
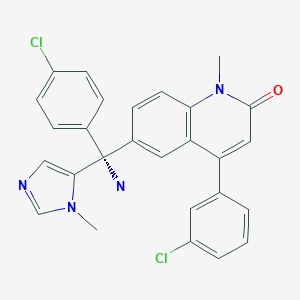

TIPIFARNIB
R-115777, NSC-702818
Categories
UNIIMAT637500A
CAS number 192185-72-1 +form
192185-68-5 (racemate)
192185-69-6 (racemic; fumarate)
192185-70-9 (racemic; diHCl)
(+)-(R)-6-[1-Amino-1-(4-chlorophenyl)-1-(1-methylimidazol-5-yl)methyl]-4-(3-chlorophenyl)-1-methylquinolin-2(1H)-one
Weight Average: 489.396
Chemical Formula C27H22Cl2N4O
PRODUCT PATENT
Tipifarnib (R-115777) is a substance that is being studied in the treatment of acute myeloid leukemia (AML) and other types of cancer. It belongs to the family of drugs called farnesyltransferase inhibitors. It is also called Zarnestra. In June 2005, the FDA issued a Not Approvable Letter for Zarnestra.
Investigated for use/treatment in colorectal cancer, leukemia (myeloid), pancreatic cancer, and solid tumors.
Drug had been granted orphan drug designation by the FDA for the treatment of AML in 2004. In 2005, the Committee for Orphan Medicinal Products of the European Medicines Agency (EMEA) adopted a positive opinion on orphan medicinal product designation for the drug. In 2014, Eiger BioPharmaceuticals licensed the product for worldwide development for the treatment of viral diseases and Kura Oncology licensed development and commercialization rights for the treatment cancer indications.
Pharmacodynamics
R115777, a nonpeptidomimetic farnesyl transferase inhibitor, suppresses the growth of human pancreatic adenocarcinoma cell lines. This growth inhibition is associated with modulation in the phosphorylation levels of signal transducers and activators of transcription 3 (STAT3) and extracellular signal-regulated kinases (ERK)
Tipifarnib (INN,[1]:213 proposed trade name Zarnestra) is a farnesyltransferase inhibitor that is being investigated in patients 65 years of age and older with newly diagnosed acute myeloid leukemia (AML). It inhibits the Ras kinase in a post-translational modification step before the kinase pathway becomes hyperactive. It inhibits prenylation of the CaaX tail motif, which allows Ras to bind to the membrane where it is active. Without this step the protein cannot function.
It is also being tested in clinical trials in patients in certain stages of breast cancer.[2] It is also investigated as a treatment for multiple myeloma.[3]
For treatment of progressive plexiform neurofibromas associated with neurofibromatosis type I, it successfully passed phase I clinical trials but was suspended (NCT00029354) in phase II.[4][5] The compound was discovered by and is under investigation by Johnson & Johnson Pharmaceutical Research & Development, L.L.C, with registration number R115777.Approval process
Tipifarnib was submitted to the FDA by Johnson & Johnson for the treatment of AML in patients aged 65 and over with a new drug application (NDA) to the FDA on January 24, 2005.
In June 2005, the FDA issued a “not approvable” letter for tipifarnib.[6]Progeria

Confocal microscopy photographs of the descending aortas of two 15-month-old progeria mice, one untreated (left picture) and the other treated with the farnsyltransferase inhibitor drug tipifarnib (right picture). The microphotographs show prevention of the vascular smooth muscle cell loss that is otherwise rampant by this age. Staining was smooth muscle alpha-actin (green), lamins A/C (red) and DAPI (blue). (Original magnification, ×40)
It was shown on a mouse model of Hutchinson–Gilford progeria syndrome that dose-dependent administration of tipifarnib can significantly prevent both the onset of the cardiovascular phenotype as well as the late progression of existing cardiovascular disease.[7]
PATENT
TIPIFARNIB BY SOLIPHARMA
WO-2018103027
Crystalline form (I, II, III and IV) of tipifarnib . Useful for the treatment and/or prevention of abnormal cell growth diseases such as lung cancer, pancreatic cancer, colon cancer, melanoma, neuroblastoma or glioma. first filing from Solipharma claiming tipifarnib which was developing by Kura Oncology , under license from Johnson & Johnson subsidiary J&JPRD (now Janssen Research & Development).

Cyclization of 3-(3-chlorophenyl)-N-phenyl-2-propenamide by means of polyphosphoric acid (PPA) at 100 °C gives 4-(3-chlorophenyl)-1,2,3,4-tetrahydroquinolin-2-one ,
Which is condensed with 4-chlorobenzoic acid by means of PPA at 140 °C to yield 6-(4-chlorobenzoyl)-4-(3-chlorophenyl)-1,2,3,4-tetrahydroquinolin-2-one
The dehydrogenation of compound by means of Br2 in bromobenzene at 160 °C affords 6-(4-chlorobenzoyl)-4-(3-chlorophenyl)quinolin-2-one,
Which is N-alkyalted with iodomethane in the presence of BnNMe3Cl and NaOH in THF to provide 6-(4-chlorobenzoyl)-4-(3-chlorophenyl)-1-methylquinolin-2-one.
Condensation of compound with 1-methylimidazole by means of BuLi in THF gives the triaryl carbinol (N-1),
Which is finally treated with NH3 in THF to afford the target Tipifarnib, R-115777 .


Scheme SHOWING COMPLICATIONS
PATENT
WO 2005105782
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2005105782
Farnesyltransf erase inhibitors block the main post-translational modification of the Ras protein, thus interfering with its localization to the inner surface of the plasma
10 membrane and subsequent activation of the downstream effectors. Although initially developed as a strategy to target Ras in cancer, farnesyltransferase inhibitors have
subsequently been acknowledged as acting by additional and more complex
mechanisms that may extend beyond Ras involving GTP-binding proteins, kinases,
centromere-binding proteins and probably other f arnesylated proteins.
15
A particular farnesyltransferase inhibitor is described in WO 97/21701, namely (R)-(+)- 6-[amino(4-chlorophenyl)(l-methyl-lH-imidazol-5-yl)methyl]-4-(3-chlorophenyl)-l- methyl-2(liϊ)-quinolinone. The absolute stereochemical configuration of the compound was not determined in the experiments described in the above-mentioned patent
20 specification, but the compound was identified by the prefix “(B)” to indicate that it was the second compound isolated from column chromatography. The compound thus obtained has been found to have the (R)-(+)-configuration. This compound will be
referred to below by its published code number Rl 15777 and has the following formula

Rl 15777 (Tipifamib) is a potent, orally active inhibitor of f arnesylprotein transferase.
It is one of the most advanced of the farnesylprotein transferase inhibitors currently
reported to be in clinical development, being one of the agents that have progressed to phase III studies.
30 Rl 15777 has been found to have very potent activity against neoplaslic diseases.
Antineoplastic activity in solid tumors, such as breast cancer, as well as in haematological malignancies, such as leukemia, have been observed. Also combination studies have been carried out demonstrating that R 115777 can be safely combined with several highly active anticancer drugs.
In WO 01/53289, the racemates (±) (4-(3-chloro-phenyl)-6-[(6-chloro-pyridin-3-yl)-(4-methoxy-benzylamino)-(3-methyl-3-f: -imidazol-4-yl)-methyl]-l-cyclopropylmethyl-liϊ-quinolin-2-one (racemate 1) and (±) 4-(3-chloro-phenyl)-6-[(6-chloro-pyridin-3-yl)-[(4-methoxy-benzylidene)-amino]-(3-methyl-3jr7-imidazol-4-yl)-methyl]-l-cyclopropylmethyl-liϊ-quinolin-2-one (racemate 2) are prepared.

racemate 1 racemate 2
After chiral molecule separation using column chromatography, either the benzylamino or the benzilidine moiety of the resulting (+) and /or (-) enantiomers are converted to an amino group under acidic conditions.
The synthesis of Rl 15777 as originally described in WO 97/21701, is presented in scheme 1.
Herein, in step 1, the intermediate 1-methyl imidazole in tetrahydrofuran, is mixed with a solution of ra-butyllithium in a hexane solvent to which is added chlorotriethylsilane (triethylsilyl chloride), followed by a further addition of ra-butyllithium in hexane, the resulting mixture being cooled to -78°C before the addition of a solution of a compound of formula (I), i.e. 6-(4-chlorobenzoyl)-4-(3-chlorophenyl)-l-methyl-2(12ϊ)-quinolinone in tetrahydrofuran. The reaction mixture is subsequently brought to room temperature, and then hydrolysed, extracted with ethyl acetate and the organic layer worked up to obtain a compound of formula (II), i.e. (±)-6-[hydroxy(4-chlorophenyl) (l-methyl-liϊ-imidazol-5-yl)methyl]-4-(3-chlorophenyl)-l-methyl-2(lia- )-quinolinone.
In step 2, the hydroxy compound of formula (II) is chlorinated with thionylchloride to form a compound of formula (III), i.e. (±)-6-[chloro(4-chlorophenyl)(l -methyl- liJ-imidazol-5-yl)methyl]-4-(3-chloroρhenyl)-l-methyl-2(li3)-quinolinone.
In step 3, the chloro compound of formula (III) is treated, with NEaL OH in
tetrahydrofuran to form the amino compound of formula (IV), i.e. (±)-6-[amino(4-chlorophenyl)(l-methyl-l -imidazol-5-yl)methyl]-4-(3-chlorophenyl)-l-methyl- 2(l/J)-quinolinone.
In step 4, the amino compound of formula (IV) is separated into its enantiomers by chiral column chromatography over Chiracel OD (25 cm; eluent: 100% ethanol; flow: 0.5 ml/rnin; wavelength: 220 nm). The pure (B)-fractions are collected and recrystallised from 2-propanol resulting in Rl 15777, the compound of formula (V).
Scheme 1
However, the procedure described in WO97/21701 has a number of disadvantages. For example, during the first step, the procedure results in the undesired formation of a corresponding compound of formula (XI), i.e. 6-[hydroxy(4-chlorophenyl) (1-methyl-lJrJ-imidazol-2-yl)methyl]-4-(3-chlorophenyl)-l-methyl-2(liϊ)-quinolinone)Jn which the imidazole ring is attached to the remainder of the molecule at the 2-position of the ring, instead of the desired 5-position. At the end of the procedure, this results in the formation of a compound of formula (XII), i.e.6-[amino(4-chlorophenyl)(l-methyl-lϊJ-imidazol-2-yl)methyl]-4-(3-chlorophenyl)-l-methyl-2(lβ -quinolinone.

(XI) CXH)
The use of n-butyllithium during the conversion of a compound of formula (I) in a compound of formula (II) is also undesirable in a commercial process in view of its pyrophoric nature and the formation of butane, a flammable gas, as the by-product. Also the carrying out of this process step, at a temperature as low as -78°C, is inconvenient and costly on a commercial scale.
Finally, the purification of compound (V) using chiral chromatography is expensive and disadvantageous in view of the large amounts of solvent needed and the specialised equipment required to perform a large scale chiral chromatography.
Another process for the synthesis of Rl 15777 as described in WO 02/072574, is presented in scheme 2.
Herein, in step 1, 1-methyl imidazole in tetrahydrofuran is mixed with a solution of n-hexyllithium in a hexane solvent to which is added tri-iso-butylsilyl chloride, followed by a further addition of n-hexyllithium in hexane. The compound of formula (I) in tetrahydrofuran is then added to the reaction mixture, keeping the temperature between -5°C and 0°C. The resulting product of formula (II) is isolated by salt formation.
In step 2, the chlorination reaction is effected by treatment of the compound of formula (II) with thionyl chloride in 1 ,3-dimethyl-2-imidazolidinone.
In step 3, the chloro compound of formula (III) is treated with a solution of ammonia in methanol. After the addition of water, the compound of formula (IV), precipitates and can be isolated.
In step 4, the compound of formula (IV) can be reacted with L-(-)-dibenzoyl tartaric acid (DBTA) to form the diastereomeric tartrate salt with formula (VI) i.e. R-(-)-6-[amino(4-chlorophenyl)(l-methyl-ljt–‘-imidazol-5-yl)methyl]-4-(3-chlorophenyl)-l-methyl-2(l Z)-quinolinone [R-(R*,RH!)]-2,3-bis(benzoyloxy)butanedioate (2:3).
Finally, in step 5, the compound of formula (VI) is treated with aqueous ammonium hydroxide, to form the crude compound of formula (V) which is then purified by recrystallisation from ethanol to the pure compound (V).

(VI) (V)
Scheme 2
However, in view of the fact that water is present during the third and the fifth step of this procedure, there is significant formation of the hydroxy compound of formula (II).
This is important because the compounds of formula (II) and (V) are difficult to separate. In order to keep the quality of the final product (V) as high as possible, it is critical to limit the formation of compound (II).
The major drawback of the above described processes is the generation of large amounts of the other enantiomer that subsequently must be recycled.
Attempts were made to develop processes that solve this problem. One of the possibilities was to enter chirality in the first step of the procedure. A first study was carried out in order to determine if the conversion of an enantiomer of the hydroxy compound of formula (II) into a compound of formula (IV) could preserve chirality. Several experimental conditions have been tested starting with an enantiomer of a compound of formula (II), but racemisation always occurred.
Another possibility was to try entering chirality by adding N-methylimidazole under the reaction conditions described herein above under steps 1 of WO97/21701 and WO 02/072574, to an N-Ct-6alkyl-(S(R))-sulfinylketimine prepared from the compound of formula (I). It turned out that the resulting N-Cι-6alkyl-(S(R))-sulfinylamide of the compound of formula (I) was in the desired R-configuration and could be used for conversion into compound (V).
These results are completely unexpected, especially in view of Shaw et al.
(Tetrahedron Letters: 42, 7173-7176). Already in 2001, Shaw et al. disclosed an asymmetric synthesis process for the production of α-aryl-α-heteroaryl alkylamines using organometallic additions to N-tert-butanesulfinyl ketimines. However, the configuration and the yield of the final enantiomer formed with this process, was depending on the configuration of the N-tert-butanesulfinyl moiety of the ketimines, the composition of the aryl and/or the heteroaryl moieties of the ketimines, as well as on the organo- and the metallic moiety of the organometallic reagent. Furthermore, the use of heteroaryllithium reagents were described in this document, as being in particular disadvantageous, in view of their instability.
Thus the present invention solves the above described problems. It provides a new process for the preparation of the compound of formula (V) without the need to recycle one of the enantiomers while minimising the formation of undesired isomers and impurities and under conditions which offer economic advantages for operation on a commercial scale.
A. Preparation of intermediates
Example AJ
a) Preparation of /V-r(4-chlorophenyl‘)((,4- -chlorophenyl’)-l-methyl-l f-quinolin-2-one’)-6-yDmethylenel-2-methyl-2-propanesulfinamide TSfR-)! (com ound 15)

Ti(OEt) (0.0122 mol) was added to a mixture of compound (I) (0.0024 mol) and (R)-(+)-2-methyl-2-propane-sulfinamide (0.0024 mol) in DCM (15ml). The mixture was stirred and refluxed for 4 days, then cooled to room temperature. Ice water was added. The mixture was filtered over celite. Celite was washed with DCM. The organic layer was extracted with saturated sodium chloride. The organic layer was separated, dried (MgS04), filtered, and the solvent was evaporated. This fraction was purified by column chromatography over silica gel (40 μm) (eluent: DCM/MeOH 98/2). The pure fractions were collected and the solvent was evaporated, yielding 0.95g of compound 15 _ (76%), melting point: 115°C.
b) Preparation of (R)-N-r(‘4-chlorophenyl1((4-(3-chlorophenyl)-l-methyl-lic/-quinoline- 2-one -6-ylVl-methyl-l/j‘-imidazole-5-yl’)methyll-2-methyl-2-propanesulfinamide rS(R)l (compound 161

(compound 16)
n-Butyllithium (1.34ml, 0.002 mol) was added dropwise at -70°C to a mixture of 1-methylimidazole (0.0021 mol) in THF (4.5ml). The mixture was stirred at -70°C for 15 minutes. Triethylsilyl chloride (0.0021 mol) was added. The mixture was stirred at -70°C for 15 minutes. n-Butyllithium (1.34ml, 0.0021 mol) was added dropwise. The mixture was stirred at -70°C for 15 minutes. A solution of compound 15 (0.0019 mol) in THF (5.5ml) was added. The mixture was stirred at -70°C for 45 minutes, poured out into ice water and extracted with EtOAc. The organic layer was separated, dried (MgS04), filtered, and the solvent was evaporated. The residue was purified by column chromatography over silica gel (15-40 m)(eluent: DCM/MeOH/ΝEUOH 95/5/0.5), yielding 0.59g (52%) of compound 16, diastereomeric excess 24%.
c) Preparation of the (B)-diastereomer (compound 18) of compound 16

(compound 18)
Compound 16 was purified by column chromatography over silica gel (15-40μm) (eluent: DCM/MeOH/NHtOH 95/5/0.5). Two fractions were collected and the solvent was evaporated, yielding 0.304g diastereomer (B) (compound 18) (27%), melting point 174°C.
Example A.2
a) Preparation of jV-r(4-chlorophenyl¥(4-(3-chlorophenyl‘)-l-methyl-l JJ-quinolin-2-one)-6-yl)methylene1-4-methylphenylsulfιnamidesulfιnamide fS(S‘)l (compound 17)

(compound 17)
Ti(OEt)4 (0.0122 mol) was added to a mixture of compound (I) (0.0123 mol) and (S)-(+)-j5-toluenesulfinamide (0.0123 mol) in DCM (80ml). The mixture was stirred and refluxed for 4 days, then cooled to room temperature. Satured sodium chloride was added. The mixture was filtered over celite. Celite was washed with DCM. The organic layer was separated, dried (MgS04), filtered, and the solvent was evaporated. A fraction was purified by column chromatography over silica gel (40 μm) (eluent: DCM MeOH 98/2). The fractions were collected and the solvent was evaporated, yielding 0.65g of pure compound 17 .
The pure compound N-[(4-chlorophenyl)((4-(3-chlorophenyl)-l-methyl-l-tf-quinolin-2-one)-6-yl)methylene]-2-methyl-2-propanesulfinamide [S(R)] can be obtained in an analogues way.
B. Preparation of final compounds
Example BJ
a Preparation of compound (V)

Hydrochloric acid in isopropanol was added to a solution of compound 16 (0.00003 mol) in methanol (0J ml). The mixture was stirred at room temperature for 30 minutes. The mixture was added to potassium carbonate (10%) on ice. The organic layer was separated, washed with a solution of saturated sodium chloride, dried (MgS04), filtered, and evaporated giving 0,017 g (100%) of compound (V), enantiomeric excess 22%, content of compound (II) < 1%.
PATENT
WO 2005105783
https://encrypted.google.com/patents/WO2005105783A1?cl=en
A. Preparation of intermediates
Example A.1
a) Preparation of N-r(4-chlorophenyl’)(l-methyl-lH-imidazol-5-yl)methylene‘)l-2- methyl-2-propanesulfinamide KSfl l (compound 25)

(compound 25) Ti(OEt)4 (0.0162 mol) was added to a mixture of (4-chlorophenyiχi-methyl-lH- imidazol-5-yl)methanone (0.0032 mol) and (R)-(+)-2-methyl-2-propane-sulfinamide (0.0032 mol) in DCE (7ml). The mixture was stirred and refluxed for 6 days, then cooled to room temperature. Ice water was added. The mixture was filtered over celite. Celite was washed with DCM. The organic layer was extracted with saturated sodium chloride. The organic layer was separated, dried (MgS04), filtered, and the solvent was evaporated. This fraction was purified by column chromatography over silica gel (40 μm) (eluent: DCM/MeOH/NH OH 97/3/0.5), yielding 0.475g of compound 25 (46%).
The compound N-[(4-chlorophenyl)(l-methyl-lH-imidazol-5-yl)methylene)]-2-methyl- 2-propanesulfinamide [(S(S)] can be obtained in an analogous way.
b) Preparation of N-r(4-chlorophenyl)((4-(3-chlorophenyl)-2-methoχy-quinoline-6- yl l-methyl-lH-imidazole-5-yl)methyn-2-methyl-2-propanesulfinamide TS(R)1 (compound 26)

(compound 26)
n-Butyllithium (0.00081 mol) in hexane, was added dropwise at -78°C to a mixture of 6-bromo-4-(3-chlorophenyl)-2-methoxy-quinoline (0.00081 mol) in THF (3 ml) under nitrogen flow. The mixture was stirred at -78°C for 30 minutes. A solution of compound 25 (0.00065 mol) in THF (0.6 ml) was added . The mixture was stirred at – 78°C for 1 hour and 30 minutes, poured out into ice water and extracted with EtOAc. The organic layer was separated, dried (MgS04), filtered, and the solvent was evaporated. This fraction was purified by column chromatography over silica gel (40μm)(eluent: DCM eOH/NB OH 97/3/0.1). The pure fractions were collected and the solvent was evaporated, yielding 0.138g (36 %) of compound 26, melting point 153°C.
The compound N-[(4-chlorophenyl)((4-(3-chlorophenyl)-2-methoxy-quinoline-6-yl)(l- methyl-lH-imidazole-5-yl)methyl]-2-methyl-2-propanesulfmamide [S(S)] can be obtained in an analogous way
c“) Preparation of (S)-l-,4-chlorophenylV l-r4-(3-chlorophenylV2-methoxy-quinoline-6- yll-l-(l-methyl-l/J-imidazole-5-yl)-methylamine (compound 27)

(compound 27) Hydrochloric acid in isopropanol was added to a solution of compound 26 (0.000018 mol) in methanol (4.2 ml). The mixture was stirred at room temperature for 30 minutes. The mixture was added to potassium carbonate (10%) on ice and extracted with ethyl acetate. The organic layer was separated, washed with a solution of saturated sodium chloride, dried (MgS0 ), filtered, and evaporated giving 0,086 g (100%) of compound 27, melting point 96°C, enantiomeric excess 88%. d) Preparation of (SV6-ramino(4-chlorophenyl¥l-methyl-l #-imidazol-5-yDmethyH-4- (3-chlorophenyD-lH)-quinorin-2-one (compound 28)

(compound 28) Compound 27 (0.00038 mol) in hydrochloric acid 3N (9.25 ml) and THF (9.25 ml), was stirred at 60°C for 24 hours and evaporated, giving 0,18 g (100%) of compound 28, melting point 210°C.
Example A.2
a) Preparation of N-r(4-chlorophenyl)(‘l-methyl-lH-imidazol-5-yl’)methylene‘)1-p-
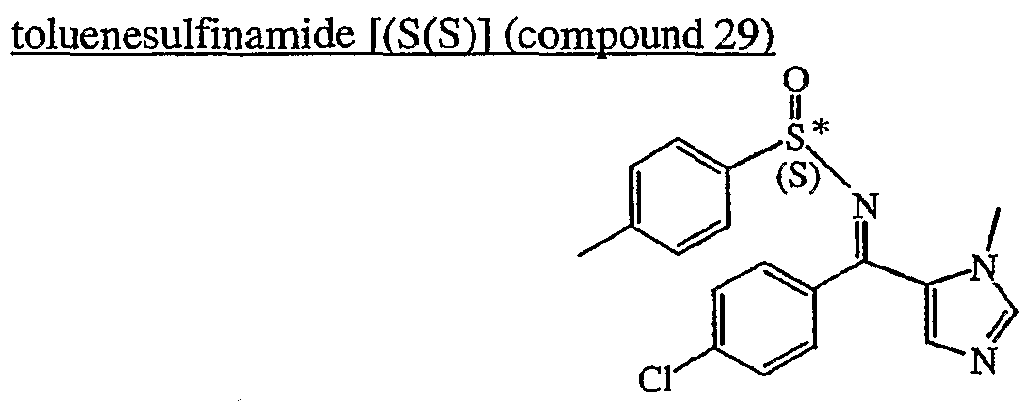
(compound 29) Ti(OEt)4 (0.0419 mol) was added to a mixture of (4-chlorophenyl)(l-methyl-lH- imidazol-5-yl)methanone (0.0084 mol) and (S)-(+)-p-_toluenesulfinamide (0.0084 mol) in DCE (18ml). The mixture was stirred and refluxed for 7 days, then cooled to room temperature. Ice water was added. The mixture was filtered over celite. Celite was washed with DCM. The organic layer was extracted with saturated sodium chloride. The organic layer was separated, dried (MgS04), filtered, and the solvent was evaporated. This fraction was purified by column chromatography over silica gel (40 μm) (eluent: DCM/MeOH/ΝHiOH 97/3/0.5), yielding 1.15 g of compound 29 (38%).
The compound N-[(4-chlorophenyl)(l-methyl-lH-imidazol-5-yl)methylene)]-p- toluenesulfinamide [(S(R)] can be obtained in an analogues way. B. Preparation of final compounds
Example B.l a) Preparation of (S)-6-ramino(4-chlorophenyl)(l-methyl-lH-imidazol-5-yl)methyll-4-

Compound 28 (0.00038 mol) was added to a solution of THF (1.8 ml) and NaOH ION (1.8 ml). BTEAC (0.0019 mol) and methyliodide (0.00076 mol) were added and the mixture was stirred for 2 hours at room temperature. EtOAc was added. The organic layer was separated, dried (MgS04), filtered, and evaporated giving 0,149 g (83%) of compound 30, enantiomeric excess 86%.
PATENT
WO 02/072574
https://encrypted.google.com/patents/WO2002072574A1?cl=en
Preparation of compound (III):
110 ml of dry tetrahydrofuran was added to 7.6 ml of 1-methylimidazole (0.0946 mole) and the resulting solution cooled to -15°C.37.8 ml of n-hexyllithium 2.5 M in n-hexane (0.0946 mole) was added, while the temperature during addition was kept between – 5°C and 0°C. After addition, the reaction mixture was stirred for 15 minutes, while cooling to -12°C. 26.2 ml of tri-w o-butylsilyl chloride (0.0964 mole) was added, while the temperature during addition was kept between -5° and 0°C. After addition, the reaction mixture was stirred for 15 minutes, while cooling to -13°C. 37.2 ml of n- hexyllithium 2.5 M in n-hexane (0.0930 mole) was added, while the temperature during addition was kept between -5°C and 0°C (some precipitation occured). After addition, the reaction mixture was stirred for 15 minutes, while cooling to -14°C. 128 ml of dry tetrahydrofuran was added to 26.22 g of 6-(4-chlorobenzoyl)-4-(3-chlorophenyl)-l- methyl-2(lH)-quinolinone (compound (II)) (0.0642 mole) and stirred until dissolution. This solution was added to the reaction mixture, while the temperature during addition was kept between -5°C and 0°C. After addition, the reaction mixture was stirred for 15 minutes between -5°C and 0°C. 128 ml of water was added to the reaction mixture, followed by the addition of 10.6 ml of acetic acid. The mixture was then heated to 40°C and stirred for 2 hours. The layers were separated and the organic layer washed with 32 ml water. 64 ml water and 7.8 ml aqueous NaOΗ 50% were added to the organic layer which was stirred for 1 hour at ambient temperature. The layers were separated and the organic layer concentrated under reduced pressure, yielding 51.08 g of a brown oil (46.6 wt% 4-(3-chlorophenyl)-6-[(4-chlorophenyl)hydroxy(l-methyl-lH-imidazol-5- yl)methyl]-l-methyl-2(lH)-quinolinone (compound HI); 75.6 % yield).
The product can be isolated via the procedures mentioned above. The resulting product was analysed by hplc using the following conditions :-
Column: Ηypersil C18-BD 3μm, 100mm x 4 mm (i.d.)
Mobile phase:
Solvent A: 0.5% NΗLjOAc
Solvent B: CΗ3CN
Gradient: Time %A %B
0 100 0
15 0 100
18 0 100 19 100 0 23 100 0 Detector: UV 254nm Solvent: DMF The product was found to have a C5:C2 ratio of 99.8:0.2. In contrast using n-butyllithium in place of n-hexyllithium, triethylsilyl chloride in place of tri-i.ro- butylsilyl chloride and conducting the process at -70°C, i.e. generally in accordance with prior art procedures discussed above, the resulting product had a C5:C2 ratio of 95:5, a significant difference in commercial terms.
Preparation of compound (IV)
A 1 liter reaction vessel was charged with 105.4 g of 4-(3-chlorophenyl)-6-[(4- chlorophenyl)hydroxy ( 1 -methyl- 1 H-imidazol-5-yl)methyl] – 1 -methyl-2( 1 H)- quinolinone hydrochloric acid salt (compound (IΗ)and 400 ml of N,N- dimethylimidazolidinone added at 22°C. The mixture was stirred vigorously for 15 minutes at 22°C and became homogeneous. 32.1 ml of thionyl chloride was added over 10 minutes to the reaction mixture, the reaction temperature rising from 22°C to 40°C. After addition of the thionyl chloride, the reaction mixture was cooled from 40°C to 22°C and stirred for three hours at the latter temperature to provide a solution of 4-(3- chlorophenyl)-6-[chloro-(4-chlorophenyl)(l-methyl-lH-imidazol-5-yl)methyl]-l- methyl-2(lH)-quinolinone (compound (IN).
Preparation of unresolved compound (I)
429 ml of ammonia in methanol 7Ν was cooled to 5°C in a 3 liter reaction vessel and the solution of compound (IN), obtained in the previous stage, added, while stirring, over 10 minutes, with an exothermic reaction, the temperature rising from 5°C to 37°C. After the addition was complete, the reaction mixture was cooled to 22°C and stirred for 20 hours. 1000ml of water was then added over 20 minutes, the addition being slightly exothermic so the reaction mixture was cooled to keep the temperature below 30°C. The mixture was then stirred for 22 hours at 22°C, the resulting precipitate filtered off and the precipitate washed three times with 100ml of water to provide a yield of 70-75% of 6-[arnino(4-chlorophenyl)-l-methyl-lH-imidazol-5-ylmethyl]-4-(3- chlorophenyl)-l-methyl-2(lH)-quinolinone. Resolution of compound (I)
a) A 3 liter reaction vessel was charged with 146.8 g of 6-[amino(4-chlorophenyl)(l- methyl-lH-imidazol-5-yl)methyl]-4-(3-chlorophenyl)-l-methyl-2(lH)-quinolinone and 301.1 g of L-(-)-dibenzoyl-tartaric acid monohydrate, 1200ml of acetone was added and the reaction mixture stirred vigorously for 10 minutes at 22°C to form a solution which was seeded with lOOmg of the final tartrate salt product (obtained from previous screening experiments) and then stirred for 22 hours at 22°C. The resulting precipitate was filtered off and the precipitate was washed twice with 75 ml of acetone and the product dried at 50°C in vacuo to yield 114.7g of R-(-)-6-[amino(4-chlorophenyl)(l- methyl-lΗ-imidazol-5-yl)methyl]-4-(3-chlorophenyl)-l-methyl-2(lΗ)-quinolinone [R- (R*,R*)]-2,3-bis(benzoyloxy)butanedioate (2:3).
b) 41.08 g of the product of stage a) and 80 ml ethanol were stirred for 15 minutes at 22°C. 12.0 ml concentrated aqueous ammonium hydroxide was added over 2 minutes, and the reaction mixture stirred for 1 hour at 25°C. 160 ml water was added over 10 minutes at 25 °C and the mixture heated to reflux and stirred at reflux for 1 hour. The reaction mixture was then cooled to 20°C and stirred for 16 hours at 20°C. The product was filtered, washed twice with 8 ml water and dried at 50°C in vacuo to yield 16.87 g of (R)-(+)-6-[amino(4-chloro-phenyl)(l-methyl-lH-imidazol-5-yl)methyl]-4-(3- chlorophenyl)-l-methyl-2(lH)-quinolinone (compound (I)).
Purification of compound (I)
265 ml of ethanol was added to 19.9g of compound (I), obtained as described in the previous stage, and the mixture warmed while stirring to reflux temperature (78 °C) and then stirred at reflux temperature for 15 minutes before cooling the solution to 75 °C. 1.0 g of activated carbon (Norit A Supra) was then added to the mixture which was stirred at reflux temperature for 1 hour, filtered while warm and the filter then washed with 20 ml warm ethanol. The filtrate and wash solvent were combined (the product spontaneously crystallizes at 48°C), and the mixture warmed to reflux temperature and concentrated by removing 203 ml of ethanol. The resulting suspension was cooled to 22°C, stirred for 18 hours at 22°C, cooled to 2°C and stirred for 5 more hours at 2°C. The precipitate was filtered and washed with 4 ml ethanol and the product dried at 50°C in vacuo to yield 17.25 g of purified compound (I) which complies with the infrared spectrum of reference material.
PAPER
Practical route to 2-quinolinones via a pd-catalyzed c-h bond Activation/C-C bond Formation/Cyclization cascade reaction
Org Lett 2015, 17(2): 222
https://pubs.acs.org/doi/10.1021/ol503292p
Practical Route to 2-Quinolinones via a Pd-Catalyzed C–H Bond Activation/C–C Bond Formation/Cyclization Cascade Reaction

Quinolinone derivatives were constructed via a Pd-catalyzed C–H bond activation/C–C bond formation/cyclization cascade process with simple anilines as the substrates. This finding provides a practical procedure for the synthesis of quinolinone-containing alkaloids and drug molecules. The utility of this method was demonstrated by a formal synthesis of Tipifarnib.
SEE https://pubs.acs.org/doi/suppl/10.1021/ol503292p/suppl_file/ol503292p_si_001.pdf

4-(3-chlorophenyl)-6-(4-chlorobenzyl)-2-quinolinone 5:

0.5 mmol 4-Amino-4′-chlorodiphenylmethane 4, 1mmol acetic anhydride and 2 mL toluene were added into the Schlenk tuble. The mixture was stirred at r.t. for 5 minutes, then 0.5 mmol TsOH•H2O, 2.5 mmol (2E)-3-(3-chlorophenyl) propenoate, 1.5 mmol Na2S2O8 and 5 mmol % Pd(OAc)2 were added into the reaction system in one time. The mixture was heated at 100 oC for 36 h and cooled down to room temperature, quenched with 50 mL saturated sodium bicarbonate solution and extracted thrice with ethyl acetate (30 mL) and the combined organic phase was dried over Na2SO4. After evaporation of the solvents the residue was purified by silica gel chromatography to afford 5 as pale yellow solid (elute: hexane-EtOAc) (180 mg, 95%).
1H NMR (400 MHz, d6-DMSO) ppm: 11.87 (s, 1H), 7.59-7.52 (m, 2H), 7.50-7.47 (m, 1H), 7.42-7.37 (m, 2H), 7.35-7.28 (m, 3H), 7.19-7.14 (m, 3H), 6.41 (s, 1H), 3.92 (s, 2H).
13C NMR (100 MHz, d6-DMSO): 161.50, 150.09, 140.52, 139.13, 138.25, 134.89, 133.85, 132.04, 131.16, 130.99, 130.95, 129.17, 128.88, 128.80, 127.94, 125.84, 122.30, 118.44, 116.55, 39.92.
HRMS (ESI) Calcd. for C22H15Cl2NO: [M + H]+ , 380.0609. Found: m/z 380.0613.
4-(3-chlorophenyl)-6-(4-chlorobenzoyl)-2-quinolinone 6:1

4-(3-chlorophenyl)-6-(4-chlorobenzyl)-2-quinolinone 5 (0.2 mmol), iodine (0.002 mmol), pyridine (0.002 mmol) and aqueous tert-butylhydroperoxide (70%, 0.5 ml) were sealed in a 5 mL tube, then stirred at 80 oC overnight. After cooling to room temperature, the mixture was purified by a short silica gel chromatography column to afford 6 as pale yellow solid (elute: DCM/acetone = 2/1) (77 mg, 98%).
1H NMR (400 MHz, d6-DMSO) ppm: 12.31 (s, 1H), 8.00 (dd, J = 8.40 Hz, 1.60 Hz, 1H), 7.76 (d, J = 8.40 Hz, 2H), 7.74 (d, J = 1.60 Hz, 1H) 7.68 (s, 1H), 7.60 (d, J = 8.40 Hz, 2H), 7.55-7.50 (m, 4H), 6.57 (s, 1H).
13C NMR (100MHz, d6-DMSO): 193.48, 161.83, 150.38, 143.00, 138.46, 137.74, 136.36, 133.92, 132.04, 131.85, 131.16, 130.20, 129.93, 129.57, 129.08, 128.99, 128.11, 123.01, 117.81, 116.74. HRMS (ESI) Calcd. for C22H13Cl2NO2: [M + H]+ , 394.0402. Found: m/z 394.0405.
Reference: 1. Zhang, J.; Wang, Z.; Wang, Y.; Wan, C.; Zheng, X.; Wang, Z. Green Chem. 2009, 11, 1973. 2. (a) Angibaud, P.; Venet, M.; Filliers, W.; Broeckx, R.; Ligny, Y.; Muller, P.; Poncelet, V.; End, D. Eur. J. Org. Chem. 2004, 479. (b) Filliers, W.; Broeckx, R.; Angibaud, P. U.S. patent, US7572916, 2009.
NMR SIMULATION


PREDICTED VALUES
1H NMR: δ 3.42 (3H, s), 3.63 (3H, s), 6.57 (1H, s), 6.67 (1H, d, J = 1.7 Hz), 7.27 (1H, dd, J = 8.3, 1.5 Hz), 7.36-7.59 (8H, 7.46 (ddd, J = 8.3, 1.5, 0.5 Hz), 7.41 (ddd, J = 8.1, 8.1, 0.5 Hz), 7.39 (ddd, J = 8.1, 1.6, 1.5 Hz), 7.49 (ddd, J = 8.1, 1.7, 1.5 Hz), 7.55 (ddd, J = 8.3, 1.6, 0.5 Hz), 7.58 (d, J = 1.7 Hz)), 7.66 (1H, dd, J = 8.3, 0.5 Hz), 7.71 (1H, dd, J = 1.5, 0.5 Hz), 7.84 (1H, ddd, J = 1.7, 1.6, 0.5 Hz).

13C NMR PREDICT



COSY PREDICT


HSQC PREDICT

References
- Jump up^ “International Nonproprietary Names for Pharmaceutical Substances (INN). Recommended International Nonproprietary Names (Rec. INN): List 46” (PDF). World Health Organization. Retrieved 16 November 2016.
- Jump up^ Sparano, JA; Moulder, S; Kazi, A; Coppola, D; Negassa, A; Vahdat, L; Li, T; Pellegrino, C; Fineberg, S; Munster, P; Malafa, M; Lee, D; Hoschander, S; Hopkins, U; Hershman, D; Wright, JJ; Kleer, C; Merajver, S; Sebti, SM (15 April 2009). “Phase II Trial of Tipifarnib plus Neoadjuvant Doxorubicin-Cyclophosphamide in Patients with Clinical Stage IIB-IIIC Breast Cancer” (PDF). Clinical Cancer Research. 15 (8): 2942–48. doi:10.1158/1078-0432.CCR-08-2658. PMC 2785076
 . PMID 19351752. Retrieved 16 November 2016.
. PMID 19351752. Retrieved 16 November 2016. - Jump up^ Alsina, M; Fonseca, R; Wilson, EF; Belle, AN; Gerbino, E; Price-Troska, T; Overton, RM; Ahmann, G; Bruzek, LM; Adjei, AA; Kaufmann, SH; Wright, JJ; Sullivan, D; Djulbegovic, B; Cantor, AB; Greipp, RP; Dalton, WS; Sebti, SM (1 May 2004). “Farnesyltransferase Inhibitor Tipifarnib Is Well Tolerated, Induces Stabilization of Disease, and Inhibits Farnesylation and Oncogenic/Tumor Survival Pathways in Patients with Advanced Multiple Myeloma” (PDF). Blood. 103 (9): 3271–7. doi:10.1182/blood-2003-08-2764. PMID 14726402. Retrieved 16 November 2016.
- Jump up^ “R115777 in Treating Patients With Advanced Solid Tumors”
- Jump up^ “R115777 to Treat Children With Neurofibromatosis Type 1 and Progressive Plexiform Neurofibromas”
- Jump up^ “Johnson & Johnson Pharmaceutical Research & Development, L.L.C. Receives Not Approvable Letter From FDA for Tipifarnib Based on Phase II Data”. PR Newswire. Jun 30, 2005. Retrieved 16 November 2016.
- Jump up^ Capell, BC; Olive, M; Erdos, MR; Cao, K; Faddah, DA; Tavarez, UL; Conneely, KN; Qu, X; San, H; Ganesh, SK; Chen, X; Avallone, H; Kolodgie, FD; Virmani, R; Nabel, EG; Collins, FS (6 October 2008). “A Farnesyltransferase Inhibitor Prevents Both the Onset and Late Progression of Cardiovascular Disease in a Progeria Mouse Model” (PDF). Proceedings of the National Academy of Sciences. 105 (41): 15902–7. doi:10.1073/pnas.0807840105. PMC 2562418
 . PMID 18838683. Retrieved 16 November 2016.
. PMID 18838683. Retrieved 16 November 2016.

|
|
| Clinical data | |
|---|---|
| Synonyms | R115777 |
| ATC code |
|
| Legal status | |
| Legal status |
|
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| IUPHAR/BPS | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEMBL | |
| Chemical and physical data | |
| Formula | C27H22Cl2N4O |
| Molar mass | 489.40 g·mol−1 |
| 3D model (JSmol) | |

| Cited Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO1997021701A1 * | Oct 16, 1996 | Jun 19, 1997 | Janssen Pharmaceutica N.V. | Farnesyl protein transferase inhibiting (imidazol-5-yl)methyl-2-quinolinone derivatives |
| WO2001051127A1 * | Jan 9, 2001 | Jul 19, 2001 | Merck & Co., Inc. | Inhibitors of prenyl-protein transferase |
| WO2001053289A1 * | Nov 29, 2000 | Jul 26, 2001 | Pfizer Products Inc. | Anticancer compound and enantiomer separation method useful for synthesizing said compound |
| WO2002020015A1 * | Aug 30, 2001 | Mar 14, 2002 | Merck & Co., Inc. | Inhibitors of prenyl-protein transferase |
| WO2002072574A1 * | Mar 5, 2002 | Sep 19, 2002 | Janssen Pharmaceutica N.V. | Process for the preparation of imidazole compounds |
| WO2002079147A2 * | Mar 26, 2002 | Oct 10, 2002 | Merck & Co., Inc. | Inhibitors of prenyl-protein transferase |
| Reference | ||
|---|---|---|
| 1 | * | SHAW A W ET AL: “Asymmetric synthesis of alpha,alpha-diaryl and alpha-aryl-alpha-heteroaryl alkylamines by organometallic additions to N-tert-butanesulfinyl ketimines” TETRAHEDRON LETTERS, ELSEVIER SCIENCE PUBLISHERS, AMSTERDAM, NL, vol. 42, no. 41, 8 October 2001 (2001-10-08), pages 7173-7176, XP004304959 ISSN: 0040-4039 cited in the application |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| US9707221 | Nov 8, 2016 | Jul 18, 2017 | Kura Oncology, Inc. | Methods of treating cancer patients with farnesyltransferase inhibitors |
//////////////////TIPIFARNIB , R-115777, типифарниб , تيبيفارنيب , 替匹法尼 , NSC-702818 , phase 3, orphan drug designation, NSC 702818, R 115777, Kura Oncology, Zarnestra, Janssen
CN1C=NC=C1[C@@](N)(C1=CC=C(Cl)C=C1)C1=CC2=C(C=C1)N(C)C(=O)C=C2C1=CC(Cl)=CC=C1