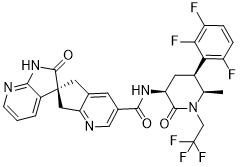


Atogepant
- Molecular FormulaC29H23F6N5O3
- Average mass603.515 Da
AGN 241689; MK 8031
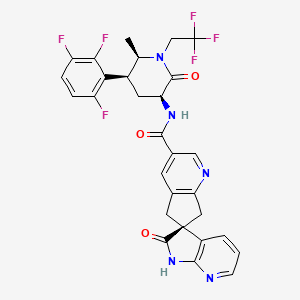
(3S)-N-[(3S,5S,6R)-6-methyl-2-oxo-1-(2,2,2-trifluoroethyl)-5-(2,3,6-trifluorophenyl)piperidin-3-yl]-2-oxospiro[1H-pyrrolo[2,3-b]pyridine-3,6′-5,7-dihydrocyclopenta[b]pyridine]-3′-carboxamide
Spiro(6H-cyclopenta(b)pyridine-6,3′-(3H)pyrrolo(2,3-b)pyridine)-3-carboxamide, 1′,2′,5,7-tetrahydro-N-((3S,5S,6R)-6-methyl-2-oxo-1-(2,2,2-trifluoroethyl)-5-(2,3,6-trifluorophenyl)-3-piperidinyl)-2′-oxo-, (3’S)-
- Originator Merck AG
- Developer Allergan
- Class Antimigraines; Monoclonal antibodies; Piperidines; Pyridines; Pyrroles; Small molecules; Spiro compounds
- Mechanism of Action Calcitonin gene-related peptide antagonists
Highest Development Phases
- Phase II/III Migraine
Most Recent Events
- 11 Jun 2018 Efficacy and adverse events data from a phase IIb/III trial in Migraine released by Allergan
- 23 Apr 2018 Allergan completes a phase II/III trial for Migraine (Prevention) in USA (PO) (NCT02848326)
- 14 Sep 2017 Chemical structure information added
The product was discovered by Merck and, in August 2015, it was licensed to Allergan for worldwide development and marketing.
Synthesis




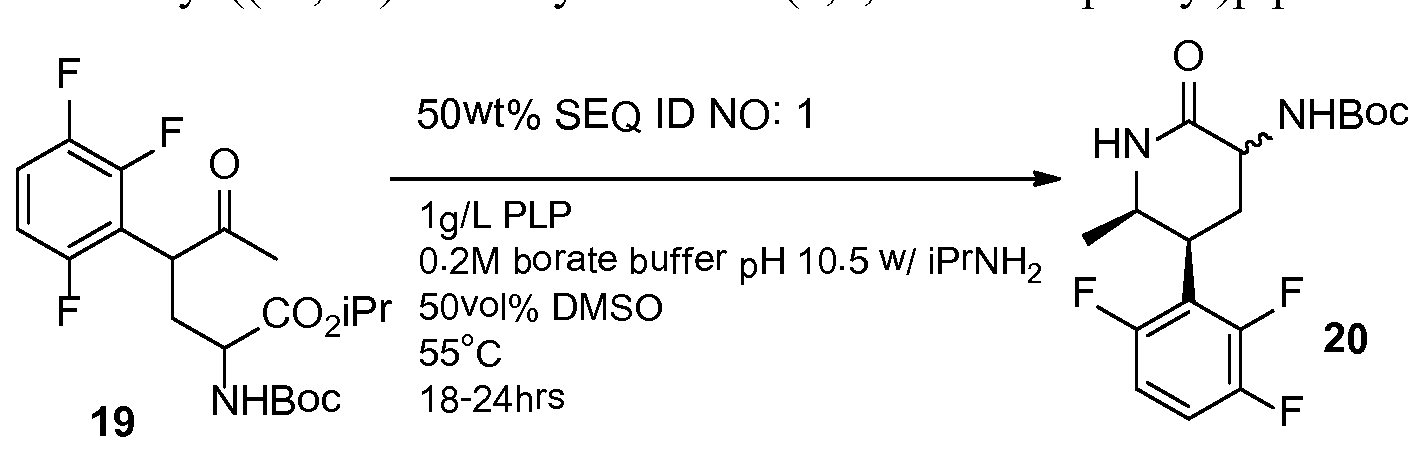





INTERMEDIATE 1


carboxylic acid
The title compound can be prepared by either Method I or Method II as described below.
Method I:
Step A: (6S)-3-Iodo-5 J-dihyc ospiro cyclopentar¾1pyrid e-6 ‘-py
one
A solution of sodium nitrite (36.1 g, 523 mmol) in water (20 mL) was added dropwise over 5 min to a solution of (6S -3-amino-5,7-dihydros iro[cyclopenta[ί)]pyridi e-6,3,– pyrrolo[2,3-0]pyridin]-2′(rH)-one (prepared according to the procedures described in
WO2008/020902, 66.0 g, 262 mmol) and -toluenesulfonic acid (149 g, 785 mmol) in acetonitrile (650 mL) at 23 °C. After stirring for 30 min, a solution of potassium iodide (109 g, 654 mmol) in water (20 mL) was added over 5 min. The resulting mixture was stirred at 23 °C for 40 min, then diluted with water (1 L) and basified by the addition of solid NaOH (33.0 g, 824 mmol) with stirring. Iodine by-product was reduced by the addition of 10% aqueous sodium thio sulfate solution and stirring for an additional 30 min. The solids were collected by filtration, washed with water, and dried under nitrogen atmosphere to give the title compound, which was used without further purification. MS: mlz = 363.9 (M + 1).
Step B: Methyl (65V2′-oxo-lΛ2 5J-tetrahydrospiroicvclopenta[6]p ridine-6.3′-pyrlΌlo[‘2. – 6]py ridine] – 3 -car boxy late
A solution of (65)-3-iodo~5 ,7-dihydrospiro[cyclopenta[&]pyridine-6,3′- pyrrolo[2,3-&]pyridin]-2′(rH)-one (51.0 g, 140 mmol), sodium acetate (23.0 g, 281 mmol) and dichloro l,l’~bis(diphenylphosphino)ferrocene palladium(II) dichloromethane adduct (2.9 g, 3.5 mmol) in MeOH (560 mL) was pressurized to 120 psi of CO at 23 °C and then heated at 80 °C for 12 h with stirring. The reaction mixture was diluted with water (1 L), and the precipitate collected by filtration, washed with water, and dried under nitrogen atmosphere to give the title compound, which was used without further purification. MS: mlz = 296.1 (M + 1).

3 -carboxylic acid
A mixture of methyl (6S)-2′-oxo-r,2′,5,7-tetrahydrospiro[cyclopenta[i)]pyridine- 6,3′-pyrrolo[2,3-&]pyridine]-3-carboxylate (30.0 g, 102 mmol) and aqueous 6 N sodium hydroxide solution (50.8 mL, 305 mmol) in MeOH (920 mL) was heated at reflux for 1 h. The mixture was allowed to cool to 23 °C before it was acidified to pH ~6 with aqueous 1 N hydrochloric acid solution, resulting in a black precipitate which was removed by filtration. The filtrate was concentrated under reduced pressure to a volume of ~100 mL and then partitioned between water (500 mL) and 2-methyltetrahydrofuran (2- eTHF, 250 mL). The aqueous layer was extracted with 2-MeTHF (5 χ 250 mL), and the combined organic layers were dried over sodium sulfate and concentrated to provide the title compound. MS: mlz ~ 282.0 (M + 1).
Method II:
Step A: Dimethyl 5-bromopyridine-2,3-dicarboxylate
Concentrated sulfuric acid (1 L, 18.7 mol) was added slowly over 10 min to a . suspension of pyridine-2,3-dicarboxylic acid (5.00 kg, 29.9 mol) in methanol (50 L), dissolving the suspension. The resulting mixture was heated at reflux for 48 h then cooled to 40 °C.
Bromine (8.0 kg, 50 mol) was added slowly over 2 h in 1-kg portions, keeping the temperature below 55 °C. The reaction mixture was then heated at 55 °C for 24 h, cooled to 50 °C and additional Br2 (4.0 kg, 25 mol) was added slowly over 1 h in 1-kg portions, keeping temperature below 55 °C. The reaction mixture was heated at 55 °C for 24 h, concentrated to a minimum volume (internal temp -30 °C, solution may occasionally foam), then diluted with isopropyl acetate (50 L) and washed with a saturated aqueous sodium sulfite solution (3 x 20 L) (final extract is ~pH 8) followed by water (20 L). The organic layer was concentrated to
approximately 15 L then diluted with heptane (40 L). The resulting slurry was stirred for 24 h at 23 °C. The solids were filtered, washed with heptane (10 L), and dried to give the title compound. Step B: (5-Bromopyridine-23-diyl)dimcthanol
Sodium borohydride (15.9g, 420 mmol) was added portionwise over 30 min to a solution of dimethyl 5-bromopyridine-2,3-dicarboxylate (20 g, 73 mmol) in ethanol (460 mL) precooled to 0 °C. A solution of calcium chloride (23.3 g, 209 mmol) in 150 mL was added slowly at 0 °C, and the reaction mixture was warmed to 23 °C and stirred overnight. Excess sodium borohydride was quenched by slow addition of aqueous 2 N HCl solution (230 mL, 460 mmol), followed by a stirring at 23 °C for 2 h. The mixture was concentrated to dryness.
Saturated aqueous sodium bicarbonate solution was added to the residue until a pH of approximately 7 was reached. The aqueous mixture was extracted with 2-methyltetrahydrofuran (4 x 200 mL). The combined organic layers were dried over sodium sulfate then treated with a solution of 4 N HC1 in dioxane (25 mL, 100 mmol). The resulting solid was filtered, washed with 2-methyltetrahydrofuran, and dried to give the title compound as a hydrochloride salt. MS: m!z = 218.1 (M + 1). Step C: (5-Bromopyridine-2,3-diyI)dimethanediyl dimethanesulfonate
A slurry of (5-bromopyridine-2,3-diyl)dimethanol hydrochloride (12.9g, 59.2 mmol) in tetrahydrofuran (400 mL) at 0 °C was treated with triethylamine (37.1 mL, 266 mmol). To the resulting mixture was added portionwise methanesulfonic anhydride (30.9 g, 177 mmol), keeping temperature below 5 °C. The reaction mixture was stirred at 0 °C for 1 h, then partitioned between saturated aqueous sodium bicarbonate solution (500 mL) and ethyl acetate (500 mL). The organic layer was washed saturated aqueous sodium bicarbonate solution, dried over magnesium sulfate, and concentrated to give the title compound. MS: m/z – 376.0 (M + 1).
Step D: 3-Bromo-r-{[2-(trimethylsilyl)ethoxy]methyl}-5,7- dihyjirpspiro [cyclop
(5-Bromopyridine-2,3-diyI)dimethanediyl dimethanesulfonate (17.0 g, 45.4 mmol) was added to a mixture of l-{[2-(trimetliylsilyl)ethoxy]methyl}-l}3-dihydro-2H- pyrrolo[2,3-&]pyridin-2-one (prepared according to the procedures described in
WO2008/020902, 14.0 g, 53.0 mmol) and cesium carbonate (49.0 g, 150 mmol) in ethanol (500 mL) 23 °C, and the resulting mixture was stirred for 20 h. The reaction mixture was
concentrated then partitioned between ethyl acetate (500 mL) and water (500 mL). The organic layer was dried over magnesium sulfate and concentrated. The residue was purified via silica gel chromatography (heptane initially, grading to 100% EtOAc) to give the title compound. MS: m/z = 448.1 (M + 1).
Step E: Methyl (6<Sf)-2′-oxo-r-{r2-(trimethylsilyl ethoxylmethyli-r,2′,5 J- tetrahydrospiro [cy clopenta[6] pyridine-6 ,3 ‘-pyrrolo [2, 3 -b]py ridinel -3 -carboxy late
A mixture of 3-bromo-r-{[2-(trimethylsilyl)ethoxy]methyl}-5,7- dihydrospiro[cyclopenta[¾]pyridine-6,3’-pyrrolo[2,3-¾pyridin]-2′(rH)-one (22.0 g, 49.3 mmol), PdCl2(dppf)»CH Cl2 (2.012g, 2.46 mmol), and sodium acetate (8.1g, 99 mmol) in in methanol (150 mL) was pressurized to 300 psi of carbon monoxide and then heated at 85 °C for 72 h. The reaction mixture was allowed to cool then concentrated. The residue was purified via silica gel chromatography (heptane initially, grading to 100% EtOAc) to give the title compound as a racemic mixture. MS: m/z – 426.1 (M +1). Resolution of the enantiomers by supercritical fluid chromatography (SFC) using a ChiralPak AD-H column and eluting with 40% ethanol in C02 (0.05% diethylamine as modifier) provided the title compound as the second enantiomer to elute.
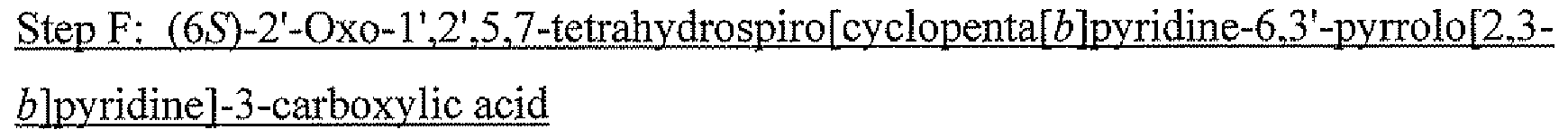
A solution of methyl (65)-2′-oxo- -{[2-(trimethylsilyl)ethoxy]methyl}-r!2′f5,7- tetrahydrospiro[cyclopenta[&]pyridine-6,3′-pyrrolo[2,3-&]pyridine]-3-carboxylate (238 g, 559 mmol) in methanol (2 L) was saturated with HCI gas, allowing temperature to increase to 55 °C. The reaction mixture was cooled to 23 °C, stirred for 20 h, then concentrated. Aqueous 10 N sodium hydroxide (400 mL, 4 mol) was added to a solution of the residue in methanol (2 L), and the resulting mixture was heated at reflux for 2 h. The solution was cooled to 23 °C and the pH was adjusted to 3 with concentrated HCI. The resulting solid was filtered, washed with water then heptane, and dried to give the title compound. MS: m!z = 282.2 (M + 1).
INTERMEDIATE 15


hydrochloride
Step A: (5SSR & 5j?,6y)-6-Methvi-l-r2.2.2-trifluoroethvn-5-(2,3.6-trifluorophenvnpiperidin-2- one
Essentially following the procedures described in Intermediate 14, but using 2,3,6-trifluorophenylboronic acid in place of 2,3,5-trifluorophenylboronic acid, the title compound was obtained. MS: m/z = 326.0 (M + 1).
Step B: GS.5S.6R & 3i?,5J?.6 ‘ -3-Azido-6-methyl-i-r2.2.2 rifluoroethyl)-5-(2.3.6- trifluorophenyl)piperidin-2-one
To a stirred solution of lithium 6w(trimethylsilyl)amide (1.0 M in THF, 4.80 mL,
4.80 mmol) in THF (20 mL) at -78 °C was added a cold (-78 °C) solution of (5S,6R & 5i?,6,S)-6- methyl-l-(2,2,2-trifluoroethyl)-5-(2,3,6-trifluorophenyl)piperidin-2-one (1.30 g, 4,00 mmol) in THF (10 mL) dropwise, keeping the internal temperature of the reaction mixture below -65 °C. The resulting mixture was stirred at -78 °C for 30 min, then a cold (-78 °C) solution of 2,4,6- triisopropylbenzenesulfonyl azide (Harmon et l. (1973) J Org. Chem. 38, 11-16) (1.61 g, 5.20 mmol) in THF (10 mL) was added dropwise, keeping the internal temperature of the reaction mixture below -65 °C. The reaction mixture was stirred at -78 °C for 30 min, then AcOH (1.05 mL, 18.4 mmol) was added. The resulting mixture was allowed to warm slowly to ambient temperature and was poured into saturated aqueous sodium bicarbonate (50 mL) and the mixture was extracted with EtOAc (2 χ 75 mL). The combined organic layers were washed with brine, then dried over sodium sulfate, filtered, and concentrated to dryness in vacuo. The crude product was purified by silica gel chromatography, eluting with a gradient of hexanes:EtOAc – 100:0 to 20:80, to give the diastereomeric azide products (3R,5Sf6R & 3S, ;5i?,65)-3-azido-6- methyl-l-(2,2,2-trifluoroethyl)-5-(2f3,5-trifluorophenyl)piperidin-2-one, which eluted second, and the title compound, which eluted first. MS: mlz = 367.1 (M+ 1).
Step C: ferf-Butyl [(3&5^6^ν6^Φν1-2-οχο-1-(2.2,2-ΐπΑηοΓθ£υΐν1 -5-ί2.3,6- trifluorophenyl)piperidin-3-yl“|carbamate
To a solution of ( S,5S,6R & 3JR,5if,6S)-3-azido-6-methyl-l-(2,2,2- trifiuoroethyl)-5-(2,3,5-trifluorophenyl)piperidin-2-one (280 mg, 0.764 mmol) and di-tert-butyl dicarbonate (217 mg, 0.994 mmol) in EtOH (5 mL) was added 10% palladium on carbon (25 mg, 0.024 mmol) and the resulting mixture was stirred vigorously under an atmosphere of hydrogen (ca. 1 atm) for 1 h. The reaction mixture was filtered through a pad of Celite® washing with EtOH, and the filtrate was concentrated in vacuo to give a crude solid. The crude product was purified by silica gel chromatography, eluting with a gradient of hexanes:EtOAc – 100:0 to 30:70, to give the racemic title compound. Separation of the enantiomers was achieved by SFC on a ChiralTech IC column, eluting with C02:MeOH:CH CN – 90:6.6:3.3, to give tert- butyl [(3i?,5i?,65)-6-methyl-2-oxo-l-(2J2,2-trifluoroemyl)-5-(2,3J6-tri¾orophenyl)piperidin-3- yl]carbamate as the first major peak, and fert-butyl [(3Sf5S,6R)-6-methyl-2-oxo-l -(2,2,2- trifluoroethyl)-5-(2,3,6-trifiuorophenyl)piperidin-3-yl]carbamate, the title compound, as the second major peak. MS: mlz = 463.2 (M + Na).
Step D: (3&5^6i?)-3-Amino-6-methyi-l-(2,2.2-trifluoroethyl)-5-(2,3,6- trifluorophenyl)piperidin-2-one hydrochloride
A solution of tert-butyl [(35′,55′,6ii)-6-methyl-2-oxo-l-(2J2,2-trifluoroethyl)-5-
(2s3,6-trifluorophenyl)piperidin-3-yl]carbamate (122 mg, 0.277 mmol) in EtOAc (10 mL) was saturated with HCl (g) and aged for 30 min. The resulting mixture was concentrated in vacuo to give the title compound. MS: mlz = 341.1 (M + 1); lH NM (500 MHz, CD3OD) δ 7.33 (qd, 1H, J- 9.3, 4.9 Hz), 7.05 (tdd, 1H, J= 9.8, 3.7, 2.2 Hz), 4.78 (dq, 1H, J= 15.4, 9.3 Hz), 4.22 (dd, 1H, J = 12.2, 6.6 Hz ), 4.06 (ddd, 1H, J- 13.3, 4.5, 2.7 Hz ), 3.97 (m, 1H), 3.73 (dq, 1H, J = 15.4, 8.8 Hz), 2.91 (qt, 1H, J- 12.7, 3.1 Hz), 2.36 (ddd, 1H, J= 12.7, 6.4, 2.0 Hz), 1.22 (d, 3H, J = 6.6 Hz).
EXAMPLE 4

f6SyN-[f3£5£6iO-6-Methyl-2-QXO-i-(2,2,,2-trifl^yl]-2′-oxo-l\2 5J~tetrahydrospiro[cyciopen^
carboxamide dihvdrochloride
To a stirred mixture of (6>$)-2′-οχο-Γ,2′,5,7- tetrahydrospirotcyclopenta[6]pyridine-6,3′-pyrroio[2,3-6]pyridine]-3-carboxylic acid (described in Intermediate 1) (264 mg, 0.939 mmol), (35′,5S’36J?)-3-amino-6-methyl-l-(2,2,2-trifluoroethyl)- 5-(2f3s6-trifluorophenyl)piperidin-2-one hydrochloride (described in Intermediate 15) (295 mg, 0.782 mmol), HOBT (144 mg, 0.939 mmol), and EDC (180 mg, 0.939 mmol) in DMF (8 mL) was added 7V,N-diisopropylethylamine (0.34 mL, 1.96 mmol), and the resulting mixture was stirred at ambient temperature for 3 h. The reaction mixture was then poured into saturated aqueous sodium bicarbonate (30 mL) and extracted with EtOAc (2 χ 40 mL). The combined organic layers were washed with brine, dried over sodium sulfate, and concentrated in vacuo. The residue was purified by silica gel chromatography, eluting with a gradient of
CH2Cl2:MeOH:NH40H – 100:0:0 to 90:10:0.1, to give the product, which was treated with HC1 in EtOAc at 0 °C to afford the title compound. HRMS: m/z = 604.1783 (M + 1), calculated m/z = 604.1778 for C29H24F6N5O3. iH NMR (500 MHz, CD3OD) δ 9.09 (s, 1H), 8.69 (s, 1H), 8.18 (dd, 1H, J = 5.9, 1.5 Hz), 7.89 (dd5 1H, J= 7.3, 1.5 Hz), 7.30 (m, 1H), 7.23 (dd, 1H, J= 7.3, 5.9 Hz), 7.03 (m, 1H), 4.78 (m, 1H), 4.61 (dd, 1H, J = 11.5, 6.6 Hz), 4.05 (dd, 1H, J= 13.8, 2.8 Hz), 3.96 (m, 1H), 3.84 (d, 1H, J= 18.6 Hz), 3.76 (d, 1H, J = 18.6 Hz), 3.73 (d, 1H, J= 17.3 Hz), (m, 1H), 3.61 (d, 1H, J = 17.3 Hz), 3.22 (m, 1H), 2.38 (m, 1H), 1.34 (d, 3H, J= 6.6 Hz).
-
Schemes 1 to 15 described below.
-
[0122]Scheme 1 illustrates a route to 3-aminopiperidinone intermediates of type 1.5 which may be used to prepare compounds of the present invention. Aryl acetone 1.1 can be alkylated using the iodoalanine derivative 1.2 under basic conditions to provide keto ester 1.3.
-
[0123]Reductive amination followed by cyclization and epimerization provides primarily cis-substituted lactam 1.4 as a racemic mixture. Chiral resolution using normal-phase liquid chromatography, for example, and removal of the Boc protecting group with HCl in EtOAc furnishes 3-aminopiperidinone 1.5 as a hydrochloride salt.
-
[0000]
![Figure US20160130273A1-20160512-C00020]()
-
[0124]An alternative sequence to 3-aminopiperidinone intermediates of type 1.5 is shown in Scheme 2. Reductive amination of keto ester 1.3 with ammonia followed by epimerization provides 2.1 as a mostly cis-substituted racemic mixture. Chiral resolution of the enantiomers provides 2.2. N-Alkylation with LiHMDS as base, for example, and an alkyl halide or epoxide affords 1.4. Removal of the Boc protecting group with HCl then affords 1.5 as a hydrochloride salt.
-
[0000]
![Figure US20160130273A1-20160512-C00021]()
-
[0125]A third method to 3-aminopiperidinone intermediates of type 1.5 is shown in Scheme 3. N-Alkylation of 5-bromo-6-methylpyridin-2(1H)-one (3.1) using cesium carbonate as base and an alkyl halide followed by nitration provides 3.2. Palladium-catalyzed cross-coupling with an aryl boronic acid then affords 3.3. Hydrogenation using platinum oxide under acidic conditions and chiral resolution of the mostly cis-substituted racemic product mixture provides 1.5 as a single enantiomer.
-
[0000]
![Figure US20160130273A1-20160512-C00022]()
-
[0126]A synthetic route to 3-aminopiperidinone intermediates of type 4.4 is shown in Scheme 4. Aryl acetonitrile 4.1 can be alkylated using the iodoalanine derivative 1.2 under basic conditions to provide cyano ester 4.2. Reductive cyclization using hydrogen and palladium hydroxide on carbon or Raney nickel, epimerization, and chiral resolution affords cis lactam 4.3 as a single enantiomer. N-Alkylation and removal of the Boc protecting group then provides 4.4 as a hydrochloride salt.
-
[0000]
![Figure US20160130273A1-20160512-C00023]()
-
[0127]Scheme 5 illustrates an alternative route to 3-aminopiperidinone intermediates of type 4.4. The arylacetonitrile 5.1 may be condensed with acrylate 5.2 at elevated temperature to give the 4-cyanobutanoate ester 5.3. Hydrogenation of nitrile 5.3 using Raney nickel catalyst and an ethanolic solution of ammonia affords the corresponding amine product, which typically cyclizes in situ to provide piperidinone 5.4. N-Alkylation of lactam 5.4 may be accomplished by a variety of methods known to those skilled in the art of organic synthesis, the exact choice of conditions being influenced by the nature of the alkylating agent, R1X. Electrophilic azidation of the resulting substituted lactam 5.5 can be accomplished using similar methodology to that described by Evans and coworkers (Evans et al. (1990) J. Am. Chem. Soc. 112, 4011-4030) to provide the azide 5.6 as a mixture of diastereoisomers, which can be separated by chromatography. The desired cis diastereomer of azide 5.6 may be reduced by catalytic hydrogenation in the presence of di-tert-butyl dicarbonate to give the corresponding Boc-protected amine 5.7, and separation of the enantiomers using chiral HPLC or SFC leads to the (3S,5S)-isomer 5.8. Finally, standard deprotection affords the desired 3-aminopiperidinone intermediate 4.4 as a hydrochloride salt.
-
[0000]
![Figure US20160130273A1-20160512-C00024]()
-
[0128]Another approach to 3-aminopiperidinone intermediates of interest, which is particularly useful for preparing 3-amino-6-methyl-5-arylpiperidin-2-ones such as 1.5, is outlined in Scheme 6. The pyridin-2(1H)-one 3.1 may be converted to the N-substituted pyridinone 6.1 by treatment with a suitable electrophile (R1X) under basic conditions. Pyridinone 6.1 can then be subjected to Suzuki-Miyaura coupling with the boronic acid 6.2, and the resulting 5-arylpyridinone 6.3 may be hydrogenated using, for example, platinum(IV) oxide catalyst to afford the corresponding 5-arylpiperidinone 6.4, which is usually obtained as predominantly the cis isomer. Further elaboration of piperidinone 6.4 may be achieved using analogous methodology to that described in Scheme 5. Specifically, electrophilic azidation followed by one-pot reduction and Boc protection leads to carbamate 6.6, and the desired enantiomer may be obtained using chiral chromatography. In some cases, the desired diastereomer of azide 6.5 may be isolated as a racemic mixture of the (3S,5S,6R)- and (3R,5R,6S)-isomers following silica gel chromatography of the crude product, and this mixture may be elaborated as outlined in Scheme 6. In other cases, it may be advantageous to take a mixture of diastereomers of azide 6.5 forward to the corresponding carbamate 6.6. The mixture of carbamate 6.6 diastereomers may be epimerized under basic conditions, such as potassium carbonate in EtOH, to afford a mixture that is significantly enriched in the desired (3S,5S,6R)- and (3R,5R,6S)-isomers, further purification may be employed to obtain the enantiomer of interest as outlined herein.
-
[0000]
![Figure US20160130273A1-20160512-C00025]()
![Figure US20160130273A1-20160512-C00026]()
-
[0129]A synthetic route to the azaoxindole pyridine acid intermediate 7.4 is shown in Scheme 7. Diazotization of aminopyridine 7.1, whose preparation is described in WO 2008/020902, followed by treatment with potassium iodide in the presence of NaNO2 provides iodide 7.2. Palladium-catalyzed carbonylation in methanol then affords ester 7.3, which may be saponified with sodium hydroxide to furnish 7.4.
-
[0000]
![Figure US20160130273A1-20160512-C00027]()
-
[0130]An alternative synthesis of the azaoxindole pyridine acid intermediate 7.4 is shown in Scheme 8. Esterification of diacid 8.1 followed by bromination provides 8.2. Reduction with sodium borohydride then furnishes diol 8.3. Alkylation of the protected azaoxindole 8.4 with the bis-mesylate produced from 8.3 affords the spirocycle 8.5. Palladium-catalyzed carbonylation in methanol followed by chiral resolution gives ester 8.6 as a single enantiomer. Removal of the SEM protecting group under acidic conditions and hydrolysis of the ester using sodium hydroxide then provides 7.4.
-
[0000]
![Figure US20160130273A1-20160512-C00028]()
-
[0131]A synthetic route to diazaoxindole carboxylic acid intermediate 9.7 is shown in Scheme 9. Esterification of acid 9.1 is followed by vinylation under palladium catalysis to afford divinyl pyridine 9.2. Ozonolysis with a borohydride reductive workup then yields diol 9.3. After mesylation and treatment with sodium choride, the resulting dichloro intermediate 9.4 can be alkylated with oxindole 9.5 under basic conditions to give spirocycle 9.6, following chiral resolution of the enantiomers. Dechlorination under buffered hydrogenation conditions and acidic deprotection affords acid 9.7.
-
[0000]
![Figure US20160130273A1-20160512-C00029]()
-
[0132]Useful derivatives of the intermediates described herein may be prepared using well-precedented methodology. One such example is illustrated in Scheme 10, in which the azaoxindole intermediate 7.4 is converted to the corresponding nitrile derivative 10.2, which may be used to prepare compounds of the present invention. Bromination of 7.4 with N-bromosuccinimide in boron trifluoride dihydrate provides the bromo derivative 10.1, which may be converted to the desired nitrile 10.2 using zinc cyanide and a palladium catalyst as shown.
-
[0000]
![Figure US20160130273A1-20160512-C00030]()
-
[0133]A synthetic route to the azaoxindole indane acid intermediate 11.17 is shown in Scheme 11. Esterification of diacid 11.1 followed by hydrogenation using palladium on carbon as a catalyst provides aniline 11.2. Dibenzylation under basic conditions with heat affords 11.3, and reduction of the diester with LiAlH4 furnishes diol 11.4. Chlorination with thionyl chloride provides benzyl chloride 11.5. Palladium-catalyzed amination of bromide 11.6 with tert-butylamine gives 11.7. Sequential treatment with n-hexyllithium and methyl chloroformate (2×) affords azaoxindole ester 11.8. Alkylation with the benzylchloride 11.5 under basic conditions in the presence of the cinchonidine-derived catalyst 11.12 (prepared via the alkylation of cinchonidine 11.10 with benzyl bromide 11.11) affords spirocycle 11.13. Deprotection of the azaoxindole using methanesulfonic acid with heat and debenzylation under standard hydrogenation conditions provides aniline 11.14. Diazotization followed by treatment with potassium iodide provides iodide 11.15. Palladium-catalyzed carbonylation in methanol then affords ester 11.16, which may be saponified with sodium hydroxide to furnish 11.17.
-
[0000]
![Figure US20160130273A1-20160512-C00031]()
-
[0134]An alternative synthesis of the azaoxindole pyridine acid intermediate 11.17 is shown in Scheme 12. Alkylation of the azaoxindole ester 11.8 with dibenzyl bromide 12.1 followed by chiral resolution of the enantiomers provides ester 12.2. Sequential deprotection of the azaoxindole using methanesulfonic acid with heat and hydrolysis of the ester provides 11.17.
-
[0000]
![Figure US20160130273A1-20160512-C00032]()
-
[0135]A synthetic route to the diazaoxindole carboxylic acid intermediate 13.4 is shown in Scheme 13. Alkylation of dibromide 12.1 with oxindole 9.5 under basic conditions and subsequent chiral resolution affords spirocycle 13.2. Dechlorination under buffered hydrogenation conditions and ester hydrolysis then affords acid 13.4.
-
[0000]
![Figure US20160130273A1-20160512-C00033]()
-
[0136]Useful derivatives of the intermediates described herein may be prepared using well-precedented methodology. One such example is illustrated in Scheme 14, in which the azaoxindole intermediate 11.17 is converted to the corresponding nitrile derivative 14.2, which may be used to prepare compounds of the present invention. Treatment of 11.17 with bromine in acetic acid provides the bromo derivative 14.1, which may be converted to the desired nitrile 14.2 using zinc cyanide and a palladium catalyst as shown.
-
[0000]
![Figure US20160130273A1-20160512-C00034]()
-
[0137]Scheme 15 illustrates conditions that can be used for the coupling of 3-aminopiperidinone intermediates, such as 15.1, and carboxylic acid intermediate 15.2, to produce, in this instance, amides 15.3. These standard coupling conditions are representative of the methods used to prepare the compounds of the present invention.
-
[0000]
![Figure US20160130273A1-20160512-C00035]()
-
[0138]The previous methods for synthesizing the lactam intermediate suffered from one or more drawbacks: racemic mixture was separated by chiral-HPLC, separation of diasteromixture by crystallization and/or use of costly PtO2. The process of the instant invention utilizes a transaminase induced dynamic kinetic resolution providing high diastereoselectivity at positions C5 and C6. N-mono-trifluoroethylation was discovered and developed. Cis and trans isomer at the alpha position of the amine was successfully controlled by crystallization in the presence of arylaldehyde derivatives. Overall, synthetic steps are shorter, practical and efficient and yield is dramatically improved.















-
- Example 1 Isopropyl 2-(tert-butoxycarbonylamino)-3-(methylsulfonyloxy)propanoate (2)
-
[0139]
![Figure US20160130273A1-20160512-C00036]()
-
[0140]To a solution of N-tert-butyl-L-serine isopropyl ester 1 (12 g, 48.5 mmol)* and methanesulfonyl chloride (4.0 ml) in dichloromethane (100 mL), triethylamine (7.2 ml) was added slowly under an ice bath. The reaction mixture was stirred at room temperature for 1 h, then 1 N HCl (40 mL) was added with stirring. The organic layer was separated, washed with 1 N HCl (40 ml) and brine (40 ml), dried over MgSO4, and concentrated in vacuo to give 2 (14.5 g, 91.9%) as a solid. 1H NMR (CDCl3, 500 MHz): δ 5.45 (s, broad, 1H), 5.13 (m, 1H), 4.62-4.47 (m, 3H), 3.04 (s, 3H), 1.48 (s, 9H), 1.31 (d, J=6.4 Hz, 6H); 13C NMR (CDCl3, 100 MHz): δ 168.0, 135.1, 80.6, 70.5, 69.1, 53.3, 37.4, 28.3, 21.7, 21.6; HRMS m/z calcd. for C12H23NO7S 348.1087 (M+Na). found 348.1097
-
[0000]* preparation of 1 was reported in J. Med. Chem., 2010, 53, 6825-6837 6825
-

Isopropyl 2-(tert-butoxycarbonylamino)-3-iodopropanoate (3)
-
-
[0141]
![Figure US20160130273A1-20160512-C00037]()
-
[0142]To a solution of 2 (392 g) in acetone (3.14 L), sodium iodide (542 g) was added. The reaction temperature went up to 29° C. from 17° C. The reaction mixture was maintained at room temperature over weekend. The mixture was filtrated and washed with MTBE. The filtrate and washings were combined and concentrated. The residue was treated with MTBE and water with a small amount of sodium thiosulfate. The organic layer was washed with water and concentrated to an oil. The oil was charged slowly into a mixture of water (2 L) and DMF (300 ml) with a small amount of seed at 5° C. The crystals were filtered and dried to give 3 (400 g, 93% yield).
-

Isopropyl 4-(4-bromophenyl)-2-(tert-butoxycarbonylamino)-5-oxohexanoate (5) and isopropyl 4-phenyl-2-(tert-butoxycarbonylamino)-5-oxohexanoate (6)
-
-
[0143]
![Figure US20160130273A1-20160512-C00038]()
-
[0144]To a solution of 4 (51.7 g, 243 mmol) in DMF (850 ml) was added 3 (88 g, 246 mmol). The resulting solution was cooled to 5° C. and Cs2CO3 (240 g) was added in one portion. The suspension was warmed to 15° C. and stirred at this temperature for 2.5 h. Additional Cs2CO3 (25 g) was charged and the mixture was stirred for additional 8 h or until HPLC analysis indicated the conversion was greater than 95%. The batch was then slowly quenched into a mixture of 2N HCl (850 mL) and MTBE (900 mL) at 5-20° C. Organic layer was separated and aqueous layer extracted with MTBE (400 mL). Combined organic layers were washed with 5% NaHCO3solution (400 mL) twice. The resulting solution containing desired product 5 (90% LC purity) was concentrated under vacuum. The residue was dissolved in isopropanol (1 L). To the solution was added K2CO3 (25 g), potassium formate (34 g) and 10% Pd/C (20 g). The mixture was warmed up to 60° C. and stirred for 2 h. The mixture was filtered after cooling to room temperature. The HPLC analysis of the filtrate indicated that the solution contained 6 (54.7 g, 95 wt %, 62% yield). The crude product was used directly in the next step without further purification. The compound 6 is a mixture of two pair of diastereomers 6-1 and 6-2, partially separable by flash chromatography on silica gel with ethyl acetate and heptane as a eluant (1:10). 6-1: 1H NMR (CDCl3, 500 MHz): δ 7.35 (m, 2H), 7.30 (m, 1H), 7.20 (m, 2H), 5.17 (br, 1H), 4.95 (m, 1H), 4.76 (br, 1H), 3.73 (m, 1H), 2.70 (br, 1H), 2.07 (s, 1H), 1.45 (s, 9H), 1.29 (d, J=6.6 Hz, 3H), 1.28 (d, J=6.6 Hz, 3H); 6-2: 1H NMR (CDCl3, 500 MHz): δ 5.12 (m, 1H), 4.70 (m, 1H), 3.27 (m, 1H), 2.80 (m 1H), 2.34 (s, 3H), 1.50 (s, 9H), 1.26 (d, J=6.6 Hz, 3H), 1.25 (d, J=6.6 Hz, 3H); HRMS m/z: cacld. for 6-1: C20H29NO5 386.1938 (M+Na). found 386.1947.
-

Isopropyl 2-((tert-butoxycarbonyl)amino)acrylate (7)
-
-
[0145]
![Figure US20160130273A1-20160512-C00039]()
-
[0146]To a solution of 1 (10.05 g, 40.6 mmol) in DMF (100 mL) was added MsCl (4.12 mL, 52.8 mmol) under ice-cooling. Triethylamine (14.16 mL, 102.0 mmol) was then added dropwise via an addition funnel over 30 min, while maintaining the reaction temperature between 0-5° C. When the addition was complete, the cooling bath was removed and the yellow heterogeneous reaction mixture was aged at room temperature under N2for overnight. The reaction mixture was diluted with ice cold water (1 L) and MTBE (1 L). The layers were separated and the aqueous layer was back-extracted with MTBE (500 mL). The organic layers were combined and washed with 1M citric acid (750 mL), water (1 L) and then 10% aqueous NaCl (1 L). The organic solution contained 7 (8.652 g, 93% yield). Solvent was switched to DMSO at <40° C. and use solution directly in next step.
-

Isopropyl 4-phenyl-2-(tert-butoxycarbonylamino)-5-oxohexanoate (6)
-
-
[0147]
![Figure US20160130273A1-20160512-C00040]()
-
[0000]Compound 6 was prepared from 7 in DMSO in the presence of 0.5 equiv. Cs2CO3 with 1.05 equiv. of phenylacetone at room temperature in 79% yield.
-

tert-Butyl(5S,6R)-6-methyl-2-oxo-5-phenylpiperidin-3-ylcarbamate (8)
-
-
[0148]
![Figure US20160130273A1-20160512-C00041]()
-
[0149]To a 5 L RBF with overhead stirring, a temperature control, a pH probe and a base addition line, was added sodiumtetraborate decahydrate (26.7 g) and DI water (1.4 L). After all solids were dissolved, isopropylamine (82.8 g) was added. The pH of the buffer was adjusted to pH 10.5 using 6 N HCl. The buffer was cooled to room temperature. Then, pyridoxal-5-phosphate (2.8 g) and SEQ ID NO: 1 (70 g) were added and slowly dissolved at room temperature.
-
[0150]An oil (197.9 g, containing 70.7 wt % keto ester 6 (140 g, 0.385 mol) were dissolved in DMSO (1.4 L). The solution was added to the flask over 5-10 min and the reaction was heated to 55° C. The pH was adjusted to 10.5 according to a handheld pH meter and controlled overnight with an automated pH controller using 8 M aqueous isopropylamine. The reaction was aged for 24 h.
-
[0151]After confirmation of >95A % conversion by HPLC, the reaction was extracted by first adding a mixture of iPA:IPAc (3:4, 2.8 L) and stirring for 20 min. The phases were separated and the aqueous layer was back extracted with a mixture of iPA:IPAc (2:8, 2.8 L). The phases were separated, the organic layers were combined and washed with DI water (0.5 L). The HPLC based assay yield in the organic layer was 8 (114.6 g) with >60:1 dr at the positions C5 and C6. The ratio of stereoisomers at position C2 was ˜1:1. The extract was concentrated and dissolved in CH2Cl2. The organic solution was washed with water then saturated aqueous NaCl, concentrated and crystallized from MTBE/n-hexane (2:3). The crystal was filtered at room temperature and washed with MTBE/n-hexane (2:3) and dried to afford a cis and trans mixture (˜1:1.2) of the lactam 8 (99.6 g, 80.0%) as crystals.
-
[0000]cis: trans (˜1:1.2) mixture but NMR integration was reported as 1:1 (for proton number counts) Mp 87-90.9° C.; 1H NMR (CDCl3, 400 MHz): δ 7.40-7.20 (m, 8H, cis and trans), 7.16-7.12 (m, 2H, cis and trans); 6.56 (broad s, 1H, trans), 6.35 (broad s, 1H, cis), 5.57 (broad d, J=4.6 Hz, 1H, cis), 5.34 (broad d, J=5.7 Hz, 1H, trans), 4.33-4.15 (m, 2H, cis and trans), 3.93 (m, 1H, trans), 3.81 (m, 1H, cis), 3.41 (dt, J=11.8, 5.0 Hz, 1H, cis), 3.29 (dt, J=8.0, 4.4 Hz, 1H, trans), 2.74 (m, 1H, cis), 2.57 (m, 1H, trans), 2.23 (ddd, J=13.5, 8.0, 4.4 Hz, trans), 2.07 (q, J=11.8 Hz, 1H, cis), 1.46 (s, 9H, cis), 1.42 (s, 9H, trans), 1.05 (d, J=6.9 Hz, 3H, trans), 0.89 (d, J=6.9 Hz, 3H, cis); 13C NMR (CDCl3, 100 MHz): δ 171.52 (cis), 171.46 (trans), 156.04 (cis or trans), 155.93 (cis or trans), 140.8 (cis), 139.9 (trans), 128.8 (trans), 128.7 (cis), 128.6 (trans), 128.1 (cis), 127.25 (trans), 127.18 (cis), 79.98 (trans), 79.91(cis), 52.4 (trans), 51.8 (broad, cis), 51.7 (cis), 49.0 (broad, trans), 42.1 (cis), 41.9 (trans), 32.4 (broad, trans), 30.1 (cis), 28.57 (cis or trans), 28.53(cis or trans), 18.3 (cis), 18.1 (broad, trans); HRMS m/z cacld. for C17H24N2O3327.1679 (M+Na). found 327.1696
-

tert-Butyl(5S,6R)-6-methyl-2-oxo-5-phenyl-1-(2,2,2-trifluoroethyl)piperidine-3-ylcarbamate (9) and tert-butyl(5S,6R)-6-methyl-2-oxo-5-phenyl-1-(2,2,2-trifluoroethyl)piperidine-3-yl(2,2,2-trifluoroethyl)carbamate (10)
-
-
[0152]
![Figure US20160130273A1-20160512-C00042]()
-
[0153]To the solution of 8 (480 g, 1.58 mol) in anhydrous THF (3.8 L) was added lithium tert-amoxide solution in heptane (512 mL, 3.1 M, 1.58 mol) over about 15 min while maintaining the reaction temperature between 15 and 20° C. The resulting solution was then cooled to a temperature between 0 and 2° C. 2,2,2-Trifluoroethyl trifluoromethanesulfonate (368 g, 1.58 mol) was added over 15 min while maintaining the reaction temperature between 0 and 3° C. The solution was agitated at 0° C. for 15 min. DMPU (300 ml) was charged to the mixture through an additional funnel over 30 min while maintaining the reaction temperature between 0 and 3° C. The resulting solution was agitated at 0° C. for 2.5 h. Another 2,2,2-trifluoroethyl trifluoromethanesulfonate (182 g, 0.79 mol) was added to the mixture over 10 min followed by another 3.1 M lithium tert-amoxide solution (104 mL) while maintaining the reaction temperature between 0 and 3° C. The batch was agitated for another 2.5 h at 0° C. The mixture was quenched into a mixture of heptane (4.8 L), water (3.4 L) and 2N HCl solution (280 mL) below 15° C. The phases were separated. The aqueous phase was extracted with heptane (4 L). The combined organic phase was washed with water (2 L). The solution was concentrated to a volume of about 1 L under vacuum between 25 and 50° C. The crude material was passed through a short silica gel plug with heptane/ethyl acetate. The resulting solution was concentrated under vacuum until distillation stopped at a temperature below 50° C., dissolved in IPAc (2 L) and used for the next processing step. The assay yield of 9 for both cis and trans isomers was 85% in the ratio of ˜8 to 1.
-
[0154]Analytically pure cis and trans isomers of 9 were isolated by chromatography on silica gel with ethyl acetate and heptane as eluant. 9 (cis): 1H NMR (CDCl3, 500 MHz): δ 7.30 (m, 5H), 5.75 (s, broad, 1H), 4.35 (m, 1H), 4.15 (m, 1H), 3.80 (m, 1H), 3.50 (m, 1H), 3.17 (m, 1H), 2.45 (m, 2H), 1.45 (s, 9H), 0.93 (d, J=6.7 Hz, 3H); 13C NMR (CDCl3, 100 MHz): δ 170.3, 155.9, 140.0, 128.6, 127.6, 127.1, 124.6 (q, J=279 Hz), 79.7, 58.7, 52.2, 45.3 (q, J=33.7 Hz), 41.9, 28.3, 27.4, 13.4; HRMS: m/z calcd for C19H25F3N2O3 387.1890 (M+H). found: 387.1899. 9 (trans): 1H NMR (CDCl3, 500 MHz): δ 7.40 (m, 2H), 7.30 (m, 3H), 5.55 (br, 1H), 4.53 (br, 1H), 4.45 (m, 1H), 3.78 (m 2H), 3.45 (m, 1H), 3.0 (m, 1H), 2.12 (m, 1H), 1.46 (s, 9H), 1.12 (d, J=7.0 Hz, 3H); 13C NMR (CDCl3, 100 MHz): δ 170.2, 155.9, 139.6, 128.7, 127.9, 127.4, 124.3 (q, J=279 Hz), 80.0, 59.6, 49.1, 46.9 (q, J=34.0 Hz), 42.1, 28.3, 25.3, 13.4; HRMS: m/z calcd for C19H25F3N2O3387.1890 (M+H). found 387.1901.
-

(3S,5S,6R)-6-Methyl-2-oxo-5-phenyl-1-(2,2,2-trifluoroethyl)piperidine-3-aminium 4-nitrobenzoate (11)
-
-
[0155]
![Figure US20160130273A1-20160512-C00043]()
-
[0156]To a solution of the crude 9 obtained from above experiment (10 g assay, 25.9 mmol) in iPAC (8 ml) was added p-toluenesulfonic acid monohydrate (6.7 g, 35.2 mmol) and the mixture was stirred at 50-60° C. for 3 hr until the reaction was completed (>99%). The solution was cooled to 15-20° C., and washed with 10% aqueous K2CO3 followed by water. The aqueous layers were re-extracted with iPAc (5 ml). The organic layers were combined and heated to 55-60° C. 4-Nitrobenzoic acid (3.9 g, 23.2 mmol) was slowly added in 20 min. The mixture was slowly cooled to room temperature. 5-Nitro-2-hydroxylbenzaldehyde (50 mg) was added and the batch was agitated for at least 12 h. The mixture was filtrated and washed with MeCN to give 11 as crystals. Optionally, a slurry in MeCN was carried out for further purification of 11. The isolated yield was 90%. Mp 205-208° C.; 1H NMR (DMSO-d6, 400 MHz): δ 8.21 (dd, J=9.0, 2.1 Hz, 2H), 8.08 (dd, J=9.0, 2.1 Hz, 2H), 7.37 (t, J=7.4 Hz, 2H), 7.28 (t, J=7.4 Hz, 1H), 7.24 (d, J=7.4 Hz, 2H), 4.65 (ddd, J=15.1, 9.7, 7.7 Hz, 1H), 3.72-3.98 (m, 3H), 3.57 (m, 1H), 2.46 (q, J=12.6 Hz, 1H), 2.25 (m, 1H), 0.90 (d, J=6.4 Hz, 3H); 19F NMR (DMSO-d6, 376 MHz): δ −69 (s); 13C NMR (DMSO-d6, 100 MHz): δ 168.7, 167.3, 148.3, 143.8, 140.1, 130.1, 128.6, 127.4, 127.0, 124.9 (q, J=280.9 Hz), 122.8, 58.7, 49.8, 44.5 (q, J=32.7 Hz), 40.6, 25.3, 13.2.
-

(5S,6R)-3-Amino-6-methyl-5-phenyl-1-(2,2,2-trifluoroethyl)piperidin-2-one (12)
-
-
[0157]
![Figure US20160130273A1-20160512-C00044]()
-
[0158]To a mixture of 8 (20.0 g, 65.7 mmol) and Na2S2O3 (0.52 g, 3.3 mmol) in THF (200 mL) was added tert-BuOLi (6.8 g, 85 mmol) at 16° C. The mixture was stirred at 16° C. for 15 min followed by addition of trifluoroethyl trifluoromethansulfonate (20.6 g, 89 mmol) in one portion. The resulting mixture was stirred for 18 h at 16° C. The reaction mixture was then quenched by addition of toluene (70 mL) followed by 0.5N HCl solution (50 mL). The aqueous layer was separated and extracted with toluene (20 mL). The combined organic layer contained 87% of 9, 6% of 10 and 6% of 8 by HPLC and yield for the desired product 9 was 87%. The organic layer was then stirred with 3N HCl solution (80 ml) and tetrabutylammoniium bromide (0.8 g) for about 3 h until HPLC analysis indicated selective removal of the Boc group in the unreacted 8 was completed. The aqueous layer was removed. The organic layer containing 9 and 10 was then concentrated under vacuum at 60° C. to remove most of solvent. The residue was dissolved in MTBE (60 mL), and 5N HCl solution (65 mL) was added. The diphasic solution was agitated vigorously at 50° C. for about 5 h until the deprotection of 9 was completed while 10 was mainly intact. After addition of heptane (30 mL) to the mixture, the organic layer was separated at 45° C. The aqueous layer was diluted with water (60 mL) and resulting aqueous and washed with heptane (30 mL) at 45° C. The aqueous solution was then mixed with MTBE (100 mL) and basified with 10 N NaOH solution until the pH of the mixture was about 10. The organic layer was separated and the aqueous layer was back-extracted with MTBE (60 mL). The combined organic layers were washed with brine (60 mL). The resulting organic solution was suitable for next reaction. The solution was contained 12 (15.6 g, 83% from 8) with 97% LC purity as a mixture of two diastereomers (cis and trans) in 4 to 1 ratio.
-

(3S,5S,6R)-6-Methyl-2-oxo-5-phenyl-1-(2,2,2-trifluoroethyl)piperidin-3-aminium 4-methylbenzoate (13)
-
-
[0159]
![Figure US20160130273A1-20160512-C00045]()
-
[0160]To a suspension of 4-methylbenzoic acid (6.8 g, 49.9 mmol) and 3,5-dichlorosalicylaldehyde (93 mg, 0.49 mmol) in MTBE (40 mL) was added a solution of 12 (13.9 g, 48.5 mmol) in MTBE (about 150 mL) over 1 h at 50° C. The resulting suspension was agitated for about 3 h at 50° C. The solids were collected by filtration after cooling to −5° C. over 1 h. The cake was washed with MTBE (50 mL). The solids were dried in a vacuum oven to give 13 (17.6 g, 86%) as crystals with 99.5% LC purity and 99.6% de. 1H NMR (DMSO-d6, 400 MHz): δ 7.85 (d, J=8.1 Hz, 2H), 7.40 (m, 2H), 7.25 (m, 5H), 6.0 (br, 3H), 4.65 (m, 1H), 3.65-3.80 (m, 2H), 3.45-3.65 (m, 2H), 2.35 (s, 3H), 2.30 (m, 1H), 2.15 (m, 1H), 0.88 (d, J=6.5 Hz, 3H); 13C NMR (DMSO-d6, 100 MHz): δ 172.4, 168.5, 142.1, 141.1, 130.9, 129.7, 129.2, 129.0, 128.0, 125.5 (q, J=279 Hz), 59.1, 51.6, 45.1 (q, J=32 Hz), 41.6, 28.0, 21.5, 13.9.
-

(S)—N-((3S,5S,6R)-6-Methyl-2-oxo-5-phenyl-1-(2,2,2-trifluoroethyl)piperidine-3-yl)-2′-oxo-1′,2′,5,7-tetrahydrospiro[cyclopenta[b]pyridine-6,3′-pyrrolo[2,3-b]pyridine]-3-carboxamide trihydrate (15)
-
-
[0161]
![Figure US20160130273A1-20160512-C00046]()
-
[0162]To a suspension of 11 (465 g, 96% wt, 0.99 mol) in iPAc (4.6 L) was added 5% aqueous K3PO4 (4.6 L). The mixture was stirred for 5 min. The organic layer was separated and washed with 5% aqueous K3PO4 (4.6 L) twice and concentrated in vacuo and dissolved in acetonitrile (1.8 L).
-
[0163]To another flask was added 14 (303 g, 91.4 wt %), acetonitrile (1.8 L) and water (1.8 L) followed by 10 N NaOH (99 mL). The resulting solution was stirred for 5 min at room temperature and the chiral amine solution made above was charged to the mixture and the container was rinsed with acetonitrile (900 mL). HOBT hydrate (164 g) was charged followed by EDC hydrochloride (283 g). The mixture was agitated at room temperature for 2.5 h. To the mixture was added iPAc (4.6 L) and organic layer was separated, washed with 5% aqueous NaHCO3 (2.3 L) followed by a mixture of 15% aqueous citric acid (3.2 L) and saturated aqueous NaCl (1.2 L). The resulting organic layer was finally washed with 5% aqueous NaHCO3 (2.3 L). The organic solution was concentrated below 50° C. and dissolved in methanol (2.3 L). The solution was slowly added to a mixture of water (6 L) and methanol (600 mL) with ˜2 g of seed crystal. And the resulting suspension was stirred overnight at room temperature. Crystals were filtered, rinsed with water/methanol (4 L, 10:1), and dried under nitrogen flow at room temperature to provide 15 (576 g, 97% yield) as trihydrate.
-
[0164]1H NMR (500 MHz, CDCl3): δ 10.15 (br s, 1H), 8.91 (br s, 1H), 8.21 (d, J=6.0 Hz, 1H), 8.16 (dd, J=5.3, 1.5 Hz, 1H), 8.01 (br s, 1H), 7.39-7.33 (m, 2H), 7.31-7.25 (m, 1H), 7.22-7.20 (m, 2H), 7.17 (dd, J=7.4, 1.6 Hz, 1H), 6.88 (dd, J=7.4, 5.3 Hz, 1H), 4.94 (dq, J=9.3, 7.6 Hz, 1H), 4.45-4.37 (m, 1H), 3.94-3.87 (m, 1H), 3.72 (d, J=17.2 Hz, 1H), 3.63-3.56 (m, 2H), 3.38-3.26 (m, 1H), 3.24 (d, J=17.3 Hz, 1H), 3.13 (d, J=16.5 Hz, 1H), 2.78 (q, J=12.5 Hz, 1H), 2.62-2.56 (m, 1H), 1.11 (d, J=6.5 Hz, 3H); 13C NMR (126 MHz, CD3CN): δ 181.42, 170.63, 166.73, 166.63, 156.90, 148.55, 148.08, 141.74, 135.77, 132.08, 131.09, 130.08, 129.66, 129.56, 128.78, 128.07, 126.25 (q, J=280.1 Hz), 119.41, 60.14, 53.07, 52.00, 46.41 (q, J=33.3 Hz), 45.18, 42.80, 41.72, 27.79, 13.46; HRMS m/z: calcd for C29H26F3N5O3 550.2061 (M+H). found 550.2059.
-

Alternative Procedure for 15
-
-
[0165]
![Figure US20160130273A1-20160512-C00047]()
-
[0166]To a suspension of 13 (10 g, 98 wt %, 23.2 mmol) in MTBE (70 mL) was added 0.6 N HCl (42 mL). The organic layer was separated and extracted with another 0.6 N HCl (8 mL). The combined aqueous solution was washed with MTBE (10 mL×3). To the resulting aqueous solution was added acetonitrile (35 mL) and 14 (6.66 g, 99 wt %). To the resulting suspension was neutralized with 29% NaOH solution to pH 6. HOPO (0.26 g) was added followed by EDC hydrochloride (5.34 g). The mixture was stirred at room temperature for 6-12 h until the conversion was complete (>99%). Ethanol (30 ml) was added and the mixture was heated to 35° C. The resulting solution was added over 2 h to another three neck flask containing ethanol (10 mL), water (30 mL) and 15 seeds (0.4 g). Simultaneously, water (70 mL) was also added to the mixture. The suspension was then cooled to 5° C. over 30 min and filtered. The cake was washed with a mixture of ethanol/water (1:3, 40 mL). The cake was dried in a vacuum oven at 40° C. to give 15 trihydrate (13.7 g, 95%) as crystals.
-

Example 2 N-Methoxy-N-methyl-2-(2,3,6-trifluorophenyl)acetamide (17)
-
-
[0167]
![Figure US20160130273A1-20160512-C00048]()
-
[0168]To a solution of DMF (58.1 mL, 750 mmol) in iPAc (951 mL) was added POCl3 (55.9 mL, 600 mmol) under ice-cooling. After aged for 1 h under ice-bath, acid 16 (95 g, 500 mmol) was added under ice-cooling. The solution was stirred under ice-cooling for 30 min. The solution was added over 30 min into a solution of K2CO3 (254 g, 1.835 mol) and NHMe(OMe)HCl (73.2 g, 750 mmol) in water (951 mL) below 8° C. After aged for 30 min below 8° C., the organic layer was separated, washed with water (500 mL) twice and sat. NaCl aq (100 mL) once, and concentrated in vacuo to afford 17 as an oil (117.9 g, 97.7 wt %, 99% yield). 1H NMR (CDCl3, 400 MHz); δ 7.05 (m, 1H), 6.82 (m, 1H), 3.86 (s, 2H), 3.76 (s, 3H), 3.22 (s, 3H); 19F NMR (CDCl3, 376.6 MHz); δ −120.4 (dd, J=15.1, 2.7 Hz), −137.9 (dd, J=20.8, 2.7 Hz), −143.5 (dd, J=20.8, 15.1 Hz); 13C NMR (CDCl3, 100 MHz); δ 169.4, 156.9 (ddd, J=244, 6.2, 2.7 Hz), 149.3 (ddd, J=249, 14.4, 8.4 Hz), 147.1 (ddd, J=244, 13.1, 3.5 Hz), 115.5 (ddd, J=19.4, 9.9, 1.5 Hz), 133.4 (dd, J=22.3, 16.4 Hz), 110.2 (ddd, J=24.8, 6.7, 4.1 Hz), 32.4 (broad), 26.6 (m); HRMS m/z calcd for C10H10F3NO2 234.0736 (M+H). found 234.0746.
-

1-(2,3,6-Trifluorophenyl)propan-2-one (18)
-
-
[0169]
![Figure US20160130273A1-20160512-C00049]()
-
[0170]A mixture of CeCl3 (438 g, 1779 mmol) and THF (12 L) was heated at 40° C. for about 2 h then cooled to 5° C. Methylmagensium chloride in THF (3 M, 3.4 L) was charged at 5-9° C. and then it was warmed up to 16° C. and held for 1 h. The suspension was re-cooled to −10 to −15° C. A solution of 17 (1.19 kg) in THF (2.4 L) was charged into the suspension over 15 min. After confirmation of completion of the reaction, the reaction mixture was transferred to a cold solution of hydrochloric acid (2 N, 8.4 L) and MTBE (5 L) in 5-10° C. The aqueous phase was separated and the organic layer was washed with aqueous 5% K2CO3 (6 L) and then 10% aqueous NaCl (5 L). The organic layer was dried over Na2SO4, concentrated to give crude 18 (917 g, >99 wt %) in 95% yield. The crude 18 was used in the next step without further purification. Analytically pure 18 was obtained by silica gel column.
-
[0171]1H NMR (CDCl3, 400 MHz); δ 7.07 (m, 1H), 6.84 (m, 1H), 3.82 (s, 2H), 2.28 (s, 3H); 19F NMR (CDCl3, 376.6 MHz); δ −120.3 (dd, J=15.3, 2.5 Hz), −137.8 (dd, J=21.2, 2.5 Hz), −143.0 (dd, J=20.2, 15.3 Hz); 13C NMR (CDCl3, 100 MHz); δ 202.2, 156.5 (ddd, J=244, 6.3, 2.9 Hz), 148.9 (ddd, J=249, 14.4, 8.6 Hz), 147.0 (ddd, J=244, 13.1, 3.5 Hz), 115.7 (ddd, J=19.4, 10.5, 1.2 Hz), 112.8 (dd, J=22.7, 17.0 Hz), 110.3 (ddd, J=24.8, 6.7, 4.1 Hz), 37.2 (d, J=1.2 Hz), 29.3.
-

Isopropyl 2-((tert-butoxycarbonyl)amino)-5-oxo-4-(2,3,6-trifluorophenyl)hexanoate (19)
-
-
[0172]
![Figure US20160130273A1-20160512-C00050]()
-
[0173]To a solution of 18 (195 g, 1.03 mol) in MTBE (1.8 L) was added zinc bromide (67 g, 0.30 mol) followed by 2 (390 g, 1.2 mol). tert-BuOLi (290 g, 3.6 mol) was then added in several portions while maintaining the reaction temperature below 40° C. The resulting mixture was stirred at 35° C. for 24 h and quenched into a mixture of 2 N HCl (5.6 L) and heptane (5 L) at 0° C. The organic layer was separated and washed with 5% aqueous NaHCO3 (5 L) twice. The resulting organic solution was concentrated under vacuum. The residue was dissolved in heptane (2 L) and the solution was concentrated again under vacuum. The resulting oil was dissolved in DMSO (2.5 L) and the solution was used in the next step without further purification. HPLC analysis indicated that the solution contained the desired product 19 (290 g, 67% yield) as the major component along with 5% of starting material 18. The analytically pure product 19 as one pair of diastereomers was isolated by chromatography on silica gel with ethyl acetate and heptane mixture as an eluant. HRMS: m/z calcd for C20H26F3NO5 418.1836 (M+H). found 418.1849.
-

tert-Butyl((5S,6R)-6-methyl-2-oxo-5-(2,3,6-trifluorophenyl)piperidin-3-yl)carbamate (20)
-
-
[0174]
![Figure US20160130273A1-20160512-C00051]()
-
[0175]To a 0.5 L cylindrical Sixfors reactor with an overhead stirring, a temperature control, a pH probe and a base addition line, was added sodiumtetraborate decahydrate (3.12 g) and DI water (163 mL). After all solids were dissolved, isopropylamine (9.63 g) was added. The pH of the buffer was adjusted to pH 10.5 using 6 N HCl. The buffer was cooled to room temperature. Then, pyridoxal-5-phosphate (0.33 g) and SEQ ID NO: 1 (8.15 g) were added and slowly dissolved at room temperature.
-
[0176]Crude keto ester 19 (23.6 g, 69 wt %, 16.3 g assay, 39 mmol) was dissolved in DMSO (163 mL) and the solution was added to the reactor over 5-10 min. Then the reaction was heated to 55° C. The pH was adjusted to 10.5 according to a handheld pH meter and controlled overnight with an automated pH controller using 8 M aqueous isopropylamine. The reaction was aged for 27.5 hours.
-
[0177]After confirmation of >95A % conversion by HPLC, the reaction was extracted by first adding a mixture of iPA: iPAc (3:4, 350 mL) and stirring for 20 min. The phases were separated and the aqueous layer was back extracted with a mixture of iPA: iPAc (2:8, 350 mL). The phases were separated. The organic layers were combined and washed with DI water (90 mL). The HPLC based assay yield in the organic layer was 20 (9.86 g, 70.5% assay yield) with >60:1 dr at the positions C5 and C6.
-

tert-Butyl((3S,5S,6R)-6-methyl-2-oxo-5-(2,3,6-trifluorophenyl)piperidin-3-yl)carbamate (21)
-
-
[0178]
![Figure US20160130273A1-20160512-C00052]()
-
[0179]A solution of crude cis and trans mixture 20 in a mixture of iPAc and iPA (1.83 wt %, 9.9 kg; 181 g assay as a mixture) was concentrated in vacuo and dissolved in 2-Me-THF (3.6 L). To the solution was added tert-BuOK (66.6 g, 0.594 mol) at room temperature. The suspension was stirred at room temperature for 2 h. The mixture was poured into water (3.5 L) and the organic layer was separated, washed with 15 wt % of aqueous NaCl (3.5 L), dried over Na2SO4, and concentrated to dryness. The residue was suspended with iPAc (275 mL) and heptane (900 mL) at 60° C. The suspension was slowly cooled down to 1° C. The solid was filtered and rinsed with iPAc and heptane (1:3), dried to afford 21 (166 g, 93 wt %; 85%) as crystals. Mp 176-179° C.; 1H NMR (CDCl3, 500 MHz): δ 7.06 (m, 1H), 6.84 (m, 1H), 5.83 (broad s, 1H), 5.58 (broad s, 1H), 4.22 (m, 1H), 3.88-3.79 (m, 2H), 2.77 (m, 1H), 2.25 (m, 1H), 1.46 (s, 9H), 1.08 (d, J=6.4 Hz, 3H); 19F NMR (CDCl3, 376 MHz): δ −117 (d, J=14 Hz), −135 (d, J=20 Hz), −142 (dd, J=20, 14 Hz); 13C NMR (CDCl3, 100 MHz): δ 171.1, 156.6 (ddd, J=245, 6.4, 2.8 Hz), 155.8, 149.3 (ddd, J=248, 14.4, 8.8 Hz), 147.4 (ddd, J=245, 14.2, 3.8 Hz), 118.0 (dd, J=19.3, 14.5 Hz), 115.9 (dd, J=19.2, 10.4 Hz), 111.0 (ddd, J=26.4, 6.0, 4.3 Hz), 79.8, 51.4, 49.5, 34.1, 29.3, 28.3, 18.0; HRMS: m/z calcd for C17H21F3N2O3 381.1396 (M+Na). found 381.1410.
-

tert-Butyl((5S,6R)-6-methyl-2-oxo-1-(2,2,2-trifluoroethyl)-5-(2,3,6-trifluorophenyl)piperidin-3-yl)carbamate (22)
-
-
[0180]
![Figure US20160130273A1-20160512-C00053]()
-
[0181]To a solution of 21 (10 g, 87% purity, 24.3 mmol) in THF (70 ml) was added tert-BuOLi (2.5 g, 31.2 mmol) at 5° C. in one portion. The solution was cooled to between 0 and 5° C. and trifluoroethyl trifluoromethanesulfonate (10.0 g, 43 mmol) was added in one portion. DMPU (7 mL) was added slowly over 15 min while maintaining the the reaction temperature below 5° C. After the mixture was stirred at 0° C. for 3 h, additional tert-BuOLi (0.9 g, 11.2 mmol) was added. The mixture was aged for an additional 90 min. The mixture was quenched with 0.2 N HCl (70 ml), followed by addition of heptane (80 ml). The organic layer was separated and aqueous layer extracted with heptane (30 ml). The combined organic layers were washed with 15% aqueous citric acid (50 mL) and 5% aqueous NaHCO3(50 mL). The solution was concentrated under vacuum at 40° C. and the resulting oil was dissolved in iPAc (30 mL). The solution was used directly in the next step without further purification. The HPLC analysis indicated that the solution contained 22 (9.8 g, 92% as cis and trans mixture in a ratio of 6.5 to 1) along with 4% of starting material 21 and 8% of a N,N′-alkylated compound. Analytically pure 22 (cis isomer) was isolated by chromatography on silica gel with ethyl acetate and heptane as an eluant. 1H NMR (CDCl3, 500 MHz): δ 7.15 (m, 1H), 6.85 (m, 1H), 5.45 (broad, s, 1H), 4.90 (m, H), 4.20 (m, 1H), 3.92 (m, 2H), 3.28 (m, 1H), 2.70 (m, 2H), 1.48 (s, 9H), 1.20 (d, J=5.9 Hz, 3H); 13C NMR (CDCl3, 100 MHz): δ 170.2, 156.9 (ddd, J=245, 6.3, 2.7 Hz), 156.0, 149.6 (ddd, J=251, 14.8, 8.8 Hz), 147.6 (ddd, J=246, 13.9, 3.6 Hz), 124.5 (q, J=281 Hz), 117.6 (dd, J=19.2, 3.7 Hz), 116.4 (dd, J=19.1, 10.4 Hz), 111.4 (ddd, J=25.8, 6.4, 4.1 Hz), 56.6, 52.8, 45.3 (q, J=34.2 Hz), 35.2, 28.7, 28.3 (br t, J=4 Hz), 14.6; HRMS: m/z calcd for C19H22F6N2O3 (M+H): 441.1607. found 441.1617.
-

(3S,5S,6R)-6-Methyl-2-oxo-1-(2,2,2-trifluoroethyl)-5-(2,3,6-trifluorophenyl)piperidin-3-aminium (S)-2-acetamido-3-phenylpropanoate (23)
-
-
[0182]
![Figure US20160130273A1-20160512-C00054]()
-
[0183]iPAc solution of 22 (529 g assayed, 1.2 mol), obtained from previous step, was diluted to 6 L with iPAc, p-toluenesulfonic acid monohydride (343 g, 1.8 mol) was added and the solution was heated to 55° C. After 4 h, the reaction completed (>99% conversion). Aqueous K2CO3 (530 g in 3 L of water) was charged into the solution after cooled to 15-25° C. The aqueous layer was separated and was back-extracted with iPAc (2 L). The iPAc solutions were combined and the total volume was adjusted to 10 L by adding iPAc. The solution was heated to 50-60° C. About 20 g of N-acetyl L-phenylalanine was added and the solution was agitated for 15 min or until solids precipitated out. The remaining N-acetyl L-phenylalanine (total 250 g, 1.2 mol) was charged slowly and 2-hydroxy-5-nitrobenzaldehyde (2 g) was charged. The suspension was agitated for 12 h at 20° C. and then cooled to 0° C. for 3 h. The suspension was filtrated, washed with iPAc three times and dried to give 23 (583 g, 89% yield) as crystals. Mp 188-190° C.; 1H NMR (DMSO-d6, 400 MHz): δ 7.96 (d, J=8.0 Hz, 1H), 7.48 (m, 1H), 7.15-7.25 (m, 6H), 4.65 (ddd, J=19.4, 15.3, 9.6 Hz, 1H), 4.33 (ddd, J=8.7, 8.4, 4.9 Hz, 1H), 3.70-3.87 (m, 3H), 3.57 (dd, J=11.5, 6.6 Hz, 1H), 3.04 (dd, J=13.7, 4.9 Hz, 1H), 2.82 (dd, J=13.7, 8.9 Hz, 1H), 2.59 (m, 1H), 2.24 (m, 1H), 2.95 (s, 3H), 1.10 (d, J=6.4 Hz, 1H); 19F NMR (DMSO-d6, 376 MHz): δ −69 (s), −118 (d, J=15 Hz), −137 (d, J=21 Hz), −142 (dd, J=21, 15 Hz); 13C NMR (DMSO-d6, 100 MHz): δ 173.6, 171.1, 168.7, 156.3 (ddd, J=243.5, 7.0, 3.1 Hz), 148.7 (ddd, J=249, 14.4, 9.1 Hz), 146.8 (ddd, J=245, 13.7, 3.1 Hz), 138.5, 129.2, 128.0, 126.1, 124.9 (q, J=280.9 Hz), 117.4.0 (dd, J=19.3, 13.8 Hz), 116.7 (dd, J=19.3, 10.6 Hz), 111.8 (ddd, J=26.0, 6.7, 3.6 Hz), 56.6, 54.3, 51.2, 44.3 (q, J=32.5 Hz), 37.2, 34.8, 26.9 (br t, J=4 Hz), 22.5, 14.1.
-

(3S,5S,6R)-6-methyl-2-oxo-1-(2,2,2-trifluoroethyl)-5-(2,3,6-trifluorophenyl)piperidin-3-aminium 2,2-diphenylacetate (25)
-
-
[0184]
![Figure US20160130273A1-20160512-C00055]()
-
[0185]To a mixture of crude material containing (5S,6R)-3-amino-6-methyl-1-(2,2,2-trifluoroethyl)-5-(2,3,6-trifluorophenyl)piperidin-2-one (24, 2.00 g, 5.88 mmol), prepared according to the same method as the previous example, and 3,5-dichloro-2-hydroxybenzaldehyde (0.011 g, 0.059 mmol) in isopropyl acetate (15.0 ml) at 55-60° C. under nitrogen was slowly added a solution of diphenylacetic acid (1.26 g, 5.88 mmol) in THF (10.0 ml) over 2 h. Upon completion of acid addition, a thick salt suspension was agitated at 55-60° C. for another 18 h and then was allowed to cool to ambient temperature. The salt was filtered and washed with isopropyl acetate. After drying at 60° C. in a vacuum oven with nitrogen purge for 8 hours, 25 (2.97 g, 91.4%) was obtained as crystals. 1H NMR (500 MHz, DMSO-d6): δ 7.48 (qd, J=9.4, 4.9 Hz, 1H), 7.32 (d, J=7.7 Hz, 4H), 7.25-7.26 (m, 4H), 7.19-7.17 (m, 3H), 6.79 (br, 3H), 4.95 (s, 1H), 4.67 (dq, J=15.3, 9.7 Hz, 1H), 3.81-3.79 (m, 3H), 3.62 (dd, J=11.6, 6.5 Hz, 1H), 2.66-2.62 (m, 1H), 2.25 (dd, J=12.9, 6.4 Hz, 1H), 1.11 (d, J=6.5 Hz, 3H); 13C NMR (100 MHz, DMSO-d6): δ 174.4, 171.8, 156.9 (ddd, J=244, 7.0, 2.5 Hz), 149.1 (ddd, J=249, 14.4, 8.5 Hz), 147.2 (ddd, J=246, 13.9, 3.2 Hz), 141.4, 129.0, 128.5, 126.7, 125.5 (q, J=281 Hz), 118.0 (dd, J=19.8, 13.8 Hz), 117.1 (dd, J=19.2, 10.6 Hz), 112.3 (ddd, J=26.1, 6.7, 3.3 Hz), 58.5, 57.1, 51.7, 44.8 (q, J=32.7 Hz), 35.3, 27.5 (br t, J=4.6 Hz), 14.5.
-

(3S,5S,6R)-6-methyl-2-oxo-1-(2,2,2-trifluoroethyl)-5-(2,3,6-trifluorophenyl)piperidin-3-aminium 1H-indole-2-carboxylate (26)
-
-
[0186]
![Figure US20160130273A1-20160512-C00056]()
-
[0187]To a mixture of crude material containing 24 (2.00 g, 5.88 mmol) and 3,5-dichloro-2-hydroxybenzaldehyde (0.011 g, 0.059 mmol) in isopropyl acetate (15.0 ml) at 55-60° C. under nitrogen was slowly added a solution of 1H-indole-2-carboxylic acid (0.96 g, 5.88 mmol) in THF (10.0 ml) over 2 hours. Upon completion of acid addition, a thick salt suspension was agitated at 55-60° C. for another 18 h and then was allowed to cool to ambient temperature. The salt was filtered and washed with isopropyl acetate. After drying at 60° C. in a vacuum oven with nitrogen purge for 8 h, 26 (2.33 g, 79.0%) was isolated as crystals. 1H NMR (500 MHz, DMSO): δ 11.40 (s, 1H), 7.56 (d, J=8.0 Hz, 1H), 7.45 (br, 3H), 7.47 (ddd, J=14.8, 10.1, 8.3 Hz, 1H), 7.41-7.40 (m, 1H), 7.16-7.14 (m, 2H), 6.98-6.97 (m, 1H), 6.87 (s, 1H), 4.69 (dq, J=15.3, 9.6 Hz, 1H), 3.84-3.81 (m, 4H), 2.76-2.71 (m, 1H), 2.34 (dd, J=12.7, 6.3 Hz, 1H), 1.13 (d, J=6.5 Hz, 3H); 13C NMR (100 MHz, DMSO-d6): δ 170.9, 164.8, 156.8 (ddd, J=244, 7.0, 2.5 Hz), 149.1 (ddd, J=249, 14.4, 8.5 Hz), 147.2 (ddd, J=246, 13.9, 3.2 Hz), 137.0, 133.5, 127.8, 125.4 (q, J=282 Hz), 123.3, 121.8, 119.7, 117.8 (dd, J=19.8, 13.8 Hz), 117.2 (dd, J=19.2, 10.6 Hz), 112.7, 112.3 (ddd, J=26.1, 6.7, 3.3 Hz), 105.1, 57.1, 51.3, 44.8 (q, J=32.7 Hz), 35.2, 26.9, 14.5.
-

N-((3S,5S,6R)-6-Methyl-2-oxo-1-(2,2,2-trifluoroethyl)-5-(2,3,6-trifluorophenyl)piperidin-3-yl)-2′-oxo-1′,2′,5,7-tetrahydrospiro[cyclopenta[b]pyridine-6,3′-pyrrolo[2,3-b]pyridine]-3-carboxamide monohydrate (28)
-
-
[0188]
![Figure US20160130273A1-20160512-C00057]()
-
[0189]To a suspension of 23 (5.0 g, 9.1 mmol) in isopropyl acetate (50 mL) was added 5% aqueous K3PO4 (50 mL). The mixture was stirred for 5 min. The organic layer was separated and washed with aqueous K3PO4 (50 mL). Solvent removed under vacuum and resulting oil (27) was dissolved in acetonitrile (20 mL). To another flask was added 14 (2.57 g), acetonitrile (40 mL), water (20 mL) and NaOH solution (10N, 0.9 mL). The solution of 27 in acetonitrile was charged to the mixture followed by HOBT monohydrate (1.5 g) and EDC hydrochloride (2.6 g). The mixture was agitated at room temperature for 4 h and HPLC analysis indicated a complete conversion. The reaction mixture was stirred with isopropyl acetate (60 mL) and the aqueous layer was removed. The organic layer was washed with 5% aqueous NaHCO3 (40 mL) followed by a mixture of 15% aqueous citric acid (40 mL) and saturated aqueous NaCl (10 mL). The resulting organic layer was finally washed with 5% aqueous NaHCO3 (40 mL). The solvent was removed under vacuum and the residue was dissolved in methanol (20 mL). The methanol solution was slowly charged into a mixture of water (50 mL) and methanol (5 mL) over 30 min with good agitation, followed by addition of water (50 mL) over 30 min. The suspension was stirred over night at room temperature. The mixture was filtered and crystals were dried in a vacuum oven for 5 h at 50° C. to give 28 (5.4 g, 95%) as monohydrate. 1H NMR (500 MHz, CD3OD): δ 8.88 (t, J=1.2 Hz, 1H), 8.15 (t, J=1.2 Hz, 1H), 8.09 (dd, J=5.3, 1.5 Hz, 1H), 7.36 (dd, J=7.4, 1.5 Hz, 1H), 7.28 (qd, J=9.3, 4.7 Hz, 1H), 7.01 (tdd, J=9.7, 3.6, 1.9 Hz, 1H), 6.96 (dd, J=7.4, 5.3 Hz, 1H), 4.80 (dq, J=15.2, 9.2 Hz, 1H), 4.56 (dd, J=11.7, 6.8 Hz, 1H), 4.03 (ddd, J=13.6, 4.2, 2.6 Hz, 1H), 3.97-3.90 (m, 1H), 3.68 (dq, J=15.3, 8.8 Hz, 1H), 3.59 (t, J=16.2 Hz, 2H), 3.35 (d, J=4.4 Hz, 1H), 3.32 (d, J=3.5 Hz, 1H), 3.21 (qt, J=12.7, 3.1 Hz, 1H), 2.38-2.32 (m, 1H), 1.34 (d, J=6.5 Hz, 3H); 13C NMR (126 MHz, CD3OD): δ 182.79, 171.48, 168.03, 166.71, 159.37 (ddd, J=244.1, 6.5, 2.1 Hz), 157.43, 150.88 (ddd, J=249.4, 14.4, 8.7 Hz), 148.96 (ddd, J=243.8, 13.7, 3.1 Hz), 148.67, 148.15, 136.84, 133.43, 131.63, 130.83, 130.48, 126.41 (q, J=280.0 Hz), 119.85, 118.89 (dd, J=19.0, 13.5 Hz), 117.77 (dd, J=19.8, 10.8 Hz), 112.80 (ddd, J=26.5, 6.5, 4.2 Hz), 58.86, 53.67, 52.87, 46.56 (q, J=33.3 Hz), 45.18, 42.06, 36.95, 27.76 (t, J=4.8 Hz), 14.11.
-

Example 3 3-Hydroxy-3-(2,3,6-trifluorophenyl)butan-2-one (30)
-
-
[0190]
![Figure US20160130273A1-20160512-C00058]()
-
[0191]To a solution of 1,2,4-trifluorobenzene (29, 49.00 g, 371 mmol) and diisopropylamine (4.23 mL, 29.7 mmol) in THF (750 mL) at −70° C. was slowly added 2.5 M of n-BuLi (156.0 ml, 390 mmol) to maintain temperature between −45 to −40° C. The batch was agitated for 30 min. To another flask, a solution of 2,3-butadione (37.7 mL, 427 mmol) in THF (150 mL) was prepared and cooled to −70° C. The previously prepared lithium trifluorobenzene solution was transferred to the second flask between −70 to −45° C. The reaction was agitated for 1 hour at −55 to −45 and then quenched by adding AcOH (25.7 mL, 445 mmol) and then water (150 mL). After warmed to room temperature, the aqueous layer was separated. The aqueous solution was extracted with MTBE (200 mL×1) and the combined organic layers were washed with brine (100 mL×1). The organic layer was concentrated at 25-35° C. The residue was flashed with heptane (100 mL×1) and concentrated to dryness and give 30 (87.94 g, 90.2 wt %, 98% yield, and >99% HPLC purity) as an oil. 1H NMR (CDCl3, 400 MHz): δ 7.16 (m, 1H), 6.86 (m, 1H), 6.88 (s, 1H), 4.59 (s, 1H), 2.22 (s, 3H), 1.84 (dd, J=4.0, 2.8 Hz, 3H); 19F NMR (CDCl3, 376.6 MHz): δ −114.6 (dd, J=14.5, 1.4 Hz), −133.6 (d, J=19.9 Hz), −141.3 (dd, J=19.9, 14.5 Hz); 13C NMR (CDCl3, 100 MHz): δ 207.4, 156.4 (ddd, J=247, 6.2, 2.9 Hz), 149.4 (ddd, J=253, 15.0, 9.0 Hz), 147.5 (ddd, J=245, 14.4, 3.3 Hz), 119.4 (dd, J=17.3, 11.7 Hz), 117.0 (ddd, J=19.3, 11.1, 1.4 Hz), 116.6 (ddd, J=26.6, 6.5, 4.1 Hz), 77.9, 25.0 (dd, J=6.5, 4.9 Hz), 23.3.
-

3-(2,3,6-Trifluorophenyl)but-3-en-2-one (31)
-
-
[0192]
![Figure US20160130273A1-20160512-C00059]()
-
[0193]The hydroxy ketone 30 (7.69 g, 35.2 mmol) and 95% H2SO4 (26.2 mL, 492.8 mmol) were pumped at 2.3 and 9.2 mL/min respectively into the flow reactor. The temperature on mixing was controlled at 22-25° C. by placing the reactor in a water bath (21° C.). The effluent was quenched into a a mixture of cold water (106 g) and heptane/IPAc (1:1, 92 mL) in a jacketed reactor cooled at 0° C.; the internal temperature of the quench solution was ˜7° C. during the reaction. The layers in the quench reactor were separated and the organic layer was washed with 10% NaH2PO4/Na2HPO4 (1:1, 50 mL). The pH of the final wash was 5-6. Solka flock (3.85 g, 50 wt %) was added to the organic solution. The resulting slurry was concentrated and solvent-switched to heptanes at 25-30° C. The mixture was filtered, rinsed with heptanes (50 mL×1). The combined filtrates were concentrated under vacuum to give 31 as an light yellow oil (6.86 g, 90 wt %, 87% yield), which solidified in a freezer. 1H NMR (CDCl3, 400 MHz): δ 7.13 (m, 1H), 6.86 (m, 1H), 6.60 (s, 1H), 6.15 (s, 1H), 2.46 (s, 3H); 19F NMR (CDCl3, 376.6 MHz): δ −117.7 (dd, J=15.0, 1.4 Hz), −135.4 (dd, J=21.4, 1.4 Hz), −42.7 (dd, J=21.4, 15.0 Hz); 13C NMR (CDCl3, 100 MHz): δ 196.3, 155.3 (ddd, J=245, 5.1, 2.9 Hz), 147.9 (ddd, J=250, 14.5, 7.8 Hz), 147.0 (ddd, J=245, 13.4, 3.7 Hz), 137.5 (d, J=1.3 Hz), 131.7, 116.6 (ddd, J=19.9, 9.7, 1.2 Hz), 116.2 (dd, J=22.6, 16.5 Hz), 110.6 (ddd, J=24.8, 6.5, 4.1 Hz), 25.8.
-

Alternative Synthesis of 3-(2,3,6-trifluorophenyl)but-3-en-2-one (31)
-
-
[0194]
![Figure US20160130273A1-20160512-C00060]()
-
[0195]A solution of 18 (3.5 g, 18.6 mmol), acetic acid (0.34 ml, 5.58 mmol), piperidine (0.37 ml, 3.72 mmol), formaldehyde (6.0 g, 37% aqueous solution) in MeCN (20 mL) was heated over weekend. The conversion was about 60%. Reaction was heated to 70° C. overnight. The mixture was concentrated and extracted with MTBE and HCl (0.5N). The organic layer was washed with aqueous K2CO3 (0.5N) and water, in turns. The organic layer was concentrated. The product was isolated by chromatography column (hexane and EtOAc), yielding 31 (2.29 g, 61.5%).
-

Isopropyl 2-((diphenylmethylene)amino)-5-oxo-4-(2,3,6-trifluorophenyl)hexanoate (32)
-
-
[0196]
![Figure US20160130273A1-20160512-C00061]()
-
[0197]Diphenylidene isopropyl glycinate (2.0 g, 7.0 mmol) and 31 (1.4 g, 7.0 mmole) were dissolved in THF (10 ml). The solution was cooled to −10° C. tert-BuOLi (0.56 g, 7.0 mmole) was charged into the solution in several portions. The reaction was warmed up to room temperature slowly and stirred overnight. After quenched by addition of aqueous NH4Cl, the solvents were removed by distillation under vacuum. The residue was subjected to silica chromatography column eluted by hexane and EtOAc yielding 32 (3.0 g, 89%) as an oil, which was directly used in the next step.
-

Isopropyl 2-((tert-butoxycarbonyl)amino)-5-oxo-4-(2,3,6-trifluorophenyl)hexanoate (19)
-
-
[0198]
![Figure US20160130273A1-20160512-C00062]()
-
[0199]Compound 32 (100 mg, 0.21 mmol) was dissolved in THF (2 ml) and the solution was cooled to −10° C. Hydrochloric acid (2N, 1 ml) was added and stirred until all starting material disappeared by TLC. The pH of the reaction was adjusted (pH.>10) by addition of aqueous K2CO3. Boc2O (68 mg, 0.31 mmole) was added into the mixture and stirred overnight. The reaction was completed checked by TLC and the product was identical to the one prepared from the iodo coupling route.
-

Isopropyl 2-((tert-butoxycarbonyl)amino)-5-oxo-4-(2,3,6-trifluorophenyl)hexanoate (19)
-
-
[0200]
![Figure US20160130273A1-20160512-C00063]()
-
[0201]To a 100 mL round bottom was charged 2-methyl THF (43.7 mL) and diisopropyl amine (4.92 mL, 34.2 mmol) and the solution was cooled to −70° C. n-BuLi (13.08 mL, 32.7 mmol) was charged dropwise during which the temperature was controlled below −45° C. The mixture was stirred at −45° C. for 0.5 h. N-Boc-glycine ester (3.58 g) was added dropwise keeping temperature between −45 to −40° C. and aged at the same temperature for 1 h.
-
[0202]The solution of 31 (2.91 g, 14.5 mmol) in 2-methyl THF (2.9 mL) was then added dropwise in the same manner at −45 to −40° C. After a 0.5-1 h age, LC analysis showed nearly complete reaction. The reaction was quenched by addition of HOAc (3.83 mL) and the mixture was warmed to −10° C. and water (11.6 mL, 4 vol) was charged at <20° C. The phase was separated, and the organic layer was washed with 16% NaCl aqueous solution (11.6 mL). Assay desired product 19 as a mixture of diastereomers in the organic solution was 5.40 g (89% yield). The organic layer was concentrated to give crude product 19, which was directly used in the next step reaction. For characterization purposes, a small sample was purified by flash chromatography (silica gel, EtOAc/hexanes=1:10) to give two diastereomers 19A and 19B. 19A as a colorless oil, 1H NMR (CD3CN, 400 MHz) δ: 7.29 (m, 1H), 7.02 (m, 1H), 5.58 (d, J=6.1 Hz, 1H), 4.91 (m, 1H), 4.19-4.05 (m, 2H), 2.79 (m, 1H), 2.05 (s, 3H), 1.84 (m, 1H), 1.41 (s, 9H), 1.23 (d, J=6.7 Hz, 3H), 1.22 (d, J=6.7 Hz, 3H); 13C NMR (CD3CN, 100 MHz) δ: 204.7, 172.4, 158.6 (ddd, J=244, 6, 3 Hz), 156.3, 149.8 (ddd, J=248, 15, 9 Hz), 148.5 (ddd, J=242, 14, 3 Hz), 118.3 (dd, J=21, 16 Hz), 117.7 (ddd, J=19, 10, 2 Hz), 112.6 (ddd, J=26, 7, 4 Hz), 80.2, 70.0, 53.5, 46.0, 32.0, 28.5, 22.0, 21.9. 19B as colorless crystals, MP 91.5-92.0° C., 1H NMR (CD3CN, 400 MHz) δ: 7.31 (m, 1H), 7.03 (m, 1H), 5.61 (d, J=8.2 Hz, 1H), 4.95 (m, 1H), 4.19 (dd, J=10.2, 5.1 Hz, 1H), 3.72 (m, 1H), 2.45-2.29 (m, 2H), 2.09 (s, 3H), 1.41 (s, 9H), 1.21 (d, J=6.3 Hz, 3H), 1.20 (d, J=6.3 Hz, 3H); 13C NMR (CD3CN, 100 MHz) δ: 205.0, 172.8, 157.9 (ddd, J=244, 7, 3 Hz), 156.5, 150.3 (ddd, J=248, 149, 9 Hz), 148.5 (ddd, J=242, 13, 4 Hz), 117.9 (dd, J=19, 10 Hz), 115.9 (dd, J=21, 15 Hz), 111.5 (ddd, J=25, 8, 4 Hz), 80.1, 69.9, 52.9, 46.5, 31.1, 28.5, 22.0, 21.9.
-

Example 4 N-Methoxy-N-methyl-2-(o-tolyl)acetamide (34)
-
-
[0203]
![Figure US20160130273A1-20160512-C00064]()
-
[0204]To a solution of NHMe(OMe).HCl (203 g, 2.1 mol) in THF (1 L), H2O (400 mL) and TEA (263 g, 2.2 mol) was added 33 (200 g, 1.3 mol) and CDI (243 g, 1.5 mol) at 0-10° C. The reaction mixture was stirred at 0-10° C. for 5 h. After HPLC showed that the reaction was complete, the mixture was filtered through celite and the filtrate was partitioned with water and EtOAc. The organic solution was dried over Na2SO4 and concentrated. The crude residual was further purified by flash chromatography on silica gel (5-10% EtOAc/PE) to give 34 (200 g, 78% yield). 1H NMR (CDCl3, 400 MHz): δ 7.17-7.13 (m, 4H), 3.75 (m, 2H), 3.66 (d, 3H), 3.11 (s, 3H), 2.20 (s, 3H), 1.63-1.55 (m, 1H); MS (ESI) m/e [M+H]+: 194.1.
-

1-(o-Tolyl)propan-2-one (35)
-
-
[0205]
![Figure US20160130273A1-20160512-C00065]()
-
[0206]A solution of CeCl3 (114.4 g, 0.45 mol) in THF (4 L) was degassed for 1 h and heated to 45-50° C. for 5 h. When the solution was cooled to −10˜−5° C., MeMgCl (193.2 g, 2.6 mol) in THF was added and the mixture was stirred for 1 h at −10˜−5° C. After amide 34 (256 g, 1.3 mol) was charged into the reaction mixture at −10˜−5° C., the mixture was stirred for 5 h at 10-20° C. After the reaction was complete monitored by LCMS, the mixture was quenched by 1M HCl, and then partitioned with water and EtOAc. The organic phase was dried over Na2SO4 and concentrated. The crude residual was further purified by flash chromatography on silica gel (2-10% EtOAc/PE) to give 35 (157 g, 80% yield). 1H NMR (CDCl3, 400 MHz): δ 7.1-6.91 (d, 4H), 3.55 (s, 3H), 2.25 (s, 3H), 2.05 (s, 3H); MS (ESI) m/e [M+H]+: 149.05.
-

Isopropyl 2-((tert-butoxycarbonyl)amino)-5-oxo-4-(o-tolyl)hexanoate (36)
-
[0207]
![Figure US20160130273A1-20160512-C00066]()
-
[0208]To a solution of 2 (181.2 g, 0.557 mol) in THF (1 L) was added TEA (84.6 g, 0.836 mol) in portions at 15-20° C. The mixture was stirred for 30 h. After the reaction was complete, the solution was concentrated to give crude 7. To a solution of 35 (82.5 g, 0.557 mol) and Cs2CO3 (91 g, 0.279 mol) in DMSO (1 L) was added slowly crude 7 in DMSO (500 mL) over 30 min at 15-20° C. The mixture was stirred for 1 h. After the reaction was complete, the mixture was partitioned with water and MTBE (5 L), and extracted with MTBE twice. The combined organic layer was dried over Na2SO4 and concentrated. The crude residual was further purified by flash chromatography on silica gel (5-10% EtOAc/PE) to give 36 (138 g, 65% yield). 1H NMR (DMSO-d6, 400 MHz): δ 7.14-7.09 (m, 3H), 7.10-6.91 (d, 1H), 4.93-4.89 (m, 1H), 4.05-3.98 (s, 3H), 2.39-2.37 (d, 3H), 1.98-1.92 (d, 3H), 1.20-1.19 (m, 9H), 1.18-1.15 (m, 6H); MS (ESI) m/e [M+H]+: 364.2

-
- (S)-1′-(tert-Butyl)-2′-oxo-1′,2′,5,7-tetrahydrospiro[cyclopenta[b]pyridine-6,3′-pyrrolo[2,3-b]pyridine]-3-carboxylic acid (59)
-
[0249]
![Figure US20160130273A1-20160512-C00088]()
-
[0250]A mixture of 58 (5.0 g, 14.5 mmol), K2CO3 (5.01 g, 36.2 mmol), Pd(OAc)2(33 mg, 0.145 mmol), 1,3-bis(dicyclohexylphosphino)propane (DCPP, 127 mg, 0.290 mmol) and water (0.522 mL, 29.0 mmol) in NMP (32 mL) was heated at 120° C. under 30 psi of CO for 24 h. After cooling to room temperature, the resulting slurry was diluted with water (100 mL). The pH was slowly adjusted to 3-4 with 2 N HCl. The slurry was aged at room temperature for 1 h, filtered, rinsed with water (40 to 50 mL), dried under oven at 60° C. to give 59 (4.64 g, 95%) as a solid. 1H NMR (DMSO-d6, 500 MHz): δ 8.90 (s, 1H), 8.19 (d, J=5.2 Hz, 1H), 7.54 (d, J=7.3 Hz, 1H,), 6.99 (dd, J=7.3, 5.2 Hz, 1H), 3.33 (m, 4H), 1.72 (s, 9H); 13C NMR (DMSO-d6, 125 MHz): δ 180.16, 167.44, 166.97, 158.07, 149.76, 146.61, 135.39, 133.09, 130.36, 128.81, 125.48, 118.44, 58.19, 51.12, 44.56, 41.24, 28.91.
-

(S)-2′-Oxo-1′,2′,5,7-tetrahydrospiro[cyclopenta[b]pyridine-6,3′-pyrrolo[2,3-b]pyridine]-3-carboxylic acid (14)
-
[0251]
![Figure US20160130273A1-20160512-C00089]()
-
[0252]To 59 (4 g, 97% wt) was charged 37% HCl (40 to 44 mL). The slurry was heated at 94° C. for up to 48 h, cooled down to room temperature. The solvent was partially removed by reducing pressure to about total 2 vol (˜4 mL water remained). The residue was diluted with water (20 mL) followed by adjusting pH to 2.6 with NaOH (3.5 N, 4.5 mL). The thick slurry was aged for 1 to 2 h, filtered, rinsed with water (2×8 mL), followed by water/acetone (1:1, 8 mL). The wet cake was dried to give compound 14 (3.1 g, 98% wt, 94%) as crystals. 1H NMR (DMSO-d6, 500 MHz): δ 13.31 (br, 1H), 11.14 (s, 1H), 8.91 (s, 1H), 8.11 (m, 2H), 7.49 (dd, J=7.3, 1.3 Hz, 1H), 6.93 (dd, J=7.3, 5.3 Hz, 1H), 3.36 (m, 4H); 13C NMR (DMSO-d6, 125 MHz): δ 181.06, 167.36, 166.95, 156.80, 149.79, 147.32, 135.37, 133.19, 130.73, 128.88, 125.50, 118.46, 51.78, 44.12, 40.70.


2′-oxo-l\2 5,7-tetrahydrospiro[cyclopenta[¾]pyridine-6,3′-pyrrolo[2,3-¾]pyridine]-3-carboxamide monohydrate (28)

To a suspension of 23 (5.0 g, 9.1 mmol) in isopropyl acetate (50 mL) was added 5% aqueous K3PO4 (50 mL). The mixture was stirred for 5 min. The organic layer was separated and washed with aqueous K3PO4 (50 mL). Solvent removed under vacuum and resulting oil (27) was dissolved in acetonitrile (20 mL). To another flask was added 14 (2.57 g), acetonitrile (40 mL), water (20 mL) and NaOH solution (10N, 0.9 mL). The solution of 27 in acetonitrile was
charged to the mixture followed by HOBT monohydrate (1.5 g) and EDC hydrochloride (2.6 g). The mixture was agitated at room temperature for 4 h and HPLC analysis indicated a complete conversion. The reaction mixture was stirred with isopropyl acetate (60 mL) and the aqueous layer was removed. The organic layer was washed with 5% aquoues NaHC03 (40 mL) followed by a mixture of 15% aqueous citric acid (40 mL) and saturated aqueous NaCl (10 mL). The resulting organic layer was finally washed with 5% aquous NaHC03 (40 mL). The solvent was removed under vacuum and the residue was dissolved in methanol (20 mL). The methanol solution was slowly charged into a mixture of water (50 mL) and methanol (5 mL) over 30 min with good agitation, followed by addition of water (50 mL) over 30 min. The suspension was stirred over night at room temperature. The mixture was filtered and crystals were dried in a vacuum oven for 5 h at 50 °C to give 28 (5.4 g, 95%) as monohydrate. Ή NMR (500 MHz, CD3OD): δ 8.88 (t, J= 1.2 Hz, 1 H), 8.15 (t, J = 1.2 Hz, 1 H), 8.09 (dd, J= 5.3, 1.5 Hz, 1 H), 7.36 (dd, J= 7.4, 1.5 Hz, 1 H), 7.28 (qd, J= 9.3, 4.7 Hz, 1 H), 7.01 (tdd, J= 9.7, 3.6, 1.9 Hz, 1 H), 6.96 (dd, J= 7.4, 5.3 Hz, 1 H), 4.80 (dq, J= 15.2, 9.2 Hz, 1 H), 4.56 (dd, J= 11.7, 6.8 Hz, 1 H), 4.03 (ddd, J= 13.6, 4.2, 2.6 Hz, 1 H), 3.97-3.90 (m, 1 H), 3.68 (dq, J= 15.3, 8.8 Hz, 1 H), 3.59 (t, J= 16.2 Hz, 2 H), 3.35 (d, J= 4.4 Hz, 1 H), 3.32 (d, J= 3.5 Hz, 1 H), 3.21 (qt, J= 12.7, 3.1 Hz, 1 H), 2.38-2.32 (m, 1 H), 1.34 (d, J= 6.5 Hz, 3 H); 13C NMR (126 MHz, CD3OD): δ 182.79, 171.48, 168.03, 166.71, 159.37 (ddd, J= 244.1, 6.5, 2.1 Hz), 157.43, 150.88 (ddd, J = 249.4, 14.4, 8.7 Hz), 148.96 (ddd, J= 243.8, 13.7, 3.1 Hz), 148.67, 148.15, 136.84, 133.43, 131.63, 130.83, 130.48, 126.41 (q, J = 280.0 Hz), 119.85, 118.89 (dd, J= 19.0, 13.5 Hz), 117.77 (dd, J= 19.8, 10.8 Hz), 112.80 (ddd, J= 26.5, 6.5, 4.2 Hz), 58.86, 53.67, 52.87, 46.56 (q, J = 33.3 Hz), 45.18, 42.06, 36.95, 27.76 (t, J= 4.8 Hz), 14.11.
This invention relates to a process for making piperidinone carboxamide indane and azainane derivatives, which are CGRP receptor antagonists useful for the treatment of migraine. This class of compounds is described in U.S. Patent Application Nos. 13/293,166 filed November 10, 2011, 13/293,177 filed November 10, 2011 and 13/293,186 filed November 10, 2011, and PCT International Application Nos. PCT/US11/60081 filed November 10, 2011 and PCT/US 11/60083 filed November 10, 2011.
CGRP (Calcitonin Gene -Related Peptide) is a naturally occurring 37-amino acid peptide that is generated by tissue-specific alternate processing of calcitonin messenger RNA and is widely distributed in the central and peripheral nervous system. CGRP is localized
predominantly in sensory afferent and central neurons and mediates several biological actions, including vasodilation. CGRP is expressed in alpha- and beta-forms that vary by one and three amino acids in the rat and human, respectively. CGRP-alpha and CGRP -beta display similar biological properties. When released from the cell, CGRP initiates its biological responses by binding to specific cell surface receptors that are predominantly coupled to the activation of adenylyl cyclase. CGRP receptors have been identified and pharmacologically evaluated in several tissues and cells, including those of brain, cardiovascular, endothelial, and smooth muscle origin.
Based on pharmacological properties, these receptors are divided into at least two subtypes, denoted CGRPi and CGRP2- Human a-CGRP-(8-37), a fragment of CGRP that lacks seven N-terminal amino acid residues, is a selective antagonist of CGRPi, whereas the linear analogue of CGRP, diacetoamido methyl cysteine CGRP ([Cys(ACM)2,7]CGRP), is a selective agonist of CGRP2- CGRP is a potent neuromodulator that has been implicated in the pathology of cerebrovascular disorders such as migraine and cluster headache. In clinical studies, elevated levels of CGRP in the jugular vein were found to occur during migraine attacks (Goadsby et al, Ann. Neurol, 1990, 28, 183-187), salivary levels of CGRP are elevated in migraine subjects between attacks (Bellamy et al., Headache, 2006, 46, 24-33), and CGRP itself has been shown to trigger migrainous headache (Lassen et al., Cephalalgia, 2002, 22, 54-61). In clinical trials, the CGRP antagonist BIBN4096BS has been shown to be effective in treating acute attacks of migraine (Olesen et al, New Engl. J. Med., 2004, 350, 1104-1110) and was able to prevent headache induced by CGRP infusion in a control group (Petersen et al., Clin. Pharmacol. Ther., 2005, 77, 202-213).
CGRP -mediated activation of the trigeminovascular system may play a key role in migraine pathogenesis. Additionally, CGRP activates receptors on the smooth muscle of intracranial vessels, leading to increased vasodilation, which is thought to contribute to headache pain during migraine attacks (Lance, Headache Pathogenesis: Monoamines, Neuropeptides, Purines and Nitric Oxide, Lippincott-Raven Publishers, 1997, 3-9). The middle meningeal artery, the principle artery in the dura mater, is innervated by sensory fibers from the trigeminal ganglion which contain several neuropeptides, including CGRP. Trigeminal ganglion stimulation in the cat resulted in increased levels of CGRP, and in humans, activation of the trigeminal system caused facial flushing and increased levels of CGRP in the external jugular vein (Goadsby et al., Ann. Neurol., 1988, 23, 193-196). Electrical stimulation of the dura mater in rats increased the diameter of the middle meningeal artery, an effect that was blocked by prior administration of CGRP(8-37), a peptide CGRP antagonist (Williamson et al., Cephalalgia, 1997, 17, 525-531). Trigeminal ganglion stimulation increased facial blood flow in the rat, which was inhibited by CGRP(8-37) (Escott et al, Brain Res. 1995, 669, 93-99). Electrical stimulation of the trigeminal ganglion in marmoset produced an increase in facial blood flow that could be blocked by the non-peptide CGRP antagonist BIBN4096BS (Doods et al, Br. J.
Pharmacol., 2000, 129, 420-423). Thus the vascular effects of CGRP may be attenuated, prevented or reversed by a CGRP antagonist.
CGRP -mediated vasodilation of rat middle meningeal artery was shown to sensitize neurons of the trigeminal nucleus caudalis (Williamson et al., The CGRP Family:
Calcitonin Gene -Related Peptide (CGRP), Amylin, and Adrenomedullin, Landes Bioscience, 2000, 245-247). Similarly, distention of dural blood vessels during migraine headache may sensitize trigeminal neurons. Some of the associated symptoms of migraine, including extracranial pain and facial allodynia, may be the result of sensitized trigeminal neurons (Burstein et al, Ann. Neurol. 2000, 47, 614-624). A CGRP antagonist may be beneficial in attenuating, preventing or reversing the effects of neuronal sensitization.
The ability of the compounds to act as CGRP antagonists makes them useful pharmacological agents for disorders that involve CGRP in humans and animals, but particularly in humans. Such disorders include migraine and cluster headache (Doods, Curr Opin Inves Drugs, 2001, 2 (9), 1261-1268; Edvinsson et al, Cephalalgia, 1994, 14, 320-327); chronic tension type headache (Ashina et al, Neurology, 2000, 14, 1335-1340); pain (Yu et al, Eur. J. Pharm., 1998, 347, 275-282); chronic pain (Hulsebosch et al, Pain, 2000, 86, 163-175);
neurogenic inflammation and inflammatory pain (Holzer, Neurosci., 1988, 24, 739-768; Delay- Goyet et al, Acta Physiol. Scanda. 1992, 146, 537-538; Salmon et al, Nature Neurosci., 2001, 4(4), 357-358); eye pain (May et al. Cephalalgia, 2002, 22, 195-196), tooth pain (Awawdeh et al, Int. Endocrin. J., 2002, 35, 30-36), non-insulin dependent diabetes mellitus (Molina et al, Diabetes, 1990, 39, 260-265); vascular disorders; inflammation (Zhang et al, Pain, 2001, 89, 265), arthritis, bronchial hyperreactivity, asthma, (Foster et al, Ann. NY Acad. Sci., 1992, 657, 397-404; Schini et al, Am. J. Physiol, 1994, 267, H2483-H2490; Zheng et al, J. Virol, 1993, 67, 5786-5791); shock, sepsis (Beer et al, Crit. Care Med., 2002, 30 (8), 1794-1798); opiate withdrawal syndrome (Salmon et al, Nature Neurosci., 2001, 4(4), 357-358); morphine tolerance (Menard et al, J. Neurosci., 1996, 16 (7), 2342-2351); hot flashes in men and women (Chen et al, Lancet, 1993, 342, 49; Spetz et al, J. Urology, 2001, 166, 1720-1723); allergic dermatitis (Wallengren, Contact Dermatitis, 2000, 43 (3), 137-143); psoriasis; encephalitis, brain trauma, ischaemia, stroke, epilepsy, and neurodegenerative diseases (Rohrenbeck et al, Neurobiol. of Disease 1999, 6, 15-34); skin diseases (Geppetti and Holzer, Eds., Neurogenic Inflammation, 1996, CRC Press, Boca Raton, FL), neurogenic cutaneous redness, skin rosaceousness and erythema; tinnitus (Herzog et al, J. Membrane Biology, 2002, 189(3), 225); inflammatory bowel disease, irritable bowel syndrome, (Hoffman et al. Scandinavian Journal of Gastroenterology, 2002, 37(4) 414-422) and cystitis. Of particular importance is the acute or prophylactic treatment of headache, including migraine and cluster headache.
The present invention describes a novel process for making piperidinone carboxamide indane and azainane derivatives, which are CGRP receptor antagonists, having less steps and improved yields as compared to previous synthetic methods for making these compounds.
Another embodiment of the invention encompasses crystalline monohydrate free base of the compound having the structure
and having the following chemical name: (S)-N-((3S,5S,6R)-6-mQthyl-2-oxo-l -(2,2,2- trifluoroethyl)-5-(2,3,6-trifluorophenyl)piperidin-3-yl)-2′-oxo- ,2′,5,7- tetrahydrospiro [cyclopenta[b]pyridine-6,3 ‘-pyrrolo [2,3 -b]pyridine] -3 -carboxamide monohydrate
EXAMPLE 2 acetamide (17)
K2C03, water
To a solution of DMF (58.1 mL, 750 mmol) in iPAc (951 mL) was added POCl3 (55.9 mL, 600 mmol) under ice-cooling. After aged for 1 h under ice-bath, acid 16 (95 g, 500 mmol) was added under ice-cooling. The solution was stirred under ice-cooling for 30 min. The solution was added over 30 min into a solution of K2CO3 (254 g, 1.835 mol) and
NHMe(OMe)HCl (73.2 g, 750 mmol) in water (951 mL) below 8 °C. After aged for 30 min below 8 °C, the organic layer was separated, washed with water (500 mL) twice and sat. NaCl aq (100 mL) once, and concentrated in vacuo to afford 17 as an oil (117.9 g, 97.7 wt%, 99% yield). ‘H NMR (CDCI3, 400 MHz); δ 7.05 (m, 1H), 6.82 (m, 1H), 3.86 (s, 2H), 3.76 (s, 3H), 3.22 (s,
3H); 19F NMR (CDCI3, 376.6 MHz); δ -120.4 (dd, J= 15.1, 2.7 Hz), -137.9 (dd, J= 20.8, 2.7 Hz), -143.5 (dd, J= 20.8, 15.1 Hz); 13C NMR (CDC13, 100 MHz); δ 169.4, 156.9 (ddd, J= 244, 6.2, 2.7 Hz), 149.3 (ddd, J= 249, 14.4, 8.4 Hz), 147.1 (ddd, J= 244, 13.1, 3.5 Hz), 115.5 (ddd, J = 19.4, 9.9, 1.5 Hz), 133.4 (dd, J= 22.3, 16.4 Hz), 110.2 (ddd, J= 24.8, 6.7, 4.1 Hz), 32.4 (broad), 26.6 (m); HRMS m/z calcd for C10H10F3NO2 234.0736 (M+H); found 234.0746 l-(2,3,6-Trifluorophenyl)propan-2-one (18)
A mixture of CeCl3 (438 g, 1779 mmol) and THF (12 L) was heated at 40 °C for about 2 h then cooled to 5 °C. Methylmagensium chloride in THF (3 M, 3.4 L) was charged at 5- 9 °C and then it was warmed up to 16 °C and held for 1 h. The suspension was re-cooled to -10 to -15 °C. A solution of 17 (1.19 kg) in THF (2.4 L) was charged into the suspension over 15 min. After confirmation of completion of the reaction, the reaction mixture was transferred to a cold solution of hydrochloric acid (2 N, 8.4 L) and MTBE (5 L) in 5-10°C. The aqueous phase was separated and the organic layer was washed with aqueous 5%> K2CO3 (6 L) and then 10%> aqueous NaCl (5 L). The organic layer was dried over Na2S04, concentrated to give crude 18 (917g, >99wt%>) in 95% yield. The crude 18 was used in the next step without further purification. Analytically pure 18 was obtained by silica gel column.
!H NMR (CDCI3, 400 MHz); δ 7.07 (m, 1H), 6.84 (m, 1H), 3.82 (s, 2H), 2.28 (s, 3H); 19F NMR (CDCI3, 376.6 MHz); δ -120.3 (dd, J= 15.3, 2.5 Hz), -137.8 (dd, J= 21.2, 2.5 Hz), -143.0 (dd, J = 20.2, 15.3 Hz); 13C NMR (CDCI3, 100 MHz); δ 202.2, 156.5 (ddd, J= 244, 6.3, 2.9 Hz), 148.9 (ddd, J= 249, 14.4, 8.6 Hz), 147.0 (ddd, J = 244, 13.1, 3.5 Hz), 115.7 (ddd, J = 19.4, 10.5, 1.2 Hz), 112.8 (dd, J= 22.7, 17.0 Hz), 110.3 (ddd, J = 24.8, 6.7, 4.1 Hz), 37.2 (d, J=1.2 Hz), 29.3. Isopropyl 2-((tert-butoxycarbonyl)amino)-5-oxo-4-(2,3,6-trifluorophenyl)hexanoate (19)
To a solution of 18 (195 g, 1.03 mol) in MTBE (1.8 L) was added zinc bromide (67 g, 0.30 mol) followed by 2 (390 g, 1.2 mol). tert-BuOLi (290 g, 3.6 mol) was then added in several portions while maintaining the reaction temperature below 40 °C. The resulting mixture was stirred at 35 °C for 24 h and quenched into a mixture of 2 N HC1 (5.6 L) and heptane (5 L) at 0 °C. The organic layer was separated and washed with 5% aqueous NaHC03 (5 L) twice. The resulting organic solution was concentrated under vacuum. The residue was dissolved in heptane (2 L) and the solution was concentrated again under vacuum. The resulting oil was dissolved in DMSO (2.5 L) and the solution was used in the next step without further purification. HPLC analysis indicated that the solution contained the desired product 19 (290 g, 67% yield) as the major component along with 5% of starting material 18. The analytically pure product 19 as one pair of diastereomers was isolated by chromatography on silica gel with ethyl acetate and heptane mixture as an eluant. HRMS: m/z calcd for C2oH26F3N05 418.1836 (M+H); found 418.1849. tert- Butyl ((55,,6i?)-6-methyl-2-oxo-5-(2,3,6-trifluorophenyl)piperidin-3-yl)carbamate (20)
To a 0.5 L cylindrical Sixfors reactor with an overhead stirring, a temperature control, a pH probe and a base addition line, was added sodiumtetraborate decahydrate (3.12 g) and DI water (163 mL). After all solids were dissolved, isopropylamine (9.63 g) was added. The pH of the buffer was adjusted to pH 10.5 using 6 N HC1. The buffer was cooled to room temperature. Then, pyridoxal-5 -phosphate (0.33 g) and SEQ ID NO: 1 (8.15 g) were added and slowly dissolved at room temperature. Crude keto ester 19 (23.6 g, 69 wt%, 16.3 g assay, 39 mmol) was dissolved in DMSO (163 mL) and the solution was added to the reactor over 5-10 min. Then the reaction was heated to 55 °C. The pH was adjusted to 10.5 according to a handheld pH meter and controlled overnight with an automated pH controller using 8 M aqueous isopropylamine. The reaction was aged for 27.5 hours.
After confirmation of >95A% conversion by HPLC, the reaction was extracted by first adding a mixture of iPA: iPAc (3:4, 350 mL) and stirring for 20 min. The phases were separated and the aqueous layer was back extracted with a mixture of iPA: iPAc (2:8, 350 mL). The phases were separated. The organic layers were combined and washed with DI water (90 mL). The HPLC based assay yield in the organic layer was 20 (9.86 g, 70.5 % assay yield) with >60:1 dr at the positions C5 and C6. tert- utyl ((35′,55′,6i?)-6-methyl-2-oxo-5-(2,3,6-trifiuorophenyl)piperidin-3-yl)carbamate (21)
A solution of crude cis and trans mixture 20 in a mixture of iPAc and iPA (1.83 wt%, 9.9 kg; 181 g assay as a mixture) was concentrated in vacuo and dissolved in 2-Me-THF (3.6 L). To the solution was added tert-BuOK (66.6 g, 0.594 mol) at room temperature. The suspension was stirred at room temperature for 2 h. The mixture was poured into water (3.5 L) and the organic layer was separated, washed with 15 wt% of aqueous NaCl (3.5 L), dried over Na2S04, and concentrated to dryness. The residue was suspended with iPAc (275 mL) and heptane (900 mL) at 60 °C. The suspension was slowly cooled down to 1 °C. The solid was filtered and rinsed with iPAc and heptane (1 :3), dried to afford 21 (166 g, 93 wt%; 85 %) as crystals. Mp 176-179 °C; 1H NMR (CDC13, 500 MHz): δ 7.06 (m, 1H), 6.84 (m, 1H), 5.83 (broad s, 1H), 5.58 (broad s, 1H), 4.22 (m, 1H), 3.88-3.79 (m, 2H), 2.77 (m, 1H), 2.25 (m, 1H), 1.46 (s, 9H), 1.08 (d, J= 6.4 Hz, 3H); 19F NMR (CDCI3, 376 MHz): δ -117 (d, J= 14 Hz), -135 (d, J= 20 Hz), -142 (dd, J= 20, 14 Hz); 13C NMR (CDC13, 100 MHz): δ 171.1, 156.6 (ddd, J = 245, 6.4, 2.8 Hz), 155.8, 149.3 (ddd, J= 248, 14.4, 8.8 Hz), 147.4 (ddd, J= 245, 14.2, 3.8 Hz), 118.0 (dd, J= 19.3, 14.5 Hz), 115.9 (dd, J= 19.2, 10.4 Hz), 111.0 (ddd, J = 26.4, 6.0, 4.3 Hz), 79.8, 51.4, 49.5, 34.1, 29.3, 28.3, 18.0; HRMS: m/z calcd for Ci7H2iF3N203 381.1396 (M+ Na); found 381.1410. tert-Butyl ((55′,6i?)-6-methyl-2-oxo-l-(2,2,2-trifluoroethyl)-5-(2,3,6-trifluorophenyl)piperidin-3- yl)carbamate (22)
To a solution of 21 (10 g, 87% purity, 24.3 mmol) in THF (70 ml) was added tert- BuOLi (2.5 g, 31.2 mmol) at 5 °C in one portion. The solution was cooled to between 0 and 5 °C and trifluoroethyl trifluoromethanesulfonate (10.0 g, 43 mmol) was added in one portion. DMPU (7 mL) was added slowly over 15 min while maintaining the the reaction temperature below 5 °C. After the mixture was stirred at 0 °C for 3 h, additional tert-BuOLi (0.9 g, 11.2 mmol) was added. The mixture was aged for an additional 90 min. The mixture was quenched with 0.2 N HC1 (70 ml), followed by addition of heptane (80 ml). The organic layer was separated and aqueous layer extracted with heptane (30 ml). The combined organic layers were washed with 15%) aquoeus citric acid (50 mL) and 5% aqueous NaHC03 (50 mL). The solution was concentrated under vacuum at 40 °C and the resulting oil was dissolved in iPAc (30 mL). The solution was used directly in the next step without further purification. The HPLC analysis indicated that the solution contained 22 (9.8 g, 92% as cis and trans mixture in a ratio of 6.5 to 1) along with 4% of starting material 21 and 8% of a N,N’-alkylated compound. Analytically pure 22 (cis isomer) was isolated by chromatography on silica gel with ethyl acetate and heptane as an eluant. 1H NMR (CDC13, 500 MHz): δ 7.15 (m, 1H), 6.85 (m, 1H), 5.45 (broad, s, 1H), 4.90 (m, H), 4.20 (m, 1H), 3.92 (m, 2H), 3.28 (m, 1H), 2.70 (m, 2H), 1.48 (s, 9H), 1.20 (d, J= 5.9 Hz, 3H); 13C NMR (CDC13, 100 MHz): δ 170.2, 156.9 (ddd, J= 245, 6.3,2.7 Hz), 156.0, 149.6 (ddd, J= 251, 14.8, 8.8 Hz), 147.6 (ddd, J= 246, 13.9,3.6 Hz), 124.5 (q, J= 281 Hz), 117.6 (dd, J = 19.2, 3.7 Hz), 116.4 (dd, J= 19.1, 10.4 Hz), 111.4 (ddd, J= 25.8, 6.4,4.1Hz), 56.6, 52.8, 45.3 (q, J= 34.2 Hz), 35.2, 28.7, 28.3 (br t, J= 4 Hz), 14.6; HRMS: m/z calcd for Ci9H22F6N203 (M+H): 441.1607; found 441.1617. (35′,55′,6i?)-6-Methyl-2-oxo-l-(2,2,2-trifluoroethyl)-5-(2,3,6-trifluorophenyl)piperi (S)-2-acetamido-3 -phenylpropanoate (23)
iPAc solution of 22 (529 g assayed, 1.2 mol), obtained from previous step, was diluted to 6 L with iPAc, /?-toluenesulfonic acid monohydride (343 g, 1.8 mol) was added and the solution was heated to 55 °C. After 4 h, the reaction completed (>99% conversion). Aqueous K2CO3 (530 g in 3 L of water) was charged into the solution after cooled to 15-25 °C. The aqueous layer was separated and was back-extracted with iPAc (2 L). The iPAc solutions were combined and the total volume was adjudted to 10 L by adding iPAc. The solution was heated to 50-60 °C. About 20 g of N-acetyl L-phenylalanine was added and the solution was agitated for 15 min or until solids precipitated out. The remaining N-acetyl L-phenylalanine (total 250 g, 1.2 mol) was charged slowly and 2-hydroxy-5-nitrobenzaldehyde (2 g) was charged. The suspension was agitated for 12 h at 20 °C and then cooled to 0 °C for 3 h. The suspension was filtrated, washed with iPAc three times and dried to give 23 (583g, 89% yield) as crystals. Mp 188 – 190 °C; 1H NMR (DMSO-de, 400 MHz): δ 7.96 (d, J= 8.0 Hz, 1H) , 7.48 (m, 1H), 7.15-7.25 (m, 6H), 4.65 (ddd, J= 19.4, 15.3, 9.6 Hz, 1H), 4.33 (ddd, J= 8.7, 8.4, 4.9 Hz, 1H), 3.70-3.87 (m, 3H), 3.57 (dd, J= 11.5, 6.6 Hz, 1H), 3.04 (dd, J= 13.7, 4.9 Hz, 1H), 2.82 (dd, J= 13.7, 8.9 Hz,lH), 2.59 (m, 1H), 2.24 (m, 1H), 2.95 (s, 3H), 1.10 (d, J= 6.4 Hz, 1H); 19F NMR (DMSO-d6, 376 MHz): δ -69 (s) , -118 (d, J= 15 Hz), -137 (d, J = 21 Hz), -142 (dd, J= 21, 15 Hz); 13C NMR (DMSO-d6, 100 MHz): δ 173.6, 171,. l, 168.7, 156.3 (ddd, J= 243.5, 7.0, 3.1 Hz), 148.7 (ddd, J= 249, 14.4, 9.1 Hz), 146.8 (ddd, J = 245, 13.7, 3.1 Hz), 138.5, 129.2, 128.0, 126.1, 124.9 (q, J= 280.9 Hz), 117.4.0 (dd, J= 19.3, 13.8 Hz), 116.7 (dd, J= 19.3, 10.6 Hz), 111.8 (ddd, J= 26.0, 6.7, 3.6 Hz), 56.6, 54.3, 51,2, 44.3 (q, J= 32.5 Hz), 37.2, 34.8, 26.9 (br t, J= 4 Hz), 22.5, 14.1.
(35′,55′,6i?)-6-methyl-2-oxo-l-(2,2,2-trifluoroethyl)-5-(2,3,6-trifluorophenyl)piperidin-3- aminium 2,2-diphenylacetate (25)
To a mixture of crude material containing (55′,6i?)-3-amino-6-methyl-l -(2,2,2- trifluoroethyl)-5-(2,3,6-trifluorophenyl)piperidin-2-one (24, 2.00 g, 5.88 mmol), prepared according to the same method as the previous example, and 3,5-dichloro-2-hydroxybenzaldehyde (0.011 g, 0.059 mmol) in isopropyl acetate (15.0 ml) at 55-60 °C under nitrogen was slowly added a solution of diphenylacetic acid (1.26 g, 5.88 mmol) in THF (10.0 ml) over 2 h. Upon completion of acid addition, a thick salt suspension was agitated at 55-60 °C for another 18 h and then was allowed to cool to ambient temperature. The salt was filtered and washed with isopropyl acetate. After drying at 60 °C in a vacuum oven with nitrogen purge for 8 hours, 25 (2.97 g, 91.4%) was obtained as crystals. 1H NMR (500 MHz, DMSO-d6): δ 7.48 (qd, J= 9.4, 4.9 Hz, 1 H), 7.32 (d, J= 7.7 Hz, 4 H), 7.25-7.26 (m, 4 H), 7.19-7.17 (m, 3 H), 6.79 (br, 3H), 4.95 (s, 1 H), 4.67 (dq, J= 15.3, 9.7 Hz, 1 H), 3.81-3.79 (m, 3 H), 3.62 (dd, J= 11.6, 6.5 Hz, 1 H), 2.66-2.62 (m, 1 H), 2.25 (dd, J= 12.9, 6.4 Hz, 1 H), 1.11 (d, J= 6.5 Hz, 3 H); 13C NMR (100 MHz, DMSO-de): δ 174.4, 171.8, 156.9 (ddd, J= 244, 7.0, 2.5 Hz), 149.1 (ddd, J= 249, 14.4, 8.5 Hz), 147.2 (ddd, J= 246, 13.9, 3.2 Hz), 141.4, 129.0, 128.5, 126.7, 125.5 (q, J= 281 Hz), 118.0 (dd, J= 19.8, 13.8 Hz), 117.1 (dd, J= 19.2, 10.6 Hz), 112.3 (ddd, J= 26.1, 6.7, 3.3 Hz), 58.5, 57.1, 51.7, 44.8 (q, J= 32.7 Hz), 35.3, 27.5 (br t, J= 4.6 Hz), 14.5.
(35′,55′,6i?)-6-methyl-2-oxo-l-(2,2,2-trifluoroethyl)-5-(2,3,6-trifluorophenyl)piperidin-3-amM lH-indole-2-carboxylate (26)
To a mixture of crude material containing 24 (2.00 g, 5.88 mmol) and 3,5-dichloro-2- hydroxybenzaldehyde (0.011 g, 0.059 mmol) in isopropyl acetate (15.0 ml) at 55-60 °C under nitrogen was slowly added a solution of lH-indole-2-carboxylic acid (0.96 g, 5.88 mmol) in THF (10.0 ml) over 2 hours. Upon completion of acid addition, a thick salt suspension was agitated at 55-60 °C for another 18 h and then was allowed to cool to ambient temperature. The salt was filtered and washed with isopropyl acetate. After drying at 60 °C in a vacuum oven with nitrogen purge for 8 h, 26 (2.33 g, 79.0%) was isolated as crystals. 1H NMR (500 MHz, DMSO): δ 11.40 (s, 1 H), 7.56 (d, J= 8.0 Hz, 1 H), 7.45 (br, 3 H), 7.47 (ddd, J= 14.8, 10.1, 8.3 Hz, 1 H), 7.41- 7.40 (m, 1 H), 7.16-7.14 (m, 2 H), 6.98-6.97 (m, 1 H), 6.87 (s, 1 H), 4.69 (dq, J= 15.3, 9.6 Hz, 1 H), 3.84-3.81 (m, 4 H), 2.76-2.71 (m, 1 H), 2.34 (dd, J= 12.7, 6.3 Hz, 1 H), 1.13 (d, J= 6.5 Hz, 3 H); 13C NMR (100 MHz, DMSO-d6): δ 170.9, 164.8, 156.8 (ddd, J= 244, 7.0, 2.5 Hz), 149.1 (ddd, J= 249, 14.4, 8.5 Hz), 147.2 (ddd, J = 246, 13.9, 3.2 Hz), 137.0, 133.5, 127.8, 125.4 (q, J = 282 Hz), 123.3, 121.8, 119.7, 117.8 (dd, J= 19.8, 13.8 Hz), 117.2 (dd, J= 19.2, 10.6 Hz), 112.7, 112.3 (ddd, J= 26.1, 6.7, 3.3 Hz), 105.1, 57.1, 51.3, 44.8 (q, J= 32.7 Hz), 35.2, 26.9, 14.5.

2′-oxo-l\2 5,7-tetrahydrospiro[cyclopenta[¾]pyridine-6,3′-pyrrolo[2,3-¾]pyridine]-3- carboxamide monohydrate (28)
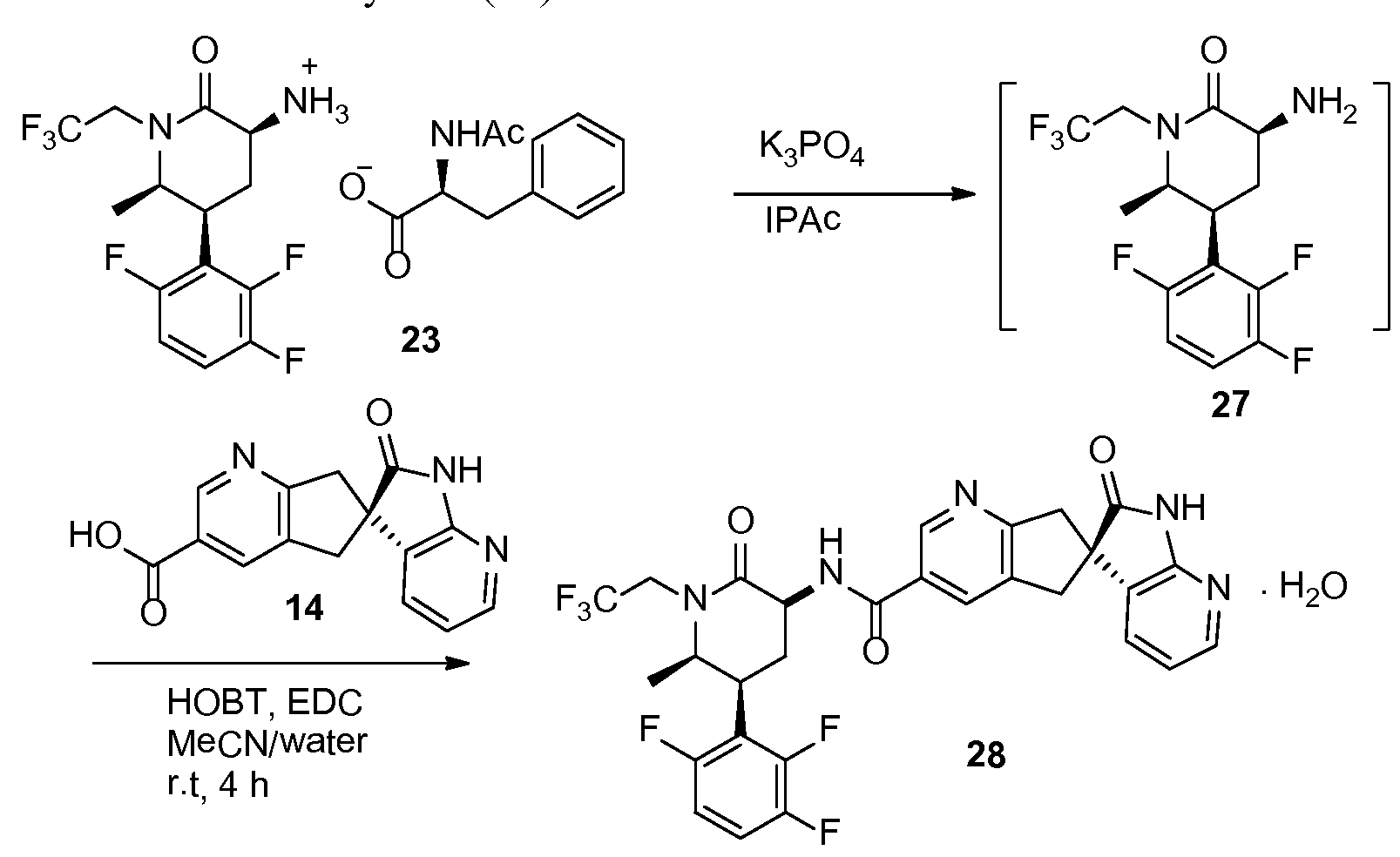
To a suspension of 23 (5.0 g, 9.1 mmol) in isopropyl acetate (50 mL) was added 5% aqueous K3PO4 (50 mL). The mixture was stirred for 5 min. The organic layer was separated and washed with aqueous K3PO4 (50 mL). Solvent removed under vacuum and resulting oil (27) was dissolved in acetonitrile (20 mL). To another flask was added 14 (2.57 g), acetonitrile (40 mL), water (20 mL) and NaOH solution (10N, 0.9 mL). The solution of 27 in acetonitrile was charged to the mixture followed by HOBT monohydrate (1.5 g) and EDC hydrochloride (2.6 g). The mixture was agitated at room temperature for 4 h and HPLC analysis indicated a complete conversion. The reaction mixture was stirred with isopropyl acetate (60 mL) and the aqueous layer was removed. The organic layer was washed with 5% aquoues NaHC03 (40 mL) followed by a mixture of 15% aqueous citric acid (40 mL) and saturated aqueous NaCl (10 mL). The resulting organic layer was finally washed with 5% aquous NaHC03 (40 mL). The solvent was removed under vacuum and the residue was dissolved in methanol (20 mL). The methanol solution was slowly charged into a mixture of water (50 mL) and methanol (5 mL) over 30 min with good agitation, followed by addition of water (50 mL) over 30 min. The suspension was stirred over night at room temperature. The mixture was filtered and crystals were dried in a vacuum oven for 5 h at 50 °C to give 28 (5.4 g, 95%) as monohydrate. Ή NMR (500 MHz, CD3OD): δ 8.88 (t, J= 1.2 Hz, 1 H), 8.15 (t, J = 1.2 Hz, 1 H), 8.09 (dd, J= 5.3, 1.5 Hz, 1 H), 7.36 (dd, J= 7.4, 1.5 Hz, 1 H), 7.28 (qd, J= 9.3, 4.7 Hz, 1 H), 7.01 (tdd, J= 9.7, 3.6, 1.9 Hz, 1 H), 6.96 (dd, J= 7.4, 5.3 Hz, 1 H), 4.80 (dq, J= 15.2, 9.2 Hz, 1 H), 4.56 (dd, J= 11.7, 6.8 Hz, 1 H), 4.03 (ddd, J= 13.6, 4.2, 2.6 Hz, 1 H), 3.97-3.90 (m, 1 H), 3.68 (dq, J= 15.3, 8.8 Hz, 1 H), 3.59 (t, J= 16.2 Hz, 2 H), 3.35 (d, J= 4.4 Hz, 1 H), 3.32 (d, J= 3.5 Hz, 1 H), 3.21 (qt, J= 12.7, 3.1 Hz, 1 H), 2.38-2.32 (m, 1 H), 1.34 (d, J= 6.5 Hz, 3 H); 13C NMR (126 MHz, CD3OD): δ 182.79, 171.48, 168.03, 166.71, 159.37 (ddd, J= 244.1, 6.5, 2.1 Hz), 157.43, 150.88 (ddd, J = 249.4, 14.4, 8.7 Hz), 148.96 (ddd, J= 243.8, 13.7, 3.1 Hz), 148.67, 148.15, 136.84, 133.43, 131.63, 130.83, 130.48, 126.41 (q, J = 280.0 Hz), 119.85, 118.89 (dd, J= 19.0, 13.5 Hz), 117.77 (dd, J= 19.8, 10.8 Hz), 112.80 (ddd, J= 26.5, 6.5, 4.2 Hz), 58.86, 53.67, 52.87, 46.56 (q, J = 33.3 Hz), 45.18, 42.06, 36.95, 27.76 (t, J= 4.8 Hz), 14.11.
EXAMPLE 3
3-Hydroxy-3-(2,3,6-trifluorophenyl)butan-2-one (30)

To a solution of 1,2,4-trifluorobenzene (29, 49.00 g, 371 mmol) and diisopropylamine (4.23 mL, 29.7 mmol) in THF (750 mL) at -70 °C was slowly added 2.5 M of ft-BuLi (156.0 ml, 390 mmol) to maintain temperature between -45 to -40 °C. The batch was agitated for 30 min. To another flask, a solution of 2,3-butadione (37.7 mL, 427 mmol) in THF (150 mL) was prepared and cooled to -70 °C. The previously prepared lithium trifluorobenzene solution was transferred to the second flask between -70 to -45 °C. The reaction was agitated for 1 hour at -55 to -45 and then quenched by adding AcOH (25.7 mL, 445 mmol) and then water (150 mL). After warmed to room temperature, the aqueous layer was seperated. The aqueous solution was extracted with MTBE (200 mL x 1) and the combined organic layers were washed with brine (100 mL x 1). The organic layer was concentrated at 25-35 °C. The residue was flashed with heptane (100 mL x 1) and concentrated to dryness and give 30 (87.94 g, 90.2 wt%, 98% yield, and >99% HPLC purity) as an oil. H NMR (CDCI3, 400 MHz): δ 7.16 (m, 1H), 6.86 (m, 1H), 6.88 (s, 1H), 4.59 (s, 1H), 2.22 (s, 3H), 1.84 (dd, J= 4.0, 2.8 Hz, 3H); 19F NMR (CDCI3, 376.6 MHz): δ -114.6 (dd, J= 14.5, 1.4 Hz), -133.6 (d, J= 19.9 Hz), -141.3 (dd, J =
19.9, 14.5 Hz); 13C NMR (CDCI3, 100 MHz): δ 207.4, 156.4 (ddd, J= 247, 6.2, 2.9 Hz), 149.4 (ddd, J= 253, 15.0, 9.0 Hz), 147.5 (ddd, J= 245, 14.4, 3.3 Hz), 119.4 (dd, J=17.3, 11.7 Hz), 117.0 (ddd, J=19.3, 11.1, 1.4 Hz), 116.6 (ddd, J= 26.6, 6.5, 4.1 Hz), 77.9, 25.0 (dd, J= 6.5, 4.9 Hz), 23.3. -(2,3,6-Trifluorophenyl)but-3-en-2-one (31)

The hydroxy ketone 30 (7.69 g, 35.2 mmol) and 95% H2S04 (26.2 mL, 492.8 mmol) were pumped at 2.3 and 9.2 mL/min respectively into the flow reactor. The temperature on mixing was controlled at 22-25 °C by placing the reactor in a water bath (21 °C). The effluent was quenched into a a mixture of cold water ( 106 g) and heptane/IP Ac ( 1 : 1 , 92 mL) in a j acketed reactor cooled at 0 °C; the internal temperature of the quench solution was ~ 7 °C during the reaction. The layers in the quench reactor were separated and the organic layer was washed with 10% NaH2P04/Na2HP04 (1 :1, 50 mL). The pH of the final wash was 5-6. Solka flock (3.85 g, 50 wt%>) was added to the organic solution. The resulting slurry was concentrated and solvent- switched to heptanes at 25-30 °C. The mixture was filtered, rinsed with heptanes (50 mL x 1). The combined filtrates were concentrated under vacuum to give 31 as an light yellow oil (6.86 g, 90 wt%, 87% yield), which solidified in a freezer. *H NMR (CDC13, 400 MHz): δ 7.13 (m, 1H), 6.86 (m, 1H), 6.60 (s, 1H), 6.15 (s, 1H), 2.46 (s, 3H); 19F NMR (CDC13, 376.6 MHz): δ -117.7 (dd, J= 15.0, 1.4 Hz), -135.4 (dd, J= 21.4, 1.4 Hz), -42.7 (dd, J= 21.4, 15.0 Hz); 13C NMR (CDCls, 100 MHz): δ 196.3, 155.3 (ddd, J= 245, 5.1, 2.9 Hz), 147.9 (ddd, J= 250, 14.5, 7.8 Hz), 147.0 (ddd, J = 245, 13.4, 3.7 Hz), 137.5 (d, J=1.3 Hz), 131.7, 116.6 (ddd, J= 19.9, 9.7, 1.2 Hz), 116.2 (dd, J= 22.6, 16.5 Hz), 110.6 (ddd, J= 24.8, 6.5, 4.1 Hz), 25.8.
Alternative synthesis of 3-(2,3,6-trifluorophenyl)but-3-en-2-one (31)

A solution of 18 (3.5 g, 18.6 mmol), acetic acid (0.34 ml, 5.58 mmol), piperidine (0.37 ml, 3.72 mmol), formaldehyde (6.0 g, 37%> aqueous solution) in MeCN (20 mL) was heated over weekend. The conversion was about 60%. Reaction was heated to 70 °C overnight. The mixtrure was concentrated and extracted with MTBE and HC1 (0.5N). The organic layer was washed with aqueous K2CO3 (0.5N) and water, in turns. The organic layer was concentrated. The product was isolated by chromatography column (hexane and EtOAc), yielding 31 (2.29 g, 61.5%).
Isopropyl 2-((diphenylmethylene)amino)-5-oxo-4-(2,3,6-trifluorophenyl)hexanoate (32)

Diphenylidene isopropyl glycinate (2.0 g, 7.0 mmol) and 31 (1.4 g, 7.0 mmole) were dissolved in THF (10 ml). The solution was cooled to -10 °C. tert- uOLi (0.56 g, 7.0 mmole) was charged into the solution in several portions. The reaction was warmed up to room temperature slowly and stirred overnight. After quenched by addition of aqueous NH4CI, the solvents were removed by distillation under vacuum. The residue was subjected to silica chromatography column eluted by hexane and EtOAc yielding 32 (3.0 g, 89 %) as an oil, which was directly used in the next step.
Isopropyl 2-((tert-butoxycarbonyl)amino)-5-oxo-4-(2,3,6-trifluorophenyl)hexanoate (19)

Compound 32 (100 mg, 0.21 mmol) was dissolved in THF (2 ml) and the solution was cooled to -10 °C. Hydrochloric acid (2N, 1 ml) was added and stirred until all starting material disappeared by TLC. The pH of the reaction was adjusted (pH.>10) by addition of aqueous K2CO3. Boc20 (68 mg, 0.31 mmole) was added into the mixture and stirred overnight. The reaction was completed checked by TLC and the product was identical to the one prepared from the iodo coupling route.
Isopropyl 2-((tert-butoxycarbonyl)amino)-5-oxo-4-(2,3,6-trifluorophenyl)hexanoate (19)

To a 100 mL round bottom was charged 2-methyl THF (43.7 mL) and diisopropyl amine (4.92 mL, 34.2 mmol) and the solution was cooled to -70 °C. n-BuLi (13.08 mL, 32.7 mmol) was charged dropwise during which the temperature was controlled below -45 °C. The mixture was stirred at -45 °C for 0.5 h. N-Boc-glycine ester (3.58 g) was added dropwise keeping temperature between -45 to -40 °C and aged at the same temperature for 1 h.
The solution of 31 (2.91 g, 14.5 mmol) in 2-methyl THF (2.9 mL) was then added dropwise in the same manner at -45 to -40 °C. After a 0.5-1 h age, LC analysis showed nearly complete reaction. The reaction was quenched by addition of HO Ac (3.83 mL) and the mixture was warmed to -10 °C and water (1 1.6 mL, 4 vol) was charged at < 20 °C. The phase was separated, and the organic layer was washed with 16% NaCl aqueous solution (11.6 mL). Assay desired product 19 as a mixture of diastereomers in the organic solution was 5.40 g (89% yield). The organic layer was concentrated to give crude product 19, which was directly used in the next step reaction. For characterization purposes, a small sample was purified by flash chromatography (silica gel, EtOAc/hexanes = 1 : 10) to give two diastereomers 19A and 19B. 19A as a colorless oil, 1H NMR (CD3CN, 400 MHz) δ: 7.29 (m, 1 H), 7.02 (m, 1 H), 5.58 (d, J = 6.1 Hz, 1 H), 4.91 (m, 1 H), 4.19-4.05 (m, 2 H), 2.79 (m, 1 H), 2.05 (s, 3 H), 1.84 (m, 1 H), 1.41 (s, 9 H), 1.23 (d, J = 6.7 Hz, 3 H), 1.22 (d, J = 6.7 Hz, 3 H); 13C NMR (CD3CN, 100 MHz) δ: 204.7, 172.4, 158.6 (ddd, J = 244, 6, 3 Hz), 156.3, 149.8 (ddd, J = 248, 15, 9 Hz), 148.5 (ddd, J = 242, 14, 3 Hz), 118.3 (dd, J = 21, 16 Hz), 117.7 (ddd, J = 19, 10, 2 Hz), 112.6 (ddd, J = 26, 7, 4 Hz), 80.2, 70.0, 53.5, 46.0, 32.0, 28.5, 22.0, 21.9. 19B as colorless crystals, MP 91.5-92.0 °C, 1H NMR (CD3CN, 400 MHz) δ: 7.31 (m, 1 H), 7.03 (m, 1 H), 5.61 (d, J = 8.2 Hz, 1 H), 4.95 (m, 1 H), 4.19 (dd, J = 10.2, 5.1 Hz, 1 H), 3.72 (m, 1 H), 2.45-2.29 (m, 2 H), 2.09 (s, 3 H), 1.41 (s, 9 H), 1.21 (d, J = 6.3 Hz, 3 H), 1.20 (d, J = 6.3 Hz, 3 H); 13C NMR (CD3CN, 100 MHz) δ: 205.0, 172.8, 157.9 (ddd, J= 244, 7, 3 Hz), 156.5, 150.3 (ddd, J= 248, 149, 9 Hz), 148.5 (ddd, J = 242, 13, 4 Hz), 117.9 (dd, J = 19, 10 Hz), 115.9 (dd, J = 21, 15 Hz), 111.5 (ddd, J = 25, 8, 4 Hz), 80.1, 69.9, 52.9, 46.5, 31.1, 28.5, 22.0, 21.9.
PATENT
https://encrypted.google.com/patents/US20120122911
[0000]

(3S,5S,6R)-3-Amino-6-methyl-1-(2,2,2-trifluoroethyl)-5-(2,3,6-trifluorophenyl)piperidin-2-one hydrochlorideStep A: (5S,6R & 5R,6S)-6-Methyl-1-(2,2,2-trifluoroethyl)-5-(2,3,6-trifluorophenyl)piperidin-2-one
Essentially following the procedures described in Intermediate 14, but using 2,3,6-trifluorophenylboronic acid in place of 2,3,5-trifluorophenylboronic acid, the title compound was obtained. MS: m/z=326.0 (M+1).
Step B: (3S,5S,6R & 3R,5R,6S)-3-Azido-6-methyl-1-(2,2,2-trifluoroethyl)-5-(2,3,6-trifluorophenyl)piperidin-2-one
To a stirred solution of lithium bis(trimethylsilyl)amide (1.0 M in THF, 4.80 mL, 4.80 mmol) in THF (20 mL) at −78° C. was added a cold (−78° C.) solution of (5S,6R & 5R,6S)-6-methyl-1-(2,2,2-trifluoroethyl)-5-(2,3,6-trifluorophenyl)piperidin-2-one (1.30 g, 4.00 mmol) in THF (10 mL) dropwise, keeping the internal temperature of the reaction mixture below −65° C. The resulting mixture was stirred at −78° C. for 30 min, then a cold (−78° C.) solution of 2,4,6-triisopropylbenzenesulfonyl azide (Harmon et al. (1973) J. Org. Chem. 38, 11-16) (1.61 g, 5.20 mmol) in THF (10 mL) was added dropwise, keeping the internal temperature of the reaction mixture below −65° C. The reaction mixture was stirred at −78° C. for 30 min, then AcOH (1.05 mL, 18.4 mmol) was added. The resulting mixture was allowed to warm slowly to ambient temperature and was poured into saturated aqueous sodium bicarbonate (50 mL) and the mixture was extracted with EtOAc (2×75 mL). The combined organic layers were washed with brine, then dried over sodium sulfate, filtered, and concentrated to dryness in vacuo. The crude product was purified by silica gel chromatography, eluting with a gradient of hexanes:EtOAc—100:0 to 20:80, to give the diastereomeric azide products (3R,5S,6R & 3S,5R,6S)-3-azido-6-methyl-1-(2,2,2-trifluoroethyl)-5-(2,3,5-trifluorophenyl)piperidin-2-one, which eluted second, and the title compound, which eluted first. MS: m/z=367.1 (M+1).
Step C: tent-Butyl [(3S,5S,6R)-6-methyl-2-oxo-1-(2,2,2-trifluoroethyl)-5-(2,3,6-trifluorophenyl)piperidin-3-yl]carbamate
To a solution of (3S,5S,6R & 3R,5R,6S)-3-azido-6-methyl-1-(2,2,2-trifluoroethyl)-5-(2,3,5-trifluorophenyl)piperidin-2-one (280 mg, 0.764 mmol) and di-tert-butyl dicarbonate (217 mg, 0.994 mmol) in EtOH (5 mL) was added 10% palladium on carbon (25 mg, 0.024 mmol) and the resulting mixture was stirred vigorously under an atmosphere of hydrogen (ca. 1 atm) for 1 h. The reaction mixture was filtered through a pad of Celite®, washing with EtOH, and the filtrate was concentrated in vacuo to give a crude solid. The crude product was purified by silica gel chromatography, eluting with a gradient of hexanes:EtOAc—100:0 to 30:70, to give the racemic title compound. Separation of the enantiomers was achieved by SFC on a ChiralTech IC column, eluting with CO2:MeOH:CH3CN—90:6.6:3.3, to give tert-butyl [(3R,5R,6S)-6-methyl-2-oxo-1-(2,2,2-trifluoroethyl)-5-(2,3,6-trifluorophenyl)piperidin-3-yl]carbamate as the first major peak, and tert-butyl [(3S,5S,6R)-6-methyl-2-oxo-1-(2,2,2-trifluoroethyl)-5-(2,3,6-trifluorophenyl)piperidin-3-yl]carbamate, the title compound, as the second major peak. MS: m/z=463.2 (M+Na).
Step D: (3S,5S,6R)-3-Amino-6-methyl-1-(2,2,2-trifluoroethyl)-5-(2,3,6-trifluorophenyl)piperidin-2-one hydrochloride
A solution of tert-butyl [(3S,5S,6R)-6-methyl-2-oxo-1-(2,2,2-trifluoroethyl)-5-(2,3,6-trifluorophenyl)piperidin-3-yl]carbamate (122 mg, 0.277 mmol) in EtOAc (10 mL) was saturated with HCl (g) and aged for 30 min. The resulting mixture was concentrated in vacuo to give the title compound. MS: m/z=341.1 (M+1); 1H NMR (500 MHz, CD3OD) δ 7.33 (qd, 1H, J=9.3, 4.9 Hz), 7.05 (tdd, 1H, J=9.8, 3.7, 2.2 Hz), 4.78 (dq, 1H, J=15.4, 9.3 Hz), 4.22 (dd, 1H, J=12.2, 6.6 Hz), 4.06 (ddd, 1H, J=13.3, 4.5, 2.7 Hz), 3.97 (m, 1H), 3.73 (dq, 1H, J=15.4, 8.8 Hz), 2.91 (qt, 1H, J=12.7, 3.1 Hz), 2.36 (ddd, 1H, J=12.7, 6.4, 2.0 Hz), 1.22 (d, 3H, J=6.6 Hz).
Example 4

(6S)—N-[(3S,5S,6R)-6-Methyl-2-oxo-1-(2,2,2-trifluoroethyl)-5-(2,3,6-trifluorophenyl)piperidin-3-yl]-2′-oxo-1′,2′,5,7-tetrahydrospiro[cyclopenta[b]pyridine-6,3′-pyrrolo[2,3-b]pyridine]-3-carboxamide dihydrochloride
To a stirred mixture of (6S)-2′-oxo-1′,2′,5,7-tetrahydrospiro[cyclopenta[b]pyridine-6,3′-pyrrolo[2,3-b]pyridine]-3-carboxylic acid (described in Intermediate 1) (264 mg, 0.939 mmol), (3S,5S,6R)-3-amino-6-methyl-1-(2,2,2-trifluoroethyl)-5-(2,3,6-trifluorophenyl)piperidin-2-one hydrochloride (described in Intermediate 15) (295 mg, 0.782 mmol), HOBT (144 mg, 0.939 mmol), and EDC (180 mg, 0.939 mmol) in DMF (8 mL) was added N,N-diisopropylethylamine (0.34 mL, 1.96 mmol), and the resulting mixture was stirred at ambient temperature for 3 h. The reaction mixture was then poured into saturated aqueous sodium bicarbonate (30 mL) and extracted with EtOAc (2×40 mL). The combined organic layers were washed with brine, dried over sodium sulfate, and concentrated in vacuo. The residue was purified by silica gel chromatography, eluting with a gradient of CH2Cl2:MeOH:NH4OH—100:0:0 to 90:10:0.1, to give the product, which was treated with HCl in EtOAc at 0° C. to afford the title compound. HRMS: m/z=604.1783 (M+1), calculated m/z=604.1778 for C29H24F6N5O3. 1H NMR (500 MHz, CD3OD) δ 9.09 (s, 1H), 8.69 (s, 1H), 8.18 (dd, 1H, J=5.9, 1.5 Hz), 7.89 (dd, 1H, J=7.3, 1.5 Hz), 7.30 (m, 1H), 7.23 (dd, 1H, J=7.3, 5.9 Hz), 7.03 (m, 1H), 4.78 (m, 1H), 4.61 (dd, 1H, J=11.5, 6.6 Hz), 4.05 (dd, 1H, J=13.8, 2.8 Hz), 3.96 (m, 1H), 3.84 (d, 1H, J=18.6 Hz), 3.76 (d, 1H, J=18.6 Hz), 3.73 (d, 1H, J=17.3 Hz), 3.72 (m, 1H), 3.61 (d, 1H, J=17.3 Hz), 3.22 (m, 1H), 2.38 (m, 1H), 1.34 (d, 3H, J=6.6 Hz).
| Cited Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| US7390798 * | Feb 9, 2005 | Jun 24, 2008 | Merck & Co., Inc. | Carboxamide spirolactam CGRP receptor antagonists |
| US20090054408 * | Sep 6, 2005 | Feb 26, 2009 | Bell Ian M | Monocyclic anilide spirolactam cgrp receptor antagonists |
| US20100160334 * | Mar 5, 2010 | Jun 24, 2010 | Bell Ian M | Tricyclic anilide spirolactam cgrp receptor antagonists |
| US20100179166 * | Jun 2, 2008 | Jul 15, 2010 | Ian Bell | Carboxamide heterocyclic cgrp receptor antagonists |
| US20120122899 * | Nov 10, 2011 | May 17, 2012 | Merck Sharp & Dohme Corp. | Piperidinone carboxamide azaindane cgrp receptor antagonists |
| US20120122900 * | Nov 10, 2011 | May 17, 2012 | Merck Sharp & Dohme Corp. | Piperidinone carboxamide azaindane cgrp receptor antagonists |
| US20120122911 * | Nov 10, 2011 | May 17, 2012 | Merck Sharp & Dohme Corp. | Piperidinone carboxamide azaindane cgrp receptor antagonists |
| Reference | ||
|---|---|---|
| 1 | * | See also references of EP2849568A4 |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| CN105037210A * | May 27, 2015 | Nov 11, 2015 | 江苏大学 | Alpha,beta-dehydrogenated-alpha-amino acid synthesis method |
| US9688660 | Oct 28, 2016 | Jun 27, 2017 | Heptares Therapeutics Limited | CGRP receptor antagonists |
| US9802935 | Oct 28, 2016 | Oct 31, 2017 | Heptares Therapeutics Limited | CGRP receptor antagonists |
| US9808457 | Oct 28, 2016 | Nov 7, 2017 | Heptares Therapeutics Limited | CGRP receptor antagonists |
| WO2006031606A2 * | Sep 9, 2005 | Mar 23, 2006 | Merck & Co., Inc. | Carboxamide spirolactam cgrp receptor antagonists |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| US9174989 | Mar 12, 2013 | Nov 3, 2015 | Merck Sharp & Dohme Corp. | Process for making CGRP receptor antagonists |
| WO2013169348A1 * | Mar 13, 2013 | Nov 14, 2013 | Merck Sharp & Dohme Corp. | Process for making cgrp receptor antagonists |
////////////////Atogepant, атогепант , أتوجيبانت , 阿托吉泮 , PHASE 3, MERCK, ALLERGAN,
CC1C(CC(C(=O)N1CC(F)(F)F)NC(=O)C2=CC3=C(CC4(C3)C5=C(NC4=O)N=CC=C5)N=C2)C6=C(C=CC(=C6F)F)F