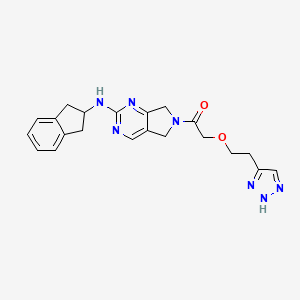


2-(2-(1H-1,2,3-triazol-5-yl)ethoxy)-1-(2-((2,3-dihydro-1H-inden-2-yl)amino)-5,7-dihydro-6Hpyrrolo[3,4-d]pyrimidin-6-yl)ethan-1-one
l-[2-(2,3-dihydro- lH-inden-2-ylamino)-5,7-dihydro-6H-pyrrolo[3,4- d]pyrimidin-6-yl]-2-[2-(lH- l ,2,3-triazol-4-yl)ethoxy]ethanone.
| 1-[2-(2,3-dihydro-1H-inden-2-ylamino)-5,7-dihydro-6H-pyrrolo[3,4-d]pyrimidin-6-yl]-2-[2-(1H-1,2,3-triazol-4-yl)ethoxy]ethanone; | |
| Molecular Formula: | C21H23N7O2 |
|---|---|
| Molecular Weight: | 405.45302 g/mol |
Scheme A


Scheme B

Scheme C

VI


Scheme E

Autotaxin is an enzyme reported to be the source of lysophosphatidic acid (LPA) which up-regulates pain-related proteins through one if its cognate receptors, LPAi. LPA is an intracellular lipid mediator which influences a multiplicity of biological and biochemical processes. Targeted inhibition of autotaxin-mediated LPA biosynthesis may provide a novel mechanism to prevent nerve injury-induced neuropathic pain.
Compounds that inhibit autotaxin are desired to offer a potential treatment option for patients in need of treatment for pain.
Pain associated with osteoarthritis (OA) is reported to be the primary symptom leading to lower extremity disability in OA patients. Over 20 million Americans have been diagnosed with OA, the most common of the arthropathies. The currently approved treatments for OA pain may be invasive, lose efficacy with long term use, and may not be appropriate for treating all patients. Additional treatment options for patients suffering from pain associated with OA are desired. Compounds that inhibit autotaxin represent another possible treatment option for patients with pain associated with OA.
U.S. Patent 7,524,852 (‘852) discloses substituted bicyclic pyrimidine derivatives as anti-inflammatory agents.
PCT/US2011/048477 discloses indole compounds as autotoxin inhibitors.
There is a need for novel compounds that provide autotaxin inhibition. The present invention provides novel compounds which are autotaxin inhibitors. The present invention provides certain novel compounds that inhibit the production of LPA.
Autotaxin inhibitor compounds are desired to provide treatments for autotaxin mediated conditions, such as pain and pain associated with OA.
PAPER

In an effort to develop a novel therapeutic agent aimed at addressing the unmet need of patients with osteoarthritis pain, we set out to develop an inhibitor for autotaxin with excellent potency and physical properties to allow for the clinical investigation of autotaxin-induced nociceptive and neuropathic pain. An initial hit identification campaign led to an aminopyrimidine series with an autotaxin IC50 of 500 nM. X-ray crystallography enabled the optimization to a lead compound that demonstrated favorable potency (IC50 = 2 nM), PK properties, and a robust PK/PD relationship.



Novel Autotaxin Inhibitors for the Treatment of Osteoarthritis Pain: Lead Optimization via Structure-Based Drug Design
http://pubs.acs.org/doi/abs/10.1021/acsmedchemlett.6b00207

Spencer Jones
Senior Research Scientist at Eli Lilly and Company
2-(2-(1H-1,2,3-triazol-5-yl)ethoxy)-1-(2-((2,3-dihydro-1H-inden-2-yl)amino)-5,7-dihydro-6Hpyrrolo[3,4-d]pyrimidin-6-yl)ethan-1-one (9)
………… Purified the resulting residue by silica gel chromatography (gradient elution: 0-9% methanol in ethyl acetate ) to give the title compound……..
1H NMR (400 MHz, CDCl3): 60:40 mixure of rotamers * indicates minor rotamer δ 8.18 (bs, 0.6H), *8.13 (bs, 0.4H), 7.49 (s, 1H), 7.21-7.09 (m, 4 H), 5.70-5.50 (m, 1H), 4.87-4.78 (m, 1H), 4.75 (s, 1.2H), *4.67 (s, 0.8H), 4.64 (s, 1.2H) *4.53 (s, 0.8H), *4.30 (s, 0.8H), 4.28 (s, 1.2H), 3.93 (t, J = 5.6 Hz, 2H), 3.43 (dd, J = 16.2, 7.1 Hz, 2H), 3.10 (t, J = 5.6 Hz, 2H), 2.89 (dd, J = 16.2, 4.9 Hz, 2H).
13C NMR (400 MHz, CDCl3): * indicates minor δ *169.3, 16 169.2, 167.0, *166.8, *162.4, 162.2, 152.8, *152.3, 141.1, 137.8, 130.9, 126.7, 124.9, 115.9, 69.8, 69.3, *69.0, 52.7, *52.5, 51.2, 49.0, *47.9, 40.1, 24.7.
LC/MS (ESI+ ): (m/z) 406 (C21H24N7O2 = (M+1)+ ).
PATENT
Example 2
Synthesis of l-[2-(2,3-dihydro- lH-inden-2-ylamino)-5,7-dihydro-6H-pyrrolo[3,4- d]pyrimidin-6-yl]-2-[2-(lH- l ,2,3-triazol-4-yl)ethoxy]ethanone.
Stir a mixture of 2-[2-(lH-triazol-5-yl)ethoxy]acetic acid 2,2,2-trifluoroacetic acid
(20.22 g; 70.90 mmol), N-(2,3-dihydro- lH-inden-2-yl)-6,7-dihydro-5H-pyrrolo[3,4- d]pyrimidin-2-amine dihydrochloride hydrate (27.99 g; 81.54 mmol) and triethylamine (98.83 mL; 709.03 mmol) in dimethylformamide (404.40 mL) at 0°C. Add a solution of 1-propanephosphonic acid cyclic anhydride (50% solution in DMF; 51.89 mL; 81.54 mmol) over 30 minutes, and stir the mixture at room temperature for 18 hours.
Concentrate the reaction mixture under reduced pressure to give a residue. Add water (200 mL) and extract the mixture with ethyl acetate (4 x 250 mL) and
dichloromethane (4 x 250 mL). Wash the combined organic layers with saturated aqueous sodium bicarbonate (2 x 100 mL) and brine (100 mL), then dry over anhydrous sodium sulfate. Filter the mixture and concentrate the solution under reduced pressure to give a red solid (25.70 g) that is slurried in ethyl acetate/methanol (9: 1 mixture; 200 mL) for 2 hours at room temperature. Filter the resulting solid and wash with cold ethyl acetate (50 mL) to give a solid (ca.18.2 g) that is re-slurried in ethyl acetate (200 mL) at reflux for 1 hour. On cooling to room temperature, stir the mixture for 1 hour and filter the resulting light pink solid.
Slurry the light pink solid in water/methanol (1 : 1 mixture; 200 mL) and heat the mixture at 50°C for 30 minutes. Add ammonium hydroxide solution (32% ; 50 mL) and continue to heat the mixture at 50°C for 30 minutes. Upon cooling to room temperature, add additional ammonium hydroxide solution (32% ; 50 mL) and continue stirring for 1 hour at room temperature. Filter the resulting light gray solid, dry and slurry again in ethyl acetate (200 mL) for 1 hour to afford a light gray solid that is filtered, washed with ethyl acetate (25 mL), and dried to give the title compound (12.42 g; 43%) as a gray solid. MS (m/z): 406 (M+l).
PATENT
US-20140200231-A1
https://www.google.com/patents/US20140200231
Scheme E

Preparation 7
Synthesis of 2-[2-(lH-triazol-5-yl)ethoxy]acetic acid.

Pressurize 1 atmosphere of hydrogen (g) to a flask containing [2-(l-benzyl-lH- l,2,3-triazol-5-yl)ethoxy]acetic acid (10.1 g; 1.00 equiv; 38.66 mmoles) and palladium (II) chloride (3 g; 16.92 mmoles; 3.00 g) in isopropyl alcohol (300 mL) and water (60 mL). Maintain the flask under a hydrogen atmosphere for 3 h, then filter through Celite™ and concentrate. Add toluene (2×50 mL) and concentrate to afford the title compound (7.96 g, 100%). ]H NMR (d6-DMSO): 2.86 (t, / = 7 Hz, 2 H), 3.65 (t, / = 7 Hz, 2 H), 3.98 (s, 2 H), 7,77 (s, 1 H), 13.4 – 13.6 (br s, 2 H).
Example 1
Synthesis of l-[2-(2,3-dihydro-lH-inden-2-ylamino)-7,8-dihydropyrido[4,3-d]pyrimidin- 6(5H)-yl]-2-[2-(lH-l,2,3-triazol-4- l)ethoxy]ethanone.

Add N-indan-2-yl-5,6,7,8-tetrahydropyrido[4,3-d]pyrimidin-2-amine (4.2 g, 15.8 mmol) to a mixture of 2-[2-(lH-triazol-5-yl)ethoxy]acetic acid (2.7 g, 15.8 mmol), 1-hydroxybenzotriazole (3.20 g, 23.7 mmol), and dimethylaminopropyl)-3- ethylcarbodiimide hydrochloride (5.44 g, 28.4 mmol) in dichloromethane (40 mL) at 25 °C. Add triethylamine (4.40 mL, 31.6 mmol) to the reaction mixture and stir for 16 h. Wash with water (2 x 50 mL) and concentrate the organic layer. Purify by silica gel column chromatography, eluting with ethyl acetate/methanol, to give the title compound (4.0 g, 60%) as a solid. MS (m/z): 420 (M + Η). Preparation 8
Synthesis of 2-chloro-l-[2-(2,3-dihydro-lH-inden-2-ylamino)-7,8-dihydropyrido[4,3- d]pyrimidin-6(5H)-yl]ethanone.

To N-indan-2-yl-5,6,7,8-tetrahydropyrido[4,3-d]pyrimidin-2-amine (11.0 g, 41.3 mmol) and triethylamine (7.48 mL, 53.7 mmol) in dichloromethane (200 mL), add 2- chloroacetyl chloride (3.61 mL, 5.13 g, 45.4 mmol) dropwise over five minutes at 23 °C. Stir for 30 minutes and pour the reaction mixture into 1 : 1 50% saturated aqueous sodium bicarbonate: dichloromethane (75 mL). Separate the organic layer from the aqueous layer and further extract the aqueous layer with dichloromethane (2 x 25 mL). Combine the organic extracts and dry over anhydrous sodium sulfate, filter, and concentrate. Dissolve the residue in chloroform (10 mL) and purify via silica gel column chromatography (gradient elution: 25% ethyl acetate in hexanes to 100% ethyl acetate) to give the title compound (9.75 g, 69%). ]H NMR (CDC13, * = minor amide rotamer) δ 2.77* (t, 2H), 2.84 (dd, 2H), 2.87 (t, 2H), 3.35 (dd, 2H), 3.76 (t, 2H), 3.85* (t, 2H), 4.12 (s, 2H), 4.52* (s, 2H), 4.57 (s, 2H), 4.72-4.82 (m, IH), 5.48-5.64 (m, IH), 7.12-7.21 (m, 4H), 8.03-8.10 (m, IH).
Preparation 9
Synthesis of 2-(but-3-yn-l-yloxy)-l-[2-(2,3-dihydro-lH-inden-2-ylamino)-7,8- dihydropyrido[4,3-d]p rimidin-6(5H)-yl]ethanone.

To sodium hydride (60 wt% in mineral oil, 1.58 g, 39.6 mmol) in tetrahydrofuran (50 mL) at 23 °C, add 3-butyn-l-ol (7.93 g, 8.59 mL, 113.2 mmol) dropwise, then stir at 23 °C for 20 minutes. Add this solution to 2-chloro-l-[2-(2,3-dihydro-lH-inden-2- ylamino)-7,8-dihydropyrido[4,3-d]pyrimidin-6(5H)-yl]ethanone (9.70 g, 28.3 mmol) in tetrahydrofuran (150 mL) at 23 °C and stir for one hour. Pour the reaction mixture into 50% saturated aqueous sodium bicarbonate solution. Separate the organic layer and further extract the aqueous layer with ethyl ether (x 2) and ethyl acetate (x 2). Combine the organic extracts and wash with brine, then dry over anhydrous sodium sulfate, filter, and concentrate. Purify the resulting crude product by silica gel column chromatography (gradient elution: 20% ethyl acetate in hexanes to 100% ethyl acetate) to give the title compound (8.16 g, 77%). MS (m/z): 377 (M + 1).
Example la
Alternative synthesis of l-[2-(2,3-dihydro- lH-inden-2-ylamino)-7,8-dihydropyrido[4,3- d]pyrimidin-6(5H)-yl]-2-[2-(lH- l,2,3-triazol-4- l)ethoxy]ethanone.

Sparge a solution of 2-(but-3-yn- l-yloxy)-l-[2-(2,3-dihydro-lH-inden-2- ylamino)-7,8-dihydropyrido[4,3-d]pyrimidin-6(5H)-yl]ethanone (8.15 g, 21.7 mmol) and L-ascorbic acid sodium salt (8.58 g, 43.3 mmol) in dimethylformamide (60 mL) and water (60 mL) with nitrogen for ten minutes, then evacuate and backfill with nitrogen three times. Add copper (II) sulfate pentahydrate (1.08 g, 4.33 mmol) and heat to 90 °C, then add azidotrimethylsilane (23.1 mL, 20.0 g, 173 mmol) dropwise and stir for one hour. Cool reaction mixture to 23 °C and pour into water (50 mL). Extract this mixture with ethyl acetate (4 x 50 mL). Combine the organic extracts and wash with saturated aqueous sodium chloride, dry over anhydrous sodium sulfate, filter, and concentrate.
Purify the resulting crude product by silica gel column chromatography (gradient elution: 0 to 10% methanol in ethyl acetate) to give the title compound (3.60 g, 40%). MS (m/z): 420 (M + 1). Preparation 10
Synthesis of tert-butyl-2-(2,3-dihydro-lH-inden-2-ylamino)-5,7-dihydro-6H-pyrrolo[3,4- d]pyrimidine-6-carboxylate.

Charge 450 rriL (2.58 mol) of N-ethyl-N-isopropylpropan-2-amine into a 15 °C solution of tert-butyl 2-chloro-5,7-dihydro-6H-pyrrolo[3,4-d]pyrimidine-6-carboxylate (220 g, 860.37 mmol) and 2,3-dihydro-lH-inden-2-amine (137.7 g, 1.03 mol) in 1- methylpyrrolidin-2-one (3.6 L). Heat the resulting mixture to 80 °C for 16 h, then cool to 30 °C and transfer the resulting mixture into 5 L of water at 25 °C. Filter the resulting solid and rinse the filter cake with water (2 x 300 rriL). Reslurry the solid in ethyl acetate (350 iriL) for 45 min at 15 °C. Filter the slurry, rinsing with 15 °C ethyl acetate ( 2 x 250 rriL), and dry to give the title compound (226 g, 75%) as an off-white solid. ‘H NMR (d6-DMSO) 1.45 (s, 9 H), 2.87 (dd, /= 7.2, 15.8 Hz, 2 H), 3.24 (dd, /= 7.2, 15.8 Hz, 2 H), 4.36 (d, 10.4 Hz, 2 H), 4.44 (d, /= 12.8 Hz, 2 H), 4.60 (m, 1 H), 7.14 (m, 2 H), 7.20 (m, 2 H), 7.55 (d, /= 6.8 Hz, 1 H), 8.27 (d, /= 7.2 Hz, 1 H).
Preparation 11
Synthesis of N-(2,3-dihydro-lH-inden-2-yl)-6,7-dihydro-5H-pyrrolo[3,4-d]pyrimidin-2- amine dihydrochloride hydrate.

Charge 670 rriL of 5 M hydrochloric acid (3.35 mol) to a solution of tert-butyl 2-
(2,3-dihydro-lH-inden-2-ylamino)-5,7-dihydro-6H pyrrolo[3,4-d]pyrimidine-6- carboxylate (226 g, 641.25 mmol) in tetrahydrofuran (2.0 L) at 17 °C, maintaining the internal temperature below 26 °C during the addition. Heat the resulting solution to 50 °C for 16 h, cool to 25 °C and dilute with 500 rriL of water and 500 mL of tert- butylmethylether. Separate the resulting layers and extract with tert-butylmethylether (3 x 1 L). Concentrate the water phase down to a reaction volume of ca. 200 mL, and filter the resulting slurry. Rinse the cake with tert-butylmethylether (2 x 200 mL) and dry to give the title product (177 g, 80%) as a light brown solid. MS (m/z): 253.2 (M-2HC1- H20+1).
Preparation 12
Syntheis of tert-butyl 2-but-3-ynox acetate.

Stir a mixture of but-3-yn-l-ol (6.00 g; 85.60 mmol), tetrabutylammonium sulfate (2.07 g; 8.54 mmol) and sodium hydroxide (40% wt/wt; 150 mL) in dichloromethane (150 mL) at 0°C. Add tert-butyl bromoacetate (19.34 mL; 128.40 mmol) dropwise and stir the mixture for 2.5 hours at room temperature. Dilute the reaction mixture with dichloromethane (200 mL) and water (100 mL), separate the layers, and further extract the aqueous layer with dichloromethane (2 x 100 mL). Wash the combined organic layers with brine (100 mL), dry over anhydrous sodium sulfate, and concentrate to afford the crude title compound as a brown oil (11.93 g). Purify the oil by silica gel column chromatography, eluting with hexane: ethyl acetate (0% to 10% mixtures) to give the title compound (11.35 g; 72%) as a colorless oil. ]H NMR (CDCI3) δ 1.48 (s, 9H), 2.00 (m, 1H), 2.52 (m, 2H), 3.67 (m, 2H), 4.01 (bs, 2H).
Preparation 13
Synthesis of tert-butyl 2-[2-(lH-triazol-5- l)ethoxy]acetate.

Stir tert-Butyl 2-but-3-ynoxyacetate (11.34 g; 61.55 mmol) and copper(I)iodide (584 mg; 3.07 mmol) in a mixture of dimethylformamide (56.70 mL) and methanol (11.34 mL) at 0°C. Add azido(trimethyl)silane (12.33 mL; 86.47 mmol) dropwise and heat the mixture at 90°C for 18 hours.
In a second batch, stir tert-butyl 2-but-3-ynoxyacetate (4.38 g; 23.77 mmol) and copper(I)iodide (226 mg; 1.19 mmol) in a mixture of dimethylformamide (22 mL) and methanol (6 mL) at 0°C. Add azido(trimethyl)silane (4.8 mL; 33.66 mmol) dropwise and the mixture heated at 90°C for 18 hours.
Upon cooling to room temperature, combine the crude products from both batches and concentrate the mixture to afford a greenish residue. Purify the crude product by filtration through a plug of silica eluting with dichloromethane: ethyl acetate (75% to 100% mixtures) to afford the title compound (14.15 g, 73%) as a colorless oil. MS (m/z): 228.15 (M+l).
Preparation 14
Synthesis of 2-[2-(lH-triazol-5-yl)ethoxy]acetic acid 2,2,2-trifluoroacetic acid.

Stir a mixture of ieri-butyl 2-[2-(lH-triazol-5-yl)ethoxy]acetate (14.15 g; 62.26 mmol) and trifluoroacetic acid (70.75 mL, 935.69 mmol) in dichloromethane (70.75 mL) for 2 hours at room temperature. Concentrate the reaction mixture under reduced pressure to provide the title compound containing additional trifluoroacetic acid (20.22 g, >100%) as a brown solid. MS (m/z): 172.05 (M+l).
Example 2
Synthesis of l-[2-(2,3-dihydro- lH-inden-2-ylamino)-5,7-dihydro-6H-pyrrolo[3,4- d]pyrimidin-6-yl]-2-[2-(lH- l ,2,3-triazol-4-yl)ethoxy]ethanone.
Stir a mixture of 2-[2-(lH-triazol-5-yl)ethoxy]acetic acid 2,2,2-trifluoroacetic acid
(20.22 g; 70.90 mmol), N-(2,3-dihydro- lH-inden-2-yl)-6,7-dihydro-5H-pyrrolo[3,4- d]pyrimidin-2-amine dihydrochloride hydrate (27.99 g; 81.54 mmol) and triethylamine (98.83 mL; 709.03 mmol) in dimethylformamide (404.40 mL) at 0°C. Add a solution of 1-propanephosphonic acid cyclic anhydride (50% solution in DMF; 51.89 mL; 81.54 mmol) over 30 minutes, and stir the mixture at room temperature for 18 hours.
Concentrate the reaction mixture under reduced pressure to give a residue. Add water (200 mL) and extract the mixture with ethyl acetate (4 x 250 mL) and
dichloromethane (4 x 250 mL). Wash the combined organic layers with saturated aqueous sodium bicarbonate (2 x 100 mL) and brine (100 mL), then dry over anhydrous sodium sulfate. Filter the mixture and concentrate the solution under reduced pressure to give a red solid (25.70 g) that is slurried in ethyl acetate/methanol (9: 1 mixture; 200 mL) for 2 hours at room temperature. Filter the resulting solid and wash with cold ethyl acetate (50 mL) to give a solid (ca.18.2 g) that is re-slurried in ethyl acetate (200 mL) at reflux for 1 hour. On cooling to room temperature, stir the mixture for 1 hour and filter the resulting light pink solid.
Slurry the light pink solid in water/methanol (1 : 1 mixture; 200 mL) and heat the mixture at 50°C for 30 minutes. Add ammonium hydroxide solution (32% ; 50 mL) and continue to heat the mixture at 50°C for 30 minutes. Upon cooling to room temperature, add additional ammonium hydroxide solution (32% ; 50 mL) and continue stirring for 1 hour at room temperature. Filter the resulting light gray solid, dry and slurry again in ethyl acetate (200 mL) for 1 hour to afford a light gray solid that is filtered, washed with ethyl acetate (25 mL), and dried to give the title compound (12.42 g; 43%) as a gray solid. MS (m/z): 406 (M+l).
Preparation 15
Synthesis of 2-chloro- l-[2-(2,3-dihydro- lH-inden-2-ylamino)-5,7-dihydro-6H- pyrrolo[3,4-d]pyrimidin-6-yl]ethanone.

Stir a suspension of N-(2,3-dihydro-lH-inden-2-yl)-6,7-dihydro-5H-pyrrolo[3,4- d]pyrimidin-2-amine dihydrochloride hydrate (14.4 g, 41.9 mmol) and triethylamine (14.3 g, 19.7 mL, 141.4 mmol) in dichloromethane (200 mL) at 23 °C for 10 minutes, then cool to -30 °C. Add 2-chloroacetyl chloride (5.49 g, 3.86 mL, 48.6 mmol) over two minutes and warm to 23 °C over 10 minutes. Add methanol (5 mL) and remove the solvent in vacuo. Slurry the crude reaction mixture in methanol (30 mL), add 50 g silica gel and remove solvent in vacuo. Load the resulting residue onto a loading column and purify via silica gel column chromatography (gradient elution: 50% ethyl acetate in hexanes to ethyl acetate to 10% methanol in ethyl acetate) to give the title compound (11.5 g, 84%). MS (m/z): 329(M+1).
Preparation 16
Synthesis of 2-(but-3-yn-l-yloxy)-l-[2-(2,3-dihydro-lH-inden-2-ylamino)-5,7-dihydro- 6H-pyrrolo[3,4-d]pyrimidin-6-yl]ethanone.

To sodium hydride (60 wt% in mineral oil, 2.06 g, 51.4 mmol) in tetrahydrofuran (86 mL) at 0 °C, add 3-butyn-l-ol (4.64 g, 5.03 mL, 64.3 mmol), then stir at 23 °C for 15 minutes. Add this solution to 2-chloro-l-[2-(2,3-dihydro-lH-inden-2-ylamino)-5,7- dihydro-6H-pyrrolo[3,4-d]pyrimidin-6-yl]ethanone (8.45 g, 25.7 mmol) in
tetrahydrofuran (86 mL) at 0 °C and stir for five minutes. Pour reaction mixture into 50% saturated aqueous sodium bicarbonate solution. Separate the organic layer and further extract the aqueous layer with ethyl ether and ethyl acetate (2 x 50 mL each). Combine the organic extracts and wash with brine, then dry over anhydrous sodium sulfate, filter, and concentrate. Combine the crude product with the crude product from a second reaction (run reaction under identical conditions and stoichiometry employing 2-chloro- 1- [2-(indan-2-ylamino)-5,7-dihydropyrrolo[3,4-d]pyrimidin-6-yl]ethanone (3.0 g, 9.1 mmol)) and purify by silica gel column chromatography (gradient elution: 25% ethyl acetate in hexanes to 100% ethyl acetate) to give the title compound (2.90 g, 23%). MS
(m/z): 363(M+1). Example 2a
Alternative synthesis of l-[2-(2,3-dihydro-lH-inden-2-ylamino)-5,7-dihydro- pyrrolo[3,4-d]pyrimidin-6-yl]-2-[2-(lH-l,2,3-triazol-4-yl)ethoxy]ethanone.

Add dimethylformamide (27 mL) and water (27 mL) to a flask containing 2-(but- 3-yn-l-yloxy)-l-[2-(2,3-dihydro-lH-inden-2-ylamino)-5,7-dihydro-6H-pyrrolo[3,4- d]pyrimidin-6-yl]ethanone (2.90 g, 8.00 mmol). Add copper (II) sulfate pentahydrate (400 mg, 1.60 mmol) and L-ascorbic acid sodium salt (3.17 g, 16.0 mmol). Evacuate flask and backfill with nitrogen (x 2), then add azidotrimethylsilane (7.37 g, 8.53 mL, 64.0 mmol) and heat the reaction to 90 °C for 70 minutes. Cool the reaction mixture to 23 °C and remove all solvent in vacuo. Suspend the residue in methanol/dichloromethane and then add silica gel and remove solvent in vacuo. Load this material onto a loading column and purify via silica gel column chromatography (gradient elution: 0-9% methanol in ethyl acetate) to give the title compound (980 mg, 30%). MS (m/z):
406(M+1).
/////////Autotaxin, LPA, osteoarthritis, tool molecule, lily, Spencer Jones, PRECLINICAL
N1(Cc2cnc(nc2C1)NC3Cc4ccccc4C3)C(=O)COCCc5cnnn5
Filed under: Preclinical drugs Tagged: Autotaxin, lily, LPA, osteoarthritis, preclinical, Spencer Jones, tool molecule







