Beta 2 adrenoceptor agonist
Chronic obstructive pulmonary disease
WO 2016027283, New patent, Indacaterol, Reddy-Cheminor Inc
A process for preparing indacaterol and salts thereof
REDDY, G Pratap; (IN).
SUNKU, Venkataiah; (IN).
BABU, Sunkaraneni Suresh; (IN)
The present invention relates to a process for preparing indacaterol or salts thereof. The process comprises of forming compound of Formula 1 by reacting compound of Formula 2 and compound of Formula 3 in the presence of a solvent to Form compound of Formula 4, 5 which on removal of the protecting groups forms compound of Formula 1.
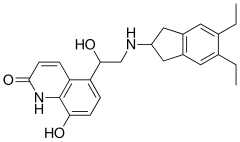
Indacaterol maleate is a beta-selective adrenoceptor agonist with potent bronchodilator activity. Indacaterol is chemically known as 5-[(R)-2-(5, 6-diethyl-indan-2- yl amino)-l-hydroxy-ethyl ]-8-hydroxy-(lH)-quinolin-2-one.
US7534890 claims a process to prepare 5-[(R)-2-(5,6-diethyl-indan-2-ylamino)- 1 -hydroxy-ethyl] -8-hydroxy-(l H)-quinolin-2-one salt. One of the key steps in the process is reacting an epoxide, such as 8-substituted oxy-5-(R)- oxiranyl-(lH)-quinoline-2-one [Formula (I)] with an amine, such as 2-amino-(5,6-diethyl)-indan to form an intermediate 5-[(R)-2-(5,6-diethyl-indan-2-ylamino)-l -hydroxy-ethyl]-8- substituted oxy-(lH)-quinolin-2-one [Formula (Ha)].

The drawback of this process is opening of epoxide ring is not regioselective and thereby resulting, in formation of substantial quantities of impurities as by products, Formula (lib) and Formula (lie) resulting in overall lower yields. The quantity of 2- amino-(5,6-diethyl)-indan used in this step is also large excess than theoretical amounts. Subsequent improvements also did not address this problem effectively.
WO 2013/132514 discloses a process to prepare Indacaterol involving the steps of treating a compound of Formula (III), wherein L is a leaving group, with the amine, 2-amino-(5,6-diethyl)-indan or its acid addition salts to obtain a compound of Formula (IV) or its acid addition salts.

Though higher yields have been claimed, the process has not overcome completely all the problems mentioned earlier.
There is a need for developing a more efficient process for preparing Indacaterol or salts thereof especially for large scale production with higher yields.
The reaction scheme of synthesis of compound of Formula 3 is represented below.

Formula 3 Formula 13 Formula 12
xample 1
Process to prepare 5-[ (R)-2-(5, 6-diethyl-indan-2-ylamino)-l-hydroxy-ethyl]-8-hydroxy-( lH)-quinolin-2-one
2-Chloro-5,6-diethylindan (4.2g) was added to a solution of 5-[(R)-(2-amino-l-hydroxy-ethyl)-8-phenylmethoxy-(lH)-quinolin-2-one (6g) in dimethylformamide (20ml) followed by addition of N,N-diisopropyl-N-ethylamine (3.6 g) and sodium iodide (lg) at room temperature and stirred for 10 minutes. The reaction mixture was heated to 90° C and the temperature was maintained at 90 °C till the completion of reaction. The reaction mass was cooled to room temperature and diluted with dichloromethane (100ml) and water (100 ml) and stirred for 30 minutes. The organic phase was separated and the aqueous layer was extracted with dichloromethane. Combined organic layer was washed with water, dried and concentrated. The resulting residue was dissolved in isopropyl alcohol under reflux and cooled slowly to obtain 5-[(R)-2-(5,6-diethyl-indan-2-ylamino)-l-hydroxy-ethyl]-8-phenylmethoxy -(lH)-quinolin-2-one, which was isolated by filtration and dried under vacuum (7.4 g). Yield: 79.3 %. Purity of the product is >95 % (HPLC).
Example 2
Process to prepare 5-[(R)-2-(5, 6-diethyl-indan-2-ylamino)-l-hydroxy-ethyl]-8-hydroxy-( lH)-quinolin-2-one
Solution of 5-[(R)-2-(5,6-diethyl-indan-2-ylamino)-l-hydroxy-ethyl]-8-phenylmethoxy-(lH)-quinolin-2-one (lOg) in methanol (100ml) and acetic acid (20ml) was hydrogenated using palladium on charcoal 5% (1.5g) until completion of the reaction. The mixture was filtered over celite and the filtrate was concentrated at 55°C under vacuum. The residue obtained was dissolved in hot methanol to precipitate 5-[(R)-2-(5,6-diethyl-indan-2-ylamino)-! -hydroxy-ethyl]-8-hydroxy-(lH)-quinolin-2-one.
Example 3
Process to prepare 5-[(R)-2-(5, 6-diethyl-indan-2-ylamino)-l-hydroxy-ethyl]-8-hydroxy-(lH)-quinolin-2-one maleate
Crude 5-[(R)-2-(5,6-diethyl-indan-2-ylamino)-l -hydroxy-ethyl]-8-hydroxy-(lH)-quinolin-2-one prepared by the process of Example 2 was added to a solution of maleic acid (2.6g) in methanol and the resulting clear solution was slowly cooled to 5° C and stirred for 2 hours at the same temperature. The slurry was filtered, washed with cold methanol and dried to obtain 5-[(R)-2-(5, 6-diethyl-indan-2-ylamino)-l-hydroxy-ethyl]-8-hydroxy-(lH)-quinolin-2-one maleate (8.8g). Yield: 83.5 %. Purity of the product is >99%. E.e. >99 %.
Example 4
Process for preparing 5-[(R)-(2-phthalimido-l-hydroxy-ethyl)-8-phenylmethoxy-(lH)-quinolin-2-one
Diisopropylethylamine (6g) was added to a solution of phthalimide (6g) in dimethylformamide (30 ml) at room temperature. To this solution, 8-(phenylmethoxy)-5-[(R)-2-bromo-l-hydroxy-ethyl]-(lH)-quinoline-2-one (11 gm) was added slowly followed by sodium iodide (1 g). The resulting mass was heated to 90°C and stirred till the completion of reaction as monitored by TLC. The reaction mass was diluted with water (200 ml) and the crude product was isolated by filtration. The wet filter cake was suspended in water (60 ml), stirred for 1 hour, filtered, washed with water to obtain 5-[(R)-(2-phthalimido-l-hydroxy-ethyl)-8-phenylmethoxy-(lH)-quinolin-2-one (10.4 gm) after drying. Yield: 80.7 %.
Method A- Process for preparing 5-[(R)-(2-amino-l-hydroxy-ethyl)-8-phenylmethoxy-(lH)-quinolin-2-one
To a solution of 5-[(R)-(2-phthalimido-l-hydroxy-ethyl)-8-phenylmethoxy-(lH)-quinolin-2-one ( 13.2 g) in a mixture of isopropanol (86 ml) and water (14 ml) sodium borohydride (4.6 g) was added slowly at room temperature and stirred overnight. Thereafter, the pH of the reaction mass was lowered to 5.5 with acetic acid, and then the reaction mass was heated to reflux for two hours. Isopropanol was distilled out under reduced pressure. The residue was diluted with ethyl acetate (120 ml) and concentrated hydrochloric acid (8 ml) was added and stirred for 15 minutes for the salts to precipitate out. The reaction mass was filtered and the salt was washed with ethyl acetate. To the clear filtrate concentrated hydrochloric acid (10 ml) was added and stirred at 5° C for 30 minutes for 5-[(R)-(2-amino-l-hydroxy-ethyl)-8-phenylmethoxy-(lH)-quinolin-2-one to separate out as hydrochloride salt. The product was isolated by filtration and dried under vacuum (8.2 g). The hydrochloride salt was dissolved in minimum amount of water and basified with sodium hydroxide solution. The product was isolated as free amine by concentrating the solution under reduced pressure and extracting the residue with isopropyl alcohol and distilling out the solvent (7.45 g). Yield 80 %.
1H-NMR (CDC13) ppm: 2.56-2.70 (m, 2H), 3.35 (s, br, 2H, exchangeable), 4.89 (m, 1H), 5.29 (s, 2H), 5.76 (s, 1H, exchangeable), 6.53 (d, 1H), 7.11-7.19 (dd, 2H), 7.29-7.36 (dd, 1H), 7.39 (d, 2H), 7.57 (d, 2H), 8.21 (d, 1H), 10.7 (s, br, 1H, exchangeable).
Method B- Process for preparing 5-[(R)-(2-amino-l-hydroxy-ethyl)-8-phenylmethoxy-( lH)-quinolin-2-one
To a solution of 5-[(R)-(2-phthalimido-l-hydroxy-ethyl)-8-phenylmethoxy-(lH)-quinolin-2-one ( 10 g) in ethanol (60 ml) hydrazine hydrate (4.8 g) was added and refluxed the mixture for about 6 hours. The solvent was distilled out under reduced pressure. To the residue, concentrated hydrochloric acid (16 ml) was added and heated to about 80°C and maintained till the completion of the reaction. The reaction mass was cooled to room temperature and filtered. The clear filtrate was basified and concentrated under reduced pressure. The product was isolated as free amine (5.8 g) by extracting with isopropyl alcohol and distilling out the solvent. Yield: 83%.
Method C
Preparation of 5-(2-benzylamino-l-hydroxy-ethyl)-8-phenylmethoxy-( lH)-quinolin-2-one 5-Acetyl-8-phenylmethoxy-(lH)-quinolin-2-one (30 g) was refluxed with selenium dioxide
(11.5 g) in a mixture of dioxane (350 ml) and water (30 ml) for 16 hours. The reaction mixture was diluted with dioxane (150 ml) and precipitated inorganic salts were removed by filtration. Clear filtrate was concentrated to about 60 ml under vacuum and diluted with methanol (100 ml). The reaction mass was cooled to 15° C and benzylamine (7.5 g) was added slowly over a period of 45 minutes and stirred at the same temperature for two hours.
The reaction mass was further cooled to 0°C and sodium borohydride (2.8 g) was added slowly over a period of one hour. Thereafter, the reaction mass was stirred at room temperature for 12 hours. The reaction mixture was concentrated under vacuum and diluted with 300 ml water and stirred at 20° C for three hours. The precipitated product was collected by filtration, washed with water followed by isopropyl ether and then dried (28.2 g) to obtain 5-(2-benzylamino-l-hydroxy-ethyl)-8-phenylmethoxy-(lH)-quinolin-2-one.
Example 5
Preparation of 5-acetyl-8-phenylmethoxy-(lH)-quinolin-2-one
To a solution of 5-acetyl-8-hydroxy-(lH)-quinolin-2-one (35 g) in dimethylformamide (175 ml) potassium carbonate (35 g) was added at room temperature and stirred for 10 minutes. To the suspension, benzylbromide (32 g) was slowly added over a period of 30 minutes and stirred for 2 hours at the same temperature for completion of reaction (monitored by TLC). The reaction mass was diluted with water (800 ml) and stirred for 20 minutes for the product to precipitate out. The product was filtered, washed with water and dried under vacuum to get the title product (48 g).
Example 6
Preparation of 5-(2-bromoacetyl)-8-phenylmethoxy-( lH)-quinolin-2-one
Boron trifluoride-diethyletherate (29 ml) was slowly added to a solution of 5-acetyl-8-phenylmethoxy-(lH)-quinolin-2-one (50 g) in dichloromethane (500 ml) at 0° C and stirred for 10 minutes at the same temperature to get a thick precipitate. The reaction mass was heated to reflux temperature and bromine solution was added (29 g in 190 ml dichloromethane) slowly over a period of 2 hours under reflux (the HBr fumes coming from the condenser was scrubbed). Thereafter, the reaction mass was refluxed for further 45 minutes. The solvent was distilled out completely under vacuum and the mass was triturated with 10% aqueous sodium carbonate solution (100 ml). The suspension was filtered, washed with water and the crude product was taken for the next stage reaction.
Example 7
Preparation of 5-(2-phthalimido-l-oxo-ethyl)-8-phenylmethoxy-(lH)-quinolin-2-one
Potassium carbonate (33.4 g) was added to a solution of phthalimide (21.73 g) in dimethylformamide (80 ml) at room temperature and stirred for 10 minutes. To this suspension, crude 5-(2-bromoacetyl)-8-phenylmethoxy-(lH)-quinolin-2-one of example 6, dissolved in dimethylformamide (120 ml), was added slowly over a period of 20 minutes. The resulting suspension was stirred at 50° C for about 1 hour for the completion of reaction as monitored by TLC. The mixture was diluted with water (800 ml) and the crude product was isolated by filtration. The wet filter cake was suspended in water (600 ml), stirred for 1 hour, filtered, washed with water and dried under vacuum to get 5-(2- phthalimido-l-oxo-ethyl)-8-phenylmethoxy-(lH)-quinolin-2-one (67.4 g). Over all yield
(after two steps): 90%.
Example 8
Preparation of 5-[(R)-(2-phthalimido-l-hydroxy-ethyl)-8-phenylmethoxy-(lH)-q inolin-2-one
To a solution of (R)-2-methyl-CBS-oxazaborolidine (1M in toluene, 4.2 ml) in dry
tetrahydrofuran (THF, 50 ml) Borane-diethylaniline (19 ml) was added slowly at – 10° C
and the contents were stirred at the same temperature for 15 minutes. A solution of 5-(2-
Phthalimido-l-oxo-ethyl)-8-phenylmethoxy-(lH)-quinolin-2-one ( 8.3 g), of example 7, in
a mixture of dry THF (50 ml) and dichloromethane (50 ml), was added slowly to the
reaction mass at – 10° C. The reaction mass was further stirred for 2 hours and then
methanol was added and the temperature was slowly raised to room temperature. Dilute
sulfuric acid (6N, 10 ml) was added to the reaction mixture and stirred for 15 minutes. The
reaction mixture was concentrated under vacuum and the crude mass was extracted with
ethyl acetate. The organic phase was washed with dilute sulfuric acid and then water. The
solvent was distilled out completely under vacuum and triturated with hexane. The
compound was isolated by filtration and dried (7.6 g). Yield: 91.1%. e.e.. >97%.
Example 9
Process of preparing 2-chloroindan
2-hydroxy indan (lOOg) was dissolved in 1, 2-dichloroethane (400 ml) and added to thionyl
chloride (125 g) slowly over a period of an hour. Temperature was maintained at less than
10° C. Thereafter, the reaction mass was slowly heated and refluxed till the completion of the reaction. The reaction was monitored by TLC. The reaction mass was cooled to room temperature and poured in to ice water, stirred for 1 hour and organic layer was separated. The aqueous layer was extracted with dichloroethane. Organic layers were combined and washed with water, sodium bicarbonate solution and dried over anhydrous sodium sulphate. Solvent was distilled out completely and the crude product was distilled under vacuum to obtain 2-chloroindan as a colorless liquid (118 g).
Example 10
Process for preparing 5-acetyl-2-chloroindan
Aluminium chloride (146 g) was added in small lots to nitromethane (500 ml) and the solution was cooled to 5° C under inert atmosphere while stirring. Acetyl chloride (84 g) was slowly added keeping the temperature at 5° C. Solution of 2-chloroindan (118 g) was slowly added in acetyl chloride (84 g) keeping temperature at 5° C. After completion of reaction, monitored by TLC, the reaction mass was poured into cold IN HC1 (2000 ml) solution and stirred for 30 minutes. The product was extracted into di-isopropyl ether. The combined organic layer was washed with water, bicarbonate solution, brine and dried over anhydrous sodium sulphate. The solvent was completely distilled out to obtain 5-acetyl-2-chloroindan as yellow waxy solid (130 g).
Example 11
Process for preparing 2-chloro-5-ethylindan
1 Liter hydrogenation vessel was charged with 50 grams of 5-acetyl-2-chloroindan, 400 ml of methanol and 10 ml of acetic acid. Palladium on charcoal 5% (5 g) was added and the reaction mass was hydrogenated until complete conversion to 2-chloro-5-ethylindan. The mixture was filtered over a bed of celite. The filtrate was concentrated under reduced pressure to obtain 2-chloro-5-ethylindan as an oily mass (42 g).
Example 12
Process for preparing 5-acetyl-2-chloro-6-ethylindan
5-acetyl-2-chloro-6-ethylindan was prepared from 2-chloro-5-ethylindan (20 g) in accordance with the procedure followed in Example 10.
Example 13
Process for preparing 2-chloro-5, 6-diethylindan
Hydrogenation of 5-acetyl-2-chloro-6-ethylindan using Palladium on charcoal adopting the procedure as reported in Example 11, gave 2-chloro-5, 6-diethylindan as a liquid. The crude product was distilled under vacuum to get colorless liquid.
1H-NMR (CDC13) ppm: 1.19-1.29 (t, 6H), 2.61-2.66 (q, 4H), 3.13-3.18 (dd, 2H), 3.36-3.41 (dd, 2H), 4.66-4.72 (m, 1H), 7.05 (s, 2H).
////////////WO 2016027283, New patent, Indacaterol, Reddy-Cheminor Inc
Filed under: PATENT, PATENTS Tagged: indacaterol, NEW PATENT, Reddy-Cheminor Inc, WO 2016027283







