
Indacaterol
QAB-149
CAS 753498-25-8 MALEATE
CAS 312753-06-3 (free base)
QAB-149 maleate
QAB-149-AFA
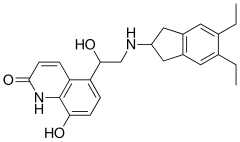
5-[2-(5,6-Diethylindan-2-ylamino)-1(R)-hydroxyethyl]-8-hydroxyquinolin-2(1H)-one maleate
R)-5-[2-[(5, 6-Diethyl-2, 3-dihydro-lH- inden-2-yl) amino]- 1 -hydroxy ethyl]-8-hydroxyquinolin-2(lH)-one, is an ultra long acting beta-adrenoceptor agonist developed by Novartis
Indacaterol (C 24 H 28 N 2 O 3 , M r = 392.49 g / mol) is chiral and is in the drug as R enantiomer and indacaterol ago. It is a derivative of 8-hydroxyquinoline and 2-aminoindan and has a certain structural similarity with other beta2-agonists , for example salbutamol . Indacaterol is lipophilic, which is a prerequisite for its long duration of action.

Indacaterol (INN) is an ultra-long-acting beta-adrenoceptor agonist[1] developed by Novartis. It was approved by the European Medicines Agency (EMA) under the trade name Onbrez Breezhaler on November 30, 2009,[2] and by the United States Food and Drug Administration (FDA), under the trade name Arcapta Neohaler, on July 1, 2011.[3] It needs to be taken only once a day,[4]unlike the related drugs formoterol and salmeterol. It is licensed only for the treatment of chronic obstructive pulmonary disease(COPD) (long-term data in patients with asthma are thus far lacking). It is delivered as an aerosol formulation through a dry powder inhaler.
Indacaterol maleate (QAB-149) is a long-acting inhaled beta2-adrenoceptor agonist. In 2008, it was filed for approval in the U.S. and the E.U. by Novartis for the treatment of chronic obstructive pulmonary disease (COPD).
In 2009, approval was granted by the EMEA and a complete response letter was assigned by the FDA.
In 2010, Novartis resubmitted an NDA seeking approval for the long-term maintenance bronchodilator treatment of airflow obstruction in adult patients with COPD, including bronchitis and/or emphysema.
In 2011, the FDA approved this indication and in 2012 the product was launched in the U.S.
The product was approved and launched in Japan in 2011 for the treatment of COPD.
In 2010, indacaterol was first launched by Novartis in Denmark and Ireland.
Clinical trials
A Phase III trial published in March 2010 examined the efficacy and safety of indacaterol in COPD patients.[5] This study, conducted in the U.S., New Zealand, and Belgium, compared indacaterol dry-powder inhaler to placebo in 416 COPD patients, mostly moderate to severe (mean FEV1 of 1.5 L). Indacaterol produced statistically improved FEV1 (both trough and AUC) and decreased use of rescue medication compared to placebo, but with safety and tolerability similar to those of placebo.
A year-long, placebo-controlled trial published in July 2010 suggests indacaterol may be significantly more effective than twice-daily formoterol in improving FEV1. There were some reductions in the need for rescue medication, but these were not significantly different; nor was there any difference in the rate of exacerbation between the 2 active treatments.[6]
A study published in October, 2011 in the European Respiratory Journal compared indacaterol with tiotropium over the study period of 12 weeks. The study found no statistical difference between the effects of the two drugs on FEV1. Indacaterol yielded greater improvements in transition dyspnoea index (TDI) total score and St. George’s Respiratory Questionnaire (SGRQ) total score.[7]
A recent Cochrane Library meta-analysis indicates that the clinical benefit in lung function from indacaterol is at least as good as that seen with twice-daily long-acting beta2-agonists. [8]
SYNTHESIS

Its synthesis is divided into two parts, a primary amine and a chiral epoxide.
Primary amine starting at 1,2 – diethyl benzene (JMC2010, 3676), two FC reaction into the ring post and then converted into oxime reduction, get four . Compound 5 obtained by Fries rearrangement 6 , phenolic hydroxyl group protected, chlorinated 7 , CBS asymmetric reduction to give the chiral secondary alcohols 8 , ring closure under alkaline conditions to obtain an epoxy compound 9 , a primary amine 4 on epoxy, to the benzyl protecting, salt to be Indacaterol Maleate.

PATENT
http://www.google.com/patents/WO2013132514A2?cl=en
Indacaterol chemically known as (R)-5-[2-[(5, 6-Diethyl-2, 3-dihydro-lH- inden-2-yl) amino]- 1 -hydroxy ethyl]-8-hydroxyquinolin-2(lH)-one, is an ultra long acting beta-adrenoceptor agonist developed by Novartis and has the following structural formula:

Indacaterol maleate is a long acting inhaled β2- agonist. Indacaterol maleate is marketed under the trade name Arcapta Neohaler in US and Onbrez in Europe.
Indacaterol maleate was disclosed in US6878721 by Novartis. The process for Indacaterol is depicted below.
Indacaterol Maleate
VII
In the above process for preparing Indacaterol maleate involves the step of reacting 8 substituted oxy-5-(R)-oxiranyl-(lH)-quinolin-2-one (III) with 2-amino- (5,6-diethyl)-indan (IV) to form a intermediate 5-[(R)-2-(5,6-diethyl-indan-2- ylamino)-l-hydroxy-ethyl]-8-substituted oxy-(lH)-quinolin-2-one (V). This epoxide ring opening is not region specific thereby along with 5-[(R)-2-(5,6- diethyl-indan-2-ylamino)- 1 -hydroxy-ethyl]-8-substituted oxy-( 1 H)-quinol intone, below mentioned products are being produced as impurities.

The above reaction mixture contains only about 60% of desired compound i.e. 5-[(R)-2-(5, 6-diethyl-indan-2-ylamino)-l-hydroxy-ethyl]-8-substituted oxy- (lH)-quinolin-2-one. The purification of this intermediate is done using silica gel chromatography which is tedious and requires large amounts of solvents, not suitable for industrial synthesis.
To overcome the above draw backs of the process for preparing Indacaterol, the patent US7534890 discloses a process that avoids the column purification by the formation of acid addition salts of intermediate (formula – IV).
Therefore, there exists a need to develop a novel process for the preparation of indacaterol maleate.
Examples
Example -1 Preparation of compound of IIIA, wherein R is Benzyl

The compound of formula IA (25 gm) was dissolved in DMSO (75 ml) and stirred for 15 min, then compound of formula IIA (0.09 mol) was added to the reaction mixture at 25 – 30°C. The triethylamine (0. 1 mol) was added to above contents slowly, following by added sodium iodide (0.03 mol) at same temperature and stirred the reaction mixture for 3 hours at same temperature. The purified water (250 ml) was added to the reaction mixture and stirred for 1.0 hour. The contents were filtered and washed with water. The wet material was dissolved in methanol (250 ml) and stirred for 30 minutes, and then water was added. The contents were stirred for lhour at 25 – 30°C and filtered to obtain the title compound. Yield: 76%
Example -2 Preparation of compound of IIIA, wherein R is Benzyl

The compound of formula IA (25 gm) was dissolved in DMSO (75 ml) and stirred for 15 min, then compound of formula IIA (0.09 mol) was added to the reaction mixture at 25 – 30°C. Potassium carbonate (0. 1 mol) was added to above contents slowly, following by added sodium iodide (0.03 mol) at same temperature and stirred the reaction mixture for 3 hours at same temperature. The purified water (250 ml) was added to the reaction mixture and stirred for 1.0 hour. The contents were filtered and washed with water. The wet material was dissolved in methanol (250 ml) and stirred for 30 minutes, and then water was added. The contents were stirred for lhour at 25 – 30 °C and filtered to obtain the title compound. Yield: 82%
Exam le -3 Preparation of compound of IIIA, wherein R is Benzyl

The compound of formula IA (25 gm) was dissolved in DMSO (75 ml) and stirred for 15 min, then compound of formula IIA (0.09 mol) was added to the reaction mixture at 25 – 30°C, then Sodium iodide (0.03 mol) was added to the reaction mixture at same temperature and stirred the reaction mixture for 3 hours at same temperature. The purified water (250 ml) was added to the reaction mixture and stirred for 1.0 hour. The contents were filtered and washed with water. The wet material was dissolved in methanol (250 ml) and stirred for 30 minutes, and then water was added. The contents were stirred for lho‘ur at 25 – 30 °C and filtered to obtain the title compound. Yield: 84%
Exam le -4 Preparation of compound of IVA, wherein R is Benzyl

The Borane-dimethyl sulfide (0.11 mol) was added at 0-5°C, followed by addition of R – (2)-Methyl CBS (0.01 mol) and stirred the contents for 10 minutes at same temperature. The compound of example-1 (20 gm) was dissolved in methylene chloride (200 ml) at same temperature and stirred the reaction mixture for 1.0 hour. The methanol was added to the reaction mixture followed by addition of 5% hydrogen peroxide (0.01 mol) at 0-5 °C and stirred the contents for 15 minutes at same temperature, gradually increased the temperature to 20- 30°C. The 6. ON sulfuric acid (10 ml) solution was added to the reaction mixture and stirred for 15 minutes.The layers were separated. The separated organic layer was washed with 2. ON sulfuric acid solution followed by washings with water, then distilled and dissolved in ethyl acetate. The contents were stirred for 1.0 hour, filtered and dried at 60°C. Yield: 85%; E.e: > 95%.
Example -5 Preparation of compound of formula VA (Indacaterol)
The compound of example-4 (10 gm) was dissolved in methanol (100 ml), followed by addition of acetic acid (50 ml) to the reaction mixture. The 5% Pd/C was added to the reaction mixture and applied hydrogen pressure 3-4 Kg/cm3‘ and then the contents were stirred for 4.0 hours at 25-30°C, filtered and distilled. The residue was dissolved in ethyl acetate, stirred for 10 min and distilled to obtain the compound. Yield: 79%
Example -6 Preparation of Indacaterol Maleate
To a methanolic solution of Indacaterol, maleic acid (0.9 mol) in methanol was slowly added at 25 -30°C and stirred the isolated compound for 2.0 hours at same temperature. The reaction mass was cooled to 0 -10°C and maintained for 2.0 hrs at same temperature. The contents were filtered, washed with methanol and dried at 60 -65 °C. Yield: 93%; E.e: >99%.
Example -7 Preparation of compound of formula IXA, wherein R and Rl is benzyl

The (Bromo compound) of formula I (25 gm) was dissolved in DMF (150 ml) and stirred the contents for 15 min. The 5,6-Diethyl indane N-benzyl amine (0.9 mol) was added to the above mixture at 25 -30°C, followed by the slow addition of triethylamine, then the reaction mixture was stirred for 5.0 min. The sodium iodide (0.01 mol) was added to the reaction mixture at same temperature and stirred for 3 hours at same temperature. The purified water was added to the reaction mixture, and then the contents were filtered and washed with water. The wet compound was dissolved in methanol then water was added to the contents and stirred for lhour at 25 -30 °C. The contents were filtered and dried the compound at 60°C. Yield: 70%.
Example -8 Preparation of compound of formula XA, wherein R and Rl is benzyl
A mixture of Borane-dimethyl sulfide (0.11 mol), R-(2)-Methyl CBS (0.01 mol) and methylene chloride was stirred for 10 minutes at 0-5 C. The compound of example-7 (20 gm) was dissolved in methylene chloride (200 ml) and was added to the reaction mixture at same temperature. The reaction mixture was stirred for 1.0 hour. The methanol was added to the reaction mixture followed by addition of 5% hydrogen peroxide (0.01 mol) at 0-5 C. Stirred the contents for 15 minutes at same temperature, gradually increased the temperature to 20-30°C. The 6. ON sulfuric acid (10 ml) solution was added to the reaction mixture and stirred for 5minutes.The layers were separated. The organic layer was washed with 2. ON sulfuric acid solution followed by washing with water. The organic layer was distilled and dissolved in ethyl acetate. Stirred the contents for 1.0 hour and filtered the compound. The compound was dried at 60°C. Yield: 80%; Purity E.e: > 95%.
Example -9 Preparation of compound of formula VA (Indacaterol)
The compound of example-8 (10 gm) was dissolved in methanol (100 ml), followed by addition of acetic acid (50 ml) to the reaction mixture. Then 5% Pd/C was added to the reaction mixture and applied hydrogen pressure 3-4 Kg/cm3 The content was stirred for 4.0 hours at 25-30°C, filtered and the filtrate was distilled. The residue was dissolved in ethyl acetate (50 ml), stirred the contents for 10 min and distilled to obtain the compound. Yield: 80%
PATENT
http://www.google.com/patents/WO2014139485A1?cl=en
WO 0075114 Al is the first to describe preparation of indacaterol ((i?)-2) (Scheme 1).

Scheme 1 The synthesis is a follow-up of the previously published method for the preparation of 8- benzyloxy-5-(i?)-oxiranyl-(lH)-quinolin-2-one, published in WO 9525104 Al.This synthesis of indacaterol ((i?)-2) was further modified un WO 04076422 Al, WO 04087668 Al and WO 05123684 A2 to be better applicable for the industrial production. A weak point of the above mentioned synthesis is the use of the expensive benzyl trichloromethyl dichloroiodate as the chlorination agent in the first step. A considerable weak point of the above mentioned synthesis is the formation of undesired side products during the reaction of 8-benzyloxy-5-(R)- oxiranyl-(lH)-quinolin-2-one with 2-amino-5,6-diethylindane (Scheme 2).

Scheme 2
Crude 5-[(i?)-2-(5,6-diethyl-indan-2-ylamino)-l-hydroxyethyl]-8-benzyloxy-(lH)-quinolin-2- one ((i?)-l) can be purified from these undesired side products by conversion to the benzoate, which is then re-crystallized, reduced with hydrogen, converted to indacaterol maleinate, which is finally re-crystallized. According to WO 04076422 Al, WO 04087668 Al and WO 05123684 A2, the yield of 5-[(i?)-2-(5,6-diethyl-indan-2-ylamino)- 1 -hydroxyethyl]-8- benzyloxy-(lH)-quinolin-2-one ((i?)-l) benzoate from 8-benzyloxy-5-(i?)-oxiranyl-(lH)- quinolin-2-one is only 67%.
Scheme 3.

The starting 8-benzyloxy-5-(2,2-dihydroxyacetyl)-lH-quinolin-2-one and 2-amino-5,6- diethylindane were prepared according to US 2004167167 Al and F. Baur et al. J. Med.
Chem. 2010, 53, 3675-3684. Example 1. Preparation of 5-[2-(5,6-diethyI ndan-2-yIamino)-l-hydroxyethyl]-8- benzyloxy-(lH)-quinoIin-2-one (1)
A mixture of 8-benzyloxy-5-(2,2-dihydroxyacetyl)-lH-quinolin-2-one (1,15 g), 2-amino-5,6- diethylindane (0.83 g) and dimethyl sulfoxide (5 ml) was stirred at 20°C for 1 h. The resulting suspension was cooled down to 0°C and methanol (5 ml) was added at this temperature. Finely triturated NaB¾ (0.39 g) was added at 0°C and the resulting clear solution was stirred at 20°C for 16 hours. Water (20 ml) was added to the mixture and the mixture was stirred at 20°C for 6 h. The product was filtered off, washed with water and air-dried. The yield was 1.68 g (98%) of beige powder.
Example 2. Preparation of 5- [2-(5,6-diethyl-indan-2-yIamino)-l -hydrox ethyl] -8- benzyloxy-(lH)-quinolin-2-one (1) A mixture of 8-benzyloxy-5-(2,2-dihydroxyacetyl)-lH-quinolin-2-one (1.95 g), 2-amino-5,6- diethylindane (1.25 g), dimethyl sulfoxide (8 ml) and acetic acid (0.05 ml) was stirred at 20°C for 2 h. The resulting suspension was cooled down to 0°C and methanol (8 ml) was added at this temperature. Finely triturated NaBH (1.13 g) was added at 0°C and the produced clear solution was stirred at 20°C for 3 h. Water (32 ml) was added to the mixture and the mixture was stirred at 20°C for 16 h. The product was filtered off, washed with water and air-dried. The yield was 2.75 g (95%) of beige powder.
Example 3. Preparation of 5-[2-(5,6-diethyI-indan-2-ylamino)-l-hydroxyethyI]-8- benzyloxy-(lH)-quinolin-2-one (1)
A mixture of 8-benzyloxy-5-(2,2-dihydroxyacetyl)-lH-quinolin-2-one (115 mg), 2-amino-5,6- diethylindane (83 mg) and dimethyl acetamide (0.5 ml) was stirred at 20°C for 1 h. The resulting suspension was cooled down to 0°C and methanol (0.5 ml) was added at this temperature. Finely triturated NaBHU (39 mg) was added at 0°C and the obtained clear solution was stirred at 20°C for 16 h. Water (2 ml) was added to the mixture and the mixture was stirred at 20°C for 6 h. The product was filtered off, washed with water and air-dried. The yield was 160 mg (94%) of beige powder. Example 4. Preparation of 5-[2-(5,6-diethyI-indan-2-ylamino)-l-hydroxyethyI]-8- benzyloxy-(lH)-quinoLm-2-one (1)
A mixture of 8-benzyloxy-5-(2,2-dihydroxyacetyl)-lH-quinolin-2-one (115 mg), 2-amino-5,6- diethylindane (83 mg) and dichloromethane (2 ml) was stirred at 20°C for 2 h. Finely triturated NaBH(OAc)3 (250 mg) was added at 20°C. The resulting mixture was stirred at 20°C for 16 h and then evaporated until dry. Water (2 ml) was added to the evaporation product and the mixture was stirred at 20°C for 6 h. The product was filtered off, washed with water and air-dried. The yield was 164 mg (96%) of beige powder.
Example 5. Preparation of 5-[2-(5,6-diethyl-indan-2-ylamino)-l-hydroxyethyl]-8- benzyloxy-(li?)-quinolin-2-one (1)
A mixture of 8-benzyloxy-5-(2,2-dihydroxyacetyl)-lH-quinolin-2-one (33 mg), 2-amino-5,6- diethylindane (21 mg) and tetrahydrofuran (1 ml) was stirred at 20°C for 1 h. The resulting suspension was cooled down to 0°C and 1 M BH3 in tetrahydrofuran (0.5 ml) was added at this temperature. The produced clear solution was stirred at 20°C for 16 h and then evaporated until dry. Water (1 ml) was added to the evaporation product and the mixture was stirred at 20°C for 6 h. The product was filtered off, washed with water and air-dried. The yield was 48 mg (99%) of beige powder.
Example 6. Preparation of 5-[2-(5,6-diethyl-indan-2-ylamino)-l-hydroxyethyl]-8- hydroxy-(l/Z)-quinolin-2-one (2) A mixture of 5-[2-(5,6-diethyl-indan-2-ylamino)- 1 -hydroxyethyl]-8-benzyloxy-(lH)-quinolin- 2-one (1) (1.21 g), ethanol (100 ml) and 5 % Pd / C (80 mg) was stirred in a hydrogen atmosphere at 20°C at the pressure of 101 kPa for 2 h. A TLC analysis of the mixture showed the pure reactant, therefore the mixture was filtered and fresh 5% Pd / C (80 mg) was added to the filtrate. The mixture was stirred in a hydrogen atmosphere at 20°C at the pressure of 101 kPa for 2 h. A TLC analysis of the mixture showed the reactant accompanied by a small amount of the product, therefore the mixture was filtered and fresh 5 % Pd / C (80 mg) was again added to the filtrate. The mixture was stirred under a hydrogen atmosphere at 40°C at the pressure of 101 kPa for 4 h. A TLC analysis of the mixture showed the pure product, therefore the mixture was hot filtered and the residue on the filter was extensively washed with hot ethanol. The filtrate was evaporated in an evaporator at a reduced pressure. The yield was 0.97 g (99%) of yellow powder. Example 7. Preparation 5-[2-(5,6-diethyl-indan-2-ylamino)-l-hydroxyethyl]-8-hydroxy- (lfl)-quinoIin-2-one (2)
A mixture of 5-[2-(5,6-diemyl-indan-2-ylammo)-l-hydroxyethyl]-8-benzyloxy-(lH)-quinolin- 2-one (1) (1,21 g), ethanol (100 ml) and Raney nickel (1 g) was stirred at 20°C for 2 h. The mixture was filtered and 5% Pd / C (0.1 g) was added to the filtrate. The mixture was stirred under a hydrogen atmosphere at 40°C at the pressure of 101 kPa at 40°C. A TLC analysis of the mixture showed the pure product, therefore the mixture was hot filtered and the residue on the filter was extensively washed with hot ethanol. The filtrate was evaporated in an evaporator at a reduced pressure. The yield was 0.96 g (98%) of yellow powder.
Example 8. Preparation of indacaterol ((R)-2)
Indacaterol ((i?)-2) was resolved from Z 5-[2-(5,6-diethyl-indan-2-ylamino)-l-hydroxyethyl]- 8-hydroxy-(lH)-quinolin-2-one (2) (0.90 g) by means of preparative HPLC. Conditions of the resolution: UV detection at 260 nm, column length 500 mm, column internal diameter 50 mm, stationary phase Chiralcel OJ (20 μηι), temperature 25°C, flow rate 120 ml/min, mobile phase A: 500 ml of hexane + 1 ml triethylamine, mobile phase B: ethanol, isocratic elution 82% A + 18% B. The fractions containing indacaterol ((R)-2) were evaporated in an evaporator at a reduced pressure. The yield was 0.44 g (49%) of white powder. HPLC enantiomeric purity 99.0% ee.
Example 9. Preparation of 5-[(R)-2-(5,6-diethyl-indan-2-ylamino)-l-hydroxyethyl]-8- benzyloxy-(lH)-quinolin-2-one ((R)-l) 5-[(i?)-2-(5,6-diethyl-indan-2-ylamino)-l-hydroxyethyl]-8-benzyloxy-(lH)-quinolin-2-one ((R)-l) was resolved from 5-[2-(5,6-diethyl-indan-2-ylamino)-l-hydroxyethyl]-8-benzyloxy- (lH)-quinolin-2-one (1) (1.00 g) by means of preparative HPLC. Conditions of the resolution: UV detection at 260 nm, column length 500 mm, column internal diameter 50 mm, stationary phase Chiralcel AS-V (20 μηι), temperature 25°C, flow rate 120 ml/min, mobile phase A: phosphate buffer (1.15 g of NH4H2P04, dissolved in 1000 ml of water, adjusted to pH 6.0 with 25% aqueous NH3), mobile phase B: acetonitrile, isocratic elution 20% A + 80% B. The fractions containing 5-[(i?)-2-(5,6-diethyl-indan-2-ylamino)-l -hydroxyethyl]-8-benzyloxy- (lH)-quinolin-2-one ((R)-l) were evaporated in an evaporator at a reduced pressure to the volume of about 50 ml. 25% aqueous NH3 was added dropwise to the resulting suspension up to pH 8-9 and the product was extracted with ethyl acetate. The combined extracts were dried with Na2S04 and evaporated in an evaporator at a reduced pressure. The yield was 0.48 g (48%) of white powder. HPLC enantiomeric purity 99.2% ee.
Example 10. Preparation of indacaterol ((R)-2)
A mixture of 5-[(i-)-2-(5,6-diethyl-indan-2-ylamino)-l-hydroxyethyl]-8-benzyloxy-(lH)- quinolin-2-one ((R)-l) (0.42 g, HPLC enantiomeric purity of 99.2% ee), ethanol (50 ml) and Raney nickel (0.5 g) was stirred at 20°C for 2 h. The mixture was filtered and 5% Pd / C (0.05 g) was added to the filtrate. The mixture was stirred under a hydrogen atmosphere at 40°C at the pressure of 101 kPa for 4 h. A TLC analysis of the mixture showed the pure product, therefore the mixture was hot filtered and the residue on the filter was extensively washed with hot ethanol. The filtrate was evaporated in an evaporator at a reduced pressure. The yield was 0.33 g (97%) of white powder. HPLC enantiomeric purity 99.0% ee.
PATENT
The compound 5-[(R)-2-(5,6-diethyl-indan-2-ylamino)-l-hydroxyethyl]-8- hydroxy-(lH)-quinolin-2-one, which is known as Indacaterol (INN), and its corresponding salts are beta-selective adrenoceptor agonists with a potent bronchodilating activity. Indacaterol is especially useful for the treatment of asthma and chronic obstructive pulmonary disease (COPD) and is sold commercially as the maleate salt. WO 00/75114 and WO 2004/076422 describe the preparation of Indacaterol for the first time through the process:

regioisomer impurity
Puri
Dep
Overall

The condensation between the indanolamine and the quinolone epoxide leads to the desired product but always with the presence of a significant amount of impurities, the most significant being the dimer impurity, which is the
consequence of a second addition of the product initially obtained with another quinolone epoxide, as well as the formation of another isomer which is the result of the addition of the indanolamine to the secondary carbon of the epoxide.
In addition, the reaction conditions to achieve the opening of the epoxide require high energies (ex. 21 of WO 00/75114) with temperatures of 110 °C or more for several hours, which favours the appearance of impurities.
WO 2004/076422 discloses the purification of the reaction mixture by the initial formation of a salt with an acid, such as tartaric acid or benzoic acid,
hydrogenation and final formation of the maleate salt. However, the yield achieved by the end of the process is only 49% overall.
It has been found that impurities of tartrate and benzoate salts can exist in the final product as a result of displacing the tartrate or benzoate with maleate without prior neutralization to Indacaterol base. In addition, WO 2004/076422 discloses that proceeding via the free base of Indacaterol is not viable due to its instability in organic solvents. WO 00/75114 does disclose a method proceeding via the Indacaterol free base, but it is not isolated in solid form.
WO 2004/076422 furthermore discloses the method for obtaining the quinolone epoxide from the corresponding a-haloacetyl compound by reduction in the presence of a chiral catalyst, such as an oxazaborolidine compound, by proceeding via the a-halohydroxy compound.
Documents WO 2007/124898 and WO 2004/013578 disclose 8-(benzyloxy)-5- [(lR)-2-bromo-l-{[tert-butyl(dimethyl)silyl]oxy}ethyl]quinolin-2(lH)-one and 8- (benzyloxy)-5-[(lR)-2-bromo-l-{tetrahydro-2H-pyran-2-yl-oxy}ethyl]quinolin- 2(lH)-one, respectively. Said documents are however not concerned with the preparation of Indacaterol. There exists, therefore, the need to develop an improved process for obtaining Indacaterol and salts thereof, which overcomes some or all of the problems associated with known methods from the state of the art. More particularly, there exists the need for a process for obtaining Indacaterol and pharmaceutically acceptable salts thereof, which results in a higher yield and/or having fewer impurities in the form of the dimer and regioisomers impurities and/or salts other than the desired pharmaceutically acceptable salt.
Examples
Example 1 – protecting the ot-halohydroxy compound of formula VI

A flask is charged with 5 ml of tetrahydrofuran (THF) and 5 ml of toluene, p- toluene sulfonic acid (0,15 mmol) and molecular sieves are added with stirring for 30 minutes. 6 mmol of butyl-vinylether and 3 mmol of 8-(phenylmethoxy)-5-((R)- 2-bromo-l-hydroxy-ethyl)-(lH)-quinolin-2-one are added. The mixture is agitated at 20/25° C until completion of the reaction, followed by filtration and distillation of the filtrate to remove the solvent. The product is obtained in quantitative yield as an oil consisting of 50% of each of the diastereomers.
^-NMR (DMSO-c/6, δ), mixture 50/50 of diastereomers: 0.61 and 0.82 (3H, t, J=7.2 Hz, CHs-Pr-O), 1.12 and 1.22 (3H, d, J=5.6 Hz, acetalic CH3), 0.90-1.40 (4H, m, CH2 + CH2), 3.20-3.80 (4H, m, CH2-OAr + CH2-Br), 4.51 and 4.82 (1H, q, J = 5.6 Hz, acetalic CH), 5.18 and 5.24 (1H, dd, J=4.0, 8.0 Hz, CH-O-acetal), 6.56 and 6.58 (1H, d, J = 10.0 Hz, H4), 7.00-7.57 (7H, m), 8.17 and 8.23 (1H, d, J = 10.0 Hz, H3), 10.71 (1H, s, NH)
13C-NMR (DMSO-c/6, δ), mixture 50/50 of diastereoisomers: 13.5 and 13.7 CH3), 18.5 and 18.8 (CH2), 19.9 and 20.0 (acetalic CH3), 30.9 and 31.4 (CH2), 36.8 and 37.3 (CH2), 63.7 and 64.2 (CH2-Br), 69.8 and 69.9 (CH2-OAr), 73.8 and 75.1 (CH- O), 97.5 and 100.4 (acetalic CH), 111.8 (CH), 116.9 and 117.2 (C), 121.2 and 122.4 (CH), 122.3 and 122.6 (CH), 127.7 and 127.8 (C), 127.8 and 127.9 (CH), 128.2 and 128.3 (CH), 128.8 and 129.1 (C), 129.4 and 129.6 (C), 136.1 and 136.5 (CH), 136.5 and 136.6 (C), 144.0 and 144.2 (C), 160.7 and 160.8 (C=0). Example 2 – protecting the ot-halohydroxy compound of formula VI

Pivaloyl chloride (0.72 g) is added to a stirred mixture of 8-(phenylmethoxy)-5- 5 ((R)-2-chloro-l-hydroxy-ethyl)-(lH)-quinolin-2-one (0.74 g), dichloromethane (15 ml) and 4-dimethylaminopyridine (0.89 g) at 20/25° C, and the reaction is stirred until all the starting material disappeared . Water (22 ml) is added and the phases are separated.
10 The organic phase is washed with 1 M HCI (22 ml) and then with water (22 ml).
The solvent is removed and the residue is crystallized from acetone to obtain 0.82 g of the product.
^-NMR (DMSO-c/6, δ) : 1.13 (9H, s, CH3), 3.92 (1H, dd, J= 4.0, 12.0 Hz, CH2-Br), 15 4.00 (1H, dd, J= 8.4, 12.0 Hz, CH2-CI), 5.28 (2H, s, Ph-CH2-0), 6.25 (1H, dd, J = 4.0, 8.4 Hz, CH-OPiv), 6.59 (1H, d, J= 10.0 Hz, H4), 7.15 (1H, d, J= 8.4 Hz, H6), 7.20 (1H, d, J= 8.4 Hz, H7), 7.27-7.30 (1H, m, Ph), 7.33-7.37 (2H, m, Ph), 7.54- 7.56 (2H, m, Ph), 8.18 (1H, d, J= 10.0 Hz, H3), 10.77 (1H, s, NH).
20 13C-NMR (DMSO-c/6, δ) : 26.7 (3 x CH3), 38.3 (C), 46.4 (CH2-CI), 69.8 (CH2-Ph), 71.3 (CH-OPiv), 111.9 (CH), 116.8 (C), 120.5 (CH), 122.9(CH), 126.0 (C), 127.8 (2 x CH), 127.9 (CH), 128.3 (2 x CH), 129.5 (C), 136.0 (C), 136.5 (CH), 144.5 (C), 160.7 (CON), 176.2 (COO). Example 3 – preparation of the compound of formula IV

A flask is charged with 2.5 ml of THF and 2.5 ml of toluene, p-toluene sulfonic 5 acid (5 mg) and molecular sieves (0.2 g) are added with stirring for 30 minutes.
1.5 ml of butyl-vinylether and 2 g of 8-(phenylmethoxy)-5-((R)-2-bromo-l- hydroxy-ethyl)-(lH)-quinolin-2-one are added . The mixture is agitated at 20/25° C until completion of the reaction. 0.015 ml of diisopropylethyl amine is added, the mixture is filtered, and the solvent is distilled off.
10
The residue is dissolved in 6 ml of dimethylformamide (DMF), 1.9 ml of
diisoproypylethyl amine, 1.2 g sodium iodide, and 1.5 g of 2-amino-5,6- diethylindane are added and the mixture is heated to 100° C. After completion of the reaction the mixture is cooled to 20/25° C, 0.4 ml of concentrated hydrochloric 15 acid and 0.4 ml of water are added, and the mixture is stirred for 30 minutes.
HPLC analysis shows the expected product with a purity of 75% and being free from the dimer and regioisomer impurities.
20 20 ml of water, 20 ml of methylene chloride, and 3 ml of 6N NaOH are added with stirring. The organic phase is separated and washed with 20 ml of water. The organic phase is distilled and the solvent is changed to ethyl acetate with a final volume of 100 ml. The mixture is heated to 70° C, 0.8 g of L-tartaric acid is added, and stirring continues for 30 minutes at 70° C. The mixture is cooled
25 slowly to 20/25° C, filtered, and washed with 8 ml of ethyl acetate to obtain 8- (phenylmethoxy)-5-[(R)-2-(5,6-diethyl-indan-2-ylamino)-l-hydroxy-ethyl]-(lH)- quinolin-2-one tartrate in 68% yield. The purity of the product is >95% by HPLC analysis. Example 4 – preparation of the compound of formula IV

A flask is charged with 19 ml of THF and 19 ml of toluene, p-toluene sulfonic acid (75 mg) and molecular sieves (1.5 g) are added and the mixture is stirred for 30 minutes. 11.2 ml of butyl-vinylether and 15 g of 8-(phenylmethoxy)-5-((R)-2- bromo-l-hydroxy-ethyl)-(lH)-quinolin-2-one are added. The mixture is agitated at 20/25° C until completion of the reaction. 0.1 ml of diisopropylethyl amine are added, the mixture is filtered, and the solvent is distilled off.
The residue is dissolved in 40 ml of butanone, 14.5 ml of diisoproypylethyl amine, 9 g sodium iodide, and 11.3 g of 2-amino-5,6-diethylindane are added and the mixture is heated to 90-100° C. After completion of the reaction the mixture is cooled to 20/25° C, 3 ml of concentrated hydrochloric acid and 3 ml of water are added, and the mixture is stirred for 30 minutes.
HPLC analysis shows the expected product with a purity of 84% and being free from the dimer and regioisomer impurities. 150 ml of water, 150 ml of methylene chloride, and 22.5 ml of 6N NaOH are added with stirring. The organic phase is separated and washed with 10 ml of water. The organic phase is distilled and the solvent is changed to isopropyl alcohol with a final volume of 300 ml. The mixture is heated to 70° C, 4.9 g of benzoic acid is added, and stirring continues for 30 minutes at 70° C. The mixture is cooled slowly to 20/25° C, filtered, and washed with 30 ml of isopropanol to obtain 8-(phenylmethoxy)-5-[(R)-2-(5,6-diethyl-indan-2-ylamino)-l-hydroxy- ethyl]-(lH)-quinolin-2-one benzoate in 59 % yield. The purity of the product is > 99 % by HPLC analysis. Example 5 – preparation of the compound of formula IV

A flask is charged with 7.5 ml of THF and 7.5 ml of toluene, p-toluene sulfonic acid (30 mg) and molecular sieves (0.6 g) are added and the mixture is stirred for 30 minutes. 4.5 ml of butyl-vinylether and 6 g of 8-(phenylmethoxy)-5-((R)-2- bromo-l-hydroxy-ethyl)-(lH)-quinolin-2-one are added. The mixture is agitated at 20/25° C until completion of the reaction. 0.040 ml of diisopropylethyl amine are added, the mixture is filtered, and the solvent is distilled off.
The residue is dissolved in 18 ml of acetonitrile (ACN), 5,8 ml of diisoproypylethyl amine, 3.6 g sodium iodide, and 4.5 g of 2-amino-5,6-diethylindane are added and the mixture is heated to 80-90° C. After completion of the reaction the mixture is cooled to 20/25° C, 1.2 ml of concentrated hydrochloric acid and 1.2 ml of water are added, and the mixture is stirred for 30 minutes. HPLC analysis shows the expected product with a purity of 89% and being free from the dimer and regioisomer impurities.
60 ml of water, 60 ml of methylene chloride, and 9 ml of 6N NaOH are added with stirring. The organic phase is separated and washed with 60 ml of water. The organic phase is distilled and the solvent is changed to isopropyl alcohol with a final volume of 120 ml. The mixture is heated to 70° C, 1.9 g of succinic acid is added, and stirring continues for 30 minutes at 70° C. The mixture is cooled slowly to 20/25° C, filtered, and washed with 12 ml of isopropanol to obtain 8- (phenylmethoxy)-5-[(R)-2-(5,6-diethyl-indan-2-ylamino)-l-hydroxy-ethyl]-(lH)- quinolin-2-one succinate in 56 % yield . The purity of the product is > 99 % by HPLC analysis. Example 6 : purification with EtOH/water

To 2.0 g of 8-(phenylmethoxy)-5-[(R)-2-(5,6-diethyl-indan-2-ylamino)-l- hydroxy-ethyl]-(lH)-quinolin-2-one, a mixture of 35 ml/g of EtOH and 5 ml/g of water are added and heated to reflux. Once this temperature is reached, benzoic acid is added (1.2 eq.) as a solution in 5 ml/g of the mixture of EtOH/water. The temperature is maintained for 30 minutes. The mixture is then cooled slowly overnight to 20-25°C. The resulting suspension is filtered and a white solid is obtained and dried in vacuum. The white solid is analyzed by HPLC to determine the chromatographic purity and by chiral HPLC to determine the enantiomeric purity, obtaining a white solid product with a proportion of enantiomeric impurity below 0.05%. No other impurities are detected.
Example 7 : purification with Acetone/water

To 2.0 g of 8-(phenylmethoxy)-5-[(R)-2-(5,6-diethyl-indan-2-ylamino)-l- hydroxy-ethyl]-(lH)-quinolin-2-one, a mixture of 35 ml/g of Acetone and 1 ml/g of water are added and heated to reflux. Once this temperature is reached, Dibenzoyl-L-tartaric monohydrate acid is added (1.2 eq.) as a solution in 5 ml/g of the mixture of Acetone /water. The temperature is maintained for 30 minutes. The mixture is then cooled slowly overnight to 20-25°C. The resulting suspension is filtered and a white solid is obtained and dried in vacuum. The white solid is analyzed by HPLC to determine the chromatographic purity and by chiral HPLC to determine the enantiomeric purity, obtaining a white solid product with a proportion of enantiomeric impurity below 0.05%. No other impurities are detected.
Example 8 : purification with EtOH/water

To 2.0 g of of 8-(phenylmethoxy)-5-[(R)-2-(5,6-diethyl-indan-2-ylamino)-l- hydroxy-ethyl]-(lH)-quinolin-2-one, a mixture of 35 ml/g of EtOH and 5 ml/g of water are added and heated to reflux. Once this temperature is reached, L Tartaric acid is added (1.2 eq.) as a solution in 5 ml/g of the mixture of
EtOH/water. The temperature is maintained for 30 minutes. The mixture is then cooled slowly overnight to 20-25°C. The resulting suspension is filtered and a white solid is obtained and dried in vacuum. The white solid is analyzed by HPLC to determine the chromatographic purity and by chiral HPLC to determine the enantiomeric purity, obtaining a white solid product with a proportion of enantiomeric impurity below 0.06%. No other impurities are detected.
Example 9 : synthesis of protected benzyl Indacaterol

A solution of sodium carbonate (0.57 kg/kg, 2 equivalents) in water (13 l/kg) is prepared in another reactor. This carbonate solution is added to the product solution from example 1, diethyl indanolamine HCI (0.72 kg/kg, 1.2 equivalents) is added and the mixture is heated and distilled at atmospheric pressure until a volume of 13 l/kg . Water (3 l/kg) is added and the mixture is distilled at atmospheric pressure until a volume of 13 l/kg . The system is placed in reflux position and reflux is maintained for 20 hours. When the reaction is complete, the mixture is cooled to 20-25°C and methylene chloride (15 l/kg) is added. The mixture is agitated, decanted, and the aqueous phase is extracted with methylene chloride (5 l/kg). The organic phases are washed with water (5 l/kg).
Example 10 – preparation of Indacaterol maleate

28 g of 8-(phenylmethoxy)-5-[(R)-2-(5,6-diethyl-indan-2-ylamino)-l-hydroxy- ethyl]-(lH)-quinolin-2-one tartrate is dissolved in a mixture of 560 ml of dichloromethane, 560 ml of water, and 30 ml of an aqueous solution of 6N sodium hydroxide under stirring . The phases are separated and the organic phase is washed with 280 ml of water. The organic phase is distilled to a final volume of 140 ml and 420 ml of methanol and 4.2 g of Pd/C (5% – 50% water) are added . The system is purged with nitrogen and subsequently with hydrogen at an overpressure of 0.3 bar and stirring until completion of the reaction. The catalyst is filtered off and the solvent is changed to isopropanol adjusting the final volume to 950 ml. The solution is heated to 70/80° C and a solution of 5.4 g maleic acid in 140 ml of isopropanol is added, maintaining the temperature between 70 and 80° C. The mixture is stirred at 70/80° C for 30 minutes and then slowly cooled to 20/25° C. The resulting suspension is filtered, the solid residue is washed with 90 ml of isopropanol and dried to obtain 18g of Indacaterol maleate (Yield : 79%). The product shows 99.6% purity by HPLC analysis.
Example 11 – Isolation of Indacaterol free base in solid form

lg of 8-(phenylmethoxy)-5-[(R)-2-(5,6-diethyl-indan-2-ylamino)-l-hydroxy- ethyl]-(lH)-quinolin-2-one tartrate is dissolved in a mixture of 20 ml of dichloromethane,20 ml of water, andl ml of an aqueous solution of 6N sodium hydroxide under stirring. The phases are separated and the organic phase is washed with 10 ml of water.
The organic phase is distilled to a final volume of 5 ml and 15 ml of methanol and 0.15 g of Pd/C (5% – 50% water) are added . The system is purged with nitrogen and subsequently with hydrogen at an overpressure of 0.3 bar and stirring until completion of the reaction.
The catalyst is filtered off and the solvent is changed to isopropanol adjusting the final volume to 8 ml. The resulting suspension is cooled to 0-5°C, filtered and the solid residue is washed with isopropanol and dried to obtain 0.47 g of Indacaterol free base (77%) showing 99.6% purity by HPLC analysis.
A sample of Indacaterol free base stored at 20-25°C is analysed one month later without showing any loss of purity. Example 12 – obtaining the maleate salt from Indacaterol free base

0.47 g of solid Indacaterol are suspended in 20 ml of isopropanol, heated to 70/80° C, and a solution of 0.15 g of maleic acid in 5 ml of isopropanol are added, maintaining the temperature between 70 and 80° C. The mixture is cooled to 0/5°C and filtration of the resulting solid affords 0.52 g of Indacaterol maleate with a purity of 99.7%.
Comparative example 13 – direct conversion to Indacaterol maleate
8-(phenylmethoxy)-5-[(R)-2-(5,6-diethyl-indan-2-ylamino)-l-hydroxy-ethyl]- (lH)-quinolin-2-one benzoate (4 g) is dissolved in acetic acid (40 ml). Pd/C (5 %, 50% wet, 0.6 g) is added and the product is hydrogenated under a hydrogen atmosphere. When the reaction is complete the catalyst is filtered off and the filtrate is vacuum distilled until a volume of 8 ml is reached.
Ethanol (40 ml) is added and the mixture is heated to 50° C. A solution of 1.2 g of maleic acid in 2.4 ml of ethanol is added and the mixture is seeded with
indacaterol maleate and then slowly cooled to 0/5° C. The solid is filtered and washed with 5 ml of ethanol and 3 ml of isopropanol to obtain 6.0 g of indacaterol maleate.
1H-NMR analysis of the solid shows the presence of acetic acid in 2-4 % by integration of the peak at δ 1.88 (400 MHz, DMSO-c/6) corresponding to acetic acid.
References
- Cazzola M, Matera MG, Lötvall J (July 2005). “Ultra long-acting beta 2-agonists in development for asthma and chronic obstructive pulmonary disease”. Expert Opin Investig Drugs 14(7): 775–83. doi:10.1517/13543784.14.7.775.PMID 16022567.
- European Public Assessment Report for Onbrez Breezhaler
- “FDA approves Arcapta Neohaler to treat chronic obstructive pulmonary disease” (Press release). U.S. Food and Drug Administration. 2011-07-01. Retrieved 2011-07-02.[1]
- Beeh KM, Derom E, Kanniess F, Cameron R, Higgins M, van As A (May 2007). “Indacaterol, a novel inhaled beta2-agonist, provides sustained 24-h bronchodilation in asthma”. Eur. Respir. J. 29 (5): 871–8. doi:10.1183/09031936.00060006.PMID 17251236.
- Feldman, G; Siler, T; Prasad, N; Jack, D; Piggott, S; Owen, R; Higgins, M; Kramer, B; Study Group, I (2010). “Efficacy and safety of indacaterol 150 mcg once-daily in COPD: a double-blind, randomised, 12-week study”. BMC pulmonary medicine10: 11. doi:10.1186/1471-2466-10-11. PMC 2848004.PMID 20211002.
- Dahl R; Chung KF; Buhl R; et al. (June 2010). “Efficacy of a new once-daily long-acting inhaled beta2-agonist indacaterol versus twice-daily formoterol in COPD”. Thorax 65 (6): 473–9.doi:10.1136/thx.2009.125435. PMID 20522841.
- R. Buhl; L.J. Dunn; C. Disdier; et al. (October 2011). “Blinded 12-week comparison of once-daily indacaterol and tiotropium in COPD”. European Respiratory Journal 38 (4): 797–803.doi:10.1183/09031936.00191810. PMID 21622587.
- http://onlinelibrary.wiley.com/doi/10.1002/14651858.CD010139.pub2/abstract;jsessionid=2E0FA3EB220BD4ADED29D7B5707FC667.f01t04
| A. BORGHESE ET AL.: “Efficient Fast Screening Methodology for Optical Resolution Agents: Solvent Effects Are Used To Affect tge Efficiency of the Resolution Process“, ORGANIC PROCESS RESEARCH & DEVELOPMENT, vol. 8, no. 3, 2004, pages 532-534, XP002725198, | ||
| 2 | * | D. BEATTIE ET AL.: “An investigation into the structure-activity relationships associated with the systematic modification of the beta2-adrenoreceptor agonist indacaterol“, BIOORGANIC & MEDICINAL CHEMISTRY LETTERS, vol. 22, 2012, pages 6280-6285, XP002724553, |
| 3 | F. BAUR ET AL. J. MED. CHEM. vol. 53, 2010, pages 3675 – 3684 | |
| 4 | * | F. BAUR ET AL.: “The Identification of Indacaterol as an Ultralong-Acting Inhaled beta2-Adrenoceptor Agonist“, JOURNAL OF MEDICINAL CHEMISTRY, vol. 53, no. 9, 2010, pages 3675-3684, XP002724552, |
| 5 | * | KRAUSE M ET AL: “Optical resolution of flavanones by high-performance liquid chromatography on various chiral stationary phases“, JOURNAL OF CHROMATOGRAPHY, ELSEVIER SCIENCE PUBLISHERS B.V, NL, vol. 514, 1990, pages 147-159, XP026539395, ISSN: 0021-9673, DOI: 10.1016/S0021-9673(01)89386-9 [retrieved on 1990-01-01] |
| 6 | * | M. NISHIKATA ET AL.: “Method for Optical Resolution of Racemic Homochlorcyclizine and Comparison of Optical Isomers in Antihistamine Activity and Pharmacokinetics“, CHEMICAL AND PHARMACEUTICAL BULLETIN, vol. 40, no. 5, 1992, pages 1341-1342, XP002725199, |
| WO1995025104A1 | Mar 3, 1995 | Sep 21, 1995 | Lee James Beeley | Novel heterocyclic ethanolamine derivatives with beta-adrenoreceptor agonistic activity |
| WO2000075114A1 | Jun 2, 2000 | Dec 14, 2000 | Novartis Ag | Beta2-adrenoceptor agonists |
| WO2004074276A1 * | Feb 13, 2004 | Sep 2, 2004 | Theravance Inc | BIPHENYL DERIVATIVES HAVING β2 ADRENERGIC RECEPTOR AGONIST AND MUSCARINIC RECEPTOR ANTAGONIST ACTIVITY |
| WO2004076422A1 | Feb 27, 2004 | Sep 10, 2004 | Olivier Lohse | Process for preparing 5-‘(r)-2-(5,6-diethyl-indian-2-ylamino)-1-hydroxy-ethyl!-8-hydroxy-(1h)-quinolin-2-one salt, useful as an adrenoceptor agonist |
| WO2004087668A1 | Apr 1, 2004 | Oct 14, 2004 | Novartis Ag | A process for the preparation of 5-(haloacetyl)-8-(substituted oxy)-(1h)-quinolin-2-ones |
| WO2005123684A2 | Jun 21, 2005 | Dec 29, 2005 | Stephan Abel | Enantioselektive preparation of quinoline derivative |
| WO2007124898A1 * | Apr 24, 2007 | Nov 8, 2007 | Almirall Lab | DERIVATIVES OF 4-(2-AMINO-1-HYDROXIETHYL)PHENOL AS AGONISTS OF THE β2 ADRENERGIC RECEPTOR |
| WO2008046598A1 * | Oct 17, 2007 | Apr 24, 2008 | Almirall Lab | DERIVATIVES OF 4-(2-AMINO-1-HYDROXYETHYL)PHENOL AS AGONISTS OF THE β2 ADRENERGIC RECEPTOR |
| WO2009106351A1 * | Feb 27, 2009 | Sep 3, 2009 | Almirall, S.A. | Derivatives of 4-(2-amino-1-hydroxyethyl) phenol as agonists of the b2 adrenergic receptor |
| EP0147719A2 * | Dec 11, 1984 | Jul 10, 1985 | Tanabe Seiyaku Co., Ltd. | Novel carbostyril derivative and process for preparing same |
| EP1405844A1 * | Jun 27, 2002 | Apr 7, 2004 | Nikken Chemicals Company, Limited | Cycloalkenone derivative |
| US20040167167 | Feb 13, 2004 | Aug 26, 2004 | Mathai Mammen | Biphenyl derivatives |
| WO2000075114A1 * | Jun 2, 2000 | Dec 14, 2000 | Novartis Ag | Beta2-adrenoceptor agonists |
| WO2002045703A2 * | Dec 3, 2001 | Jun 13, 2002 | Bernard Cuenoud | Mixtures or organic compounds for the treatmentof airway diseases |
| WO2004076422A1 * | Feb 27, 2004 | Sep 10, 2004 | Olivier Lohse | Process for preparing 5-‘(r)-2-(5,6-diethyl-indian-2-ylamino)-1-hydroxy-ethyl!-8-hydroxy-(1h)-quinolin-2-one salt, useful as an adrenoceptor agonist |
| WO2004087668A1 * | Apr 1, 2004 | Oct 14, 2004 | Novartis Ag | A process for the preparation of 5-(haloacetyl)-8-(substituted oxy)-(1h)-quinolin-2-ones |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2014154841A1 * | Mar 27, 2014 | Oct 2, 2014 | Laboratorios Lesvi, S.L. | Process for the manufacture of (r)-5-[2-(5,6-diethylindan-2-ylamino)-1-hydroxyethyl]-8-hydroxy-(1h)-quinolin-2-one |
 |
|
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
5-[2-[(5,6-Diethyl-2,3-dihydro-1H-inden-2-yl)amino]-1-hydroxyethyl]-8-hydroxyquinolin-2(1H)-one
|
|
| Clinical data | |
| Trade names | Onbrez, Arcapta |
| AHFS/Drugs.com | International Drug Names |
| Licence data |
|
| Pregnancy category |
|
| Routes of administration |
Inhalation |
| Legal status | |
| Identifiers | |
| CAS Number | 312753-06-3  |
| ATC code | R03AC18 |
| PubChem | CID 6433117 |
| IUPHAR/BPS | 7455 |
| ChemSpider | 5293751 |
| UNII | 8OR09251MQ |
| KEGG | D09318 |
| ChEBI | CHEBI:68575  |
| ChEMBL | CHEMBL1095777 |
| Chemical data | |
| Formula | C24H28N2O3 |
| Molar mass | 392.490 g/mol |

//////
O=C4/C=C\c1c(c(O)ccc1[C@@H](O)CNC3Cc2cc(c(cc2C3)CC)CC)N4
Filed under: Uncategorized Tagged: indacaterol, Indacaterol Maleate, QAB-149







