
Daclatasvir

Status: Launched 2014 (EU, Japan)
Originator: Bristol-Myers Squibb


Daclatasvir (USAN[1]) (formerly BMS-790052, trade name Daklinza) is a drug for the treatment of hepatitis C (HCV). It is was developed by Bristol-Myers Squibb and was approved in Europe on 22 August 2014.
Daclatasvir inhibits the HCV nonstructural protein NS5A.[2][3] Recent research suggests that it targets two steps of the viral replication process, enabling rapid decline of HCV RNA.[4]
Daclatasvir has been tested in combination regimens with pegylated interferon and ribavirin,[5] as well as with other direct-acting antiviral agents including asunaprevir[6][7][8][9] and sofosbuvir.[10][11]
It is on the World Health Organization’s List of Essential Medicines, a list of the most important medications needed in a basic health system.[12]

EUROPEAN MEDICINES AGENCY ADVISES ON COMPASSIONATE USE OF DACLATASVIR
- The first compassionate-use opinion for a hepatitis C treatment was adopted by the CHMP in October 2013.
- Sofosbuvir, which is part of this compassionate-use opinion, received a positive opinion from the CHMP recommending granting of a marketing authorisation at its November 2013 meeting.
- Daclatasvir is developed by Bristol-Myers Squibb and sofosbuvir is developed by Gilead.
|
1-6-2012
|
Anti-Viral Compounds
|
|
|
2-13-2009
|
CRYSTALLINE FORM OF METHYL ((1S)-1-(((2S)
-2-(5-(4′-(2-((2S)-1((2S)-2-((METHOXYCARBONYL)AMINO)-3-METHYLBUTANOYL)-2-PYRROLIDINYL) -1H-IMIDAZOL-5-YL)-4-BIPHENYLYL)-1H-IMIDAZOL-2-YL)-1-PYRROLIDINYL)CARBONYL) -2-METHYLPROPYL)CARBAMATE DIHYDROCHLORIDE SALT |
Synthesis
EXAMPLES

A 1 L, 3-neck round bottom flask, fitted with a nitrogen line, overhead stirrer and thermocouple, was charged with 20 g (83.9 mmol, 1 equiv) 1,1′-(biphenyl-4,4′-diyl)diethanone, 200 mL CH2Cl2 and 8.7 mL (27.1 g, 169.3 mmol, 2.02 quiv) bromine. The mixture was allowed to stir under nitrogen for about 20 hours under ambient conditions. The resulting slurry was charged with 200 mL CH2Cl2 and concentrated down to about 150 mL via vacuum distillation. The slurry was then solvent exchanged into THF to a target volume of 200 mL via vacuum distillation. The slurry was cooled to 20-25° C. over 1 hour and allowed to stir at 20-25° C. for an additional hour. The off-white crystalline solids were filtered and washed with 150 mL CH2Cl2. The product was dried under vacuum at 60° C. to yield 27.4 g (69.2 mmol, 82%) of the desired product : 1H NMR (400 MHz, CDCl3) δ 7.95-7.85 (m, 4H), 7.60-7.50 (m, 4H), 4.26 (s, 4H); 13C NMR (100 MHz, CDCl3) 6 191.0, 145.1, 133.8, 129.9, 127.9, 30.8; IR (KBr, cm−1) 3007, 2950, 1691, 1599, 1199; Anal calcd for C16H12Br2O2: C, 48.52; H, 3.05; Br, 40.34. Found: C, 48.53; H, 3.03; Br, 40.53 HRMS calcd for C16H13Br2O2 (M+H; DCI+): 394.9282. Found: 394.9292. mp 224-226° C.

A 500 mL jacketed flask, fitted with a nitrogen line, thermocouple and overhead stirrer, was charged with 20 g (50.5 mmol, 1 equiv) of Compound 2, 22.8 g (105.9 moles, 2.10 equiv) 1-(tert-butoxycarbonyl)-L-proline and 200 mL acetonitrile. The slurry was cooled to 20° C. followed by the addition of 18.2 mL (13.5 g, 104.4 mmol, 2.07 equiv) DIPEA. The slurry was warmed to 25° C. and allowed to stir for 3 hours. The resulting clear, organic solution was washed with 3×100 mL 13 wt % aqueous NaCl. The rich acetonitrile solution was solvent exchanged into toluene (target volume=215 mL) by vacuum distillation until there was less than 0.5 vol % acetonitrile.

The toluene solution of Compound 3 was charged with 78 g (1.011 moles, 20 equiv) ammonium acetate and heated to 95-100° C. The mixture was allowed to stir at 95-100° C. for 15 hours. After reaction completion, the mixture was cooled to 70-80° C. and charged with 7 mL acetic acid, 40 mL n-butanol, and 80 mL of 5 vol % aqueous acetic acid. The resulting biphasic solution was split while maintaining a temperature >50° C. The rich organic phase was charged with 80 mL of 5 vol % aqueous acetic acid, 30 mL acetic acid and 20 mL n-butanol while maintaining a temperature >50° C. The resulting biphasic solution was split while maintaining a temperature >50° C. and the rich organic phase was washed with an additional 80 mL of 5 vol % aqueous acetic acid. The rich organic phase was then solvent exchanged into toluene to a target volume of 215 mL by vacuum distillation. While maintaining a temperature >60° C., 64 mL methanol was charged. The resulting slurry was heated to 70-75° C. and aged for 1 hour. The slurry was cooled to 20-25° C. over 1 hour and aged at that temperature for an additional hour. The slurry was filtered and the cake was washed with 200 mL 10:3 toluene:methanol. The product was dried under vacuum at 70° C., resulting in 19.8 g (31.7 mmol, 63%) of the desired product: 1H NMR (400 MHz, DMSO-d6) δ 13.00-11.00 (s, 2H), 7.90-7.75 (m, 4H), 7.75-7.60 (m, 4H), 7.60-7.30 (s, 2H), 4.92-4.72 (m, 2H), 3.65-3.49 (m, 2H), 3.49-3.28 (m, 2H), 2.39-2.1 (m, 2H), 2.10-1.87 (m, 6H), 1.60-1.33 (s, 8H), 1.33-1.07 (s, 10H); 13C NMR (100 MHz, DMSO-d6) δ 154.1, 153.8, 137.5, 126.6, 125.0, 78.9, 78.5, 55.6, 55.0, 47.0, 46.7, 33.7, 32.2, 28.5, 28.2, 24.2, 23.5; IR (KBr, cm−1) 2975, 2876, 1663, 1407, 1156, 1125; HRMS calcd for C36H45N6O4 (M+H; ESI+): 625.3502. Found: 625.3502. mp 190-195° C. (decomposed).

To a 250 mL reactor equipped with a nitrogen line and overhead stirrer, 25.0 g of Compound 4 (40.01 mmol, 1 equiv) was charged followed by 250 mL methanol and 32.85 mL (400.1 mmol, 10 equiv) 6M aqueous HCl. The temperature was increased to 50° C. and agitated at 50° C. for 5 hours. The resulting slurry was cooled to 20-25° C. and held with agitation for about 18 hours. Filtration of the slurry afforded a solid which was washed successively with 100 mL 90% methanol/water (V/V) and 2×100 mL of methanol. The wet cake was dried in a vacuum oven at 50° C. overnight to give 18.12 g (31.8 mmol, 79.4%) of the desired product.
Recrystallization of Compound 5
To a 250 mL reactor equipped with a nitrogen line and an overhead stirrer, 17.8 g of Compound 5 from above was charged followed by 72 mL methanol. The resulting slurry was agitated at 50° C. for 4 hours, cooled to 20-25° C. and held with agitation at 20-25° C. for 1 hour. Filtration of the slurry afforded a crystalline solid which was washed with 60 mL methanol. The resulting wet cake was dried in a vacuum oven at 50° C. for 4 days to yield 14.7 g (25.7 mmol, 82.6%) of the purified product: 1H NMR (400 MHz, DMSO-d6) δ 10.5-10.25 (br, 2H), 10.1-9.75 (br, 2H), 8.19 (s, 2H), 7.05 (d, J=8.4, 4H), 7.92 (d, J=8.5, 4H), 5.06 (m, 2H), 3.5-3.35 (m, 4H), 2.6-2.3 (m, 4H), 2.25-2.15 (m, 2H), 2.18-1.96 (m, 2H); 13C NMR (100 MHz, DMSO-d6) δ 156.6, 142.5, 139.3, 128.1, 127.5, 126.1, 116.9, 53.2, 45.8, 29.8, 24.3; IR (KBr, cm−1) 3429, 2627, 1636, 1567, 1493, 1428, 1028. Anal calcd for C26H32N6Cl4: C, 54.75; H, 5.65; Cl, 24.86; Adjusted for 1.9% water: C, 53.71; H, 5.76; N, 14.46; Cl, 24.39. Found: C, 53.74; H, 5.72; N, 14.50; Cl, 24.49; KF=1.9. mp 240° C. (decomposed).

A 1 L jacketed flask equipped with a nitrogen line and an overhead stirrer was sequentially charged with 100 mL acetonitrile, 13.69 g (89.4 mmol, 2.5 equiv) hydroxybenzotriazole hydrate, 15.07 g (86 mmol, 2.4 equiv) N-(methoxycarbonyl)-L-valine, 16.46 g (85.9 mmol, 2.4 equiv) 1-(3-dimethyaminopropyl)-3-ethylcarbodiimide hydrochloride and an additional 100 mL acetonitrile. The resulting solution was agitated at 20° C. for 1 hour and charged with 20.4 g (35.8 mmol, 1 equiv) of purified Compound 5. The slurry was cooled to about 0° C. and 18.47 g (142.9 mmol, 4 equiv) diisopropylethylamine was added over 30 minutes while maintaining a temperature below 10° C. The solution was slowly heated to 15° C. over 3 hours and held at 15° C. for 12 hours. The resulting solution was charged with 120 mL 13 wt % aqueous NaCl and heated to 50° C. for 1 hour. After cooling to 20° C., 100 mL of isopropyl acetate was added. The biphasic solution was filtered through a 0.45 μm filter and the mixture split. The rich organic phase was washed with 2×240 mL of a 0.5 N NaOH solution containing 13 wt % NaCl followed by 120 mL 13 wt % aqueous NaCl. The mixture was then solvent exchanged into isopropyl acetate by vacuum distillation with a target volume of 400 mL. The resulting hazy solution was cooled to 20° C. and filtered through a 0.45 μm filter. The clear solution was then solvent exchanged into ethanol by vacuum distillation with a target volume of 140 mL. While maintaining a temperature of 50° C., 66.4 mL (82.3 mmol, 2.3 equiv) of 1.24M HCl in ethanol was added. The mixture was then charged with 33 mg (0.04 mmol, 0.001 equiv) of seed crystals of Compound (I) (see preparation below) and the resulting slurry was stirred at 50° C. for 3 hours. The mixture was cooled to 20° C. over 1 hour and aged at that temperature for an additional 22 hours. The slurry was filtered and the wet cake was washed with 100 mL of 2:1 acetone:ethanol. The solids were dried in a vacuum oven at 70° C. to give 22.15 g (27.3 mmol, 76.3%) of the desired product.

A solution of Compound (I) was prepared by dissolving 3.17 g of Compound (I) from above in 22 mL methanol. The solution was passed through a 47 mm Cuno Zeta Carbon® 53SP filter at ˜5 psig at a flow rate of ˜58 mL/min. The carbon filter was rinsed with 32 mL of methanol. The solution was concentrated down to 16 mL by vacuum distillation. While maintaining a temperature of 40-50° C., 15.9 mL acetone and 5 mg of seed crystals of Compound (I) (see procedure below) were added. The resulting slurry was then charged with 32 mL acetone over 30 minutes. The slurry was held at 50° C. for 2 hours, cooled to 20° C. over about 1 hour and held at 20° C. for about 20 hours. The solids were filtered, washed with 16 mL 2:1 acetone:methanol and dried in a vacuum oven at 60° C. to give 2.14 g (67.5%) of purified Compound (I):
1H NMR (400 MHz, DMSO-d6, 80° C.): 8.02 (d, J=8.34 Hz, 4 H), 7.97 (s, 2 H), 7.86 (d, J=8.34 Hz, 4 H), 6.75 (s, 2 H), 5.27 (t, J=6.44 Hz, 2 H), 4.17 (t, J=6.95 Hz, 2 H), 3.97-4.11 (m, 2 H), 3.74-3.90 (m, 2 H), 3.57 (s, 6 H), 2.32-2.46 (m, 2 H), 2.09-2.31 (m, 6 H), 1.91-2.07 (m, 2 H), 0.88 (d, J=6.57 Hz, 6 H), 0.79 (d, J=6.32 Hz, 6 H);
13C NMR (75 MHz, DMSO-d6): δ 170.9, 156.9, 149.3, 139.1, 131.7, 127.1, 126.5, 125.9, 115.0, 57.9, 52.8, 51.5, 47.2, 31.1, 28.9, 24.9, 19.6, 17.7;
IR (neat, cm−1): 3385, 2971, 2873, 2669, 1731, 1650.
Anal. Calcd for C40H52N8O6Cl2: C, 59.18; H, 6.45; N, 13.80; Cl, 8.73. Found C, 59.98; H, 6.80; N, 13.68; Cl, 8.77. mp 267° C. (decomposed).
Preparation of Seed Crystals of Compound (I)
A 250 mL round-bottom flask was charged with 6.0 g (10.5 mmol, 1 equiv) Compound 5, 3.87 g (22.1 mmol, 2.1 equiv) N-(methoxycarbonyl)-L-valine, 4.45 g (23.2 mmol, 2.2 equiv) 1-(3-dimethyaminopropyl)-3-ethylcarbodiimide hydrochloride, 0.289 g (2.14 mmol, 0.2 equiv) 1-hydroxybenzotriazole, and 30 mL acetonitrile. The resulting slurry was then charged with 7.33 mL (42.03 mmol, 4 equiv) diisopropylethylamine and allowed to stir at 24-30° C. for about 18 hours. The mixture was charged with 6 mL of water and heated to 50° C. for about 5 hours. The mixture was cooled and charged with 32 mL ethyl acetate and 30 mL water. The layers were separated and the rich organic layer was washed with 30 mL of 10 wt % aqueous NaHCO3, 30 mL water, and 20 mL of 10 wt % aqueous NaCl. The rich organic layer was then dried over MgSO4, filtered, and concentrated down to a residue. The crude material was then purified via flash chromatography (silica gel, 0-10% methanol in dichloromethane) to provide the free base of Compound (I).
The free-base of Compound (I) (0.03 g) was dissolved in 1 mL isopropanol at 20° C. Anhydrous HCl (70 μL, dissolved in ethanol, approximately 1.25M concentration) was added and the reaction mixture was stirred. To the solution was added methyl tert-butyl ether (1 mL) and the resulting slurry was stirred vigorously at 40° C. to 50° C. for 12 hours. The crystal slurry was cooled to 20° C. and filtered. The wet cake was air-dried at 20° C. A white crystalline solid (Form N-2 of Compound (I)) was obtained.
…………………
Daclatasvir synthesis: WO2009020828A1

Procedure:
Step a: A 1 L, 3 -neck round bottom flask, fitted with a nitrogen line, overhead stirrer and thermocouple, was charged with 20 g (83.9 mmol, 1 equiv) 1,1′-(biphenyl-4,4′-diyl)diethanone, 200 mL Dichloromethane and 8.7 mL (27.1g, 169.3 mmol, 2.02 equiv) bromine. The mixture was allowed to stir under nitrogen for about 20 hours under ambient conditions. The resulting slurry was charged with 200 mL Dichloromethane and concentrated down to about 150 mL via vacuum distillation. The slurry was then solvent exchanged into THF to a target volume of 200 mL via vacuum distillation. The slurry was cooled to 20-25 0C over 1 hour and allowed to stir at 20-25 0C for an additional hour. The off-white crystalline solids were filtered and washed with 150 mL Dichloromethane. The product was dried under vacuum at 60 0C to yield 27.4 g (69.2 mmol, 82%) of the desired product: 1H NMR (400 MHz, CDCl3) d 7.95-7.85 (m, 4H), 7.60-7.50 (m, 4H), 4.26 (s, 4H); 13C NMR 100 MHz, CDCl3) d 191.0, 145.1, 133.8, 129.9, 127.9, 30.8; IR (KBr, cm-1) 3007, 2950, 1691, 1599, 1199; Anal calcd for C16H12Br2O2: C, 48.52; H, 3.05; Br, 40.34. Found: C, 48.53; H, 3.03; Br, 40.53. HRMS calcd for C16H12Br2O2 (M + H; DCI+): 394.9282. Found: 394.9292. mp 224-226 0C.
Step b: A 500 mL jacketed flask, fitted with a nitrogen line, thermocouple and overhead stirrer, was charged with 20 g (50.5 mmol, 1 equiv) of Compound 2, 22.8 g (105.9 moles, 2.10 equiv) 1-(tert-butoxycarbonyl)-L-proline and 200 mL acetonitrile. The slurry was cooled to 20 0C followed by the addition of 18.2 mL (13.5 g, 104.4 mmol, 2.07 equiv) DIPEA. The slurry was warmed to 25 0C and allowed to stir for 3 hours. The resulting clear, organic solution was washed with 3 x 100 mL 13 wt% aqueous NaCl. The rich acetonitrile solution was solvent exchanged into toluene (target volume = 215 mL) by vacuum distillation until there was less than 0.5 vol% acetonitrile.
Step c: The toluene solution of Compound 3 was charged with 78 g (1.011 moles, 20 equiv) ammonium acetate and heated to 95-100 0C. The mixture was allowed to stir at 95-100 0C for 15 hours. After reaction completion, the mixture was cooled to 70- 80 0C and charged with 7 mL acetic acid, 40 mL n-butanol, and 80 mL of 5 vol% aqueous acetic acid. The resulting biphasic solution was split while maintaining a temperature > 50 0C. The rich organic phase was charged with 80 mL of 5 vol% aqueous acetic acid, 30 mL acetic acid and 20 mL n-butanol while maintaining a temperature > 50 0C. The resulting biphasic solution was split while maintaining a temperature > 50 0C and the rich organic phase was washed with an additional 80 mL of 5 vol% aqueous acetic acid. The rich organic phase was then solvent exchanged into toluene to a target volume of 215 mL by vacuum distillation. While maintaining a temperature > 60 0C, 64 mL methanol was charged. The resulting slurry was heated to 70-75 0C and aged for 1 hour. The slurry was cooled to 20-25 0C over 1 hour and aged at that temperature for an additional hour. The slurry was filtered and the cake was washed with 200 mL 10:3 toluene:methanol. The product was dried under vacuum at 70 0C, resulting in 19.8 g (31.7 mmol, 63%) of the desired product: 1H NMR (400 MHz, DMSO-^) d 13.00-11.00 (s, 2H), 7.90-7.75 (m, 4H), 7.75-7.60 (m, 4H), 7.60-7.30 (s, 2H), 4.92-4.72 (m, 2H), 3.65-3.49 (m, 2H), 3.49-3.28 (m, 2H), 2.39-2.1 (m, 2H), 2.10-1.87 (m, 6H), 1.60-1.33 (s, 8H), 1.33-1.07 (s, 10H); 13C NMR (100 MHz, DMSO-?fe) d 154.1, 153.8, 137.5, 126.6, 125.0, 78.9, 78.5, 55.6, 55.0, 47.0, 46.7, 33.7, 32.2, 28.5, 28.2, 24.2, 23.5; IR (KBr, cm-1) 2975, 2876, 1663, 1407, 1156, 1125; HRMS calcd for C36H45N6O4 (M + H; ESI+): 625.3502. Found: 625.3502. mp 190-195 0C (decomposed).
Step d: To a 250 mL reactor equipped with a nitrogen line and overhead stirrer, 25.0 g of Compound 4 (40.01 mmol, 1 equiv) was charged followed by 250 mL methanol and 32.85 mL (400.1 mmol, 10 equiv) 6M aqueous HCl. The temperature was increased to 50 0C and agitated at 50 0C for 5 hours. The resulting slurry was cooled to 20-25 0C and held with agitation for about 18 hours. Filtration of the slurry afforded a solid which was washed successively with 100 mL 90% methanoI/water (WV) and 2 x 100 mL of methanol. The wet cake was dried in a vacuum oven at 50 0C overnight to give 18.12 g (31.8 mmol, 79.4%) of the desired product.
CUT PASTE…….WO2009020825

Preparation of Compound (I)
A 1 L jacketed flask equipped with a nitrogen line and an overhead stirrer was sequentially charged with 100 mL acetonitrile, 13.69 g (89.4 mmol, 2.5 equiv) hydroxybenzotriazole hydrate, 15.07 g (86 mmol, 2.4 equiv) N-(methoxycarbonyl)- L-valine, 16.46 g (85.9 mmol, 2.4 equiv) l-(3-dimethyaminopropyl)-3- ethylcarbodiimide hydrochloride and an additional 100 mL acetonitrile. The resulting solution was agitated at 20 0C for 1 hour and charged with 20.4 g (35.8 mmol, 1 equiv) of purified Compound 7. The slurry was cooled to about 0 0C and 18.47 g (142.9 mmol, 4 equiv) diisopropylethylamine was added over 30 minutes while maintaining a temperature below 10 0C. The solution was slowly heated to 15 0C over 3 hours and held at 15 0C for 12 hours. The resulting solution was charged with 120 mL 13 wt% aqueous NaCl and heated to 50 0C for 1 hour. After cooling to 20 0C, 100 mL of isopropyl acetate was added. The biphasic solution was filtered through a 0.45 μm filter and the mixture split. The rich organic phase was washed with 2 x 240 mL of a 0.5 Ν NaOH solution containing 13 wt% NaCl followed by 120 mL 13 wt% aqueous NaCl. The mixture was then solvent exchanged into isopropyl acetate by vacuum distillation with a target volume of 400 mL. The resulting hazy solution was cooled to 20 0C and filtered through a 0.45 μm filter. The clear solution was then solvent exchanged into ethanol by vacuum distillation with a target volume of 140 mL. While maintaining a temperature of 50 0C, 66.4 mL (82.3 mmol, 2.3 equiv) of 1.24M HCl in ethanol was added. The mixture was then charged with 33 mg (0.04 mmol, 0.001 equiv) of seed crystals of Compound (I) (see preparation below) and the resulting slurry was stirred at 50 0C for 3 hours. The mixture was cooled to 20 0C over 1 hour and aged at that temperature for an additional 22 hours. The slurry was filtered and the wet cake was washed with 100 mL of 2: 1 acetone:ethanol. The solids were dried in a vacuum oven at 70 0C to give 22.15 g (27.3 mmol, 76.3%) of the desired product.

Alternative Preparation of Compound (I)
A jacketed reactor equipped with a mechanical agitator, a thermocouple and a nitrogen inlet was sequentially charged with 10 L acetonitrile, 0.671 kg (4.38 moles, 2.50 equiv) 1-hydroxybenzotriazole, 0.737 kg (4.21 moles, 2.40 equiv) N- (methoxycarbonyl)-L-valine and 0.790 kg (4.12 moles, 2.35 equiv) l-(3- dimethyaminopropyl)-3-ethylcarbodiimide hydrochloride. The mixture was agitated at 200C for 1 hour, cooled to 5 0C and charged with 1 kg (1.75 moles, 1.00 equiv) Compound 7. While maintaining a temperature < 10 0C, 0.906 kg (7.01 moles, 4 equiv) diisopropylethylamine was added. The mixture was heated to 15-20 0C over 2 hours and agitated for an additional 15 hours. After the reaction was complete, the mixture was washed once with 6.0 L 13 wt% aqueous NaCl, twice with 6.1 L (6.12 moles, 3.5 equiv) 1.0 M aqueous NaOH containing 13 wt% NaCl and once with 6.0 L 13 wt% aqueous NaCl. Water was then removed from the rich organic solution via azeotropic distillation. The mixture was cooled to 20 0C, agitated for 1 hour and filtered. The rich organic solution was then solvent exchanged into EtOH via vacuum distillation to a target volume of 5 L. While maintaining a temperature of 50 0C, 3.2 L (4.0 moles, 2.3 equiv) 1.25M HCl in EtOH was charged. The mixture was seeded with 1.6 g Compound (I) (see preparation below) and agitated at 50 0C for 3 hours. The resulting slurry was cooled to 20 0C and agitated for at least 3 hours. The product was collected by filtration and washed with 5 L 2: 1 acetone:
EtOH to give 1.29 kg (ca. 90 wt% product) of wet crude product. A reactor equipped with an overhead agitator, nitrogen inlet and thermocouple was charged with 1.11 kg of the above crude product and 7 L methanol. The resulting solution was treated with Cuno Zeta Carbon (TM) 55SP. The carbon was washed with 15 L MeOH and the combined filtrate and wash was concentrated down to 4 L via vacuum distillation. The concentrated solution was charged with 5 L acetone and seeded with 1.6 g Compound (I) (see preparation below) while maintaining a temperature of 50 0C. An additional 10 L acetone was charged and the resulting slurry was stirred at 50 0C for 3 hours. The slurry was cooled to 20 0C and allowed to agitate at 200C for 3 hours. The product was collected by filtration, washed with 5 L 2: 1 acetone: EtOH and dried under vacuum at 50-60 0C to give 0.900 kg (1.11 moles, 74% adjusted) of Compound (I)-

Carbon Treatment and Recrystallization of Compound (I) A solution of Compound (I) was prepared by dissolving 3.17 g of Compound (I) from above in 22 mL methanol. The solution was passed through a 47mm Cuno Zeta Carbon 53SP filter at ~5 psig at a flow rate of~58mL/min. The carbon filter was rinsed with 32 mL of methanol. The solution was concentrated down to 16 mL by vacuum distillation. While maintaining a temperature of 40-50 0C, 15.9 mL acetone and 5 mg of seed crystals of Compound (I) (see procedure below) were added. The resulting slurry was then charged with 32 mL acetone over 30 minutes. The slurry was held at 50 0C for 2 hours, cooled to 20 0C over about 1 hour and held at 20 0C for about 20 hours. The solids were filtered, washed with 16 mL 2: 1 acetone:methanol and dried in a vacuum oven at 60 0C to give 2.14 g (67.5%) of purified Compound (I):
1H NMR (400 MHz, DMSO-έfc, 80 0C): 8.02 (d, J=8.34 Hz, 4 H), 7.97 (s, 2 H), 7.86 (d, J=8.34 Hz, 4 H), 6.75 (s, 2 H), 5.27 (t, J=6.44 Hz, 2 H), 4.17 (t, J=6.95 Hz, 2 H), 3.97 – 4.11 (m, 2 H), 3.74 – 3.90 (m, 2 H), 3.57 (s, 6 H), 2.32 – 2.46 (m, 2 H), 2.09 – 2.31 (m, 6 H), 1.91 – 2.07 (m, 2 H), 0.88 (d, J=6.57 Hz, 6 H), 0.79 (d, J=6.32 Hz, 6 H);
13C NMR (75 MHz, DMSO-έfc): δ 170.9, 156.9, 149.3, 139.1, 131.7, 127.1, 126.5, 125.9, 115.0, 57.9, 52.8, 51.5, 47.2, 31.1, 28.9, 24.9, 19.6, 17.7;
IR (neat, cm“1): 3385, 2971, 2873, 2669, 1731, 1650.
Anal. Calcd for C40H52N8O6Cl2: C, 59.18; H, 6.45; N, 13.80; Cl, 8.73. Found C, 59.98; H, 6.80; N, 13.68; Cl, 8.77. mp 267 0C (decomposed).
Characteristic diffraction peak positions (degrees 2Θ + 0.1) @ RT, based on a high quality pattern collected with a diffractometer (CuKa) with a spinning capillary with 2Θ calibrated with a NIST other suitable standard are as follows: 10.3, 12.4, 12.8, 13.3, 13.6, 15.5, 20.3, 21.2, 22.4, 22.7, 23.7
Daclatasvir faces problems in USA
The US-FDA in 2014 issued a complete response letter for NS5A inhibitor daclatasvir saying it was unable to approve the drug because the marketing application was for its use in tandem with asunaprevir, an NS3/NS4A protease inhibitor discontinued in the US by BMS for commercial reasons. Daclatasvir is already on the market in Europe-where it is sold as Daklinza-and also in Japan where it was approved alongside asunaprevir in July as the country’s first all-oral HCV therapy. However, a delay in the large US market is clearly a major setback for BMS’ ambitions in hepatitis therapy.
To make the matter worse, US FDA has rescinded breakthrough therapy designation status from Bristol-Myers Squibb for Daclatasvir for the treatment of hepatitis C virus infection in Feb 2015.
………………..
PAPER
Makonen, B.; et. al. Hepatitis C Virus NS5A Replication Complex Inhibitors: The Discovery of Daclatasvir. J Med Chem 2014, 57(5), 2013–2032.
http://pubs.acs.org/doi/abs/10.1021/jm401836p
……………………..
PATENT
http://www.google.com/patents/WO2008021927A2?cl=en
Example 24-23

methyl ((lS)-l-(((2S)-2-(5-(4′-(2-((2S)-l-((2S)-2-((methoxycarbonyl)amino)-3- methylbutanoyl)-2-pyrrolidinyl)-lH-imidazol-5-yl)-4-biphenylyl)-lH-imidazol-2-yl)-
1 -pyrrolidinyl) carbonyl) -2-methylpropyl) carbamate
A 50 mL flask equipped with a stir bar was sequentially charged with 2.5 mL acetonitrile, 0.344 g (2.25 mmol, 2.5 equiv) hydroxy benzotriazole hydrate, 0.374 g (2.13 mmol, 2.4 equiv) N-(methoxycarbonyl)-L-valine, 0.400 g (2.09 mmol, 2.4 equiv) 1 -(3 -dimethyaminopropyl)-3-ethylcarbodiimide hydrochloride and an additional 2.5 mL acetonitrile. The resulting solution was agitated at 20 0C for 1 hour and charged with 0.501 g (0.88 mmol, 1 equiv) Example A-le-4. The slurry was cooled to about 0 0C and 0.45 g (3.48 mmol, 4 equiv) diisopropylethylamine was added over 30 minutes while maintaining a temperature below 10 0C. The solution was slowly heated to 15 0C over 3 hours and held at 15 0C for 16 hours. The temperature was increased to 20 0C and stirred for 3.25 hours. The resulting solution was charged with 3.3 g of 13 wt% aqueous NaCl and heated to 50 0C for 1 hour. After cooling to 20 0C, 2.5 mL of isopropyl acetate was added. The rich organic phase was washed with 2 x 6.9 g of a 0.5 N NaOH solution containing 13 wt% NaCl followed by 3.3 g of 13 wt% aqueous NaCl. The mixture was then solvent exchanged into isopropyl acetate by vacuum distillation to a target volume of 10 mL. The resulting hazy solution was cooled to 20 0C and filtered through a 0.45 μm filter. The clear solution was then solvent exchanged into ethanol by vacuum distillation with a target volume of 3 mL. 1.67 mL (2.02 mmol, 2.3 equiv) of 1.21 M HCl in ethanol was added. The mixture was then stirred at 25 0C for 15 hours. The resulting slurry was filtered and the wet cake was washed with 2.5 mL of 2: 1 acetone:ethanol. The solids were dried in a vacuum oven at 50 0C to give 0.550 g (0.68 mmol, 77 %) of the desired product.
RecrystalHzation of Example 24-23
A solution of Example 24-23 prepared above was prepared by dissolving 0.520 g of the above product in 3.65 mL methanol. The solution was then charged with 0.078 g of type 3 Cuno Zeta loose carbon and allowed to stir for 0.25 hours. The mixture was then filtered and washed with 6 ml of methanol. The product rich solution was concentrated down to 2.6 mL by vacuum distillation. 7.8 mL acetone was added and allowed to stir at 25 0C for 15 h. The solids were filtered, washed with 2.5 mL 2: 1 acetone:ethanol and dried in a vacuum oven at 70 0C to give 0.406 g (57.0%) of the desired product as white crystals: 1H NMR (400 MHz, OMSO-d6, 80 0C): 8.02 (d, J=8.34 Hz, 4 H), 7.97 (s, 2 H), 7.86 (d, J=8.34 Hz, 4 H), 6.75 (s, 2 H), 5.27 (t, J=6.44 Hz, 2 H), 4.17 (t, J=6.95 Hz, 2 H), 3.97 – 4.11 (m, 2 H), 3.74 – 3.90 (m, 2 H), 3.57 (s, 6 H), 2.32 – 2.46 (m, 2 H), 2.09 – 2.31 (m, 6 H), 1.91 – 2.07 (m, 2 H), 0.88 (d, J=6.57 Hz, 6 H), 0.79 (d, J=6.32 Hz, 6 H); 13C NMR (75 MHz, DMSO- d6): δ 170.9, 156.9, 149.3, 139.1, 131.7, 127.1, 126.5, 125.9, 115.0, 57.9, 52.8, 51.5, 47.2, 31.1, 28.9, 24.9, 19.6, 17.7; IR (neat, cm“1): 3385, 2971, 2873, 2669, 1731, 1650. Anal. Calcd for C40H52N8O6Cl2: C, 59.18; H, 6.45; N, 13.80; Cl, 8.73. Found C, 59.98; H, 6.80; N, 13.68; Cl, 8.77. mp 267 0C (decomposed). Characteristic diffraction peak positions (degrees 2Θ ± 0.1) @ RT, based on a high quality pattern collected with a diffractometer (CuKa) with a spinning capillary with 2Θ calibrated with a NIST other suitable standard are as follows: 10.3, 12.4, 12.8, 13.3, 13.6, 15.5, 20.3, 21.2, 22.4, 22.7, 23.7
………………
Bioorganic & Medicinal Chemistry Letters (2015), 25(16), 3147-3150
http://www.sciencedirect.com/science/article/pii/S0960894X15005995

Synthetic route for the preparation of the target compounds 8a–8y. Reagents and conditions: (a) Br2, CH2Cl2, rt, overnight, 86%; (b) N-Boc-l-proline, MeCN, Et3N, rt, 2 h, 98%; (c) NH4OAc, toulene, 130 °C, 15 h, 85%; (d) 6 N HCl, MeOH, 50 °C, 4 h, 87%; (e) HATU, N-(methoxycarbonyl)-l-valine, DIPEA, rt, 14 h, 83%; (f) RCOCl, TEA, CH2Cl2, rt, 3 h, 64–87%.
Dimethyl((2S,2’S)-((2S,2’S)-2,2′-(5,5′-([1,1′-biphenyl]-4,4′-diyl)bis(1H-imidazole-
5,2-diyl))bis(pyrrolidine-2,1-diyl))bis(3-methyl-1-oxobutane-2,1-
diyl))dicarbamate 7……………FREE BASE
To a solution of 5 (90 mg, 0.181 mmol), N-me-thoxycarbonyl-l-valine 6 (92 mg,0.525 mmol) and DIPEA (0.18 mL, 1.03 mmol) in DMF (5 mL) was added HATU(165.5 mg, 0.434 mmol). The resulting reaction was allowed to stir at room temperature for 15 h, the reaction mixture was filtered and the residue was partitioned between EtOAc and H2O, The aqueous phase was extracted with EtOAc, and the combined organic phase was dried (MgSO4), filtered, and concentrated in vacuo. The residue was purified by flash chromatography (silica gel; 5% Methanol /CH2Cl2) to
afford 7 (0.11 g, 83 %)as white solid.
1H NMR (DMSO-d6, 500 MHz) δ: 11.56 (s, 2H), 7.69-7.48 (m, 8H), 7.26-7.03 (m, 4H), 5.24-5.05 (m, 2H), 4.09-4.04 (m, 2H), 3.85-3.75 (m, 4H), 3.58 (s, 6H), 2.24-1.98 (m, 10H), 0.87 (d, J = 3.6 Hz, 12H).
Anal. calcd. (%) for C40H50N8O6: C 65.02, H 6.82, N 15.17; found: C 65.20, H 6.79, N 15.31.
ESI-MS m/z: 739.5 (M+H)+.
……………..
1H NMR PREDICT



………………….
13C NMR PREDICT



COSY PREDICT

Patents












Click on images to view






Click on images to view
 |
|
| Names | |
|---|---|
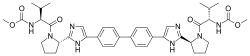
| IUPAC name
Methyl [(2S)-1-{(2S)-2-[4-(4’-{2-[(2S)-1-{(2S)-2-[(methoxycarbonyl)amino]-3-methylbutanoyl}-2-pyrrolidinyl]-1H-imidazol-4-yl}-4-biphenylyl)-1H-imidazol-2-yl]-1-pyrrolidinyl}-3-methyl-1-oxo-2-butanyl]carbamate
|
|
| Other names
BMS-790052
|
|
| Identifiers | |
1009119-64-5  |
|
| ATC code | J05AX14 |
| ChEBI | CHEBI:82977 |
| ChEMBL | ChEMBL2023898 ChEMBL2303621 |
| ChemSpider | 24609522 |
| Jmol-3D images | Image |
| Properties | |
| C40H50N8O6 | |
| Molar mass | 738.89 g·mol−1 |
References
- 1 Statement on a Nonproprietary Name Adopted by the USAN Council
- 2 Gao, Min; Nettles, Richard E.; Belema, Makonen; Snyder, Lawrence B.; Nguyen, Van N.; Fridell, Robert A.; Serrano-Wu, Michael H.; Langley, David R.; Sun, Jin-Hua; O’Boyle, Donald R., II; Lemm, Julie A.; Wang, Chunfu; Knipe, Jay O.; Chien, Caly; Colonno, Richard J.; Grasela, Dennis M.; Meanwell, Nicholas A.; Hamann, Lawrence G. (2010). “Chemical genetics strategy identifies an HCV NS5A inhibitor with a potent clinical effect”. Nature 465 (7294): 96–100. doi:10.1038/nature08960. PMID 20410884.
- 3 Bell, Thomas W. (2010). “Drugs for hepatitis C: unlocking a new mechanism of action”. ChemMedChem 5 (10): 1663–1665. doi:10.1002/cmdc.201000334. PMID 20821796.
- 4 Modeling shows that the NS5A inhibitor daclatasvir has two modes of action and yields a shorter estimate of the hepatitis C virus half-life. Guedj, J et al. Proceedings of the National Academy of Sciences. February 19, 2013.
- 5 AASLD: Daclatasvir with Pegylated Interferon/Ribavirin Produces High Rates of HCV Suppression. Highleyman, L. HIVandHepatitis.com. 6 December 2011.
- 6Preliminary Study of Two Antiviral Agents for Hepatitis C Genotype 1. Lok, A et al. New England Journal of Medicine. 366(3):216-224. January 19, 2012.
- 7“Bristol-Myers’ Daclatasvir, Asunaprevir Cured 77%: Study”. Bloomberg. Apr 19, 2012.
- 8AASLD: Daclatasvir plus Asunaprevir Rapidly Suppresses HCV in Prior Null Responders. Highleyman, L. HIVandHepatitis.com. 8 November 2011.
- 9High rate of response to BMS HCV drugs in harder-to-treat patients – but interferon-free prospects differ by sub-genotype. Alcorn, K. Aidsmap.com. 12 November 2012.
- 10AASLD 2012: Sofosbuvir + Daclatasvir Dual Regimen Cures Most Patients with HCV Genotypes 1, 2, or 3. Highleyman, L. HIVandHepatitis.com. 15 November 2012.
- 11Mark Sulkowski et al. (January 16, 2014). “Daclatasvir plus Sofosbuvir for Previously Treated or Untreated Chronic HCV Infection”. New England Journal of Medicine. doi:10.1056/NEJMoa1306218.
- 12“www.who.int” (PDF).
| WO2004005264A2 * | 7 Jul 2003 | 15 Jan 2004 | Axxima Pharmaceuticals Ag | Imidazole compounds for the treatment of hepatitis c virus infections |
| WO2008021927A2 * | 9 Aug 2007 | 21 Feb 2008 | Squibb Bristol Myers Co | Hepatitis c virus inhibitors |
| WO2008021928A2 * | 9 Aug 2007 | 21 Feb 2008 | Squibb Bristol Myers Co | Hepatitis c virus inhibitors |
| WO2008021936A2 * | 9 Aug 2007 | 21 Feb 2008 | Squibb Bristol Myers Co | Hepatitis c virus inhibitors |
//////////
Filed under: FDA 2015, Uncategorized Tagged: BMS 790052, Bristol-Myers Squibb, daclatasvir, Даклатасвир, 达拉他韦, FDA 2015, Hepatitis C Virus, Treatment of hepatitis C, داكلاتاسفير







