
Zavegepant
ザベジェパント;
- 1337918-83-8
- as HCl: 1414976-20-7
C36H46N8O3 BASE
638.8 g/mol BASE
- Vazegepant
- BMS-742413
- BHV-3500
FDA APPR 3/9/2023Zavzpret
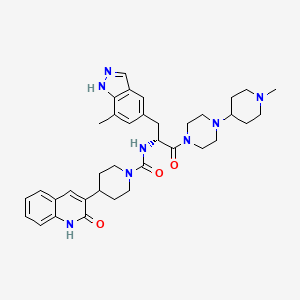
N-[(2R)-3-(7-methyl-1H-indazol-5-yl)-1-[4-(1-methylpiperidin-4-yl)piperazin-1-yl]-1-oxopropan-2-yl]-4-(2-oxo-1H-quinolin-3-yl)piperidine-1-carboxamide
ZAVZPRET is indicated for the acute treatment of migraine with or without aura in adults.
The recommended dose of ZAVZPRET is 10 mg given as a single spray in one nostril, as needed. The maximum dose that may be given in a 24-hour period is 10 mg (one spray). The safety of treating more than 8 migraines in a 30-day period has not been established, Nasal spray: 10 mg of zavegepant per device. Each unit-dose nasal spray device delivers a single spray containing 10 mg of zavegepant.
ZAVZPRET (zavegepant) nasal spray contains zavegepant hydrochloride, a calcitonin generelated peptide receptor antagonist. Zavegepant hydrochloride is described chemically as (R)-N- (3-(7-methyl-1H-indazol-5-yl)-1-(4-(1-methylpiperidin-4-yl) piperazin-1-yl)-1-oxopropan-2-yl)- 4-(2-oxo-1,2-dihydroquinolin-3-yl) piperidine-1-carboxamide hydrochloride and its structural formula is:

Its molecular formula is C36H46N8O3․HCl, representing a molecular weight of 675. 28 g/mol. Zavegepant free base has a molecular weight of 638.82 g/mol. Zavegepant hydrochloride is a white to off-white powder, freely soluble in water, and has pKa values of 4.8 and 8.8. Each unit-dose ZAVZPRET device for nasal administration delivers 10 mg of zavegepant (equivalent to 10.6 mg of zavegepant hydrochloride) in a buffered aqueous solution containing dextrose, hydrochloric acid, sodium hydroxide, and succinic acid in water for injection. The solution has a pH of 5.3 to 6.7.
Active ingredients in ZAVZPRET: zavegepant Inactive ingredients in ZAVZPRET: dextrose, hydrochloric acid, sodium hydroxide, and succinic acid in water for injection.
Zavegepant, sold under the brand name Zavzpret, is a medication used for the treatment of migraine.[1] Zavegepant is a calcitonin gene-related peptide receptor antagonist.[1] It is sprayed into the nose.[1] It is sold by Pfizer.[1]
The most common adverse reactions include taste disorders, nausea, nasal discomfort, and vomiting.[1]
Zavegepant was approved for medical use in the United States in March 2023.[1][2][3]
Medical usesZavegepant is a Calcitonin Gene-related Peptide Receptor Antagonist. The mechanism of action of zavegepant is as a Calcitonin Gene-related Peptide Receptor Antagonist.
Zavegepant is indicated for the acute treatment of migraine with or without aura in adults.[1]
Zavegepant is an antagonist of the calcitonin gene-related peptide (CGRP) receptor currently in phase 3 trials in an intranasal formulation for the treatment of migraine. If FDA approved, it will join other previously-approved “-gepant” drugs [rimegepant] and [ubrogepant] as an additional treatment alternative for patients with migraine, particularly those for whom traditional triptan therapy has proven ineffective. On April 15th, 2020, a phase 2 clinical trial (NCT04346615: Safety and Efficacy Trial of Vazegepant Intranasal for Hospitalized Patients With COVID-19 Requiring Supplemental Oxygen) began to investigate the use of intranasally administered zavegepant to combat the acute respiratory distress syndrome (ARDS) sometimes seen in patients with COVID-19. Acute lung injury activates the release of CGRP, which plays a role in the development of ARDS – CGRP antagonists, then, may help to blunt the significant inflammation associated with COVID-19. The clinical trial is expected to complete in September 2020.
Zavegepant is a highly soluble small molecule calcitonin gene related peptide (CGRP) receptor antagonist, with potential analgesic and immunomodulating activities. Upon administration, zavegepant targets, binds to and inhibits the activity of CGRP receptors located on mast cells in the brain. This may inhibit neurogenic inflammation caused by trigeminal nerve release of CGRP. In addition, by blocking the CGRP receptors located in smooth muscle cells within vessel walls, zavegepant inhibits the pathologic dilation of intracranial arteries. Zavegepant, by blocking the CGRP receptors, also suppresses the transmission of pain by inhibiting the central relay of pain signals from the trigeminal nerve to the caudal trigeminal nucleus. Altogether, this may relieve migraine. As CGRP receptors induce the release of pro-inflammatory mediators, such as interleukin-6 (IL-6), from inflammatory cells, zavegepant may prevent an IL-6-mediated inflammatory response. Zavegepant may also inhibit the CGRP-mediated induction of eosinophil migration and the stimulation of beta-integrin-mediated T cell adhesion to fibronectin at the site of inflammation, and may abrogate the CGRP-mediated polarization of the T cell response towards the pro-inflammatory state characterized by Th17 and IL-17. This may improve lung inflammation and oxygenation, prevent edema, and further lung injury. CGRP, a 37 amino-acid peptide expressed in and released from a subset of polymodal primary sensory neurons of the trigeminal ganglion and nerve fibers projecting to the airways and by pulmonary neuroendocrine cells, plays an important role in pain transmission, inflammation, and neurogenic vasodilatation. It is released upon acute lung injury and upregulation of transient receptor potential (TRP) channels.
SYN’
Synthesis of a CGRP Receptor Inhibitor
Publication Date: 2013
Publication Name: Synfacts
Azepino-indazoles as calcitonin gene-related peptide (CGRP) receptor antagonists
- PMID: 33096162Publication Date: 2021-01-01Journal: Bioorganic & medicinal chemistry lettersDiscovery of (R)-N-(3-(7-methyl-1H-indazol-5-yl)-1-(4-(1-methylpiperidin-4-yl)-1-oxopropan-2-yl)-4-(2-oxo-1,2-dihydroquinolin-3-yl)piperidine-1-carboxamide (BMS-742413): a potent human CGRP antagonist with superior safety profile for the treatment of migraine through intranasal delivery
PMID: 23632269Publication Date: 2013-06-01Journal: Bioorganic & medicinal chemistry letters

////////

AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
Patent
https://patents.google.com/patent/US20120245356A1/en
Patent
WO 2022165291
https://patents.google.com/patent/WO2022165291A1/en
Migraine is a chronic and debilitating disorder characterized by recurrent attacks lasting four to 72 hours with multiple symptoms, including typically one-sided, pulsating headaches of moderate to severe pain intensity that are associated with nausea or vomiting, and/or sensitivity to sound (phonophobia) and sensitivity to light (photophobia). Migraines are often preceded by transient neurological warning symptoms, known as auras, which typically involve visual disturbances such as flashing lights, but may also involve numbness or tingling in parts of the body. Migraine is both widespread and disabling. The Migraine Research Foundation ranks migraine as the world’s third most prevalent illness, and the Global Burden of Disease Study 2015 rates migraine as the seventh highest specific cause of disability worldwide. According to the Migraine Research Foundation, in the United States, approximately 36 million individuals suffer from migraine attacks. While most sufferers experience migraine attacks once or twice per month, more than 4 million people have chronic migraine, defined as experiencing at least 15 headache days per month, of which at least eight are migraine, for more than three months. Others have episodic migraine, which is characterized by experiencing less than 15 migraine days per month. People with episodic migraine may progress to chronic migraine over time. Migraine attacks can last four hours or up to three days. More than 90% of individuals suffering from migraine attacks are unable to work or function normally during a migraine attack, with many experiencing comorbid conditions such as depression, anxiety and insomnia. Also, those suffering from migraine often have accompanying nausea and have an aversion to consuming food or liquids during an attack.
CGRP (calcitonin gene-related peptide) is a 37 amino acid neuropeptide, which belongs to a family of peptides that includes calcitonin, adrenomedullin and amylin. In humans, two forms of CGRP (a-CGRP and 0-CGRP) exist and have similar activities. They vary by three amino acids and exhibit differential distribution. At least two CGRP receptor subtypes may also account for differential activities. The CGRP receptor is located within pain-signaling pathways, intracranial arteries and mast cells and its activation is thought to play a causal role in migraine pathophysiology. For example, research and clinical studies have shown: serum levels of CGRP are elevated during migraine attacks, infusion of intravenous CGRP produces persistent pain in migraine sufferers and non-migraine sufferers, and treatment with anti-migraine drugs normalizes CGRP activity.
Currently, clinicians use a number of pharmacologic agents for the acute treatment of migraine. A study published by the American Headache Society in 2015 concluded that the medications deemed effective for the acute treatment of migraine fell into the following classes: triptans, ergotamine derivatives, non-steroidal anti-inflammatory drugs (“NSAIDs”), opioids and combination medications. The current standard of care for the acute treatment of migraine is prescription of triptans, which are serotonin 5-HT IB/ID receptor agonists. Triptans have been developed and approved for the acute treatment of migraine over the past two decades. The initial introduction of triptans represented a shift toward drugs more selectively targeting the suspected pathophysiology of migraine. While triptans account for almost 80% of anti-migraine therapies prescribed at office visits by healthcare providers, issues such as an incomplete effect or headache recurrence remain important clinical limitations. In fact, only about 30% of patients from clinical trials are pain free at two hours after taking triptans. In addition, triptans are contraindicated in patients with cardiovascular disease, cerebrovascular disease, or significant risk factors for either because of potential systemic and cerebrovascular vasoconstriction from the 5-HT IB -mediated effects. Also, according to a January 2017 study published in the journal Headache, an estimated 2.6 million migraine sufferers in the United States have a cardiovascular event, condition or procedure that limits the potential of triptans as a treatment option.
Accordingly, there remains a significant unmet medical need for a novel migraine-specific medication that provides enhanced patient benefits compared to existing therapies.
Possible CGRP involvement in migraine has been the basis for the development and clinical testing of a number of compounds, including for example, advanced clinical candidates rimegepant (BHV-3000) and zavegepant (BHV-3500), which are developed by Biohaven Pharmaceutical Holding Company Ltd., New Haven, CT.
Zavegepant (also known as vazegepant) is a third generation, high affinity, selective and structurally unique small molecule CGRP receptor antagonist having the following formula I:

I
Zavegepant is described, for example, in WO 03/104236 published December 18, 2003 and US 8,481,546 issued July 9, 2013, which are incorporated herein in their entireties by reference.
While zavegepant is a highly soluble molecule, its bioavailability characteristics may render it challenging to prepare the drug in an oral dosage form. Enhancing the bioavailability of zavegepant and other CGRP inhibitors by different administration routes would therefore be desirable.
Calcitonin gene-related peptide (CGRP) is widely distributed in nociceptive pathways in human peripheral and central nervous system and its receptors are also expressed in pain pathways. While CGRP is involved in migraine pathophysiology, its role in non-headache pain has not been quite clear. There remains a need for new medicines to treat various pain disorders in patients in need thereof.
Scheme 1


Scheme 3

Scheme 4

tert-butyl 4-(2-methoxy-2-oxoethylidene)piperidine-l -carboxylate. Sodium hydride in mineral oil (60%, 7.92 g, 198.02 mmoles) was washed with hexanes then suspended in dimethylformamide (220 mL). The mixture was cooled to 0°C. Trimethyl phosphonoacetate (29.0 mL, 189.82 mmoles) was added dropwise to the stirred reaction mixture. After 20 min at 0°C, a solution of A-/c/7-butoxycarbonyl-4-pi peri done (30.41 g, 152.62 mmoles) in dimethylformamide (80 mL) was added to the mixture dropwise. The reaction was stirred at room temperature for 3 h and then diluted with diethyl ether (650 mL). The mixture was washed once with water and the aqueous layer was extracted once with diethyl ether. The combined organic layers were washed 4 times with water and the aqueous phase was discarded. The organic phase was washed with brine and dried over magnesium sulfate, filtered, and concentrated to dryness. The title compound was obtained as a white solid in 92% yield. 1 H- NMR (300 MHz, CDCh): 5 = 5.68 (s, 1 H), 3.66 (s, 3 H), 3.40-3.51 (m, 4 H), 2.90 (t, J= 5.49, 2 H), 2.25 (t, J= 5.49, 2 H), 1.44 (s, 9 H).

ed-butyl 4-(2-methoxy-2-oxoethyl)piperidine-l -carboxylate. A solution of tert-butyl 4- (2-methoxy-2-oxoethylidene)piperidine-l -carboxylate (35.71 g, 140 mmoles) in a mixture of 1 : 1 ethyl acetate/methanol (220 mL) was carefully treated with 50% wet 10% palladium on carbon (3.3 g). The reaction vessel was charged with 55 psi of hydrogen gas and the mixture was shaken on a Parr apparatus at room temperature for 16 h. The reaction mixture was then filtered to remove the catalyst and the filtrate concentrated in vacuo. The title compound was obtained as a clear colorless oil in 97% yield. ‘H-NMR (300 MHz, CDCh): 5 = 4.04 (d, J= 10.25, 2 H), 3.64 (s, 3 H), 2.68 (t, J= 12.44, 2 H), 2.21 (d, J= 6.95, 2 H), 1.98-1.77 (m, 1 H), 1.64 (d, J= 13.54, 2 H), 1.41 (s, 9 H), 1.25-0.99 (m, 2 H).

4-[2-Hydroxy-l-methoxycarbonyl-2-(2-nitro-phenyl)-ethyl]-piperidine-l-carboxylic acid tert-butyl ester. A A-diisopropylamine (4.40 mL, 31.3 mmoles) was dissolved in tetrahydrofuran (50 mL). The mixture was cooled to -78°C. Butyllithium (2.5 M in hexanes, 12.4 mL, 31 mmoles) was added dropwise to the stirred solution. After stirring at -78°C for 30 min, a solution of tert-butyl 4-(2-methoxy-2-oxoethyl)piperidine-l -carboxylate (6.65 g, 25.8 mmoles) in tetrahydrofuran (15 mL) was added dropwise to the mixture. Stirring was continued at -78°C for 1 h. A solution of 2-nitrobenzaldehyde (3.90 g, 25.8 mmoles) in tetrahydrofuran (20 mL) was then added to the mixture dropwise, and then stirring was continued at -78°C for a further 2.5 h. The reaction was quenched with cold aqueous ammonium chloride and then diluted with water. The mixture was extracted twice with ethyl acetate and the aqueous phase was discarded. The material was dried (magnesium sulfate) filtered, and concentrated to dryness. Silica gel chromatography afforded the desired product in 94% yield as light yellow foam. MS m/e (M- C4H8+H)+= 353.1.

4-(4-Hydroxy-2-oxo-l , 2, 3, 4-tetrahydro-quinolin-3-yl)-piperidine-l -carboxylic acid tertbutyl ester. In a 3 neck flask fitted with a nitrogen inlet, thermometer, and a mechanical stirrer, 4-[2-hydroxy-l -methoxy carbonyl-2-(2-nitro-phenyl)-ethyl]-piperidine-l -carboxylic acid tertbutyl ester (9.93 g, 24.3 mmoles) was dissolved in acetic acid (1.75 moles, 100 mL). Iron powder (8.90 g, 159 mmoles) was added to the vessel with stirring. The stirred mixture was slowly heated to 80°C for 30 min and then cooled to room temperature. It was then diluted with ethyl acetate and filtered through a pad of celite. Solids were washed with 20% methanol/ethyl acetate, and then with methanol. The filtrate was concentrated and the residue partitioned between ethyl acetate and aqueous sodium bicarbonate. The layers were separated. The resulting aqueous phase was extracted twice with ethyl acetate. The organic layers were combined. The mixture was washed twice with water and the aqueous phase was discarded. The material was dried (magnesium sulfate) filtered, and concentrated to dryness. Silica gel chromatography afforded the title compound as light yellow foam in 77% yield. MS m/e (M-H)’ = 345.1.

3-(Piperidin-4-yl)quinolin-2(lH) hydrochloride . A stirred solution of 4-(4-hydroxy-2- oxo-l,2,3,4-tetrahydro-quinolin-3-yl)-piperidine-l-carboxylic acid tert-butyl ester (5.60 g, 16.2 mmoles) in ethyl acetate (70 mL) was treated with HC1 in dioxane (4N, 40 mmoles, 10 mL). The mixture was stirred at room temperature for 45 min. More HC1 in dioxane (4N, 120 mmoles, 30 mL) was then added and stirring was continued at room temperature for 16 h. The resulting solid was collected by filtration and washed with ethyl acetate. It was then suspended in 5% water-isopropanol (100 mL) and the mixture was warmed to reflux and stirred for 20 min. The mixture was cooled to room temperature and stirred at room temperature for 16 h. The solid was collected by filtration, washed with isopropanol, and dried under high vacuum. The title compound was obtained as white solid in 75% yield. ‘H-NMR (DMSO-de) 5 11.85 (s, 1 H), 9.02 (bs, 1 H), 8.88 (bs, 1 H), 7.70 (t, J= 3.81 Hz, 2 H), 7.53 – 7.30 (d, J= 8.24 Hz, 1 H), 7.17 (t, J= 7.48 Hz, 2 H), 3.36 (d, J= 12.51 Hz, 2 H), 3.10 – 2.94 (m, 3 H), 2.01 (d, J= 13.43 Hz, 2 H), 1.87 – 1.73 (m, 2 H); MS m/e (M+H)+ = 229.0.

4-Iodo-2,6-dimethylbenzenamine hydrochloride . To a suspension of sodium bicarbonate (126 g, 1.5 moles) and 2,6-dimethylaniline (61.5 mL, 500 mmoles) in methanol (700 mL) was added iodine monochloride (1.0 M in dichloromethane, 550 mL, 550 mmoles) at room temperature over 1 h. After addition was complete, stirring was continued for 3 h. The reaction was filtered to remove excess sodium bicarbonate and the solvent removed in vacuo. The residue was re-dissolved in diethyl ether (1.5 L) and treated with hydrochloric acid (2M in ether, 375 mL, 750 mmoles). The resulting suspension was stored in the freezer (-15°C) overnight. The solid was filtered and washed with diethyl ether until it became colorless, to give 126.5 g (89%) as a grey-green powder. ‘H-NMR (DMSO-de) 5 2.33 (s, 6 H), 7.48 (s, 2 H), 9.05 (bs, 3 H); 13C-NMR (DMSO-de) 5 17.4, 91.5, 133.1, 131.2, 136.9.

Methyl 2 -(benzyloxy carbonyl) acrylate . To a flame dried three-neck round bottom flask, fitted with a mechanical stirrer, was added (S)-methyl 2-(benzyloxycarbonyl)-3- hydroxypropanoate (129 g, 509 mmoles), anhydrous dichloromethane (2 L), and methanesulfonyl chloride (49.3 mL, 636 mmoles). The mixture was cooled to -15°C, and treated with tri ethylamine (213 mL, 1527 mmoles), dropwise, to ensure the temperature of the reaction mixture did not exceed 0°C. The addition of the first equivalent of triethylamine was exothermic. After addition of tri ethylamine, the mixture was stirred at 0°C for 30 min. The cooling bath was removed and the mixture stirred at room temperature for 1.5 h. The reaction was quenched by addition of methanol (21 mL). The mixture was washed with 0.5% aqueous potassium bisulfate until the washings were pH 5, then saturated sodium bicarbonate, and brine, dried over sodium sulfate, and concentrated. Flash chromatography (silica gel, 1 :9 ethyl acetate/hexanes) gave I l l g (92%) as a viscous colorless oil, which crystallized upon standing. ’H-NMR (DMSO-de) 5 3.71 (s, 3 H), 5.10 (s, 2 H), 5.60 (s, 1 H), 5.76 (s, 1 H), 7.39-7.35 (m, 5 H), 8.96 (s, 1 H); 13C-NMR (DMSO-de) 5 52.3, 65.9, 127.8, 128.1, 128.3, 128.8, 133.3, 136.3, 153.5, 163.7.

(Z)-Methyl 3-(4-amino-3,5-dimethylphenyl)-2-(benzyloxycarbonyl) acrylate. A 2 L round bottom flask was charged 4-iodo-2,6-dimethylbenzenamine hydrochloride salt (55 g, 194 mmoles), methyl 2-(benzyloxycarbonyl)acrylate (59.2 g, 252 mmoles), tetrabutylammonium chloride (59.2 g, 213 mmoles), palladium (II) acetate (4.34 g, 19.4 mmoles), and tetrahydrofuran (1.2 L, degassed by a flow of nitrogen for 30 min). The mixture was stirred so that a suspension was formed and then degassed by a flow of nitrogen for 30 min. Triethylamine (110 mL, 789 mmoles) was added and the resulting mixture was heated at reflux for 3 h. After cooling to room temperature, the reaction mixture was filtered through a pad of celite, washed with tetrahydrofuran (2 x 100 mL), and concentrated. The residue was dissolved in di chloromethane, washed with water (3X) and brine (2X), dried over sodium sulfate, and concentrated. Flash chromatography (silica gel, using 1 :9 ethyl acetate/dichloromethane) gave a tan solid. The solid was recrystallized from warm methanol (210 mL) and water (100 mL). The mixture was held at room temperature overnight, then at 0°C for 2 h, and finally at -15°C for 2 h. The resulting solid was filtered, washed with ice cold 1 : 1 methanol/water, and dried under high vacuum overnight to give 44.7 g (65%) as a light tan solid which was a mixture of ZZE isomers (73 :27). ’H-NMR (DMSO-de) 5, 2.05 (s, 6 H), 3.61 (s, 0.8 H), 3.68 (s, 2.2 H), 5.00 (s, 0.54 H), 5.13 (s, 1.46 H), 5.24 (s, 2 H), 7.40-7.21 (m, 8 H), 8.51 (s, 0.27 H), 8.79 (s, 0.73 H); 13C-NMR (DMSO-de) 5 17.8, 51.7, 65.3, 119.4, 120.0, 120.3, 127.3, 127.7, 128.3, 130.9, 135.8, 137.2, 146.9, 154.7, 166.0.

(R)-Methyl 3-(4-amino-3,5-dimethylphenyl)-2-(benzyloxycarbonyl)propanoate. A flame- dried 2 L Parr hydrogenation bottle was charged with (Z)-methyl 3-(4-amino-3,5- dimethylphenyl)-2-(benzyloxycarbonyl)acrylate (84.5 g, 239 mmoles), di chloromethane (300 mL), and methanol (300 mL). The bottle was swirled so that a light brown suspension was formed. The mixture was degassed using a flow of nitrogen for 30 min. To this was quickly added (-)-l,2-bis((2A,5A)-2,5-diethylphospholano)-bezene(cyclooctadiene) rhodium (I) tetrafluoroborate ([(2A,5A)-Et-DuPhosRh]BF4) (2.11 g, 3.20 mmoles). The bottle was immediately attached to a Parr Hydrogenator. After 5 cycles of hydrogen (60 psi) and vacuum, the bottle was pressurized to 65 psi and the suspension was agitated at room temperature for 16 h. The reaction had become homogeneous. The reaction mixture was concentrated, and the resulting residue purified by flash chromatography (silica gel, 1 :9 ethyl acetate/dichloromethane) to give 82.9 g (98%). ‘H-NMR (DMSO-de) 5 2.04 (s, 6 H), 2.65 (dd, J= 13.4, 9.8 Hz, 1H), 2.82 (dd, J= 13.7, 5.2 Hz, 1 H), 3.62 (s, 3 H), 4.15-4.10 (m, 1H), 4.41 (s, 2 H), 5.00 (s, 2 H), 6.68 (s, 2 H), 7.37-7.28 (m, 5 H), 7.70 (d, J= 7.9 Hz, 1 H); 13C-NMR (DMSO-de) 5 17.7, 35.9, 51.7, 56.1, 65.3, 120.4, 124.0, 127.5, 127.7, 128.2, 128.3, 136.9, 142.6, 155.9, 172.5.

(R)-Methyl 2-(benzyloxycarbonyl)-3-(7-methyl-lH-indazol-5-yl)propanoate. (R)-Methyl 3-(4-amino-3,5-dimethylphenyl)-2-(benzyloxycarbonyl)propanoate (50.0 g, 140 mmoles) was weighed into a flame-dried 5 L three neck round bottom flask, followed by the addition of toluene (2.4 L) and glacial acetic acid (120 mL, 2.1 moles). The mixture was mechanically stirred to form a clear solution, and then potassium acetate (103 g, 1.05 moles) was added. To the resulting white suspension, z.w-amyl nitrite (20.7 mL, 154 mmoles) was added dropwise at room temperature, and the resulting mixture was stirred at room temperature for 16 h. Saturated sodium bicarbonate (I L) was added, followed by the careful addition of solid sodium bicarbonate to neutralize the acetic acid. The mixture was extracted with a mixture of di chloromethane (2 L) and brine (1.5 L). After separation, the aqueous layer was extracted with di chloromethane (500 mL). The combined organic layers were dried over anhydrous sodium sulfate and filtered. Solvents were removed to afford a tan solid, which was washed with hexanes (2 L) and toluene (150 mL). The solid was recrystallized from hot acetone (260 mL) and hexanes (700 mL). The slightly cloudy mixture was allowed to cool to room temperature slowly, then to 0°C for 1.5 h, and finally to -15°C for 1.5 h. The resulting solid was filtered and washed with ice-cold acetone/hexanes (1 : 1, 200 mL) to afford 39.1 g (76% yield). Analytical HPLC showed >98% UV purity. The enantiomeric excess (ee) was determined to be 99.8% (conditions: Chiralpak AD column, 4.6 x 250 mm, 10 pm; A = ethanol, B = 0.05% diethylamine/heptane; 85%B @1.0 mL/min. for 55 min. The retention times for R was 44.6 min and for S was 28.8 min). ‘H-NMR (DMSO-de) 5 2.48 (s, 3 H), 2.93 (dd, J= 13.4, 10.7 Hz, 1H), 3.10 (dd, J= 13.7, 4.9 Hz, 1H), 3.63 (s, 3H), 4.32-4.27 (m, 1 H), 4.97 (s, 2 H), 7.03 (s, 1 H), 7.24-7.22 (m, 2 H), 7.29 -7.27 (m, 3 H), 7.41 (s, 1 H), 7.83 (d, J= 8.2 Hz, 1H), 7.99 (s, 1H), 13.1 (s, 1 H); 13C-NMR (DMSO-de) 5 16.7, 36.5, 51.8, 56.0, 65.3, 117.6, 119.6, 122.7, 127.2, 127.4, 127.6, 128.2, 129.3, 133.4, 136.8, 139.2, 155.9, 172.4. Mass spec.: 368.16 (MH)+.

(R)-Methyl 2-amino-3-(7-methyl-lH-indazol-5-yl)propanoate. A Parr hydrogenation bottle was charged with (R)-methyl 2-(benzyloxycarbonyl)-3-(7-methyl-lH-indazol-5- yl)propanoate (11.0 g, 29.9 mmoles) and methanol (75 mL). The suspension was purged with nitrogen and treated with palladium (10% on charcoal, 700 mg). The bottle was shaken under hydrogen (15 psi) overnight. The mixture was filtered through a pad of celite to remove the catalyst. Concentration of the eluent gave 7.7 g (quant.) as an oil which was used without further purification. XH-NMR (CD3OD) 5 2.54 (s, 3 H), 2.98 (dd, J= 13.5, 7.0 Hz, 1 H), 3.09 (dd, J= 13.5, 5.9 Hz, 1 H), 3.68 (s, 3 H), 3.75 (dd, J= 7.0, 6.2 Hz, 1 H), 7.01 (s, 1 H), 7.39 (s, 1 H), 7.98 (s, 1 H). Mass spec.: 232.34 (M-H)’.

(R)-methyl 3-(7-methyl-lH-indazol-5-yl)-2-(4-(2-oxo-l,2-dihydroquinolin-3- yl)piperidine-l-carboxamido)propanoate. To a solution of (R)-methyl 2-amino-3-(7-methyl-lH- indazol-5-yl)propanoate hydrochloride (7.26 g, 27.0 mmoles) in dimethylformamide (50 mL) at room temperature was added N, A’-disuccinimidyl carbonate (7.60 g, 29.7 mmoles) followed by triethylamine (11.29 mL, 81 mmoles). The resulting mixture was stirred for 30 min and treated with 3-(piperidin-4-yl)quinolin-2(lH)-one (6.77 g, 29.9 mmoles) in portions. The reaction was allowed to stir for 24 h. The mixture was concentrated, dissolved in ethyl acetate, and washed sequentially with water, brine, and 0.5 N HC1 (2X). The organic phase was dried over magnesium sulfate, filtered, and concentrated. The resulting residue was purified by flash chromatography (silica gel, 20: 1 ethyl acetate/methanol) to give 11.9 g (78%). 1 H-NMR (CD3OD) 5 13.0 (s, 1 H), 11.8 (s, 1 H), 7.98 (s, 1 H), 7.63 (d, J= 7.6 Hz, 1 H), 7.57 (s, 1 H), 7.45 – 7.41 (m, 2 H), 7.27 (d, J= 8.2Hz, 1 H), 7.16 (t, J= 7.9 Hz, 1 H), 7.03 (s, 1 H), 6.85 (d, J= 7.9 Hz, 1 H), 4.31 – 4.26 (m, 1 H), 4.10 – 4.08 (m, 2 H), 3.60 (s, 3 H), 3.07 – 3.01 (m, 2 H), 2.93 – 2.88 (m, 1 H), 2.77 – 2.67 (m, 2 H), 2.48 (s, 3 H), 1.78 – 1.72 (m, 2 H), 1.34 – 1.26 (m, 2 H). Mass spec.: 488.52 (MH)+.

(R)-3-(7-methyl-lH-indazol-5-yl)-2-(4-(2-oxo-l,2-dihydroquinolin-3-yl)piperidine-l- carboxamido)propanoic acid. A solution of (R)-methyl 3-(7-methyl-lH-indazol-5-yl)-2-(4-(2- oxo-1, 2-dihydroquinolin-3-yl)piperidine-l-carboxamido)propanoate_(5.50 g, 11.3 mmoles) in tetrahydrofuran (50 mL) and methanol (10 mL) was cooled to 0°C. To this was added a cold (0°C) solution of lithium hydroxide monohydrate (0.95 g, 22.6 mmoles) in water (20 mL), dropwise over 15 min. The reaction was stirred at room temperature for additional 3 h. The mixture was concentrated to remove the organic solvents. The resulting residue was dissolved in a minimum amount of water, cooled to 0°C, and treated with cold (0°C) IN HC1 until pH 2 was attained. The resulting solid was collected by filtration, washed with cold water and ether, and then dried overnight under high vacuum to give 5.0 g (94%) as a white solid. ’H-NMR (DMSO- d6) 5 13.05 (bs, 1 H), 11.77 (s, 1 H), 7.98 (s, 1 H), 7.62 (d, J= 8.0 Hz, 1 H), 7.55 (s, 1 H), 7.44 (d, J= 8.2Hz, 1 H), 7.42 (s, 1 H), 7.27 (d, J= 8.2 Hz, 1 H), 7.16 (t, J= 7.6 Hz, 1 H), 7.05 (s, 1 H), 6.65 (d, J= 7.9 Hz, 1 H), 4.27 – 4.22 (m, 1 H), 4.10 – 4.07 (m, 2 H), 3.12 – 3.07 (m, 1 H), 3.03 – 2.99 (m, 1 H), 2.93 – 2.88 (m, 1 H), 2.77 – 2.66 (m, 2 H), 2.47 (s, 3 H), 1.77 – 1.74 (m, 2 H), 1.34 – 1.27 (m, 2 H). Mass spec.: 474.30 (MH)+.

(R)-N-(3-(7-methyl-lH-indazol-5-yl)-l-(4-(l-methylpiperidin-4-yl)piperazin-l-yl)-l- oxopropan-2-yl)-4-(2-oxo-l,2-dihydroquinolin-3-yl)piperidine-l-carboxamide (I). A flask was charged with (R)-3-(7-methyl-lH-indazol-5-yl)-2-(4-(2-oxo-l,2-dihydroquinolin-3- yl)piperidine-l-carboxamido)propanoic acid (2.9 g, 6.11 mmoles), triethylamine (3.00 mL, 21.5 mmoles), l-(l-methylpiperidin-4-yl)piperazine (1.23 g, 6.72 mmoles), and dimethylformamide (10 mL). The resulting solution was treated with 2-(lH-benzotriazole-l-yl)-l, 1,3,3- tetramethyluronium tetrafluoroborate (2.26 g, 7.03 mmoles) in portions. The reaction was allowed to stir at room temperature overnight. The mixture was concentrated under vacuum to remove dimethylformamide. The crude product was dissolved in 7% methanol in di chloromethane and purified by flash chromatography using 7% methanol in di chloromethane containing 2% of aqueous ammonium hydroxide as eluent. The pure fractions were collected and solvent was removed under vacuum. The desired product was crystallized from hot acetone to give the compound having Formula I in 77% yield. Analytical HPLC showed 99.0 % UV purity at 230 nm. The enantiomeric excess (ee) was determined to be >99.9% (conditions: Chiralpak AD column, 4.6 x 250 mm, 10 pm; eluent: 70% (0.05% diethylamine)/heptane/30%ethanol; @1.0 mL/min. for 45 min. The retention times were 18.7 min for R and 28.1 min for S). ‘H-NMR (500 MHz, DMSO-de) 5 ppm 13.01 (s, 1 H), 11.76 (s, 1 H), 7.96 (s, 1 H), 7.62 (d, J= 7.10 Hz, 1 H), 7.60 (s, 1 H), 7.42 (m, 1 H), 7.36 (s, 1 H), 7.26 (d, J = 8.25 Hz, 1 H), 7.14 (m, 1 H), 7.00 (s, 1 H), 6.69 (d, J= 8.25 Hz, 1 H), 4.78 (q, J= 7.79 Hz, 1 H), 4.14 (d, J= 12.37 Hz, 2 H), 3.54 (dd, J= 9.16, 4.58 Hz, 1 H), 3.24 (m, 1 H), 3.11 (m, 1 H), 2.97 (m, 1 H), 2.89 (m, 2 H), 2.69 (m, 4 H), 2.32 (m, 1 H), 2.21 (m, 1 H), 2.07 (m, 4 H), 1.95 (t, J= 8.25 Hz, 1 H), 1.87 (m, J= 11.28, 11.28, 3.55, 3.44 Hz, 1 H), 1.76 (t, J= 12.03 Hz, 2 H), 1.68 (t, J= 11.11 Hz, 2 H), 1.53 (t, J= 8.25 Hz, 1 H), 1.32 (m, 4 H), 1.16 (m, 2 H); 13C-NMR (DMSO-de) 5 16.80, 27.30, 30.51, 30.51, 30.67, 35.50, 38.04, 41.74, 44.00, 44.16, 45.35, 45.78, 48.14, 48.39, 51.45, 54.76, 54.76, 60.61, 114.53, 117.79, 119.29, 119.34, 121.57, 122.78, 127.46, 127.79, 129.29, 129.79, 133.31, 133.72, 136.98, 137.41, 139.12, 156.50, 161.50, 170.42.
Accurate mass analysis: m/z 639.3770, [MH]+, A = -0.2 ppm. Optical rotation: -27.36° @ 589 nm, concentration = 4.71 mg/mL in methanol. DESCRIPTION AND DOSAGE FORM
The physical and chemical properties of zavegepant (BHV-3500) drug substance mono-hydrochloride salt form are provided in Table 1.
Table 1 Physical and Chemical Properties
Biohaven number BHV-3500
Molecular formula C36H47CIN8O3
Molecular weight 675.26 (HO salt); 638.82 (free base)
Appearance White to off-white powder
Melting point ~178°C pH-solubility profile 105 mg/mL at pH = 8.2 and > 300 mg/mL at lower pH pKa 4.8 and 8.8 logD 1.21

Patent
US2022401439Bioorg Med Chem Lett
. 2021 Jan 1;31:127624.
doi: 10.1016/j.bmcl.2020.127624. Epub 2020 Oct 21.
References
- ^ Jump up to:a b c d e f g h https://www.accessdata.fda.gov/drugsatfda_docs/label/2023/216386s000lbl.pdf
- ^ https://www.accessdata.fda.gov/drugsatfda_docs/appletter/2023/216386Orig1s000ltr.pdf
- ^ “Pfizer’s Zavzpret (Zavegepant) Migraine Nasal Spray Receives FDA Approval” (Press release). 10 March 2023.
Further reading
- Croop R, Madonia J, Stock DA, Thiry A, Forshaw M, Murphy A, Coric V, Lipton RB (October 2022). “Zavegepant nasal spray for the acute treatment of migraine: A Phase 2/3 double-blind, randomized, placebo-controlled, dose-ranging trial”. Headache. 62 (9): 1153–1163. doi:10.1111/head.14389. PMC 9827820. PMID 36239038.
- Noor N, Angelette A, Lawson A, Patel A, Urits I, Viswanath O, et al. (2022). “A Comprehensive Review of Zavegepant as Abortive Treatment for Migraine”. Health Psychology Research. 10 (3): 35506. doi:10.52965/001c.35506. PMC 9239361. PMID 35774914.
- Scuteri D, Tarsitano A, Tonin P, Bagetta G, Corasaniti MT (November 2022). “Focus on zavegepant: the first intranasal third-generation gepant”. Pain Management. 12 (8): 879–885. doi:10.2217/pmt-2022-0054. PMID 36189708. S2CID 252681912.
External links
- Clinical trial number NCT04571060 for “Randomized Trial in Adult Subjects With Acute Migraines” at ClinicalTrials.gov
- Clinical trial number NCT03872453 for “Acute Treatment Trial in Adult Subjects With Migraines” at ClinicalTrials.gov
| Clinical data | |
|---|---|
| Trade names | Zavzpret |
| Other names | BHV-3500 |
| License data | US DailyMed: Zavegepant |
| Routes of administration | Nasal |
| Drug class | Calcitonin gene-related peptide receptor antagonist |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only[1] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 1337918-83-8as HCl: 1414976-20-7 |
| PubChem CID | 53472683as HCl: 134819878 |
| DrugBank | DB15688 |
| ChemSpider | 30814207 |
| UNII | ODU3ZAZ94Jas HCl: 000QCM6HAL |
| KEGG | D11898as HCl: D11899 |
| ChEMBL | ChEMBL2397415as HCl: ChEMBL4650220 |
| Chemical and physical data | |
| Formula | C36H46N8O3 |
| Molar mass | 638.817 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
////////FDA 2023, APPROVALS 2023, Vazegepant, BMS-742413, BHV-3500, ザベジェパント , Zavegepant, ZAVZPRET, BMS
PFIZERCC1=CC(=CC2=C1NN=C2)CC(C(=O)N3CCN(CC3)C4CCN(CC4)C)NC(=O)N5CCC(CC5)C6=CC7=CC=CC=C7NC6=O
