WO 2016024284, New Patent, MIRABEGRON, Wanbury Ltd

WANBURY LTD. [IN/IN]; BSEL tech park, B wing, 10th floor, sector 30A opp. Vashi Railway Station, Vashi Navi Mumbai 400703 Maharashtra (IN)
DR. NITIN SHARADCHANDRA PRADHAN; (IN).
DR. NILESH SUDHIR PATIL; (IN).
DR. RAJESH RAMCHANDRA WALAVALKAR; (IN).
MR. NILESH SUBHASH KULKARNI; (IN).
MR. SANTOSH NAMDEV RAWOOL; (IN).
MR. PURUSHOTTAM EKANATH AWATE; (IN)

LEFT , DR K CHANDRAN, DIRECTOR WANBURY
MR ASOK SHINKAR
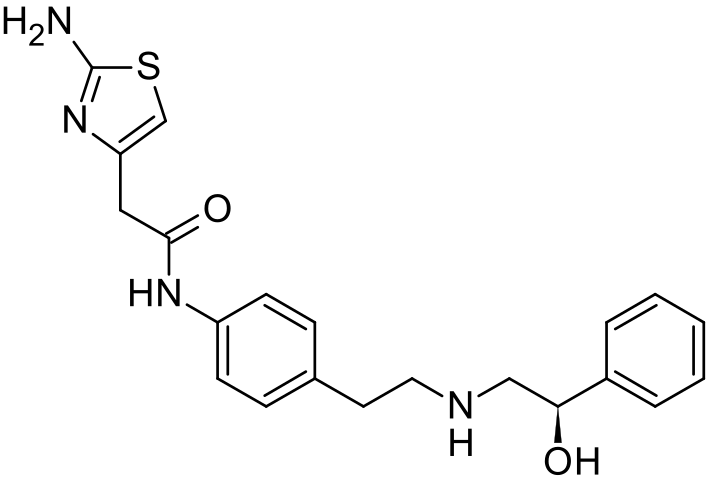
The present invention relates to a novel process for preparation of Mirabegron of Formula (I) using intermediates of Formula (II), (IIIa), (Illb) and (IV).
The present invention relates to a process for preparation of Mirabegron of Formula
(I).

Formula (I)
The present invention further relates to the preparation of Mirabegron of Formula (I) by using compounds of Formula (II), (Ilia), (Illb) and (IV)

Formula (II)

Formula (IlIa) Formula (Illb)

Formula (IV)
Furthermore, the present invention relates to process for preparation of compound of Formula (II), (Ilia), (Illb) and (IV).
Background of the invention:
Mirabegron is chemically known as 2-amino-N-[4-[2-[[(2R)-2-hydroxy-2-phenylethyl]amino]ethyl]phenyl]-4-thiazoleactamide and is marketed under trade name Myrbetiq.
Mirabegron is a drug used for treatment of overactive bladder. It was first disclosed in US 6,346,532, wherein (R)-Styrene oxide is reacted with 4-nitrophenyl ethyl amine hydrochloride to obtain (R)-l- phenyl-2-[[2-(4-nitrophenyl)ethyl]amino]ethanol, the later is then protected with BOC anhydride and subjected to reduction in the presence of Pd/C to yield N-[2-(4-Aminophenyl)ethyl]-N-[(2R)-2-hydroxy-2-phenylethyljcarbamic acid tert-butyl ester. Thus formed compound was then coupled with (2-amino-l,3-thiazol-4yl) acetic acid to obtain BOC protected Mirabegron which is de-protected to give Mirabegron hydrochloride.
The synthetic route proposed in US 6,346,532 is presented in Scheme-I.
Scheme-I

The major draw-backs of the presented synthetic scheme are as follows:
1. Less atomic efficiency
2. Low yield and extensive impurities formations
3. Use of expensive and sensitive protecting agents
4. Column chromatographic techniques for purifications of intermediates.
One more synthetic route for the preparation of Mirabegron have been proposed US 6,346,532, however it is not exemplified.
US 7,342,117 disclose a process for preparation of Mirabegron. The process involves the step of condensation of 4-nitrophenyl ethylamine and (R)- mandelic acid in presence of tri ethylamine, hydroxybentriazole and l-(3-dimethylaminopropyl)-3-ethyl carbodiimide in N,N-dimethylformamide to obtain compound of Formula (A). The second step involves conversion of compound of Formula (A) to compound of Formula (B) in presence of l,3-dimethyl-2-imidazolidone and borontetrahydro fluoride in tetrahydrofuran. In third step, compound of Formula (B) is subjected to reduction using 10% palladium-carbon in methanol to afford (R)-2-[[2′-(4-aminophenyl)-ethyl amino] -1-phenylethanol (Formula IV), which was further condensed with 2-aminothiazol-4-yl acetic acid in presence of l-(3-dimethylaminopropyl)-3 -ethyl carbodiimide and hydrochloric acid in water to obtain Mirabegron of Formula (I). The schematic representation is as Scheme-II

Another patent application CN103193730, discloses a novel process for preparation of Mirabegron wherein the amino group of 2-aminothiazole-5-acetic acid is protected with a protecting group and is condensed with 4-amino phenyl ethanol to obtain an intermediate (A); which on further oxidation yields intermediate (B). The intermediate B is subjected to reductive amination with (R)-2-amino-l -phenyl ethanol and deprotection, simultaneously to yield Mirabegron. The schematic representation is as Scheme-Ill.

Formula (I)
Scheme-Ill
Other references wherein process for preparation of Mirabegron are disclosed CN103387500 and CN103232352.
Most of the prior art reported for preparation of Mirabegron uses expensive and sensitive protecting agents thereby making process less feasible on industrial scale. Furthermore, the yield and purity of Mirabegron obtained by the processes known in art is not satisfactory. It is well known fact that pharmaceutical products like Mirabegron should have high purity due to the therapeutic advantages and also due to the stringent requirements of regulatory agencies. The purity requirements can be fulfilled either by avoiding the formation of by-products during the process or by purifying the end product of the process. The inventors of present invention have skillfully developed the process to provide Mirabegron with unachieved level of purity. Furthermore, the process of present invention is simple, industrially viable, and economic and avoids unfavorable reaction conditions.
![]()
According to present invention, the process for preparation of compound of Formula (IV), is depicted in Scheme IV

The present invention further relates to a process for preparation of Mirabegron of Formula (I)
The schematic reaction scheme of Mirabegron according to present invention is depicted in Scheme-V.
 Wherein R is -OH or -CI
Wherein R is -OH or -CI
The detail of the invention provided in the following examples is given by the way of illustration only and should not be construed to limit the scope of the present invention.
EXAMPLES
Example 1: Preparation of [2-(formylamino)-l,3-thiazol-4-yl]acetyl chloride; Formula (V); wherein R is -CI
20g of [2-(formylamino)-l,3-thiazol-4-yl]acetic acid was added to 250 ml of methylene dichloride and the mixture was cooled to -10°C followed by lot wise addition of 25g of phosphorous pentachloride. The mixture stirred while maintaining temperature of -10°C for 2-3 hours. After confirming completion of reaction, the product was filtered out, washed with methylene dichloride and dried to obtain 24g (Yield: 92%) of compound of Formula (V); wherein R is -CI
Example 2: Preparation of 4-nitrophenyl-[2-(formylamino)-l,3-thiazol-4-yl]acetate; Formula (IlIa)
2g of p-nitrophenol was added to 40ml of methylene chloride and 4.963g of potassium carbonate, the mixture was cooled to 10-15°C followed by lot wise addition of 3.95g of compound of Formula (V) of example 1. After confirming completion of reaction, 5.87g (Yield: 99%) of compound of Formula (Ilia) was isolated. The obtained compound has been identified by;
HNMR(D20 Exchange)
8.614 (S,lH),7.359(d,2H),8.119(d,2H),6.561(S,lH),3.765(S,2H).
Example 3: Preparation of (2-amino-l,3-thiazol-4-yl)acetyl chloride; Formula (VI); wherein R is -CI
5g of (2-amino-l,3-thiazol-4-yl)acetic acid was added to 50 ml of methylene dichloride with few drops of dimethylformamide and 6g of oxalyl chloride at temperature ranging from 0-5°C. the mixture was maintained at 0-5°C for 4-5 hours and after completion of reaction, solid mass was filtered out, washed with methylene dichloride and dried to afford 5g (Yield: 89%) of compound of Formula (VI); wherein R is -CI
Example 4: Preparation of 4-nitrophenyl-(2-amino-l,3-thiazol-4-yl)acetate; Formula (Illb)
2g of p-nitrophenol was added to 40ml of methylene chloride and 4.96g of potassium carbonate, and the mixture was cooled to 10-15 °C followed by lot wise addition of 3.95g of compound of Formula (VI) prepared in example 3. After confirming completion of reaction, 6.18g (Yield: 99%) of 4-nitrophenyl-(2-amino-l,3-thiazol-4-yl)acetate of Formula (Illb) was isolated.
The obtained compound has been identified by
HNMR ( D2O Exchange)
7.359(d,2H),8.1 19(d,2H),6.425(S,lH).3.775(S,2H).
Example 5: In-situ preparation of (lR)-2-[[2-(4-aminophenyl)ethyl]amino]-l-phenylethanol or its hydrochloride salt, of Formula (IV)
Step I – Preparation of (2R)-2-hydroxy-N-[2-(4-nitrophenyl)ethyl]-2-phenylethanamide of Formula (IX)
(R)-2-hydroxy-2-phenylacetic acid (75g), triethylamine (50g), hydroxybenzotriazole (HOBt) (33.3g) and l-(3-dimethylaminopropyl)-3-ethyl carbodiimide hydrochloride (EDC.HC1) (50g) were added to a mixture of 2-(4-nitrophenyl)ethylamine hydrochloride (100g) in Ν,Ν-dimethylformamide (375ml) at 25-30°C. The mixture was stirred for 30 minutes followed by addition of another lot of HOBt (33.3g) and EDC.HC1 (50g) in reaction mixture. The reaction mixture was maintained at 25-30°C for 15 hours under stirring. After completion of reaction, water (1850ml) was added to the reaction mixture and stirred. Subsequently, ethyl acetate (1500ml) was added to the reaction mixture at 25-30°C and stirred. The organic phase was separated from aqueous phase, and was washed sequentially with 1M HC1 solution, 20%aqueous potassium carbonate solution and water. The organic solvent was distilled out under reduced pressure to obtain residue comprising of (2R)-2-hydroxy-N-[2-(4-nitrophenyl)ethyl] -2 -phenyl ethanamide of Formula (IX)
Step II – Preparation of (2R)-2-hydroxy-N-[2-(4-aminophenyl)ethyl]-2-phenylethanamide of Formula (X)
The residue from step I, methanol (740ml) and Raney Nickel (14.8g) were charged into an autoclave vessel, 10 kg/cm2 hydrogen gas pressure was applied to the reaction mixture at 25-30°C and the mixture was maintained under stiring 6 hours. Reaction mixture filtered through hyflo bed. Distilled off the solvent completely from the filtrate under reduced pressure to obtain residue comprising (2R)-2-hydroxy-N-[2-(4-aminophenyl)ethyl]-2-phenylethanamide of Formula (X)
Step III – Preparation of (lR)-2-[[2-(4-aminophenyl)ethyl]amino]-l-phenylethanol dihydrochloride salt, of Formula (IV)
The residue of step II was added in tetrahydrofuran (665ml) and the mixture was cooled to -5 to 0°C. To this cooled mixture was then successively added sodium borohydride (56.26g) and BF3-diethyl ether (466g), and the mixture was stirred for 15 minutes. The temperature of reaction mixture was gradually increased to 50-55°C and was maintained under stirring for 5 hours. After completion of reaction, the reaction mixture was cooled to 0-5°C and 50% sodium hydroxide solution was added till pH is basic. The temperature of reaction mixture is then raised to 25-30°C followed by addition of ethyl acetate (500ml). The organic layer was separated and subjected to distillation to afford a residue. To the residue was added isopropyl alcohol (665ml) and mixture was refluxed for 30 minutes. The mixture was then allowed to cool to 40-45°C, isopropyl alcohol hydrochloride (200ml) was added till pH acidic and mixture was stirred for 2 hours to afford precipitate. The precipitate was filtered out and washed with isopropyl alcohol. The wet cake thus obtained was added to 20% aqueous sodium hydroxide solution (till pH basic) followed by addition of dichloromethane (500ml). The organic layer was separated from aqueous layer and was subjected to distillation under reduced pressure to obtain residue. The residue was taken in toluene (500ml), heated to 55-60°C for 30 minutes and cooled to 10-15°C. The precipitate obtained was filtered, washed with toluene and to the wet cake afforded was added isopropyl alcohol (665ml). The mixture was refluxed for 30 minutes and then cooled to 50-55°C. At 50-55°C slowly isopropyl alcohol hydrochloride (200ml) till pH acidic was added and mixture was stirred for 2 hours to obtain precipitate. The precipitate was filtered out, washed with isopropyl alcohol and dried to get (lR)-2-[[2-(4-aminophenyl)ethyl]amino]-l-phenylethanol dihydrochloride salt, of Formula (IV)
Yield-70%
HPLC Purity: 98%
Example 6: Alternate method for preparation of (2R)-2-hydroxy-N-[2-(4-nitrophenyl)ethyl]-2-phenylethanamide of Formula (IX)
Step I – A mixture of (R)-2-hydroxy-2-phenylacetic acid (lOg), dichloromethane (50ml) and triethylamine (24ml) was cooled to 0-5°C and slowly para-toluene sulfonyl chloride (12.53g) was added to it. The temperature of reaction mixture was raised to 25-30°C and maintained for 12 hours. After completion of reaction, water (100ml) was added to the reaction mixture and the mixture was stirred for 15 minutes. The organic phase was separated and distills out completely under reduced pressure to obtain [(R)-2-hydroxy -2-phenyl acetic tosyl ester].
Yield-56%
Step II – 2-(4-nitrophenyl)ethylamine hydrochloride (6g) was added to dichloromethane (50ml) and stirred for 30 minutes at 25-30°C. The mixture was
then cooled to 0-5 °C and triethylamine (13ml) was added. To say cooled mixture was then slowly added a mixture of (R)-2-hydroxy -2-phenyl acetic tosyl ester (lOg) and dichloromethane (50ml). The temperature of reaction mixture was then raised to reflux temperature and maintained for 5 hours. After completion of reaction, water (50ml) was added to the reaction mixture and the mixture was stirred for 15 minutes. The organic phase was separated and distill out completely under reduced pressure to obtain (R)-2-hydroxy-N-[2-(4-nitrophenyl) ethyl]-2-phenylacetamide
Yield-70%, Purity-96%
Example 7: Preparation of compound of Formula (II) from compound of Formula (V); wherein R is -OH
1.58g of [2-(formylamino)-l,3-thiazol-4-yl]acetic acid of Formula (V) was added solution of (1R )-2-{[2-(4-aminophenyl)ethyl]amino}-l-phenylethanol of Formula (IV) in water (2g of Formula (IV) in 50ml water) followed by addition of 0.66g concentrated hydrochloric acid and 3.27g of l-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride. The mixture was stirred at 25-30°C for 0.5 hours. After completion of reaction, pH was adjusted to 8-9 using aqueous saturated solution of sodium carbonate. The solid precipitated out was filtered, washed with water and dried to obtain 2.1g of compound of Formula (II). (Yield: 72%) The obtained compound has been identified by HNMR
2.502(m,4H),2.599(m,2H),3.685(S,2H),4.9(S, NH protons),7.01(m, 10H, aromatic), 8.54(S,1H), 10.0(S, -OH proton),
HNMR(D20 Exchange) 2.502(m,4H),2.60(m,2H),4.57(m,lH),7.0(m, 10H, aromatic), 8.43(S,1H)
Example 8: Preparation of compound of Formula (II) from compound of Formula (V); wherein R is -CI
lOg of ( 1R)-2-{[2-(4-aminophenyl)ethyl]amino}-l-phenylethanol of Formula (IV) (prepared by methods known in prior art/ as given in example 5), was added to 150ml of acetonitrile with 16.17g of potassium carbonate and the mixture was cooled to 10-15°C. 18.8g of Formula (V) of example 1 was added to above mixture at 10-15°C in lot wise. After completion of reaction, the reaction mixture was concentrated under vacuum and 90ml of water was added for isolation. The product was then filtered out, washed with water and dried to obtain 72g (Yield: 70%) of compound of Formula (II).
Example 9: Preparation of compound of Formula (II) from compound of Formula (IlIa)
5.87g of compound of Formula (IlIa) was added to 40 ml of methylene dichloride with 2.36 g of potassium carbonate and 3.67g of ( 1))-2-{[2-(4-aminophenyl)ethyl]amino}-l-phenylethanol (Formula-IV ; prepared by methods known in prior art/ as given in example 5) . The mixture was stirred at 25-30°C for 1 hour. After completion of reaction, the reaction mixture was concentrated followed by addition of 60 ml of water to isolate lg of compound of Formula (II).
Example 10: Insitu preparation of compound of Formula (II) without isolation of compound of Formula (IlIa)
2g of p-nitrophenol was added to 40 ml of methylene chloride with 4.963g of potassium carbonate, and the mixture was cooled to 10-15°C followed by lot wise addition of 3.95g of [2-(formylamino)-l,3-thiazol-4-yl]acetyl chloride of Formula (V) of example 1. After confirming complete formation of compound of Formula (Ilia), 2.36g of potassium carbonate and 3.67g of (1R)-2-{[2-(4-aminophenyl)ethyl]amino}-1 -phenyl ethanol of Formula (IV) (prepared by methods known in prior art/ as given in example 5) was added insitu, and the mixture was stirred at 25-30°C for 1 hour. After completion of reaction, the reaction mixture was concentrated followed by addition of 60 ml of water to isolate lg of compound of Formula (II).
Example 11: Preparation of Mirabegron from compound of Formula (II)
To 2g of compound of Formula (II) was added 30ml of 10% sodium hydroxide and the mixture was stirred at 55-60°C for 3 hours. After completion of reaction, the mixture was cooled to 25-30°C and the solid obtained was filtered, washed with water and dried to yield 1.3g of Mirabegron. (Yield: 70%)
Example 12: Preparation of Mirabegron from compound of Formula (Illb)
6.18g of 4-nitrophenyl-(2-amino-l,3-thiazol-4-yl)acetate was added to 40ml of methylene dichloride with 2.36g of potassium carbonate and 3.65g of (1R)-2-{ [2-(4-aminophenyl)ethyl]amino}-l-phenylethanol of Formula (IV) (prepared by methods known in prior art/ as given in example 5), and the mixture was stirred at 25-30°C for 1 hour. After completion of reaction, solid was filtered out, washed with methylene dichlrode and dried to yield lg of Mirabegron of Formula (I).
Example 13: Insitu preparation of Mirabegron without isolation of compound of Formula (Illb)
To 40ml of methylene chloride was added 2g of p-nitrophenol and 4.96g of potassium carbonate, and the mixture was cooled to 10-15°C followed by lot wise addition of 3.95g of compound of Formula (VI) prepared in example 3. After confirming complete formation of compound of Formula (Illb), 2.36g of potassium carbonate and 3.65g of (1R)-2-{[2-(4-aminophenyl)ethyl]amino}-l-phenylethanol of Formula (IV) (prepared by methods known in prior art/ as given in example 5) was added insitu, and the mixture was stirred at 25-30°C for 1 hour. After completion of reaction, After completion of reaction, solid was filtered out, washed with methylene dichlrode and dried to yield lg of Mirabegron of Formula (I).
Example 14: Preparation of Mirabegron from compound of Formula (VI); wherein R is -CI
To 20ml of acetone was added 2g of (l/?)-2-{[2-(4-aminophenyl)ethyl]amino}-l-phenylethanol of Formula (IV) and 2.15g of potassium carbonate, and the mixture was cooled to 10-15°C followed by addition of (2-amino-l,3-thiazol-4-yl)acetyl chloride of Formula (VI). After completion of reaction, acetone was concentrated under vacuum and 90ml of water was added for for isolation. The product was then filtered out, washed with water and dried to obtain 2g (Yield: 70%) of Mirabegron.



/////WO-2016024284, WO 2016024284, New Patent, MIRABEGRON, Wanbury Ltd
Filed under: PATENT, PATENTS, Uncategorized Tagged: Mirabegron, NEW PATENT, Wanbury, WO 2016024284







