
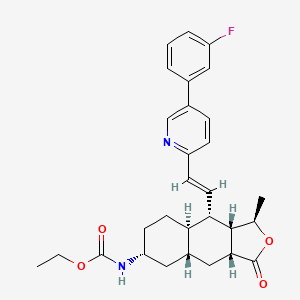
VORAPAXAR
Thrombosis, Antiplatelet Therapy, PAR1 Antagonists , MERCK ..ORIGINATOR
Ethyl N-[(3R,3aS,4S,4aR,7R,8aR,9aR)-4-[(E)-2-[5-(3-fluorophenyl)-2-pyridyl]vinyl]-3-methyl-1-oxo-3a,4,4a,5,6,7,8,8a,9,9a-decahydro-3H-benzo[f]isobenzofuran-7-yl]carbamate
Ethyl ((1R,3aR,4aR,6R,8aR,9S,9aS)-9-((1E)-2-(5-(3-fluorophenyl)pyridin-2-yl)ethenyl)- 1-methyl-3-oxododecahydronaphtho(2,3-c)furan-6-yl)carbamate
Carbamic acid, ((1R,3aR,4aR,6R,8aR,9S,9aS)-9-((1E)-2-(5-(3-fluorophenyl)-2- pyridinyl)ethenyl)dodecahydro-1-methyl-3-oxonaphtho(2,3-c)furan-6-yl)-, ethyl ester
618385-01-6 CAS NO FREE FORM
CAS Number: 705260-08-8 SULPHATE
Has antiplatelet activity.
- UNII-ZCE93644N2
- Zontivity
Registered – 2015 MERCK Thrombosis
Vorapaxar (formerly SCH 530348) is a thrombin receptor (protease-activated receptor, PAR-1) antagonist based on the natural product himbacine. Discovered by Schering-Plough and currently being developed by Merck & Co., it is an experimental pharmaceutical treatment for acute coronary syndrome chest pain caused by coronary artery disease.[1]
In January 2011, clinical trials being conducted by Merck were halted for patients with stroke and mild heart conditions.[2] In a randomized double-blinded trial comparing vorapaxar with placebo in addition to standard therapy in 12,944 patients who had acute coronary syndromes, there was no significant reduction in a composite end point of death from cardiovascular causes, myocardial infarction, stroke, recurrent ischemia with rehospitalization, or urgent coronary revascularization. However, there was increased risk of major bleeding.[3]
A trial published in February 2012, found no change in all cause mortality while decreasing the risk of cardiac death and increasing the risk of major bleeding.[4]
SCH-530348 is a protease-activated thrombin receptor (PAR-1) antagonist developed by Schering-Plough and waiting for approval in U.S. for the oral secondary prevention of cardiovascular events in patients with a history of heart attack and no history of stroke or transient ischemic attack. The drug candidate is being investigated to determine its potential to provide clinical benefit without the liability of increased bleeding; a tendency associated with drugs that block thromboxane or ADP pathways. In April 2006, SCH-530348 was granted fast track designation in the U.S. for the secondary prevention of cardiovascular morbidity and mortality outcomes in at-risk patients.
Vorapaxar was recommended for FDA approval on January 15, 2014.[5]
Vorapaxar is a protease-activated thrombin receptor (PAR-1) antagonist developed by Schering-Plough (now, Merck & Co.) and approved in the U.S. in 2014 for the reduction of thrombotic cardiovascular events in patients with a history of myocardial infarction or with peripheral arterial disease. However, in 2018 Aralez discontinued U.S. commercial operations. In 2015, the product was approved in the E.U. for the reduction of atherothrombotic events in adult patients with a history of myocardial infarction. In April 2006, vorapaxar was granted fast track designation in the U.S. for the secondary prevention of cardiovascular morbidity and mortality outcomes in at-risk patients. In 2016, Aralez Pharmaceuticals acquired the U.S. and Canadian rights to the product pursuant to an asset purchase agreement entered into between this company and Merck & Co.
Merck & Co (following its acquisition of Schering-Plough) has developed and launched vorapaxar (Zontivity; SCH-530348; MK-5348), an oral antagonist of the thrombin receptor (protease-activated receptor-1; PAR1); the product is marketed in the US by Aralez Pharmaceuticals
WO-03089428, published in October 2003, claims naphtho[2,3-c]furan-3-one derivatives as thrombin receptor antagonists. WO-03033501 and WO-0196330, published in April 2003 and December 2001, respectively, claim himbacine analogs as thrombin receptor antagonists. WO-9926943 published in June 1999 claims tricyclic compounds as thrombin receptor antagonists
 VORAPAXAR
VORAPAXAR17 JAN 2014
FDA advisory panel votes to approve Merck & Co’s vorapaxar REF 6
https://www.accessdata.fda.gov/drugsatfda_docs/nda/2014/204886Orig1s000ChemR.pdf
Zontivity (vorapaxar) tablets NDA 204886



VORAPAXAR SULPHATE

CAS Number: 705260-08-8 SULPHATE
Molecular Formula: C29H33FN2O4.H2O4S
Molecular Weight: 590.7
Chemical Name: Ethyl [(1R,3aR,4aR,6R,8aR,9S,9aS)-9-[(1E)-2-[5-(3-fluorophenyl)pyridin-2- yl]ethenyl]-1-methyl-3-oxododecahydronaphtho[2,3-c]furan-6-yl]carbamate sulfate
Synonyms: Carbamic acid, [(1R,3aR,4aR,6R,8aR,9S,9aS)-9-[(1E)-2-[5-(3-fluorophenyl)-2- pyridinyl]ethenyl]dodecahydro-1-methyl-3-oxonaphtho[2,3-c]furan-6-yl]-,ethyl ester,sulfate; SCH-530348
Vorapaxar Sulfate (SCH 530348) a thrombin receptor (PAR-1) antagonist for the prevention and treatment of atherothrombosis.
POLYMORPH
U.S.Pat. No. 7,304,078 discloses Vorapaxar base. U.S.Pat. No. 7,235,567 discloses Polymorph I and II of vorapaxar sulphate
CN 106478608 provides a crystalline polymorph A
EMA
Atherosclerosis and ischemic cardiovascular (CV) diseases like coronary artery disease (CAD) are progressive systemic disorders in which clinical events are precipitated by episodes of vascular thrombosis. Patients with an established history of atherothrombotic or athero-ischemic disease are at particular risk of future cardiac or cerebral events, and vascular death. Anti-thrombotic therapy options in patients with stable atherosclerosis are not well-established. Long-term therapies to effectively modulate the key components responsible for atherothrombosis in secondary prevention of ischemic CV disease are therefore required. Vorapaxar is a first – in – class selective antagonist of the protease-activated receptor 1 (PAR-1), the primary thrombin receptor on human platelets, which mediates the downstream effects of this critical coagulation factor in hemostasis and thrombosis. Thrombin-induced platelet activation has been implicated in a variety of cardiovascular disorders including thrombosis, atherosclerosis, and restenosis following percutaneous coronary intervention (PCI). As an antagonist of PAR-1, vorapaxar blocks thrombin-mediated platelet aggregation and thereby has the potential to reduce the risk of atherothrombotic complications of coronary disease. The applicant has investigated whether a new class of antiplatelet agents, PAR-1 antagonists, can further decrease the risk of cardiovascular events in a population of established atherothrombosis when added to standard of care, in secondary prevention of ischemic diseases. The following therapeutic indication has been submitted for vorapaxar: Vorapaxar is indicated for the reduction of atherothrombotic events in patients with a history of MI. Vorapaxar has been shown to reduce the rate of a combined endpoint of cardiovascular death, MI, stroke, and urgent coronary revascularization. Vorapaxar will be contraindicated in patients with a history of stroke or TIA. The indication sought in the current application is supported by the efficacy results of the TRA 2P-TIMI, which is considered the pivotal trial for this indication. During the procedure, the applicant requested the possibility of extending the indication initially sought for, to extend it to the population of PAD patients. This request was discussed at the CHMP and not accepted by the Committee.
Introduction The finished product is presented as immediate release film-coated tablets containing 2.5 mg of vorapaxar sulfate as active substance per tablet, corresponding to 2.08 mg vorapaxar. Other ingredients are: lactose monohydrate, microcrystalline cellulose (E460), croscarmellose sodium (E468), povidone (E1201) , magnesium stearate (E572), hypromellose (E464), titanium dioxide (E171), triacetin (glycerol triacetate) (E1518), iron oxide yellow (E172), as described in section 6.1 of the SmPC. The product is available in Aluminium–Aluminium blisters (Alu-Alu) as described in section 6.5 of the SmPC.
General information The chemical name of the active substance vorapaxar sulfate is ethyl[(1R,3aR,4aR,6R,8aR,9S,9aS)- -9-{(1E)-2-[5-(3-fluorophenyl)pyridin-2-yl]ethen-1-yl}-1-methyl-3-oxododecahydronaphtho[2,3-c] furan-6-yl]carbamate sulfate, corresponding to the molecular formula C29H33FN2O4 • H2SO4 and has a relative molecular mass 590.7. It has the following structure:

The structure of the active substance has been confirmed by mass spectrometry, infrared spectroscopy, 1H- and 13C-NMR spectroscopy and X-ray crystallography, all of which support the chemical structure elemental analysis. It appears as a white to off-white, slightly hygroscopic, crystalline powder. It is freely soluble in methanol and slightly soluble in ethanol and acetone but insoluble to practically insoluble in aqueous solutions at pH above 3.0. The highest solubility in aqueous solution can be achieved at pH 1.0 or in simulated gastric fluids at pH 1.4. The dissociation constant of vorapaxar sulfate was determined to be pKa = 4.7 and its partition coefficient LogP was determined to be 5.1. Vorapaxar sulfate contains seven chiral centers and a trans double bond. The seven chiral centres are defined by the manufacturing process of one of the intermediates in the vorapaxar synthesis and potential enantiomers are controlled by appropriate specifications. The cis-isomer of the double bond is controlled by a highly stereo-specific process reaction resulting in non-detectable levels of cis-isomer impurity. The cis-isomer impurity is controlled in one of the intermediates as an unspecified impurity. A single crystalline stable anhydrous form has been observed.
GENERAL INTRODUCTION
SIMILAR NATURAL PRODUCT
+ HIMBACINE


Himbacine is an alkaloid muscarinic receptor antagonist displaying more potent activity associated with M2 and M2 subtypes over M1 or M3. Observations show himbacine bound tightly to various chimeric receptors in COS-7 cells as well as possessed the ability to bind to cardiac muscarinic receptors allosterically. Recent studies have produced series of thrombin receptor (PAR1) antagonists derived from himbacine Himbacine is an inhibitor of mAChR M2 and mAChR M4.
| Physical State: | Solid |
| Derived from: | Australian pine Galbulimima baccata |
| Solubility: | Soluble in ethanol (50 mg/ml), methanol, and dichloromethane. Insoluble in water. |
| Storage: | Store at -20° C |
| Melting Point: | 132-134 °C |
| Boiling Point: | 469.65 °C at 760 mmHg |
| Density: | 1.08 g/cm3 |
| Refractive Index: | n20D 1.57 |
| Optical Activity: | α20/D +51.4º, c = 1.01 in chloroform |
| Application: | An alkaloid muscarinic receptor antagonist |
| CAS Number: | 6879-74-9 |
| Molecular Weight: | 345.5 |
| Molecular Formula: | C22H35NO2 |
General scheme:

PATENT
WO 2006076415
WO 2006076452
WO 2003089428
US 6063847
CN 107540564
WO 2008005344
CN 106749138
PATENT
Example 1:
[0027] The steel shed amide (300mg, 7. 93mmol) and 15 blood THF was added to 100 blood Ξ jar. The starting material II (2.OOg, 5. 89mmol) was dissolved in 15mL of THF dropwise via pressure-equalizing dropping funnel to the reaction system, the process temperature will produce a large number of bubbles -2 ~ 0 ° C, in the process, Lan mix of about 0.1 until no bubbles generate. THF solution containing 13 Blood Ship (0.75 Yap, 2. 95mmol) is transferred to a pressure-equalizing dropping funnel. It was slowly added dropwise to the reaction system. After the completion of dropwise continue to embrace mix ratio. After the treatment, at 0 ° C under 0.8 blood, Imol / L 1 fat slowly dropped into the embrace mixed reaction system, after adding the right amount of water, acetic acid extraction. The combined organic phase with Imol / L of 0H (17mLX3) washing the organic phase coating. Tu brine, dried over anhydrous sulfate steel, 25 ° C under reduced pressure to spin dry to give 1. 75g light yellow oil, yield 91%.
[0028] After the content was determined using the external standard method, first prepared by a qualified reference determine its content, W this as a standard substance, measuring the external standard method to get the content of 99%.
[0029] Zan NMR: (400MHz, CD3CN):… 5 46 of r, 1H), 4 70 (td, 1H), 4 03 based 2H), 3 69-3 57 (m, 2 Η).. , 3. 45-3. 32 (based, IH), 2. 77 (br, IH), 2. 61-2. 51 (m, IH), 2. 49-2. 39 (m, 1 field, 2 30 of r IH), 2 .12-1. 92 (m, IH), 1. 87 (dt, IH), 1. 81-1. 72 (m, IH), 1. 61-1. 50 ( …. m, IH), 1 48 (d, 3H), 1 23-1 09 (m, 7H), 1. 05-0 90 (m, 2H);
[0030] MS (ES +) m / z: 326. 24 [M + + field.
[Cited 00] Example 2:
[003 cited the steel shed amide (312mg, 8. 25mmol) and 16 blood THF was added to the lOOmL Ξ jar. The starting material II (2.OOg, 5. 89mmol) was dissolved in 15mL of THF dropwise via pressure-equalizing dropping funnel to the reaction system, the process temperature will produce a large number of bubbles -2 ~ -5 ° C, in the process and takes about 45min mix until no bubbles generate. The 13 ships of blood containing 60g, 2. 36mmol) in THF solution was transferred to a pressure-equalizing dropping funnel. It was slowly added dropwise to the reaction system. After the completion of dropwise continue to embrace mix ratio. After the treatment, at 0 ° C under 0.8 blood, Imol / L 1 fat slowly dropped into the embrace mixed reaction system, after adding the right amount of water, acetic acid extraction. The combined organic phase with llmol / L of 0H (17mLX3) washing the organic phase coating. Tu brine, dried over anhydrous sulfate steel, 25 ° C under reduced pressure to spin dry to give 1. 65g light yellow oil.
[0033] Determination of Reference Example 1 in an amount of 98.7%.
[0034] MS (ES +) m / z: 326. 24 [M + + field.
[003 cited Example 3:
[0036] 50 single jar of blood, condenser. Intermediate inb (l.〇〇g, 3. 07mmol) was dissolved in 10ml of dichloromethane burn during and after the blood was added to a 50-port flask, make dioxide of 32g, 3.68mmol), the reaction of reflux. After completion of the reaction by TLC, cooled to 20 ~ 25 ° C after suction filtration, the filter cake rinsed with methylene burning (the X3 3 blood), at 30 ° CW and the filtrate was concentrated to dryness. To the residue was added 5 blood acetic acid, at 20 ~ 25 ° C after mixing 0. embrace of suction, the resulting cake was vacuum dried at 30 ° C 10 ~ 12h. Give 0. 87g of white solid.
[0037] Electric NMR: (400MHz, CD3CN):. 9 74 oriented 1H), 5 40 of r, 1H), 4 77-4.66 (m, 1H), 4 09-3 98 (m, 2H…. ), 3. 49-3. 37 (m, IH), 2. 75-2. 64 (m, 2H), 2. 55-2. 48 (m, IH), 1. 95-1. 87 (m , 2H), 1. 89-1 .77 (m, 2H), 1. 61-1. 49 (m, IH), 1. 32-1. 13 (m, 9H), 1. 08-0. 82 (m, 2H);
[0038] MS (ES +) m / z: 324. 33 [M + + field.
PATENT
CN 106478608 crystal
https://patents.google.com/patent/CN106478608A/en
The present invention provides a crystalline polymorph A one kind of the compound of formula I:

In another embodiment, the present invention provides a method of preparing a crystalline polymorph of compound A I,

Which comprising, a) the compound II is dissolved in acetonitrile and stirred to form a mixture; b) heating the mixture to 50 ° C ~ 70 ° C; c) adding sulfuric acid to the heated mixture; d) evaluating the temperature was lowered to 0 ° C ~ 20 ° C, seeded and stirred to precipitate crystals.
Preparation [0042] A crystalline polymorph of the compound of Example 1 I

Compound II (1. 0g) was dissolved in 5. 0ml of acetonitrile, stirred and heated to 50 ° C ~ 70 ° C was added and this temperature was added 1.2ml 2N H2S04 / acetonitrile solution and then lowering the temperature of the system to 15 ° C ~ 20 ° C, the system was added to the appropriate amount of seed crystals and stirred for 2h, the precipitated solid was filtered and the cake washed twice with 2. 5ml of acetonitrile to give a white solid, the white solid was placed under 40 ° C desolventizing 2 hours and then dried at 80 ° C for vacuo to give a white solid 0. 83 g, 69. 3% yield, HPLC:. 99 94%. A powder X-ray diffraction spectrum shown in Figure 1, a DSC endothermic curve shown in Figure 2, which HPLC profile shown in Fig.
PATENT
https://patents.google.com/patent/CN106478608A/en
PATENT
WO 2009093972 synthesis
https://encrypted.google.com/patents/WO2009093972A1?cl=ko&hl=en&output=html_text
Clip
Vorapaxar sulfate (Zontivity)
Merck Sharp & Dohme successfully obtained approval in the EU in 2014 for vorapaxar sulfate, marketed as Zontivity. The drug is a first-in-class thrombin receptor (also referred to as a protease-activated or PAR-1) antagonist which, when used in conjunction with antiplatelet therapy, has been shown to reduce the chance of
myocardial infarction and stroke, particularly in patients with a history of cardiac events.277
Antagonism of PAR-1 allows for thrombin-mediated fibrin deposition while blocking thrombinmediated platelet activation.277 Although a variety of papers and patents describe the synthesis of vorapaxar sulfate (XXXVII),278–282 a combination of two patents describe the largest-scale synthesis reported in the literature, and this is depicted in Scheme 52.


Retrosynthetically, the drug can be divided into olefination partners 306 and 305.283,284 Lactone 305
is further derived from synthons 300 and 299, which are readily prepared from commercially available starting materials. Dienyl acid 300 was constructed in two steps starting from commercial vinyl bromide 307, which first undergoes a Heck reaction with methacrylate (308) followed by saponification of the ester to afford the desired acid 300 in 71% over two steps (Scheme 53).
The synthesis of alcohol 299 begins with tetrahydropyranyl (THP) protection of enantioenriched alcohol 295 to afford butyne 297 (Scheme 52). Lithiation of this system followed by trapping with (benzyloxy)chloroformate and Dowex work-up to remove the protective functionality provided acetyl ester 298. Hydrogenation of the alkyne with Lindlar’s catalyst delivered cis-allylic alcohol 299 in 93% yield. Acid 300 was then esterified with alcohol 299 by way of a 1,3-dicyclohexylcarbodiimide (DCC) coupling and, upon heating in refluxing xylenes, an intramolecular Diels–
Alder reaction occurred. Subsequent subjection to DBU secured the tricyclic system 301 in 38% over three steps as a single enantiomer.
Diastereoselective hydrogenation reduced the olefin with concomitant benzyl removal to give key fragment 302. Next, acidic revelation of the ketone followed by reductive amination with ammonium formate delivered primary amines 303a/303b as a mixture of diastereomers. These amines were then converted to the corresponding carbamates, and resolution by means of recrystallization yielded 50% of 304 as the desired diastereomer. Acid 304
was treated with oxalyl chloride and the resulting acid chloride was reduced to aldehyde 305 in 66% overall yield. Finally, deprotonation of phosphonate ester 306 (whose synthesis is described in Scheme 54) followed by careful addition of 305 and acidic quench delivered vorapaxar sulfate (XXXVII) in excellent yield over the
two-step protocol.
The preparation of vorapaxar phosponate ester 306 (Scheme 54)commenced from commercial sources of 5-(3-fluorophenyl)-2-methylpyridine (310). Removal of the methyl proton with LDA followed by quench with diethyl chlorophosphonate resulted in phosponate ester 306.

277. Frampton, J. E. Drugs 2015, 75, 797.
278. Chackalamannil, S.; Wang, Y.; Greenlee, W. J.; Hu, Z.; Xia, Y.; Ahn, H.; Boykow,G.; Hsieh, Y.; Palamanda, J.; Agans-Fantuzzi, J.; Kurowski, S.; Graziano, M.;Chintala, M. J. Med. Chem. 2008, 51, 3061.
279. Sudhakar, A.; Kwok, D.; Wu, G. G.; Green, M. D. WO Patent 2006076452A2,2006.
280. Wu, G. G.; Sudhakar, A.; Wang, T.; Ji, X.; Chen, F. X.; Poirier, M.; Huang, M.;Sabesan, V.; Kwok, D.; Cui, J.; Yang, X.; Thiruvengadam, T.; Liao, J.; Zavialov, I.;Nguyen, H. N.; Lim, N. K. WO Patent 2006076415A2, 2006.
281. Yong, K. H.; Zavialov, I. A.; Yin, J.; Fu, X.; Thiruvengadam, T. K. US Patent20080004449A1, 2008.
282. Chackalamannil, S.; Clasby, M.; Greenlee, W. J.; Wang, Y.; Xia, Y.; Veltri, E.;Chelliah, M. WO Patent 03089428A1, 2003.
283. Thiruven-Gadam, T. K.; Wang, T.; Liao, J.; Chiu, J. S.; Tsai, D. J. S.; Lee, H.; Wu,W.; Xiaoyong, F. WO Patent 2006076564A1, 2006.
284. Chackalamannil, S.; Asberon, T.;Xia, Y.; Doller, D.; Clasby, M. C.; Czarniecki,M. F. US Patent 6,063,847, 2000.
PRODUCT PATENT
SYNTHESIS
Inventor Samuel ChackalamannilMartin C. ClasbyWilliam J. GreenleeYuguang WangYan XiaEnrico P. VeltriMariappan ChelliahWenxue Wu
Original Assignee Schering Corporation
Priority date 2002-04-16
THE EXACT BELOW COMPD IS 14
Example 2
Step 1 :

Phosphonate 7, described in US 6,063,847, (3.27 g, 8.1 mmol) was dissolved in THF (12 ml) and C(O)Oled to 0 °C, followed by addition of 2.5 M n- BuLi (3.2 ml, 8.1 mmol). The reaction mixture was stirred at 0 °C for 10 min and warmed up to rt. A solution of aldehyde 6, described in US 6,063,847, in THF (12 ml) was added to the reaction mixture. The reaction mixture was stirred for 30 min. Standard aqueous work-up, followed by column chromatography (30-50% EtOAc in hexane) afforded product 8. 1HNMR (CDCI3): δ 0.92-1.38 (m, 31 H), 1.41 (d, J= 6 Hz, 3H), 1.40-1.55 (m, 2H), 1.70-1.80 (m, 2H), 1.81-1.90 (m, 2H), 2.36 (m, 2H), 2.69 (m, 1 H), 3.89 (m, 4H), 4.75 (m, 1 H), 6.28-6.41 (m, 2H), 7.05-7.15 (m, 2H), 8.19 (br s, 1 H). Step 2:

Compound 8 (2.64 g, 4.8 mmol) was dissolved in THF (48 ml). The reaction mixture was C(O)Oled to 0 °C followed by addition of 1 M TBAF (4.8 ml). The reaction mixture was stirred for 5 min followed by standard aqueous work-up. Column chromatography (50% EtOAc/hexane) afforded product 9 (1.9 g, 100%). 1HNMR (CDCI3): δ 1.15-1.55 (m, 6H), 1.41 (d, J= 6 Hz, 3H), 1.70-1.82 (m, 3H), 1.85-1.90 (m, 1 H), 2.36 (m, 2H), 2.69 (m, 1 H), 3.91 (m, 4H), 4.75 (m, 1 H), 6.18- 6.45 (m, 2H), 7.19 (br s, 2H), 8.19 (br s, 1 H). Step 3:

To a solution of compound 9 (250 mg, 0.65 mmol) in pyridine (5 ml) C(O)Oled to 0 °C was added Tf2O (295 μL, 2.1 mmol). The reaction mixture was stirred overnight at rt. Standard aqueous work-up followed by column chromatography afforded product 10 (270 mg, 80%). 1HNMR (CDCI3): δ 1.15-1.55 (m, 6H), 1.41 (d, J= 6 Hz, 3H), 1.70-1.82 (m, 3H), 1.85-1.90 (m, 1 H), 2.36 (m, 2H), 2.69 (m, 1 H), 3.91 (m, 4H), 4.75 (m, 1 H), 6.42-6.68 (m, 2H), 7.25 (m, 1 H), 7.55 (m, 1 H), 8.49 (d, J= 2.8 Hz, 1 H).

Compound 10 (560 mg, 1.1 mmol), 3-fluorophenyl boronic acid (180 mg, 1.3 mmol) and K2CO3 (500 mg, 3.6 mmol) were mixed with toluene (4.4 ml), H2O (1.5 ml) and EtOH (0.7 ml) in a sealed tube. Under an atmosphere of N2, Pd(Ph3P)4 (110 mg, 0.13 mmol) was added. The reaction mixture was heated at 100 °C for 2 h under N2. The reaction mixture was C(O)Oled down to rt, poured to EtOAc (30 ml) and washed with water (2X20 ml). The EtOAc solution was dried with NaHCO3 and concentrated at reduced pressure to give a residue. Preparative TLC separation of the residue (50% EtOAc in hexane) afforded product 11 (445 mg, 89%). 1HNMR (CDCI3): δ 1.15-1.59 (m, 6H), 1.43 (d, J= 6 Hz, 3H), 1.70-1.79 (m, 2H), 1.82 (m, 1H), 1.91 (m, 2H), 2.41 (m, 2H), 2.69 (m, 1 H), 3.91 (m, 4H), 4.75 (m, 1 H), 6.52-6.68 (m, 2H), 7.15 (m, 1 H), 7.22 (m, 2H), 7.35 (m, 1 H), 7.44 (m, 1 H), 7.81 (m, 1 H), 8.77 (d, J= 1.2 Hz, 1 H). Step 5:

Compound 11 (445 mg, 0.96 mmol) was dissolved in a mixture of acetone (10 ml) and 1 N HCI (10 ml). The reaction mixture was heated at 50 °C for 1 h.
Standard aqueous work-up followed by preparative TLC separation (50% EtOAc in hexane) afforded product 12 (356 mg, 89%). 1HNMR (CDCI3): δ 1.21-1.45 (m, 2H), 1.47 (d, J= 5.6 Hz, 3H), 1.58-1.65 (m, 2H), 2.15 (m, 1 H), 2.18-2.28 (m, 2H), 2.35- 2.51 (m, 5H), 2.71 (m, 1 H), 4.79 (m, 1 H), 6.52-6.68 (m, 2H), 7.15 (m, 1 H), 7.22 (m, 2H), 7.35 (m, 1 H), 7.44 (m, 1 H), 7.81 (m, 1 H), 8.77 (d, J= 1.2 Hz, 1 H). Step 6:

Compound 12 (500 mg, 4.2 mmol) was dissolved in EtOH (40 ml) and CH2CI2 (15 ml) NH3 (g) was bubbled into the solution for 5 min. The reaction mixture was C(O)Oled to 0 °C followed by addition of Ti(O/‘Pr)4 (1.89 ml, 6.3 mmol). After stirring at 0 °C for 1 h, 1 M TiCI (6.3 ml, 6.3 mmol) was added. The reaction mixture was stirred at rt for 45 min and concentrated to dryness under reduced pressure. The residue was dissolved in CH3OH (10 ml) and NaBH3CN (510 mg, 8 mmol) was added. The reaction mixture was stirred overnight at rt. The reaction mixture was poured to 1 N NaOH (100 ml) and extracted with EtOAc (3x 100 ml). The organic layer was combined and dried with NaHC03. Removal of solvent and separation by PTLC (5% 2 M NH3 in CH3OH/ CH2CI2) afforded β-13 (spot 1 , 30 mg, 6%) and α-13 (spot 2, 98 mg, 20%). β-13: 1HNMR (CDCI3): δ 1.50-1.38 (m, 5H), 1.42 (d, J= 6 Hz, 3H), 1.51-1.75 (m, 5H), 1.84 (m, 2H), 2.38 (m, 1 H), 2.45 (m, 1 H), 3.38 (br s, 1 H), 4.78 (m, 1 H), 6.59 (m, 2H), 7.15 (m, 1 H), 7.26 (m, 2H), 7.36 (m, 1 H), 7.42 (m, 1 H), 7.82 (m, 1 H), 8.77 (d, J= 2 Hz, 1 H). α-13:1HNMR (CDCI3): δ 0.95 (m, 2H), 1.02-1.35 (m, 6H), 1.41 (d, J= 6 Hz, 3H), 1.82-1.95 (m, 4H), 2.37 (m; 2H), 2.69 (m, 2H), 4.71 (m, 1 H), 6.71 (m, 2H), 7.11 (m, 1 H), 7.25 (m, 2H), 7.38 (m, 1 H), 7.42 (m, 1 H), 7.80 (m, 1 H), 8.76 (d, J= 1.6 Hz, 1 H). Step 7:
Compound α-13 (300 mg, 0.71 mmol) was dissolved in CH2CI2 (10 ml) followed by addition of Et3N (0.9 ml). The reaction mixture was C(O)Oled to 0 °C and ethyl chloroformate (0.5 ml) was added. The reaction mixture was stirred at rt for 1 h. The reaction mixture was directly separated by preparative TLC (EtOAc/ hexane, 1 :1) to give the title compound (14) VORAPAXAR (300 mg, 86%). MS m/z 493 (M+1).
HRMS Calcd for C29H34N2O4F (M+1 ): 493.2503, found 493.2509.
PATENT
SYNTHESIS 1
http://www.google.com/patents/WO2006076564A1
VORAPAXAR= COMPD A
Example 6 – Preparation of Compound A

To a three-neck flask equipped with an agitator, thermometer and nitrogen inertion was added 7A (13.0 g), THF (30 mL). The mixture was cooled to below -200C after which lithium diisopropylamide (2M, 20 mL) was slowly added. The reaction mixture was agitated for an additional hour (Solution A). To another flask was added 6 (10.0 g) and THF (75 mL) . The mixture was stirred for about 30 minutes and then slowly transferred into the solution A while maintaining the temperature below 200C. The mixture was stirred at below -200C for an additional hour before quenching the reaction by adding 20 mL of water. The reaction mixture was warmed to 00C and the pH was adjusted to about 7 by addition of 25% HaSO4 (11 mL). The mixture was further warmed to 200C and then diluted with 100 mL of ethyl acetate and 70 mL of water. The two phases that had formed were separated and the aqueous layer was extracted with 50 mL of ethyl acetate. The solvents THF and ethyl acetate were then replaced with ethanol, and the Compound A was precipitated out as a crystalline solid from ethanol with seeding at 35 to 4O0C. After cooling to O0C, the suspension was stirred for an additional hour and then the product was filtered and washed with cold ethanol. The product was dried at 50 – 600C under vacuum to provide an off-white solid. VORAPAXAR
Yield: 12.7 g, (90%). m.p. 104.90C (DSC onset point).
1H NMR (CDCl3) δ 8.88 (d, J = 2.4 Hz, IH), 8.10 (dd, J = 8.2, 2.4 Hz, IH), 7.64 (IH), 7.61 (d, J = 8.8 Hz, IH), 7.55 (m, J = 8.2, 6.2 Hz, IH), 7.51 (d, J = 8.0 Hz, IH), 7.25 (dt, J = 9.0, 2.3 Hz, IH), 7.08 (d, J = 8.0 Hz, IH), 6.68 (dd, J = 15.4, 9.4 Hz, IH), 6.58 (d, J = 9.6 Hz, IH), 4.85 (dd, J = 14.2, 7.2 Hz, IH), 3.95 (dd, J = 14.2, 7.1 Hz, 2H), 3.29 (m, IH), 2.66 (m, J = 12.0, 6.4 Hz, IH), 2.33 (m, 2H), 1.76 (m, 4H), 1.30 (d, J = 5.6 Hz, 3H), 1.19 (m, 4H), 1.14 (t, J = 7.2 Hz, 3H), 0.98 (m, IH), 0.84 (m, IH). MS (EI) m/z: calcd. 492, found 492.
BISULPHATE SALT
Example 7 – Preparation of an Acid Salt (bisulfate) of Compound A:
Compound IA (5 g) was dissolved in about 25 mL of acetonitrile.
The solution was agitated for about 10 minutes and then heated to about 50 0C. About 6 mL of 2M sulfuric acid in acetonitrile was added into the heated reaction mixture. The solid salt of Compound A precipitated out during the addition of sulfuric acid in acetonitrile. After addition of sulfuric acid solution, the reaction mixture was agitated for 1 hour before cooling to room temperature. The precipitated solid was filtered and washed with about 30 mL of acetonitrile. The wet solid was dried under vacuum at room temperature for 1 hour and at 80 0C for about 12 hours to provide about 5 g white solid (yield 85%). m.p. 217.0 0C. 1H NMR (DMSO) 9.04 (s, IH), 8.60 (d, J = 8.1 Hz, IH), 8.10 (d, J = 8.2 Hz, IH), 7.76 (d, J = 10.4, IH), 7.71 (d, J = 7.8 Hz, IH), 7.60 (dd, J = 8.4, 1.8 Hz, IH), 7.34 (dd, 8.4, 1.8 Hz, IH), 7.08 (d, J = 8.0 Hz, IH), 7.02 (m, IH), 6.69 (d, J = 15.8 Hz, IH), 4.82 (m, IH), 3.94 (dd, J = 14.0, 7.0 Hz, 2H), 3.35 (brs, IH), 2.68 (m, IH), 2.38 (m, 2H), 1.80-1.70 (m, 4H), 1.27 (d, J = 5.8 Hz, 3H), 1.21 (m, 2H), 1.13 (t, J = 7.0 Hz, 3H), 0.95 (m, IH, 0.85 (m, IH). MS (EI) m/z calcd. 590, found 492.
INTERMEDIATE 6
Example 5- Preparation of Compound 6

To a three-neck flask equipped with an agitator, thermometer and nitrogen inert were added the crude product solution of Compound 5 (containing about 31 g. of Compound 5 in 300 mL solution) and anhydrous DMF (0.05 mL). After the mixture was agitated for 5 minutes, oxalyl chloride (12.2 mL) was added slowly while maintaining the batch temperature between 15 and 25°C. The reaction mixture was agitated for about an hour after the addition and checked by NMR for completion of reaction. After the reaction was judged complete, the mixture was concentrated under vacuum to 135 mL while maintaining the temperature of the reaction mixture below 300C. The excess oxalyl chloride was removed completely by two cycles of vacuum concentration at below 500C with replenishment of toluene (315 mL) each time, resulting in a final volume of 68 mL. The reaction mixture was then cooled to 15 to 25°C, after which THF (160 mL) and 2,6-lutidine (22 mL) were added. The mixture was agitated for 16 hours at 20 to 25°C under 100 psi hydrogen in the presence of dry 5% Pd/C (9.0 g). After the reaction was judged complete, the reaction mixture was filtered through celite to remove catalyst. More THF was added to rinse the hydrogenator and catalyst, and the reaction mixture was again filtered through celite. Combined filtrates were concentrated under vacuum at below 25°C to 315 mL. MTBE (158 mL) and 10% aqueous solution of phosphoric acid (158 mL) were added for a thorough extraction at 100C to remove 2,6- lutidine. Then phosphoric acid was removed by extracting the organic layer with very dilute aqueous sodium bicarbonate solution (about 2%), which was followed by a washing with dilute brine. The organic solution was concentrated atmospherically to a volume of 90 mL for solvent replacement. IPA (315 mL) was added to the concentrated crude product solution. The remaining residual solvent was purged to <_ 0.5% of THF (by GC) by repeated concentration under vacuum to 68 mL, with replenishment of IPA (315 mL) before each concentration. The concentrated (68 mL) IPA solution was heated to 50°C, to initiate crystallization. To this mixture n-heptane (68 mL) was added very slowly while maintaining the batch temperature at 50°C. The crystallizing mixture was cooled very slowly over 2.5 hours to 25°C. Additional n- heptane (34 mL) was added very slowly into the suspension mixture at 250C. The mixture was further cooled to 200C, and aged at that temperature for about 20 hours. The solid was filtered and washed with a solvent mixture of 25% IPA in n-heptane, and then dried to provide
19.5 g of a beige colored solid of Compound 6. (Yield: 66%) m.p. 169.30C. IH NMR (CD3CN) δ 9.74 (d, J = 3.03 Hz, IH), 5.42 (br, IH), 4.69 (m, IH), 4.03 (q, J = 7.02 Hz, 2H), 3.43 (qt, J = 3.80, 7.84 Hz, IH), 2.67 (m, 2H), 2.50 (dt, J = 3.00, 8.52 Hz, IH), 1.93 (d, J = 12.0 Hz, 2H), 1.82 (dt, J = 3.28, 9.75 Hz, 2H), 1.54 (qd, J = 3.00, 10.5 Hz, IH), 1.27 (d, J = 5.97 Hz, 3H), 1.20 (m, 6H), 1.03 – 0.92 (m, 2H). MS (ESI) m/z (M++1): calcd. 324, found 324.
INTERMEDIATE 7A
Example 4 – Preparation of Compound 7A
+ 1-Pr2NLi + (EtO)2POCI – + LiCI


7A
To a 10 L three-necked round bottomed flask equipped with an agitator, thermometer and a nitrogen inlet tube, was added 20Og of
Compound 8 (1.07 mol, from Synergetica, Philadelphia, Pennsylvania). THF (1000 mL) was added to dissolve Compound 8. After the solution was cooled to -80 0C to -50 0C, 2.0 M LDA in hexane/THF(1175 mL, 2.2 eq) was added while maintaining the batch temperature below -50 0C. After about 15 minutes of agitation at -800C to -50 0C, diethyl chlorophosphate (185 mL, 1.2 eq) was added while maintaining the batch temperature below -50 0C. The mixture was agitated at a temperature from -800C to – 50 0C for about 15 minutes and diluted with n-heptane (1000 mL). This mixture was warmed up to about -35 0C and quenched with aqueous ammonium chloride (400 g in 1400 mL water) at a temperature below -10 0C. This mixture was agitated at -150C to -10 0C for about 15 minutes followed by agitation at 150C to 25 0C for about 15 minutes. The aqueous layer was split and extracted with toluene (400 mL). The combined organic layers were extracted with 2N hydrochloric acid (700 mL) twice. The product-containing hydrochloric acid layers were combined and added slowly to a mixture of toluene (1200 mL) and aqueous potassium carbonate (300 g in 800 mL water) at a temperature below 30 0C. The aqueous layer was extracted with toluene (1200 mL). The organic layers were combined and concentrated under vacuum to about 600 ml and filtered to remove inorganic salts. To the filtrate was added n-heptane (1000 ml) at about 55 0C. The mixture was cooled slowly to 40 0C, seeded, and cooled further slowly to -10 0C. The resulting slurry was aged at about -10 0C for 1 h, filtered, washed with n- heptane, and dried under vacuum to give a light brown solid (294 g, 85% yield), m.p. 52 0C (DSC onset point).1H NMR (CDCl3) δ 8.73 (d, J = 1.5 Hz, IH), 7.85 (dd, Ji = 8.0 Hz, J2 = 1.5 Hz, IH), 7.49 (dd, Ji = 8.0 Hz, J2 = 1.3 Hz, IH), 7.42 (m, IH), 7.32 (d, J = 7.8 Hz, IH), 7.24 (m, IH), 7.08 (dt, Ji = 8.3 Hz, J2 = 2.3 Hz, IH), 4.09 (m, 4H), 3.48 (d, J = 22.0 Hz, 2H), 1.27 (t, J = 7.0 Hz, 6H). MS (ESI) for M+H calcd. 324, found 324.
Example 3 – Preparation of Compound 5:

4 5
To a three-necked round bottomed flask equipped with an agitator, thermometer and a nitrogen inlet tube was added a solution of Compound 4 in aqueous ethanol (100 g active in 2870 ml). The solution was concentrated to about 700 ml under reduced pressure at 350C to 40°C to remove ethyl alcohol. The resultant homogeneous mixture was cooled to 200C to 300C and its pH was adjusted to range from 12 to 13 with 250 ml of 25% sodium hydroxide solution while maintaining the temperature at 20-300C. Then 82 ml of ethyl chloroformate was slowly added to the batch over a period of 1 hour while maintaining the batch temperature from 200C to 300C and aged for an additional 30 minutes. After the reaction was judged complete, the batch was acidified to pH 7 to 8 with 10 ml of concentrated hydrochloric acid (37%) and 750 ml of ethyl acetate. The pH of the reaction mixture was further adjusted to pH 2 to 3 with 35% aqueous hydrochloric acid solution. The organic layer was separated and the aqueous layer was extracted again with 750 ml of ethyl acetate. The combined organic layers were washed twice with water (200 ml) . Compound 5 was isolated from the organic layer by crystallization from ethyl acetate and heptane mixture (1: 1 mixture, 1500 ml) at about 700C to 80 0C. The solid was filtered at 500C to 60 °C, washed with heptane and then dried to provide an off-white solid (yield 50%). m.p. 197.7°C. 1HNMR (CD3CN) δ 5.31 (brs, IH), 4.67 (dt, J = 16.1, 5.9 Hz, IH), 4.03 (q, J = 7.1 Hz, 2H), 3.41 (m, IH), 2.55 – 2.70 (m, 2H), 1.87 – 1.92 (m, IH), 1.32 – 1.42 (m, IH), 1.30 (d, J = 5.92 Hz, 3H), 1.30 – 1.25 (m, 6H), 0.98 (qt, J = 15.7, 3.18 Hz, 2H). MS (ESI) M+l m/z calculated 340, found 340.
Example 2 – Preparation of Compound 4;

3 4
7.4 kg of ammonium formate was dissolved in 9L of water at 15- 250C, and then cooled to 0-100C. 8.9 kg of Compound 3 was charged at 0-150C followed by an addition of 89L of 2B ethyl alcohol. The batch was cooled to 0-50C 0.9 kg of 10% Palladium on carbon (50% wet) and 9 L of water were charged. The batch was then warmed to 18-280C and agitated for 5 hours, while maintaining the temperature between 18-28 0C. After the reaction was judged complete, 7 IL of water was charged. The batch was filtered and the wet catalyst cake was then washed with 8OL of water. The pH of the filtrate was adjusted to 1-2 with 4N aqueous hydrochloric acid solution. The solution was used in the next process step without further isolation. The yield is typically quantiative. m.p. 216.40C. IH NMR (D2O+1 drop HCl) δ 3.15 (m, IH), 2.76 (m, IH), 2.62 (m, IH), 2.48 (dd,J-5.75Hz, IH), 1.94 (m, 2H), 1.78 (m, 2H), 1.38 (m, 2H), 1.20 (m, 6H), 1.18 (m, IH), 0.98 (q,J=2.99Hz, IH).
Example 1 – Preparation of Compound 3

2B 3
To a reactor equipped with an agitator, thermometer and nitrogen, were added about 10.5 kg of 2B, 68 L of acetone and 68 L of IN aqueous hydrochloric acid solution. The mixture was heated to a temperature between 50 and 600C and agitated for about 1 hour before cooling to room temperature. After the reaction was judged complete, the solution was concentrated under reduced pressure to about 42 L and then cooled to a temperature between 0 and 50C. The cooled mixture was agitated for an additional hour. The product 3 was filtered, washed with cooled water and dried to provide an off-white solid (6.9 kg, yield 76%). m.p. 2510C. Η NMR (DMSO) δ 12.8 (s, IH), 4.72 (m, J = 5.90 Hz, IH), 2.58 (m, 2H), 2.40 (m, J = 6.03 Hz, 2H), 2.21 (dd, J = 19.0, 12.8 Hz, 3H), 2.05 (m, IH), 1.87 (q, J = 8.92 Hz, IH), 1.75 (m, IH), 1.55 (m, IH), 1.35 (q, J = 12.6 Hz, IH), 1.27 (d, J = 5.88 Hz, 3H). MS (ESI) M+l m/z calcd. 267, found 267.
NOTE
Compound 7A may be prepared from Compound 8 by treating Compound 8 with diethylchlorophosphate:

Compound 8 may be obtained by the process described by Kyoku, Kagehira et al in “Preparation of (haloaryl)pyridines,” (API Corporation, Japan). Jpn. Kokai Tokkyo Koho (2004). 13pp. CODEN: JKXXAF JP
2004182713 A2 20040702. Compound 8 is subsequently reacted with a phosphate ester, such as a dialkyl halophosphate, to yield Compound 7A. Diethylchlorophosphate is preferred. The reaction is preferably conducted in the presence of a base, such as a dialkylithium amide, for example diisopropyl lithium amide.
Paper
J Med Chem 2008, 51(11): 3061
http://pubs.acs.org/doi/abs/10.1021/jm800180e
The discovery of an exceptionally potent series of thrombin receptor (PAR-1) antagonists based on the natural product himbacine is described. Optimization of this series has led to the discovery of 4 (SCH 530348), a potent, oral antiplatelet agent that is currently undergoing Phase-III clinical trials for acute coronary syndrome (unstable angina/non-ST segment elevation myocardial infarction) and secondary prevention of cardiovascular events in high-risk patients.
Ethyl [(3aR,4aR,8aR,9aS)-9(S)-[(E)-2-[5-(3-fluorophenyl)-2-
pyridinyl]ethenyl]dodecahydro-1(R)-methyl-3-oxonaphtho[2,3-c]furan-6(R)-yl]carbamate (4).
4 (300 mg, 86%). MS m/z 493 (M+1).
HRMS Calcd for C29H34N2O4F
(M+1): 493.2503, found 493.2509; mp125 °C;
[]D20 6.6 (c 0.5, MeOH).
1HNMR (CDCl3):
http://pubs.acs.org/doi/suppl/10.1021/jm800180e/suppl_file/jm800180e-file002.pdf
0.88-1.18 (m, 5 H), 1.22-1.30 (m, 3 H), 1.43 (d, J = 5.85 Hz, 3 H), 1.88-2.10 (m, 4 H), 2.33-2.42 (m, 2 H),
2.75-2.67 (m, 1 H), 3.52-3.60 (m, 1 H), 4.06-4.14 (m, 2 H), 4.54-4.80 (m, 1 H), 4.71-4.77 (m, 1 H),
6.55-6.63 (m, 2 H), 7.07-7.12 (m, 1 H), 7.26-7.29 (m, 2 H), 7.34 (d, J = 8.05 Hz, 1 H), 7.41-7.46 (m, 1 H), 7.80-7.82 (m, 1 H), 8.76-8.71 (m, 1 H).
PATENT
IN 201621010411
An improved process for preparation of Vorapaxar intermediates and a novel polymorphic form of Vorapaxar
ALEMBIC PHARMACEUTICALS LIMITED
Vorapaxar Sulfate is indicated for the reduction of thrombotic cardiovascular events in patients with a history of myocardial infarction (MI) or with peripheral arterial disease (PAD). ZONTIVITY has been shown to reduce the rate of a combined endpoint of cardiovascular death, MI, stroke, and urgent coronary revascularization (UCR).
According to present invention Vorapaxar sulfate is synthesized from compound of formula 1.

wherein R1 and R2 are each independently selected from the group consisting of H, alkyl, alkenyl, alkynyl, alkoxy, cycloalkyl, aryl, alkylaryl, arylalkyl, and heteroaryl groups. Process for the preparation of compound of formula 1 is disclosed in U.S Pat. No. 7,605,275. It disclosed preparation of compound of formula 1 via cyclization of compound 2 in presence of solvent selected from xylene, N-methylpyrrolidinone, Dimethylsulfoxide, diphenyl ether, dimethylacetamide. This cyclization step takes approximately 6-8 hrs.
There is need to develop a process which takes less time for cyclization step to prepare compound of formula 1. Therefore, our scientist works tenaciously to develop process which takes approximately 1-2 hrs for cyclization of compound 1.

5 According to present invention Vorapaxar sulfate is synthesized from intermediate compound of formula-II.

Formula-II Compound of formula-II is critical intermediate in the preparation of Vorapaxar Sulfate.
10 Patent WO2006076415 discloses the process of preparation of above Formula-II in example 7, in which purification/crystallisation step involves treating the reaction mixture having compound of Formula-II with an ethanol/water mixture followed by azeotropic distillation of the mixture. This process yielded formula-II with low yields and with low purities. WO2009055416 (page 9, second paragraph) discloses that use of various solvent systems for
15 formula-II purification such as Methyl-tert-Butyl Ether (MTBE) and various solvent/antisolvent systems, for example, ethylacetate/heptane and toluene/heptane and by using these solvent systems, compound of formula-II are obtained as oil. These oils did not yield a reduced impurity profile in synthesis of the compound of Formula II, nor provide an improvement in the quality of the product compound of Formula II.
20 The inventors surprisingly found that using the process according to the invention provides formula-II with improved yield and high purity. Further, present invention provides a process for the preparation of novel crystalline form of Vorapaxar base. The present invention also relates to novel impurity and process for its preparation.
U.S.Pat. No. 7,304,078 discloses Vorapaxar base. U.S.Pat. No. 7,235,567 discloses Polymorph I and II of vorapaxar sulphate
Example 1- Preparation of compound 1a:

Process A: 5.0 g of compound 2a was suspended in 10.0 ml silicone oil at room temperature. The reaction mixture was then heated to 125°C and stirred for 30 min. Then reaction mass was further heated up to 150°C and stirred for 30 min. After completion of reaction, the reaction mass was cooled to 50-60°C and 25 ml of cyclohexane was added to the reaction mass. The reaction mass was cooled slowly up to room temperature and stirred for 30 min.
15 The precipitated product was filtered off and washed with 5.0 ml Cyclohexane. Wet solid was suspended in mixture of 45.0 ml isopropyl alcohol and 20.0 ml denatured ethanol at 40-45°C and further epimerized with 0.17 ml DBU. The crystallized solid was filtered off with suction, washed with mixture of 1.5 ml Isopropyl alcohol and 0.67 ml denatured ethanol and dried.
20 Process B: 5.0 g of compound 2a was suspended in 10.0 ml paraffin oil at room temperature. The reaction mixture was then heated to 125°C and stirred for 30 min. Then reaction mass was further heated up to 150°C and stirred for 30 min. After completion of reaction, the reaction mass was cooled to 50-60°C and 25 ml of cyclohexane was added to the reaction mass. The reaction mass was cooled slowly up to room temperature and stirred for 30 min.
25 The precipitated product was filtered off and washed with 5.0 ml Cyclohexane. Wet solid was suspended in mixture of 45.0 ml isopropyl alcohol and 20.0 ml denatured ethanol at 40-45°C and further epimerized with 0.17 ml DBU. The crystallized solid was filtered off with suction, washed with mixture of 1.5 ml Isopropyl alcohol and 0.67 ml denatured ethanol and dried. Yield: 4.3 g
Process C: 5.0 g of compound 2a was charged in reaction vessel at room temperature. The solid was then heated to 125°C and stirred for 30 min. Then reaction mass was further heated up to 150°C and stirred for 30 min. After completion of reaction, the reaction mass was cooled to 50-60°C and was added mixture of 45.0 ml isopropyl alcohol and 20.0 ml
5 denatured ethanol at 50-60°C. This was cooled to 40-45°C and further epimerized with 0.17 ml DBU. The crystallized solid was filtered off with suction, washed with mixture of 1.5 ml Isopropyl alcohol and 0.67 ml denatured ethanol and dried. Yield: 4.5 g Example 2: Preparation of Intermediate (Formula-II) of vorapaxar
10 Example 2(a): 50.0g of 1,3,3a,4,4a,5,6,7,8,9a-Decahydro-3-methyl-7-nitro-1-oxo-N,Ndiphenylnaphtho[2,3-c]furan-4-carboxamide compound was suspended in 300.0 ml THF, 15 g 10% Pd/C (50% wet) and 200 ml Process water at room temperature. The reaction mixture was heated to 45°C and drop wise formic acid (35 ml) was added and then stirred for 15 hrs. After completion of reaction, the reaction mass was cooled to 25-30°C and 100 ml THF was
15
added and pH was made acidic with 2M sulfuric acid solution. The reaction mass was filtered and washed with 150 ml THF, 150 ml water. Organic and aqueous layer were separated and aqueous layer was extracted with THF. Organic layers were combined and washed with water. The organic layer was cooled up to 5-10°C, 20 ml of TEA and 13 ml of Ethyl chloro formate were added. The reaction mass was stirred for 30 min. After completion of reaction,
20
reaction mass was washed with 2M sulfuric acid solution and distilled out reaction mass completely under vacuum. Acetonitrile (50 ml) was added to residue and heated up to 40- 45°C. Cooled the reaction mass up to 25-30°C and filtered the solid. Purity: 94-96% Example 2(b): Crystallization with Acetonitrile Acetonitrile (50 ml) was added to above obtained solid and heated to 40-45°C. Cooled the
25 reaction mass slowly up to 25-30°C and then up to 5-10°C. The reaction mass was stirred and the solid was filtered. XRD: Fig-1 Purity: 98-99% Example 2(c): Crystallization with Ethyl acetate To the solid obtained in example-1(a) Ethyl acetate (30 ml) was added. The reaction mass was heated up to 70-75°C and stirred for 10-15 min. The reaction mass was cooled slowly up 30 to 25-30°C and then up to 5-10°C. The reaction mass was stirred for 30 min. The solid was filtered and washed with Ethyl acetate. XRD: Fig-2 Purity: 98-99%
Example 3: Preparation of Amorphous Form of Vorapaxar base Vorapaxar base (10.0 g) was dissolved in 500 ml of 40% Ethyl acetate in Cyclohexane. The solvent was then completely removed under vacuum at 45-50o C to give a solid. Yield: 9.8 g
Example 3 (a): Preparation of crystalline vorapaxar base 5 (2-{[Ethyl (ethylperoxy)phosphory]methyl}-5-(3-fluorophenyl)pyridine) (10 g) was dissolved in THF (30ml) at 25±5°C under Nitrogen. Cool the reaction mass up to -30 to – 50°C. Add drop wise LDA (2.0 M solution in THF). After 1 hr add drop wise (N- [(1R,3aR,4aR,6R,8aR,9S,9aS)-9-formyl dodecahydro-1-methyl-3-oxonaphtho[2,3-c]furan-6- yl]-ethyl ester Carbamic acid) solution (10 g dissolved in 70 ml THF). After completion of 10 reaction mass quench the reaction mass to sulphuric acid solution. Separate the layers and distilled out organic layer under vacuum get foamy residue. (purity 82%) Add MIBK (10 ml) in above residue and stir it at 40-50°C till clear solution. Add drop wise n-Heptane (10 ml) and stir the reaction mass for 30 min. Gradually cool the reaction mass up to 25-30°C. Stir the reaction mass for 24 hrs. Filter the solid and washed it with n-Heptane (5.0 ml). Dry the 15 solid. Yield: 7.0 g. XRD: Fig-3 purity 96%
Example 3(b): Preparation of crystalline vorapaxar base Vorapaxar advance intermediate (2-{[Ethyl (ethylperoxy)phosphory]methyl}-5-(3- fluorophenyl)pyridine) (10 g) was dissolved in THF (30ml) at 25±5°C under Nitrogen. Cool the reaction mass up to -30 to -50°C. Add drop wise LDA (2.0 M solution in THF). After a 1
20 hr add drop wise VORA-Aldehyde (N-[(1R,3aR,4aR,6R,8aR,9S,9aS)-9-formyl dodecahydro1-methyl-3-oxonaphtho[2,3-c]furan-6-yl]-ethyl ester Carbamic acid) solution (10 g dissolved in 70 ml THF). After completion of reaction mass quench the reaction mass to sulphuric acid solution. Separate the layers and distilled out organic layer under vacuum get foamy residue (purity 82%). Add MTBE (10 ml) in above residue and stir it at 40-50°C till clear solution.
25 Add drop wise n-Heptane (30 ml) and stir the reaction mass for 30 min. Gradually cool the reaction mass up to 25-30°C. Stir the reaction mass for 24 hrs. Filter the solid and washed it with n-Heptane (5.0 ml). Dry the solid. Yield: 8.5.0 g. XRD: Fig-4 purity 97%
References
- Samuel Chackalamannil; Wang, Yuguang; Greenlee, William J.; Hu, Zhiyong; Xia, Yan; Ahn, Ho-Sam; Boykow, George; Hsieh, Yunsheng et al. (2008). “Discovery of a Novel, Orally Active Himbacine-Based Thrombin Receptor Antagonist (SCH 530348) with Potent Antiplatelet Activity”. Journal of Medicinal Chemistry 51 (11): 3061–4.doi:10.1021/jm800180e. PMID 18447380.
- Merck Blood Thinner Studies Halted in Select Patients, Bloomberg News, January 13, 2011
- Tricoci et al. (2012). “Thrombin-Receptor Antagonist Vorapaxar in Acute Coronary Syndromes”. New England Journal of Medicine 366 (1): 20–33.doi:10.1056/NEJMoa1109719. PMID 22077816.
- Morrow, DA; Braunwald, E; Bonaca, MP; Ameriso, SF; Dalby, AJ; Fish, MP; Fox, KA; Lipka, LJ; Liu, X; Nicolau, JC; Ophuis, AJ; Paolasso, E; Scirica, BM; Spinar, J; Theroux, P; Wiviott, SD; Strony, J; Murphy, SA; TRA 2P–TIMI 50 Steering Committee and, Investigators (Apr 12, 2012). “Vorapaxar in the secondary prevention of atherothrombotic events.”. The New England Journal of Medicine 366 (15): 1404–13. doi:10.1056/NEJMoa1200933.PMID 22443427.
- “Merck Statement on FDA Advisory Committee for Vorapaxar, Merck’s Investigational Antiplatelet Medicine”. Merck. Retrieved 16 January 2014.
- http://www.forbes.com/sites/larryhusten/2014/01/15/fda-advisory-panel-votes-in-favor-of-approval-for-mercks-vorapaxar/
- SCH-530348 (Vorapaxar) is an investigational candidate for the prevention of arterial thrombosis in patients with acute coronary syndrome and peripheral arterial disease. “Convergent Synthesis of Both Enantiomers of 4-Hydroxypent-2-ynoic Acid Diphenylamide for a Thrombin Receptor Antagonist Sch530348 and Himbacine Analogues.” Alex Zaks et al.: Adv. Synth. Catal. 2009, 351: 2351-2357 Full text;
- Discovery of a novel, orally active himbacine-based thrombin receptor antagonist (SCH 530348) with potent antiplatelet activity
J Med Chem 2008, 51(11): 3061
- Stu Borman (2005). “Hopes Ride on Drug Candidates: Researchers reveal potential new medicines for thrombosis, anxiety, diabetes, and cancer”. Chemical & Engineering News 83 (16): 40–44.
PATENTS
- WO 2003089428
- WO 2006076452
- US 6063847
- WO 2006076565
- WO 2008005344
- WO2010/141525
- WO2008/5353
- US2008/26050
- WO2006/76564 mp, nmr
|
3-21-2012
|
EXO-SELECTIVE SYNTHESIS OF HIMBACINE ANALOGS
|
|
|
10-14-2011
|
EXO- AND DIASTEREO- SELECTIVE SYNTHESIS OF HIMBACINE ANALOGS
|
|
|
8-3-2011
|
Exo- and diastereo-selective syntheses of himbacine analogs
|
|
|
3-18-2011
|
COMBINATION THERAPIES COMPRISING PAR1 ANTAGONISTS WITH NAR AGONISTS
|
|
|
8-11-2010
|
Exo-selective synthesis of himbacine analogs
|
|
|
6-4-2010
|
SYNTHESIS Of DIETHYLPHOSPHONATE
|
|
|
5-12-2010
|
THROMBIN RECEPTOR ANTAGONISTS
|
|
|
3-31-2010
|
Synthesis of diethyl{[5-(3-fluorophenyl)-pyridine-2yl]methyl}phosphonate
|
|
|
12-4-2009
|
Local Delivery of PAR-1 Antagonists to Treat Vascular Complications
|
|
|
12-2-2009
|
SYNTHESIS OF HIMBACINE ANALOGS
|
|
10-21-2009
|
Exo- and diastereo- selective syntheses of himbacine analogs
|
|
|
6-31-2009
|
Synthesis of 3-(5-nitrocyclohex-1-enyl) acrylic acid and esters thereof
|
|
|
6-3-2009
|
Synthesis of himbacine analogs
|
|
|
1-23-2009
|
METHODS AND COMPOSITIONS FOR TREATING CARDIAC DYSFUNCTIONS
|
|
|
9-26-2008
|
REDUCTION OF ADVERSE EVENTS AFTER PERCUTANEOUS INTERVENTION BY USE OF A THROMBIN RECEPTOR ANTAGONIST
|
|
|
2-8-2008
|
IMMEDIATE-RELEASE TABLET FORMULATIONS OF A THROMBIN RECEPTOR ANTAGONIST
|
|
|
1-32-2008
|
SOLID DOSE FORMULATIONS OF A THROMBIN RECEPTOR ANTAGONIST
|
|
|
12-5-2007
|
Thrombin receptor antagonists
|
|
|
11-23-2007
|
THROMBIN RECEPTOR ANTAGONISTS
|
|
|
8-31-2007
|
THROMBIN RECEPTOR ANTAGONISTS AS PROPHYLAXIS TO COMPLICATIONS FROM CARDIOPULMONARY SURGERY
|
|
8-31-2007
|
CRYSTALLINE POLYMORPH OF A BISULFATE SALT OF A THROMBIN RECEPTOR ANTAGONIST
|
|
|
6-27-2007
|
Crystalline polymorph of a bisulfate salt of a thrombin receptor antagonist
|
|
|
8-4-2006
|
Preparation of chiral propargylic alcohol and ester intermediates of himbacine analogs
|
|
|
9-31-2004
|
Methods of use of thrombin receptor antagonists
|
| US6063847 * | Nov 23, 1998 | May 16, 2000 | Schering Corporation | Thrombin receptor antagonists |
| US6326380 * | Apr 7, 2000 | Dec 4, 2001 | Schering Corporation | Thrombin receptor antagonists |
| US20030216437 * | Apr 14, 2003 | Nov 20, 2003 | Schering Corporation | Thrombin receptor antagonists |
| US20040176418 * | Jan 9, 2004 | Sep 9, 2004 | Schering Corporation | Crystalline polymorph of a bisulfate salt of a thrombin receptor antagonist |
| WO2011128420A1 | Apr 14, 2011 | Oct 20, 2011 | Sanofi | Pyridyl-vinyl pyrazoloquinolines as par1 inhibitors |
//////////////fast track designation , VORAPAXAR, FDA 2014, EU 2016, Zontivity, NDA 204886, MERCK, VORAPAXAR SULPHATE
CCOC(=O)NC1CCC2C(C1)CC3C(C2C=CC4=NC=C(C=C4)C5=CC(=CC=C5)F)C(OC3=O)C