![Skeletal formula of (1S,4S,7Z,10S,16E,21R)-7-ethylidene-4,21-diisopropyl-2-oxa-12,13-dithia-5,8,20,23-tetrazabicyclo[8.7.6]tricos-16-ene-3,6,9,19,22-pentone](http://upload.wikimedia.org/wikipedia/commons/thumb/7/71/Romidepsin_structure_%282%29.svg/200px-Romidepsin_structure_%282%29.svg.png)
| Romidepsin; Depsipeptide; FK228; Chromadax; FR901228; Istodax; | |
| Molecular Formula: | C24H36N4O6S2 |
|---|---|
| Molecular Weight: | 540.69584 g/mol |
CAS 128517-07-7
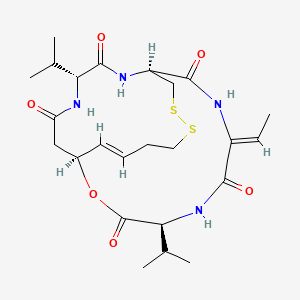
(1S,4S,7Z,10S,16E,21R)-7-ethylidene-4,21-di(propan-2-yl)-2-oxa-12,13-dithia-5,8,20,23-tetrazabicyclo[8.7.6]tricos-16-ene-3,6,9,19,22-pentone
(E)-N-(2-amino-4-fluorophenyl)-4-((3-(pyridin-3-yl)acrylamido)methyl)benzamide
Romidepsin, also known as Istodax, is an anticancer agent used in cutaneous T-cell lymphoma (CTCL) and other peripheral T-cell lymphomas (PTCLs). Romidepsin is a natural product obtained from the bacteria Chromobacterium violaceum, and works by blocking enzymes known as histone deacetylases, thus inducing apoptosis.[1] It is sometimes referred to as depsipeptide, after the class of molecules to which it belongs. Romidepsin is branded and owned by Gloucester Pharmaceuticals, now a part of Celgene.[2]
Romidepsin, a histone deacetylase inhibitor, originally developed by Fujisawa (now Astellas Pharma), causes cell cycle arrest,
differentiation, and apoptosis in various cancer cells.
In 2004, the FDA granted fast-track designation for romidepsin as monotherapy for the treatment of cutaneous T-cell lymphoma (CTCL) in patients who have relapsed following, or become refractory to, other systemic therapies. The FDA designated romidepsin as an orphan drug and it was approved in 2009 for this indication and it was commercialized in 2010. In 2007, another fast-track designation was granted for the product as monotherapy of previously treated peripheral T-cell lymphoma.
Romidepsin (FR901228) was originally discovered and isolated from the fermentation broth of Chromobacterium violaceum No. 968. It was identified through efforts in the search for novel agents which selectively reverse the morphological phenotype of Ras oncogene-transformed cells since the Ras signaling pathway plays a critical role in cancer development. Therefore, the drug could also have multiple molecular targets for its anticancer activity besides HDAC.
FR901228 is a bicyclic depsipeptide which is structurally unrelated to any known class of cyclic peptides with an unusual disulfide bond connecting a thiol and D-cysteine.
This drug is commercially produced by fermentation; however its interesting and novel structure warrants examination of its synthesis within the context of this review
Romidepsin is a histone deacetylase (HDAC) inhibitor.HDACs catalyze the removal of acetyl groups from acetylated lysine residues in histone and non-histone proteins, resulting in the modulation of gene expression.
Romidepsin is indicated for treatment of cutaneous T-cell lymphoma (CTCL) in patients who have received at least
one prior systemic therapy; treatment of peripheral T-cell lymphoma (PTCL) in patients who have received at least
one prior therapy.
Available as an injection, containing 10 mg of romidepsin and recommended dose is 14 mg/m2 administered intravenously over a 4-hour period on days 1, 8, and 15 of a 28-day cycle until disease progression or unacceptable toxicity.

History
Romidepsin was first reported in the scientific literature in 1994, by a team of researchers from Fujisawa Pharmaceutical Company (now Astellas Pharma) in Tsukuba, Japan, who isolated it in a culture of Chromobacterium violaceum from a soil sample obtained inYamagata Prefecture.[3] It was found to have little to no antibacterial activity, but was potently cytotoxic against several human cancercell lines, with no effect on normal cells; studies on mice later found it to have antitumor activity in vivo as well.[3]
The first total synthesis of romidepsin was accomplished by Harvard researchers and published in 1996.[4] Its mechanism of actionwas elucidated in 1998, when researchers from Fujisawa and the University of Tokyo found it to be a histone deacetylase inhibitorwith effects similar to those of trichostatin A.[5]

Clinical trials
Phase I studies of romidepsin, initially codenamed FK228 and FR901228, began in 1997.[6] Phase II and phase III trials were conducted for a variety of indications. The most significant results were found in the treatment of cutaneous T-cell lymphoma (CTCL) and other peripheral T-cell lymphomas (PTCLs).[6]
In 2004, romidepsin received Fast Track designation from the FDA for the treatment of cutaneous T-cell lymphoma, and orphan drugstatus from the FDA and the European Medicines Agency for the same indication.[6] The FDA approved romidepsin for CTCL in November 2009[7] and approved romidepsin for other peripheral T-cell lymphomas (PTCLs) in June 2011.[8]
Mechanism of action
Romidepsin acts as a prodrug with the disulfide bond undergoing reduction within the cell to release a zinc-binding thiol.[3][9][10] The thiol reversibly interacts with a zinc atom in the binding pocket of Zn-dependent histone deacetylase to block its activity. Thus it is anHDAC inhibitor. Many HDAC inhibitors are potential treatments for cancer through the ability to epigenetically restore normal expression of tumor suppressor genes, which may result in cell cycle arrest, differentiation, and apoptosis.[11]

Adverse effects
The use of romidepsin is uniformly associated with adverse effects.[12] In clinical trials, the most common were nausea and vomiting,fatigue, infection, loss of appetite, and blood disorders (including anemia, thrombocytopenia, and leukopenia). It has also been associated with infections, and with metabolic disturbances (such as abnormal electrolyte levels), skin reactions, altered taste perception, and changes in cardiac electrical conduction.[12]
CLIP
http://www.scielo.br/scielo.php?script=sci_arttext&pid=S0103-50532012001200003

CLIP
Romidepsin was first isolated from the fermentation broth of Chromobacterium Violaceum WB968 in a nutrient
medium. Sterilized of 1% glucose and 1% bouillon solution were incubated with Chromobacterium Violaceum WB968, followed by further incubation with 1% glucose solution, 1% bouillon solution and adekanol gave the target romidepsin after extraction, silica gel chromatography and recrystallization.[Okuhara, M.; Goto, T.; Hori, Y. et al. US4977138A, 1990.]



The synthetic route was initiated by the deprotection L-(Fmoc)Thr-L-Val-OMe 1, subsequently coupled with
N-Alloc-S-Trt-D-Cys, followed by tosylation and then elimination to produce tripeptide 3 in the yield of 63.7% over four steps. The N-Alloc deprotection of 3 and then coupling with N-Fmoc-D-Valine were proceeded to provide tetrapeptide 4, which was subsequently removed Fmoc group to afford relative tetrapeptide 5 in 83.0% yield from compound 3. Condensation of 5 with β-hydroxy mercapto acid 6 was carried out by treating with benzotriazol-1-yl-oxy-tris-(dimethylamino)-phosphonium hexafluorphosphate (BOP) to give relative amide 7, and sequential hydrolysis yielded corresponding acid, which was performed by Mitsunobu macrolactonization
to produce depsipeptide 8 in 17.5% yield over three steps. Finally, romidepsin was obtained in the presence of iodine
in 81.0% yield and the overall yield of 7.5%.
The synthesis of intermediate β-hydroxy mercapto acid 6 commenced with the commercially available methyl 3,3-dimethoxypropionate 9. Nucleophilic addition of 9 with N,O-dimethylhydroxylamine provided Weinreb amide 10, followed by addition with lithium acetylide to give propargylic ketone 12 in the yield of 50.2% over two steps. Noyori’s asymmetric hydrogenation of ketone 12 provided (E)-alkene 14, which was removed the silyl group and then substituted with paratoluensulfonyl chloride to yield tosylate 15 in 40.6% yield across three steps. The dimethyl acetal of 15 was hydrolyzed to corresponding aldehyde by using lithium tetrafluoroborate,
which was immediately oxidized to relative carboxylic acid by applying Pinnick oxidation conditions. The trityl mercaptan was introduced by tosylate displacement to provide 6 in 65.0% yield over three steps and the overall yield of 13.3%.[2]
REF Greshock, T. J.; Johns, D. M.; Noguchi, Y., et al. Org. Lett. 2008, 10 (4), 613-616.
CLIP
Romidepsin (Istodax)
Romidepsin, a histone deacetylase inhibitor, originally developed by Fujisawa (now Astellas Pharma), causes cell cycle arrest,
differentiation, and apoptosis in various cancer cells.111 In 2004, the FDA granted fast-track designation for romidepsin as monotherapy for the treatment of cutaneous T-cell lymphoma (CTCL) in patients who have relapsed following, or become refractory
to, other systemic therapies. The FDA designated romidepsin as an orphan drug and it was approved in 2009 for this indication
and it was commercialized in 2010. In 2007, another fast-track designation was granted for the product as monotherapy of
previously treated peripheral T-cell lymphoma. Romidepsin (FR901228) was originally discovered and isolated from the fermentation
broth of Chromobacterium violaceum No. 968. It was identified through efforts in the search for novel agents which
selectively reverse the morphological phenotype of Ras oncogene-transformed cells since the Ras signaling pathway plays a
critical role in cancer development. Therefore, the drug could also have multiple molecular targets for its anticancer activity besides
HDAC.112 FR901228 is a bicyclic depsipeptide which is structurally unrelated to any known class of cyclic peptides with an unusual
disulfide bond connecting a thiol and D-cysteine. This drug is commercially produced by fermentation; however its interesting
and novel structure warrants examination of its synthesis within the context of this review.113,114 The synthesis of romidepsin
described is based on the total synthesis reported by the Williams115 and Simon groups (Scheme 20).116
L-Valine methyl ester (134) was coupled to N-Fmoc-L-threonine in the presence of the BOP reagent in 95% yield. The N-Fmoc protecting group was removed with Et2NH and the corresponding free amine was coupled to N-alloc-(S-triphenylmethyl)-D-cysteine with 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDCI) and HOBT in DMF and CH2Cl2 to yield the tripeptide 135 in good yield. The threonine residue of tripeptide 135 was then subjected to dehydrating conditions to give alkene 136 in 95% yield. The N-alloc protecting group of the dehydrated tripeptide 136 was removed with palladium and tin reagents and the corresponding free amine was subsequently coupled with N-Fmoc-D-valine to give tetrapeptide 137 in 83% yield. After removal of the N-Fmoc protecting group of compound 137 with Et2NH amine 138 was obtained in quantitative yield. The acid coupling partner 145 for
amine 138 was prepared as follows: methyl 3,3-dimethoxypropionate (139) was converted to its corresponding Weinreb amide by standard conditions and reacted with lithium acetylide 140 to give propargylic ketone 141 in 75% yield. Noyori’s asymmetric reduction of ketone 141 using ruthenium catalyst 142 gave the (R)-propargylic alcohol in 98% ee. This was followed by Red-Al reduction of the alkyne to selectively yield (E)-alkene 143 in 58% yield for the two steps. Liberation of the primary alcohol
with tetrabutylammonium fluoride (TBAF) followed by selective tosylation gave 144 in 70% yield in two steps. Hydrolysis of the dimethyl acetal of 144 with LiBF4 was followed by a Pinnick oxidation to give the corresponding carboxylic acid. The tosylate was displaced with trityl mercaptan in the presence of tert-butyl alcohol to give allylic alcohol 145 in 65% yield for the three steps.
Aminoamide 138 was then coupled to acid 145 using BOP to give peptide 146 in quantitative yield. The methyl ester of compound 146 was hydrolyzed with lithium hydroxide to provide the free carboxylic acid which underwent macrolactonization under Mitsunobu conditions in the presence of diisopropyl azodicarboxylate (DIAD) and triphenylphosine to give macrocycle 147 in 24% yield.
Finally, the disulfide linkage was formed by treating bis-tritylsulfane 147 with iodine in methanol at room temperature to give romidepsin (XIII) in 81% yield.


111 Bertino, E. M.; Otterson, G. A. Expert Opin. Invest. Drugs 2011, 20, 1151.
112. Furumai, R.; Matsuyama, A.; Kobashi, N.; Lee, K.-H.; Nishiyama, M.; Nakajima,
H.; Tanaka, A.; Komatsu, Y.; Nishino, N.; Yoshida, M.; Horinouchi, S. Cancer
Res. 2002, 62, 4916.
113. Verdine, G. L.; Vrolijk, N. H.; Bertel, S. WO 2008083288 A2, 2008.
114. Verdine, G. L.; Vrolijk, N. H. WO 2008083290 A1, 2008.
115. Greshock, T. J.; Johns, D. M.; Noguchi, Y.; Williams, R. M. Org. Lett. 2008, 10,
613.
116. Li, K. W.; Wu, J.; Xing, W.; Simon, J. A. J. Am. Chem. Soc. 1996, 118, 7237.
CLIP
http://pubs.rsc.org/en/content/articlelanding/2009/np/b817886k#!divAbstract

Williams’ improved synthesis of FK228.


Williams’ synthesis of the FK228 amide isostere (74).
References
- Jump up^ “Romidepsin”. National Cancer Institute. Retrieved2009-09-11.
- Jump up^ “Romidepsin”. Gloucester Pharmaceuticals. Retrieved2009-09-11.
- ^ Jump up to:a b c Ueda H, Nakajima H, Hori Y, et al. (March 1994). “FR901228, a novel antitumor bicyclic depsipeptide produced byChromobacterium violaceum No. 968. I. Taxonomy, fermentation, isolation, physico-chemical and biological properties, and antitumor activity”. Journal of Antibiotics. 47 (3): 301–10.doi:10.7164/antibiotics.47.301. PMID 7513682.
- Jump up^ Li KW, Wu J, Xing W, Simon JA (July 1996). “Total synthesis of the antitumor depsipeptide FR-901,228”. Journal of the American Chemical Society. 118 (30): 7237–8. doi:10.1021/ja9613724.
- Jump up^ Nakajima H, Kim YB, Terano H, Yoshida M, Horinouchi S (May 1998). “FR901228, a potent antitumor antibiotic, is a novel histone deacetylase inhibitor”. Experimental Cell Research. 241(1): 126–33. doi:10.1006/excr.1998.4027. PMID 9633520.
- ^ Jump up to:a b c Masuoka Y, Shindoh N, Inamura N (2008). “Histone deacetylase inhibitors from microorganisms: the Astellas experience”. In Petersen F, Amstutz R. Natural compounds as drugs. 2. Basel: Birkhäuser. pp. 335–59. ISBN 978-3-7643-8594-1. Retrieved on November 8, 2009 through Google Book Search.
- Jump up^ http://chembl.blogspot.com/2009/11/new-drug-approvals-pt-xxiii-romidepsin.html
- Jump up^http://www.accessdata.fda.gov/scripts/cder/drugsatfda/index.cfm?fuseaction=Reports.MonthlyApprovalsAll
- Jump up^ Shigematsu, N.; Ueda, H.; Takase, S.; Tanaka, H.; Yamamoto, K.; Tada, T. (1994). “FR901228, a novel antitumor bicyclic depsipeptide produced by Chromobacterium violaceum No. 968. II. Structure determination.”. J. Antibiot. 47 (3): 311–314.doi:10.7164/antibiotics.47.311. PMID 8175483.
- Jump up^ Ueda, H.; Manda, T.; Matsumoto, S.; Mukumoto, S.; Nishigaki, F.; Kawamura, I.; Shimomura, K. (1994). “FR901228, a novel antitumor bicyclic depsipeptide produced by Chromobacterium violaceum No. 968. III. Antitumor activities on experimental tumors in mice.”. J. Antibiot. 47 (3): 315–323.doi:10.7164/antibiotics.47.315. PMID 8175484.
- Jump up^ Greshock, Thomas J.; Johns, Deidre M.; Noguchi, Yasuo; Williams, Robert M. (2008). “Improved Total Synthesis of the Potent HDAC Inhibitor FK228 (FR-901228)”. Organic Letters.10 (4): 613–616. doi:10.1021/ol702957z. PMC 3097137
 .PMID 18205373.
.PMID 18205373. - ^ Jump up to:a b [No authors listed] (October 2014). “ISTODEX Label Information (updated to October 2014)” (PDF). U.S. Food and Drug Administration.
 .
.External links
|
|
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
(1S,4S,7Z,10S,16E,21R)-7-ethylidene-4,21-diisopropyl-2-oxa-12,13-dithia-5,8,20,23-tetrazabicyclo[8.7.6]tricos-16-ene-3,6,9,19,22-pentone
|
|
| Clinical data | |
| Trade names | Istodax |
| MedlinePlus | a610005 |
| License data |
|
| Pregnancy category |
|
| Routes of administration |
Intravenous infusion |
| Legal status | |
| Legal status |
|
| Pharmacokinetic data | |
| Bioavailability | Not applicable (IV only) |
| Protein binding | 92–94% |
| Metabolism | Hepatic (mostly CYP3A4-mediated) |
| Biological half-life | 3 hours |
| Identifiers | |
| CAS Number | 128517-07-7  |
| ATC code | none |
| PubChem | CID 5352062 |
| IUPHAR/BPS | 7006 |
| UNII | CX3T89XQBK |
| ChEBI | CHEBI:61080 |
| ChEMBL | CHEMBL1213490 |
| Synonyms | FK228; FR901228; Istodax |
| Chemical data | |
| Formula | C24H36N4O6S2 |
| Molar mass | 540.695 g/mol |
//////////fast-track designation, Romidepsin, Depsipeptide, FK228, Chromadax, FR901228, Istodax, FDA 2009, Fujisawa, Astellas Pharma, 128517-07-7
CC=C1C(=O)NC(C(=O)OC2CC(=O)NC(C(=O)NC(CSSCCC=C2)C(=O)N1)C(C)C)C(C)C
Filed under: Uncategorized Tagged: 128517-07-7, ASTELLAS PHARMA, Chromadax, Depsipeptide, Fast Track Designation, FDA 2009, FK228, FR901228, Fujisawa, Istodax, Romidepsin







