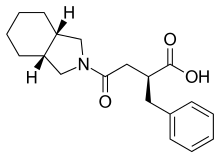
- MF C19H25NO3
- MW 315.407 Da
Mitiglinide (INN, trade name Glufast) is a drug for the treatment of type 2 diabetes.[1]
Mitiglinide belongs to the meglitinide class of blood glucose-lowering drugs and is currently co-marketed in Japan by Kissei and Takeda. The North America rights to mitiglinide are held by Elixir Pharmaceuticals. Mitiglinide has not yet gained FDA approval.
Pharmacology
Mitiglinide is thought to stimulate insulin secretion by closing the ATP-sensitive K(+) K(ATP) channels in pancreatic beta-cells.

Dosage
Mitiglinide is delivered in tablet form.

| Molecular Weight | 333.42 |
| Formula | C19H27NO4 |
| CAS Number | 207844-01-7 |
Mitiglinide calcium hydrate




The condensation of dimethyl succinate (I) with benzaldehyde (II) by means of NaOMe in refluxing methanol followed by hydrolysis with NaOH in methanol/water gives 2-benzylidenesuccinic acid (III). Compound (III) is treated with refluxing Ac2O, yielding the corresponding anhydride (IV), which by reaction with cis-perhydroisoindole (V) in toluene affords the monoamide (VI). This amide is reduced with H2 over a chiral Rhodium catalyst and treated with (R)-1-phenylethylamine (VII) to provide the chiral salt (VIII) as a single diastereomer isolated by crystallization. Finally, this salt is treated first with aqueous NH4OH and then with aqueous CaCl2.

he optical resolution of racemic 2-benzylsuccinic acid (XV) using the chiral amines (R)-1-phenylethylamine (VII), (R)-1-(1-naphthyl)ethylamine (XIV) or (S)-1-phenyl-2-(4-tolyl)ethylamine (XVI) is carried out by fractional crystallization of the corresponding diastereomeric salts and treatment with 2N HCl, providing the desired enantiomer 2(S)-benzylsuccinic acid (XVII). Reaction of (XVII) with SOCl2 gives the corresponding acyl chloride (XVIII), which is treated with 4-nitrophenol (XIX) and TEA in dichloromethane to yield the activated diester (XX). The regioselective reaction of (XX) with cis-perhydroisoindole (V) in dichloromethane affords the monoamide (XXI), which by reaction with HCl and methanol provides the corresponding methyl ester (XXII). This ester is hydrolyzed with NaOH to the previously described chiral succinamic acid (XIII), which is finally converted into its calcium salt.

PATENT
https://www.google.com/patents/WO2009047797A2?cl=en
Perhydroisoindole derivative, (S)-mitiglinide of formula I is a potassium channel antagonist for the treatment of type 2 diabetes mellitus and is chemically known as (5)-2-benzyl-3-(cis-hexahydro-2- isoindolinylcarbonyl) propionic acid.
Formula I

It has potent oral hypoglycemic activity and is structurally different from the sulphonylureas, although it stimulates calcium influx by binding to the sulphonylurea receptor on pancreatic β-cells and closing K+ ATP channels. Perhydroisoindole derivatives including (S)-mitiglinide and salts thereof were first disclosed in US patent 5,202,335. This patent discloses preparation of (S)-mitiglinide by the reaction of (5)-3-benzyloxycarbonyl-4-phenylbutyric acid with cis-hexahydroisoindoline in the presence of N- methylmorpholine and isobutyl chloroformate followed by debenzylation with palladium on carbon in ethyl acetate to yield (5)-mitiglinide as viscous oil. (S)-Mitiglinide is isolated as its hemi calcium salt using calcium chloride in water which is further recrystallized with diisopropyl ether. Melting point of calcium salt of mitiglinide calcium dihydrate salt is herein reported as 179-185 0C. (S)-Mitiglinide prepared by the above process is obtained in low yields. Further, the synthetic method described in the patent does not enable the desired regioselectivity. Extensive purification steps are required to obtain the desired compound, which makes the process unattractive from industrial point of view. US patent 6,133,454 discloses a process for the preparation of (S)-mitiglinide by reacting dimethyl succinate with benzaldehyde in methanolic medium, to yield a diacid which is converted to corresponding anhydride and is further reacted with the perhydroisoindole to yield 2-[(cis- perhydroisomdol^-ytycarbonylmethyl^-phenylacrylic acid which is then subjected to catalytic hydrogenation using the complex rhodium/(2S,4S)-N-butoxycarbonyl-4-diphenylphosphino-2-diphenyl- phosphino-methylpyrrolidine (Rh/(S,S) BPPM) as asymmetric hydrogenation catalyst, followed by conversion to pharmaceutically acceptable salt of (S)-mitiglinide. The above patent utilizes ruthenium complex which is expensive, carcinogenic and toxicity, hence not recommended for industrial scale. European patent publication no. EP 0967204 discloses the preparation of mitiglinide by deprotecting benzyl-(S)-2-benzyl-3-(cis-hexahydro-2-isoindolinyl-carbonyl) propionate and converting the same to calcium dihydrate salt in crystalline form using calcium chloride, water and ethanol. The crystals of calcium salt are further recrystallized using ethanol and water. But the patent is silent about the crystalline form of mitiglinide calcium.
It will be appreciated by those skilled in the art that perhydroisoindole derivative, (S)-mitiglinide of formula I contains a chiral centre and therefore exists as enantiomers. Optically active compounds have increasingly gained importance since the technologies to develop optically active compounds in high purity have considerably improved. Obtaining asymmetric molecules has traditionally involved resolving the desired molecule from a racemic mixture using a chiral reagent, which is not profitable as it increases the cost and processing time. Alternatively, desired enantiomer can be obtained by selective recrystallization of one enantiomer. However such a process is considered inefficient, in that product recovery is often low, purity is uncertain and more than 50% of the material is lost. Enantiomers can also be resolved chromatographically, although the large amount of solvent required for conventional batch chromatography is cost prohibitive and results in the preparation of relatively dilute products. Limited throughput volumes also often make batch chromatography impractical for large-scale production. Even so, it is a common experience for those skilled in the art to find chiral separation of certain chiral mixtures to be inefficient or ineffective, thereby resulting in the efforts towards development of newer methodologies for asymmetric synthesis.
It would be of significant advantage to obtain (.S)-mitiglinide by development of reaction conditions necessary for productive manufacture of the required (5)-enantiomer, substantially free of the unwanted (R)-enantiomer, in large quantities that meet acceptable pharmaceutical standards. It is the property of the solid compounds to exist in different polymorphic form. By the term polymorphs mean to include different physical forms, crystal forms, crystalline/liquid crystalline/non-crystalline (amorphous) forms. This has especially become very interesting after observing that many antibiotics, antibacterials, tranquilizers etc, exhibit polymorphism and some/one of the polymorphic forms of a given drug exhibit superior bio-availability and consequently show much higher activity compared to other polymorphs. It has also been disclosed that the amorphous forms in a number of drugs exhibit different dissolution characteristics and in some cases different bioavailability patterns compared to the crystalline form [Konne T., Chem. Pharm. Bull. 38, 2003 (1990)]. The solubility of a material is also influenced by its solid-state properties, and it has been suggested that the solubility of an amorphous compound is 10 to 1600 times higher than that of its most stable crystalline structures (Bruno C. Hancock and Michael Parks, ‘What is the true solubility advantage for amorphous pharmaceuticals’, Pharmaceutical Research 2000, Apr; 17(4):397-404). Thus it can be concluded that amorphous products are in general more soluble and often show improved absorption in humans.
Thus, there is a widely recognized need for developing a stable polymorph, which would further offer advantages over crystalline forms in terms of better dissolution and the availability profiles. Also none of the prior art references disclose amorphous form of mitiglinide calcium. Thus present invention provides amorphous form of mitiglinide calcium.
It is also required that the final API like mitiglinide whether in the amorphous form or crystalline form must be free from the other impurities including the unwanted enantiomer, these can be side product and by product of the reaction, degradation products and starting materials. Impurities in final API are undesirable and in extreme cases, might even be harmful to a patient being treated with a dosage form containing the API. Therefore impurities introduced during commercial manufacturing processes must be limited to very small amounts and are preferably substantially absent. These limits are less than about 0.15 percent by weight of each identified impurity and 0.10 % by weight of unidentified and/or uncharacterized impurities. After the manufacture of APIs, the purity of the products, such as (S)- mitiglinide calcium dihydrate is required before commercialization, and in the manufacture of formulated pharmaceuticals. Therefore, pharmaceutical active compounds must be either free from these impurities or contain the impurities in acceptable limits. There is also a need for the isolation, characterization and identification of the impurities and their use as reference markers and reference standard. Thus, the present invention meets the need in the art for a novel, efficient and industrially advantageous process for providing optically pure perhydroisoindole derivatives, particularly (iS)-mitiglinide, which is unique with respect to its simplicity, scalability and involves controlling the steps of the reaction so that predominantly the desired (S)-enantiomer is produced in high yields and purity. The present invention also provides substantially pure (S)-mitiglinide and salts thereof having novel amide impurity in acceptable limit or free from this impurity.
Example 1: Preparation of (R) 4-benzyl-3-(3-phenylpropionv0-oxazolidin-2-one To a solution of (R)-4-benzyloxazolidin-2-one (50 g), 4-dimethylaminopyridine (4.85 g), 3-phenyl propionic acid (55.08 g) in dichloromethane (375 ml) under nitrogen atmosphere at 0-5 0C, dicyclohexylcarbodiimide (975.65 g) was added. The temperature was slowly raised to 25-30 0C and stirring was continued until no starting material was left as was confirmed by thin layer chromatography. Dicyclohexylurea formed during the reaction was filtered, washed with dichloromethane (200 ml) and the filtrate was washed with saturated solution of sodium bicarbonate (500 ml). The solution was dried over sodium sulphate and solvent was distilled off to obtained crude product which was purified from methanol (200 ml) at 10-15 °C and washed with methanol (50 ml) to obtain 81.0 g of the title compound. Example 2: Preparation of 3(5)-benzyl-4-(4-(J?)-benzyl-2-oxo-oxazolidin-3-yl)-4-oxo-butyrϊc acid tert-butyl ester
To a solution of (/?)-4-benzyl-3-(3-phenyl-propionyl)-oxazolidin-2-one (150 g) in anhydrous tetrahydrofuran (1.5 It) was added a solution of sodium hexamethyldisilazane (462 ml, 36-38% solution in tetrahydrofuran) with stirring at -85 to -95 0C for 60 minutes. Tert-butyl bromo acetate (137.5 g) in tetrahydrofuran (300 ml) was added to reaction mass and then stirred to 60 minutes at -85 to -95 0C. After completion of the reaction (monitored by TLC), the reaction mixture was poured into ammonium chloride solution (10%, 2.0 It) and extracted with ethyl acetate (2×750 ml). The combined organic layer was washed with demineralized water (1×750 ml) and dried over sodium sulphate. The solvent was evaporated under reduced pressure to obtain oily residue which was stirred with mixture of n-hexane (100 ml) and isopropyl alcohol (100 ml) at Oto -50C, filtered and dried under vacuum to obtain 153.12 g of title compound having chemical purity 99.41%, chiral purity 99.91% by HPLC, [α]D 20: (-)97.52° (c = 1, CHCl3) and M.P. : 117.1-118.20C.
Example 3: Preparation of 3(5)-benzyl-4-(4(i?)-benzyl-2-oxo-oxazolidin-3-yl)-4-oxobutyric acid Trifluoroacetic acid (100 g) was added to a solution of 3(5)-benzyl-4-(4-(/?)-benzyl-2-oxo-oxazolidin-3- yl)-4-oxobutyric acid tert-butyl ester (100 g) in dichloromethane (700 ml) at 25 0C and mixture was stirred further for about 12 hours ( when TLC indicated reaction to be complete). The reaction mixture was poured in to ammonium chloride solution (10%, 500 ml). The dichloromethane layer was separated and aqueous layer was extracted with dichloromethane (2 x 250 ml). The combined organic layer was dried over sodium sulphate and evaporated under reduced pressure to obtain title compound. The crude product was recrystallized from a mixture of ethyl acetate: n-hexane (1:4, 500 ml) to obtain 78.75g of the title compound having purity 99.56% by HPLC and M.P.: 145.9-146.40C.
Example 4: Preparation of (2S)-2-benzyl-l-((4R)-4-benzyl-2-oxo-oxazolidin-3-vI)-4-(hexahydro- isoindolin-2-yl)-butane-l,4-dione
To a solution of 3(5)-benzyl-4-(4-(/?)-benzyl-2-oxo-oxazolidin-3-yl)-4-oxo-butyric acid (50 g) in anhydrous dichloromethane (1.25 It) was added triethylamine (50 ml) with stirring at -20 to -30 0C and the stirred for 15 minutes. A solution of isobutylchloroformate (37.50g) in anhydrous dichloromethane (50 ml) was added at -20 to -30 0C and stirred for 60 minutes. Thereafter, a solution of cis- hexahydroisoindoline (32.50 g) in anhydrous dichloromethane (50 ml) was slowly added by maintaining temperature -20 to -300C. After the completion of the reaction (monitored by HPLC), the mixture was successively washed with 0.5N hydrochloric acid solution (500 ml), brine (300 ml) and dried over sodium sulphate. The solvent was evaporated under reduced pressure to obtain 102.0 g of the title compound having purity 94.39% by HPLC.
Example 5: Purification of r2S)-2-benzyl-l-((4R)-4-benzyl-2-oxo-oxazolidin-3-yl)-4-(hexahydro- isoindolin-2-vD-butane-l,4-dione
To the crude (2S)-2-benzyl-l-((4R)-4-benzyl-2-oxo-oxazolidin-3-yl)-4-(hexahydro-isoindolin-2-yl)- butane- 1,4-dione (51.0 g) was added methanol (150 ml) and the mixture was stirred for 5 hours at 0 to 5 0C. Solid that precipitated out was filtered, slurry washed with cold methanol (25 ml) and dried at 45 -50 0C under vacuum to obtain 28.80 g of pure title compound as a crystalline solid having purity of 99.71% by HPLC and M. P.: 104.1-105.70C.
Example 6: Preparation of calcium salt of (-SVmitiglinide. Step-1: Preparation of (-SVmitiglinide
(2S)-2-Benzyl- 1 -((4R)-4-benzyl-2-oxo-oxazolidin-3-yl)-4-(hexahydro-isoindolin-2-yl)-butane- 1 ,4-dione (28.0 g) was dissolved in tetrahydrofuran (196 ml) and a mixture of lithium hydroxide monohydrate (3.51 g) in demineralized water (56 ml) and hydrogen peroxide (40% solution, 5.5 ml) was added with stirring at 0 to 5 0C over a period of 30 minutes. The reaction mixture was further stirred at 0 to 5 0C till the completion of the reaction. After the completion of the reaction (monitored by TLC), the reaction was quenched with the addition of cooled sodium meta-bisulphate solution (25%, 168 ml) at 0 to 10 0C. The reaction mixture was extracted with ethyl acetate (2×112 ml), the layers were separated and the aqueous layer was discarded. The HPLC analysis of the aqueous layer shows 0.77% of amide impurity. The ethyl acetate layer was then extracted with aqueous ammonia solution (4%, 2×40 ml). The layers were separated and the aqueous layer was further extracted with ethyl acetate (2×280 ml). Combined ethyl acetate layer was discarded. This aqueous layer (280 ml) was used as such in the next stage. The aqueous layer display purity 96.19 % by HPLC and amide impurity 0.04% by HPLC. Step-2: Preparation of calcium salt of dSVmitiglinide
To the above stirred solution of (S)-mitiglinide in water and ammonia(280 ml), methanol (168 ml) was added, followed by calcium chloride (4.48 g) dissolved in demineralized water (56 ml) at ambient temperature and the mixture was stirred for 2 hours. The resulting precipitate was filtered, successively slurry washed with water (3 x 140 ml) and acetone (2 x 70 ml) and dried at 450C -500C under vacuum to obtain 16.1 g of title compound having purity 99.67% by HPLC and amide impurity 0.01% by HPLC. The title product was re-precipitated from a mixture of methanol and water and dried to obtain pure title compound.
Example 7: Preparation of (.SVmitiglinide
To a solution of (2S)-2-benzyl-l-((4R)-4-benzyl-2-oxo-oxazolidin-3-yl)-4-(hexahydro-isoindolin-2-yl)- butane- 1,4-dione (50 g) in tetrahydrofuran (350 ml) was added a solution of lithium hydroxide monohydrate (8.65 g) in demineralized water (100 ml) and hydrogen peroxide (30% w/w, 40 ml) with stirring at 5 to 10 0C over a period of 15 minutes. After the completion of reaction, sodium meta- bisulphate solution (40%, 500 ml) was added to the reaction mixture and the mixture was extracted with ethyl acetate (2 x 250 ml). The organic layer was dried over sodium sulphate and evaporated under vacuum to obtain 45.5 g of title compound having 35 % of R-benzyl oxozolidin-2-one as impurity. Example 8: Purification of (.S)-mitiglinide
Aqueous ammonia solution (4%, 300 ml) was added to the crude (5)-mitiglinide (30 g) and stirred. The reaction mixture was washed with ethyl acetate (3 x 300 ml). Thereafter the reaction mixture was acidified to pH 1 to 2 with IN hydrochloric acid solution (250 ml) and extracted with ethyl acetate (2 x 150 ml). The layers were separated and ethyl acetate layer was washed with demineralized water (2 x 150 ml), dried over sodium sulphate and then evaporated under reduced pressure to obtain 16.2 g of pure (5)-mitiglinide having purity 95.55% by HPLC Example 9: Preparation of calcium salt of (S)-mitiglinide
To a solution of (<S)-mitiglinide (15 g) in water (150 ml) and aqueous ammonia solution (25%, 15 ml) at 25 to 30 0C, a solution of calcium chloride (7.5 g) in demineralized water (37.5 ml) was added. The mixture was stirred for 1 hour to precipitate the calcium salt of (5)-mitiglinide dihydrate. The resulting precipitate was filtered, slurry washed with water (3 x 150ml) and dried at 45 to 50 0C to obtain 13.25 g of the title compound having purity of 98.84% by HPLC. Example 10: Purification of calcium salt of (5)-mitiglinide
(iS)-mitiglinide calcium (10 g) was dissolved in dimethylformamide (100 ml). This is followed by the addition of demineralized water (500 ml) at 25 to 30 0C. The mixture was stirred for 30 minutes. The precipitated solid was filtered, washed with water (10x 50ml) and dried at 45 to 50 0C under vacuum to obtain 8g of pure title compound as a crystalline solid having purity of 99.62% by HPLC. Example 11: Preparation of amorphous mitiglinide calcium
Crystalline mitiglinide calcium (2.0 g) was dissolved in tetrahydrofuran (20 ml) and filtered to remove undissolved and suspended particles. The solvent was then evaporated under vacuum to obtain a powder which was then dried under vacuum at 40-600C to obtain 1.70 g of the title compound. Example 12: Preparation of amorphous mitiglinide calcium
Crystalline mitiglinide calcium (2.0 g) was dissolved in dichloromethane (30 ml) and filtered to remove undissolved and suspended particles. The solvent was then evaporated under vacuum to obtain a powder which was then dried under vacuum at 40-600C to obtain 1.64 g of the title compound. Example 13: Preparation of amorphous mitiglinide calcium
Mitiglinide (2.0 g) was dissolved in methanol (20 ml) and methanolic ammonia (5.0 ml) solution was added to it. The solution was stirred at 25-30 0C and calcium chloride (1.5 g) dissolved in methanol was mixed with the solution of mitiglinide and ammonia in methanol and the solution was filtered to remove the suspended particles. The solvent was then evaporated under vacuum to obtain a powder which was then dried under vacuum at 40-600C to obtain 1.9 g of the title compound. Example 14: Preparation of amorphous mitiglinide calcium
Mitiglinide (2.0 g) was dissolved in dichloromethane (20 ml) and aqueous ammonia (3.6 ml, 25 % solution) was added to it. The solution was stirred at 25-300C and solid calcium chloride (1.5 g) was mixed with the solution of mitiglinide and ammonia in dichloromethane and the solution warmed at 30 – 35 0C. The solution was washed with water (2 xlO ml) and the clear solution was dried over sodium sulfate, filtered and evaporated under vacuum and finally dried at under vacuum at 40-60 0C to obtain 1.75 g of the title compound.
Example 15: Preparation of amorphous mitiglinide calcium
Crystalline mitiglinide calcium dihydrate (2.0 g) was dissolved in ethyl acetate (30 ml) and filtered to remove undissolved and suspended particles. Approimately. 60 % of the solvent was distilled off under vacuum to obtain a stirrable solution. The solution was then cooled to 15-2O0C, mixed with n-heptane (20 ml) and the mixture was stirred for 30 minutes. The resulting solid was filtered, washed with n-heptane and dried under vacuum at 45-600C to yield 1.72 g of the title compound. Example 16: Preparation of amorphous mitiglinide calcium
Crystalline mitiglinide calcium (2.Og) was dissolved in dichloromethane (30 ml) and filtered to remove undissolved and suspended particles. Approximately 60 % of the solvent was distilled off under vacuum to obtain a stirrable solution. The solution was then cooled to 15-200C and mixed with diisopropyl ether (20 ml). The mixture was stirred for 30 minutes and the resulting solid was filtered, washed with diisopropyl ether and dried under vacuum at 45-600C to obtain 1.70 g of the title compound. Example 17: Preparation of amorphous mitiglinide calcium
Mitiglinide (2.0 g) was dissolved in dichloromethane (20 ml) and aqueous ammonia (3.6 ml, 25 % solution) solution was added to it. The solution was stirred at 25-30 0C and mixed with solid calcium chloride (1.5 g) and the solution warmed at 30-35 0C and stirred for 30 minutes. The solution was washed with water (2 x 10 ml) and the clear solution was dried over sodium sulfate, and filtered. Approximately 60% of the solvent was distilled off under vacuum and the resulting viscous oil was cooled to 10-15 0C and mixed with diisopropyl ether (50 ml). The reaction mixture was stirred for 30-35 minutes and the resulting solid was filtered and dried at 40-600C to obtain 1.75 g of the title compound. Example 18: Conversion of amorphous mitiglinide calcium into crystalline mitiglinide calcium A suspension of amorphous mitiglinide calcium in diisopropyl ether (30 ml) was stirred for 2 hours at 25- 300C, filtered and dried under vacuum at 45-600C to obtain crystalline form of mitiglinide calcium. Example 19: Preparation of crystalline mitiglinide calcium
To a solution of mitiglinide (2.5 g) in water (2.5 ml), aqueous ammonia solution (approx 25%, 4.0 ml) and acetonitrile (2.5 ml) at 10-150C, calcium chloride (1.32 g) dissolved in demineralized water (15 ml) was added. The mixture was stirred for 2 hours. The resulting precipitate was filtered, slurry washed with water (3 x 25 ml) and acetone (2 x 5 ml) and dried at 45-500C under vacuum to obtain 2.12 g of title compound having purity: 99.72 % by HPLC.
Example 20: Preparation of crystalline mitiglinide calcium
To a solution of mitiglinide (2.5 g) in water (2.5 ml), aqueous ammonia solution (approx 25%, 4.0 ml) and tetrahydrofuran (2.5 ml) at 10-150C, calcium chloride (1.32 g) dissolved in demineralized water (15 ml) was added. The mixture was stirred for 2 hours. The resulting precipitate was filtered, slurry washed with water (3 x 25 ml) and acetone (2 x 5 ml) and dried at 45-500C under vacuum to obtain 1.95 g of title compound having purity: 99.52 % by HPLC.
Example 21; Preparation of crystalline mitiglinide calcium
To a solution of mitiglinide (30.0 g) in water (300 ml), aqueous ammonia solution (approx 25%, 48 ml) and acetone (300 ml) at 10-150C, calcium chloride (15.8 g) dissolved in demineralized water (180 ml) was added. The mixture was stirred for 2 hours. The resulting precipitate was filtered, slurry washed with water (3 x 300 ml) and acetone (2 x 60 ml) and dried at 45-500C under vacuum to obtain 24.32 g of title compound having purity: 99.42 % by HPLC.
Example 22: Preparation of crystalline mitiglinide calcium
To a solution of mitiglinide (3.0 g) in water (30 ml), aqueous ammonia solution (approx 25%, 4.8 ml) and isopropyl alcohol (300 ml) at 10-150C, calcium chloride (1.58 g) dissolved in demineralized water
(18 ml) was added. The mixture was stirred for 2 hours. The resulting precipitate was filtered, slurry washed with water (3 x 30 ml) and acetone (2 x 6 ml) and dried at 45-500C under vacuum to obtain 1.92 g of title compound having purity: 99.65 % by HPLC.
Example 23: Preparation of (2S)-2-benzyWV-((lR)-l-benzyl-2-hydroxy-ethyl)-4-(hexahvdro- isoindolin-2-yl)-4-oxo-buryramide
To a solution of (2S)-2-benzyl-l-((4R)-4-benzyl-2-oxo-oxazolidin-3-yl)-4-(hexahydro-isoindolin-2-yl)- butane-l,4-dione (20.0 g) in tetrahydrofuran (140 ml), a solution of lithium hydroxide monohydrate
(3.43 g,) in demineralized water (40 ml) was added and the reaction mixture was refluxed for 4 hours till the completion of the reactions (monitored by thin layer chromatography). After the completion of the reaction, the reaction mixture was poured into demineralized water (100 ml) and extracted with ethyl acetate (2 x 80 ml). The combined organic layer was washed with water (80 ml) and dried over sodium sulphate. The solvent was evaporated under reduced pressure to give residue which was stirred in isopropyl alcohol at 0-5 0C for 5 hours. The mixture was filtered and then dried at 40-45 0C under vacuum to obtain 12.48 g of title compound having purity 99.77 % by HPLC. Melting point = 77 – 800C.
PAPER
An Effective and Convenient Method for the Preparation of KAD-1229
Helvetica Chimica ActaVolume 87, Issue 8, Version of Record online: 27 AUG 2004

PAPER
asian journal of chemistry asian journal of chemistry
(S)-Mitiglinide calcium dihydrate is designated chemically … Identification, Synthesis and Characterization of Impurities of (S)-Mitiglinide Calcium Dihydrate………http://www.asianjournalofchemistry.co.in/(X(1))/User/ViewFreeArticle.aspx?ArticleID=26_9_51

PATENT
https://www.google.com/patents/CN104311471A?cl=en
Mitiglinide calcium (mitiglinide calcium), the chemical name (2S) -2_ benzyl-3- (cis – hexahydro-2-isoindoline-carbonyl) propionic acid calcium salt dihydrate , for the treatment of type II diabetes. Kissei by Japanese pharmaceutical company research and development, and for the first time on sale in Japan in May 2004. Mitiglinide calcium is the second repaglinide, nateglinide after the first three columns MAG urea drugs, are ATP-dependent potassium channel blocker, is a derivative of phenylalanine, and its mechanism Similar sulfonylureas, but a faster onset of action and short half-life, is conducive to reducing postprandial blood glucose in diabetic patients, and avoid continuous glucose-induced low blood sugar, with the “in vitro pancreas” reputation.
郑德强 etc. on “Food and Drug” magazine was first disclosed the synthesis of calcium Mitiglinide, this method dimethyl succinate and benzaldehyde for raw materials, Stobble condensation, hydrolysis, dehydration anhydride, cis – perhydro isoindole reduced to give racemic acid after condensation, and then split, and salt get Mitiglinide calcium. Specific synthetic route the following equation. The method is relatively complex, in the preparation process to generate half of the unwanted enantiomer, which will waste a lot of cis – perhydro isoindole, and in the preparation of cis – to use science as a whole hydride hydrogen isoindole time reducing agent, the operation is more complicated, the cost is relatively high, and the chiral amine as a resolving agent split, the yield is low.

The patent discloses a CN201010573666 diethyl succinate and benzaldehyde, condensation occurs Stobble sodium ethoxide in ethanol and then hydrolyzed benzylidene succinic acid, succinic acid benzylidene get by catalytic hydrogenation DL-2-benzyl succinic acid, DL-2-benzyl succinic acid by (R) – a chiral amine resolving to give (S) -2- benzyl succinic acid, (S) -2- benzyl succinic acid anhydride to generate its role in the acetic anhydride, and the resulting acid anhydride and cis – hexahydro isoindole reaction of Mitiglinide acid, calcium chloride and ammonia most 后米格列奈 acid reacts with calcium Mitiglinide dihydrate. The synthesis route following formula. This method effectively avoids the expensive intermediate cis – perhydro isoindole waste, reduce costs, but still amounted to a six-step synthesis route much so that the reagent type, long cycle, low yield, and direct use in the synthesis process Sodium block protonated reagent preparation sodium methylate, generate a lot of flammable hydrogen gas, limiting the industrial application of the method.
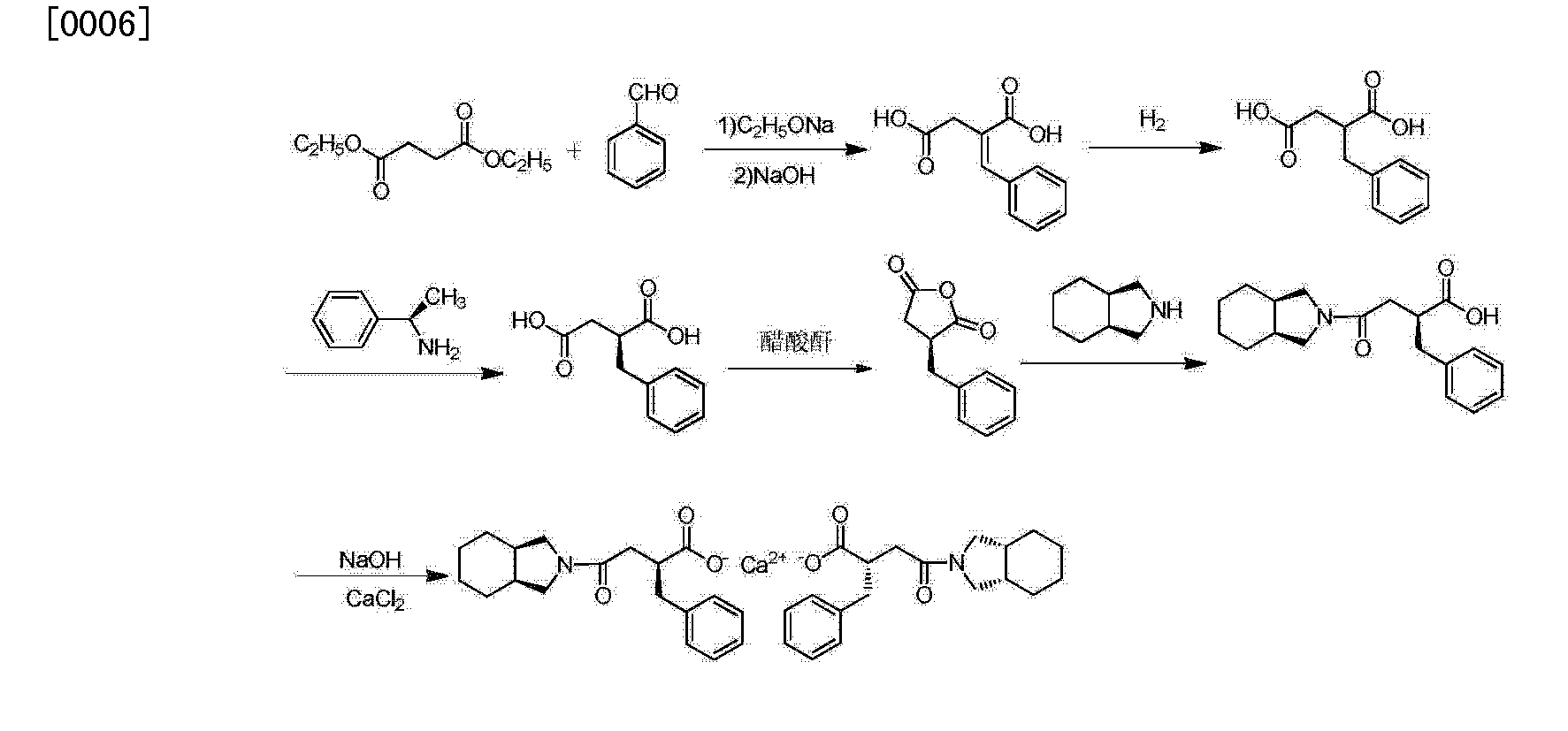
The present invention solves is to overcome the existing routes that exist in step lengthy reagent variety, low yield, long cycle, high cost, not suitable for industrial production shortcomings. The present invention provides the following formula preparation process route mitiglinide calcium, organic solvent for this preparation method uses less synthesis process is simple, high yield, good purity, suitable for industrial production.

An improved Mitiglinide calcium industrialized preparation method comprises the following steps: Step 1: Preparation of 2-benzylidene succinic acid; 2 steps: (S) prepared _2_ section succinic acid; Step 3: 2- (S) – section group _4_ oxo – (cis – perhydro isoindol-2-yl) butyric acid; Step 4: Preparation Mitiglinide calcium. Characterized in that: in step 1, using commercially available reagents protonated organic bases, protonation process using an organic alkali solution was slowly feeding methods. Step 2 chiral asymmetric reduction. Step 3 fails anhydride using direct selective amidation. Step 4 beating impurities using an aqueous solvent, prepared mitiglinide calcium dihydrate purification method.
The preparation step 1, using a commercially available organic bases as sodium methoxide or sodium ethoxide protonation agent. As optimization program, feeding method using sodium methoxide or sodium ethoxide solution formulated as the corresponding alcohol and the corresponding dialkyl succinate protonating a nucleophilic substitution reaction.
The preparation method described in Step 2, the use of Ru with BINAP homogeneous catalyst Ru (OAc) 2 [(S) -BINAP] as a chiral asymmetric synthesis of chiral reducing reagent.
The steps of the preparation method 3, using ethyl acetate as a reaction solvent, acid binding agent triethylamine do, imidazole and thionyl chloride selective amidation reagent, for cis – perhydro isoindole conduct Selective condensation title intermediate.
The step of preparing said 4, mitiglinide calcium crude product was slurried in 95% ethanol by suction, after simple preparation of high purity mitiglinide calcium dihydrate.
More specifically, the industrialized Mitiglinide calcium preparation, the following steps: Step 1: Preparation of succinate 2_ Benzylidene

Sodium methoxide (sodium ethoxide) was dissolved in methanol (ethanol), was added dropwise to dimethyl succinate (ethyl) ester, was heated at reflux for 30min, benzaldehyde was added dropwise under reflux, stirring at reflux completed the dropwise 3~5h, drops adding an aqueous solution of 4N NaOH dropwise Bi refluxed 4~6h, cooled to room temperature, adjusted with 6N HCl San PH 2, a solid precipitated, centrifuged, and dried to give the title intermediate 1. Step 2: Preparation of (S) -2- acid, benzyl butyl

Intermediate 1, methanol, and Ru (OAc) 2 [(S) -BINAP] into the reactor, the reactor with N2 the replacement air after heating to 50 ° C, a hydrogen pressure through 10h, cooled, filtered, The filtrate was concentrated to dryness to give the title intermediate 2. Step 3: 2- (S) – benzyl-4-oxo – (cis – perhydro isoindol-2-yl) butyric acid

Ethyl acetate was added to the reactor, triethylamine, imidazole and Intermediate 2, was stirred and cooled to -15~-5 ° C, was added dropwise thionyl chloride addition was complete, the -15 ° C~_5 ° C Under continued stirring 6h, a solution of cis – perhydro isoindole, drip completed, stirred at room temperature overnight, the reaction mixture was added IN hydrochloric acid, stirred Ih, separation, and the organic layer was washed with sodium hydroxide solution to extract IN The combined aqueous layer was washed with a small amount of ethyl acetate, the aqueous layer was adjusted with IN hydrochloric acid and the PH = 3, the aqueous layer was extracted with ethyl acetate, the organic layers combined, washed with water and saturated brine, and the organic layer was dried over anhydrous Na2SO4, filtered and the filtrate concentrated under reduced pressure to obtain the objective compound 3 billion Step 4: Preparation of calcium Mitiglinide

The 3 was dissolved in ethanol, was added 2N sodium hydroxide solution, after mixing the solution was added dropwise a 10% aqueous solution of calcium chloride, the reaction mixture was stirred vigorously 3~5h, ice-cooled, filtered, the filter cake with 95% ethanol beating crystallization, filtration, and dried in vacuo to give the title compound I.
Accordingly, the present invention is a method for preparing mitiglinide calcium has the following advantages:
1, Step 1, using commercially available sodium methylate (sodium ethanol) instead of sodium block as a proton agent, effectively avoid the risk of sodium block formed during the reaction a lot of flammable hydrogen gas, industrial production safer. Another use dropping protonated reagent feeding method can effectively avoid succinic acid alkyl ester of two methylene groups are protonated and reduce the incidence of side effects, so that the yield increased by nearly 20%.
2, Step 2, the selective reduction of chiral reagent (S) -BINAP instead of the original route after the first split reduction method, not only simplifies the reaction step, but low yield while avoiding split It leads to the risk of an increase in cost.
3, Step 3, the fixed selective amidation reaction conditions instead of the original first into anhydride after amidation reaction that simplifies the reaction steps to reduce the unit operations, shortening the production cycle, improve production efficiency.
4, Step 4, by using an aqueous solution of calcium Mitiglinide ethanol refining crude beating, then dried under reduced pressure to control the moisture content and reduce the difficulty of the operation, more conducive to industrial production.DETAILED DESCRIPTION The following examples further illustrate the invention, but the present invention is not limited thereto. Example One Step I: Preparation 2_ benzylidene succinic acid Sodium methoxide (9kg) and methanol (48L) into the 100L reactor, stirring to dissolve, into the high slot 50L. The dimethyl succinate (20kg) into the 200L reaction vessel, heated to reflux, methanol was added dropwise a solution of fast high tank of sodium methoxide, refluxed for reaction completion dropwise 30min, was added dropwise under reflux benzaldehyde (10. 9kg) dropwise with stirring at reflux completed 3~5h, HPLC detection benzaldehyde completion of the reaction, a solution of aqueous 4N NaOH (38L), Bi dropwise refluxed 4~6h, cooled to room temperature, 2, adjusted with 6N HCl and the precipitated solid was San PH, centrifugation, and dried in vacuo to give a pale yellow solid 19kg, i.e. an intermediate, yield 90%. Step 2: Preparation of (S) -2- butyric acid benzyl 200L detecting a high pressure hydrogenation reactor airtight, Intermediate I (19kg), methanol (95L) containing 5% Ru (0Ac) 2 [(S ) -BINAP] molecular sieve (SBA-15) supported catalyst (0. 95kg, homemade) into the reactor, purge the inside of the reactor with N2 atmosphere, followed by heating to 50 ° C, atmospheric pressure hydrogen-10h, cooled, filtered and the filtrate was concentrated to dryness under reduced pressure, the resulting solid was recrystallized from ethyl acetate and dried in vacuo to give an off-white solid 15. 5kg, i.e. intermediate 2, yield 81%, chiral purity 90. 5% θ. θ .. Step 3: 2- (S) – benzyl-4-oxo – (cis – perhydro isoindol-2-yl) butyric acid in 500L reaction vessel was charged with ethyl acetate (225L), triethylamine (1.8kg), imidazole (9. 8kg) and Intermediate 2 (15kg), stirred and cooled to -KTC, was added dropwise thionyl chloride (17. 2kg), the addition was complete, the -KTC~-5 ° C under Stirring was continued for 6h, a solution of cis – perhydro isoindole (9kg), drip completed, the reaction was stirred at room temperature for 18h, the reaction mixture was added IN HCl (150L) was stirred Ih, liquid separation, the organic layer was washed with IN sodium hydroxide solution (100LX3) extracted aqueous layers were combined, washed with ethyl acetate (50L) with, water layer was washed with IN of hydrochloric acid adjusted to PH = 3, the aqueous layer was extracted with ethyl acetate (IOOmLX 3), the combined organic layers , saturated brine (50LX 3) was washed, and the organic layer was dried over anhydrous Na2SO4, filtered, and the filtrate was concentrated under reduced pressure to give an oil 19. 8kg, i.e. Intermediate 3 Yield: 87%. Step 4: Preparation of mitiglinide calcium Intermediate 3 (. 19 8kg) and absolute ethanol (99L) into the 200L reactor, and stirred to dissolve, was added 2N sodium hydroxide solution (35L), minutes after mixing Batch into the high slot. The 500L reaction vessel was added 5% aqueous calcium chloride solution (155L), stirring was added dropwise a solution of the high slot, dropwise with vigorous stirring the reaction completion 3~5h, centrifuged, the cake was washed with 95% ethanol (99L) was recrystallized beating, centrifugation and dried in vacuo (50 ° C / 0. 09MPa), to give the title compound I 16. lkg, yield 73%.
PATENT
https://www.google.com/patents/CN102424664A?cl=en
Mitiglinide calcium Phenylalanine belong chiral compound synthesis routes according to different methods of constructing chiral center has the following three synthetic process:
① split method Document: CN 102101838A, CN 1844096, etc.)
Document: CN 102101838A, CN 1844096, etc.)

In this method, diethyl succinate and benzaldehyde by Mobbe condensation, hydrolysis, dehydration anhydride, and after cis-hydrogenated isoindole condensation is reduced to give racemic acid, and then split, and salt to give Mitiglinide calcium. The first method step condensation reaction impurities, product separation and purification difficult, finally resolving the yield is low. This method is also a lack atom economy.
② asymmetric hydrogenation Document tetrahedron Letters, 1987,28 (17), 1905-1908; Tetrahedron Letters, 1989,30 (6), 735-738)

[0027] This method requires expensive rhodium complexes (Rh, (2S, 4Q-N_-butoxycarbonyl-4-diphenylphosphino _2_ diphenylphosphino-2-diphenylphosphino methylpyrrolidine alkyl), making the production cost is greatly improved, and the need for high-pressure hydrogenation reaction, is not conducive to industrial production.
③ chiral method Document: CN 1680321A)

The method uses phenylalanine as chiral starting materials, after diazotization, nucleophilic substitution, high temperature decarboxylation and condensation reaction product. Wherein the decarboxylation temperature is too low yield, making the overall process costs.
DISCLOSURE
The object of the present invention is to provide a simple, effective and easy-to-operate preparation Mitiglinide calcium.
The present invention provides a process for the preparation of calcium Mitiglinide, the synthesis route is as follows:

Step 1: D- phenylalanine in the acid hydrolysis of formula (¾ 2- hydroxy acid;
Step 2: formula (¾ 2- hydroxy acid under basic conditions to give protected hydroxyl sulfonate of formula (¾-hydroxyphenyl propionic acid ester;
Step 3: The formula (¾-hydroxyphenyl propionic acid ester in the acid-catalyzed carboxyl ester-protected formula (4) phenylalanine methyl sulfonate carboxylate;
Step 4: cis-hydrogen isoindole synthesis formula (6) perhydro isoindole halide;
Step 5: Under alkaline conditions, the formula ⑷ formula (6) nucleophilic substitution reaction formula (5) Mitiglinide acid
Step 6: Under alkaline conditions, the formula (¾ Mitiglinide ester hydrolysis to the calcium salt of formula (1) Mitiglinide calcium.
Preferably, the specific steps include:
Step 1: (D) – phenylalanine hydrolysis in a strong acid of formula (2) 2-hydroxyphenyl propionic acid
In (D) – phenylalanine as a starting material, in the presence of a strong acid such as sulfuric acid, _5 ° C _5 ° C hydrolysis, to give Formula (2) 2-hydroxyphenyl propionic acid White solid.
Step 2: The formula (¾ 2- hydroxy acid under basic conditions to protect the hydroxyl group sulfonic acid ester of formula (¾-hydroxyphenyl propionic acid ester
2-hydroxyphenyl propionic acid in an organic base such as triethylamine or pyridine, or an inorganic base such as sodium bicarbonate, sodium carbonate or potassium carbonate effect, p-hydroxybenzoic acid ester protecting performed, the protecting group used is an aliphatic or aromatic sulfonic acid group such as mesylate, tosylate or p-toluenesulfonic acid group, a sulfonic acid group is preferably methyl group or p-toluenesulfonic acid.
Step 3: Protect formula formula (¾-hydroxyphenyl propionic acid ester in the acid-catalyzed carboxyl ester group (4) benzenepropanoic
MitigIinide1 (I) carboxylic acid ester sulfonate
In the catalytic acid carboxyl benzenepropanoic acid ester group protection, the use of alcohol may be fatty alcohols or aromatic alcohols, preferably ethanol, t-butanol or benzyl alcohol.
Step 4: cis-hydrogen isoindole synthesis formula (6) perhydro isoindole halide
In the synthesis of perhydro isoindole halide in the haloacetyl halide can be used chloroacetyl chloride, bromoacetyl chloride or bromoacetyl bromide, chloroacetyl chloride is preferred.
Step 5: Under alkaline conditions, (4) and (6) a nucleophilic substitution reaction formula (¾ Mitiglinide acid
Under the conditions of a strong base, such as sodium alkoxide such as sodium ethoxide or sodium methylate, perhydro isoindole halide and phenylalanine sulfonate nucleophilic substitution reaction Mitiglinide ethyl reaction temperature of -10 ° C -25 ° c, preferably 0 ° C.
Step 6: Under alkaline conditions, the formula (¾ Mitiglinide ester hydrolysis to the calcium salt of formula (1) calcium Mitiglinide
Ethyl mitiglinide under basic conditions such as sodium hydroxide, potassium hydroxide, or an amine (ammonia) in the presence of an aqueous solution of calcium chloride, and hydrolyzed as calcium salt, in aqueous solution under conditions of heavy alcohol crystallization, high purity mitiglinide calcium.
The present invention and the prior art comparison, has the following advantages:
1, to find an innovative high-yield process for preparing calcium Mitiglinide route, a total yield of 47%;
2, with respect to the routing methods reported in the literature, the optical yield doubled, ee greater than 99%;
3. The process route of the raw materials are cheap, readily available, avoiding costly chiral resolving agents or the use of a catalyst;
4. The process route mild conditions, high temperature decarboxylation overcome the harsh reaction conditions.
In the present invention, (D) – phenylalanine as a starting material, after diazotization, a hydroxyl group and a carboxyl group protected, nucleophilic substitution, hydrolysis and other reactions prepared mitiglinide calcium, high yield. The present invention provides a process used by a wide range of raw materials, low prices, the total yield of 47%, optical purity greater than 99%, and mild reaction conditions, the reaction process is simple, avoid the literature, such as split, high-pressure hydrogenation method low yield, long reaction steps and other shortcomings, but also to overcome the harsh conditions of high temperature reaction deacidification, etc. for preparation and production of calcium Mitiglinide provides a new choice.
The process route mild conditions, high temperature decarboxylation overcome the harsh reaction conditions.
In the present invention, (D) – phenylalanine as a starting material, after diazotization, a hydroxyl group and a carboxyl group protected, nucleophilic substitution, hydrolysis and other reactions prepared mitiglinide calcium, high yield. The present invention provides a process used by a wide range of raw materials, low prices, the total yield of 47%, optical purity greater than 99%, and mild reaction conditions, the reaction process is simple, avoid the literature, such as split, high-pressure hydrogenation method low yield, long reaction steps and other shortcomings for Mitiglinide calcium preparation and production of a new choice.
Preferably, in the above embodiment, each step may be the following alternative, the embodiment can achieve the same advantageous effects to a third embodiment of embodiment:
Step 1: (D) – phenylalanine in the acid hydrolysis of formula (¾ 2- hydroxy acid
In (D) – phenylalanine as a starting material, in the presence of sulfuric acid, -50C _5 ° C hydrolysis, to give Formula O) 2-hydroxyphenyl propionic acid White solid.
Step 2: formula (¾ 2- hydroxy acid under basic conditions to give protected hydroxyl sulfonate of formula C3) hydroxyphenyl propionic acid ester
2-hydroxyphenyl propionic acid in an organic base such as triethylamine or pyridine, or an inorganic base such as sodium bicarbonate, sodium carbonate or potassium carbonate effect, p-hydroxybenzoic acid ester protecting performed, the protecting group used is an aliphatic or aromatic sulfonic acid group such as mesylate, tosylate or p-toluenesulfonic acid group, a sulfonic acid group is preferably methyl group or p-toluenesulfonic acid.
Step 3: Formula C3) hydroxyphenyl propionic acid ester in the acid-catalyzed carboxyl ester-protected formula (4) phenylalanine methyl sulfonate carboxylate [0118] In the acid-catalyzed, styrene-acrylic acid ester-protected carboxy, the use of alcohol may be fatty alcohols or aromatic alcohols, preferably ethanol, t-butanol or benzyl alcohol.
Step 4: cis-hydrogen isoindole synthesis formula (6) perhydro isoindole halide
In the synthesis of perhydro isoindole halide in the haloacetyl halide can be used chloroacetyl chloride, bromoacetyl chloride or bromoacetyl bromide, chloroacetyl chloride is preferred.
Step 5: Under alkaline conditions, the formula ⑷ formula (6) nucleophilic substitution reaction formula (5) Mitiglinide acid
Under the conditions of a strong base, such as sodium alkoxide such as sodium ethoxide or sodium methylate, perhydro isoindole halide and phenylalanine sulfonate nucleophilic substitution reaction Mitiglinide ethyl reaction temperature of -10 ° C -25 ° c, preferably 0 ° C.
Step 6: Under alkaline conditions, the formula (¾ Mitiglinide ester hydrolysis to the calcium salt of formula (1) calcium Mitiglinide
Ethyl mitiglinide under basic conditions such as sodium hydroxide, potassium hydroxide, or an amine (ammonia) in the presence of an aqueous solution of calcium chloride, and hydrolyzed as calcium salt, in aqueous solution under conditions of heavy alcohol crystallization, high purity mitiglinide calcium.
Patent
https://www.google.com/patents/CN103724253A?cl=en
bis [(2s) -2- benzyl-3- (cis – hexahydro isoindole-2-carbonyl) propionic acid] monocalcium dihydrate (mitiglinide calcium), the formula C38H48CaN206.2Η20 English called Mitiglinide Calcium Hydrate, structural formula (I) as

Mitiglinide Calcium is synthesized by Japan Orange Health Pharmaceutical Co., Ltd., in April 2004 in Japan, for through diet and exercise therapy can effectively control high blood sugar in type II diabetes patients.Mitiglinide calcium is the second repaglinide, nateglinide third after the United States and Glenn urea drugs belong phenylalanine derivatives. By closing ΑΤΡ Mitiglinide calcium-dependent pancreatic β cell membrane Κ channel, resulting in the Ca flow, increase intracellular Ca concentration of extracellular vesicles containing threshing leaving insulin, thereby stimulating the secretion of insulin.And only when the meal will be rapid and transient stimulates the pancreas to secrete insulin, sulphonylureas with the traditional Compared to the rapid onset and short duration of action, inhibition of postprandial hyperglycemia characteristic of type II diabetes, to avoid low blood sugar react, early first- and mild diabetes treatment, and well tolerated.
According to the literature and patent reports, prepared Mitiglinide calcium are the following methods.
Method I: 2_ (S) _ benzyl succinic acid as raw material, amides, reduction, calcium salt formation Mitiglinide this method, although fewer steps, but the chiral compound materials, expensive , the production cost is high, not suitable for industrial production. References: Sorbera LA, Leeson PA, Castaner RM, et al.Mitiglinidecalcium (KAD-1229) [J] .Drugs Future, 2000,25 (10):. 1034-1042 [0007] Method Two: succinate methyl ester with benzaldehyde for raw materials, Stobble condensation, hydrolysis, dehydration anhydride, cis – perhydro isoindole after condensation is reduced to give racemic acid, and then split into calcium salts and the like have Mitiglinide. This method is relatively complex and condensation reaction impurities, product separation and purification difficult, costly, and chiral separation time yield is low.[Reference: Zheng Dejiang, Liu Wentao, Wu Lihua synthetic calcium Mitiglinide [J] Food and Drug, 2007,9 (11): 13-15]
Method three: dimethyl succinate and benzaldehyde for raw materials, Stobbe condensation, reduction, split, with p-nitrophenol and dicyclohexyl carbodiimide activated calcium salt formation Mitiglinide This production cost is relatively high, and used column chromatography, suitable for industrial production. References: Synthesis Technology Zhang Hongmei Chen meritorious, Cao Xiaohui Mitiglinide of [J], modern chemicals, 2008,28 (8): 56-59.]
Example 1:
The cis – hexahydro-isoquinoline (250.4g, 2mol), anhydrous potassium carbonate (304.0g, 2.2mol), methylene burn (1000ml) was added to the reaction flask, keeping the temperature 0-5 ° C with vigorous stirring, dropwise acetyl chloride (271.0g, 2.4mol) in dichloromethane (500ml) solution, drip completed, room temperature 2.5h, point board monitoring, reaction complete, additional water 1000ml, organic layer was separated, water (1000ml), saturated brine (1000ml), dried over anhydrous sodium sulfate overnight, dichloromethane was distilled off under reduced pressure to give cis -N- chloroacetyl hexahydro isoindole (2) 357.4g oil close Rate: 88.6%.
The cis -N- chloroacetyl hexahydro isoindole (302.5g, 1.5mol), N_ within phenylpropionyl camphor sulfonamide (573.0g, 1.65mol), 70% sodium hydride (56.6g, 1.65 mol), Ν, Ν- dimethylformamide (900ml) was added to the reaction flask, at 50 ° C, the reaction was stirred vigorously 12h, to give the alkylated product, placed to room temperature before use.
100ml of water was slowly dropped to the above-mentioned system, drip complete, lithium hydroxide (39.5g, 1.65mol), tetrahydrofuran (600ml), at 0-5 ° C under a 30% solution of hydrogen peroxide solution 680ml, drop Albert, was transferred to the reaction was continued at room temperature for 18h, point board monitoring, reaction complete, additional water 1200ml, adjusting the pH to about 2_3, extracted with dichloromethane (900ml X 3), the combined organic phases with saturated brine (1500ml) wash, overnight over anhydrous sodium sulfate, the solvent was distilled off under reduced pressure to give a viscous liquid, to which was added ethyl acetate 250ml, stirred at room temperature, suction filtered, the filter cake with ethyl acetate (150ml) and dried to give (2s) – 2-benzyl-3- (cis – hexahydro isoindole-2-carbonyl) – propionic acid (6) as a white solid 231.8g, two steps yield: 49%. Compound 6 (230g, 0.73mol), water 1150ml, added to the reaction flask. After the whole solution, was added 2mol / L sodium hydroxide solution, 400ml, stirred at rt for 30min, was slowly added dropwise with vigorous stirring chloride (162.0g, 1.46mol) in water (320ml) solution dropwise was completed, the reaction was continued for 1.5h, filtration, water (200ml X 2) washing the filter cake to give a white solid, 60 ° C and dried under reduced pressure to 3h, the filter cake with 95% ethanol (2300ml) recrystallized Mitiglinide calcium (I) 430g, yield: 83.6%, mp: 178 ~ 183 ° C, FAB-MS: m / z316 [M + l] +; [α] D20 = + 5.45 ° (C = 1, methanol) [Document: m.ρ.: 179 ~ 185Ό, [α] d20 = + 5.64 ° (C = L 0, methanol)]; purity: 99.8% [HPLC normalization method : Column C18, mobile phase L OOmol / L potassium dihydrogen phosphate buffered saline – acetonitrile-water (20:35: 30) (adjusted pH = 2.10); detection wavelength 210nm]; iH-NMlUCDCldOOM), δ: 1.1 ~ 1.5 (16Η, m), 1.8 ~ 2.4 (6Η, m), 2.5 ~ 3.1 (14Η, m) 3.3 ~
3.8 (6H, m) 7.4 ~ 7.6 (10H, m); Elemental analysis (%):. C64.68, Η7.35, Ν3.94, Theory: C64.75, Η7.44, Ν3.97 yield : 36.05%, a purity of 99.8%.

PAPER
WEI HUANG,等: “Novel Convenient Synthesis of Mitiglinide“, 《SYNTHETIC COMMUNICATIONS》, vol. 37, no. 13, 3 July 2007 (2007-07-03), pages 2153 – 2157, XP055079498, DOI: doi:10.1080/00397910701392590
http://www.tandfonline.com/doi/abs/10.1080/00397910701392590
Abstract: A novel convenient synthesis of the hypoglycemic agent mitiglinide was developed. (2S)-4-[(3aR,7aS)-Octahydro-2H-isoindol-2-yl]-4-oxo-2-benzyl-butanoic acid (6) was prepared by selective hydrolysis of ethyl 4-[(3aR,7aS)-octahydro-2Hisoindol-2-yl]-4-oxo-2-benzyl-butanoate (5) using a-chymotrypsin; the latter was prepared by a novel facile route from (3aR,7aS)-octahydro-2H-isoindole. The overall yield was 25.6%.
Keywords: a-chymotrypsin, mitiglinide, synthesis
Mitiglinide (calcium bis[(2S)-4-[(3aR,7aS)-octahydro-2H–isoindol-2-yl]-4oxo-2-benzylbutanoate]dihydrate) is a novel oral hypoglycemic agent. It inhibits the adenosine triphosphate (ATP)-sensitive potassium channels in pancreatic b-cells and stimulates insulin release like sulfonylureas,[1] but has a rapid onset and short-lasting hypoglycemic effect as compared with the latter.
Mitiglinide has been synthesized by several related methods that involve optical resolution,[2] asymmetric synthesis,[2a,3] and diasteroselective alkylation using chiral auxiliary.[4]
In a previous article,[2] two optical resolution methods of the key compound racemic acid 4 were reported. One of them involves esterification with optically active alcohols, which are separated into the diastereomers by column chromatogeaphy and hydrolyzed. Only the diastereomeric (S)-Nbenzyl mandelamide ester could be separated; the overall yield was 28%,
The alternative method was optical resolution by optically active bases. The best result was 30.8% yield and 97% ee when using (R)-1-(1-naphthyl)-ethylamine as a base. In this article, we have developed a new optical resolution method of racemic ester 5 by a-chymotrypsin in 45.3% yield; the optical purity of (S)-acid (6) determined by chiral-phase high performance liquid chromatography (HPLC) on Sumichiral
OA3300 was 99.2% ee, and, the method can be used for scale-up preparation.
The synthesis of free acid 6 is shown in Scheme 1. (3aR,7aS)-Octahydro2H-isoindole was chloroacetylated in the presence of Et3N to afford (3aR, 7aS)-2-(chloro-acetyl)-octahydro-2H-isoindole (2), which was condensed with diethyl benzylmalonate followed by hydrolysis and decarbonylation to obtain 4-[(3aR,7aS)-octahydro-2H-isoindol-2-yl]-4-oxo-2-benzyl-butanoic acid (4). The overall yield of the three-step synthesis was 62.9%. The racemic acid (4) was esterified with SOCl2/EtOH to give the corresponding racemic ester (5). The (R)-ester was selectively hydrolyzed by a-chymotrypsin to separate out the (S)-ester, which was subjected to hydrolysis, giving 6.
The overall yield was 28.5% [based on (3aR,7aS)-octahydro-2H-isoindole].
Compound 6 was treated with calcium chloride and 25% ammonium hydroxide to give mitiglinide; after recrystallization from 95% EtOH, the pure product was obtained in 90% yield.
Patent
https://www.google.com/patents/WO2009047797A2?cl=en
EXAMPLES
Example 1: Preparation of (R) 4-benzyl-3-(3-phenylpropionv0-oxazolidin-2-one To a solution of (R)-4-benzyloxazolidin-2-one (50 g), 4-dimethylaminopyridine (4.85 g), 3-phenyl propionic acid (55.08 g) in dichloromethane (375 ml) under nitrogen atmosphere at 0-5 0C, dicyclohexylcarbodiimide (975.65 g) was added. The temperature was slowly raised to 25-30 0C and stirring was continued until no starting material was left as was confirmed by thin layer chromatography. Dicyclohexylurea formed during the reaction was filtered, washed with dichloromethane (200 ml) and the filtrate was washed with saturated solution of sodium bicarbonate (500 ml). The solution was dried over sodium sulphate and solvent was distilled off to obtained crude product which was purified from methanol (200 ml) at 10-15 °C and washed with methanol (50 ml) to obtain 81.0 g of the title compound. Example 2: Preparation of 3(5)-benzyl-4-(4-(J?)-benzyl-2-oxo-oxazolidin-3-yl)-4-oxo-butyrϊc acid tert-butyl ester
To a solution of (/?)-4-benzyl-3-(3-phenyl-propionyl)-oxazolidin-2-one (150 g) in anhydrous tetrahydrofuran (1.5 It) was added a solution of sodium hexamethyldisilazane (462 ml, 36-38% solution in tetrahydrofuran) with stirring at -85 to -95 0C for 60 minutes. Tert-butyl bromo acetate (137.5 g) in tetrahydrofuran (300 ml) was added to reaction mass and then stirred to 60 minutes at -85 to -95 0C. After completion of the reaction (monitored by TLC), the reaction mixture was poured into ammonium chloride solution (10%, 2.0 It) and extracted with ethyl acetate (2×750 ml). The combined organic layer was washed with demineralized water (1×750 ml) and dried over sodium sulphate. The solvent was evaporated under reduced pressure to obtain oily residue which was stirred with mixture of n-hexane (100 ml) and isopropyl alcohol (100 ml) at Oto -50C, filtered and dried under vacuum to obtain 153.12 g of title compound having chemical purity 99.41%, chiral purity 99.91% by HPLC, [α]D 20: (-)97.52° (c = 1, CHCl3) and M.P. : 117.1-118.20C.
Example 3: Preparation of 3(5)-benzyl-4-(4(i?)-benzyl-2-oxo-oxazolidin-3-yl)-4-oxobutyric acid Trifluoroacetic acid (100 g) was added to a solution of 3(5)-benzyl-4-(4-(/?)-benzyl-2-oxo-oxazolidin-3- yl)-4-oxobutyric acid tert-butyl ester (100 g) in dichloromethane (700 ml) at 25 0C and mixture was stirred further for about 12 hours ( when TLC indicated reaction to be complete). The reaction mixture was poured in to ammonium chloride solution (10%, 500 ml). The dichloromethane layer was separated and aqueous layer was extracted with dichloromethane (2 x 250 ml). The combined organic layer was dried over sodium sulphate and evaporated under reduced pressure to obtain title compound. The crude product was recrystallized from a mixture of ethyl acetate: n-hexane (1:4, 500 ml) to obtain 78.75g of the title compound having purity 99.56% by HPLC and M.P.: 145.9-146.40C.
Example 4: Preparation of (2S)-2-benzyl-l-((4R)-4-benzyl-2-oxo-oxazolidin-3-vI)-4-(hexahydro- isoindolin-2-yl)-butane-l,4-dione
To a solution of 3(5)-benzyl-4-(4-(/?)-benzyl-2-oxo-oxazolidin-3-yl)-4-oxo-butyric acid (50 g) in anhydrous dichloromethane (1.25 It) was added triethylamine (50 ml) with stirring at -20 to -30 0C and the stirred for 15 minutes. A solution of isobutylchloroformate (37.50g) in anhydrous dichloromethane (50 ml) was added at -20 to -30 0C and stirred for 60 minutes. Thereafter, a solution of cis- hexahydroisoindoline (32.50 g) in anhydrous dichloromethane (50 ml) was slowly added by maintaining temperature -20 to -300C. After the completion of the reaction (monitored by HPLC), the mixture was successively washed with 0.5N hydrochloric acid solution (500 ml), brine (300 ml) and dried over sodium sulphate. The solvent was evaporated under reduced pressure to obtain 102.0 g of the title compound having purity 94.39% by HPLC.
Example 5: Purification of r2S)-2-benzyl-l-((4R)-4-benzyl-2-oxo-oxazolidin-3-yl)-4-(hexahydro- isoindolin-2-vD-butane-l,4-dione
To the crude (2S)-2-benzyl-l-((4R)-4-benzyl-2-oxo-oxazolidin-3-yl)-4-(hexahydro-isoindolin-2-yl)- butane- 1,4-dione (51.0 g) was added methanol (150 ml) and the mixture was stirred for 5 hours at 0 to 5 0C. Solid that precipitated out was filtered, slurry washed with cold methanol (25 ml) and dried at 45 -50 0C under vacuum to obtain 28.80 g of pure title compound as a crystalline solid having purity of 99.71% by HPLC and M. P.: 104.1-105.70C.
Example 6: Preparation of calcium salt of (-SVmitiglinide. Step-1: Preparation of (-SVmitiglinide
(2S)-2-Benzyl- 1 -((4R)-4-benzyl-2-oxo-oxazolidin-3-yl)-4-(hexahydro-isoindolin-2-yl)-butane- 1 ,4-dione (28.0 g) was dissolved in tetrahydrofuran (196 ml) and a mixture of lithium hydroxide monohydrate (3.51 g) in demineralized water (56 ml) and hydrogen peroxide (40% solution, 5.5 ml) was added with stirring at 0 to 5 0C over a period of 30 minutes. The reaction mixture was further stirred at 0 to 5 0C till the completion of the reaction. After the completion of the reaction (monitored by TLC), the reaction was quenched with the addition of cooled sodium meta-bisulphate solution (25%, 168 ml) at 0 to 10 0C. The reaction mixture was extracted with ethyl acetate (2×112 ml), the layers were separated and the aqueous layer was discarded. The HPLC analysis of the aqueous layer shows 0.77% of amide impurity. The ethyl acetate layer was then extracted with aqueous ammonia solution (4%, 2×40 ml). The layers were separated and the aqueous layer was further extracted with ethyl acetate (2×280 ml). Combined ethyl acetate layer was discarded. This aqueous layer (280 ml) was used as such in the next stage. The aqueous layer display purity 96.19 % by HPLC and amide impurity 0.04% by HPLC. Step-2: Preparation of calcium salt of dSVmitiglinide
To the above stirred solution of (S)-mitiglinide in water and ammonia(280 ml), methanol (168 ml) was added, followed by calcium chloride (4.48 g) dissolved in demineralized water (56 ml) at ambient temperature and the mixture was stirred for 2 hours. The resulting precipitate was filtered, successively slurry washed with water (3 x 140 ml) and acetone (2 x 70 ml) and dried at 450C -500C under vacuum to obtain 16.1 g of title compound having purity 99.67% by HPLC and amide impurity 0.01% by HPLC. The title product was re-precipitated from a mixture of methanol and water and dried to obtain pure title compound.
Example 7: Preparation of (.SVmitiglinide
To a solution of (2S)-2-benzyl-l-((4R)-4-benzyl-2-oxo-oxazolidin-3-yl)-4-(hexahydro-isoindolin-2-yl)- butane- 1,4-dione (50 g) in tetrahydrofuran (350 ml) was added a solution of lithium hydroxide monohydrate (8.65 g) in demineralized water (100 ml) and hydrogen peroxide (30% w/w, 40 ml) with stirring at 5 to 10 0C over a period of 15 minutes. After the completion of reaction, sodium meta- bisulphate solution (40%, 500 ml) was added to the reaction mixture and the mixture was extracted with ethyl acetate (2 x 250 ml). The organic layer was dried over sodium sulphate and evaporated under vacuum to obtain 45.5 g of title compound having 35 % of R-benzyl oxozolidin-2-one as impurity. Example 8: Purification of (.S)-mitiglinide
Aqueous ammonia solution (4%, 300 ml) was added to the crude (5)-mitiglinide (30 g) and stirred. The reaction mixture was washed with ethyl acetate (3 x 300 ml). Thereafter the reaction mixture was acidified to pH 1 to 2 with IN hydrochloric acid solution (250 ml) and extracted with ethyl acetate (2 x 150 ml). The layers were separated and ethyl acetate layer was washed with demineralized water (2 x 150 ml), dried over sodium sulphate and then evaporated under reduced pressure to obtain 16.2 g of pure (5)-mitiglinide having purity 95.55% by HPLC Example 9: Preparation of calcium salt of (S)-mitiglinide
To a solution of (<S)-mitiglinide (15 g) in water (150 ml) and aqueous ammonia solution (25%, 15 ml) at 25 to 30 0C, a solution of calcium chloride (7.5 g) in demineralized water (37.5 ml) was added. The mixture was stirred for 1 hour to precipitate the calcium salt of (5)-mitiglinide dihydrate. The resulting precipitate was filtered, slurry washed with water (3 x 150ml) and dried at 45 to 50 0C to obtain 13.25 g of the title compound having purity of 98.84% by HPLC. Example 10: Purification of calcium salt of (5)-mitiglinide
(iS)-mitiglinide calcium (10 g) was dissolved in dimethylformamide (100 ml). This is followed by the addition of demineralized water (500 ml) at 25 to 30 0C. The mixture was stirred for 30 minutes. The precipitated solid was filtered, washed with water (10x 50ml) and dried at 45 to 50 0C under vacuum to obtain 8g of pure title compound as a crystalline solid having purity of 99.62% by HPLC. Example 11: Preparation of amorphous mitiglinide calcium
Crystalline mitiglinide calcium (2.0 g) was dissolved in tetrahydrofuran (20 ml) and filtered to remove undissolved and suspended particles. The solvent was then evaporated under vacuum to obtain a powder which was then dried under vacuum at 40-600C to obtain 1.70 g of the title compound. Example 12: Preparation of amorphous mitiglinide calcium
Crystalline mitiglinide calcium (2.0 g) was dissolved in dichloromethane (30 ml) and filtered to remove undissolved and suspended particles. The solvent was then evaporated under vacuum to obtain a powder which was then dried under vacuum at 40-600C to obtain 1.64 g of the title compound. Example 13: Preparation of amorphous mitiglinide calcium
Mitiglinide (2.0 g) was dissolved in methanol (20 ml) and methanolic ammonia (5.0 ml) solution was added to it. The solution was stirred at 25-30 0C and calcium chloride (1.5 g) dissolved in methanol was mixed with the solution of mitiglinide and ammonia in methanol and the solution was filtered to remove the suspended particles. The solvent was then evaporated under vacuum to obtain a powder which was then dried under vacuum at 40-600C to obtain 1.9 g of the title compound. Example 14: Preparation of amorphous mitiglinide calcium
Mitiglinide (2.0 g) was dissolved in dichloromethane (20 ml) and aqueous ammonia (3.6 ml, 25 % solution) was added to it. The solution was stirred at 25-300C and solid calcium chloride (1.5 g) was mixed with the solution of mitiglinide and ammonia in dichloromethane and the solution warmed at 30 – 35 0C. The solution was washed with water (2 xlO ml) and the clear solution was dried over sodium sulfate, filtered and evaporated under vacuum and finally dried at under vacuum at 40-60 0C to obtain 1.75 g of the title compound.
Example 15: Preparation of amorphous mitiglinide calcium
Crystalline mitiglinide calcium dihydrate (2.0 g) was dissolved in ethyl acetate (30 ml) and filtered to remove undissolved and suspended particles. Approimately. 60 % of the solvent was distilled off under vacuum to obtain a stirrable solution. The solution was then cooled to 15-2O0C, mixed with n-heptane (20 ml) and the mixture was stirred for 30 minutes. The resulting solid was filtered, washed with n-heptane and dried under vacuum at 45-600C to yield 1.72 g of the title compound. Example 16: Preparation of amorphous mitiglinide calcium
Crystalline mitiglinide calcium (2.Og) was dissolved in dichloromethane (30 ml) and filtered to remove undissolved and suspended particles. Approximately 60 % of the solvent was distilled off under vacuum to obtain a stirrable solution. The solution was then cooled to 15-200C and mixed with diisopropyl ether (20 ml). The mixture was stirred for 30 minutes and the resulting solid was filtered, washed with diisopropyl ether and dried under vacuum at 45-600C to obtain 1.70 g of the title compound. Example 17: Preparation of amorphous mitiglinide calcium
Mitiglinide (2.0 g) was dissolved in dichloromethane (20 ml) and aqueous ammonia (3.6 ml, 25 % solution) solution was added to it. The solution was stirred at 25-30 0C and mixed with solid calcium chloride (1.5 g) and the solution warmed at 30-35 0C and stirred for 30 minutes. The solution was washed with water (2 x 10 ml) and the clear solution was dried over sodium sulfate, and filtered. Approximately 60% of the solvent was distilled off under vacuum and the resulting viscous oil was cooled to 10-15 0C and mixed with diisopropyl ether (50 ml). The reaction mixture was stirred for 30-35 minutes and the resulting solid was filtered and dried at 40-600C to obtain 1.75 g of the title compound. Example 18: Conversion of amorphous mitiglinide calcium into crystalline mitiglinide calcium A suspension of amorphous mitiglinide calcium in diisopropyl ether (30 ml) was stirred for 2 hours at 25- 300C, filtered and dried under vacuum at 45-600C to obtain crystalline form of mitiglinide calcium. Example 19: Preparation of crystalline mitiglinide calcium
To a solution of mitiglinide (2.5 g) in water (2.5 ml), aqueous ammonia solution (approx 25%, 4.0 ml) and acetonitrile (2.5 ml) at 10-150C, calcium chloride (1.32 g) dissolved in demineralized water (15 ml) was added. The mixture was stirred for 2 hours. The resulting precipitate was filtered, slurry washed with water (3 x 25 ml) and acetone (2 x 5 ml) and dried at 45-500C under vacuum to obtain 2.12 g of title compound having purity: 99.72 % by HPLC.
Example 20: Preparation of crystalline mitiglinide calcium
To a solution of mitiglinide (2.5 g) in water (2.5 ml), aqueous ammonia solution (approx 25%, 4.0 ml) and tetrahydrofuran (2.5 ml) at 10-150C, calcium chloride (1.32 g) dissolved in demineralized water (15 ml) was added. The mixture was stirred for 2 hours. The resulting precipitate was filtered, slurry washed with water (3 x 25 ml) and acetone (2 x 5 ml) and dried at 45-500C under vacuum to obtain 1.95 g of title compound having purity: 99.52 % by HPLC.
Example 21; Preparation of crystalline mitiglinide calcium
To a solution of mitiglinide (30.0 g) in water (300 ml), aqueous ammonia solution (approx 25%, 48 ml) and acetone (300 ml) at 10-150C, calcium chloride (15.8 g) dissolved in demineralized water (180 ml) was added. The mixture was stirred for 2 hours. The resulting precipitate was filtered, slurry washed with water (3 x 300 ml) and acetone (2 x 60 ml) and dried at 45-500C under vacuum to obtain 24.32 g of title compound having purity: 99.42 % by HPLC.
Example 22: Preparation of crystalline mitiglinide calcium
To a solution of mitiglinide (3.0 g) in water (30 ml), aqueous ammonia solution (approx 25%, 4.8 ml) and isopropyl alcohol (300 ml) at 10-150C, calcium chloride (1.58 g) dissolved in demineralized water
(18 ml) was added. The mixture was stirred for 2 hours. The resulting precipitate was filtered, slurry washed with water (3 x 30 ml) and acetone (2 x 6 ml) and dried at 45-500C under vacuum to obtain 1.92 g of title compound having purity: 99.65 % by HPLC.
Example 23: Preparation of (2S)-2-benzyWV-((lR)-l-benzyl-2-hydroxy-ethyl)-4-(hexahvdro- isoindolin-2-yl)-4-oxo-buryramide
To a solution of (2S)-2-benzyl-l-((4R)-4-benzyl-2-oxo-oxazolidin-3-yl)-4-(hexahydro-isoindolin-2-yl)- butane-l,4-dione (20.0 g) in tetrahydrofuran (140 ml), a solution of lithium hydroxide monohydrate
(3.43 g,) in demineralized water (40 ml) was added and the reaction mixture was refluxed for 4 hours till the completion of the reactions (monitored by thin layer chromatography). After the completion of the reaction, the reaction mixture was poured into demineralized water (100 ml) and extracted with ethyl acetate (2 x 80 ml). The combined organic layer was washed with water (80 ml) and dried over sodium sulphate. The solvent was evaporated under reduced pressure to give residue which was stirred in isopropyl alcohol at 0-5 0C for 5 hours. The mixture was filtered and then dried at 40-45 0C under vacuum to obtain 12.48 g of title compound having purity 99.77 % by HPLC. Melting point = 77 – 800C.
PATENT
https://www.google.com/patents/CN102101838A?cl=en
Mitiglinide calcium (mitiglinide calcium), the chemical name (2S) _2_ benzyl _3_ (cis – hexahydro _2_ isoindolinyl-carbonyl) propionate dihydrate by Japanese pharmaceutical company developed Kissei ATP-dependent potassium channel blockers, 2004 for the first time in Japan for the treatment of type II diabetes.
Mitiglinide calcium is the second repaglinide, nateglinide after the first three columns MAG urea drugs, is a derivative of phenylalanine, which acts like mechanism sulfonylurea, but faster onset and the short half-life, is conducive to reducing postprandial blood glucose in diabetic patients, but also to avoid low blood sugar caused by continuous glucose, with “in vitro pancreas” reputation.
In recent years, synthetic methods as described in patent application number: Patent 200510200127 9, the synthesis process first synthesized racemic (±) 2_-benzyl-3- (cis – hexahydro iso-indole-2. carbonyl) propionic acid, and then split to give (2S) -2- benzyl-3- (cis – hexahydro isoindole-2-carbonyl) propionic acid, not a lot of waste material along _ hexahydro isoindole, and Chiral separation is not high.
DISCLOSURE
The technical problem to be solved by the present invention is to provide a material savings along _ hexahydro isoindole, and preparation of a high degree of chiral separation.
To solve the above technical problem, the technical solution of the present invention is employed as a method for preparing mitiglinide calcium, comprising the steps of:
Step 1 Synthesis, benzylidene succinic acid
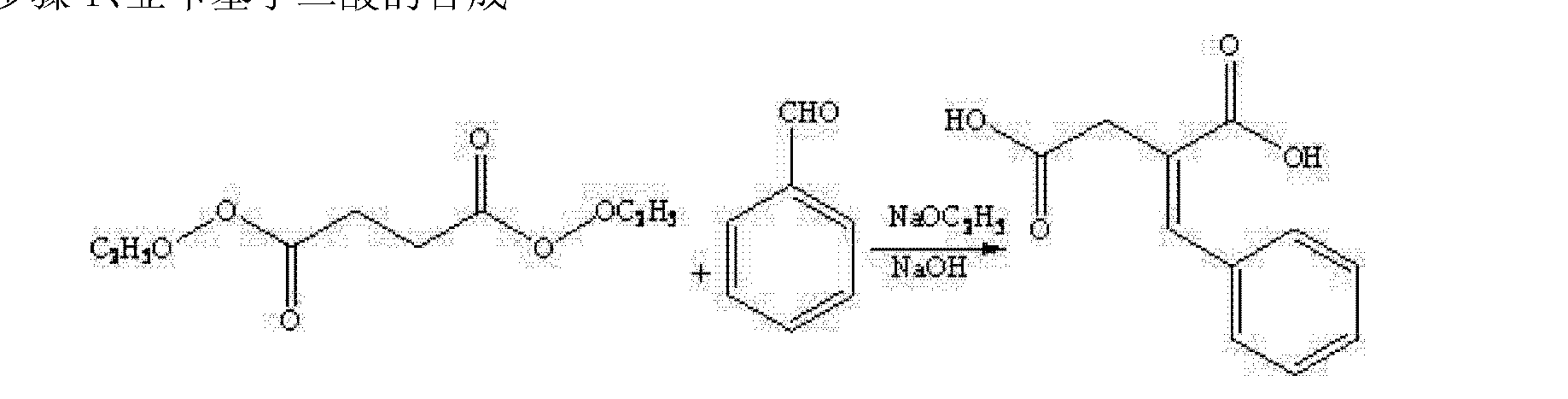
With stirring, was added sodium metal in absolute ethanol, under an inert gas, the solution was heated to reflux with stirring, reflux for 45 fly 0 minutes, under reflux before the dropwise addition of benzaldehyde, and then added dropwise diethyl succinate esters, reaction stirring was continued for 2 to 3 hours, slowly reducing the LC-Ms detection, the ratio of formaldehyde starting material benzene, cooled to room temperature, after use 5 (T55wt% aqueous solution of NaOH to adjust the PH San 13.0, and then heated at reflux;. Γ4 hours, cooled to at room temperature, keeping the reaction solution temperature <25 ° C, pH adjusted with concentrated hydrochloric San 2.0, filtration, recrystallization cold tetrahydrofuran, wherein the molar ratio of sodium metal with benzaldehyde and diethyl succinate is: 0.3 … ~ 0 5: 1 2~1 5: 1; Step 2 synthesis, benzyl butyl acid

The benzylidene succinic acid into the reactor, 10% Pd / C and ethanol, evacuated, and then replaced with hydrogen three times, introducing hydrogen, atmospheric hydrogenation reaction 12~15 hours, the reaction solution suction After the filtrate was evaporated to dryness under reduced pressure, the resulting solid was recrystallized from ethyl acetate, wherein the mass ratio of benzylidene succinic acid with 10% Pd / C is 1: 0 0 15 ^ 20.
3 Synthesis [0006] step, (S) -2- acid, benzyl butyl

Benzyl succinic acid dissolved in methanol was added dropwise with stirring (R) – a chiral amine, stirred at room temperature 2 hours wide, and the precipitated solid was filtered and the solid dispersed in water, under stirring 6 mol / mL hydrochloric acid adjusted ρΗ = 1 (Γ2.0, stirred for 30 minutes, the solid by suction filtration, dried, and wherein the benzyl succinic acid (R) – chiral amine molar ratio of 1: 0~2 5 2;…
Said (R) – a chiral amine (R) -I- phenylethylamine, (R) -I- naphthylethylamine or (R) -I- phenyl-2-p-amine;
4 (S) synthesis step, -2-benzyl succinic anhydride

Reactor, has added (S) -2- benzyl succinic acid and acetic anhydride, at 7 5,0 ° C reaction 1 to 2 hours, isopropyl ether low temperature crystallization after cooling, heavy with ethyl acetate crystallization, wherein (S) -2- molar ratio of benzyl succinic acid and acetic anhydride: 1: 7 · 0 to 7 · 5;
Step 5, (2S) -2- benzyl-3- (cis – hexahydro isoindole-2-carbonyl) propionic acid Synthesis

Stirring, S- benzyl succinic anhydride is dissolved in dichloromethane, control the internal temperature <0 V, a solution of cis _ hexahydro isoindole, dropping it, keeping the internal temperature at <0! : Continue stirring for 2 to 3 hours, the reaction in 2 (T25 ° C 10~15 hours, concentrated to give a pale yellow viscous material, wherein the (S) -2- benzyl succinic anhydride and cis – hexahydro isoindole molar ratio of 1: 2 (Γ2 5; step 6, mitiglinide calcium synthesis.

To the reactor was added (2S) -2- benzyl-3- (cis – hexahydro isoindole-2-carbonyl) propionic acid, water and concentrated ammonia, stir until completely dissolved, a solution of anhydrous calcium chloride aqueous solution, gradually precipitated white solid was added dropwise and then stirred at room temperature 12~ after 15 hours, suction filtered, the filter cake washed with water, dried to give a white solid, i.e. Mitiglinide calcium crude, obtained crude product with methanol and water (volume Than 0.5 4~0 5: 1) and recrystallized as a white solid Mitiglinide calcium;
Beneficial effects: The invention provides a method for preparing calcium Mitiglinide not only saves raw material cis – hexahydro isoindole, and chiral separation is high.
Embodiment 1
Step 1 Synthesis, benzylidene succinic acid
Under stirring, sodium metal (1.7 g, 0. 072 mol) was added absolute ethanol (50 mL), and under argon, the solution was heated to reflux with stirring, reflux for 50 minutes under reflux before the dropwise addition of benzene Formaldehyde (23 mL, 0. 183 mol), and then added dropwise diethyl succinate (50 mL, 0. 275 mol), stirring was continued for 2.5 hours the reaction, reducing the slow LC-Ms detection, the ratio of formaldehyde starting material benzene , was cooled to room temperature, with 55wt.% aqueous NaOH solution adjusting pH ≥ 13.0, and then heated at reflux for 3 hours, cooled to room temperature, the reaction solution temperature maintained <25 ° C, with concentrated hydrochloric pH≤2.0, leaching, cryogenic tetrahydrofuran recrystallization, yield = 81.3%;
Step 2 synthesis, benzyl butyl acid
The benzylidene succinic acid (23. 7 g, 0. 114 mol) into the reactor, then add 10% Pd / C (4. 7g) and anhydrous ethanol (300 mL), evacuated, then Hydrogen replacement three times, introducing hydrogen, hydrogenated at atmospheric pressure for 14 hours, the reaction solution after filtration, evaporated to dryness under reduced pressure, the resulting solid was recrystallized from ethyl acetate, yield: 98% 9; step 3, (S). -2-butyric acid benzyl
Benzyl succinic acid (31. 2 g, 0. 156 mol) was dissolved in methanol (500 mL), and added dropwise with stirring (R) -I- phenylethylamine (41.2 g, 0. 343 mol), room temperature stirred for 1.5 hours, the precipitated solid was filtered, the solid dispersion to water (100 mL) and stirred at with 6 mol / mL hydrochloric acid adjusted ρΗ = 1. (Γ2. 0, stirred for 30 minutes, the solid was suction filtered, and dried Yield 87. 3%; 4 (S) synthesis step, -2-benzyl succinic anhydride
Reactor, has added (S) -2- benzylbutyl acid (27. 8 g, 0. 132 mol) and acetic anhydride (88 mL, 0. 964 mol), at 7 5,0 ° C for 1 hours, cooled and added to isopropyl ether (150 mL) low temperature crystallization, after recrystallization from ethyl acetate, yield: 73% 9;.
Under – (hexahydro-isoindole-2-carbonyl cis) acid synthesis stirring S- benzyl succinic anhydride (12. 7 g, 0 Step 5, (2S) -2- benzyl-3. 067 mol) was dissolved in dichloromethane (250 mL), to control the internal temperature <0 ° C, a solution of cis – hexahydro isoindole (18. 5 g, 0 154 mol), the addition was complete, maintaining the internal temperature in <0! : Continue stirring for 2.5 hours, the reaction in 2 (T25 ° C 12 hours, concentrated to give a pale yellow viscous material, yield: 83 1%; Step 6 Synthesis Mitiglinide calcium.
To the reactor was added (2S) -2- benzyl-3- (cis – hexahydro isoindole-2-carbonyl) propionic acid (. 28. 7 g, 0 091 mol), water (150 mL), and concentrated aqueous ammonia (12 mL), stirring until completely dissolved, a solution of anhydrous calcium chloride (12. 1 g, 0.109 mol) water (100 mL) solution was gradually precipitated white solid was added dropwise at room temperature and then stirred for 13 End hours, filtration, washing the filter cake, and drying to give a white solid, crude Mitiglinide calcium, derived from crude methanol and water (volume ratio 0.5 4~0 5: 1) and recrystallized as a white solid MIG Chennai column calcium, yield: 87.3%.
Second Embodiment
Example A similar experimental method steps 1 through 6 was carried out except in step 3, using (R) -1- naphthyl-amine (61. 4 g, 0. 359 mol) substituted (R) -I- phenylethylamine Other operating homogeneous reaction similar to this step of the synthesis yield: 87.3%.
Third Embodiment
Example A similar experimental method steps 1 through 6 was carried out except in step 3, using (R) -I- phenyl-2-p-tolyl-ethylamine (90. 2 g, 0. 374 mol) substituent (R ) -I- phenethylamine, other homogeneous reaction procedure similar to the synthesis yield of this step:. 83 4% ο
References
- Malaisse WJ (October 2008). “Mitiglinide: a rapid- and short-acting non-sulfonylurea insulinotropic agent for the treatment of type 2 diabetic patients”. Expert Opin Pharmacother. 9 (15): 2691–8. doi:10.1517/14656566.9.15.2691. PMID 18803455.
External links
- Elixir Pharmaceuticals – website of the U.S. rights holder for mitiglinide.
| Cited Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| EP0507534A1 * | Mar 30, 1992 | Oct 7, 1992 | Kissei Pharmaceutical Co., Ltd. | Succinic acid compounds |
| EP0967204A1 * | Jan 22, 1998 | Dec 29, 1999 | Kissei Pharmaceutical Co Ltd | Process for producing benzylsuccinic acid derivatives |
| US6133454 * | Jul 1, 1998 | Oct 17, 2000 | Adir Et Compagnie | Method for preparing a substituted perhydroisoindole |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| CN102898348A * | Sep 8, 2012 | Jan 30, 2013 | 迪沙药业集团有限公司 | Preparation method for Mitiglinide calcium |
| CN102898348B * | Sep 8, 2012 | Sep 2, 2015 | 迪沙药业集团有限公司 | 一种米格列奈钙的制备方法 |
| CN103450069A * | Jun 24, 2013 | Dec 18, 2013 | 山西大同大学 | Preparation method of mitiglinide calcium |
| CN103724253A * | Dec 11, 2013 | Apr 16, 2014 | 苑振亭 | Preparation method for Mitiglinide calcium hydrate |
| CN103724253B * | Dec 11, 2013 | Jun 15, 2016 | 苑振亭 | 一种米格列奈钙的制备方法 |
| CN102659562A * | May 9, 2012 | Sep 12, 2012 | 山东铂源药业有限公司 | Synthesis method of mitiglinide calcium intermediate |
| CN102898348A * | Sep 8, 2012 | Jan 30, 2013 | 迪沙药业集团有限公司 | Preparation method for Mitiglinide calcium |
| CN102898348B * | Sep 8, 2012 | Sep 2, 2015 | 迪沙药业集团有限公司 | 一种米格列奈钙的制备方法 |
| CN103450069A * | Jun 24, 2013 | Dec 18, 2013 | 山西大同大学 | Preparation method of mitiglinide calcium |
| CN103709092A * | Nov 4, 2013 | Apr 9, 2014 | 河北科技大学 | High purity mitiglinide calcium preparation method |
| CN103709092B * | Nov 4, 2013 | Jul 6, 2016 | 河北科技大学 | 米格列奈钙的制备方法 |
| CN104311471A * | Sep 23, 2014 | Jan 28, 2015 | 山东省药学科学院 | Improved mitiglinide calcium industrialized preparation method |
| CN1616427A * | Nov 13, 2003 | May 18, 2005 | 中国科学院上海药物研究所 | New method for preparing medicine mitiglinide for treating diabetes |
| CN101270074A * | Mar 21, 2007 | Sep 24, 2008 | 北京德众万全药物技术开发有限公司 | Method for preparing high purity mitiglinide calcium |
| CN101492411A * | Jan 22, 2008 | Jul 29, 2009 | 北京华禧联合科技发展有限公司 | Improved method for preparation of mitiglinide |
| WO2009047797A2 * | Oct 7, 2008 | Apr 16, 2009 | Ind-Swift Laboratories Limited | Process for the preparation of perhydroisoindole derivative |
| Reference | ||
|---|---|---|
| 1 | * | 张永亮,等: “米格列奈合成方法研究“, 《化工中间体》, no. 1, 31 December 2009 (2009-12-31), pages 16 – 22 |
| Cited Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| CN101492411A * | Jan 22, 2008 | Jul 29, 2009 | 北京华禧联合科技发展有限公司 | Improved method for preparation of mitiglinide |
| WO2005030719A1 * | Sep 24, 2004 | Apr 7, 2005 | Les Laboratoires Servier | Novel method for preparing cis-octahydro-isoindole |
| Reference | ||
|---|---|---|
| 1 | * | WEI HUANG,等: “Novel Convenient Synthesis of Mitiglinide“, 《SYNTHETIC COMMUNICATIONS》, vol. 37, no. 13, 3 July 2007 (2007-07-03), pages 2153 – 2157, XP055079498, DOI: doi:10.1080/00397910701392590 |
| 2 | * | 张永亮,等: “米格列奈合成方法研究“, 《化工中间体》, no. 1, 31 January 2009 (2009-01-31), pages 16 – 22 |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| CN103709092A * | Nov 4, 2013 | Apr 9, 2014 | 河北科技大学 | High purity mitiglinide calcium preparation method |
| CN103709092B * | Nov 4, 2013 | Jul 6, 2016 | 河北科技大学 | 米格列奈钙的制备方法 |
| EP0507534A1 * | Mar 30, 1992 | Oct 7, 1992 | Kissei Pharmaceutical Co., Ltd. | Succinic acid compounds |
| EP0967204A1 * | Jan 22, 1998 | Dec 29, 1999 | Kissei Pharmaceutical Co Ltd | Process for producing benzylsuccinic acid derivatives |
| US6133454 * | Jul 1, 1998 | Oct 17, 2000 | Adir Et Compagnie | Method for preparing a substituted perhydroisoindole |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| CN102898348A * | Sep 8, 2012 | Jan 30, 2013 | 迪沙药业集团有限公司 | Preparation method for Mitiglinide calcium |
| CN102898348B * | Sep 8, 2012 | Sep 2, 2015 | 迪沙药业集团有限公司 | 一种米格列奈钙的制备方法 |
| CN103450069A * | Jun 24, 2013 | Dec 18, 2013 | 山西大同大学 | Preparation method of mitiglinide calcium |
| CN103724253A * | Dec 11, 2013 | Apr 16, 2014 | 苑振亭 | Preparation method for Mitiglinide calcium hydrate |
| CN103724253B * | Dec 11, 2013 | Jun 15, 2016 | 苑振亭 | 一种米格列奈钙的制备方法 |
|
|
| Systematic (IUPAC) name | |
|---|---|
|
(2S)-2-benzyl-4-[(3aR,7aS)-octahydro-2H-isoindol- 2-yl]-4-oxobutanoic acid
|
|
| Clinical data | |
| AHFS/Drugs.com | International Drug Names |
| Routes of administration |
oral |
| Identifiers | |
| CAS Number | 145375-43-5  |
| ATC code | A10BX08 (WHO) |
| PubChem | CID 121891 |
| DrugBank | DB01252  |
| ChemSpider | 108739 |
| UNII | D86I0XLB13 |
| KEGG | D01854 |
| ChEMBL | CHEMBL471498 |
| Chemical data | |
| Formula | C19H25NO3 |
| Molar mass | 315.41 g/mol |
/////////207844-01-7, 145525-41-3, KAD-1229, S-21403, MITIGLINIDE, Glufast, Kissei, 145375-43-5
Quality Control & MSDS
O=C(O)[C@@H](Cc1ccccc1)CC(=O)N3C[C@H]2CCCC[C@H]2C3
Filed under: Uncategorized Tagged: 145375-43-5, 145525-41-3, 207844-01-7, Glufast, KAD-1229, Kissei, MITIGLINIDE, S-21403



























