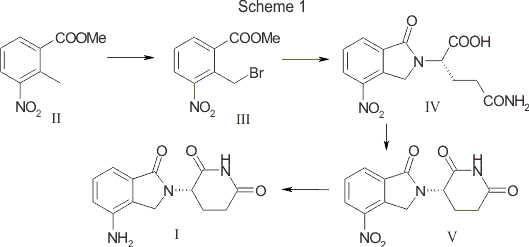
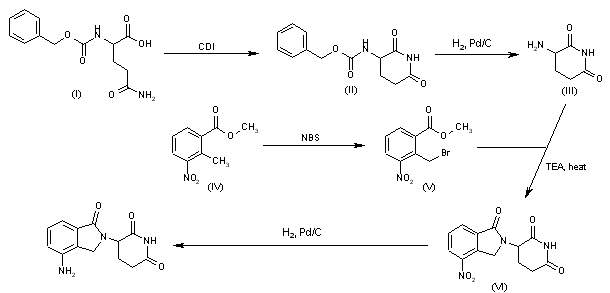
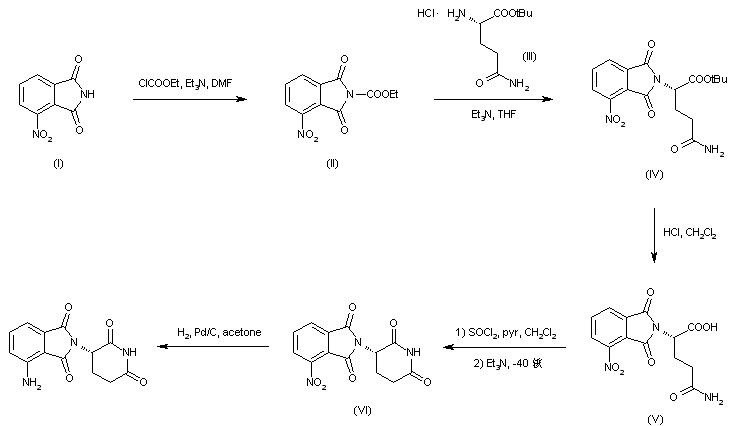
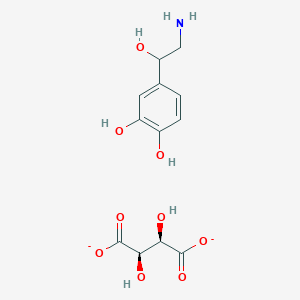
Fenfluramine
- DEA No. 1670
- S 768
2020/12/18, FDA APPROVED, Fintepla
Fenfluramine hydrochloride
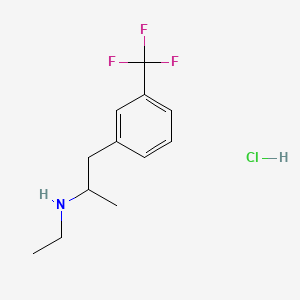
| Formula | C12H16F3N. HCl |
|---|---|
| CAS | 404-82-0458-24-2 (FREE) |
| Mol weight | 267.7183 |
Antiobesity
| Efficacy | Appetite suppressant |
|---|---|
| Disease | Dravet syndrome |
(+-)-Fenfluramine hydrochloride
Racemic fenfluramine hydrochloride
Fenfluramine hydrochloride [USAN]
1-(m-Trifluoromethylphenyl)-2-(ethylamino)propane hydrochloride
Research Code:ZX-008
MOA:Serotonin agonist
Indication:Dravet syndrome
Company:Zogenix (Originator)
Synonyms of Fenfluramine [INN:BAN]
- (+-)-Fenfluramine
- BRN 4783711
- dl-Fenfluramine
- DL-Fenfluramine
- EINECS 207-276-3
- Fenfluramina
- Fenfluramina [DCIT]
- Fenfluramine
- Fenfluraminum
- Fenfluraminum [INN-Latin]
- HSDB 3080
- Obedrex
- Pesos
- Ponderax PA
- Rotondin
- S 768
- UNII-2DS058H2CF
mp 160-161 °C, ethyl acetate US 3198834
nmr Salsbury, Jonathon S.; Magnetic Resonance in Chemistry 2005, V43(11), P910-917 C
IR BIORAD: Infrared spectral data from the Bio-Rad/Sadtler IR Data Collection was obtained from Bio-Rad Laboratories, Philadelphia, PA (US).FenfluramineCAS Registry Number: 458-24-2CAS Name:N-Ethyl-a-methyl-3-(trifluoromethyl)benzeneethanamineAdditional Names:N-ethyl-a-methyl-m-(trifluoromethyl)phenethylamine; 2-ethylamino-1-(3-trifluoromethylphenyl)propaneManufacturers’ Codes: S-768Molecular Formula: C12H16F3NMolecular Weight: 231.26Percent Composition: C 62.32%, H 6.97%, F 24.65%, N 6.06%Literature References: Prepn: L. G. Beregi et al.,FRM1658; eidem,US3198833 (1963, 1965 both to Sci. Union et Cie Soc. Franc. Recherche Méd.). Prepn of optical isomers: eidem,US3198834 (1965 to Sci. Union et Cie Soc. Franc. Recherche Med.). Pharmacology: Presse Med.71, 181 (1963). Pharmacology and toxicity of isomers and racemate: J. C. Le Douarec et al.,Arch. Int. Pharmacodyn. Ther.161, 206 (1966). Pharmacokinetics: S. Caccia et al.,Eur. J. Clin. Pharmacol.29, 221 (1985). Clinical trial of dextrofenfluramine in refractory obesity: N. Finer et al.,Curr. Ther. Res.38, 847 (1985). Comprehensive review: Pinder et al.,Drugs10, 241-323 (1975).Properties: bp12 108-112°. LD50 i.p. in mice: 144 mg/kg (US3198833).Boiling point: bp12 108-112°Toxicity data: LD50 i.p. in mice: 144 mg/kg
Derivative Type: HydrochlorideCAS Registry Number: 404-82-0Trademarks: Acino (IMA); Adipomin (Streuli); Obedrex (Beta); Pesos (Valeas); Ponderal (Servier); Ponderax (Selpharm); Ponderex (Robins); Pondimin (Robins); Rotondin (Casasco)Molecular Formula: C12H16F3N.HClMolecular Weight: 267.72Percent Composition: C 53.84%, H 6.40%, F 21.29%, N 5.23%, Cl 13.24%Properties: Crystals from ethanol + ether, mp 166°.Melting point: mp 166°
Derivative Type:d-FormCAS Registry Number: 3239-44-9Additional Names: Dexfenfluramine; dextrofenfluramineProperties: [a]D25 +9.5° (c = 8 in ethanol). LD50 orally in rats: 114.6 mg/kg (Le Douarec).Optical Rotation: [a]D25 +9.5° (c = 8 in ethanol)Toxicity data: LD50 orally in rats: 114.6 mg/kg (Le Douarec)
Derivative Type:d-Form hydrochlorideCAS Registry Number: 3239-45-0Trademarks: Adifax (Servier); Glypolix (Stroder); Isomeride (Ardix); Redux (Wyeth-Ayerst)Properties: Crystals from ethyl acetate, mp 160-161°.Melting point: mp 160-161°
Derivative Type:l-FormCAS Registry Number: 37577-24-5Properties: [a]D25 -9.6° (c = 8 in ethanol). LD50 orally in rats: 195 mg/kg (Le Douarec).Optical Rotation: [a]D25 -9.6° (c = 8 in ethanol)Toxicity data: LD50 orally in rats: 195 mg/kg (Le Douarec)
Derivative Type:l-Form hydrochlorideCAS Registry Number: 3616-78-2Properties: Crystals from ethyl acetate, mp 160-161°.Melting point: mp 160-161°
NOTE: This is a controlled substance: 21 CFR, 1308.14.Therap-Cat: Anorexic.Keywords: Anorexic.
A centrally active drug that apparently both blocks serotonin uptake and provokes transport-mediated serotonin release.
Fenfluramine Hydrochloride has been filed an IND application with the FDA in USA to initiate phase III trials by Brabant Pharma (acquired by Zogenix in 2014) for the treatment of dravets syndrome (also known as severe myoclonic epilepsy of infancy, SMEI), this compound has been granted orphan drug designation in Europe and U.S..
Fenfluramine Hydrochloride was launched in 1963 by Servier in France and in 1973 by Wyeth (now a wholly owned subsidiary of Pfizer) in US for the treatment of obesity. However, it was withdrawn from the market in 1997 due to heart disease.
Dravet syndrome is a pediatric encephalopathy that typically manifests within the first year of life following exposure to elevated temperatures. It is characterized by recurrent pharmacoresistant seizures, which increase in frequency and severity with disease progression. Concomitantly with these seizures, patients typically display delayed development and neurocognitive impairment.6,9,10,11 Fenfluramine is a serotonergic phenethylamine originally used as an appetite suppressant until concerns regarding cardiotoxicity in obese patients lead to its withdrawal from the market in 1997.6,12,13 Through its ability to modulate neurotransmission, fenfluramine has reemerged as an effective therapy against pharmacoresistant seizures, such as those involved in Dravet syndrome.3,5,8
Fenfluramine was granted initial FDA approval in 1973 prior to its withdrawal; it was granted a new FDA approval on June 25, 2020, for treatment of Dravet syndrome patients through the restricted FINTEPLA REMS program. It is currently sold under the name FINTEPLA® by Zogenix INC.16
Fenfluramine, sold under the brand name Fintepla, is a medication used for the treatment of seizures associated with Dravet syndrome in people age two and older.[2][3]
The most common adverse reactions include decreased appetite; drowsiness, sedation and lethargy; diarrhea; constipation; abnormal echocardiogram; fatigue or lack of energy; ataxia (lack of coordination), balance disorder, gait disturbance (trouble with walking); increased blood pressure; drooling, salivary hypersecretion (saliva overproduction); pyrexia (fever); upper respiratory tract infection; vomiting; decreased weight; risk of falls; and status epilepticus.[2]
Dravet syndrome is a pediatric encephalopathy that typically manifests within the first year of life following exposure to elevated temperatures. It is characterized by recurrent pharmacoresistant seizures, which increase in frequency and severity with disease progression. Concomitantly with these seizures, patients typically display delayed development and neurocognitive impairment.6,9,10,11 Fenfluramine is a serotonergic phenethylamine originally used as an appetite suppressant until concerns regarding cardiotoxicity in obese patients lead to its withdrawal from the market in 1997.6,12,13 Through its ability to modulate neurotransmission, fenfluramine has reemerged as an effective therapy against pharmacoresistant seizures, such as those involved in Dravet syndrome.3,5,8
Fenfluramine was granted initial FDA approval in 1973 prior to its withdrawal; it was granted a new FDA approval on June 25, 2020, for treatment of Dravet syndrome patients through the restricted FINTEPLA REMS program. It is currently sold under the name FINTEPLA® by Zogenix INC.16
Medical uses
Fenfluramine is indicated for the treatment of seizures associated with Dravet syndrome in people age two and older.[2][3]
Dravet syndrome is a life-threatening, rare and chronic form of epilepsy.[2] It is often characterized by severe and unrelenting seizures despite medical treatment.[2]
Adverse effects
The U.S. Food and Drug Administration (FDA) fenfluramine labeling includes a boxed warning stating the drug is associated with valvular heart disease (VHD) and pulmonary arterial hypertension (PAH).[2] Because of the risks of VHD and PAH, fenfluramine is available only through a restricted drug distribution program, under a risk evaluation and mitigation strategy (REMS).[2] The fenfluramine REMS requires health care professionals who prescribe fenfluramine and pharmacies that dispense fenfluramine to be specially certified in the fenfluramine REMS and that patients be enrolled in the REMS.[2] As part of the REMS requirements, prescribers and patients must adhere to the required cardiac monitoring with echocardiograms to receive fenfluramine.[2]
At higher therapeutic doses, headache, diarrhea, dizziness, dry mouth, erectile dysfunction, anxiety, insomnia, irritability, lethargy, and CNS stimulation have been reported with fenfluramine.[4]
There have been reports associating chronic fenfluramine treatment with emotional instability, cognitive deficits, depression, psychosis, exacerbation of pre-existing psychosis (schizophrenia), and sleep disturbances.[4][5] It has been suggested that some of these effects may be mediated by serotonergic neurotoxicity/depletion of serotonin with chronic administration and/or activation of serotonin 5-HT2A receptors.[5][6][7][8]
Heart valve disease
The distinctive valvular abnormality seen with fenfluramine is a thickening of the leaflet and chordae tendineae. One mechanism used to explain this phenomenon involves heart valve serotonin receptors, which are thought to help regulate growth. Since fenfluramine and its active metabolite norfenfluramine stimulate serotonin receptors, this may have led to the valvular abnormalities found in patients using fenfluramine. In particular norfenfluramine is a potent inhibitor of the re-uptake of 5-HT into nerve terminals.[9] Fenfluramine and its active metabolite norfenfluramine affect the 5-HT2B receptors, which are plentiful in human cardiac valves. The suggested mechanism by which fenfluramine causes damage is through over or inappropriate stimulation of these receptors leading to inappropriate valve cell division. Supporting this idea is the fact that this valve abnormality has also occurred in patients using other drugs that act on 5-HT2B receptors.[10][11]
According to a study of 5,743 former users conducted by a plaintiff’s expert cardiologist, damage to the heart valve continued long after stopping the medication.[12] Of the users tested, 20% of women, and 12% of men were affected. For all ex-users, there was a 7-fold increase of chances of needing surgery for faulty heart valves caused by the drug.[12]
Overdose
In overdose, fenfluramine can cause serotonin syndrome and rapidly result in death.[13][14]
Pharmacology
Pharmacodynamics
Fenfluramine acts primarily as a serotonin releasing agent.[15][16] It increases the level of serotonin, a neurotransmitter that regulates mood, appetite and other functions.[15][16] Fenfluramine causes the release of serotonin by disrupting vesicular storage of the neurotransmitter, and reversing serotonin transporter function.[17] The drug also acts as a norepinephrine releasing agent to a lesser extent, particularly via its active metabolite norfenfluramine.[15][16] At high concentrations, norfenfluramine, though not fenfluramine, also acts as a dopamine releasing agent, and so fenfluramine may do this at very high doses as well.[15][16] In addition to monoamine release, while fenfluramine binds only very weakly to the serotonin 5-HT2 receptors, norfenfluramine binds to and activates the serotonin 5-HT2B and 5-HT2C receptors with high affinity and the serotonin 5-HT2A receptor with moderate affinity.[18][19] The result of the increased serotonergic and noradrenergic neurotransmission is a feeling of fullness and reduced appetite.
The combination of fenfluramine with phentermine, a norepinephrine–dopamine releasing agent acting primarily on norepinephrine, results in a well-balanced serotonin–norepinephrine releasing agent with weaker effects of dopamine release.[15][16]
| Drug | NE | DA | 5-HT | Type | Ref |
|---|---|---|---|---|---|
| Fenfluramine | 739 | >10,000 | 79.3–108 | SRA | [20][15][16] |
| D-Fenfluramine | 302 | >10,000 | 51.7 | SNRA | [20][15] |
| L-Fenfluramine | >10,000 | >10,000 | 147 | SRA | [15][21] |
| Norfenfluramine | 168–170 | 1,900–1,925 | 104 | SNRA | [15][16] |
| Phentermine | 39.4 | 262 | 3,511 | NDRA | [20] |
Pharmacokinetics
The elimination half-life of fenfluramine has been reported as ranging from 13 to 30 hours.[4] The mean elimination half-lives of its enantiomers have been found to be 19 hours for dexfenfluramine and 25 hours for levfenfluramine.[13] Norfenfluramine, the major active metabolite of fenfluramine, has an elimination half-life that is about 1.5 to 2 times as long as that of fenfluramine, with mean values of 34 hours for dexnorfenfluramine and 50 hours for levnorfenfluramine.[13]
Chemistry
Fenfluramine is a substituted amphetamine and is also known as 3-trifluoromethyl-N-ethylamphetamine.[13] It is a racemic mixture of two enantiomers, dexfenfluramine and levofenfluramine.[13] Some analogues of fenfluramine include norfenfluramine, benfluorex, flucetorex, and fludorex.
History
Fenfluramine was developed in the early 1960s and was introduced in France in 1963.[13] Approximately 50 million Europeans were treated with fenfluramine for appetite suppression between 1963 and 1996.[13] Fenfluramine was approved in the United States in 1973.[13] The combination of fenfluramine and phentermine was proposed in 1984.[13] Approximately 5 million people in the United States were given fenfluramine or dexfenfluramine with or without phentermine between 1996 and 1998.[13]
In the early 1990s, French researchers reported an association of fenfluramine with primary pulmonary hypertension and dyspnea in a small sample of patients.[13] Fenfluramine was withdrawn from the U.S. market in 1997 after reports of heart valve disease[22][23] and continued findings of pulmonary hypertension, including a condition known as cardiac fibrosis.[24] It was subsequently withdrawn from other markets around the world. It was banned in India in 1998.[25]
Fenfluramine was an appetite suppressant which was used to treat obesity.[13] It was used both on its own and, in combination with phentermine, as part of the anti-obesity medication Fen-Phen.[13]
In June 2020, fenfluramine was approved for medical use in the United States with an indication to treat Dravet syndrome.[2][26]
The effectiveness of fenfluramine for the treatment of seizures associated with Dravet syndrome was demonstrated in two clinical studies in 202 subjects between ages two and eighteen.[2] The studies measured the change from baseline in the frequency of convulsive seizures.[2] In both studies, subjects treated with fenfluramine had significantly greater reductions in the frequency of convulsive seizures during the trials than subjects who received placebo (inactive treatment).[2] These reductions were seen within 3–4 weeks, and remained generally consistent over the 14- to 15-week treatment periods.[2]
The U.S. Food and Drug Administration (FDA) granted the application for fenfluramine priority review and orphan drug designations.[2][27][28] The FDA granted approval of Fintepla to Zogenix, Inc.[2]
On 15 October 2020, the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) adopted a positive opinion, recommending the granting of a marketing authorization for the medicinal product Fintepla, intended for the treatment of seizures associated with Dravet syndrome.[29] Fenfluramine was approved for medical use in the European Union in December 2020.[3]
Society and culture
Recreational use
Unlike various other amphetamine derivatives, fenfluramine is reported to be dysphoric, “unpleasantly lethargic“, and non-addictive at therapeutic doses.[30] However, it has been reported to be used recreationally at high doses ranging between 80 and 400 mg, which have been described as producing euphoria, amphetamine-like effects, sedation, and hallucinogenic effects, along with anxiety, nausea, diarrhea, and sometimes panic attacks, as well as depressive symptoms once the drug had worn off.[30][31][32] At very high doses (e.g., 240 mg, or between 200–600 mg), fenfluramine induces a psychedelic state resembling that produced by lysergic acid diethylamide (LSD).[32][33] Indirect (via induction of serotonin release) and/or direct activation of the 5-HT2A receptor would be expected to be responsible for the psychedelic effects of the drug at sufficient doses.
Research
Under the development code ZX008, the pharmaceutical company Zogenix is studying fenfluramine’s potential to treat seizures.[34] Clinical trials have studied the use of fenfluramine in patients with Dravet syndrome.[35] Results of a Phase III clinical trial showed a 64% reduction in seizures.[36]Route 1
Reference:1. J. Org. Chem.1979, 44, 3580-3583.Route 2
Reference:1. EP0810195A1.
2. Chem. Ind. Times 2002, 16, 33-34.Route 3
Reference:1. ACS Symp. Ser. 2009, 1003, 165-181.
ref
BE 609630
FR 1658 M 19630218
US 3198834
DE 1593595
US 3769319
NL 7215548
Ger. (East) (1974), DD 108971
EP 3170807
SYN
US20170174613

PATENT
US 20170174613
| Step 4.2: Crystallization of Fenfluramine Hydrochloride |
| Procedure: Charge Fenfluramine.HCl (crude) (1.00 wt, 1.0 eq.) and TBME (10.0 vol, 7.4 wt) to the vessel and commence stirring. Heat the suspension to reflux (50 to 58° C.). Charge ethanol (5.0 vol, 3.9 wt) maintaining the temperature at 50 to 58° C. Addition time 20 minutes. Stir at 50 to 58° C. for 5 to 10 minutes and check for dissolution. Stir the solution at 50 to 58° C. for 5 to 10 minutes, targeting 54 to 58° C. Clarify the reaction mixture through a 0.1 μm in-line filter at 54 to 58° C., followed by a line rinse with TBME (1 vol, 0.7 wt). Cool the solution to 48 to 50° C. Charge Fenfluramine.HCl, code FP0188 (0.01 wt). Check for crystallization. Allow the suspension to cool to 15 to 20° C., target 17° C. over 5 to 5.5 hours at an approximately constant rate. Stir the mixture at 15 to 20° C., target 17° C. for 2 to 3 hours. Filter the mixture and wash the filter-cake with clarified TBME (2×3.0 vol, 2×2.2 wt) at 5 to 15° C. Dry the solid at up to 40° C. until the TBME content is <0.5% w/w TBME and the ethanol content is <0.5% w/w EtOH by 1H-NMR analysis. 4 to 8 hours. Determine the w/w assay of the isolated Fenfluramine.HCl by 1H-NMR analysis. |
| Yields and Profiles: The yield for the stage 4 Demonstration batch is summarized in Table 1E below. Input: 750.0 g uncorr. Fenfluramine.HCl crude (1.00 eq, 1.00 wt uncorr.) for input calculation. FIG. 3 shows an exemplary HPLC chromatogram of a crystallized fenfluramine hydrochloride sample (210 nm UV absorbance). |
PATENT
US 20180208543
| Fenfluramine, i.e., 3-trifluoromethyl-N-ethylamphetamine, has the following chemical structure: |
| The marketing of fenfluramine as a pharmaceutical active ingredient in the United States began in 1973 and was used in a therapy in combination with phentermine to prevent and treat obesity. Anyway, in 1997 fenfluramine was withdrawn from the market in the United States and immediately thereafter in other countries, since its use was associated with the onset of cardiac fibrosis and pulmonary hypertension. As a consequence of this event, the pharmaceutical compounds containing this active ingredient were withdrawn from the market. However, fenfluramine, even after its exit from the market, has continued to attract scientific interest, as will become apparent from the discussion presented hereinafter. |
| In the literature, over the years, numerous syntheses or processes have been reported for preparing fenfluramine or its dextrorotatory enantiomer dexfenfluramine or an analog containing a highly electron-attractor group on the aromatic ring as in the fenfluramine molecule (see for example Pentafluorosulfanyl Serotonin Analogs: Synthesis, Characterization, and Biological Activity, John T. Welch and Dongsung Lim Chapter 8, pp 165-181 DOI: 10.1021/bk-2009-1003.ch008). Many of these synthesis paths are long and provide for multiple synthesis steps that can include reagents that are dangerous or scarcely environment-friendly and are therefore scarcely convenient for an industrial synthesis. Hereinafter, any reference to “fenfluramine” is understood to reference the racemic form, i.e., (RS)-N-ethyl-1-[3-(trifluoromethyephenyl]propan-2-amine. |
| To the best of the knowledge of the inventors, the first method for fenfluramine synthesis reported in the literature dates back to 1962 and is referenced in patent BE609630 and in similar patents U.S. Pat. No. 3,198,833 and FR1324220. All the synthesis methods reported in these patents provide for numerous synthesis steps. By way of example, one of the methods provides for the transformation into oxime of a ketone, 1-(3-trifluoromethyephenyl-propan-2-one, as shown here: |
| The oxime is then hydrogenated in the presence of Raney nickel catalyst so as to yield the corresponding primary amine, which is acetylated subsequently with ethanoic anhydride before being converted into fenfluramine by reduction with lithium aluminum hydride. |
| As can be seen, the final step of this chemical process provides for the use of lithium aluminum hydride and the persons skilled in the art will acknowledge that the use of this reagent should be avoided, if possible, on an industrial level, since it is extremely flammable and is the source of accidents. Furthermore, lithium is a potentially neurotoxic metal and therefore its use should be avoided where possible. Furthermore, the Raney nickel catalyst is used in the oxime reduction step and can contaminate the final active ingredient; the use of hydroxylamine also entails problems of toxicity for workers assigned to production. |
| A further disadvantage of this process is, as already mentioned earlier, the number of steps, not only because a large number of synthesis steps entails a reduction of the overall yield of active ingredient, but also because each synthesis step in principle can generate impurities and a larger number of steps can therefore entail a higher number of impurities in the final active ingredient. Many of these impurities, furthermore, due to their structural similarity to fenfluramine, are difficult to eliminate and remove from a fenfluramine preparation. One impurity for example that can be formed in the process described above and is difficult to eliminate is the following: |
| This impurity, which is a primary amine, shares physical-chemical properties that are similar to fenfluramine and therefore, like fenfluramine, it can form a hydrochloride salt by treatment with hydrochloric acid and thus contaminate the active ingredient fenfluramine hydrochloride. Furthermore, this impurity—as a free base—has a boiling point that is similar to that of fenfluramine (73° C. vs. 89° C. at 6 mmHg respectively), and therefore its elimination by distillation also can be problematic. |
| The process described above can in principle generate other impurities, which are listed in FIG. 1. |
| EP 0441160 claims a synthesis in 5 steps of dexfenfluramine, dextrorotatory enantiomer of fenfluramine. This synthesis can be adapted easily to produce fenfluramine instead of its dextrorotatory enantiomer simply by performing the first reduction step with a non-chiral reducing agent. In the first step, in fact: |
(MOL) (CDX) a ketone, 1-(3-trifluoromethyl)phenyl-propan-2-one, is first reduced to the corresponding alcohol in the presence of yeast, D-glucose, ethanol and water. Then the alcohol is converted into the tosylate in the second step:
(MOL) (CDX)
| This reaction occurs in the presence of triethylamine and tosyl chloride in methylene chloride as solvent. After purification, the tosylate is converted to fenfluramine by means of three successive steps: |
| In the first of these three steps, the tosylate is converted into an azide intermediate by reaction with sodium azide in dimethylformamide. The azide intermediate is then hydrogenated in the presence of a catalyst, palladium on carbon. Finally, the resulting primary amine is converted into fenfluramine by reaction with acetaldehyde and sodium borohydride. |
| Persons skilled in the art may see easily that this process is not desirable from an industrial standpoint due to reasons related to environmental risk, safety and costs. For example, the sodium azide used in the process is a notoriously explosive compound and its use at the industrial level is dangerous. Furthermore, palladium is an expensive material and its use in the process entails an increase in the production costs of fenfluramine. Furthermore, palladium can contaminate the finished active ingredient. |
| In another method for the synthesis of dexfenfluramine in 3-4 steps, reported by Goument et al. in Bulletin of the Chemical Society of France (1993), 130, p. 450-458, 3-bromobenzotrifluoride is subjected to a Grignard reaction with enantiopure 1,2-propylene-epoxide to yield 1-[3-(trifluoromethyl)phenyl]propan-2-ol as shown hereafter: |
| If this reaction is performed with racemic 1,2-propylene-epoxide, the synthesis can be adapted to the preparation of fenfluramine. |
| The alcohol thus obtained is first transformed into trifluoromethyl sulfonate by reaction with trifluoromethanesulfonic anhydride and then treated with ethylamine to yield fenfluramine, as shown in the diagram hereinafter: |
| In this article, the authors acknowledge that the main byproducts of the reaction are isomer alkenes having the following chemical structures: |
| The process proposed by Goument et al. is not interesting from the industrial standpoint for a series of reasons. First of all, it is known that the use of Grignard reagents, especially on an industrial scale, is problematic, because these compounds are often pyrophoric and corrosive. Furthermore, 1,2-propylene epoxide is a suspected carcinogenic compound. Finally, the formation of the three isomer alkenes as byproducts listed above is a disadvantage of the process. In the article, Goument presents methods for activation of the intermediate alcohol which are alternative to trifluoromethylsulfonate, for example by converting it to chloride (via thionyl chloride) or to mesylate (via mesyl chloride), but these process variations share the same disadvantages as the main process analyzed above. |
| In addition to the methods with multiple synthesis steps discussed so far in detail, the literature reports other methods or processes for producing fenfluramine or dexfenfluramine. In general, persons skilled in the art acknowledge that the syntheses in the literature for producing dexfenfluramine sometimes can be applied to the preparation of fenfluramine simply by replacing the initial materials and/or enantiopure reagents with the corresponding racemates while maintaining the reaction conditions. For example, patents that present long synthesis methods in multiple steps are the following: |
| DE1593595 and U.S. Pat. No. 3,769,319 |
| NL7215548 |
| EP810195 and EP882700 (dexfenfluramine) |
| EP0301925 (dexfenfluramine) |
| Other examples of preparation of fenfluramine, taken from non-patent literature, are the following: |
| Synthesis, November 1987, p. 1005-1007 |
| J. Org. Chem, 1991, 56, p. 6019 |
| Tetrahedron, 1994, 50(1), p. 171 |
| Bull. Soc. Chim. France, 1993, 130(4), p. 459-466 (dexfenfluramine) |
| Chirality, 2002, 14(4), p. 325-328 (dexfenfluramine) |
| Without analyzing in detail the individual methods described in these patents or articles, it can be stated in summary that all these methods are not attractive and interesting from the industrial standpoint because these are processes with many synthesis steps or because the initial materials described therein are not easily available and therefore have to be prepared separately, with a further expenditure of time and with further costs, or because they provide for the use of reagents that are dangerous/explosive/toxic or because they entail the use of catalysts based on heavy metals that can contaminate the final active ingredient. |
| One should consider that in the literature there are methods for the preparation of fenfluramine that did not provide for long syntheses and multiple steps but are shorter and consist of one or two steps. These processes, which therefore would be more interesting from the industrial standpoint, have other specific disadvantages, as will become apparent in detail hereinafter. For example, in the literature there is a first group of articles or patents that describe the reaction between 1-(3-trifluoromethyl)phenyl-propan-2-one and ethylamine in the presence of hydrogen gas and of a transition metal as catalyst: |
| In particular, in Huagong Shikan, 2002, 16(7), p. 33, the reaction is performed with hydrogen gas (2.9-3.38 atm), at 65-75° C., for 9 hours, in the presence of Raney nickel. Likewise, in patent DD108971 (1973), Raney nickel and hydrogen gas and methanol are used as solvent to perform this reaction. |
| In HU55343, instead, a similar reaction in one step is performed with hydrogen gas in the presence of another transition metal catalyst, such as palladium on carbon. |
| Although these three methods describe short single-step processes, they have the disadvantage of the use of hydrogen gas. As is known to persons skilled in the art, hydrogen gas is a dangerous gas due to the inherent danger of forming explosive mixtures with air and must be used by expert personnel in expensive facilities dedicated to its use and built with special precautions. Despite being used in purpose-built facilities, the use of hydrogen at the industrial level is inherently dangerous and to be avoided if possible. Another danger element that is shared by the processes described above is the fact that the reactions are performed under pressure. The third industrial disadvantage then arises from the use of heavy metal catalysts, which have a high cost and therefore increase the overall cost of the final active ingredient and may then contaminate the active ingredient fenfluramine even after filtration of the catalyst and purification of said active ingredient. |
| Analysis of the background art shows, however, that an attempt has been made to devise a process for the production or synthesis of fenfluramine that is short (one or two steps) and does not entail the use of hydrogen gas or of catalysts based on nickel or palladium or the like. In particular, for example, Synthesis 1987, 11, p. 1005, and then DECHEMA Monographien (1989), 112 (Org. Elektrochem.—Angew. Elektrothermie), 367-74, present a method for the synthesis of fenfluramine which starts from 1-(3-trifluoromethyl)phenyl-propan-2-one, which is made to react with ethylamine in great excess, in an electrochemical process, which uses a mercury cathode in a water/ethanol solution with pH 10-11. One obtains fenfluramine with 87% yield. This process has some drawbacks from an industrial standpoint: it is a process of the electrochemical type and therefore requires special equipment which is scarcely widespread, dedicated cells and reactors, and it is not possible to use the classic multipurpose reactors available in the pharmaceutical industry. Furthermore, the use of mercury at the industrial level poses severe environment safety problems, requiring constant health monitoring on workers who manage the equipment and systems for the management and destruction of wastewater that are particularly onerous; finally, mercury can be transferred from the cathode to the reaction environment and therefore to the active ingredient, and this obviously is to be considered very dangerous due to the accumulation of the metal in human beings; small traces of mercury are very toxic. |
| Another method for fenfluramine synthesis in a single step is the one presented in J. Org. Chem, 1979, 44(20), p. 3580. Here the reaction is described between an alkene derivative and ethylamine in the presence of sodium borohydride and mercury nitrate: |
| Again, this process is not interesting from an industrial standpoint since it has the same problems, if not even greater ones, related to the use of mercury (used here as a water-soluble salt) discussed previously. The complication introduced in this process with the use of mercury nitrate together with sodium borohydride highlights the level of innovation of the synthesis path found here. |
| In the past, therefore, it has not been possible to provide a process for synthesizing fenfluramine in a small number of steps by using modern reducing agents that are commonly and easily used. Indeed, while Gaodeng Xuexiao Huaxue Xuebao, 9(2), 1988, p. 134-139, describes and exemplifies the synthesis of 2-N-ethyl-1-phenyl propane by means of (1) the treatment of the precursor ketone with ethylamine followed by (2) sodium cyanoborohydride as reducing agent, Xuexiao Huaxue Xuebao provides no example for fenfluramine. Moreover, for the latter, Xuexiao Huaxue Xuebao indicates a melting point for the hydrochloride of 161° C., a data item that matches the value indicated in the literature initially (see BE609630); these facts prove thats fenfluramine synthesis with cyanoborohydride was not performed, otherwise one cannot explain why the author did not transcribe, in the document, the example of a product that at the time was very important. It should be noted in fact that 1-phenyl propan-2-one and 1-(3-trifluoromethyl)phenyl-propan-2-one can have different reactivities to reductive amination due to the presence of a highly electron-attractor-trifluoromethyl group, hence the need for an example to demonstrate its feasibility. The use of cyanoborohydride shares some disadvantages with other methods discussed in the preceding paragraphs. The excellent selectivity for reductive aminations of this reagent is highly appreciated, but its application can be less advantageous with respect to other reducing systems in the synthesis of fenfluramine, where the latter is intended for therapeutic application in human beings. The reasons for this are the possible contamination of the finished pharmaceutical active ingredient with cyanide ions, the toxicity of the reagent itself and finally the danger of its use. It is known to persons skilled in the art that sodium cyanoborohydride can release hydrocyanic acid if the pH of the reaction environment is acid enough and it is known that hydrocyanic acid is a powerful poison, since it competes with oxygen for hemoglobin coordination. As a consequence of this, particular care must be taken in its use and in the disposal of the production wastewater, which can be contaminated by cyanides. Not least, one must consider that the cost of sodium cyanoborohydride is considerable. |
| To conclude, it can be seen that more than 50 years after the publication of its first synthesis dated 1962, there are still numerous disadvantages or limitations in the synthesis paths developed in the past decades in the literature for the preparation of fenfluramine. |
| Moreover, recently there has been renewed pharmaceutical interest in the fenfluramine molecule, since the possibility of its therapeutic use in severe disorders of infancy has appeared in the medical literature. For example, mention can be made of Ceulemans et al., Epilepsia, 53(7), pages 1131 to 1139, 2012. |
| According to a certain part of medical literature, fenfluramine might therefore be interesting as a medication in a chronic therapy for the treatment of symptoms of epilepsy and other correlated severe disorders. |
| Based on recent medical developments, therefore, the need exists for a synthesis method that is better than the existing ones and can overcome in particular the disadvantages of the processes that are present in the literature. Particularly important, in view of use in chronic therapies for children such as epilepsy and other severe disorders, it would be fundamentally important to identify a path for synthesis of the active ingredient fenfluramine that does not entail the use of heavy metals and/or transition metals, which in a chronic therapy might accumulate in the body of the patients over the years, with severe consequences on health. |
| More generally, it is desirable to identify a synthesis path that uses reagents from which (or from the transformation products of which) it is then possible to easily purify fenfluramine. |
| It would be equally desirable to identify a synthesis path that comprises a small number of synthesis steps and uses reagents that are widely commercially available and easy to use. |
| At the same time, the new identified synthesis path should avoid if possible the formation of byproducts. |
DESCRIPTION OF THE FIGURES
| FIG. 1: impurities generated theoretically by means of the reagents used in the first fenfluramine synthesis according to BE609630. |
| FIG. 2: DSC of crude fenfluramine hydrochloride, obtained by reduction with sodium cyanoborohydride according to test 12 (table B) of the description that follows. |
| FIG. 3: DSC of fenfluramine hydrochloride recrystallized from 2-butanol as in example 2b (reduction with sodium borohydride). |
SUMMARY OF THE INVENTION
| The inventors of the present application have found surprisingly that the aim and objects indicated above are achieved by a new method for the synthesis of fenfluramine or of a pharmaceutically acceptable salt thereof, comprising the transformation of a ketone having the structure (I): |
(MOL) (CDX) wherein R 1 is CF 3, with ethylamine or with a salt thereof, and with a reducing agent chosen from the group consisting of alkaline cation or ammonium borohydride, alkaline cation or ammonium triacetoxyborohydride and alkaline cation or ammonium cyanoborohydride, in which the alkaline cation is always different from lithium cation and mixtures thereof, to yield fenfluramine, optionally followed by the transformation of the obtained fenfluramine into a pharmaceutically acceptable salt.
| Furthermore, the inventors of the present invention have also discovered a new preparation of fenfluramine, which can be obtained by means of the method described hereinafter, and new pharmaceutical compositions that contain it. |
Example 1
Synthesis of Fenfluramine
| A suspension of sodium hydroxide (34.62 g-0.866 mol, 3.5 eq) in 170 mL of methanol, under mechanical agitation, receives the addition, drop by drop, over the course of 30 minutes, of a solution of ethylamine hydrochloride (70.59 g-0.866 mol, 3.5 eq) in 165 mL of methanol, followed by 1-(3-trifluoromethyl)phenyl-propan-2-one (50 g-0.247 mol). The mixture is left under agitation at 20° C. for 4.5 hours, then cooling to 0° C. is performed and a solution of sodium borohydride (9.36 g-0.247 mol) in 19 mL of sodium hydroxide 1M in water is then added drop by drop, keeping the temperature below 10° C. The reaction is then left under agitation at 20° C. for another 2 hours. Once the reaction is complete, 270 mL of methanol are removed at a reduced pressure at 40° C. and then 200 mL of water are added and the mixture is extracted with heptane (200 mL). The aqueous phase is eliminated and the organic phase is washed with water (200 mL×3). The organic phase is concentrated at 50° C. at reduced pressure to yield free base fenfluramine as colorless oil. Yield: 72%; purity: 77%—as listed in test 3 of table A above. |
Example 2
Purification of Fenfluramine
| Purification of free base fenfluramine can be performed in two ways: |
| distillation of the free base |
| crystallization of the fenfluramine hydrochloride salt |
| Depending on the degree of purity that is desired, both purification processes are performed in sequence (distillation first and then crystallization), or only one of the two purification processes is performed. |
Example 2a
Distillation
| Free base fenfluramine (10 g), prepared as in Example 1, is distilled under reduced pressure with a distillation column of the Vigreux type: the distillation heads are eliminated, the fraction that is distilled at 89-90° C. at 6 mmHg, which is the active ingredient fenfluramine (8.5 g) with a high degree of purity, is collected. |
Example 2b
Conversion into Hydrochloride Salt and Crystallization
| Crude fenfluramine, prepared as in Example 1, or purified fenfluramine as in Example 2a, is dissolved in 125 mL of ethyl acetate, and cooling is performed to 0° Celsius under agitation. 272 mL of a solution of 1M HCl in ethyl acetate are added drop by drop at 0° C. The precipitate that forms is filtered and washed with ethyl acetate (125 mL×2) to yield approximately 55 g of solid fraction. The solid fraction is crystallized by 2-butanol (260 mL), keeping the solid for 22 hours at 3° C. under slow agitation before filtering it. Filtering is performed and washing is performed with cold 2-butanol. The solid fraction, fenfluramine hydrochloride, is dried in a vacuum stove, yielding 51.7 g of product. A DSC of the resulting product is shown in FIG. 3. |
PAPER
Journal of Organic Chemistry (1979), 44(20), 3580-3.J. Org. Chem. 1979, 44, 20, 3580–3583
Publication Date:September 1, 1979
https://doi.org/10.1021/jo01334a031https://pubs.acs.org/doi/abs/10.1021/jo01334a031
PATENT
https://patents.google.com/patent/US20170174613A1/en
- Fenfluramine is an amphetamine drug that was once widely prescribed as an appetite suppressant to treat obesity. Fenfluramine is devoid of the psychomotor stimulant and abuse potential of D-amphetamine and interacts with the 5-hydroxytryptamine (serotonin, 5-HT) receptors to release 5-HT from neurons. Fenfluramine has been investigated as having anticonvulsive activity in the treatment of Dravet Syndrome, or severe myoclonic epilepsy in infancy, a rare and malignant epileptic syndrome. This type of epilepsy has an early onset in previously healthy children.
- [0003]
Anorectic treatment with fenfluramine has been associated with the development of cardiac valvulopathy and pulmonary hypertension, including the condition cardiac fibrosis which led to the withdrawal of fenfluramine from world-wide markets. Interaction of fenfluramine’s major metabolite norfenfluramine with the 5-HT2B receptor is associated with heart valve hypertrophy. In the treatment of epilepsy, the known cardiovascular risks of fenfluramine are weighed against beneficial anticonvulsive activity.


- [0097]
Chemical Abstract Service (CAS) Registry Number (RN): 404-82-0 (HCl Salt), 458-24-2 (Parent Free Base) - [0098]
Chemical Name: N-ethyl-α-methyl-3-(trifluoromethyl)-benzeneethanamine hydrochloride (1:1). Other Names: Fenfluramine HCl, DL-Fenfluramine, (±)-Fenfluramine - [0099]
Structure of Hydrochloride Salt: - [0100]
Stereochemistry: Fenfluramine HCl has one chiral center and is being developed as the racemate and contains dexfenfluramine and levofenfluramine - [0101]
Molecular Formula of hydrochloride salt: C12H16F3N.HCl - [0102]
Molecular Mass/Weight: 267.72 g/mol
2. General Properties
- [0103]
Table 1 summarizes the chemical and physical properties of Fenfluramine HCl. - TABLE 1 General Properties of Fenfluramine HCl Drug Substance Property Result Appearance (color, White to off-white powder physical form) DSC (melting 170° C. (melt/sublimation) point)a TGA Onset 147° C. 0.03% at 150° C. 91% at 220° C. (evaporation) pKa (water) 10.15-10.38 Solubility (mg/mL) Resultant pH 25° C. 37° C. Solubility pH 6.69 (water) 54.13 71.22 (Aqueous) pH 1.73 buffer 25.34 53.68 pH 3.43 buffer 29.50 61.97 pH 6.41 buffer 37.42 95.60 0.9% NaCl (water) 22.98 — Solvent Solubility 25° C. (mg/mL) Solubility (Organic Ethanol 150 Solvents) Dichloromethane 30-35 Ethyl Acetate, 1-5 mg Tetrahydrofuran, Toluene, Acetonitrile UV Absorption Maxima: 210, 265 nm Solution pH (water) 6.69 Hygroscopicity @30% RH: ~0.05% (Dynamic Vapor @60% RH: ~0.07% Sorption (DVS) @90% RH: ~0.20%a) Polymorphism Fenfluramine HCl has been consistently isolated as a single crystalline Form 1 as determined by DSC and x-ray powder diffraction (XRPD) Solvation/Hydration Fenfluramine HCl is isolated as a nonhydrated, nonsolvated solid Solution Stability 8 weeks @ pH 6.7 phosphate buffer medium at 40° C. and 60° C. using concentrations of 0.5, 2.5 and 5.0 mg/ml. All conditions, no new impurities >0.1% by HPLC. Solid Stability 8 weeks @ 40° C., 60° C. and 80° C. 7 days at 150° C. All conditions, no new impurities >0.1% by HPLC.
3. Synthesis of Fenfluramine Drug Substance
- [0104]
Scheme 3.1 shows a 2-step route of synthesis used to manufacture initial clinical supplies of Fenfluramine HCl from ketone (2). The batch size is 4 kg performed in laboratory glassware (kilo lab). No chromatography is required and the process steps are amenable to scale-up. In process 1 there is one isolated intermediate Fenfluramine Free Base (1) starting from commercially supplied 1-(3-(trifluoromethyl)phenyl) acetone (Ketone 2). All steps are conducted under cGMPs starting from Ketone (2). - [0105]
Scheme 3.2 shows a 4-step route of synthesis to Fenfluramine HCl that can be used for commercial supply. Route 2 utilizes the same 2-step process used by Route 1 to convert Ketone (2) to Fenfluramine HCl with the exception that Ketone (2) is synthesized under cGMP conditions starting from 3-(Trifluoromethyl)-phenyl acetic acid (Acid 4). Bisulfate Complex (3) is an isolatable solid and can be purified before decomplexation to Ketone (2). In-situ intermediates which are oils are shown in brackets. Batch sizes of 10 Kg are performed. Commercial batch sizes of 20 kg are performed in fixed pilot plant equipment. Steps 1-2 of Scheme 3.2 to manufacture Ketone (2) have been demonstrated on a 100 g scale to provide high purity ketone (2) of >99.8% (GC & HPLC). Conversion of Ketone (2) to Fenfluramine using either Route 1 or 2 has provided similar purity profiles. - Starting materials are designated by enclosed boxes. Bracketed and non bracketed compounds respectively indicate proposed in-situ and isolated intermediates. NMI=N-Methyl Imidazole.
4.1. Narrative Description (Route 1)
- [0106]
Step 1: Reductive Amination (Preparation of Fenfluramine Free Base 1) - [0107]
A solution of ethylamine, water, methanol, and 1-(3-(trifluoromethyl)phenyl) acetone (Ketone 2) was treated with sodium triacetoxyborohydride and stirred for 16 h at 25° C. at which time HPLC analysis (IPC-1; In Process Control No. 1) showed the reaction to be complete and sodium hydroxide solution was added until pH>10. Toluene was added and the phases separated, and the aqueous phase (IPC-2) and organic phase (IPC-3) are checked for remaining Fenfluramine and Fenfluramine alcohol and the organic phase was reduced. Purified water was added and the pH adjusted to <2 using conc. HCl and the phases were separated. The aqueous phase was washed with toluene and the toluene phase (IPC-4) and the aqueous phase (IPC-5) was checked for Fenfluramine and Fenfluramine alcohol content. The aqueous phase containing product is pH adjusted to >10 using sodium hydroxide solution. The basic aqueous phase was extracted with MTBE until removal of Fenfluramine from the aqueous phase was observed by HPLC (<0.5 mg/ml) (IPC-6). The organic phase was dried over sodium sulfate and filtered. The filtrate was concentrated in vacuo to give the intermediate product Fenfluramine Free Base 1 as a pale yellow oil tested per specifications described herein which showed by NMR the material to contain 2.93% toluene giving an active yield of 88.3% with a purity of 98.23% by HPLC (0.67% Fenfluramine alcohol). - [0108]
Step 2: Salt Formation (Preparation of Fenfluramine HCl) - [0109]
To a flask was charged ethanol and acetyl chloride. The solution was stirred slowly overnight before ethyl acetate was added. The HCl in ethyl acetate solution formed was polish filtered into a clean carboy and retained for later use. To a vessel was added Fenfluramine free base 1 and MTBE. The Fenfluramine solution in MTBE was collected in two carboys before the vessel was cleaned and checked for particulate residue. The Fenfluramine solution was polish filtered into a vessel and cooled and HCl in ethyl acetate solution was added giving a final pH of 6-7. The batch was stirred for 1 h and filtered. The product was dried under vacuum at 40° C. The product (96.52% yield) was tested per IPC-7 had a purity of 99.75% by HPLC and GC headspace analysis showed MTBE (800 ppm) and EtOAc (150 ppm) to be present. The product was then tested per specifications shown herein.
4.2. Narrative Description (Route 2)
- [0110]
Step 1: Preparation of Ketone Bisulfite Adduct - [0111]
Procedure: Charge acetic anhydride, (2.8 vol, 3.0 wt, 5.0 eq.) to a vessel and commence stirring. Cool the solution to −5 to 5° C., targeting −4° C. Charge 1-methylimidazole, (0.2 vol, 0.21 wt, 0.5 eq.) to the mixture at −5 to 5° C. Caution: very exothermic. If necessary, adjust the temperature to 0 to 5° C. Charge ZX008 acid, (1.00 wt, 1.0 eq.) to the mixture at 0 to 5° C. Caution: exothermic. Stir the mixture at 0 to 5° C. until ≦2.1% area ZX008 acid by HPLC analysis, typically 7 to 9 hours. Charge 15% w/w sodium chloride solution (2.0 vol) to the mixture at 0 to 5° C., 60 to 90 minutes. Caution: very exothermic which will be slightly delayed. Warm the mixture to 18 to 23° C. over 45 to 60 minutes and continue stirring for a further 30 to 45 minutes at 18 to 23° C. Charge TBME, (5.0 vol, 3.7 wt) to the mixture and stir for 10 to 15 minutes at 18 to 23° C. Separate the aqueous layer and retain the organic layer. Back-extract the aqueous layer with TBME, (2×3.0 vol, 2×2.2 wt) at 18 to 23° C. retaining each organic layer. Adjust the pH of the combined organic layer to pH 6.5 to 9.0, targeting 7.0 by charging 20% w/w sodium hydroxide solution (5.3 to 8.3 vol) at 18 to 23° C. Caution: exothermic. Separate the aqueous layer and retain the organic layer. Wash the organic layer with 4% w/w sodium hydrogen carbonate solution (2×3.0 vol) at 18 to 23° C. Determine the residual ZX008 acid content in the organic layer by HPLC analysis, pass criterion ≦0.10% area ZX008 acid. Wash the organic layer with purified water, (2×3.0 vol) at 18 to 23° C. Concentrate the organic layer under reduced pressure to ca. 2 vol at 40 to 45° C., targeting 43° C. - [0112]
Determine the w/w assay of ZX008 ketone (WIP) in the mixture by 1H-NMR analysis for information only and calculate the contained yield of ZX008 ketone (WIP) in the mixture. Note: This step can be removed from the process since the process is robust and consistently delivers 80 to 90% th yield. The achieved yield was factored into the charges of the subsequent steps. - [0113]
Charge n-heptane, (4.0 vol, 2.7 wt) to the mixture at 40 to 45° C., targeting 43° C. Concentrate the mixture to ca. 2 vol at 40 to 45° C., targeting 43° C. Determine the TBME content in the mixture by 1H-NMR analysis, (pass criterion ≦5.0% w/w TBME vs. ZX008 ketone). Charge n-heptane, (2.4 vol, 1.6 wt) at 40 to 45° C., targeting 43° C., vessel A. To vessel B, charge sodium metabisulfite, (0.82 wt, 0.88 eq.) at 18 to 23° C. To vessel B, charge a solution of sodium hydrogen carbonate, (0.16 wt, 0.4 eq.) in purified water, code RM0120 (2.0 vol) at 18 to 23° C. followed by a line rinse with purified water, code RM0120 (0.4 vol) at 18 to 23° C. Caution: gas evolution. Heat the contents of vessel B to 40 to 45° C., targeting 43° C. Charge the contents from vessel A to vessel B followed by a line rinse with n-heptane, (0.8 vol, 0.5 wt) at 40 to 45° C., targeting 43° C. Stir the mixture for 1 to 1.5 hours at 40 to 45° C., targeting 43° C. Charge n-heptane, code RM0174 (3.2 vol, 2.2 wt) to the mixture with the temperature being allowed to cool to 18 to 45° C. at the end of the addition. Cool the mixture to 18 to 23° C. at approximately constant rate over 45 to 60 minutes. Stir the mixture at 18 to 23° C. for 1.5 to 2 hours. - [0114]
Sample the mixture to determine the residual ZX008 ketone content by 1H-NMR analysis, (pass criterion ≦10.0% mol, target 5.0% mol ZX008 ketone vs. ZX008 ketone bisulfite adduct). Filter the mixture and slurry wash the filter-cake with n-heptane, (2×2.0 vol, 2×1.4 wt) at 18 to 23° C. Dry the solid at up to 23° C. until the water content is <10.0% w/w water by KF analysis according to AKX reagent. At least 16 hours. Determine the w/w assay of the isolated ZX008 ketone bisulfite adduct by 1H-NMR analysis and calculate the contained yield of ZX008 ketone bisulfite adduct. - [0115]
Yields and Profiles: The yield for the stage 1 Demonstration batch is summarized Table below. Input: 1700.0 g uncorr., acid, 99.50% area (QC, HPLC), 2-isomer not detected, 4-isomer 0.02% area, RRT1.58 (previously not observed) 0.48% area as per the preparative method. The analytical data is summarized in Table 1A below. - TABLE 1A Table for isolated yields for step 1 Demonstration batch Corr. % area Reference Corr. Yield % w/w (HPLC, number Input Output (% th)** (1H-NMR)* QC) Comments Batch A1 1700.0 g 1500.1 g 89.1 45.0 —.— Crude ketone as TBME sol. Batch A2 1500.1 g 1716.1 77.8 76.0 98.15 Bisulfite adduct only 67.3 Overall product
- [0116]
Step 2: Preparation of Ketone - [0117]
Procedure: Charge toluene, (5.0 vol, 4.3 wt), and purified water, (5.0 vol) to the vessel and commence stirring. If necessary, adjust the temperature to 18 to 23° C. and charge ZX008 ketone bisulfite adduct, (1.00 wt corrected for % w/w assay) to the mixture at 18 to 23° C. Charge 20% w/w sodium hydroxide solution to the mixture at 18 to 23° C. adjusting the pH of the mixture to pH 8.0 to 12.0, targeting 9.0 (0.5 to 1.0 vol). - Separate the lower aqueous layer and retain the top organic layer. Wash the organic layer with purified water, (3.0 vol) at 18 to 23° C. Concentrate the organic layer under reduced pressure to ca. 2 vol at 45 to 50° C., targeting 48° C. Charge methanol, (5.0 vol, 4.0 wt) to the mixture at 45 to 50° C., targeting 48° C. Re-concentrate the mixture under reduced pressure to ca. 2 vol at 45 to 50° C., targeting 48° C. Repeat steps 7 and 8 once before continuing with step 9. Cool the mixture to 18 to 23° C. Clarify the mixture into a tared, suitably-sized drum followed by a methanol (1.0 vol, 0.8 wt) line rinse at 18 to 23° C. Determine the w/w assay of ZX008 ketone (WIP) in the mixture by 1H-NMR analysis and calculate the contained yield of ZX008 ketone (WIP) in the mixture. Determine the toluene content in the mixture by 1H-NMR analysis.
- [0118]
Yields and Profiles: The yield for the step 2 Demonstration batch is summarized in Table 1B below. Input: 1200.0 g corr. Ketone bisulfite adduct, 76.0% w/w assay (NMR, using DMB as internal standard in d6-DMSO), (1.00 eq, 1.00 wt corr. for w/w assay) for input calculation. - TABLE 1B Table for isolated yields for step 2 Demonstration batch % w/w % area Corr. Corr. Corr. Yield (1H- (HPLC, Input Output (% th) NMR)* QC) Comments 1200.0 g 858.15 g 108.3 25.5 99.31 Purified ketone
- [0119]
Step 3: Preparation of Fenfluramine HCl Crude - [0120]
Procedure: Charge the ZX008 ketone (corr. for assay, 1.00 wt, 1.00 eq. isolated as solution in MeOH in stage 2) to a vessel. Charge methanol, code RM0036 (5.0 vol, 4.0 wt) to the mixture at 18 to 23° C. Cool the solution to 0 to 5° C. Charge 70 wt % aqueous ethylamine solution (1.3 vol, 1.6 wt, 4.0 eq) to the mixture at 0 to 10° C., over 15 to 30 minutes, followed by a line rinse with methanol (1.0 vol, 0.8 wt). Warm the mixture to 15 to 20° C. and stir the mixture for a further 60 to 70 minutes at 15 to 20° C. Adjust the mixture to 15 to 18° C. if required, targeting 15° C. Charge sodium triacetoxyborohydride (2.4 wt, 2.25 eq.) to the mixture in approximately 10 portions, keeping the mixture at 15 to 20° C., targeting 17° C. Addition time 1.5 to 2 hours. Caution: Exothermic. Stir the mixture at 15 to 20° C. until complete by HPLC analysis, pass criterion ≦3.0% area ZX008 ketone, typically 2 to 3 hours. Adjust the pH of the mixture to pH>12 by charging 20% w/w aqueous sodium hydroxide solution (5.0 to 6.0 vol) to the mixture at 15 to 40° C. Addition time 10 to 30 minutes. Caution: Exothermic. If necessary, adjust the temperature to 18 to 23° C. Extract the mixture with toluene (3×3.0 vol, 3×2.6 wt) at 18 to 23° C., retaining and combining the top organic layer after each extraction. Wash the combined organic layer with purified water, (1.0 vol) at 18 to 23° C. Heat the mixture to 40 to 50° C., targeting 48° C. Concentrate the mixture under reduced pressure at constant volume maintaining ca. 5 vol by charging the organic layer at approximately the same rate as the distillation rate at 40 to 50° C., targeting 48° C. Cool the mixture to 18 to 23° C. Charge purified water (10.0 vol) to the mixture at 18 to 23° C. Adjust the pH of the mixture to 0.1<pH<1.5 at 18 to 23° C. by charging concentrated hydrochloric acid, 0.5 vol. Do not delay from this step until neutralization. - [0121]
Separate the layers at 18 to 23° C. retaining the bottom aqueous layer. Wash the aqueous layer with toluene, (3.0 vol, 2.6 wt) at 18 to 23° C. retaining the aqueous layer. Adjust the pH of the aqueous layer to pH>12 by charging 20% w/w sodium hydroxide solution at 18 to 23° C. 0.8 to 0.9 vol. Caution: Exothermic. Charge TBME, code RM0002 (2.0 vol, 1.5 wt) to the basic aqueous layer. Separate the layers at 18 to 23° C. retaining the top organic layer. Back-extract the aqueous layer with TBME (2×2.0 vol, 2×1.5 wt) at 18 to 23° C. retaining the organic layers. Wash the combined organic layer with purified water, (2×1.0 vol) at 18 to 23° C. Concentrate the combined organic layers under reduced pressure at 40 to 50° C., targeting 48° C. to ca. 3 vol. Determine the residual toluene content of the mixture by 1H-NMR analysis. Sample for determination of residual water content by KF analysis, AKX reagent. Charge TBME (8.7 vol, 6.4 wt) to the mixture at 40 to 50° C. Cool the solution to 0 to 5° C., targeting 2° C. Charge concentrated hydrochloric acid (0.54 vol, 0.46 wt) maintaining the temperature <15° C. Caution: Exothermic. Line rinse with TBME (1.0 vol, 0.7 wt). If necessary, adjust the temperature to 0 to 10° C. and stir the mixture at 0 to 10° C. for a further 2 to 3 hours. Filter the mixture and wash the filter-cake with TBME (2×4.4 vol, 2×3.3 wt) at 0 to 10° C. Dry the solid at up to 40° C. until the TBME content is <0.5% w/w TBME by 1H-NMR analysis. 4 to 8 hours. - [0122]
Yields and Profiles: The yield for the step 3 Demonstration batch is summarized in Table 1C below. Input: 856.8 g corr. Ketone, 44.2% w/w assay (NMR, using TCNB as internal standard in CDCl3), (1.00 eq, 1.00 wt corr. for w/w assay) for input calculation. FIG. 2 and Table 1D shows an exemplary HPLC chromatogram of a crude preparation of fenfluramine hydrochloride (210 nm UV absorbance). - TABLE 1C Table for isolated yields for step 3 Demonstration batch Corr. % area Reference Corr. Corr. Yield % w/w (HPLC, number Input Output (% th) (1H-NMR)* QC) Comments Batch A1 856.8 g 836.31 g 85.3 44.2 99.15 Fenfluramine free base (in situ intermediate) Batch A2 880.7 84.0 based 99.5 100.00 Fenfluramine•HCl on ketone crude (step 3 an bisulfite d 4.1) adduct (77.6 based on purified ketone)
- TABLE 1D Purity of crude fenfluramine hydrochloride by HPLC (see FIG. 2) Processed Channel Descr. DAD AU Ch 1 Sample 210, Bw 4 Peak Results USP USP USP Name RT RelRT Area Height Tailing Resolution Plate Count EP s/n % Area 1 NorFenfluramine 7.46 2 2-Fenfluramine 7.68 3 Fenfluramine 8.67 1.000 3789064 778178 1.7 70796 2549.8 99.15 4 4-Fenfluramine 8.95 5 11 34 1.308 6073 1449 1.2 23.5 215529 3 8 0.16 6 ZX008 acid 12.93 7 Fenfluramine alcohol 14.16 1.633 15266 2972 1.3 24.8 215040 8.7 0.40 8 ZX008 ketone 14.83 9 Fenfluramine acetamide 15.55 10 TOLUENE 15 75 11 15.92 1.836 4110 1122 2.7 0.11 12 16.60 1.915 6861 1630 1.5 451209 4.3 0.18 Sum 3821374 100.00
- [0123]
Step 4.2: Crystallization of Fenfluramine Hydrochloride - [0124]
Procedure: Charge Fenfluramine.HCl (crude) (1.00 wt, 1.0 eq.) and TBME (10.0 vol, 7.4 wt) to the vessel and commence stirring. Heat the suspension to reflux (50 to 58° C.). Charge ethanol (5.0 vol, 3.9 wt) maintaining the temperature at 50 to 58° C. Addition time 20 minutes. Stir at 50 to 58° C. for 5 to 10 minutes and check for dissolution. Stir the solution at 50 to 58° C. for 5 to 10 minutes, targeting 54 to 58° C. Clarify the reaction mixture through a 0.1 μm in-line filter at 54 to 58° C., followed by a line rinse with TBME (1 vol, 0.7 wt). Cool the solution to 48 to 50° C. Charge Fenfluramine HCl, code FP0188 (0.01 wt). Check for crystallization. Allow the suspension to cool to 15 to 20° C., target 17° C. over 5 to 5.5 hours at an approximately constant rate. Stir the mixture at 15 to 20° C., target 17° C. for 2 to 3 hours. Filter the mixture and wash the filter-cake with clarified TBME (2×3.0 vol, 2×2.2 wt) at 5 to 15° C. Dry the solid at up to 40° C. until the TBME content is <0.5% w/w TBME and the ethanol content is <0.5% w/w EtOH by 1H-NMR analysis. 4 to 8 hours. Determine the w/w assay of the isolated Fenfluramine.HCl by 1H-NMR analysis. - [0125]
Yields and Profiles: The yield for the stage 4 Demonstration batch is summarized in Table 1E below. Input: 750.0 g uncorr. Fenfluramine HCl crude (1.00 eq, 1.00 wt uncorr.) for input calculation. FIG. 3 shows an exemplary HPLC chromatogram of a crystallized fenfluramine hydrochloride sample (210 nm UV absorbance). - TABLE 1E Table for isolated yields for stage 4 Demonstration batch Uncorr. Uncorr. Uncorr. Yield HPLC (% area, Input Output (% th) QC) Comments 750.0 g 608.0 81.1 100.00* Fenfluramine•HCl
PATENT
https://patents.google.com/patent/EP3170807A1/en
- Fenfluramine, i.e., 3-trifluoromethyl-N-ethylamphetamine, has the following chemical structure:
- [0003]
The marketing of fenfluramine as a pharmaceutical active ingredient in the United States began in 1973 and was used in a therapy in combination with phentermine to prevent and treat obesity. However, in 1997 fenfluramine was withdrawn from the market in the United States and immediately thereafter in other countries, since its ingestion was associated with the onset of cardiac fibrosis and pulmonary hypertension. As a consequence of this event, the pharmaceutical compounds containing this active ingredient were withdrawn from the market. However, fenfluramine, even after its exit from the market, has continued to attract scientific interest, as will become apparent from the discussion presented hereinafter. - [0004]
In the literature, over the years, numerous syntheses or processes have been reported for preparing fenfluramine or its dextrorotatory enantiomer dexfenfluramine or an analog containing a highly electron-attractor group on the aromatic ring as in the fenfluramine molecule (see for example Pentafluorosulfanyl Serotonin Analogs: Synthesis, Characterization, and Biological Activity, John T. Welch and Dongsung Lim Chapter 8, pp 165-181 DOI: 10.1021/bk-2009-1003.ch008). Many of these synthesis paths are long and foresee multiple stages or synthesis steps that can include reagents that are dangerous or scarcely environment-friendly and are therefore scarcely convenient for an industrial synthesis. Hereinafter, any reference to “fenfluramine” is understood to referto the racemic form, i.e, (RS)-N-ethyl-1-[3-(trifluoromethyl)phenyl]propan-2-amine. - [0005]
To the best of the knowledge of the inventors, the first method for fenfluramine synthesis reported in the literature dates back to 1962 and is referenced in patent BE609630 and in analogous patents US3198833 and FR1324220 . All the synthesis methods reported in these patents provide for numerous synthesis steps. By way of example, one of the methods provides for the transformation into oxime of a ketone, 1-(3-trifluoromethyl)phenyl-propan-2-one, as shown here: - [0006]
The oxime is then hydrogenated in the presence of Raney nickel catalyst so as to yield the corresponding primary amine, which is acetylated subsequently with ethanoic anhydride before being converted into fenfluramine by reduction with lithium aluminum hydride. - [0007]
As can be seen, the final step of this chemical process provides for the use of lithium aluminum hydride and the persons skilled in the art will acknowledge that the use of this reagent should be avoided, if possible, on an industrial level, since it is extremely flammable and is the source of accidents. Furthermore, lithium is a potentially neurotoxic metal and therefore its use should be avoided where possible. Furthermore, the Raney nickel catalyst is used in the oxime reduction step and can contaminate the final active ingredient; the use of hydroxylamine also entails problems of toxicity for workers assigned to production. - [0008]
A further disadvantage of this process is, as already mentioned earlier, the number of steps, not only because a large number of synthesis steps entails a reduction of the overall yield of active ingredient, but also because each synthesis step in principle can generate impurities and a larger number of steps can therefore entail a higher number of impurities in the final active ingredient. Many of these impurities, furthermore, due to their structural similarity to fenfluramine, are difficult to eliminate and remove from a fenfluramine preparation. One impurity for example that can be formed in the process described above and is difficult to eliminate is the following: - [0009]
This impurity, which is a primary amine, shares physical-chemical properties that are similar to fenfluramine and therefore, like fenfluramine, it can form a hydrochloride salt by treatment with hydrochloric acid and thus contaminate the active ingredient fenfluramine hydrochloride. Furthermore, this impurity – as a free base – has a boiling point that is similar to that of fenfluramine (73°C vs. 89°C at 6 mmHg respectively), and therefore its elimination by distillation also can be problematic. - [0010]
The process described above can in principle generate other impurities, which are listed in Figure 1 . - [0011]
EP 0441160 claims a synthesis in 5 steps of dexfenfluramine, dextrorotatory enantiomer of fenfluramine. This synthesis can be adapted easily to produce fenfluramine instead of its dextrorotatory enantiomer simply by performing the first reduction step with a non-chiral reducing agent. In the first step, in fact:a ketone, 1-(3-trifluoromethyl)phenyl-propan-2-one, is first reduced to the corresponding alcohol in the presence of yeast, D-glucose, ethanol and water. Then the alcohol is converted into the tosylate in the second step: - [0012]
This reaction occurs in the presence of triethylamine and tosyl chloride in methylene chloride as solvent. After purification, the tosylate is converted to fenfluramine by means of three successive steps: - [0013]
In the first of these three steps, the tosylate is converted into an azide intermediate by reaction with sodium azide in dimethylformamide. The azide intermediate is then hydrogenated in the presence of a catalyst, palladium on carbon. Finally, the resulting primary amine is converted into fenfluramine by reaction with acetaldehyde and sodium borohydride. - [0014]
Persons skilled in the art may see easily that this process is not desirable from an industrial standpoint due to reasons related to environmental risk, safety and costs. For example, the sodium azide used in the process is a notoriously explosive compound and its use at the industrial level is dangerous. Furthermore, palladium is an expensive material and its use in the process entails an increase in the production costs of fenfluramine. Furthermore, palladium can contaminate the finished active ingredient. - [0015]
In another method for the synthesis of dexfenfluramine in 3-4 steps, reported by Goument et al. in Bulletin of the Chemical Society of France (1993), 130, p. 450-458, 3-bromobenzotrifluoride is subjected to a Grignard reaction with enantiopure 1,2-propylene-epoxide to yield 1-[3-(trifluoromethyl)phenyl]propan-2-ol as shown hereafter: - [0016]
If this reaction is performed with racemic 1,2-propylene-epoxide, the synthesis can be adapted to the preparation of fenfluramine. - [0017]
The alcohol thus obtained is first transformed into trifluoromethyl sulfonate by reaction with trifluoromethanesulfonic anhydride and then treated with ethylamine to yield fenfluramine, as shown in the diagram hereinafter: - [0018]
In this article, the authors acknowledge that the main byproducts of the reaction are isomer alkenes having the following chemical structures: - [0019]
The process proposed by Goument et al. is not interesting from the industrial standpoint for a series of reasons. First of all, it is known that the use of Grignard reagents, especially on an industrial scale, is problematic, because these compounds are often pyrophoric and corrosive. Furthermore, 1,2-propylene epoxide is a suspected carcinogenic compound. Finally, the formation of the three isomer alkenes as byproducts listed above is a disadvantage of the process. In the article, Goument presents methods for activation of the intermediate alcohol which are alternative to trifluoromethylsulfonate, for example by converting it to chloride (via thionyl chloride) or to mesylate (via mesyl chloride), but these process variations share the same disadvantages as the main process analyzed above. - [0020]
In addition to the methods with multiple synthesis steps discussed so far in detail, the literature reports other methods or processes for producing fenfluramine or dexfenfluramine. In general, persons skilled in the art acknowledge that the syntheses in the literature for producing dexfenfluramine sometimes can be applied to the preparation of fenfluramine simply by replacing the initial materials and/or enantiopure reagents with the corresponding racemates while maintaining the reaction conditions. For example, patents that present long synthesis methods in multiple steps are the following: - [0021]
Other examples of preparation of fenfluramine, taken from non-patent literature, are the following:- Synthesis, Nov.1987, p. 1005-1007
- J.Org.Chem, 1991, 56, p. 6019
- Tetrahedron, 1994, 50(1), p. 171
- Bull. Soc. Chim. France, 1993, 130(4), p. 459-466 (dexfenfluramine)
- Chirality, 2002, 14(4), p. 325-328 (dexfenfluramine)
- [0022]
Without analyzing in detail the individual methods described in these patents or articles, it can be stated in summary that all these methods are not attractive and interesting from the industrial standpoint because these are processes with many synthesis steps or because the initial materials described therein are not easily available and therefore have to be prepared separately, with a further expenditure of time and with further costs, or because they provide for the use of reagents that are dangerous/explosive/toxic or because they entail the use of catalysts based on heavy metals that can contaminate the final active ingredient. - [0023]
One should consider that in the literature there are methods for the preparation of fenfluramine that do not provide for long syntheses and multiple steps but are shorter and consist of one or two steps. These processes, which therefore would be more interesting from the industrial standpoint, have other specific disadvantages, as will become apparent in detail hereinafter. For example, in the literature there is a first group of articles or patents that describe the reaction between 1-(3-trifluoromethyl)phenyl-propan-2-one and ethylamine in the presence of hydrogen gas and of a transition metal as catalyst: - [0024]
In particular, in Huagong Shikan, 2002, 16(7), p. 33, the reaction is performed with hydrogen gas (2.9 – 3.38 atm), at 65-75°C, for 9 hours, in the presence of Raney nickel. Likewise, in patent DD108971 (1973), Raney nickel and hydrogen gas and methanol are used as solvent to perform this reaction. - [0025]
In HU55343 , instead, a similar reaction in one step is performed with hydrogen gas in the presence of another transition metal catalyst, such as palladium on carbon. - [0026]
Although these three methods describe short single-step processes, they have the disadvantage of the use of hydrogen gas. As is known to persons skilled in the art, hydrogen gas is a dangerous gas due to the inherent danger of forming explosive mixtures with air and must be used by expert personnel in expensive facilities dedicated to its use and built with special precautions. Despite being used in purpose-built facilities, the use of hydrogen at the industrial level is inherently dangerous and to be avoided if possible. Another danger element that is shared by the processes described above is the fact that the reactions are performed under pressure. The third industrial disadvantage then arises from the use of heavy metal catalysts, which have a high cost and therefore increase the overall cost of the final active ingredient and -on the other hand- may contaminate the active ingredient fenfluramine even after filtration of the catalyst and purification of said active ingredient. - [0027]
Analysis of the background art shows, however, that an attempt has been made to devise a process for the production or synthesis of fenfluramine that is short (one or two steps) and does not entail the use of hydrogen gas or of catalysts based on nickel or palladium or the like. In particular, for example, Synthesis 1987, 11, p. 1005, and then DECHEMA Monographien (1989), 112 (Org. Elektrochem.–Angew. Elektrothermie), 367-74, present a method for the synthesis of fenfluramine which starts from 1-(3-trifluoromethyl)phenyl-propan-2-one, which is made to react with ethylamine in great excess, in an electrochemical process, which uses a mercury cathode in a water/ethanol solution with pH 10-11. One obtains fenfluramine with 87% yield. This process has some drawbacks from an industrial standpoint: it is a process of the electrochemical type and therefore requires special equipment which is scarcely widespread, dedicated cells and reactors, and it is not possible to use the classic multipurpose reactors available in the pharmaceutical industry. Furthermore, the use of mercury at the industrial level poses severe environment safety problems, requiring constant health monitoring on workers who manage the equipment and systems for the management and destruction of wastewater that are particularly onerous; finally, mercury can be transferred from the cathode to the reaction environment and therefore to the active ingredient, and this obviously is to be considered very dangerous due to the accumulation of the metal in human beings; small traces of mercury are very toxic. - [0028]
Another method for fenfluramine synthesis in a single step is the one presented in J.Org.Chem, 1979, 44(20), p. 3580. Here the reaction is described between an alkene derivative and ethylamine in the presence of sodium borohydride and mercury nitrate: - [0029]
Again, this process is not interesting from an industrial standpoint since it has the same problems, if not even greater ones, related to the use of mercury (used here as a water-soluble salt) discussed previously. The complication introduced in this process with the use of mercury nitrate together with sodium borohydride highlights the level of innovation of the synthesis path found here. - [0030]
In past years, therefore, it has not been possible to provide a process for synthesizing fenfluramine in a small number of steps by using modern reducing agents that are commonly and easily used. Indeed, while Gaodeng Xuexiao Huaxue Xuebao, 9(2), 1988, p. 134-139, describes and exemplifies the synthesis of 2-N-ethyl-1-phenyl propane by means of (1) the treatment of the precursor ketone with ethylamine followed by (2) sodium cyanoborohydride as reducing agent, Xuexiao Huaxue Xuebao provides no example for fenfluramine. Moreover, for the latter, Xuexiao Huaxue Xuebao indicates a melting point for the hydrochloride of 161°C, a data item that matches the value indicated in the literature initially (see BE609630 ); these facts prove that fenfluramine synthesis with cyanoborohydride was not performed, otherwise one cannot explain why the author did not transcribe, in the document, the example of a product that at the time was very important. It should be noted in fact that 1-phenyl propan-2-one and 1-(3-trifluoromethyl)phenyl-propan-2-one can have different reactivities to reductive amination due to the presence of a highly electron-attractor -trifluoromethyl group, hence the need for an example to demonstrate its feasibility. The use of cyanoborohydride shares some disadvantages with other methods discussed in the preceding paragraphs. The excellent selectivity for reductive aminations of this reagent is highly appreciated, but its application can be less advantageous with respect to other reducing systems in the synthesis of fenfluramine, where the latter is intended for therapeutic application in human beings. The reasons for this are the possible contamination of the finished pharmaceutical active ingredient with cyanide ions, the toxicity of the reagent itself and finally the danger of its use. It is known to persons skilled in the art that sodium cyanoborohydride can release hydrocyanic acid if the pH of the reaction environment is acid enough and it is known that hydrocyanic acid is a powerful poison, since it competes with oxygen for hemoglobin coordination. As a consequence of this, particular care must be taken in its use and in the disposal of the production wastewater, which can be contaminated by cyanides. Not least, one must consider that the cost of sodium cyanoborohydride is considerable. - [0031]
To conclude, it can be seen that more than 50 years after the publication of its first synthesis dated 1962, there are still numerous disadvantages or limitations in the synthesis paths developed in the past decades in the literature for the preparation of fenfluramine. - [0032]
Moreover, recently there has been renewed pharmaceutical interest in the fenfluramine molecule, since the possibility of its therapeutic use in severe disorders of infancy has appeared in the medical literature. For example, mention can made of Ceulemans et al., Epilepsia, 53(7), pages 1131 to 1139, 2012. - [0033]
According to a certain part of medical literature, fenfluramine might therefore be interesting as a medication in a chronic therapy for the treatment of symptoms of epilepsy and other correlated severe disorders. - [0034]
Based on recent medical developments, therefore, the need exists for a synthesis method that is better than the existing ones and can overcome in particular the disadvantages of the processes that are present in the literature. Particularly important, in view of use in chronic therapies for children such as epilepsy and other severe disorders, it would be fundamentally important to identify a path for synthesis of the active ingredient fenfluramine or of isomers thereof and/or analogs thereof that does not entail the use of heavy metals and/or transition metals, which in a chronic therapy might accumulate in the body of the patients over the years, with severe consequences on health. - [0035]
More generally, it is desirable to identify a synthesis path that uses reagents from which (or from the transformation products of which) it is then possible to easily purify fenfluramine (or isomers and/or analogs thereof). - [0036]
It would be equally desirable to identify a synthesis path that comprises a small number of synthesis steps and uses reagents that are widely commercially available and easy to use. - [0037]
At the same time, the new identified synthesis path should avoid if possible the formation of byproducts.
EXAMPLES
- [0082]
The present invention is exemplified by, but not limited to, the following examples:
Example 1 – Synthesis of fenfluramine
- [0083]
A suspension of sodium hydroxide (34.62 g – 0.866 mol, 3.5 eq) in 170 mL of methanol, under mechanical agitation, receives the addition, drop by drop, over the course of 30 minutes, of a solution of ethylamine hydrochloride (70.59 g – 0.866 mol, 3.5 eq) in 165 mL of methanol, followed by 1-(3-trifluoromethyl)phenyl-propan-2-one (50 g – 0.247 mol). The mixture is left under agitation at 20°C for 4.5 hours, then cooling to 0°C is performed and a solution of sodium borohydride (9.36 g – 0.247 mol) in 19 mL of sodium hydroxide 1M in water is then added drop by drop, keeping the temperature below 10°C. The reaction is then left under agitation at 20°C for another 2 hours. Once the reaction is complete, 270 mL of methanol are removed at a reduced pressure at 40°C and then 200 mL of water are added and the mixture is extracted with heptane (200 mL). The aqueous phase is eliminated and the organic phase is washed with water (200 mL x 3). The organic phase is concentrated at 50°C at reduced pressure to yield free base fenfluramine as colorless oil. Yield: 72%; purity: 77% – as listed in test 3 of table A above.
Example 2 – Purification of fenfluramine
- [0084]
Purification of free base fenfluramine can be performed in two ways: - distillation of the free base
- crystallization of the fenfluramine hydrochloride salt
- [0085]
Depending on the degree of purity that is desired, both purification processes are performed in sequence (distillation first and then crystallization), or only one of the two purification processes is performed.
Example 2a – Distillation:
- [0086]
Free base fenfluramine (10 g), prepared as in Example 1, is distilled under reduced pressure with a distillation column of the Vigreux type: the distillation heads are eliminated, the fraction that is distilled at 89-90°C at 6 mmHg, which is the active ingredient fenfluramine (8.5 g) with a high degree of purity, is collected.
Example 2b – Conversion into hydrochloride salt and crystallization:
- [0087]
Crude fenfluramine, prepared as in Example 1, or purified fenfluramine as in Example 2a, is dissolved in 125 mL of ethyl acetate, and cooling is performed to 0°Celsius under agitation. 272 mL of a solution of 1M HCl in ethyl acetate are added drop by drop at 0°C. The precipitate that forms is filtered and washed with ethyl acetate (125 mL x 2) to yield approximately 55 g of solid fraction. The solid fraction is crystallized by 2-butanol (260 mL), keeping the solid for 22 hours at 3°C under slow agitation before filtering it. Filtering is performed and washing is performed with cold 2-butanol. The solid fraction, fenfluramine hydrochloride, is dried in a vacuum stove, yielding 51.7 g of product. A DSC of the resulting product is shown in Figure 3 .
CLIP
- Synthetic Method of Dexfenfluramine hydrochloride
- (CAS NO.: ), with its systematic name of (S)-N-Ethyl-alpha-methyl-m-(trifluoromethyl)phenethylamine hydrochloride, could be produced through many synthetic methods.Following is one of the synthesis routes:
![Systematic Method of Dexfenfluramine hydrochloride]() The action of d-camphoric acid on (rac)-fenfluramine (I) affords the camphorate of (+)-fenfluramine (II). After purification of this salt by crystallization, sodium hydroxide in methylene chloride is added, forming (+)-fenfluramine (III) after removal of camphoric acid. Finally, the action of hydrogen chloride in methyl cyclohexane on (+)-fenfluramine produces the corresponding salt: (+)-fenfluramine hydrochloride.
The action of d-camphoric acid on (rac)-fenfluramine (I) affords the camphorate of (+)-fenfluramine (II). After purification of this salt by crystallization, sodium hydroxide in methylene chloride is added, forming (+)-fenfluramine (III) after removal of camphoric acid. Finally, the action of hydrogen chloride in methyl cyclohexane on (+)-fenfluramine produces the corresponding salt: (+)-fenfluramine hydrochloride.
 The action of d-camphoric acid on (rac)-fenfluramine (I) affords the camphorate of (+)-fenfluramine (II). After purification of this salt by crystallization, sodium hydroxide in methylene chloride is added, forming (+)-fenfluramine (III) after removal of camphoric acid. Finally, the action of hydrogen chloride in methyl cyclohexane on (+)-fenfluramine produces the corresponding salt: (+)-fenfluramine hydrochloride.
The action of d-camphoric acid on (rac)-fenfluramine (I) affords the camphorate of (+)-fenfluramine (II). After purification of this salt by crystallization, sodium hydroxide in methylene chloride is added, forming (+)-fenfluramine (III) after removal of camphoric acid. Finally, the action of hydrogen chloride in methyl cyclohexane on (+)-fenfluramine produces the corresponding salt: (+)-fenfluramine hydrochloride.PAPER
https://www.designer-drug.com/pte/12.162.180.114/dcd/chemistry/fenfluramine.html
Fenfluramine 1 is the active ingredient of a obesity drug acting on the digestion of carbohydrates, the activity being restricted mainly to the S enantiomer [1, 2], which can be obtained by separation of the diastereoisomers [3] or by preferential crystallisation of derivates, which were identified of being conglomerates [4]. Only two syntheses of optical active fenfluramine have been described until now: one by stereoselective reduction of the imine derived from the ketone 2 and (R) or (S)-alpha-phenylethylamine [5], the other starting from (S)-alanine [6]. Two recent publications [7, 8] about the synthesis of (S)-fenfluramine via the intermediate alcohol (S)-3 (scheme 1) made us publish our previous results [9]. Through yeast reduction of the ketone 2, the authors obtain the alcohol (S)-3, the configuration of which they inverse in three steps. The alcohol (R)-3, via the intermediate tosylate (R)-4a and further the azide (S)-5 leads to the amine (S)-6 after reduction and finally to the (S)-fenfluramine (S)-1 after reductive amination in presence of acetaldehyde:
The (S)-fenfluramine is such obtained in 7 steps starting from the alcohol (S)-3 or in 4 steps from the alcohol (R)-3.
In this article we present a new way of preparing the two enantiomers of the alcohol 3, a new two step synthesis of (S)-fenfluramine starting from the alcohol (R)-3, a one step synthesis of (S)-fenfluramine starting from the azide (S)-5, which doesn’t pass over the intermediate primary amine (S)-6 and finally a much faster process (3 steps) of preparing (S)-fenfluramine starting from the alcohol (S)-3.
Results and discussion
Synthesis of 1-[3-(trifluoromethyl)phenyl]propan-2-ol (R)-3
The racemic alcohol is seldom mentioned. It’s one of the metabolites of fenfluramine in the human body, secreted in urine [10]. It can also be obtained by metabolic transformations of an oxime by different kinds of microorganisms [11]. It was used as an intermediate for the synthesis of a family of anorexics [12] and a family of antispasmodic and psychotherapeutic agents [13]. It was obtained by the reaction of methyloxirane 7 with the magnesium compound 8, with a yield of 50%. This same reaction was described earlier as being little regioselective [14], a fact we observed too [9].
The only synthesis of the optically active alcohol 3 is the reduction of the ketone 2 with yeast as described above. One abtains the S enantiomer, the R enantiomer is obtained by inversion.
On our part we used the condensation of the commercial [15] methyloxirane (R)-7, of which many syntheses are known [16], with the magnesium compound 8 and cuprous chloride [17, 18].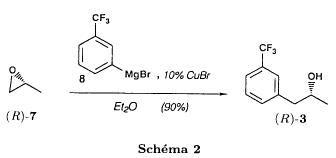
The yield is about 90% and the reaction very selective (purity GC: 93%). The optical purity of the methyloxirane 7 was determined by 1H-NMR in presence of the europium complex Eu(hfc)3 [19]. The optical purity of the alcohol (R)-3 was obtained by 1H-NMR and HPLC over silica of the Mosher derivate [20]. The comparison of these values show that the chiral centre is preserved. This procedure has the advantage of allowing us the preparation of the alcohol (S)-3 with the same reaction, because the methyloxirane (S)-7 is also commercially available and multiple syntheses are known [21].
Two step synthesis of the fenfluramine (S)-1 starting from the alcohol (R)-3
With the goal of obtaining the simplest procedure we have studied at first the transformation of the alcohol (R)-3 into fenfluramine (S)- 1 in two steps via the intermediate of the easily obtained sulfonates (R)-4: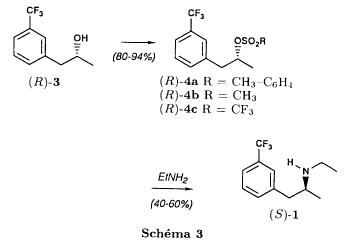
The substitution of the mesitylate (R)-4b and the tosylate (R)-4a with ethylamine was realised with medium yields always between 40 and 50% in spite of the large number of conditions tested: solvents (DMSO, DMF, ethanol, ethylamine), different dilutions (in proportions from 1 to 5) and temparatures from 50 to 160°C (with different times of contact). With the triflate (R)-4c the yield of the substitution is 60% but under non comparable conditions (-20°C in acetonitrile) because of its higher reactivity. In all cases the non aminated, and thus easily separated, byproducts are mainly the alkenes 9, 10Z and 10E (10E >> 10Z > 9).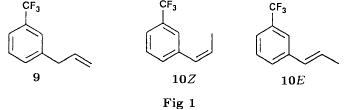
The enantiomeric purity of the amine (S)-1 is analysed by HPLC chromatography through silica of the camphanylated derivate [22] and compared to the previously analysed alcohol (R)-3: we have thus shown that the optical centre is conserved during the nucleophilic substitution. One had indeed to fear that due to the participation of the aromatic ring as neighbour group there could be partial or complete racemisation with an phenonium ion as intermediate. With the results obtained, which match with the literature [23-26], one can suppose that the trifluoromethyl group in meta position is sufficiently deactivating the aromatic ring in order to prevent participation in the substitution. We probably have thus in our case a pure nucleophilic SN2 substitution in competition with an elimination reaction. We believe that this elimination reaction is due to the simultaneous nucleophilic and basic properties of the ethylamine.
Although the yields are medium, this method has the advantage of being relatively fast because it permits to prepare fenfluramine (S)-1 starting from the alcohol (R)-2 in two steps instead of four [7, 8]. As far as we know it was never mentioned in literature.
Synthesis of fenfluramine (S)-1 from 2-azido-1-[3-(trifluoromethyl)phenyl]propane (S)-5
The substitution of the mesylate (R)-4b by sodium azide (scheme 4), an only slightly basic nucleophile compared to ethylamine, forms no elimination side products. One obtains the optically pure azide (S)-5 with a yield of 95%.
The enantiomeric purity couldn’t be directly analysed on the azide (S)-5. Only for analytical purposes did we reduce it into the amine (S)- 6. Among the numerous methods for the reductions of azides to amines mentioned in the literature [27] we chose the catalytic hydrogenation with 5% Pd on calcium carbonate at standard temperature and pressure [27f]. The HPLC analysis through silica column of the champhanyl derivate [22] of the amine (S)-6 such obtained shows that the enantiomeric centre was totally inverted during the substitution when compared to the enantiomeric purity of the alcohol (R)-3.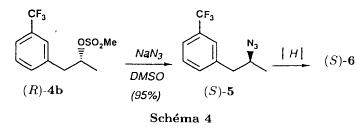
The reductive amination of the amine 6 in presence of acetaldehyde is known for a long time [28]. It was used recently in the works listed in the introduction [7, 8]. On our part, we propose another synthetic route for fenfluramine (S)-1 starting from the azide (S)-5 which does not go via the primary amine (S)-6 (Schema 5).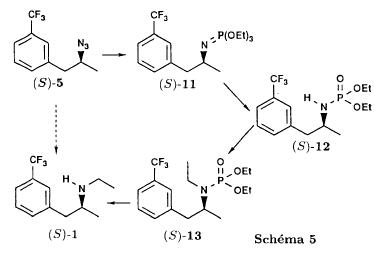
The reaction of Staudinger, reacting a stoichiometric quantity of triethylphosphite on the azide (S)-5 in THF at room temperature [29], gives quantitative yields of the phosphorimide 11 in 48 hours. It’s total conversion into the phosphoramide 13, by reacting with ethyl iodide [30] could not be realised [9]. We always obtained different mixtures of the phosphoramides 12 and 13 (referential compounds prepared from the amines 6 and 1). We also noted that the phosphorimide 11 can’t be isolated. When the solvent is evaporated, a partly transformation into the phosphoramide 12 takes place. This transformation is completed in less then 2h by simple heating to 100°C under argon after evaporation of the solvent. Because the phosphorimides are strongly basic compounds, we believe that an intramolecular arrangement of the phosphorimide, pictured in Schema 6, takes place.
Having the phosphoramide 12, we investigated the alkylation into the phosphoramide 13 in DMF at room temperature [31, 32]: one deprotonates with sodium hydride then alkylates with diethyl sulfate. After treatment with hydrogen bromide [33], one obtains fenfluramine 1 with a yield of 85% and a purity of 97% (GC).
With the goal of simplifying the reaction scheme by avoiding the isolation of the intermediates we have again studied the transformation 5 -> 11 -> 12 in DMF (Schema 7). First, we noted that the reaction of Staudinger can be directly realised in this solvent. Thereafter we pinned down the transformation of the phosphorimide 11 into the phosphoramide 12 by reaction with water [34]. One then proceeds as described above. The transformation is thus performed without isolation of a single intermediate with a yield of 83%.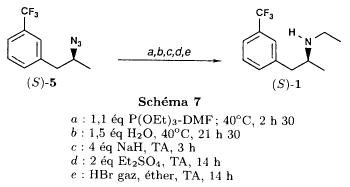
HPLC analysis on silica column of the camphanyl derivate of the amine (S)-1 [22] shows that the optical centre is conserved during the whole transformation.
Synthesis via the intermediate 2-chloro-1-[3-(trifluoromethyl)phenyl]propane 14
The yeast reduction of the ketone 2 gives the alcohol (S)-3, of which the authors have inverted the configuration to get the pharmacological active S enantiomer of fenfluramine [7, 8]. Independent research, using the epoxidation method of Sharpless [9, 35] lead us too to the alcohol (S)- 3 which we tried to convert into fenfluramine (S)-1 using a different method. The reaction scheme we kept uses the chloride 14 and proceeds via two inversions of the optical centre (scheme 8). Not owning enough alcohol (S)-3 during the studies, we tested the principle starting with the alcohol (R)-3, produced earlier, and studied the transformation into the azide (R)-5 (scheme 8), the latter being able to lead to (R)-fenfluramine using different methods, like the one outlined above: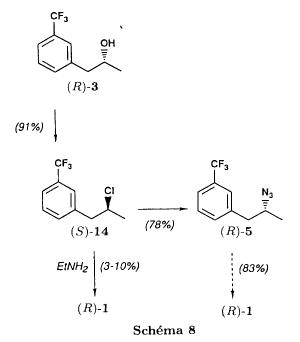
It is well known that the action of thionylchloride on an optically active alcohol gives the corresponding chlorine derivate, with inversion of the configuration in presence of bases and with retention of the configuration in the other case. We have performed the reaction with a catalytical amount of pyridine. One thus obtains the chloride (S)-14 with 91% yield and a purity of 91% (GC): it contains 9% of the elimination products 9, 10Z and 10E which are not separable by chromatography on silica.
The direct substitution of the chloride 14 with ethylamine with similar conditions to those used for the mesylate (R)-4b (EtNH2, DMSO, 110°C, 5h30 or EtNH2 (solvent and reactant), 140°C, 5h), gives mainly the elimination products. The yield of fenfluramine is below 10%.
By action of sodium azide in DMSO, on the other hand, one obtains the azide (R)-5 with a yield of 78%, the elimination products formed here or in the last step can be removed by chromatography on silica. HPLC analysis on silica of the camphanyl derivate of the amine (R)-6 [22] obtained by catalytic reduction of the azide (R)-5 has confirmed the double inversion without racemisation after comparison with the starting alcohol (R)-3. Then the fenfluramine (R)-1 is prepared without racemisation with a 83% yield starting from the azide (R)-5 like detailed above.
This procedure with two inversions allows to transform the alcohol 3 in the azide 5 with the same configuration in two steps with a global yield (non optimised) of 70% and without racemisation. It’s thus preferred over the recently published one [7, 8], which needs 5 steps for a lower global yield (55%) and in addition features an epimerisation of 10% [8]. It’s a promising way to fenfluramine (S)-1 starting from the alcohol (S)- 3.
References
- ^ “Fintepla- fenfluramine solution”. DailyMed. Retrieved 8 January 2021.
- ^ Jump up to:a b c d e f g h i j k l m n o p q “FDA Approves New Therapy for Dravet Syndrome”. U.S. Food and Drug Administration (FDA) (Press release). 25 June 2020. Retrieved 25 June 2020.
![]() This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ Jump up to:a b c d “Fintepla EPAR”. European Medicines Agency. Retrieved 8 January2021.
- ^ Jump up to:a b c d Richard C. Dart (2004). Medical Toxicology. Lippincott Williams & Wilkins. pp. 874–. ISBN 978-0-7817-2845-4. Archived from the original on 2018-05-09.
- ^ Jump up to:a b James O’Donnell (Pharm. D.); Gopi Doctor Ahuja (2005). Drug Injury: Liability, Analysis, and Prevention. Lawyers & Judges Publishing Company. pp. 276–. ISBN 978-0-913875-27-8.
- ^ Integrating the Neurobiology of Schizophrenia. Academic Press. 27 February 2007. pp. 142–. ISBN 978-0-08-047508-0.
- ^ The Pharmacology of Corticotropin-releasing Factor (CRF). Effects on Sensorimotor Gating in the Rat. 2006. pp. 12–. ISBN 978-0-549-53661-1.
- ^ Christian P. Muller; Barry Jacobs (30 December 2009). Handbook of the Behavioral Neurobiology of Serotonin. Academic Press. pp. 630–. ISBN 978-0-08-087817-1.
- ^ Vickers SP, Dourish CT, Kennett GA (2001). “Evidence that hypophagia induced by d-fenfluramine and d-norfenfluramine in the rat is mediated by 5-HT2C receptors”. Neuropharmacology. 41 (2): 200–9. doi:10.1016/s0028-3908(01)00063-6. PMID 11489456. S2CID 23374227.
- ^ Roth, B. L. (2007). “Drugs and Valvular Heart Disease”. New England Journal of Medicine. 356 (1): 6–9. doi:10.1056/NEJMp068265. PMID 17202450.
- ^ Rothman, R. B.; Baumann, M. H. (2009). “Serotonergic Drugs and Valvular Heart Disease”. Expert Opinion on Drug Safety. 8 (3): 317–329. doi:10.1517/14740330902931524. PMC 2695569. PMID 19505264.
- ^ Jump up to:a b Dahl, C. F.; Allen, M. R.; Urie, P. M.; Hopkins, P. N. (2008). “Valvular Regurgitation and Surgery Associated with Fenfluramine Use: An Analysis of 5743 Individuals”. BMC Medicine. 6: 34. doi:10.1186/1741-7015-6-34. PMC 2585088. PMID 18990200.
- ^ Jump up to:a b c d e f g h i j k l m Donald G. Barceloux (3 February 2012). Medical Toxicology of Drug Abuse: Synthesized Chemicals and Psychoactive Plants. John Wiley & Sons. pp. 255–262. ISBN 978-1-118-10605-1. Archived from the original on 9 May 2018.
- ^ Mann SC, Caroff SN, Keck PE, Lazarus A (20 May 2008). Neuroleptic Malignant Syndrome and Related Conditions. American Psychiatric Pub. pp. 111–. ISBN 978-1-58562-744-8.
- ^ Jump up to:a b c d e f g h i Rothman RB, Clark RD, Partilla JS, Baumann MH (2003). “(+)-Fenfluramine and its major metabolite, (+)-norfenfluramine, are potent substrates for norepinephrine transporters”. J. Pharmacol. Exp. Ther. 305 (3): 1191–9. doi:10.1124/jpet.103.049684. PMID 12649307. S2CID 21164342.
- ^ Jump up to:a b c d e f g Setola V, Hufeisen SJ, Grande-Allen KJ, Vesely I, Glennon RA, Blough B, Rothman RB, Roth BL (2003). “3,4-methylenedioxymethamphetamine (MDMA, “Ecstasy”) induces fenfluramine-like proliferative actions on human cardiac valvular interstitial cells in vitro”. Mol. Pharmacol. 63 (6): 1223–9. doi:10.1124/mol.63.6.1223. PMID 12761331. S2CID 839426.
- ^ Nestler, E. J. (2001). Molecular Neuropharmacology: A Foundation for Clinical Neuroscience. McGraw-Hill.
- ^ Giuseppe Di Giovanni; Vincenzo Di Matteo; Ennio Esposito (2008). Serotonin-dopamine Interaction: Experimental Evidence and Therapeutic Relevance. Elsevier. pp. 393–. ISBN 978-0-444-53235-0.
- ^ Fitzgerald LW, Burn TC, Brown BS, Patterson JP, Corjay MH, Valentine PA, Sun JH, Link JR, Abbaszade I, Hollis JM, Largent BL, Hartig PR, Hollis GF, Meunier PC, Robichaud AJ, Robertson DW (2000). “Possible role of valvular serotonin 5-HT(2B) receptors in the cardiopathy associated with fenfluramine”. Mol. Pharmacol. 57 (1): 75–81. PMID 10617681.
- ^ Jump up to:a b c Rothman RB, Baumann MH, Dersch CM, Romero DV, Rice KC, Carroll FI, Partilla JS (January 2001). “Amphetamine-type central nervous system stimulants release norepinephrine more potently than they release dopamine and serotonin”. Synapse. 39 (1): 32–41. doi:10.1002/1098-2396(20010101)39:1<32::AID-SYN5>3.0.CO;2-3. PMID 11071707.
- ^ Rothman RB, Baumann MH (2002). “Therapeutic and adverse actions of serotonin transporter substrates”. Pharmacol. Ther. 95 (1): 73–88. doi:10.1016/s0163-7258(02)00234-6. PMID 12163129.
- ^ Connolly, H. M.; Crary, J. L.; McGoon, M. D.; Hensrud, D. D.; Edwards, B. S.; Edwards, W. D.; Schaff, H. V. (1997). “Valvular Heart Disease Associated with Fenfluramine-Phentermine”. New England Journal of Medicine. 337 (9): 581–588. doi:10.1056/NEJM199708283370901. PMID 9271479.
- ^ Weissman, N. J. (2001). “Appetite Suppressants and Valvular Heart Disease”. The American Journal of the Medical Sciences. 321 (4): 285–291. doi:10.1097/00000441-200104000-00008. PMID 11307869. S2CID 46466276.
- ^ FDA September 15, 1997. FDA Announces Withdrawal Fenfluramine and Dexfenfluramine (Fen-Phen) Archived 2013-07-19 at the Wayback Machine
- ^ “Drugs banned in India”. Central Drugs Standard Control Organization, Dte.GHS, Ministry of Health and Family Welfare, Government of India. Archived from the original on 2015-02-21. Retrieved 2013-09-17.
- ^ “FDA Approves FINTEPLA (fenfluramine) for the Treatment of Seizures Associated with Dravet Syndrome” (Press release). Zogenix Inc. 25 June 2020. Retrieved 25 June 2020 – via GlobeNewswire.
- ^ “Fenfluramine Orphan Drug Designations and Approvals”. U.S. Food and Drug Administration (FDA). 24 December 1999. Retrieved 25 June 2020.
- ^ “Fenfluramine Orphan Drug Designations and Approvals”. U.S. Food and Drug Administration (FDA). 24 December 1999. Retrieved 25 June 2020.
- ^ “Fintepla: Pending EC decision”. European Medicines Agency (EMA). 16 October 2020. Retrieved 16 October 2020. Text was copied from this source which is © European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^ Jump up to:a b John Calvin M. Brust (2004). Neurological Aspects of Substance Abuse. Butterworth-Heinemann. pp. 117–. ISBN 978-0-7506-7313-6.
- ^ United States. Congress. Senate. Select Committee on Small Business. Subcommittee on Monopoly and Anticompetitive Activities (1976). Competitive problems in the drug industry: hearings before Subcommittee on Monopoly and Anticompetitive Activities of the Select Committee on Small Business, United States Senate, Ninetieth Congress, first session. U.S. Government Printing Office. pp. 2–.
- ^ Jump up to:a b Drug Addiction II: Amphetamine, Psychotogen, and Marihuana Dependence. Springer Science & Business Media. 27 November 2013. pp. 258–. ISBN 978-3-642-66709-1.
- ^ Drug Addiction II: Amphetamine, Psychotogen, and Marihuana Dependence. Springer Science & Business Media. 27 November 2013. pp. 249–. ISBN 978-3-642-66709-1.
- ^ “Zogenix ends enrolment in second Phase lll trial of ZX008”. Drug Development Technology. Kable. 1 February 2018. Archived from the original on 2018-02-04. Retrieved 3 February 2018.
- ^ “A Two-Part Study to Investigate the Dose-Ranging Safety and Pharmacokinetics, Followed by the Efficacy and Safety of ZX008 (Fenfluramine Hydrochloride) Oral Solution as an Adjunctive Therapy in Children ≥2 Years Old and Young Adults With Dravet Syndrome”. ClinicalTrials.gov. Archived from the original on 29 September 2017. Retrieved 9 May 2018.
- ^ “Zogenix drug cuts seizure rate in patients with severe epilepsy”. statnews.com. 29 September 2017. Archived from the original on 30 April 2018. Retrieved 9 May 2018.
Further reading
- Gustafsson BI, Tømmerås K, Nordrum I, Loennechen JP, Brunsvik A, Solligård E, Fossmark R, Bakke I, Syversen U, Waldum H (March 2005). “Long-term serotonin administration induces heart valve disease in rats”. Circulation. 111 (12): 1517–22. doi:10.1161/01.CIR.0000159356.42064.48. PMID 15781732.
- Welch, J. T.; Lim, D. S. (2007). “The Synthesis and Biological Activity of Pentafluorosulfanyl Analogs of Fluoxetine, Fenfluramine, and Norfenfluramine”. Bioorganic & Medicinal Chemistry. 15 (21): 6659–6666. doi:10.1016/j.bmc.2007.08.012. PMID 17765553.
External links
- “Fenfluramine”. Drug Information Portal. U.S. National Library of Medicine.
- “Fenfluramine hydrochloride”. Drug Information Portal. U.S. National Library of Medicine.
- Inchem.org – Fenfluramine hydrochloride
| Clinical data | |
|---|---|
| Trade names | Fintepla |
| Other names | ZX008 |
| AHFS/Drugs.com | Professional Drug Facts |
| MedlinePlus | a620045 |
| License data | US DailyMed: Fenfluramine |
| Pregnancy category | AU: B2 |
| Routes of administration | By mouth |
| ATC code | A08AA02 (WHO) N03AX26 (WHO) |
| Legal status | |
| Legal status | US: Schedule IV [1][2]EU: Rx-only [3] |
| Pharmacokinetic data | |
| Elimination half-life | 13–30 hours[4] |
| Identifiers | |
| IUPAC name[show] | |
| CAS Number | 458-24-2 |
| PubChem CID | 3337 |
| IUPHAR/BPS | 4613 |
| DrugBank | DB00574 |
| ChemSpider | 3220 |
| UNII | 2DS058H2CF |
| KEGG | D07945 C06996 |
| ChEBI | CHEBI:5000 |
| ChEMBL | ChEMBL87493 |
| CompTox Dashboard (EPA) | DTXSID4023044 |
| ECHA InfoCard | 100.006.616 |
| Chemical and physical data | |
| Formula | C12H16F3N |
| Molar mass | 231.262 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| Chirality | Racemic mixture |
| SMILES[hide]FC(F)(C1=CC(CC(C)NCC)=CC=C1)F | |
| InChI[hide]InChI=1S/C12H16F3N/c1-3-16-9(2)7-10-5-4-6-11(8-10)12(13,14)15/h4-6,8-9,16H,3,7H2,1-2H3 Key:DBGIVFWFUFKIQN-UHFFFAOYSA-N |
CLIP
http://www.inchem.org/documents/pims/pharm/pim938.htm
////////////Fenfluramine, 塩酸フェンフルラミン , dravet, AHR-3002, ZX-008, Fintepla
CCNC(C)CC1=CC(=CC=C1)C(F)(F)F.Cl
PATENT
https://patents.google.com/patent/US20170174613A1/en
- [0093]
Many general references providing commonly known chemical synthetic schemes and conditions useful for synthesizing the disclosed compounds are available (see, e.g., Smith and March, March’s Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, Fifth Edition, Wiley-Interscience, 2001; or Vogel, A Textbook of Practical Organic Chemistry, Including Qualitative Organic Analysis, Fourth Edition, New York: Longman, 1978). - [0094]
Compounds as described herein can be purified by any purification protocol known in the art, including chromatography, such as HPLC, preparative thin layer chromatography, flash column chromatography and ion exchange chromatography. Any suitable stationary phase can be used, including normal and reversed phases as well as ionic resins. In certain embodiments, the disclosed compounds are purified via silica gel and/or alumina chromatography. See, e.g., Introduction to Modern Liquid Chromatography, 2nd Edition, ed. L. R. Snyder and J. J. Kirkland, John Wiley and Sons, 1979; and Thin Layer Chromatography, ed E. Stahl, Springer-Verlag, New York, 1969. - [0095]
During any of the processes for preparation of the subject compounds, it may be necessary and/or desirable to protect sensitive or reactive groups on any of the molecules concerned. This may be achieved by means of conventional protecting groups as described in standard works, such as J. F. W. McOmie, “Protective Groups in Organic Chemistry”, Plenum Press, London and New York 1973, in T. W. Greene and P. G. M. Wuts, “Protective Groups in Organic Synthesis”, Third edition, Wiley, New York 1999, in “The Peptides”; Volume 3 (editors: E. Gross and J. Meienhofer), Academic Press, London and New York 1981, in “Methoden der organischen Chemie”, Houben-Weyl, 4th edition, Vol. 15/1, Georg Thieme Verlag, Stuttgart 1974, in H.-D. Jakubke and H. Jescheit, “Aminosauren, Peptide, Protein”, Verlag Chemie, Weinheim, Deerfield Beach, and Basel 1982, and/or in Jochen Lehmann, “Chemie der Kohlenhydrate: Monosaccharide and Derivate”, Georg Thieme Verlag, Stuttgart 1974. The protecting groups may be removed at a convenient subsequent stage using methods known from the art. - [0096]
The subject compounds can be synthesized via a variety of different synthetic routes using commercially available starting materials and/or starting materials prepared by conventional synthetic methods. A variety of examples of synthetic routes that can be used to synthesize the compounds disclosed herein are described in the schemes below.
Example 11. Fenfluramine Nomenclature & Structure
- [0097]
Chemical Abstract Service (CAS) Registry Number (RN): 404-82-0 (HCl Salt), 458-24-2 (Parent Free Base) - [0098]
Chemical Name: N-ethyl-α-methyl-3-(trifluoromethyl)-benzeneethanamine hydrochloride (1:1). Other Names: Fenfluramine HCl, DL-Fenfluramine, (±)-Fenfluramine - [0099]
Structure of Hydrochloride Salt: - [0100]
Stereochemistry: Fenfluramine HCl has one chiral center and is being developed as the racemate and contains dexfenfluramine and levofenfluramine - [0101]
Molecular Formula of hydrochloride salt: C12H16F3N.HCl - [0102]
Molecular Mass/Weight: 267.72 g/mol
2. General Properties
- [0103]
Table 1 summarizes the chemical and physical properties of Fenfluramine HCl. - TABLE 1 General Properties of Fenfluramine HCl Drug Substance Property Result Appearance (color, White to off-white powder physical form) DSC (melting 170° C. (melt/sublimation) point)a TGA Onset 147° C. 0.03% at 150° C. 91% at 220° C. (evaporation) pKa (water) 10.15-10.38 Solubility (mg/mL) Resultant pH 25° C. 37° C. Solubility pH 6.69 (water) 54.13 71.22 (Aqueous) pH 1.73 buffer 25.34 53.68 pH 3.43 buffer 29.50 61.97 pH 6.41 buffer 37.42 95.60 0.9% NaCl (water) 22.98 — Solvent Solubility 25° C. (mg/mL) Solubility (Organic Ethanol 150 Solvents) Dichloromethane 30-35 Ethyl Acetate, 1-5 mg Tetrahydrofuran, Toluene, Acetonitrile UV Absorption Maxima: 210, 265 nm Solution pH (water) 6.69 Hygroscopicity @30% RH: ~0.05% (Dynamic Vapor @60% RH: ~0.07% Sorption (DVS) @90% RH: ~0.20%a) Polymorphism Fenfluramine HCl has been consistently isolated as a single crystalline Form 1 as determined by DSC and x-ray powder diffraction (XRPD) Solvation/Hydration Fenfluramine HCl is isolated as a nonhydrated, nonsolvated solid Solution Stability 8 weeks @ pH 6.7 phosphate buffer medium at 40° C. and 60° C. using concentrations of 0.5, 2.5 and 5.0 mg/ml. All conditions, no new impurities >0.1% by HPLC. Solid Stability 8 weeks @ 40° C., 60° C. and 80° C. 7 days at 150° C. All conditions, no new impurities >0.1% by HPLC.
3. Synthesis of Fenfluramine Drug Substance
- [0104]
Scheme 3.1 shows a 2-step route of synthesis used to manufacture initial clinical supplies of Fenfluramine HCl from ketone (2). The batch size is 4 kg performed in laboratory glassware (kilo lab). No chromatography is required and the process steps are amenable to scale-up. In process 1 there is one isolated intermediate Fenfluramine Free Base (1) starting from commercially supplied 1-(3-(trifluoromethyl)phenyl) acetone (Ketone 2). All steps are conducted under cGMPs starting from Ketone (2). - [0105]
Scheme 3.2 shows a 4-step route of synthesis to Fenfluramine HCl that can be used for commercial supply. Route 2 utilizes the same 2-step process used by Route 1 to convert Ketone (2) to Fenfluramine HCl with the exception that Ketone (2) is synthesized under cGMP conditions starting from 3-(Trifluoromethyl)-phenyl acetic acid (Acid 4). Bisulfate Complex (3) is an isolatable solid and can be purified before decomplexation to Ketone (2). In-situ intermediates which are oils are shown in brackets. Batch sizes of 10 Kg are performed. Commercial batch sizes of 20 kg are performed in fixed pilot plant equipment. Steps 1-2 of Scheme 3.2 to manufacture Ketone (2) have been demonstrated on a 100 g scale to provide high purity ketone (2) of >99.8% (GC & HPLC). Conversion of Ketone (2) to Fenfluramine using either Route 1 or 2 has provided similar purity profiles. - Starting materials are designated by enclosed boxes. Bracketed and non bracketed compounds respectively indicate proposed in-situ and isolated intermediates. NMI=N-Methyl Imidazole.
4.1. Narrative Description (Route 1)
- [0106]
Step 1: Reductive Amination (Preparation of Fenfluramine Free Base 1) - [0107]
A solution of ethylamine, water, methanol, and 1-(3-(trifluoromethyl)phenyl) acetone (Ketone 2) was treated with sodium triacetoxyborohydride and stirred for 16 h at 25° C. at which time HPLC analysis (IPC-1; In Process Control No. 1) showed the reaction to be complete and sodium hydroxide solution was added until pH>10. Toluene was added and the phases separated, and the aqueous phase (IPC-2) and organic phase (IPC-3) are checked for remaining Fenfluramine and Fenfluramine alcohol and the organic phase was reduced. Purified water was added and the pH adjusted to <2 using conc. HCl and the phases were separated. The aqueous phase was washed with toluene and the toluene phase (IPC-4) and the aqueous phase (IPC-5) was checked for Fenfluramine and Fenfluramine alcohol content. The aqueous phase containing product is pH adjusted to >10 using sodium hydroxide solution. The basic aqueous phase was extracted with MTBE until removal of Fenfluramine from the aqueous phase was observed by HPLC (<0.5 mg/ml) (IPC-6). The organic phase was dried over sodium sulfate and filtered. The filtrate was concentrated in vacuo to give the intermediate product Fenfluramine Free Base 1 as a pale yellow oil tested per specifications described herein which showed by NMR the material to contain 2.93% toluene giving an active yield of 88.3% with a purity of 98.23% by HPLC (0.67% Fenfluramine alcohol). - [0108]
Step 2: Salt Formation (Preparation of Fenfluramine HCl) - [0109]
To a flask was charged ethanol and acetyl chloride. The solution was stirred slowly overnight before ethyl acetate was added. The HCl in ethyl acetate solution formed was polish filtered into a clean carboy and retained for later use. To a vessel was added Fenfluramine free base 1 and MTBE. The Fenfluramine solution in MTBE was collected in two carboys before the vessel was cleaned and checked for particulate residue. The Fenfluramine solution was polish filtered into a vessel and cooled and HCl in ethyl acetate solution was added giving a final pH of 6-7. The batch was stirred for 1 h and filtered. The product was dried under vacuum at 40° C. The product (96.52% yield) was tested per IPC-7 had a purity of 99.75% by HPLC and GC headspace analysis showed MTBE (800 ppm) and EtOAc (150 ppm) to be present. The product was then tested per specifications shown herein.
4.2. Narrative Description (Route 2)
- [0110]
Step 1: Preparation of Ketone Bisulfite Adduct - [0111]
Procedure: Charge acetic anhydride, (2.8 vol, 3.0 wt, 5.0 eq.) to a vessel and commence stirring. Cool the solution to −5 to 5° C., targeting −4° C. Charge 1-methylimidazole, (0.2 vol, 0.21 wt, 0.5 eq.) to the mixture at −5 to 5° C. Caution: very exothermic. If necessary, adjust the temperature to 0 to 5° C. Charge ZX008 acid, (1.00 wt, 1.0 eq.) to the mixture at 0 to 5° C. Caution: exothermic. Stir the mixture at 0 to 5° C. until ≦2.1% area ZX008 acid by HPLC analysis, typically 7 to 9 hours. Charge 15% w/w sodium chloride solution (2.0 vol) to the mixture at 0 to 5° C., 60 to 90 minutes. Caution: very exothermic which will be slightly delayed. Warm the mixture to 18 to 23° C. over 45 to 60 minutes and continue stirring for a further 30 to 45 minutes at 18 to 23° C. Charge TBME, (5.0 vol, 3.7 wt) to the mixture and stir for 10 to 15 minutes at 18 to 23° C. Separate the aqueous layer and retain the organic layer. Back-extract the aqueous layer with TBME, (2×3.0 vol, 2×2.2 wt) at 18 to 23° C. retaining each organic layer. Adjust the pH of the combined organic layer to pH 6.5 to 9.0, targeting 7.0 by charging 20% w/w sodium hydroxide solution (5.3 to 8.3 vol) at 18 to 23° C. Caution: exothermic. Separate the aqueous layer and retain the organic layer. Wash the organic layer with 4% w/w sodium hydrogen carbonate solution (2×3.0 vol) at 18 to 23° C. Determine the residual ZX008 acid content in the organic layer by HPLC analysis, pass criterion ≦0.10% area ZX008 acid. Wash the organic layer with purified water, (2×3.0 vol) at 18 to 23° C. Concentrate the organic layer under reduced pressure to ca. 2 vol at 40 to 45° C., targeting 43° C. - [0112]
Determine the w/w assay of ZX008 ketone (WIP) in the mixture by 1H-NMR analysis for information only and calculate the contained yield of ZX008 ketone (WIP) in the mixture. Note: This step can be removed from the process since the process is robust and consistently delivers 80 to 90% th yield. The achieved yield was factored into the charges of the subsequent steps. - [0113]
Charge n-heptane, (4.0 vol, 2.7 wt) to the mixture at 40 to 45° C., targeting 43° C. Concentrate the mixture to ca. 2 vol at 40 to 45° C., targeting 43° C. Determine the TBME content in the mixture by 1H-NMR analysis, (pass criterion ≦5.0% w/w TBME vs. ZX008 ketone). Charge n-heptane, (2.4 vol, 1.6 wt) at 40 to 45° C., targeting 43° C., vessel A. To vessel B, charge sodium metabisulfite, (0.82 wt, 0.88 eq.) at 18 to 23° C. To vessel B, charge a solution of sodium hydrogen carbonate, (0.16 wt, 0.4 eq.) in purified water, code RM0120 (2.0 vol) at 18 to 23° C. followed by a line rinse with purified water, code RM0120 (0.4 vol) at 18 to 23° C. Caution: gas evolution. Heat the contents of vessel B to 40 to 45° C., targeting 43° C. Charge the contents from vessel A to vessel B followed by a line rinse with n-heptane, (0.8 vol, 0.5 wt) at 40 to 45° C., targeting 43° C. Stir the mixture for 1 to 1.5 hours at 40 to 45° C., targeting 43° C. Charge n-heptane, code RM0174 (3.2 vol, 2.2 wt) to the mixture with the temperature being allowed to cool to 18 to 45° C. at the end of the addition. Cool the mixture to 18 to 23° C. at approximately constant rate over 45 to 60 minutes. Stir the mixture at 18 to 23° C. for 1.5 to 2 hours. - [0114]
Sample the mixture to determine the residual ZX008 ketone content by 1H-NMR analysis, (pass criterion ≦10.0% mol, target 5.0% mol ZX008 ketone vs. ZX008 ketone bisulfite adduct). Filter the mixture and slurry wash the filter-cake with n-heptane, (2×2.0 vol, 2×1.4 wt) at 18 to 23° C. Dry the solid at up to 23° C. until the water content is <10.0% w/w water by KF analysis according to AKX reagent. At least 16 hours. Determine the w/w assay of the isolated ZX008 ketone bisulfite adduct by 1H-NMR analysis and calculate the contained yield of ZX008 ketone bisulfite adduct. - [0115]
Yields and Profiles: The yield for the stage 1 Demonstration batch is summarized Table below. Input: 1700.0 g uncorr., acid, 99.50% area (QC, HPLC), 2-isomer not detected, 4-isomer 0.02% area, RRT1.58 (previously not observed) 0.48% area as per the preparative method. The analytical data is summarized in Table 1A below. - TABLE 1A Table for isolated yields for step 1 Demonstration batch Corr. % area Reference Corr. Yield % w/w (HPLC, number Input Output (% th)** (1H-NMR)* QC) Comments Batch A1 1700.0 g 1500.1 g 89.1 45.0 —.— Crude ketone as TBME sol. Batch A2 1500.1 g 1716.1 77.8 76.0 98.15 Bisulfite adduct only 67.3 Overall product
- [0116]
Step 2: Preparation of Ketone - [0117]
Procedure: Charge toluene, (5.0 vol, 4.3 wt), and purified water, (5.0 vol) to the vessel and commence stirring. If necessary, adjust the temperature to 18 to 23° C. and charge ZX008 ketone bisulfite adduct, (1.00 wt corrected for % w/w assay) to the mixture at 18 to 23° C. Charge 20% w/w sodium hydroxide solution to the mixture at 18 to 23° C. adjusting the pH of the mixture to pH 8.0 to 12.0, targeting 9.0 (0.5 to 1.0 vol). - Separate the lower aqueous layer and retain the top organic layer. Wash the organic layer with purified water, (3.0 vol) at 18 to 23° C. Concentrate the organic layer under reduced pressure to ca. 2 vol at 45 to 50° C., targeting 48° C. Charge methanol, (5.0 vol, 4.0 wt) to the mixture at 45 to 50° C., targeting 48° C. Re-concentrate the mixture under reduced pressure to ca. 2 vol at 45 to 50° C., targeting 48° C. Repeat steps 7 and 8 once before continuing with step 9. Cool the mixture to 18 to 23° C. Clarify the mixture into a tared, suitably-sized drum followed by a methanol (1.0 vol, 0.8 wt) line rinse at 18 to 23° C. Determine the w/w assay of ZX008 ketone (WIP) in the mixture by 1H-NMR analysis and calculate the contained yield of ZX008 ketone (WIP) in the mixture. Determine the toluene content in the mixture by 1H-NMR analysis.
- [0118]
Yields and Profiles: The yield for the step 2 Demonstration batch is summarized in Table 1B below. Input: 1200.0 g corr. Ketone bisulfite adduct, 76.0% w/w assay (NMR, using DMB as internal standard in d6-DMSO), (1.00 eq, 1.00 wt corr. for w/w assay) for input calculation. - TABLE 1B Table for isolated yields for step 2 Demonstration batch % w/w % area Corr. Corr. Corr. Yield (1H- (HPLC, Input Output (% th) NMR)* QC) Comments 1200.0 g 858.15 g 108.3 25.5 99.31 Purified ketone
- [0119]
Step 3: Preparation of Fenfluramine HCl Crude - [0120]
Procedure: Charge the ZX008 ketone (corr. for assay, 1.00 wt, 1.00 eq. isolated as solution in MeOH in stage 2) to a vessel. Charge methanol, code RM0036 (5.0 vol, 4.0 wt) to the mixture at 18 to 23° C. Cool the solution to 0 to 5° C. Charge 70 wt % aqueous ethylamine solution (1.3 vol, 1.6 wt, 4.0 eq) to the mixture at 0 to 10° C., over 15 to 30 minutes, followed by a line rinse with methanol (1.0 vol, 0.8 wt). Warm the mixture to 15 to 20° C. and stir the mixture for a further 60 to 70 minutes at 15 to 20° C. Adjust the mixture to 15 to 18° C. if required, targeting 15° C. Charge sodium triacetoxyborohydride (2.4 wt, 2.25 eq.) to the mixture in approximately 10 portions, keeping the mixture at 15 to 20° C., targeting 17° C. Addition time 1.5 to 2 hours. Caution: Exothermic. Stir the mixture at 15 to 20° C. until complete by HPLC analysis, pass criterion ≦3.0% area ZX008 ketone, typically 2 to 3 hours. Adjust the pH of the mixture to pH>12 by charging 20% w/w aqueous sodium hydroxide solution (5.0 to 6.0 vol) to the mixture at 15 to 40° C. Addition time 10 to 30 minutes. Caution: Exothermic. If necessary, adjust the temperature to 18 to 23° C. Extract the mixture with toluene (3×3.0 vol, 3×2.6 wt) at 18 to 23° C., retaining and combining the top organic layer after each extraction. Wash the combined organic layer with purified water, (1.0 vol) at 18 to 23° C. Heat the mixture to 40 to 50° C., targeting 48° C. Concentrate the mixture under reduced pressure at constant volume maintaining ca. 5 vol by charging the organic layer at approximately the same rate as the distillation rate at 40 to 50° C., targeting 48° C. Cool the mixture to 18 to 23° C. Charge purified water (10.0 vol) to the mixture at 18 to 23° C. Adjust the pH of the mixture to 0.1<pH<1.5 at 18 to 23° C. by charging concentrated hydrochloric acid, 0.5 vol. Do not delay from this step until neutralization. - [0121]
Separate the layers at 18 to 23° C. retaining the bottom aqueous layer. Wash the aqueous layer with toluene, (3.0 vol, 2.6 wt) at 18 to 23° C. retaining the aqueous layer. Adjust the pH of the aqueous layer to pH>12 by charging 20% w/w sodium hydroxide solution at 18 to 23° C. 0.8 to 0.9 vol. Caution: Exothermic. Charge TBME, code RM0002 (2.0 vol, 1.5 wt) to the basic aqueous layer. Separate the layers at 18 to 23° C. retaining the top organic layer. Back-extract the aqueous layer with TBME (2×2.0 vol, 2×1.5 wt) at 18 to 23° C. retaining the organic layers. Wash the combined organic layer with purified water, (2×1.0 vol) at 18 to 23° C. Concentrate the combined organic layers under reduced pressure at 40 to 50° C., targeting 48° C. to ca. 3 vol. Determine the residual toluene content of the mixture by 1H-NMR analysis. Sample for determination of residual water content by KF analysis, AKX reagent. Charge TBME (8.7 vol, 6.4 wt) to the mixture at 40 to 50° C. Cool the solution to 0 to 5° C., targeting 2° C. Charge concentrated hydrochloric acid (0.54 vol, 0.46 wt) maintaining the temperature <15° C. Caution: Exothermic. Line rinse with TBME (1.0 vol, 0.7 wt). If necessary, adjust the temperature to 0 to 10° C. and stir the mixture at 0 to 10° C. for a further 2 to 3 hours. Filter the mixture and wash the filter-cake with TBME (2×4.4 vol, 2×3.3 wt) at 0 to 10° C. Dry the solid at up to 40° C. until the TBME content is <0.5% w/w TBME by 1H-NMR analysis. 4 to 8 hours. - [0122]
Yields and Profiles: The yield for the step 3 Demonstration batch is summarized in Table 1C below. Input: 856.8 g corr. Ketone, 44.2% w/w assay (NMR, using TCNB as internal standard in CDCl3), (1.00 eq, 1.00 wt corr. for w/w assay) for input calculation. FIG. 2 and Table 1D shows an exemplary HPLC chromatogram of a crude preparation of fenfluramine hydrochloride (210 nm UV absorbance). - TABLE 1C Table for isolated yields for step 3 Demonstration batch Corr. % area Reference Corr. Corr. Yield % w/w (HPLC, number Input Output (% th) (1H-NMR)* QC) Comments Batch A1 856.8 g 836.31 g 85.3 44.2 99.15 Fenfluramine free base (in situ intermediate) Batch A2 880.7 84.0 based 99.5 100.00 Fenfluramine•HCl on ketone crude (step 3 an bisulfite d 4.1) adduct (77.6 based on purified ketone)
- TABLE 1D Purity of crude fenfluramine hydrochloride by HPLC (see FIG. 2) Processed Channel Descr. DAD AU Ch 1 Sample 210, Bw 4 Peak Results USP USP USP Name RT RelRT Area Height Tailing Resolution Plate Count EP s/n % Area 1 NorFenfluramine 7.46 2 2-Fenfluramine 7.68 3 Fenfluramine 8.67 1.000 3789064 778178 1.7 70796 2549.8 99.15 4 4-Fenfluramine 8.95 5 11 34 1.308 6073 1449 1.2 23.5 215529 3 8 0.16 6 ZX008 acid 12.93 7 Fenfluramine alcohol 14.16 1.633 15266 2972 1.3 24.8 215040 8.7 0.40 8 ZX008 ketone 14.83 9 Fenfluramine acetamide 15.55 10 TOLUENE 15 75 11 15.92 1.836 4110 1122 2.7 0.11 12 16.60 1.915 6861 1630 1.5 451209 4.3 0.18 Sum 3821374 100.00
- [0123]
Step 4.2: Crystallization of Fenfluramine Hydrochloride - [0124]
Procedure: Charge Fenfluramine.HCl (crude) (1.00 wt, 1.0 eq.) and TBME (10.0 vol, 7.4 wt) to the vessel and commence stirring. Heat the suspension to reflux (50 to 58° C.). Charge ethanol (5.0 vol, 3.9 wt) maintaining the temperature at 50 to 58° C. Addition time 20 minutes. Stir at 50 to 58° C. for 5 to 10 minutes and check for dissolution. Stir the solution at 50 to 58° C. for 5 to 10 minutes, targeting 54 to 58° C. Clarify the reaction mixture through a 0.1 μm in-line filter at 54 to 58° C., followed by a line rinse with TBME (1 vol, 0.7 wt). Cool the solution to 48 to 50° C. Charge Fenfluramine HCl, code FP0188 (0.01 wt). Check for crystallization. Allow the suspension to cool to 15 to 20° C., target 17° C. over 5 to 5.5 hours at an approximately constant rate. Stir the mixture at 15 to 20° C., target 17° C. for 2 to 3 hours. Filter the mixture and wash the filter-cake with clarified TBME (2×3.0 vol, 2×2.2 wt) at 5 to 15° C. Dry the solid at up to 40° C. until the TBME content is <0.5% w/w TBME and the ethanol content is <0.5% w/w EtOH by 1H-NMR analysis. 4 to 8 hours. Determine the w/w assay of the isolated Fenfluramine.HCl by 1H-NMR analysis. - [0125]
Yields and Profiles: The yield for the stage 4 Demonstration batch is summarized in Table 1E below. Input: 750.0 g uncorr. Fenfluramine HCl crude (1.00 eq, 1.00 wt uncorr.) for input calculation. FIG. 3 shows an exemplary HPLC chromatogram of a crystallized fenfluramine hydrochloride sample (210 nm UV absorbance). - TABLE 1E Table for isolated yields for stage 4 Demonstration batch Uncorr. Uncorr. Uncorr. Yield HPLC (% area, Input Output (% th) QC) Comments 750.0 g 608.0 81.1 100.00* Fenfluramine•HCl
5. In-Process Controls
- [0126]
Table 2 summarizes the in-process controls (IPCs) by IPC number as cited in the narrative procedures above used for Process 1. - TABLE 2 In-Process Controls Performed during Process 1 Critical IPC Synthesis Process No. Step Sample Description Method Acceptance Criteria 1 1 Reaction Reaction HPLC NMT 3.0% Ketone (1) Mixture Completion 2 1 Extraction Purity HPLC Report percent Aqueous Fenfluramine Free Base and Phase Fenfluramine Alcohol 3 1 Extraction Purity HPLC Report percent Organic Fenfluramine Free Base and Phase Fenfluramine Alcohol 4 1 Extraction Purity HPLC Report percent Organic Fenfluramine Free Base and Phase Fenfluramine Alcohol 5 1 Extraction Purity HPLC NLT 98.0% Fenfluramine Aqueous HCl Phase LT 1.0% Fenfluramine Alcohol 6 1 Extraction Purity HPLC Report percent result of Aqueous Fenfluramine HCl Phase Fenfluramine Alcohol 7 2 Reaction Purity 1H-NMR Residual Solvents by 1H- Mixture NMR Ethanol NMT 0.50% w/w Ethyl Acetate NMT 0.50% w/w Methanol NMT 0.50% w/w Toluene NMT 0.50% w/w MTBE NMT 0.50% w/w
6. Starting Materials
- [0127]
This section provides information and specification controls for the starting materials used to produce clinical supplies of fenfluramine per the routes shown herein. - TABLE 3 Starting Materials via the Route 1 Chemical Name Code [CAS. No.] Name Structure Source Step 1-(3- (Trifluoromethyl)- phenylacetone [21906-39-8] Ketone (1)Fluorochem 1 Ethyl Amine Ethyl EtNH2 Alfa Aesar 1 (70% in water) Amine [75-04-7]
- TABLE 4 Starting Materials via Route 2 Chemical Name Code [CAS. No.] Name Structure Source Step 3-(Trifluoromethyl)- phenylacetic acid [351-35-9] Acid (1a)To be determined 1 Acetic Anhydride [108-24-7] Acetic AnhydrideVarious 1 Ethyl Amine Ethyl EtNH2 Various 3 (70% in water) Amine [75-04-7]
- [0128]
Table 5 provides a list of the intermediates for the Route 2 synthesis. Both routes share the same intermediate Fenfluramine Free Base (1). Fenfluramine Free Base (1) was treated as an isolated intermediate in the Route 1 process however the Route 2 process uses fixed equipment where both Ketone (2) and Fenfluramine Free Base 1, both non-isolatable oils, are telescoped as a solution and controlled as in-situ intermediates. The Bisulfate Complex (3) is isolated as a solid thus is amenable to treatment as an isolated intermediate and released as such. Crude Fenfluramine HCl can be isolated as an intermediate before recrystallization. - [0129]
A Specification and Testing Strategy for Intermediates is used. Additional tests and acceptance criteria are be added based upon review of data from the primary stability batches and process validation critical parameter studies. Analytical reference standards are used in full characterization of each intermediate. HPLC methods to determine assay and impurities are the same as the drug substance release method and are validated for Accuracy, Precision: Repeatability, Intermediate Precision, Selectivity/Specificity, Detection limit, Quantitation limit, Linearity, Range, and Robustness. - TABLE 5 In-Situ and Isolated Intermediates Chemical Name [CAS No] Code Name Step No. Control Structure Bisulfate Complex of Ketone 1 Bisulfate Complex (3) Step 1 Isolated (Solid)1-(3- (Trifluoromethyl)- phenylacetone [21906-39-8] Ketone (2) Step 2 In-Situ (oil)Fenfluramine Free Base [458-24-2] Fenfluramine Free Base (1) Step 3 In-Situ (oil)Fenfluramine HCl [404-82-0] Crude Fenfluramine HCl Step 4 Isolated (Solid)
7. Characterization
- [0130]
Physiochemical Characteristics of Drug Substance. - [0131]
Fenfluramine HCl is developed as a single polymorph Form 1. A polymorphism and pre-formulation study has been conducted. Under a wide range of solvents and conditions crystalline material is produced of the same polymorph Form 1 based on a well-defined XRPD pattern and a consistent reproducible endotherm by DSC analysis. A summary of the chemophysical properties of Fenfluramine HCl from this study is provided below. Tabulated data includes example diffractograms, DSCs, and micrographs. - [0132]
The input Fenfluramine HCl (from precipitative isolation) was characterized to provide reference data and also to determine if the salt was of the same form as that identified from previous salt formations. The XRPD pattern of the salt reveals a crystalline solid that visually matches the reflection patterns obtained from formal crystallization of Fenfluramine HCl and has been arbitrarily termed Form 1. Comparison of the μATR-FTIR data for the salt from various batches gave profiles that had a 99.95% match. - [0133]
Thermal data analysis matched previous data obtained with only one major endotherm on the DSC thermograph peaking at 172.3° C. that matches the beginning of potential decomposition shown in a TGA thermograph. This also matches the reported melting point quoted for the reference standard. - [0134]
Isolation of the amorphous form has been shown to be difficult, with attempts using three common methods (rapid solvent evaporation, anti-solvent precipitation and lyophilization) all yielding highly crystalline solids that very closely share the same XRPD pattern of the input Form 1. - [0135]
Stability analysis of the salt after one week at 40° C./0% RH, three weeks at 40° C./75% RH, and under photostability conditions revealed that the input Form 1 has been maintained with no new impurities observed at 0.1% threshold. - [0136]
Results from DSC heat cycling analysis of Fenfluramine HCl are comparable to results generated when the material was held at 170° C. No crystallization event was noted and the amorphous was not generated but rather Form 1 was returned. - [0137]
Holding Fenfluramine HCl at approximately 170° C. for several hours causes a melt and evaporation event to take place with recombination and cooling to provide a white solid. Analysis of the white solid by XRPD, DSC and 1H NMR indicates no change in chemical or physical form, purity, or dissociation. - [0138]
Forced degradation studies carried out have proven Fenfluramine HCl to be stable under a range of conditions. Thermal modulation of Fenfluramine HCl repeatedly yielded the input Form 1.
8. Impurities
- [0139]
Impurities in a drug substance can be organic impurities (process impurities or drug substance-related degradants), inorganic impurities (salt residues or metals) and residual solvents; some of these impurities must be evaluated as to whether or not they are genotoxic agents. These impurities are taken into consideration and controlled in Fenfluramine HCl preparation by using either compendia or validated analytical methods per the specifications or by separate “for information only” testing. The following sections address the actual and potential impurities in Fenfluramine HCl. - [0140]
Actual Impurities and the Qualification of Synthesis Batch - [0141]
No impurities reported in cGMP drug substance batches intended for use in humans have exceeded the ICHQ3A qualification thresholds of 0.15% (Table 8). All impurities >0.1% are identified and handled as described in ICH Q3A unless they are genotoxic impurities. - [0142]
Process Impurities - [0143]
Table 6 lists the known potential impurities arising from the route of synthesis. All of these impurities are controlled to below ICHQ3A qualification threshold of 0.15% by either process changes and/or control of starting material input purities. - TABLE 6 Fenfluramine HCl Known Potential Process Impurities (Route 1) Observed Observed in in Development cGMP Name PLC Batches Batches [Cas. No.] Source (RRT) ≧0.10%1) ≧0.10%1) Ketone (2) Starting RRT No No [351-35-9] Material or 0.89 Intermediate Fenfluramine By-product RRT Yes No Alcohol 1.60 [621-45-4] Norfenfluramine By-product RRT Yes Yes [1886-26-6] 1.67 2-Fenfluramine Starting RRT No No [172953-70-7] Material 0.89 (isomer) 4-Fenfluramine Starting RRT Yes Yes [1683-15-4] Material 1.02 (isomer) N-(3- By-product RRT Yes Yes (trifluoromethyl)- 0.53-0.57 benzyl)ethanamine [90754-95-3] 1)ICH Q3A Identification threshold. The Reporting threshold (LOQ) for the HPLC method is 0.05%.
- [0144]
Degradation Impurities - [0145]
No change in impurity profile is observed upon long-term storage based on forced degradation studies under the ICH Q1A(R2) conditions of heat (solid, solution), acid, base, oxidizing, and ICH Q1B photostability conditions (solid, solution). Fenfluramine HCL is stable for 7 days as a solid at 150° C. (99.90 parent area %), as a solution in water-acetonitrile at 70° C. (99.73 parent area %), as a solution in acid, base, or photosensitizing conditions at ambient. Only oxidizing conditions (peroxide conditions) produced degradation of Fenfluramine HCl to 94.42% after 1 day producing several new related substances at −1% each consistent by LC-MS with +16 oxidation by-products - [0146]
Organic Volatiles/Residual Solvents - [0147]
Table 11 in the Batch Analysis section summarizes the solvents used in the process and the resulting amounts found in drug substance. All solvents used in the GMP steps are controlled at ICH Q3A limits using a suitably qualified Head-Space (HS) GC method. - [0148]
Inorganic Impurities - [0149]
Heavy Metals conform to either USP <231> or ICP method USP <233> as well as ICH Q3D. - [0150]
Genotoxic Impurities - [0151]
The ICH guidelines Q3A and Q3B are not sufficient to provide guidance on impurities that are DNA-reactive. The European Medicines Agency (EMA) guideline (2006) “Guideline on the Limits of Genotoxic Impurities” (EMA 2006) and the ICH Guideline M7 (2014) “Assessment and Control of DNA Reactive (Mutagenic) Impurities in Pharmaceuticals to Limit Potential Carcinogenic Risk” (ICH Guideline M7) are taken into consideration in controlling for potential genotoxic impurities. The diazonium route to prepare ketone (2) described in FIG. 5 has a disadvantage due to the potential formation of genotoxic intermediates shown as boxed compounds (e.g., N-hydroxyaryl, N-nitrosamine and Nitro compound). Muller et al. (Regulatory Toxicology and Pharmacology 44 (2006) 198-211) list potential functional alert groups that can be genotoxic. Safety guidances and regulations indicate that analysis of a process and identification of potential genotoxic agents, and control of such impurities at sub 10 parts per million levels is critical for safety. Often removal of such impurities and/or demonstrating their absence is costly and time consuming and sometimes difficult to achieve technically. For these reasons, selecting synthetic routes that circumvent the potential for such toxic intermediates is important. Because of the potential problems with the diazo route discussed above, as well as potential safety issues using diazo (shock-sensitive) intermediates, as well as the lower purity profiles with this route, this route is less preferred than the preferred route to ketone (2) starting from Nitrile (5). This route produces no potential genotoxic agents and leads to high purity Ketone (2) after isolation by distillation or via the bisulfite salt adduct—hydrolysis sequence. - [0152]
Additionally, attempts to remove isomer by-products present in commercial supplies of Aniline were unsuccessful whereas crystallization the Acid (4) resulting from hydrolysis of the nitrile (5) provides crystalline Acid (4) which can be purified to remove isomers early in synthesis. Removing impurities and/or isomers early in a synthesis is preferred if it is known such impurities track to final product, as the need to crystallize a final product at the end of a synthesis is more costly in losses and impacts cost of goods more greatly than removing early in synthesis before raw materials are invested along the process. - TABLE 7 Potential Impurities in Fenfluramine Synthesis Synthesis Route No. Compound Route 1 Route 2 CAS. No. 1No Starting Material [351-35-9] 2Starting Material Intermediate [21906-39-8] 4No Intermediate Not Available 5Potential Impurity Potential Impurity [621-45-4] 6Potential Impurity Potential Impurity [1886-26-6] 7Potential Impurity Potential Impurity [172953-70-7] 8Potential Impurity Potential Impurity [1683-15-4] 9Potential Impurity Potential Impurity [90754-95-3]
- TABLE 8 Batch Analyses of Fenfluramine HCl Drug Substance Test Batch 1 Batch 2 Batch 3 Batch 4 Appearance* White solid White solid White solid White solid Identification: FTIR* a) a Conforms Conforms Identification: 1H-NMR Conforms Conforms Conforms Conforms Identification: 13C-NMR Conforms Conforms Conforms Conforms Identification: MS Conforms Conforms Conforms Conforms Purity (HPLC area %) 99.57 99.77 b) b Assay (w/w %)* 99.49 100.37 100.79 100.13 Anhydrous Basis (HPLC) Impurities 2-Fenfluramine ND ND ND ND (HPLC 4-Fenfluramine) 0.16 0.15 0.11 0.12 area %) Fenfluramine Alcohol ND ND ND ND 1-((3-trifluoromethyl)phenyl)acetone ND ND ND ND Acetamide 0.27 ND ND ND N-(3-(trifluoromethyl)- ND 0.08 0.07 0.13 benzyl)ethanamine (RRT 0.53-0.57) Total 0.43 0.23 0.19 0.25 Residual Solvents Methanol ND ND ND ND (GC): ppm Ethanol ND ND ND ND MTBE 597 844 472 800 Ethyl Acetate 115 164 79 150 Toluene 4 7 ND ND Residue on Ignition (w/w %) 0.01 0.02 0.04 ND Heavy Metals (as Pb) <10 ppm <10 ppm b b Heavy Metals ICP (ppm) As a a <0.1 <0.1 Cd a a 0.1 0.1 Hg a a <0.1 <0.1 Pb a a 0.2 <0.4 Co a a <0.1 0.1 Mo a a <0.1 <0.1 Se a a <0.1 <0.1 V a a <0.1 <0.1 Water Determination* 0.21 0.08 0.02 0.03 (Karl Fischer) Chloride content by titration 13.19 13.09 12.92 12.93 XRPD* Form 1 Form 1 Form 1 Form 1 Differential Scanning Onset 169.42° C. 169.23° C. 169.85° C. 168.70° C. Calorimetry (DSC)* Peak 172.82° C. 171.55° C. 172.22° C. 171.97° C. Particle Size % Volume mean (D) a 11 11 19 Malvern (μm) D10 a 1 1 1 D50 a 5 7 9 D90 a 17 26 32 Microbial Total aerobic a a LT 100 CFU/g LT 100 CFU/g Limits Tests* microbial Count USP <61> Total yeast and a a LT 100 CFU/g LT 100 CFU/g molds count USP <62> Absence of a a Absent Absent Pathogens a)These tests were added to the specifications recently thus only recent lots have been tested using this test. b)These tests have been dropped from the specifications thus only historical lots have been tested using this test.
Example 3Method for Hydrolysis of Nitrile (5) to Acid
- [0153]
- TABLE 9 Step Operation 1. Charge 3-(trifluoromethyl)phenyl acetonitrile (1.0 eq., 1.00 wt) and purified water (5.0 vol) to a vessel and commence stirring. 2. Dissolve sodium hydroxide (1.1 wt, 5.0 eq.) in purified water (4.0 vol) at up to 40° C. in a suitable make-up vessel. Caution very exothermic. 3. Charge the aqueous sodium hydroxide solution to the mixture from step 1 at up to 40° C. followed by a line rinse with purified water, code RM0120 (1.0 vol) at up to 40° C. 4. Heat the mixture to 75 to 85° C., target 80° C. over 1 to 2 hours. 5. Heat the mixture at 80° C. until ≦0.1% area nitrile by HPLC analysis, expected 4 to 6 hours. 6. Cool the mixture to 18 to 23° C. 7. Adjust the pH of the mixture to pH ≦2 by charging 6M HCl (expected 7.0 vol) to the mixture at 18 to 23° C. Caution exothermic. 8. Stir the mixture for 15 to 30 minutes at 18 to 23° C. 9. Filter and wash the filter-cake with purified water (2 × 5.0 vol) at 18 to 23° C. 10. Slurry wash the filter-cake with n-heptane, code RM (2 × vol) at 0 to 5° C. 11. Dry the isolated solid at up to 45° C. until the water content is ≦.0.2% w/w by KF analysis according to MET/AN/0163, AKX-reagent. 12. Crystallization of crude stage 1 acid (1.00 wt for input calculation) 13. Charge the crude stage 1 acid (1.00 wt), ethyl acetate (0.75 vol) and n-heptane (10.5 vol) to a vessel and commence stirring. 14. Heat the mixture to 50° C. to achieve dissolution. 15. Cool the mixture to 5° C. and age at 5° C. for at least 30 mins. 16. Filter and wash the filter-cake with n-heptane (2 × 5.0 vol). 17. Dry the isolated solid at up to 45° C. until the residual solvent content by 1H-NMR analysis is ≦.0% w/w EtOAc and ≦.0% w/w n-heptane. Expected yield: 60 to 90% th uncorr. 68 to 103% w/w Expected purity: 93.00 to 99% area by HPLC
Example 4Evaluation of Minor Components Formed During Dakin-West Reaction in Preparation of Ketone (2)
- [0154]
The impurities formed during the Dakin-West chemistry and their subsequent removal using the distillation or via isolation of the product ketone as the bisulfite salt are described. The two major impurities found are shown below. - [0155]
Table 10 shows a table of analytical data for crude Ketone (2) isolated from Dakin-West reaction before and after bisulfite purification. In entry 1 crude Ketone isolated directly from the Dakin-West step (pre-bisulfite treatment) is 61.66% purity (e.g. about 62%) and contains 1.98% (e.g., about 2%) and 4.64% (e.g., about 5%) respectively of impurities having RRTs 1.20 and 1.34, which are proposed to be the acetate and dimer impurities (e.g., depicted above), respectively. In entry 2 which is post bisulfite treatment these are other impurities are removed leading to an overall purity of 95.55% (e.g., about 96%). Other entries shown in Table 10 provide other examples of this impurity enhancement by bisulfite treatment of crude Dakin-West ketone. The last two entries use pure Fluorchem ketone as input to the salt formation step and re-isolation of ketone thus illustrating that the salt formation and re-isolation does not produce any impurities itself. Additionally use of bicarbonate extraction procedure during reaction workup provides an improvement in purity of the resulting composition as it serves to remove any unreacted acid. Crude Ketone (2) made by the Diazo route showed similar improvements in purity when treated with bisulfite and isolated. - TABLE 10 Analytical purity data for crude Ketone (2) isolated from Dakin-West reaction before and after bisulfite purification. RRT is relative retention time (min) in chromatogram. RRT 0.93 1.00 1.009 1.06 Entry 0.85 Aniline 0.95 0.99 Ketone 1.004 Nitrile 1.02 Acid 1.10 1.15 1.34 1.38 1 1.38 1.76 0.04 0.49 61.66 nd 0.29 0.29 0.26 1.98 0.66 4.64 0.14 2 0.82 nd nd nd 95.53 0.31 0.14 nd 0.23 0.01 0.10 0.43 0.27 3 nd nd nd nd 77.82 nd nd nd nd 3.12 0.01 7.76 6.16 4 nd nd nd nd 98.82 nd 0.63 nd nd nd 0.02 0.30 0.22 5 0.08 nd nd 0.05 72.02 nd 0.02 nd nd 7.11 0.04 3.58 10.33 6 nd nd nd nd 99.49 nd 0.02 nd nd nd 0.02 0.11 0.24 7 0.15 0.23 nd nd 98.35 nd nd nd nd nd nd nd 0.24 8 nd nd nd nd 99.84 nd nd nd nd nd nd nd nd Entry 1 (Crude ketone from Route 1); Entry 2 (Ketone Route 1 post bisulfite release); Entry 3 (Crude ketone using crude acid); Entry 4 (Ketone using crude acid Post bisulfite); Entry 5 (Crude ketone using cryst. acid); Entry 6 (Crude ketone using cryst. acid post bisulfite); Entry 7 (Crude ketone using cryst. acid); Entry 8 (Fluorochem ketone); Entry 9 (Fluorochem ketone post bisulfite).
Example 5
- [0156]
- Additional Method for Preparation of 1-(3-trifluoromethyl)phenyl-propan-2-one
- [0157]
35 mL of water and 45 g of 37% (w/w) aqueous hydrochloric acid are put in a flask equipped with stirrer and dropping funnel. 24.25 Grams (0.151 moles) of m-trifluoromethylaniline are added after having cooled to 10 degree C. with an ice bath and then, at 5 degree C., an aqueous solution containing 12.43 g (0.180 moles) of sodium nitrite in 150 ml of water is slowly added. The reaction mixture is stirred for 30 minutes and then is poured during 30 minutes into a mixture made by 90 ml of water, 1.35 g (0.014 moles) of cuprous chloride, 2.30 g (0.013 moles) of cupric chloride dihydrate, 50 ml of acetone, 40.8 g (0.300 moles) of sodium acetate trihydrate and 23 g (0.230 moles) of isopropenyl acetate while keeping the reaction temperature at 30 degree C. After further 30 minutes of stirring, the reaction mixture is brought to 20 degree C., 50 ml of methylene chloride are added and the two layers are separated. - The aqueous layer is discarded while the organic layer is concentrated under vacuum until an oil is obtained which is treated with 35 g of sodium metabisulfite, 70 ml of water and 150 ml of heptane under stirring at room temperature for 12 hours. The suspension is filtered, the bisulfite complex is washed on the filter with 50 ml of heptane and then suspended in a two-phase mixture made by 100 ml of methylene chloride and 150 ml of a 10% (w/v) aqueous solution of sodium hydroxide. The layers are separated after one hour of stirring at room temperature, the aqueous phase is discarded while the organic layer is washed with water and evaporated under vacuum to give pure ketone.







































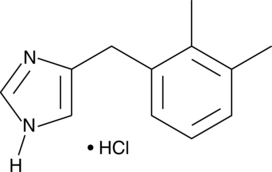






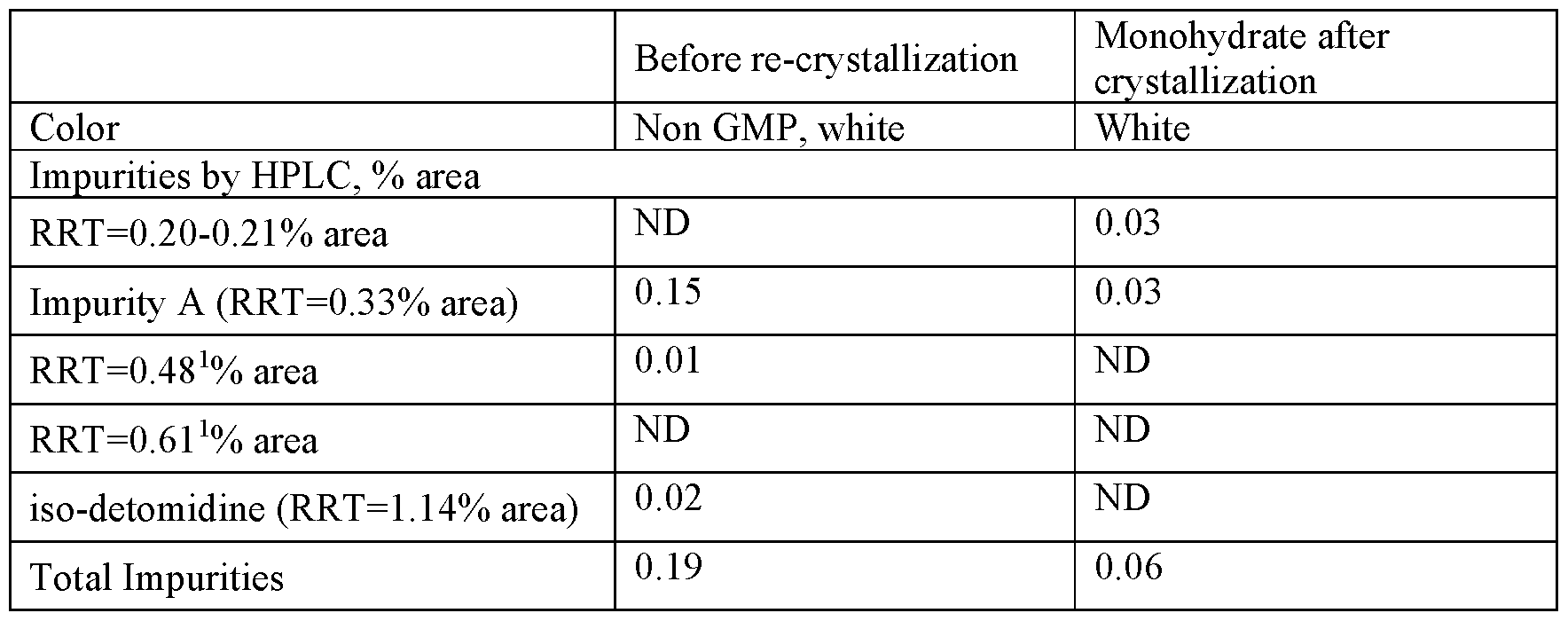















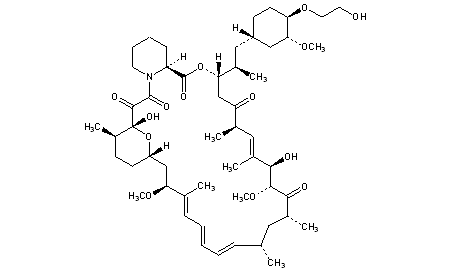









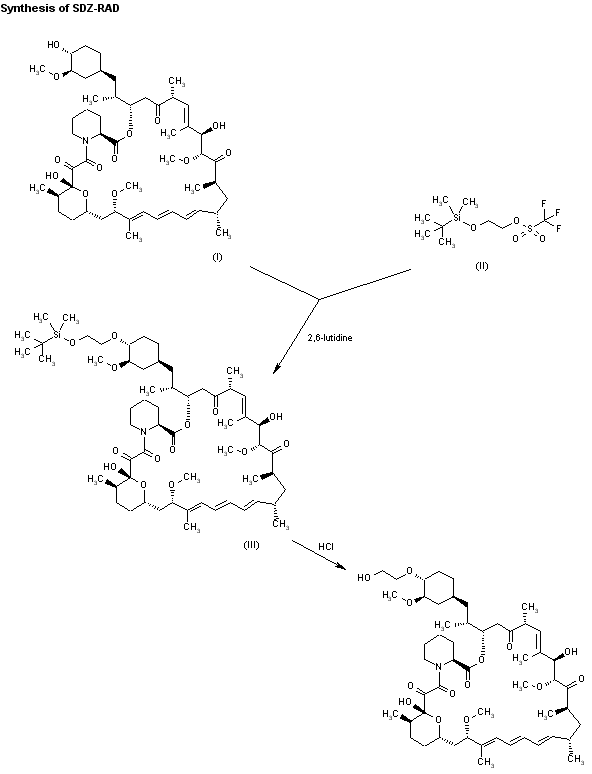
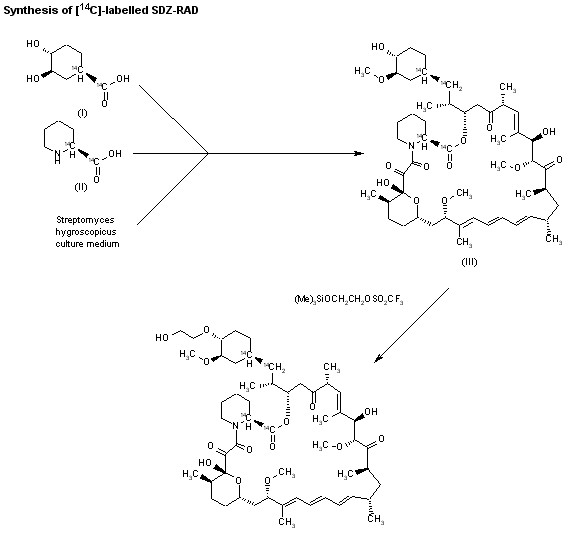
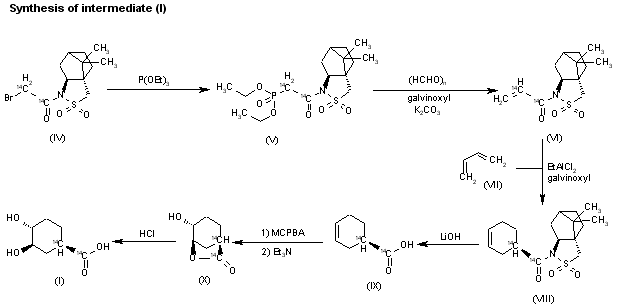
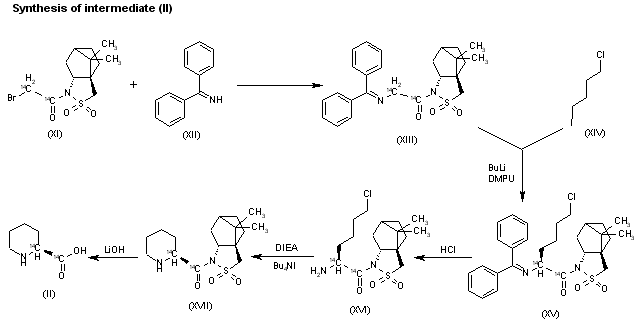









 (USA) and Certican
(USA) and Certican















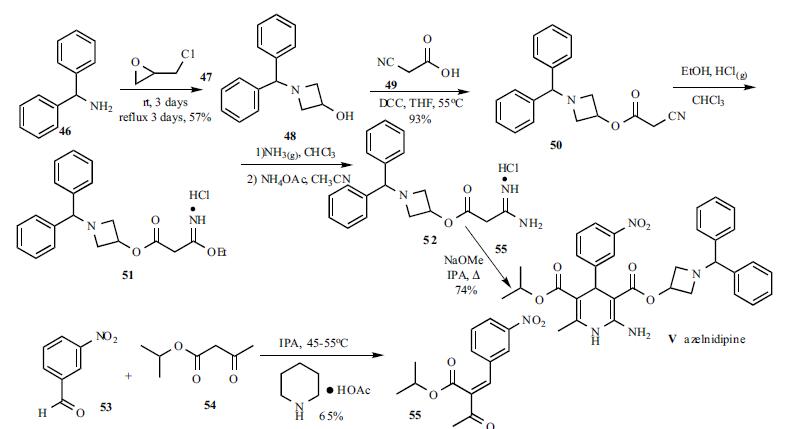






































 .HCl
.HCl




















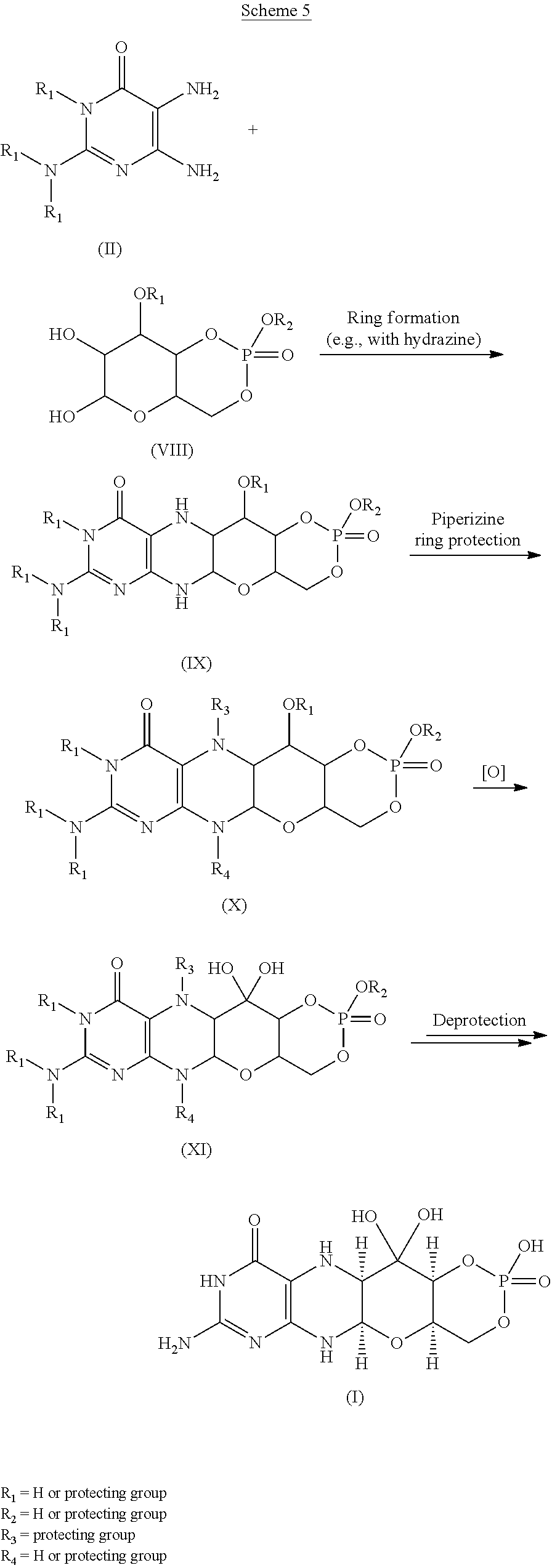










 Lisocabtagene maraleucel (liso-cel; JCAR017; Anti-CD19 CAR T-Cells) is an investigational chimeric antigen receptor (CAR) T-cell therapy designed to target CD19, [1][2] which is a surface glycoprotein expressed during normal B-cell development and maintained following malignant transformation of B cells. [3][4][5] Liso-cel CAR T-cells aim to target and CD-19 expressing cells through a CAR construct that includes an anti-CD19 single-chain variable fragment (scFv) targeting domain for antigen specificity, a transmembrane domain, a 4-1BB costimulatory domain hypothesized to increase T-cell proliferation and persistence, and a CD3-zeta T-cell activation domain. [1][2][6][7][8][9] The defined composition of liso-cel may limit product variability; however, the clinical significance of defined composition is unknown. [1][10] Image Courtesy: 2019/2020 Celgene/Juno Therapeutics / Bristol Meyers Squibb.
Lisocabtagene maraleucel (liso-cel; JCAR017; Anti-CD19 CAR T-Cells) is an investigational chimeric antigen receptor (CAR) T-cell therapy designed to target CD19, [1][2] which is a surface glycoprotein expressed during normal B-cell development and maintained following malignant transformation of B cells. [3][4][5] Liso-cel CAR T-cells aim to target and CD-19 expressing cells through a CAR construct that includes an anti-CD19 single-chain variable fragment (scFv) targeting domain for antigen specificity, a transmembrane domain, a 4-1BB costimulatory domain hypothesized to increase T-cell proliferation and persistence, and a CD3-zeta T-cell activation domain. [1][2][6][7][8][9] The defined composition of liso-cel may limit product variability; however, the clinical significance of defined composition is unknown. [1][10] Image Courtesy: 2019/2020 Celgene/Juno Therapeutics / Bristol Meyers Squibb.