
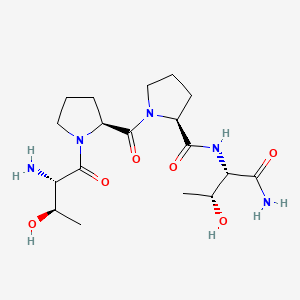
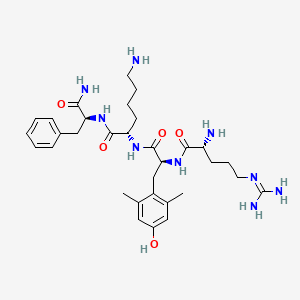
RAPASTINEL
- Molecular Formula C18H31N5O6
- Average mass 413.469 Da
L-threonyl-L-prolyl-L-prolyl-L-threoninamide
(2S)-1-[(2S)-1-[(2S,3R)-2-amino-3-hydroxybutanoyl]pyrrolidine-2-carbonyl]-N-[(2S,3R)-1-amino-3-hydroxy-1-oxobutan-2-yl]pyrrolidine-2-carboxamide
UNII-6A1X56B95E; 117928-94-6; 6A1X56B95E
BV-102; GLYX13, GLYX-13, in phase 3 clinical trials
Originator Northwestern University
- Developer Allergan; Naurex
- Class Amides; Antidepressants; Neuropsychotherapeutics; Oligopeptides; Small molecules
- Mechanism of Action NR2B N-Methyl-D-Aspartate receptor agonists
Highest Development Phases
- Phase III Major depressive disorder
- Discontinued Bipolar depression; Neuropathic pain
Most Recent Events
- 01 Jan 2017 Allergan initiates enrolment in a phase III trial for Major depressive disorder (Adjunctive treatment) in USA (IV, Injection) (NCT03002077)
- 21 Dec 2016 Allergan plans a phase III trial for Major depressive disorder (Adjunctive treatment) in USA (IV, Injection) (NCT03002077)
- 01 Nov 2016 Phase-III clinical trials in Major depressive disorder (Adjunctive treatment, Prevention of relapse) in USA (IV) (NCT02951988)
![Image result for RAPASTINEL]()

It is disclosed that GLYX-13 (Rapastinel) acts as NMDA receptor partial agonist, useful for treating neurodegenerative disorders such as stroke-related brain cell death, convulsive disorders, and learning and memory. See WO2015065891 , claiming peptidyl compound. Naurex , a subsidiary of Allergan is developing rapastinel (GLYX-13) (in phase3 clinical trials), a rapid-acting monoclonal antibody-derived tetrapeptide and NMDA receptor glycine site functional partial agonist as well as an amidated form of NT-13, for treating depression.
Rapastinel (INN) (former developmental code names GLYX-13, BV-102) is a novel antidepressant that is under development by Allergan (previously Naurex) as an adjunctive therapy for the treatment of treatment-resistant major depressive disorder.[1][2] It is a centrally active, intravenously administered (non-orally active) amidated tetrapeptide (Thr-Pro-Pro-Thr-NH2) that acts as a selective, weak partial agonist (mixed antagonist/agonist) of an allosteric site of the glycine site of the NMDA receptor complex (Emax ≈ 25%).[1][2]The drug is a rapid-acting and long-lasting antidepressant as well as robust cognitive enhancer by virtue of its ability to both inhibit and enhance NMDA receptor-mediated signal transduction.[1][2]
On March 3, 2014, the U.S. FDA granted Fast Track designation to the development of rapastinel as an adjunctive therapy in treatment-resistant major depressive disorder.[3] As of 2015, the drug had completed phase II clinical development for this indication.[4] On January 29, 2016, Allergan (who acquired Naurex in July 2015) announced that rapastinel had received Breakthrough Therapydesignation from the U.S. FDA for adjunctive treatment of major depressive disorder.
Rapastinel belongs to a group of compounds, referred to as glyxins (hence the original developmental code name of rapastinel, GLYX-13),[5] that were derived via structural modification of B6B21, a monoclonal antibody that similarly binds to and modulates the NMDA receptor.[2][6][7] The glyxins were invented by Joseph Moskal, the co-founder of Naurex.[5] Glyxins and B6B21 do not bind to the glycine site of the NMDA receptor but rather to a different regulatory site on the NMDA receptor complex that serves to allosterically modulate the glycine site.[8] As such, rapastinel is technically an allosteric modulator of the glycine site of the NMDA receptor, and hence is more accurately described as a functional glycine site weak partial agonist.[8]
In addition to its antidepressant effects, rapastinel has been shown to enhance memory and learning in both young adult and learning-impaired, aging rat models.[9] It has been shown to increase Schaffer collateral–CA1 long-term potentiation in vitro. In concert with a learning task, rapastinel has also been shown to elevate gene expression of hippocampal NR1, a subunit of the NMDA receptor, in three-month-old rats.[10] Neuroprotective effects have also been demonstrated in Mongolian Gerbils by delaying the death of CA1, CA3, and dentate gyrus pyramidal neurons under glucose and oxygen-deprived conditions.[11] Additionally, rapastinel has demonstrated antinociceptive activity, which is of particular interest, as both competitive and noncompetitive NMDA receptor antagonists are ataxic at analgesic doses, while rapastinel and other glycine subunit ligands are able to elicit analgesia at non-ataxic doses.[12]
Apimostinel (NRX-1074), an analogue of rapastinel with the same mechanism of action but dramatically improved potency, is being developed by the same company as a follow-on compound to rapastinel.
CN 104109189,




PAPER
Tetrahedron Letters (2017), 58(16), 1568-1571
http://www.sciencedirect.com/science/article/pii/S0040403917303015
Novel silaproline (Sip)-incorporated close structural mimics of potent antidepressant peptide drug rapastinel (GLYX-13)
Highlights
- •
-
Structural mimics of rapastinel comprising silaproline is reported.
- •
-
Sip introduction is expected to improve its pharmacokinetic profiles.
- •
-
Standard peptide coupling strategy in the solution-phase is utilized for synthesis.
Abstract
Rapastinel (GLYX-13) is a C-amidated tetrapeptide drug under clinical development for adjunctive treatment of major depressive disorder (MDD). Rapastinel features two consecutive proline residues centered at the peptide sequence (Thr-Pro-Pro-Thr-NH2), which are detrimental to its biological activity. In this communication, we report the synthesis of very close structural analogues of rapastinel comprising silaproline (Sip) as proline surrogate. By virtue of its enhanced lipophilicity and metabolic stability, Sip introduction in the native rapastinel sequence is expected to improve its pharmacokinetic profiles.
Graphical abstract
This paper reports the synthesis of silaproline (Sip)-incorporated close structural mimics of potent antidepressant peptide drug rapastinel (GLYX-13).

PATENT
Depression is the most common neuropsychiatric diseases, seriously affecting people’s health. In China With accelerated pace of life, increasing the incidence of depression was significantly higher social pressure.
[0003] Drug therapy is the primary means of treatment of depression. The main treatment drugs, including tricyclic antidepressants such as imipramine, amitriptyline and the like; selective serotonin reuptake inhibitors such as fluoxetine, sertraline and the like; serotonin / norepinephrine dual uptake inhibitors such as venlafaxine, duloxetine. However, commonly used drugs slow onset, usually takes several weeks to months, and there is not efficient and toxicity obvious shortcomings.
[0004] GLYX-13 is a new antidepressant, Phase II clinical study is currently underway. It does this by regulating the brain NMDA (N_ methyl -D- aspartate) receptors play a role, and none of them have serious side effects such as ketamine and R-rated, such as hallucinations and schizophrenia and so on.GLYX-13 can play a strong, fast and sustained antidepressant effects, the onset time of less than 24 hours, and the sustainable average of 7 days. As a peptide drug, GLYX-13 was well tolerated and safe to use.
[0005] GLYX-13 is a tetrapeptide having the sequence structure Thr-Pro-Pro-Thr, which is a free N-terminal amino group, C terminal amide structure. GLYX-13 synthesis methods include traditional methods of two solid-phase peptide synthesis and liquid phase peptide synthesis, because of its short sequence, the amount of solid phase synthesis of amino acids, high cost, and difficult to achieve a lot of preparation. A small amount of liquid phase amino acids, high yield can be prepared in large quantities.
The present invention can be further described by the following examples.
Preparation of r-NH2; [0013] Example 1 Four peptide H-Thr-Pr〇-P; r〇-Th
[0014] 1.1 threonine carboxyl amidation (H-Thr-NH2)
[0015] 500ml three flask was added Boc-Thr (tBu) -0H20g (0.073mol), anhydrous tetrahydrofuran (THF) 150ml, stirring to dissolve the solid. Ice-salt bath cooled to -10 ° C~_15 ° C, was added N- methylmorpholine 8ml, then l〇ml isobutyl chloroformate, keeping the temperature not higher than -10 ° C, after the addition was complete retention low temperature reaction 10min, then adding ammonia 20ml, ice bath reaction 30min, then at room temperature the reaction 8h. The reaction was stopped, water 300ml, 200ml ethyl acetate was added to extract the precipitate, washed with water 3 times.Dried over anhydrous sodium sulfate 6h. Filtered, and then the solvent was distilled off under reduced pressure to give a white solid 16. 6g, 83% yield.
[0016] The above product was dissolved in 50ml of trifluoroacetic acid or 2N hydrochloric acid / ethyl acetate solution was reacted at room temperature lh, the solvent was distilled off to give a white solid, i.e. amidated carboxyl threonine trifluoroacetic acid / hydrochloric acid salt H- Thr-NH 2. HC1.
[0017] 1.2 Pro – Preparation of threonine dipeptide fragment H-Pr〇-Thr-NH2 of
[0018] 500ml flask was added Boc-Pr〇 three-0H20g (0. 093mol), in anhydrous tetrahydrofuran (TH F) 200ml, stirring to dissolve solids, cooled to ice-salt bath -l〇 ° C~-15 ° C, added N- methylmorpholine 11ml, then dropwise isobutyl 13ml, keeping the temperature not higher than -10 ° C, keep it cool after the addition was complete the reaction 10min. H-Thr-NH2. HC114. 5g dissolved in 50ml of tetrahydrofuran, was added N- methyl morpholine 11ml. The above solution was added to the reaction mixture, the low temperature reaction 30min, then at room temperature the reaction 8h. The reaction was stopped, water 300ml, 200ml ethyl acetate was added to extract the precipitate, washed with water 3 times. Dried over anhydrous sodium sulfate 6h. Filtered and then evaporated under reduced pressure to give a white solid 25.7g, 82% yield.
[0019] The above product was dissolved in 100ml of 2N trifluoroacetic acid or hydrochloric acid / ethyl acetate solution was reacted at room temperature lh, the solvent was distilled off to give a white solid, i.e., proline – threonine dipeptide hydrochloride salt of H-Pr〇 -Thr-NH 2. HC1.
[0020] The above product was dissolved in 100ml of pure water, sodium carbonate solution was added to adjust the PH value, the precipitated white solid was filtered and dried in vacuo to give the desired product proline – threonine dipeptide fragment H-Pr square-Thr- NH223g.
Protected threonine [0021] 1.3 – Preparation of dipeptide fragment Boc-Thr (tBu) -Pr〇-0H of
[0022] Boc-Thr (tBu) -0H20g (0 · 073mol) was dissolved in dry tetrahydrofuran (THF) 150ml, stirring to dissolve the solid.Ice-salt bath cooled to -10 G~-15 ° C, was added N- methylmorpholine 8ml, then dropwise isobutyl 10ml, maintained at a temperature no higher than -10 ° C, kept cold reaction After dropping 10min. Proline methyl ester hydrochloride
PAPER
Journal of Medicinal Chemistry (1989), 32(10), 2407-11.
Threonylprolylprolylthreoninamide (HRP-7). The synthesis of HRP-7 was begun with 3 g of p-methylbenzhydrylamine-resin containing 1.41 mmol of attachment sites. The protected tetrapeptidyl-resin (1.63 g) was subjected to HF cleavage. Radioactivity was found in the 1% acetic acid extract (77%) and in the 5% extract (24%). These solutions were combined and lyophilized. Crude peptide (309 mg, 97%) was gel filtered on Sephadex G-15 (1.1 X 100 cm). Peptide eluting between 34 and 46 mL was pooled and lyophilized to yield 294 mg (95%, overall yield 92%) of homogeneous HRP-7.
PATENT
PATENT
WO 2017136348
Process for synthesizing dipyrrolidine peptide compounds (eg GLYX-13) is claimed.
An N-methyl-D-aspartate (NMDA) receptor is a postsynaptic, ionotropic receptor that is responsive to, inter alia, the excitatory amino acids glutamate and glycine and the synthetic compound NMDA. The NMDA receptor (NMDAR) appears to controls the flow of both divalent and monovalent ions into the postsynaptic neural cell through a receptor associated channel and has drawn particular interest since it appears to be involved in a broad spectrum of CNS disorders. The NMDAR has been implicated, for example, in neurodegenerative disorders including stroke-related brain cell death, convulsive disorders, and learning and memory.
NMDAR also plays a central role in modulating normal synaptic transmission, synaptic plasticity, and excitotoxicity in the central nervous system. The NMDAR is further involved in Long-Term Potentiation (LTP), which is the persistent strengthening of neuronal connections that underlie learning and memory The NMDAR has been associated with other disorders ranging from hypoglycemia and cardiac arrest to epilepsy. In addition, there are preliminary reports indicating involvement of NMDA receptors in the chronic neurodegeneration of Huntington’s, Parkinson’s, and Alzheimer’s diseases. Activation of the NMDA receptor has been shown to be responsible for post-stroke convulsions, and, in certain models of epilepsy, activation of the NMDA receptor has been shown to be necessary for the generation of seizures. In addition, certain properties of NMDA receptors suggest that they may be involved in the information-processing in the brain that underlies consciousness itself. Further, NMDA receptors have also been implicated in certain types of spatial learning.
[0003] In view of the association of NMDAR with various disorders and diseases, NMDA-modulating small molecule agonist and antagonist compounds have been developed for therapeutic use. NMDA receptor compounds may exert dual (agonist/antagonist) effect on the NMDA receptor through the allosteric sites. These compounds are typically termed “partial agonists”. In the presence of the principal site ligand, a partial agonist will displace some of the ligand and thus decrease Ca flow through the receptor. In the absence of the principal site ligand or in the presence of a lowered level of the principal site ligand, the partial agonist acts to increase Ca++ flow through the receptor channel.







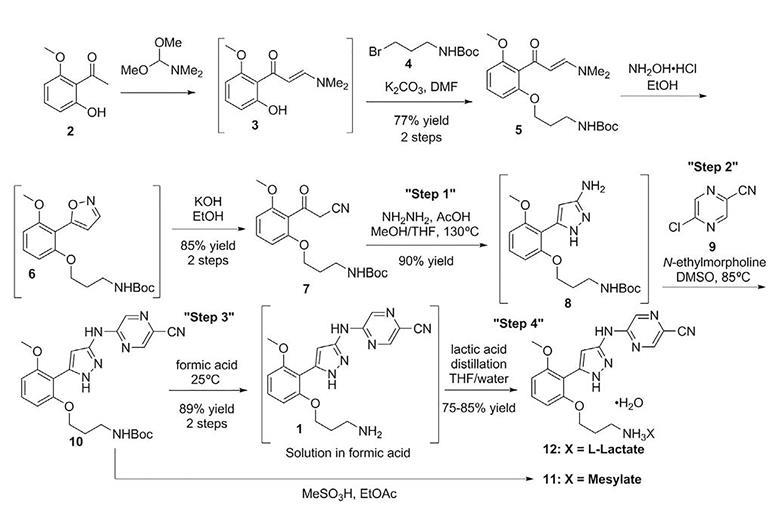
Example 2: Synthesis of GLYX-13
[00119] GLYX-13 was prepared as follows, using intermediates KSM-1 and KSM-2 produced in Example 1. The synthetic route for the same is provided in Figure 2.
Stage A – Preparation of (S)-N-((2S, 3R)-l-amino-3-hydroxy-l-oxobutan-2-yl)-l-((S)-pyrrolidine-2-carbonyl) pyrrolidine-2-carboxamide (Compound XI)
[00120] In this stage, KSM -1 was reacted with 10%Pd/C in presence of methanol to produce a compound represented by Formula XI. The reaction was optimized and performed up to 4.0 kg scale in the production plant and observed consistent quality (>80% by HPLC%PA) and yields (80% to 85%).
[00121] The reaction scheme involved in this method is as follows:

[00122] Raw materials used for this method are illustrated in Table 7 as follows:
Table 7.

[00123] In stage A, 10% Palladium on Carbon (w/w, 50% wet) was charged into the pressure reactor at ambient temperature under nitrogen atmosphere. KSM-1 was dissolved in methanol in another container and sucked into above reactor under vacuum. Hydrogen pressure was maintained at 45-60 psi at ambient temperature for over a period of 5-6 hrs. Progress of the reaction mixture was monitored by HPLC for KSM-1 content; limit is not more than 5%.
Hyflow bed was prepared with methanol (Lot-II). The reaction mass was filtered through nutsche filter under nitrogen atmosphere and bed was washed with Methanol Lot-Ill. Filtrate was transferred into the reactor and distilled completely under reduced pressure at below 50 °C (Bath temperature) to get the syrup and syrup material was unloaded into clean and dry container and samples were sent to QC for analysis.
[00124] From the above reaction(s), 1.31 kg of compound represented by Formula XI was obtained with a yield of 89.31% and with a purity of 93.63%).
Stage B – Preparation of Benzyl (2S, 3R)-l-((S)-2-((S)-2-((2S, 3R)-I-amino-3-hydroxy-I- oxobutan-2-ylcarbamoyl) pyrrolidine-! -carbonyl) pyrrolidin-1 -yl)-3-hydroxy-l -oxobutan-2- ylcarbamate (Compound XII)
[00125] In this stage the compound represented by Formula XI obtained above was reacted with KSM-2 to produce a compound represented by Formula XII. This reaction was optimized and scaled up to 3.0 kg scale in the production plant and obtained 25% to 28% yields with UPLC purity (>95%).
[00126] The reaction scheme is as follows:

[00127] Raw materials used for this method are illustrated in Table 8 as follows:
Table 8.


[00128] Stage B: ethanol was charged into the reactor at 20 to 35 °C. Compound represented by Formula XI was charged into the reactor under stirring at 20 to 35 °C and reaction mass was cooled to -5 to 0°C. EDC.HC1 was charged into the reaction mass at -5 to 0 °C and reaction mass, was maintained at -5 to 0 °C for 10-15 minutes. N-Methyl morpholine was added drop wise to the above reaction mass at -5 to 0 °C and reaction mass was maintained at -5 to 0 °C for 10-15 minutes.
[00129] KSM-2 was charged into the reactor under stirring at -5 to 0 °C and reaction mass was maintained at -5 to 0 °C for 3.00 to 4.00 hours. The temperature of the reaction mass was raised to 20 to 35 °C and was maintained at 20 to 35 °C for 12 – 15 hours under stirring. (Note:
Monitor the reaction mass by HPLC for Stage A content after 12.0 hours and thereafter every 2.0 hours. The content of stage A should not be more than 2.0%). Ethanol was distilled out completely under vacuum at below 50 °C (Hot water temperature) and reaction mass was cooled to 20 to 35 °C. Water Lot-1 was charged into the residue obtained followed by 10% DCM-Isopropyl alcohol (Mixture of Dichloromethane Lot-1 & Isopropyl alcohol Lot-1 prepared in a cleaned HDPE container) into the reaction mass at 20 – 35 °C.
[00130] Both the layers were separated and the aqueous layer was charged into the reactor. 10%) DCM-Isopropyl alcohol (Mixture of Dichloromethane Lot-2 & Isopropyl alcohol Lot-2 prepared in a cleaned HDPE container) was charged into the reaction mass at 20 to 35 °C. Both the layers were separated and the aqueous layer was charged back into the reactor. 10%> IDCM-isopropyl alcohol (Mixture of Dichloromethane Lot-3 & Isopropyl alcohol Lot-3 prepared in a cleaned HDPE container) was charged into the reaction mass at 20 to 35 °C. Both the layers were separated and the aqueous layer was charged back into the reactor. 10%> DCM-Isopropyl alcohol (Mixture of Dichloromethane Lot-4 & Isopropyl alcohol Lot-4 prepared in a cleaned HDPE container) was charged into the reaction mass at 20 to 35 °C and separated both the layers. The above organic layers were combined and potassium hydrogen sulfate solution (Prepare a solution in a HDPE container by dissolving Potassium hydrogen sulfate Lot-1 in water Lot-2) was charged into the reaction mass at 20 to 35 °C. Separated both the layers and charged back organic layer into the reactor. Potassium hydrogen sulfate solution (Prepared a solution in a HDPE container by dissolving Potassium hydrogen sulfate Lot-2 in water Lot-3) was charged into the reaction mass at 20 to 35 °C. Separated both the layers and the organic layer was dried over Sodium sulfate and distilled out the solvent completely under vacuum at below 45 °C (Hot water temperature).
[00131] The above crude was absorbed with silica gel (100-200mesh) Lot-1 in
dichloromethane. Prepared the column with silica gel (100-200 mesh) Lot-2, and washed the silica gel bed with from Dichloromethane Lot-5 and charged the adsorbed compound into the column. Eluted the column with 0-10% Methanol Lot-1 in Dichloromethane Lot-5 and analyzed fractions by HPLC. Solvent was distilled out completely under vacuum at below 45 °C (Hot water temperature). Methyl tert-butyl ether Lot-1 was charged and stirred for 30 min. The solid was filtered through the Nutsche filter and washed with Methyl tert-butyl ether Lot-2 and
samples were sent to QC for complete analysis. (Note: If product quality was found to be less than 95%, column purification should be repeated).
[00132] From the above reaction(s), 0.575 kg of compound represented by Formula XII was obtained with a yield of 17% and with a purity of 96.28%).
Stage C – Preparation of Benzyl (S)-N-((2S, 3R)-l-amino-3-hydroxy-l-oxobutan-2-yl)-l-((S)-l- ((2R, 3R)-2-amino-3-hydroxybutanoyl) pyrrolidine-2 carbonyl) pyrrolidine-2-carboxamide (GLYX-13)
[00133] In this reaction step the compound of Formula XII obtained above was reacted with 10%oPd in presence of methanol to produce GLYX-13. This reaction was optimized and performed up to 2.8 kg scale in the production plant and got 40% to 45% of yields with UPLC purity >98%.
[00134] The reaction scheme involved in this method is as follows:


i
[00135] Raw materials used for this method are illustrated in Table 9 as follows:
Table 9.



30 Nitrogen cylinder – – – – – 31 Hydrogen cylinder – – – – –
[00136] In an exemplary embodiment of stage C, 10% Palladium Carbon (50% wet) was charged into the pressure reactor at ambient temperature under nitrogen atmosphere. Compound of Formula XII was dissolved in methanol in a separate container and sucked into the reactor under vacuum. Hydrogen pressure was maintained 45-60 psi at ambient temperature over a period of 6-8 hrs. Progress of the reaction was monitored by HPLC for stage-B (compound represented by Formula XII) content (limit is not more than 2%). If HPLC does not comply continue the stirring until it complies. Prepared the hyflow bed with methanol (Lot-II) and the reaction mass was filtered through hyflow bed under nitrogen atmosphere, and the filtrate was collected into a clean HDPE container. The bed was washed with Methanol Lot-Ill and the filtrate was transferred into the Rota Flask and distilled out the solvent completely under reduced pressure at below 50°C (Bath temperature) to get the crude product. The material was unloaded into clean HDPE container under Nitrogen atmosphere.
[00137] Neutral Alumina Lot-1 was charged into the above HDPE container till uniform mixture was formed. The neutral Alumina bed was prepared with neutral alumina Lot-2 and dichloromethane Lot-1 in a glass column. The neutral Alumina Lot-3 was charged and
Dichloromethane Lot-2 into the above prepared neutral Alumina bed. The adsorbed compound was charged into the column from op.no.11. The column was eluted with Dichloromethane Lot-2 and collect 10 L fractions. The column was eluted with Dichloromethane Lot-3 and collected 10 L fractions. The column was eluted with Dichloromethane Lot-4 and Methanol Lot-4 (1%) and collected 10 L fractions. The column was eluted with Dichloromethane Lot-5 and Methanol Lot-5 (2%) and collected 10 L fractions. The column was eluted with Dichloromethane Lot-6 and Methanol Lot-6 (3%) and collected 10 L fractions. The column was eluted with
Dichloromethane Lot-7 and Methanol Lot-7 (5%). and collected 10 L fractions. The column was eluted with Dichloromethane Lot-8 and Methanol Lot-8 (8%). and collected 10 L fractions. The column was eluted with Dichloromethane Lot-9 and Methanol Lot-9 (10%) and collected 10 L fractions. Fractions were analyzed by HPLC (above 97% purity and single max impurity >0.5% fractions are pooled together)
[00138] Ensured the reactor is clean and dry. The pure fractions were transferred into the reactor.
[00139] The solvent was distilled off completely under vacuum at below 45 °C (Hot water temperature). The material was cooled to 20 to 35°C. Charged Dichloromethane Lot- 10 and Methanol Lot- 10 into the material and stirred till dissolution. Activated carbon was charged into the above mixture at 20 to 35°C and temperature was raised to 45 to 50 °C.
[00140] Prepared the Hyflow bed with Hyflow Lot-2 and Methanol Lot-11 Filtered the reaction mass through the Hy-flow bed under nitrogen atmosphere and collect the filtrate into a clean FIDPE container. Prepared solvent mixture with Dichloromethane Lot-11 and Methanol Lot- 12 in a clean FIDPE container and washed Nutsche filter with same solvent. Charged filtrate in to Rota evaporator and distilled out solvent under vacuum at below 50°C. Dry the compound in Rota evaporator for 5 to 6 hours at 50°C, send sample to QC for Methanol content (residual solvent) which should not be more than 3000 ppm. The material was cooled to 20 to 35 °C and the solid material was unloaded into clean and dry glass bottle. Samples were sent to QC for complete analysis.
[00141] From the above reaction(s), 0.92 kg of Glyx-13 was obtained with a yield of 43.5% and with a purity of 99.73%.
| Cited Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| US5166136 * | Jul 25, 1989 | Nov 24, 1992 | Glaxo Group Limited | Spirolactam containing peptides |
| US6025471 * | Jun 3, 1998 | Feb 15, 2000 | Deghenghi; Romano | Diazaspiro, azepino and azabicyclo therapeutic peptides |
| US6541453 * | Jun 3, 1997 | Apr 1, 2003 | Syngenta Limited | Peptide derivatives |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2012149389A3 * | Apr 27, 2012 | May 8, 2014 | Northwestern University | Methods of treating alzheimer’s disease, huntington’s disease, autism, or other disorders |
| WO2013063120A3 * | Oct 24, 2012 | Oct 16, 2014 | Northwestern University | Nmda receptor modulators and uses thereof |
| WO2014120783A1 * | Jan 29, 2014 | Aug 7, 2014 | Naurex, Inc. | Spiro-lactam nmda receptor modulators and uses thereof |
| WO2014120784A1 * | Jan 29, 2014 | Aug 7, 2014 | Naurex, Inc. | Spiro-lactam nmda receptor modulators and uses thereof |
| WO2014120786A1 * | Jan 29, 2014 | Aug 7, 2014 | Naurex, Inc. | Spiro-lactam nmda receptor modulators and uses thereof |
| WO2014120789A1 * | Jan 29, 2014 | Aug 7, 2014 | Naurex, Inc. | Spiro-lactam nmda receptor modulators and uses thereof |
| WO2014120800A1 * | Jan 29, 2014 | Aug 7, 2014 | Naurex, Inc. | Spiro-lactam nmda receptor modulators and uses thereof |
| WO2017093354A1 | Nov 30, 2016 | Jun 8, 2017 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Nmdar antagonists for the treatment of diseases associated with angiogenesis |
| CN103974712A * | Apr 27, 2012 | Aug 6, 2014 | 西北大学 | Methods of treating alzheimer’s disease, huntington’s disease, autism, or other disorders |
| CN104321071A * | Oct 24, 2012 | Jan 28, 2015 | 西北大学 | Nmda受体调节剂及其用途 |
| EP2542254A1 * | Feb 11, 2011 | Jan 9, 2013 | Northwestern University | Secondary structure stabilized nmda receptor modulators and uses thereof |
| EP2542254A4 * | Feb 11, 2011 | Jul 10, 2013 | Univ Northwestern | Secondary structure stabilized nmda receptor modulators and uses thereof |
| EP2771021A4 * | Oct 24, 2012 | Dec 23, 2015 | Univ North Western | Nmda receptor modulators and uses thereof |
| US8492340 | Sep 10, 2012 | Jul 23, 2013 | Northwestern University | Methods of treating depression and other related diseases |
| US8673843 | Jun 18, 2012 | Mar 18, 2014 | Northwestern University | NMDA receptors modulators and uses thereof |
| US8951968 | Apr 5, 2012 | Feb 10, 2015 | Northwestern University | Methods of treating depression and other related diseases |
| US9101612 | Feb 11, 2011 | Aug 11, 2015 | Northwestern University | Secondary structure stabilized NMDA receptor modulators and uses thereof |
| US9149501 | Jul 9, 2013 | Oct 6, 2015 | Northwestern University | Methods of treating depression and other related diseases |
| US9198948 | Jul 9, 2013 | Dec 1, 2015 | Northwestern University | Methods of treating depression and other related diseases |
| US9340576 | Jun 4, 2013 | May 17, 2016 | Northwestern University | Methods of treating depression and other related diseases |
| US9504670 | Nov 4, 2015 | Nov 29, 2016 | Aptinyx, Inc. | Spiro-lactam NMDA receptor modulators and uses thereof |
| US9512133 | Oct 10, 2013 | Dec 6, 2016 | Northwestern University | NMDA receptor modulators and uses thereof |
| US9512134 | Jan 29, 2014 | Dec 6, 2016 | Aptinyx, Inc. | Spiro-lactam NMDA receptor modulators and uses thereof |
| US9579304 | Feb 22, 2016 | Feb 28, 2017 | Aptinyx, Inc. | Spiro-lactam NMDA receptor modulators and uses thereof |
| US9593145 | May 14, 2015 | Mar 14, 2017 | Northwestern University | Secondary structure stabilized NMDA receptor modulators and uses thereof |
| US9708335 | Jan 29, 2014 | Jul 18, 2017 | Apytinyx Inc. | Spiro-lactam NMDA receptor modulators and uses thereof |
| US20140107037 * | Apr 27, 2012 | Apr 17, 2014 | Northwestern University | Methods of treating alzheimer’s disease, huntington’s disease, autism, or other disorders |
| US20150051262 * | Oct 24, 2012 | Feb 19, 2015 | Northwestern University | Nmda receptor modulators and uses thereof |
| US20160122359 * | Jan 29, 2014 | May 5, 2016 | Naurex, Inc. | Spiro-lactam nmda receptor modulators and uses thereof |
See also
References
- ^ Jump up to:a b c Hashimoto K, Malchow B, Falkai P, Schmitt A (August 2013). “Glutamate modulators as potential therapeutic drugs in schizophrenia and affective disorders”. Eur Arch Psychiatry Clin Neurosci. 263 (5): 367–77. PMID 23455590. doi:10.1007/s00406-013-0399-y.
- ^ Jump up to:a b c d Moskal JR, Burgdorf JS, Stanton PK, Kroes RA, Disterhoft JF, Burch RM, Amin Khan M (2016). “The Development of Rapastinel (Formerly GLYX-13); a rapid acting and long lasting antidepressant”. Curr Neuropharmacol. PMID 26997507.
- Jump up^ FDA Grants Fast Track Designation to Naurex’s Rapid-Acting Novel Antidepressant GLYX-13 http://www.prnewswire.com/news-releases/fda-grants-fast-track-designation-to-naurexs-rapid-acting-novel-antidepressant-glyx-13-248174561.html
- Jump up^ http://naurex.com/wp-content/uploads/2014/12/Naurex_P2b_Data_Press_Release_FINAL_Approved.pdf
- ^ Jump up to:a b Burgdorf, Jeffrey; Zhang, Xiao-lei; Weiss, Craig; Matthews, Elizabeth; Disterhoft, John F.; Stanton, Patric K.; Moskal, Joseph R. (2011). “The N-methyl-d-aspartate receptor modulator GLYX-13 enhances learning and memory, in young adult and learning impaired aging rats”. Neurobiology of Aging. 32 (4): 698–706. ISSN 0197-4580. PMC 3035742
 . PMID 19446371. doi:10.1016/j.neurobiolaging.2009.04.012.
. PMID 19446371. doi:10.1016/j.neurobiolaging.2009.04.012. - Jump up^ Haring R, Stanton PK, Scheideler MA, Moskal JR (1991). “Glycine-like modulation of N-methyl-D-aspartate receptors by a monoclonal antibody that enhances long-term potentiation”. J. Neurochem. 57 (1): 323–32. PMID 1828831. doi:10.1111/j.1471-4159.1991.tb02131.x.
- Jump up^ Moskal JR, Kuo AG, Weiss C, Wood PL, O’Connor Hanson A, Kelso S, Harris RB, Disterhoft JF (2005). “GLYX-13: a monoclonal antibody-derived peptide that acts as an N-methyl-D-aspartate receptor modulator”. Neuropharmacology. 49 (7): 1077–87. PMID 16051282. doi:10.1016/j.neuropharm.2005.06.006.
- ^ Jump up to:a b Burch RM, Amin Khan M, Houck D, Yu W, Burgdorf J, Moskal JR (2016). “NMDA Receptor Glycine Site Modulators as Therapeutics for Depression: Rapastinel has Antidepressant Activity without Causing Psychotomimetic Side Effects”. Curr Neuropharmacol. PMID 26830963.
- Jump up^ Burgdorf, Jeffrey; Zhang, Xiao-lei; Weiss, Craig; Matthews, Elizabeth; Disterhoft, John F.; Stanton, Patric K.; Moskal, Joseph R. (2011). “The N-methyl-d-aspartate receptor modulator GLYX-13 enhances learning and memory, in young adult and learning impaired aging rats”. Neurobiology of Aging. 32 (4): 698–706. PMC 3035742
 . PMID 19446371. doi:10.1016/j.neurobiolaging.2009.04.012.
. PMID 19446371. doi:10.1016/j.neurobiolaging.2009.04.012. - Jump up^ Moskal, Joseph R.; Kuo, Amy G.; Weiss, Craig; Wood, Paul L.; O’Connor Hanson, Amy; Kelso, Stephen; Harris, Robert B.; Disterhoft, John F. (2005). “GLYX-13: A monoclonal antibody-derived peptide that acts as an N-methyl-d-aspartate receptor modulator”. Neuropharmacology. 49 (7): 1077–87. PMID 16051282. doi:10.1016/j.neuropharm.2005.06.006.
- Jump up^ Stanton, Patric K.; Potter, Pamela E.; Aguilar, Jennifer; Decandia, Maria; Moskal, Joseph R. (2009). “Neuroprotection by a novel NMDAR functional glycine site partial agonist, GLYX-13”. NeuroReport. 20 (13): 1193–7. PMID 19623090. doi:10.1097/WNR.0b013e32832f5130.
- Jump up^ Wood, Paul L.; Mahmood, Siddique A.; Moskal, Joseph R. (2008). “Antinociceptive action of GLYX-13: An N-methyl-D-aspartate receptor glycine site partial agonist”. NeuroReport. 19(10): 1059–61. PMID 18580579. doi:10.1097/WNR.0b013e32830435c9.

External links
- Rapastinel – AdisInsight
- Rapastinel (GLYX-13) – Naurex, Inc.
- Rapastinel Receives FDA Breakthrough Therapy Designation – Allergan plc. press release
 |
|
 |
|
| Clinical data | |
|---|---|
| Pregnancy category |
|
| ATC code |
|
| Legal status | |
| Legal status | |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| ChemSpider | |
| Chemical and physical data | |
| Formula | C18H31N5O6 |
| Molar mass | 413.47 g/mol |
| 3D model (JSmol) | |
/////////////RAPASTINEL, BV-102, GLYX-13, PEPTIDE, phase 3, рапастинел , راباستينيل , 雷帕替奈
CC(C(C(=O)N1CCCC1C(=O)N2CCCC2C(=O)NC(C(C)O)C(=O)N)N)O
Filed under: Phase3 drugs Tagged: BV-102, 雷帕替奈, GLYX-13, рапастинел, peptide, PHASE 3, RAPASTINEL, راباستينيل








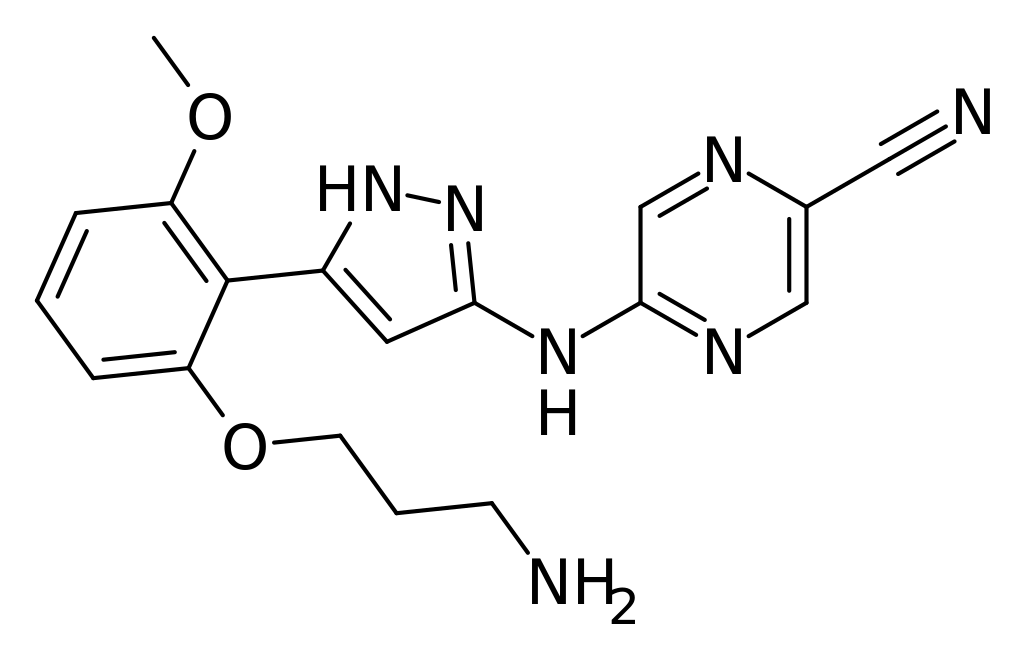


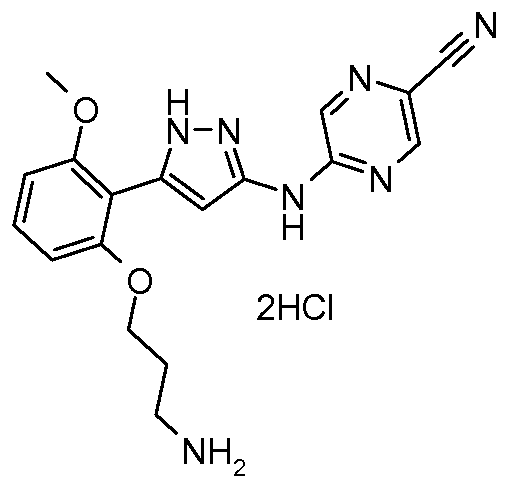
 2100300-72-7 CAS
2100300-72-7 CAS









































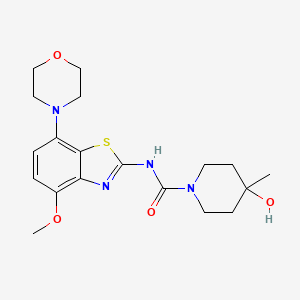





























































 Next generation β-lactamase inhibitors recently approved or in clinical trials. A. Avibactam. B. Relebactam. C. Vaborbactam.
Next generation β-lactamase inhibitors recently approved or in clinical trials. A. Avibactam. B. Relebactam. C. Vaborbactam.








 Moxalactam synthesis
Moxalactam synthesis









![[1,2,4]Triazolo[5,1-c][1,2,4]triazin-4(1H)-one, 7-(methylthio)-3-nitro-.png](http://pubchem.ncbi.nlm.nih.gov/image/imgsrv.fcgi?cid=3113817&t=l)

















































































































































































 (Ref. 0001)Optical rotation
(Ref. 0001)Optical rotation






















