-binding protein 5 in Enterococcus faecalis.[12]
Medical uses
In the US, ceftobiprole is indicated for the treatment of adults with Staphylococcus aureus bloodstream infections (bacteremia) including those with right-sided infective endocarditis;[4] adults with acute bacterial skin and skin structure infections;[4] and people with community-acquired bacterial pneumonia.[4]
Microbiology
Ceftobiprole has shown in vitro antimicrobial activity against a broad range of Gram-positive and Gram-negative pathogens. Among the Gram-positive pathogens, ceftobiprole has demonstrated good in vitro activity against methicillin-resistant Staphylococcus aureus, methicillin-susceptible Staphylococcus aureus and coagulase-negative staphylococci, as well as against strains of methicillin-resistant Staphylococcus aureus with reduced susceptibility to linezolid, daptomycin or vancomycin.[13] Ceftobiprole has also displayed potent activity against Streptococcus pneumoniae (including penicillin-sensitive, penicillin-resistant and ceftriaxone-resistant strains) and Enterococcus faecalis, but not against Enterococcus faecium. For Gram-negative pathogens, ceftobiprole has shown good in vitro activity against Haemophilus influenzae (including both ampicillin-susceptible and ampicillin-non-susceptible isolates), Pseudomonas aeruginosa and strains of Escherichia coli, Klebsiella pneumoniae and Proteus mirabilis that do not produce extended-spectrum β-lactamases (ESBL). Like all other cephalosporins, ceftobiprole was inactive against strains that produce extended-spectrum β-lactamases.[14]
The efficacy of ceftobiprole has been demonstrated in two large randomized, double-blind, phase 3 clinical trials in patients with hospital-acquired and community-acquired pneumonia. Ceftobiprole was non-inferior to ceftazidime plus linezolid in the treatment of hospital-acquired pneumonia (excluding ventilator-acquired pneumonia) and non-inferior to ceftriaxone with or without linezolid in the treatment of community-acquired pneumonia.[15][16]
Pharmacology
Ceftobiprole medocaril
Ceftobiprole is the active moiety of the prodrug ceftobiprole medocaril and is available for intravenous treatment only. It is mainly excreted via the kidney.[17]
Society and culture
Legal status
500 mg powder
Ceftobiprole has been approved for the treatment of adults with hospital acquired pneumonia (excluding ventilator-acquired pneumonia) and community-acquired pneumonia in twelve European countries, Canada, and Switzerland.[18]
In February 2010, the Committee for Medicinal Products for Human Use of the European Medicines Agency adopted a negative opinion, recommending the refusal of the marketing authorization for the medicinal product Zeftera, intended for treatment of complicated skin and soft-tissue infections in adults. The company that applied for authorization is Janssen-Cilag International N.V. The applicant requested a re-examination of the opinion. After considering the grounds for this request, the CHMP re-examined the opinion, and confirmed the refusal of the marketing authorization in June 2010.[19]
Processes for producing ceftobiprole medocaril are known per se. What the processes known from the prior art have in common is that, starting from 7-aminocephalosporanic acid, a large number of intermediates have to be produced, isolated and purified in order to obtain ceftobiprole medocaril of the general formula (1) in sufficient purity.
The compound of the general formula (1) is known per se and is described, for example, in WO 99/65920. It can be used for the treatment and prophylaxis of bacterial infectious diseases, especially infectious diseases caused by methicillin-resistant Staphylococcus Aureus strains.
WO 99/65920 describes, as the last step in the production process of ceftobiprole Medocaril, a reaction in which the Medocaril prodrug unit is introduced into a compound of the general formula (2).
The compound of the general formula (2) is also known per se and has been described, for example, in EP 0 849 269 A1. The compound of the general formula (2) is prepared according to EP 0 849 269 A1 starting from (2R,6R,7R)-te rt. B u toxyc abonylamin o-3-formyl-8-oxo-5-thia-1 -azabicyclo[4.2.0]oct-3-ene-2-carboxylic acid benzhydryl ester by Wittig reaction with (1 ‘-allyloxycarbonyl-2- oxo-[1,3’]bipyrrolidinyl-3-yl)-triphenylphosphonium bromide. The resulting Δ2 reaction product is reisomerized to the desired Δ3 isomer by sulfoxidation and subsequent reduction and then deprotected from the benzhydryl ester with trifluoroacetic acid. The acylation in position 7 occurs by reaction with (Z)-(5-amino-[1,2,4]-thiadiazol-3-yl)-trityloxyiminothioacetic acid S-benzothiazol-2-yl ester. The compound of the general formula (2) is then obtained by removing the protective groups.
In EP 1 067 131 A1 the formation of the ylide in toluene or a mixture of toluene and dichloromethane is tert by adding alkali. Butylate in tetrahydrofuran, which allows the base to be added as a solution. The reaction of the ylide with the corresponding aldehyde is described at a reaction temperature of -70 0 C.
EP 0 841 339 A1 relates to cephalosporin derivatives and processes for their production. WO 95/29182 also discloses intermediates for the production of cephalosporins.
WO 01/90111 describes a further production of ceftobiprole Medocaril in several stages starting from desacetyl-7-aminocephalosporanic acid by acylation with (Z)-(5-amino-[1,2,4]-thiadiazol-3-yl)- trityloxyiminothioacetic acid S-benzothiazol-2-yl ester in N,N-dimethylformamide, followed by in situ esterification with diphenyldiazomethane in dichloromethane to give the corresponding benzohydryl ester, which is precipitated and isolated by adding hexane. In the next step, this product is oxidized to the corresponding aldehyde using TEMPO/NaOCI in dichloromethane/water or with Braunstein in tetrahydrofuran/dichloromethane. The next reaction step involves the Wittig reaction to the 3-vinyl-substituted derivative, in which the reaction takes place in dichloromethane/toluene/tetrahydrofuran at -78°C. The crude product is stirred with ethanol and made from dichloromethane/tert. Butyl methyl ether recrystallized or purified chromatographically. According to the method disclosed in WO 01/901 11, the Wittig reaction is carried out at low temperatures of -80 to -70 0 C in a complex solvent mixture of dichloromethane, toluene and tetrahydrofuran. This leads to significant disadvantages when carrying out the reaction on a production scale, since regeneration of the process solvents is difficult.
5 , 1 4 g of 7-amino-3-formyl-ceph-3-em-4-carboxy I at was dissolved in 2 7 , 8 m bis(trimethylsilyl)acetamide and 50 ml propylene oxide. 16.8 g of (1 R/S,3’R)-(1′-tert-butyloxycarbonyl-2-oxo-[1,3′]bipyrrolidinyl-3-yl)-triphenylphosphonium bromide (EP1067131, WO02/14332) slowly added in portions. Stirring was continued at 1 ° C until the starting material had reacted and then the crystalline precipitate was added
Nitrogen atmosphere filtered off and washed with 50 ml cyclohexane/bis(tirmethylsilyl)acetamide 99.5/0.5. After drying under vacuum, the desired product was obtained in silylated form.
The material was dissolved in 100ml dichloromethane and at 0 0 C with 50ml 3%
NaHCC> 3 solution added. The phases were separated, the organic phase was washed with 30 ml of water and the combined water phases were adjusted to pH 3.5 with 3% H 3 PO 4 after activated carbon treatment. The crystalline precipitate was filtered, washed with water and dried under vacuum.
10.28 g of 7-amino-3-formyl-ceph-3-em-4-carboxylate were dissolved in 55.6 ml of bis(trimethylsilyl)acetamide and 100 ml of propylene oxide. 33.6 g of (1R/S, 3’R)-(1′-tert. Butyloxycarbonyl-2-oxo-[1,3′]bipyrrolidinyl-3-yl)-triphenylphosphonium bromide (EP1067131, WO02.) were then added at 0 0 C /14332) slowly added in portions over 22 hours. The mixture was stirred at 1° C. until the starting material had reacted and then the reaction mixture was cooled to -20 ° C. The crystalline precipitate was filtered off under a nitrogen atmosphere and washed in portions with 180 ml of cyclohexane/bis(trimethylsilyl)acetamide 99.5/0.5. After drying under vacuum, the bissilylated
1.0g (6R,7R)-7-Trimethylsilylamino-3[E-(R)-1′-(5-tert.butyloxycarbonyl)-2-oxo-[I.Slbipyrrolidinyl-S-ylidenemethyO-δ-oxo-δ -thia-i-aza-bicyclo^^.Oloct^-ene^- carboxylic acid trimethylsilyl esters were dissolved in 10ml dichloromethane and a solution of 300mg dicyclohexylamine in 1ml EtOH and 10ml ethyl acetate was added. The precipitate was filtered off, washed with ethyl acetate and dried in vacuo.
3.0g (6R,7R)-7-Amino-3[E-(R)-1′-(5-tert-butyloxycarbonyl)-2-oxo-[1,3′]bipyrrolidinyl-3-ylidenemethyl]-8 -oxo-5-thia-1-aza-bicyclo[4.2.0]oct-2-ene-2-carboxylic acid in silylated form was dissolved in 150 ml of dichloromethane at 0°. 600 μl of DMF/water 5/1 and 1.8 ml of bis(trimethylsilyl) acetamide and 2.29 g of 2-trityloxyimino-2-(5-amino-1,2,4-thiadiazol-3-yl) acetic acid chloride were then added Hydrochoride (J. Antibiotics 37:557 – 571, 1984) was added in portions.
After 3 hours at 0°, the mixture was poured into 30 ml MeOH/120 ml water and the methylene chloride phase was separated off. The organic phase was concentrated to 66g and 25ml of trifluoroacetic acid was added. After 10 minutes, 1.5 ml triethylsilane and 10 ml water were added and the mixture was cooled to -15 ° C. The organic phase was separated off and again with 6 ml
Washed trifluoroacetic acid/water 1/1. The combined aqueous phases were diluted to 150 ml with water and filtered through an adsorber resin column with XAD-1600. After washing out the column with water, elution was carried out with water/acetonitrile 85/15. The product-containing fractions were concentrated in vacuo and allowed to stand at 0° for post-crystallization. The crystalline
Product was filtered off, washed with water and dried under vacuum.
Auswaage: 2,66g
4.2 Variant B:
7.4g (6R,7R)-7-Amino-3[E-(R)-1′-(5-tert-butyloxycarbonyl)-2-oxo-[1,3′]bipyrrolidinyl-3-ylidenemethyl]-8 -oxo-5-thia-1-aza-bicyclo[4.2.0]oct-2-ene-2-carboxylic acid was dissolved at 0° in 781 ml of dichloromethane with the addition of 6.7 ml of triethylamine. 8.65 g of 2-trityloxyimino-2-(5-amino-1,2,4-thiadiazol-3-yl)-acetic acid chloride hydrochloride were then added in portions. After the starting material had reacted, the mixture was poured into 500 ml of water and the methylene chloride phase was separated off. The organic phase was dried over Na 2 SC> 4 and concentrated in vacuo.
The residue was dissolved in 148 ml of dichloromethane and 4.5 ml of triethylsilane and 74 ml of trifluoroacetic acid were added at room temperature. After 30 minutes, 222 ml of dichloromethane and 222 ml of water were added and the mixture was cooled to -20 0 C. The organic phase was separated off and washed again with a mixture of 37 ml of trifluoroacetic acid and 148 ml of water. The combined aqueous phases were diluted with water to 364 ml, filtered through an adsorber resin and eluted with acetonitrile/water 15/85.
The filtrate was concentrated to 35g on a Rotavapor, filtered and washed with water.
After drying in a vacuum, 4.5 g of the sample was obtained.
6.0g (6R,7R)-7-Amino-3[E-(R)-1′-(5-tert-butyloxycarbonyl)-2-oxo-[1,3′]bipyrrolidinyl-3-ylidenemethyl]-8 -oxo-5-thia-1-aza-bicyclo[4.2.0]oct-2-ene-2-carboxylic acid in silylated form was dissolved in 300 ml of dichloromethane at 0°. 1200 μl of DMF/water 5/1 and 8.1 ml of bis(tirmethylsilyl)acetamide were then added as well as 5.3g of 2-trityloxyimino-2-(5-amino-1,2,4-thiadiazol-3-yl)acetic acid chloride Hydrochoride (J. Antibiotics 37:557 – 571, 1984) added in portions. The mixture was then poured into 60 ml MeOH/240 ml water and the methylene chloride phase was separated off. The organic phase was concentrated to 48g and 1.5 ml of triethylsilane was added. After adding 50ml
Trifluoroacetic acid was stirred at room temperature for 60 min, 20 ml of water was added and the mixture was cooled to -15°C. The organic phase was separated off and washed again with 20 ml trifluoroacetic acid/water 1/1. The combined aqueous phases were diluted to 500 ml with water and treated with 2.0 g of activated carbon. After filtration, the solution was concentrated in vacuo.
The residue was diluted to 50 ml with water and adjusted to pH 6.9 with saturated NaHCO 3 solution. The mixture was stirred at 0 0 C for 2 hours, filtered and the precipitate washed with water.
0, 5 3 g ( 6 R , 7 R )-7-[(Z)-2-(5-amino-[1,2,4]thiadiazol-3-yl)-2-hydroxyimino-acetylamino]-8- oxo-3-[(E)-(R)-2-oxo-[1,3′]bipyrrolidinyl-3-ylidenemethyl]-5-thia-1 – aza-bicyclo[4.2.0]oct-2-ene- 2 carboxylic acid were dissolved in 5 ml of dimethyl sulfoxide and 0.27 g of carbonic acid (5-methyl-2-oxo-[1,3]dioxol-4-ylmethyl)-4-nitrophenyl ester were added and stirred at room temperature. A solution of sodium ethyl hexanoate in 30 ml of acetone was added for precipitation. The precipitate was filtered and washed with acetone.
^ Zhanel GG, Lam A, Schweizer F, Thomson K, Walkty A, Rubinstein E, et al. (2008). “Ceftobiprole: a review of a broad-spectrum and anti-MRSA cephalosporin”. American Journal of Clinical Dermatology. 9 (4): 245–254. doi:10.2165/00128071-200809040-00004. PMID18572975. S2CID24357533.
^ Farrell DJ, Flamm RK, Sader HS, Jones RN (April 2014). “Activity of ceftobiprole against methicillin-resistant Staphylococcus aureus strains with reduced susceptibility to daptomycin, linezolid or vancomycin, and strains with defined SCCmec types”. International Journal of Antimicrobial Agents. 43 (4): 323–327. doi:10.1016/j.ijantimicag.2013.11.005. PMID24411474.
^ Nicholson SC, Welte T, File TM, Strauss RS, Michiels B, Kaul P, et al. (March 2012). “A randomised, double-blind trial comparing ceftobiprole medocaril with ceftriaxone with or without linezolid for the treatment of patients with community-acquired pneumonia requiring hospitalisation”. International Journal of Antimicrobial Agents. 39 (3): 240–246. doi:10.1016/j.ijantimicag.2011.11.005. PMID22230331.
^“Zeftera (previously Zevtera) EPAR”. European Medicines Agency (EMA). 18 February 2010. Retrieved 6 April 2024. Text was copied from this source which is copyright European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
External links
Clinical trial number NCT03138733 for “Ceftobiprole in the Treatment of Patients With Staphylococcus Aureus Bacteremia” at ClinicalTrials.gov
Clinical trial number NCT03137173 for “Ceftobiprole in the Treatment of Patients With Acute Bacterial Skin and Skin Structure Infections” at ClinicalTrials.gov
Clinical trial number NCT00326287 for “Ceftobiprole in the Treatment of Patients With Community-Acquired Pneumonia” at ClinicalTrials.gov
Clinical trial number NCT03439124 for “Ceftobiprole in the Treatment of Pediatric Patients With Pneumonia” at ClinicalTrials.gov
////////Ceftobiprole, BAL-9141, BAL-9141-000, BAL-9141000, BAL9141-000, RO 63-9141, RO-63-9141, RO-639141, fda 2024, zevtera, approvals 2024, Ceftobiprole medocaril sodium salt
4/23/2024 FDA APROVED, To treat relapsed or refractory pediatric low-grade glioma, Ojemda
AMG 2112819
BIIB 024
BIIB-024
BIIB024
DAY 101
DAY-101
DAY101
MLN 2480
MLN-2480
MLN2480
TAK 580
TAK-580
TAK580
Tovorafenib, sold under the brand name Ojemda, is a medication used for the treatment of glioma.[1] It is a kinase inhibitor.[1]
The most common adverse reactions include rash, hair color changes, fatigue, viral infection, vomiting, headache, hemorrhage, pyrexia, dry skin, constipation, nausea, dermatitis acneiform, and upper respiratory tract infection.[2] The most common grade 3 or 4 laboratory abnormalities include decreased phosphate, decreased hemoglobin, increased creatinine phosphokinase, increased alanine aminotransferase, decreased albumin, decreased lymphocytes, decreased leukocytes, increased aspartate aminotransferase, decreased potassium, and decreased sodium.[2]
It was approved for medical use in the United States in April 2024,[1][2][3][4] and is the first approval of a systemic therapy for the treatment of people with pediatric low-grade glioma with BRAF rearrangements, including fusions.[2]
Medical uses
Tovorafenib is indicated for the treatment of people six months of age and older with relapsed or refractory pediatric low-grade glioma harboring a BRAF fusion or rearrangement, or BRAF V600 mutation.[1][2]
History
Efficacy was evaluated in 76 participants enrolled in FIREFLY-1 (NCT04775485), a multicenter, open-label, single-arm trial in participants with relapsed or refractory pediatric low-grade glioma harboring an activating BRAF alteration detected by a local laboratory who had received at least one line of prior systemic therapy.[2] Participants were required to have documented evidence of radiographic progression and at least one measurable lesion.[2] Participants with tumors harboring additional activating molecular alterations (e.g., IDH1/2 mutations, FGFR mutations) or with a known or suspected diagnosis of neurofibromatosis type 1 were excluded.[2] Participants received tovorafenib based on body surface area (range: 290 to 476 mg/m2, up to a maximum dose of 600 mg) once weekly until they experienced disease progression or unacceptable toxicity.[2] The US Food and Drug Administration (FDA) granted the application for tovorafenib priority review, breakthrough therapy, and orphan drug designations.[2]
^World Health Organization (2022). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 88”. WHO Drug Information. 36 (3). hdl:10665/363551.
Clinical trial number NCT04775485 for “A Study to Evaluate DAY101 in Pediatric and Young Adult Patients With Relapsed or Progressive Low-Grade Glioma and Advance Solid Tumors (FIREFLY-1)” at ClinicalTrials.gov
Berdazimer sodium, sold under the brand name Zelsuvmi, is a medication used for the treatment for molluscum contagiosum.[1] Berdazimer sodium is a nitric oxide releasing agent.[1] It is a polymer formed from sodium 1-hydroxy-3-methyl-3-(3-(trimethoxysilyl)propyl)-1-triazene-2-oxide and tetraethyl silicate.[2]
Berdazimer sodium was approved for medical use in the United States in January 2024.[3][4][5]
Medical uses
Berdazimer sodium is indicated for the topical treatment of molluscum contagiosum.[1]
Pharmacology
Mechanism of action
Berdazimer sodium is a nitric oxide releasing agent.[1] The mechanism of action for the treatment of molluscum contagiosum is unknown.[1]
Pharmacodynamics
The pharmacodynamics of berdazimer sodium are unknown.[1]
Society and culture
Legal status
Berdazimer sodium was approved for medical use in the United States in January 2024.[4]
Berdazimer is a polymeric substance consisting of a polysiloxane backbone (Si-O-Si bonds) with covalently bound N-diazeniumdiolate nitric oxide (NO) donors. It releases NO through exposure to proton donors like water, which will degrade the N-diazeniumdiolate entity.2 Berdazimer was previously investigated as a potential treatment for molluscum contagiosum, a viral cutaneous infection mainly affecting children, sexually active adults, and immunocompromised patients. It is one of the 5 most prevalent skin diseases in the world and the third-most common viral skin infection in children.3 Previously, the first line treatment for molluscum contagiosum was surgical excision, although it poses challenges such as repeated doctor visits, post-surgical scarring and skin discoloration, and fear and anxiety in the pediatric population.3
On Jan 05, 2024, the FDA approved berdazimer under the brand name ZELSUVMI for the treatment of adult and pediatric molluscum contagiosum, and it is the first drug to be approved for this condition. This decision is based on positive results demonstrated in 2 Phase 3 trials, B-SIMPLE 4 and B-SIMPLE 2, where reduced lesion counts were observed with once-a-day uses of berdazimer.5
^World Health Organization (2018). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 79”. WHO Drug Information. 32 (1). hdl:10665/330941.
Further reading
Pera Calvi I, R Marques I, Cruz SA, Mesquita YL, Padrao EM, Souza RM, et al. (2023). “Safety and efficacy of topical nitric oxide-releasing berdazimer gel for molluscum contagiosum clearance: A systematic review and meta-analysis of randomized controlled trials”. Pediatric Dermatology. 40 (6): 1060–1063. doi:10.1111/pde.15419. PMID37721050. S2CID262045499.
Han H, Smythe C, Yousefian F, Berman B (February 2023). “Molluscum Contagiosum Virus Evasion of Immune Surveillance: A Review”. Journal of Drugs in Dermatology. 22 (2): 182–189. doi:10.36849/JDD.7230. PMID36745361. S2CID256613906.
Clinical trial number NCT04535531 for “A Phase 3 Molluscum Contagiosum Efficacy and Safety Study (B-SIMPLE4)” at ClinicalTrials.gov
Clinical trial number NCT03927703 for “A Phase 3 Efficacy & Safety of SB206 & Vehicle Gel for the Treatment of MC (B-SIMPLE2)” at ClinicalTrials.gov
Clinical trial number NCT03927716 for “A Phase 3 Randomized Parallel Group Study Comparing the Efficacy & Safety of SB206 & Vehicle Gel in the Treatment of MC (B-SIMPLE1)” at ClinicalTrials.gov
CAS Name: 1-[[(6R,7R)-7-[[(2Z)-(2-Amino-4-thiazolyl)(methoxyimino)acetyl]amino]-2-carboxy-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-en-3-yl]methyl]-1-methylpyrrolidinium inner salt
Additional Names: 1-[[(6R,7R)-7-[2-(2-amino-4-thiazolyl)glyoxylamido]-2-carboxy-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-en-3-yl]methyl]-1-methylpyrrolidinium hydroxide inner salt 72-(Z)-2-(O-methyloxime); 7-[(Z)-2-(2-aminothiazol-4-yl)-2-methoxyiminoacetamido]-3-(1-methylpyrrolidinio)methyl-3-cephem-4-carboxylate
Manufacturers’ Codes: BMY-28142
Molecular Formula: C19H24N6O5S2
Molecular Weight: 480.56
Percent Composition: C 47.49%, H 5.03%, N 17.49%, O 16.65%, S 13.34%
Literature References: Semisynthetic, fourth generation cephalosporin antibiotic. Prepn: S. Aburaki et al.,DE3307550; eidem,US4406899 (both 1983 to Bristol-Myers); and antibacterial activity: T. Naito et al.,J. Antibiot.39, 1092 (1986). In vitro comparative antimicrobial spectrum: N. J. Khan et al.,Antimicrob. Agents Chemother.26, 585 (1984); and b-lactamase stability: H. C. Neu et al.,J. Antimicrob. Chemother.17, 441 (1986). HPLC determn in plasma and urine: R. H. Barbhaiya et al.,Antimicrob. Agents Chemother.31, 55 (1987). Clinical evaluations in infection: N. Clynes et al.,Diagn. Microbiol. Infect. Dis.12, 257 (1989); S. Oster et al.,Antimicrob. Agents Chemother.34, 954 (1990). Review of clinical pharmacokinetics: M. P. Okamoto et al.,Clin. Pharmacokinet.25, 88-102 (1993).
Properties: Colorless powder, mp 150° (dec). uv max (pH 7 phosphate buffer): 235, 257 nm (e 16700, 16100).
Melting point: mp 150° (dec)
Absorption maximum: uv max (pH 7 phosphate buffer): 235, 257 nm (e 16700, 16100)
Derivative Type: Sulfate
Molecular Formula: C19H24N6O5S2.H2SO4
Molecular Weight: 578.64
Percent Composition: C 39.44%, H 4.53%, N 14.52%, O 24.89%, S 16.62%
Properties: mp 210° (dec). uv max (pH 7 phosphate buffer): 236, 258 nm (e 17200, 16900).
Melting point: mp 210° (dec)
Absorption maximum: uv max (pH 7 phosphate buffer): 236, 258 nm (e 17200, 16900)
Derivative Type: Hydrochloride monohydrate
CAS Registry Number: 123171-59-5
CAS Name: 1-[[(6R,7R)-7-[[(2Z)-(2-Amino-4-thiazolyl)(methoxyimino)acetyl]amino]-2-carboxy-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-en-3-yl]methyl]-1-methylpyrrolidinium chloride monohydrochloride monohydrate
FDA APPROVED 2/22/2024, To treat complicated urinary tract infections, Exblifep
BMY 28142
BMY-28142
Cefepime is a fourth-generation cephalosporinantibiotic. Cefepime has an extended spectrum of activity against Gram-positive and Gram-negativebacteria, with greater activity against both types of organism than third-generation agents. A 2007 meta-analysis suggested when data of trials were combined, mortality was increased in people treated with cefepime compared with other β-lactam antibiotics.[1] In response, the U.S. Food and Drug Administration (FDA) performed their own meta-analysis which found no mortality difference.[2]
Cefepime is a broad-spectrum cephalosporin antibiotic and has been used to treat bacteria responsible for causing pneumonia and infections of the skin and urinary tract. Some of these bacteria include Pseudomonas, Escherichia, and Streptococcus species. The following represents MIC susceptibility data for a few medically significant microorganisms:[7]
Escherichia coli: ≤0.007 – 128 μg/ml
Pseudomonas aeruginosa: 0.06 – >256 μg/ml
Streptococcus pneumoniae: ≤0.007 – >8 μg/ml
Chemistry
The combination of the syn-configuration of the methoxyiminomoiety and the aminothiazole moiety confers extra stability to β-lactamase enzymes produced by many bacteria. The N–methylpyrrolidine moiety increases penetration into Gram-negative bacteria. These factors increase the activity of cefepime against otherwise resistant organisms including Pseudomonas aeruginosa and Staphylococcus aureus.
Semisynthetic, fourth generation cephalosporin antibiotic. Prepn: S. Aburaki et al., DE 3307550; eidem, US 4406899 (both 1983 to Bristol-Myers); and antibacterial activity: T. Naito et al., J. Antibiot. 39, 1092 (1986).
Trade names
Following expiration of the Bristol-Myers Squibb patent,[] cefepime became available as a generic and is now] marketed by numerous companies worldwide under tradenames including Neopime (Neomed), Maxipime, Cepimax, Cepimex, and Axepim.
^ Yahav D, Paul M, Fraser A, Sarid N, Leibovici L (May 2007). “Efficacy and safety of cefepime: a systematic review and meta-analysis”. The Lancet. Infectious Diseases. 7 (5): 338–348. doi:10.1016/S1473-3099(07)70109-3. PMID17448937.
^ “Cefepime (maxipime), large spectrum 4th generation cephalosporin, resistant to beta-lactamases]”.
^World Health Organization (2019). Executive summary: the selection and use of essential medicines 2019: report of the 22nd WHO Expert Committee on the selection and use of essential medicines. Geneva: World Health Organization. hdl:10665/325773. WHO/MVP/EMP/IAU/2019.05. License: CC BY-NC-SA 3.0 IGO.
^ Chapman TM, Perry CM (2003). “Cefepime: a review of its use in the management of hospitalized patients with pneumonia”. American Journal of Respiratory Medicine. 2 (1): 75–107. doi:10.1007/bf03256641. PMID14720024.
Aprocitentan, sold under the brand name Tryvio, is a medication used to treat hypertension (high blood pressure).[1] It is developed by Idorsia.[2] It is taken by mouth.[1]
Aprocitentan is indicated for the treatment of hypertension in combination with other antihypertensive drugs, to lower blood pressure in adults who are not adequately controlled on other medications.[1]
Data from animal reproductive toxicity studies with other endothelin-receptor agonists indicate that use is contraindicated in pregnant women.[1]
Mechanism of action
Aprocitentan is an endothelin receptor agonist that inhibits the protein endothelin-1 from binding to endothelin A and endothelin B receptors.[1][4] Endothelin-1 mediates various adverse effects via its receptors, such as inflammation, cell proliferation, fibrosis, and vasoconstriction.[1]
Society and culture
Economics
Aprocitentan is developed by Idorsia, which sold it to Janssen and purchased the rights back in 2023, for US$343 million.[6]
Legal status
Aprocitentan was approved for medical use in the United States in March 2024.[1]
In April 2024, the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency adopted a positive opinion, recommending the granting of a marketing authorization for the medicinal product Jeraygo, intended for the treatment of resistant hypertension in adults.[7] The applicant for this medicinal product is Idorsia Pharmaceuticals Deutschland GmbH.[7]
The following example was prepared according to the procedures described below. All compounds were characterized by 1H-NMR (300 MHz) and occasionally by 13C-NMR (75 MHz) (Varian Oxford, 300 MHz; chemical shifts are given in ppm relative to the solvent used; multiplicities: s=singlet, d=doublet, t=triplet; m=multiplet), by LC-MS (Finnigan Navigator with HP 1100 Binary Pump and DAD, column: 4.6×50 mm, Develosil RP Aqueous, 5 μm, 120 A, gradient: 5-95% acetonitrile in water, 1 min, with 0.04% trifluoroacetic acid, flow: 4.5 ml/min), t R is given in min; by TLC (TLC-plates from Merck, Silica gel 60 F 254) and occasionally by melting point.
Preparation A: Benzylsulfamide Potassium Salt
A.i. Benzylsulfamide
Chlorosulfonylisocyanate (14.14 g) was dissolved in DCM (50 mL) and cooled to 0° C. A solution of t-BuOH (9.6 mL) in DCM (50 mL) was added within 30 min. Stirring was continued for additional 30 min at rt. The solution thus obtained was then added at 0° C. within 1 h to a solution of benzylamine (10.7 g) and TEA (15.32 mL) in DCM (200 mL). Stirring is continued for 10 h at rt. The mixture was concentrated in vacuo, taken up in EA (500 mL) and washed with water (2×40 mL) and brine (30 mL), dried over MgSO 4, filtered. The filtrate was concentrated in vacuo and the crude material was crystallized from EA and dried under HV to give N-benzyl-N′-tert-butoxycarbonyl sulfamide (13.68 g).
This material was dissolved in dioxane (20 ml) and 4 M HCl in dioxane (120 mL) was added within 1 h at rt. The mixture was stirred for 8 h before the solvent was evaporated and the residue dried under HV to give benzylsulfamide as an off-white powder (9.47 g).
To a solution of benzylsulfamide (17.98 g) in McOH (300 mL) was carefully added potassium tert-butylate (10.8 g). The mixture was stirred at rt for 15 min before the solvent was evaporated. The remaining residue was dried under HV to give benzylsulfamide potassium salt as an off-white powder (21.73 g).
To a solution of 4-bromophenylacetic acid (50 g) in methanol (250 ml) was added dropwise thionyl chloride (34.2 mL) while the temperature of the reaction mixture was kept at 0-5° C. Upon complete addition cooling was stopped and the mixture was allowed to warm to rt. Stirring was continued for 75 min before the solvent was removed in vacuo. The yellow oil was dissolved in benzene and again concentrated. The residue was dissolved in EA, washed with water, brine, 2 N aq. Na 2CO 3, and again brine. The org. extract was dried over MgSO 4, filtered, concentrated and dried under HV at 85° C. for 30 min to give the expected product as a yellow oil (52.4 g).
At 40° C., a solution of intermediate B.i (52 g) in THF (100 mL) was carefully added over a period of 40 min to a suspension of NaH (15.6 g) in dry THF (450 mL). Stirring was continued for 70 min without heating and the temperature dropped to 27° C. The evolution of gas stopped before dimethylcarbonate (76.42 mL) was added dropwise while the temperature of the mixture was maintained at 29-31° C. Stirring was continued for 22 h at rt. The mixture was cooled to −10° C. and then carefully neutralized to pH 6-7 with aq. HCl before bulk of the THF was removed in vacuo. The residue was dissolved in EA (700 mL), washed 3 times with 1 N aq. HCl-solution and once with brine, dried over MgSO 4. Most of the EA was evaporated before Hex was added. The product crystallised overnight at 4° C. The crystals were collected, washed with Hex and dried to give the expected product as pale yellow crystals (45.9 g).
A solution of intermediate B.ii (11.73 g) in MeOH (100 mL) was added at 0° C. to a solution of sodium (2.83 g) in MeOH (100 mL). The mixture was stirred for 18 h at rt before formamidine hydrochloride (4.10 g) was added. The suspension was stirred at rt for 4 h. The solvent was removed and the residue was suspended in 10% aq. citric acid (100 mL) and stirred for 10 min. The white precipitate was collected, washed with 10% aq. citric acid, water, evaporated three times from cyclohexane and dried under HV at 40° C. to give 5-(4-bromophenyl)-pyrimidine-4,6-diol as a pale beige powder (9.90 g).
LC-MS: t R=0.62 min, [M+H] +=266.89/268.89 (Br-isotopes).
B.iv. 5-(4-bromo-phenyl)-4,6-dichloro-pyrimidine
To a suspension of 5-(4-bromophenyl)-pyrimidine-4,6-diol (9.90 g) in POCl 3 (130 mL) was carefully added N,N-dimethylaniline (13.5 mL). The mixture is heated to 130° C. for 2 h. The dark brown solution is concentrated in vacuo and the residue was poured into ice/water. The suspension is diluted with 2 N HCl and water and stirred for 20 min. The precipitate that formed is collected and washed with water. The solid material is dissolved in EA, washed with 1 N aq. HCl and brine. The org. phase is dried over MgSO 4 and evaporated. The material is further purified by column chromatography on silica gel eluting with Hex:EA 95:5 to 1:1 followed by crystallisation from Hex/EA at −20° C. to give 4,6-dichloro-5-(4-bromophenyl)-pyrimidine as pale yellow crystals (8.3 g).
A solution of 5-(4-bromophenyl)-4,6-dichloro-pyrimidine (4.00 g, 13.2 mmol) and benzylsulfamide potassium salt (7.38 g, 32.9 mmol) in DMSO (30 mL) was stirred at it for 24 h before being diluted with a 10% aq. citric acid solution (200 mL). The suspension that formed was filtered. The collected solid was washed well with water and dried under HV at 40° C. for 48 h to give the expected product as a white powder (6.15 g).
t-BuOK (18.5 g, 164.5 mmol) was added portionwise to a suspension of intermediate 1.i (7.46 g, 16.4 mmol) in ethylene glycol (50 mL). The mixture became warm and thick and was diluted with DME (75 mL). The mixture was stirred at 95° C. for 24 h before it was cooled to rt, diluted with water (50 mL) and a 10% aq. citric acid solution (250 mL). The milky suspension was extracted with EA (2×300 mL). The combined org. extracts were dried over MgSO 4, filtered and the filtrate was concentrated. The remaining crystalline solid was suspended in MeOH, collected, washed well with MeOH and dried under HV to give the expected product as a white crystalline powder (6.49 g).
To a solution of intermediate 1.ii (6.49 g, 13.5 mmol) in THF (120 mL) was added carefully NaH (1.77 g, 40.6 mmol, 55% dispersion in mineral oil). The mixture was stirred for 10 min before 2-chloro-5-bromo-pyrimidine (3.93 g, 20.3 mmol) was added. The mixture was diluted with DMF (15 mL) and then stirred at rt for 20 min. The mixture was heated to 60° C. and stirred for 3 h before being again cooled to rt. The reaction was quenched with water and 10% aq. citric acid solution (250 mL) and the mixture was extracted with EA (2×300 mL). The org. extracts were washed with water, combined, dried over MgSO 4, filtered and the solvent of the filtrate was evaporated. The crude product was crystallised from MeOH/ether. The crystalline material was collected, washed with additional MeOH/ether and dried under HV to give the expected product as a white powder (6.47 g).
A solution of borontribromide (25.5 mL, 1 M in DCM) was slowly added to a solution of intermediate 1.iii (6.50 g, 10.2 mmol) in chloroform (250 mL). The mixture became turbid and an oily residue separated. The mixture was stirred at rt. Another portion of BBr 3 solution (5 mL) was added after 6, 24, and 33 h. After the last addition of BBr 3, the beige suspension was stirred vigorously for additional 2 h before being carefully quenched with MeOH. The mixture became slightly warm and clear. The solution was washed with cold water (0° C., 2×150 mL). The washings were extracted back with DCM. The combined org. extracts were again washed with water, dried over MgSO 4, filtered and concentrated. The crude product was purified by CC on silica gel eluting with heptane:EA 1:1 followed by crystallisation from DCM. The purified crystalline product was dried under HIV at 45° C. for 48 h to give the expected product as a white, crystalline powder (1.62 g).
^ Jump up to:ab“Jeraygo EPAR”. European Medicines Agency. 25 April 2024. Archived from the original on 30 April 2024. Retrieved 27 April 2024. Text was copied from this source which is copyright European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
Further reading
Mahfooz K, Najeed S, Tun HN, Khamosh M, Grewal D, Hussain A, et al. (July 2023). “New Dual Endothelin Receptor Antagonist Aprocitentan in Hypertension: A Systematic Review and Meta-Analysis”. Current Problems in Cardiology. 48 (7): 101686. doi:10.1016/j.cpcardiol.2023.101686. PMID36893968.
Lazertinib is an oral, third-generation, epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor (TKI).2,3 Lazertinib was first approved in South Korea on January 18, 2021, for the treatment of EGFR T790M mutation-positive non-small cell lung cancer (NSCLC) with EGFR mutations.1 It was approved by the FDA on August 19, 2024.5 Lazertinib is used alone or in combination with other chemotherapeutic agents.4
The most common adverse reactions include rash, nail toxicity, infusion-related reactions (amivantamab), musculoskeletal pain, edema, stomatitis, venous thromboembolism, paresthesia, fatigue, diarrhea, constipation, COVID-19 infection, hemorrhage, dry skin, decreased appetite, pruritus, nausea, and ocular toxicity.[2]
Lazertinib was approved for medical use in South Korea in January 2021,[4][5] and in the United States in August 2024.[2][6]
Medical uses
Lazertinib is indicated in combination with amivantamab for the first-line treatment of adults with locally advanced or metastatic non-small cell lung cancer with epidermal growth factor receptor exon 19 deletions or exon 21 L858R substitution mutations.[2
History
Efficacy was evaluated in MARIPOSA (NCT04487080), a randomized, active-controlled, multicenter trial of 1074 participants with exon 19 deletion or exon 21 L858R substitution mutation-positive locally advanced or metastatic non-small cell lung cancer and no prior systemic therapy for advanced disease.[2] Participants were randomized (2:2:1) to receive lazertinib in combination with amivantamab, osimertinib monotherapy, or lazertinib monotherapy (an unapproved regimen for non-small cell lung cancer) until disease progression or unacceptable toxicity.[2]
Society and culture
Legal status
Lazertinib was approved for medical use in the United States in August 2024.[2]Names
^World Health Organization (2018). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 79”. WHO Drug Information. 32 (1). hdl:10665/330941.
External links
Clinical trial number NCT04487080 for “A Study of Amivantamab and Lazertinib Combination Therapy Versus Osimertinib in Locally Advanced or Metastatic Non-Small Cell Lung Cancer (MARIPOSA)” at ClinicalTrials.gov
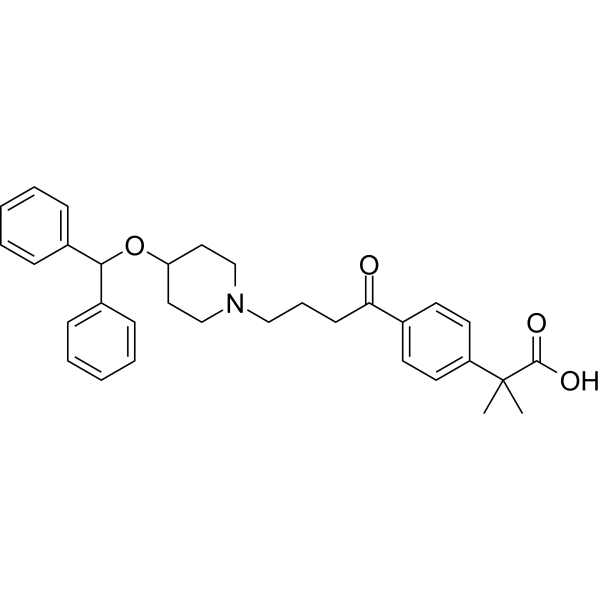


























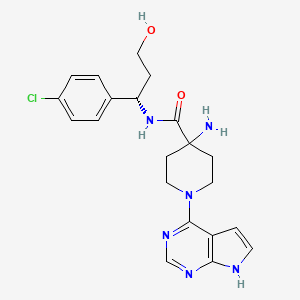
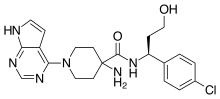
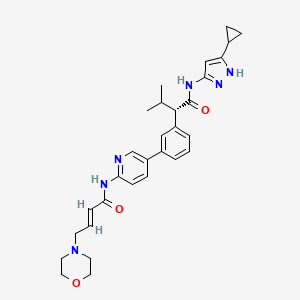





















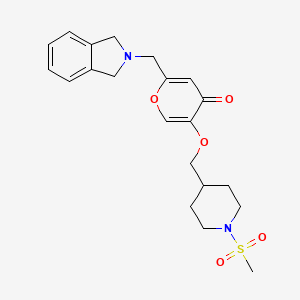
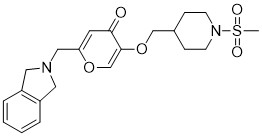








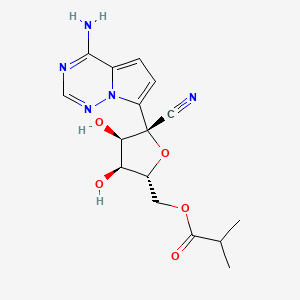




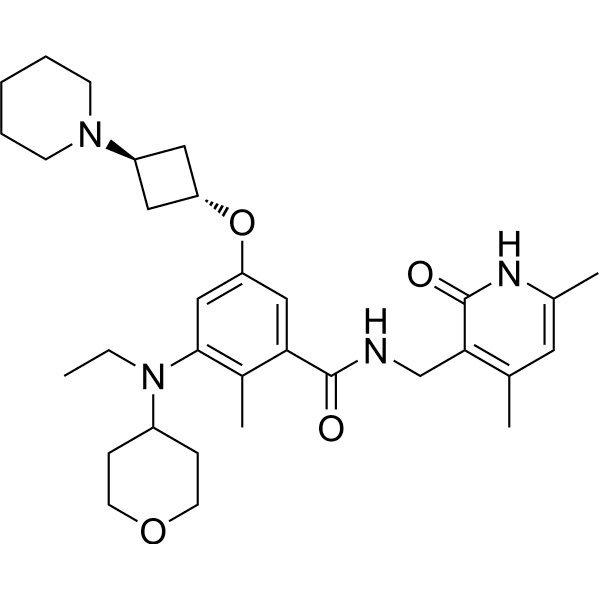





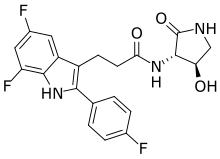
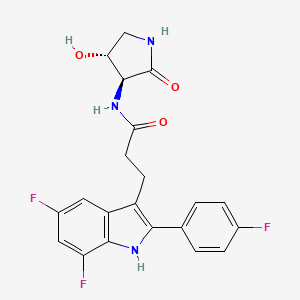













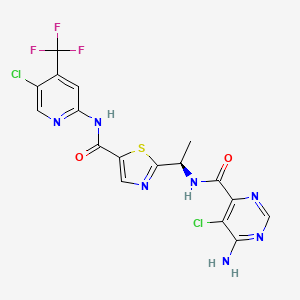






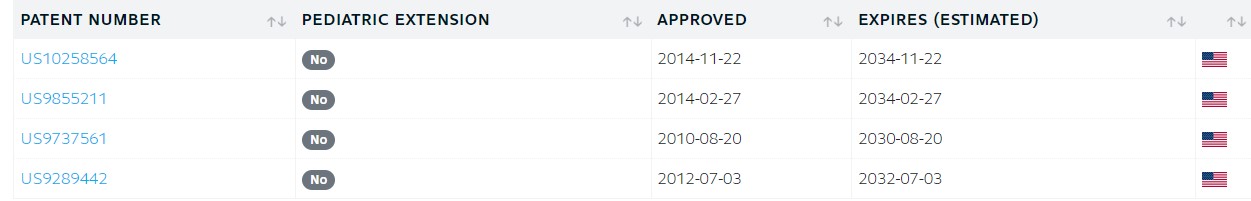 References
References