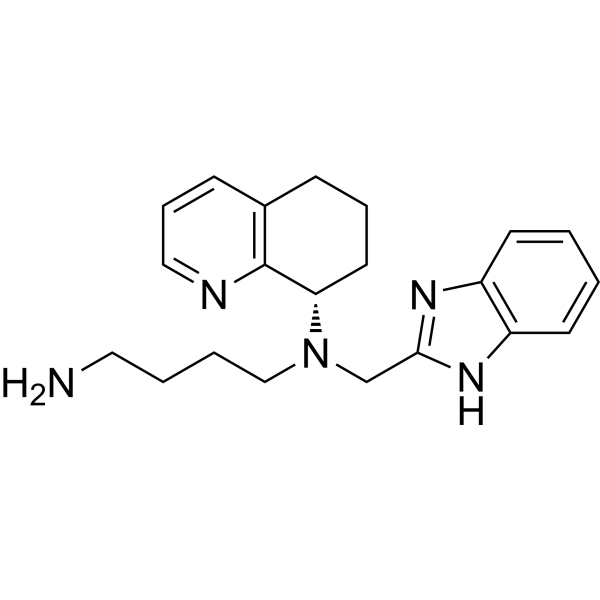
MAVORIXAFOR
AMD 070
N1-(1H-BENZIMIDAZOL-2-YLMETHYL)-N1-((S)-5,6,7,8-TETRAHYDROQUINOLIN-8-YL)-BUTANE-1,4-DIAMINE
Mavorixafor (AMD-070) is a potent, selective and orally available CXCR4 antagonist, with an IC50 value of 13 nM against CXCR4 125I-SDF binding, and also inhibits the replication of T-tropic HIV-1 (NL4.3 strain) in MT-4 cells and PBMCs with an IC50 of 1 and 9 nM, respectively.
| Molecular Weight | 349.47 |
|---|---|
| Appearance | Solid |
| Formula | C21H27N5 |
| CAS No. | 558447-26-0 |
| SMILES | NCCCCN(CC1=NC2=C(N1)C=CC=C2)[C@@H]3C4=C(CCC3)C=CC=N4 |
PHASE 2
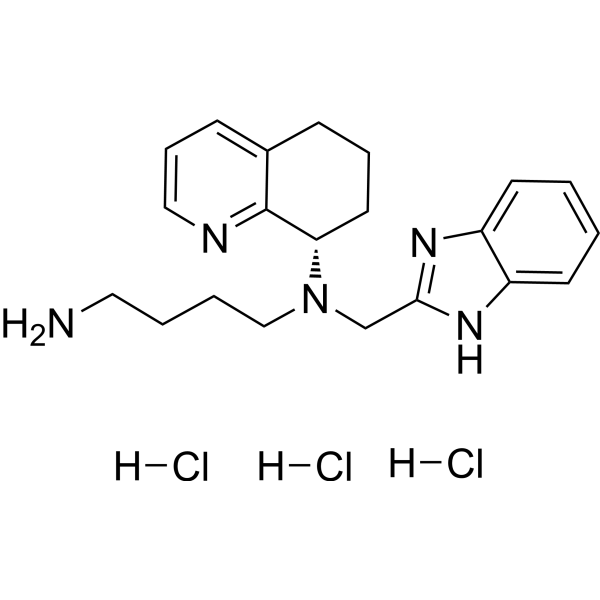
Mavorixafor trihydrochloride
| Molecular Weight | 458.86 |
|---|---|
| Appearance | Solid |
| Formula | C21H30Cl3N5 |
| CAS No. | 2309699-17-8 |
| SMILES | [H]Cl.[H]Cl.[H]Cl.NCCCCN(CC1=NC2=CC=CC=C2N1)[C@@H]3C4=NC=CC=C4CCC3 |

AMD-070 is a small molecule drug candidate that belongs to a new investigational class of anti-HIV drugs known as entry (fusion) inhibitors. Approximately 76% of HIV-patients with measurable viral load are infected with a strain of virus that is resistant to one or more classes of antiretroviral agents, thus reducing treatment options. Unlike many existing HIV drugs that target the virus after it has infected a healthy cell, AMD-070 blocks the virus from entering a healthy cell, thus preventing the replication process. AMD-070 targets the CXCR4 receptor on HIV and prevents the virus from entering and infecting healthy cells. AMD-070 is specific for the CXCR4 receptor and does not interact with any other chemokine receptors in vitro. AMD-070 strongly inhibits viral infection by all CXCR4 using virus (including virus using CXCR4 alone and/or virus using CXCR4 and CCR5) in vitro. AMD-070 is orally bioavailable in animals, it has suitable PK and toxicity profile for oral dosing. AMD-070 shows additive or synergistic effects in vitro in combination with other known anti-HIV agents. AMD-070 is active against CXCR4 using HIV strains that are resistant to existing antiretroviral therapies in vitro, reveals potent anti-HIV activity against CXCR4-using laboratory strains and clinical isolates. MD-070 had been in phase II clinical trials by Genzyme for the treatment of HIV infection. However, this research has been discontinued. AMD-070 has been studied in Phase I/II clinical trials for the treatment of Renal cell carcinoma and Phase I clinical trials for the treatment of malignant melanoma and solid tumours.
PAPER
https://pubs.acs.org/doi/10.1021/acs.oprd.2c00076
Org. Process Res. Dev. 2022, 26, 6, 1831–1836
A novel and practical synthesis of mavorixafor (1) is reported. The novelty of this synthetic route is the use of 8-chloro-5,6,7,8-tetrahydroquinoline (9) and 1,4-diaminobutane as the materials, instead of 8-amino-5,6,7,8-tetrahydroquinoline (4) and N,N-diprotected aminobutyraldehyde (6a or 6b). The preparation of (S)-8-(4-aminobutylamino)-5,6,7,8-tetrahydroquinoline (13) by resolution with N-acetyl-l-leucine was first achieved. Then the one-pot synthesis of 1 from 13 involving protection, condensation, and subsequent hydrolysis was successfully developed. In addition, the final product with a satisfactory purity (>99.5%, detected by both achiral and chiral HPLC) was obtained by a simple operation (salification) without column chromatographic purification.





NEW PAT
Scheme I


Mavorixafor


EXEMPLIFICATION
Example 1: Synthesis of Sulfonate adduct F-2d:
Scheme V:
1) AcOH, NaCI, water 1) Na 2 S 2 O 5 , THF, water
2) n-Heptane, THF 2) THF/n-heptane, acetonitrile
Step 1C Step 1 D


Step 1A: Preparation of Dl
Charge diethyl-4-aminobutyl acetal (E) (1.00 wt, 1.00 eq) to vessel A. Charge acetonitrile (10.0 vol, 7.8 wt) and adjust temperature to 20°C. Heat the mixture to 40°C. Concentrate the reaction mixture to 6.0 vol under reduced pressure at 35 to 45°C.
[0098] Acetonitrile filler (5.0 vol, 3.9wt) at 35 to 45°C. Concentrate the reaction mixture to 6.0 vol under reduced pressure 35 to 45°C. This step is repeated once as described below.
[0099] Acetonitrile filler (5.0 vol, 3.9wt) at 35 to 45°C. Concentrate the reaction mixture to 6.0 vol under reduced pressure at 35 to 45°C. Cool to 20°C.
[00100] Charge di-tert-butyl dicarbonate (1.1 eq, 1.5 wt) to a drum, followed by acetonitrile (0.4 vol, 0.3 wt) and agitate until fully dissolved. Concentrate the reaction mixture to 6.0 vol under reduced pressure at 35 to 45°C.
[00101] Charge this di-tert-butyl dicarbonate solution in acetonitrile to vessel A maintaining 20°C. Charge acetonitrile (1.5 vol, 1.1 wt) to the solution as a line rinse and stir at 20°C for 30 to 60 min..
[00102] Charge 4-dimethylaminopyridine (0.076 wt, 0.10 eq) to the vessel A at 20°C. Heat the solution to 40°C. Concentrate the reaction mixture to 5.0 vol under reduced pressure. Charge acetonitrile (5.0 vol, 3.9 wt) to the solution. Concentrate the reaction mixture to 5.0 vol under reduced pressure.
[00103] Take the resulting solution of Dl into next reaction without isolation.
Step IB: Preparation of Cl
[00104] Charge acetonitrile (2.0 vol, 1.6 wt) at 35 to 45°C to vessel A containing solution of D-1 from Step 1A.
[00105] Charge di-tert-butyl dicarbonate (1.4 eq, 1.9 wt) to a drum, followed by acetonitrile (10.0 vol, 7.8 wt) and agitate until fully dissolved. Charge this di-tert-butyl dicarbonate solution to vessel A, 2 to 6 h while distilling under vacuum at 35 to 45°C maintaining the volume of the reaction at 7.0 vol. Load acetonitrile (3.0 vol, 2.4 wt) over 20 to 40 min. as a line rinse while distilling under vacuum at 35 to 45°C, maintaining the volume of the reaction at 7.0 vol.
[00106] Charge di-tert-butyl dicarbonate, (0.14 eq, 0.19 wt) to a drum, followed by acetonitrile (1.0 vol, 0.74 wt) and agitate until fully dissolved. Charge this di-tert-butyl dicarbonate solution to vessel A over 20 to 40 min.. Charge acetonitrile (0.3 vol, 0.24 wt) over 10 to 20 min as a line rinse while distilling under vacuum at 35 to 45°C, maintaining the volume of the reaction at 7.0 vol.
[00107] Concentrate the reaction mixture to 5.0 vol distilling under vacuum at 35 to 45°C.
[00108] Charge n-heptane, (7.5 vol, 5.1 wt) to the reaction mixture, and concentrate the reaction mixture to 5.0 vol under reduced pressure at 40°C. This step is repeated once as described below.
[00109] Charge n-heptane, (7.5 vol, 5.1 wt) to the reaction mixture, and concentrate the reaction mixture to 5.0 vol under reduced pressure at 40°C.
[00110] Charge decolorizing, activated charcoal (0.2 wt) to the solution and stir for 1 to 2 h at 40°C. Filter the reaction mixture at 40°C. Charge n-heptane, (2.0 vol, 1.4 wt) to the reactor vessel and stir for 5 to 15 min. at 20°C before charging to the filter as a line rinse. Combine the filtrate and wash, and as required adjust to 20°C.
[00111] Take the resulting solution of Cl into next reaction without isolation.
Step 1C: Preparation of Bl
[00112] Charge 15% v/v acetic acid (2.0 vol) caution gas evolution, to vessel A containing solution of Cl from Step IB, maintaining the temperature at 20°C and stir for 10 min. at 20°C. Allow the phases to separate for 15 min. at 20°C. Discharge the aqueous phase to waste, retaining the organic phase in vessel A. This step is repeated once as described below.
[00113] Charge 15% v/v acetic acid (2.0 vol) maintaining 20°C and stir for 10 min. at 20°C. Allow the phases to separate for 15 min. at 20°C. Discharge the aqueous phase to waste, retaining the organic phase in vessel A.
[00114] Adjust the reaction to 30°C. Charge 4% w/w sodium chloride solution (2.1 vol) to the vessel maintaining the temperature at 30°C. Charge glacial acetic acid (4.1 vol, 4.3 wt) to the vessel maintaining 30°C. Stir the reaction mixture for 2 h maintaining the temperature at 30°C.
[00115] Charge purified water, (6.0 vol) at 30°C. Stir the contents for 5 to 10 min. at 30°C, and separate the phases, retaining the upper organic phase in vessel A. Charge the lower aqueous phase to vessel B.
[00116] Charge purified water (4.0 vol) at 30°C and stir for 5 to 10 min. maintaining the temperature at 30°C. Separate the phases at 30°C, retaining the upper organic phase in vessel A. Charge the lower aqueous phase to vessel B.
[00117] Adjust the temperature to 30°C of vessel B containing combined aqueous phases. Charge n-heptane, (2.0 vol, 1.4 wt) to vessel B and stir for 5 to 10 min. maintaining the temperature at 30°C. Separate the phases at 30°C, over 15 min.. Charge the upper organic phase to vessel A and recharge the lower aqueous phase to vessel B. This step is repeated two additional times as described below.
[00118] Charge n-heptane, (2.0 vol, 1.4 wt) to vessel B and stir for 5 to 10 min. maintaining the temperature at 30°C. Separate the phases at 30°C, over 15 min.. Charge the upper organic phase to vessel A and recharge the lower aqueous phase to vessel B.
[00119] Charge n-heptane, (2.0 vol, 1.4 wt) to vessel B and stir for 5 to 10 min. maintaining the temperature at 30°C. Separate the phases at 30°C, over 15 min., discharge the lower aqueous phase to waste and charge the upper organic layer to vessel A.
[00120] Concentrate the combined organic phases in vessel A to 3.0 vol at 10 to 20°C under reduced pressure. Offload the solution to new HDPE drum(s) and line rinse with n-heptane (0.5
vol, 0.4 wt) at 20°C. Homogenize the drum and store as “Bl solution in n-heptane,” and take into next reaction without isolation.
Step ID: Preparation of F-2d
[00121] Calculate a new 1.00 wt based on the above assay.
[00122] Charge “Bl solution in n-heptane” from Step 1C (1.00 wt, 1.00 eq, corrected for w/w assay, ca. 3.0 vol), into an appropriate vessel. THF load (3.0 vol, 2.7 wt). Heat the reaction mixture to 40°C.
[00123] Charge purified water, (0.02 vol, 0.02 wt) followed by sodium metabisulphite, (0.125 eq, 0.08 wt) as a solid via the charge hole at 40°C. Stir the resulting mixture for 30 to 35 min. at 40°C. This step was repeated four additional times to add the reagent in five portions total, as detailed below.
[00124] Charge purified water, (0.02 vol, 0.02 wt) followed by sodium metabisulphite, (0.125 eq, 0.08 wt) as a solid via the charge hole at 40°C. Stir the resulting mixture for 30 to 35 min. at 40°C.
[00125] Charge purified water, (0.02 vol, 0.02 wt) followed by sodium metabisulphite, (0.125 eq, 0.08 wt) as a solid via the charge hole at 40°C. Stir the resulting mixture for 30 to 35 min. at 40°C.
[00126] Charge purified water, (0.02 vol, 0.02 wt) followed by sodium metabisulphite, (0.125 eq, 0.08 wt) as a solid via the charge hole at 40°C. Stir the resulting mixture for 30 to 35 min. at 40°C.
[00127] Charge purified water, (0.02 vol, 0.02 wt) followed by sodium metabisulphite, (0.125 eq, 0.08 wt) as a solid via the charge hole at 40°C. Stir the resulting mixture for 36 hours at 40°C.
[00128] Cool the reaction mixture to 20°C over 3 to 4 h at a target constant rate. Filter the reaction mixture at 20°C on a 1-2 pm cloth.
[00129] Wash the solid with a pre-mixed mixture of THF (0.5 vol, 0.5 wt) and n-heptane (0.5 vol, 0.3 wt) maintaining the temperature at 20°C. This step was repeated an additional three times, as detailed below.
[00130] Wash the solid with n-heptane, (2.0 vol, 1.4 wt) as a line rinse and apply to the filtercake at 20°C.
[00131] Wash the solid with n-heptane, (2.0 vol, 1.4 wt) as a line rinse and apply to the filtercake at 20°C.
[00132] Wash the solid with acetonitrile, (2.0 vol, 1.6 wt) as a line rinse and apply to the filtercake at 20°C.
[00133] Dry the solid at 38°C under a flow of nitrogen for 12 h.
[00134] Determine residual solvent content. Pass criteria acetonitrile <2.0% w/w, n-heptane <2.0% w/w and tetrahydrofuran <2.0% w/w.
[00135] Yield of compound F-2d: 52-69%.
[00136] ‘H NMR (400 MHz, d 6 -DMSO): 8 5.22 (s, 1H), 3.77 (s, 1H), 3.45 (t, 2H), 1.70 (m, 2H), 1.44 (m, 20H) ). 13 C NMR (400 MHz, d 6 -DMSO): 8 152.6, 83.2, 82.0, 46.5, 29.6, 28.1, 26.0. FTIR (wavenumber, cm’ 1 ) 3294, 1721, 1738, 1367, 1233, 1180, 1135, 1109, 1045.
Example 2: Synthesis of F-3a:
Scheme VI:


Step 2A: Preparation of Gl
[00137] Charge J, (1.00 wt, 1.00 eq) to vessel A. Charge purified water, (1.0 vol, 1.0 wt) to vessel A and as necessary adjust the temperature to 20°C. Charge concentrated hydrochloric acid, (4.0 eq, 3.0 vol, 3.6 wt) to vessel A maintaining the temperature at 20°C. Line rinse with purified water, (0.5 vol, 0.5 wt) maintaining the contents of vessel A at 15 to 25°C.
[00138] Charge chloroacetic acid, (1.3 wt, 1.5 eq) and purified water, (1.0 vol, 1.0 wt) to vessel B and as necessary, adjust the temperature to 20°C. Stir until fully dissolved, expected 10 to 20 min.
[00139] Charge the chloroacetic acid solution to vessel A maintaining the temperature of vessel A at 20°C. Line rinse vessel A with purified water, (0.5 vol, 0.5 wt) at 15 to 25°C and charge to vessel B at 20°C. Heat the reaction mixture to 80°C. Stir the reaction mixture at 80°C for 20 h.
[00140] Cool the reaction mixture to 10°C over 1.5 h. Load 47% w/w potassium phosphate solution (6.0 vol) over 60 min. targeting a constant rate maintaining 10°C. Adjust the pH of the reaction mixture by charging 47% w/w potassium phosphate solution to pH 7.0 maintaining the reaction temperature at 10°C. Expected charge is 2.0 to 3.5 vol 47% w/w potassium phosphate solution.
[00141] Stir the slurry for >30 min. maintaining 10°C and rechecking the pH, pass criterion pH 7.0. Filter the reaction mixture through 20 pm cloth at 10°C. Wash the filter-cake with purified water, (1.0 vol, 1.0 wt) at 10°C. This step is repeated additional three times as described below.
[00142] Slurry wash the filter-cake in the reactor vessel with purified water, (10.0 vol, 10.0 wt) for 45 min. to 90 min. at 10°C. Filter the mixture through 20 pm cloth at 10°C.
[00143] Slurry wash the filter-cake in the reactor vessel with purified water, (10.0 vol, 10.0 wt) for 45 min. to 90 min. at 10°C. Filter the mixture through 20 pm cloth at 10°C.
[00144] Slurry wash the filter-cake in the reactor vessel with purified water, (10.0 vol, 10.0 wt) for 45 min. to 90 min. at 10°C. Filter the mixture through 20 pm cloth at 10°C.
[00145] Wash the filter-cake with purified water, (1.0 vol, 1.0 wt) at 10°C. The filter-cake was washed with purified water additional five times as described below.
[00146] Wash the filter-cake with purified water, (1.0 vol, 1.0 wt) at 10°C.
[00147] Wash the filter-cake with acetonitrile, (2×1.3 vol, 2×1.0 wt) at 10°C.
[00148] Dry the filter-cake on the filter under vacuum and strong nitrogen flow through the filter cake at 20°C until the water content is <15.0% w/w by Karl-Fisher analysis.
[00149] Dry the filter-cake on the filter under vacuum and strong nitrogen flow through the filter cake at 30°C until the water content is <5.0% w/w by Karl-Fisher analysis.
[00150] Dry the filter-cake on the filter under vacuum and strong nitrogen flow through the filter cake at 50°C until the water content is <1.0% w/w by Karl-Fisher analysis.
[00151] Yield of compound Gl: about 75%.
Step 2B: Preparation of F-3a
Charge di-/c/7-butyl dicarbonate, (1.85 wt, 1.4 eq) to vessel A followed by N,N-dimethylformamide, (2.6 wt, 2.7 vol) and stir at 20°C for 20 min. until dissolution achieved. Add A,A-diisopropylethylamine, (0.08 wt, 0.11 vol, 0.1 eq) to contents of vessel A at 20°C. Heat the contents of vessel A to 40°C.
[00153] Charge Gl, (1.00 wt) to vessel B followed by YW-di methyl form am ide, (5.2 wt, 5.5 vol) and adjust to 14°C.
[00154] Charge the Gl/DMF solution from vessel B to vessel A over 5 h at 40°C, at an approximately constant rate. Line rinse with Y,Y-di methyl form am ide, (0.4 wt, 0.4 vol), maintaining vessel A at 40°C. Stir the resulting reaction mixture at 40°C for 16 h.
[00155] Charge decolorizing charcoal activated, (0.20 wt). Adjust the mixture to 40°C and stir at 40°C for 60 to 90 min..
[00156] Clarify (filter) the reaction mixture into vessel B at 40°C. Charge N,N-dimethylformamide, (0.9 wt, 1.0 vol) via vessel A and filter at 40°C. Charge purified water, (3.5 vol) to the combined filtrates, over 60 min., maintaining the temperature at 40°C. As required, cool the mixture to 35°C over 30 to 60 min..
[00157] Filler F-3a, (0.02 wt) as seed material at 35°C. Stir at 34°C for 1.5 h then check for crystallization. Cool slurry to 30°C over 40 min.
[00158] Filler F-3a, (0.02 wt) as seed material at 30°C. Stir at 30°C for 1.5 h then check for crystallization.
[00159] Cool slurry at 20°C over 3.5 h at a targeted constant rate. Stir at 20°C for 3 hours. Charge purified water, (1.0 vol), maintaining the temperature at 20°C over 60 min..
Stir at 20°C for 3 hours.
[00160] Cool slurry to 2°C over 2.5 h. Stir at 2°C for 2.5 hours. Filter through 20 pm cloth and pull dry until no further filtrate passes. Wash the solid with pre-mixed Y,Y-di methyl form am ide / purified water, (2.0 vol, 1:2 v:v) at 2°C. Wash the solid with purified water, (2 x 3.0 vol) at 2°C. Dry under vacuum at 28°C until KF <0.2% w/w, and Y,Y-di methyl form am ide <0.4% w/w.
[00161] Yield of compound F-3a: 62-70%.
Example 3: Synthesis of Mavorixafor:
Scheme VI:


nce
Step 3A: Preparation of imine Q-1
[00162] To vessel A charge purified water, (8.7 vol, 8.7 wt) followed by potassium phosphate, (5.52 eq, 5.3 wt) portion-wise and cool to 15°C. Charge tetrahydrofuran, (4.3 vol, 3.8 wt) and n-heptane, (2.2 vol, 1.5 wt) to vessel A and cool the biphasic mixture to 0°C. Charge Fl, (1.00 eq, 1.00 wt) to the vessel in 2 portions maintaining 0°C.
[00163] Charge F-2d, (1.10 eq, 1.95 wt) to the vessel in 4 portions maintaining 0°C, ensuring portions are spaced by 10 min.. Stir the resulting biphasic mixture for 1.5 h at 0°C. Allow the layers to separate for 45 min. at 0°C before separating the layers. Retain the upper organic phase within vessel A.
[00164] Take the resulting solution of Ql into next reaction without isolation.
Step 3B: Preparation of amine P-1
[00165] To vessel B, charge tetrahydrofuran, (6.0 vol, 5.3 wt) and adjust to 15°C. Charge zinc chloride, (1.5 eq, 0.92 wt) to vessel B in 4 portions, maintaining 10 to 30°C. Adjust the reaction mixture in vessel B to 15°C. Stir the mixture at 15°C for 1 hour. Charge sodium borohydride,(1.0 eq, 0.17 wt) to vessel B in 2 portions maintaining 15°C. Cool the reaction mixture in vessel B to 15°C. Stir the mixture for 1 hour maintaining 15°C. Cool the reaction mixture in vessel B to -5°C.
[00166] Cool the retained organic solution of Ql in vessel A, from Step 3A, to -5°C.
[00167] Charge the organic solution in vessel A into vessel B over 1 to 2 h maintaining -5°C. Charge tetrahydrofuran, (1.0 vol, 0.9 wt) to vessel A as a line rinse and adjust to -5°C. Transfer the contents of vessel A to vessel B maintaining -5°C.
[00168] Stir the resulting reaction mixture in vessel B for 1.5 h maintaining -5°C.
[00169] Charge purified water, (4.5 vol, 4.5 wt) and glacial acetic acid, (1.0 eq, 0.27 wt, 0.26 vol) to the cleaned vessel A and cool to 0°C. Charge the contents of vessel B to vessel A over 1 to 2 h maintaining 0°C. Charge tetrahydrofuran, (1.0 vol, 0.9 wt) to vessel B as a vessel rinse, cool to 0°C and transfer to vessel A maintaining 0°C.
[00170] Warm the resulting mixture in vessel A to 30°C. Stir the resulting mixture in vessel A at 30°C for 1 h. Allow the layers to settle for 15 min. at 30°C before separating the layers. Retain the upper organic phase.
[00171] Cool the retained organic phase to 15°C. Charge to the vessel 25% w/w ammonia solution (3.0 vol) at 10 to 30°C. Cool the reaction mixture to 20°C. Charge to the vessel 25% w/w ammonium chloride solution (3.0 vol) at 20°C and stir for 1 h. Separate the layers for 15 min. at 20°C, retain the upper organic phase. Wash the retained organic phase with 10% w/w sodium chloride solution (3.0 vol) at 20°C for 10 min.. Allow the layers to settle for 10 min. at 20°C before separating and retaining the upper organic phase within the vessel.
[00172] Charge tert-butyl methyl ether, (0.5 vol, 0.4 wt) to the organic phase. Cool the mixture to 5°C. Adjust the pH of the reaction mixture to pH 5 with hydrochloric acid aqueous solution (expected ca. 9.0 vol) over 1 h at a targeted constant rate at 5°C. Stir the mixture at 5°C for 45 min.. Measure the pH of the aqueous phase to confirm the value is pH 5.
[00173] Charge sodium chloride, (2.1 wt) to the reaction mixture at 5°C and stir the mixture until everything is dissolved. Adjust the temperature of the reaction mixture to 20°C. Separate the layers at 20°C and retain the organic phase within the vessel. Tetrahydrofuran charge, (1.5 vol, 1.3 wt) maintaining 20°C.
[00174] Charge to the vessel 24% w/w sodium chloride solution (7.5 vol) at 20°C and stir for 10 min.. Separate the layers at 20°C and retain the organic phase in the vessel. This step is repeated additional one more time as described below.
[00175] Charge to the vessel 24% w/w sodium chloride solution (7.5 vol) at 20°C and stir for 10 min.. Separate the layers at 20°C and retain the organic phase in the vessel.
[00176] Heat the retained organic phase to 35°C and concentrate the mixture to 6.0 vol under reduced pressure maintaining 35°C.
[00177] Tetrahydrofuran charge, (15.0 vol, 13.2 wt) maintaining 35°C. Concentrate the mixture to 6.0 vol under reduced pressure maintaining 35°C.
[00178] Tetrahydrofuran charge, (15.0 vol, 13.2 wt) maintaining 35°C. Concentrate the mixture to 11.0 vol under reduced pressure maintaining 35°C.
[00179] Cool the mixture to -5°C. Load tert-butyl methyl ether, (10.0 vol, 7.4 wt) over 1 h maintaining -5°C. Stir the mixture at -5°C for 1.5 hours. Filter the solid on 1 to 2 pm filter cloth at -5°C. Wash the solid with pre-mixed tetrahydrofuran, (1.9 vol, 1.7 wt) and tert-butyl methyl ether, (3.1 vol, 1.9 wt) at -5°C as a displacement wash.
[00180] Wash the solid with tert-butyl methyl ether, (5.0 vol, 3.7 wt) at -5°C.
[00181] Dry the solid on the filter under a flow of nitrogen at 23°C.
[00182] Yield of compound P-1: 76-87%.
Step 3C: Preparation of compound 0-1
[00183] Charge purified water, (2.0 vol, 2.0 wt) followed by potassium phosphate, (3.3 eq, 1.54 wt), carefully portion-wise, maintaining <15°C, to vessel A. Charge toluene, (4.5 vol, 3.9 wt) to the vessel maintaining <15°C. As necessary, adjust the temperature to 10°C.
[00184] Charge P-1, (1.00 eq, 1.00 wt) to the vessel in two portions maintaining 10°C. Stir the reaction mixture at 10°C for 15 min..
[00185] Load F-3a, (1.1 eq, 0.64 wt) in 4 equal portions ensuring portions are spaced by 10 min. at 10°C.
[00186] Tetrabutylammonium iodide (TBAI) filler (0.20 eq, 0.16 wt). Heat the reaction mixture to 40°C. Stir the reaction mixture at 40°C for 30 h.
[00187] Charge pre-mixed 2-mercaptoacetic acid, (0.40 eq, 0.08 wt, 0.06 vol), and toluene, (0.5 vol, 0.4 wt) over 20 min. to Vessel A at 40°C. Line rinse with toluene, (0.5 vol, 0.4 wt) at 40°C. Adjust the temperature of the reaction mixture to 50°C. Stir the mixture at 50°C for 2.5 hours.
[00188] Adjust the temperature of Vessel A to 20°C. Charge purified water, (3.0 vol, 3.0 wt) maintaining 20°C. Stir the reaction mixture at 20°C for 15 min. and transfer to a new, clean HDPE container. Line/vessel rinse with toluene, (0.5 vol, 0.4 wt) at 20°C. Clarify (filter) the reaction mixture via a 1 pm filter at 20°C into clean Vessel A. Wash the vessel and the filter with toluene, (0.5 vol, 0.4 wt) at 20°C. Allow the layers to separate for 15 min. at 20°C, retaining the upper organic layer (organic layer 1).
[00189] Wash the aqueous layer with toluene, (2.5 vol, 2.2 wt) at 20°C for 15 min.. Allow the layers to separate for 15 min. at 20°C. Retain the upper organic layer (organic layer 2).
[00190] Combine the organic layer 1 and organic layer 2 and adjust the temperature to 20°C. Wash the combined organic layers with 10% w/w sodium chloride solution (5.0 vol) at 20°C for 15 min.. Allow the layers to settle for 15 min. at 20°C. Retain the upper organic layer.
[00191] Take the resulting solution of Ol into next reaction without isolation.
Step 3D: Preparation of compound Kl
[00192] Charge n-butanol, (2.4 wt, 3.0 vol) to vessel B and adjust to 5°C. Charge concentrated sulfuric acid, (1.1 wt, 5.0 eq, 0.6 vol) slowly to Vessel B maintaining <15°C. Line rinse with toluene, (0.4 wt, 0.5 vol) maintaining <15°C. Adjust the temperature of Vessel B to 25°C.
[00193] Heat the n-butanol/ sulfuric acid solution in Vessel B to 55°C. Charge the organic layer from Vessel A (from Step 3C) to the butanol/ sulfuric acid solution in Vessel B over 60 to 90 min. maintaining 55°C. Charge toluene, (1.3 wt, 1.5 vol) to Vessel A as a line rinse and transfer to Vessel B maintaining 55°C. Stir the contents of Vessel B at 55°C for 1.5 h.
[00194] Stir the mixture in Vessel B for 4.5 h at 55°C. Cool the contents of Vessel B to 20°C over 10 h. Filter the slurry over 1-2 pm filter cloth under nitrogen at 20°C. Wash the filter cake with pre-mixed toluene, (3.5 wt, 4.0 vol) and n-butanol, (1.0 vol, 0.8 wt) at 20°C. Wash the filter cake with toluene, (4.3 wt, 5.0 vol) at 20°C. Dry the solid at 30°C under vacuum.
[00195] Correct the output weight for assay. Expected 50-55% w/w.
[00196] Yield of compound K1: 89-92%.
Step 3E: Preparation of Mavorixafor Drug Substance
[00197] Charge Kl, (1.00 eq, 1.00 wt, corrected for HPLC assay) in vessel A followed by nitrogen-purged purified water, (2.0 wt, 2.0 vol) and if necessary, adjust the temperature to 20°C. Charge nitrogen-purged toluene, (12.0 wt, 14.0 vol) to the solution maintaining 20°C. Charge nitrogen-purged n-butanol, (0.8 wt, 1.0 vol) to the solution maintaining 20°C. Heat the biphasic mixture to 30°C. Charge nitrogen-purged 3.0 M aqueous sodium hydroxide solution (6.2 eq, 5.9 vol) maintaining 30°C. Check the pH (expected 12 to 13). Adjust the pH of the aqueous layer to pH 10.0 with nitrogen-purged 0.3 M sulfuric acid solution (expected up to 2.5 vol) maintaining 30°C. Stir the mixture at 30°C for 45 min..
[00198] Measure the pH to confirm the value is pH 10.0.
[00199] Allow the layers to settle at 30°C for 30 min. and separate the layers retaining the organic phase in the vessel, and discharge the aqueous layer into a separate container (container C).
[00200] Charge pre-mixed toluene, (4.1 wt, 4.7 vol) and n-butanol, (0.24 wt, 0.3 vol) to a separate vessel; heat the contents to 30°C and charge the aqueous layer from container C. As required adjust the temperature to 30°C and stir for 5 to 10 min. at 30°C. Allow the phases to separate for 10 to 15 min. at 30°C. Discharge the aqueous phase to waste and combine the organic phase to the organic phase in vessel A.
[00201] Charge nitrogen-purged purified water, (2.0 wt, 2.0 vol) to the organic layer maintaining the temperature at 30°C and stir for 5 to 10 min. at 30°C. Allow the phases to separate for 10 to 15 min. at 30°C. Discharge the aqueous phase to waste retaining the organic phase in the vessel. Heat the retained organic solution to 40°C. Concentrate the resulting organic phase to 7.0 vol by vacuum distillation at 40°C.
[00202] Charge nitrogen -purged toluene, (13.0 wt, 15.0 vol) to the mixture and concentrate the solution 7.0 vol by vacuum distillation at 40°C. This step is repeated additional one time as described below.
[00203] Charge nitrogen -purged toluene, (13.0 wt, 15.0 vol) to the mixture and concentrate the solution 7.0 vol by vacuum distillation at 40°C.
[00204] Charge nitrogen-purged toluene, (7.0 wt, 8.0 vol) to the mixture at 40°C, heat to 55°C and clarify the hot reaction mixture under nitrogen via a 1 pm filter.
[00205] Charge clarified nitrogen-purged toluene, (1.7 wt, 2.0 vol) to the mixture as a line and vessel rinse at 40°C. Concentrate the solution to 7.0 vol by vacuum distillation at 40°C. At the end of the distillation the product is expected to have precipitated. Heat the mixture to 63°C.
[00206] Adjust the temperature to 60.5°C. This batch will be referred to as the main batch.
[00207] Load seed material, (0.02 wt) to a new clean container. Charge clarified nitrogen-purged toluene, (0.09 wt, 0.10 vol) to this seed material and gently shake.
[00208] Seed the main batch with the slurry maintaining the temperature at 60.5 ± 2°C. Stir the reaction at the 60.5± 2°C for 1 hour.
[00209] Cool to 40°C for 2.5 h. Stir the reaction at 40°C for 1 hour.
[00210] Cool to 30°C over 2 h.. Stir the reaction at 30°C for 1 h.
[00211] Cool to 25°C 50 min. Stir the reaction at 25°C over 2 hours.
[00212] Cool to 2°C over 4 h. Stir the mixture for 12 hours at 2°C.
[00213] Filter the mixture at 2°C over 1 to 2 pm cloth. Wash the filter cake with clarified nitrogen-purged toluene, (2.0 vol, 1.7 wt) at 2°C. Dry the filter cake under vacuum and a flow of nitrogen for 1.5 h.
[00214] Dry the solid at 40°C under vacuum and a flow of nitrogen until drying specification is achieved.
[00215] Yield of the final compound mavorixafor: 72%.
[00216] When toluene is used as the recrystallization solvent, optionally with a dissolution aid such butanol or methanol, for maxorixa for recrystallization, advantages were found compared to using dichloromethane and isopropyl acetate. We have found that these solvents do not react with the API, and accordingly we believe that this change has caused the significant reduction of impurities A (imine), B (N-formyl) and C (acetamide) that we have observed.
[00217] In some embodiments, the mavorixafor composition included 7000, 6000, 5000, 4500, 4450, 4000, 3500, 3000, 2500, 2000, 1750, 1700, 1650, 1600, 1550, 1500, 1450, 1400, 1400, 1400, 1400 gold 50 ppm of toluene or less. In some embodiments, the mavorixafor composition comprises a detectable amount of toluene. In some embodiments, the mavorixafor composition comprises from a detectable amount of toluene to 1350 ppm of toluene.

////////

AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
| Description | Mavorixafor (AMD-070) is a potent, selective and orally available CXCR4 antagonist, with an IC50 value of 13 nM against CXCR4 125I-SDF binding, and also inhibits the replication of T-tropic HIV-1 (NL4.3 strain) in MT-4 cells and PBMCs with an IC50 of 1 and 9 nM, respectively. |
|---|---|
| IC50 & Target[1] | 125I-SDF-CXCR413 nM (IC50)HIV-1 (NL4.3 strain)1 nM (IC50, in MT-4 cells)HIV-1 (NL4.3 strain)9 nM (IC50, in PBMCs)HIV-1 (NL4.3 strain)3 nM (IC90, in MT-4 cells)HIV-1 (NL4.3 strain)26 nM (IC90, in PBMCs) |
| In Vitro | Mavorixafor (AMD-070) is a potent and orally available CXCR4 antagonist, with an IC50 value of 13 nM against CXCR4 125I-SDF binding, and also inhibits the replication of T-tropic HIV-1 (NL4.3 strain) in MT-4 cells and PBMCs with an IC50 of 1 and 9 nM, respectively. Mavorixafor (AMD-070) shows no effect on other chemokine receptors (CCR1, CCR2b, CCR4, CCR5, CXCR1, and CXCR2)[1]. Mavorixafor (AMD-070) (6.6 µM) significantly suppresses the anchorage-dependent growth, the migration and matrigel invasion of the B88-SDF-1 cells[2].MCE has not independently confirmed the accuracy of these methods. They are for reference only. |
| In Vivo | Mavorixafor (AMD-070) (2 mg/kg, p.o.) significantly reduces the number of metastatic lung nodules in mice, and lowers the expression of human Alu DNA in mice, without body weight loss[2].MCE has not independently confirmed the accuracy of these methods. They are for reference only. |
| Clinical Trial | NCT NumberSponsorConditionStart DatePhaseNCT00089466National Institute of Allergy and Infectious Diseases (NIAID)|AIDS Clinical Trials GroupHIV InfectionsNovember 2004Phase 1|Phase 2NCT02667886X4 PharmaceuticalsClear Cell Renal Cell CarcinomaJanuary 2016Phase 1|Phase 2NCT02823405X4 PharmaceuticalsMelanomaSeptember 15, 2016Phase 1NCT00361101Genzyme, a Sanofi Company|SanofiHIV Infections|X4 Tropic VirusOctober 2005Phase 1NCT03005327X4 PharmaceuticalsWHIM SyndromeDecember 2016Phase 2NCT04274738X4 PharmaceuticalsWaldenstrom´s MacroglobulinemiaApril 30, 2020Phase 1NCT04154488X4 PharmaceuticalsNeutropeniaOctober 16, 2020Phase 1NCT03995108X4 PharmaceuticalsWHIM SyndromeOctober 17, 2019Phase 3NCT05103917Abbisko Therapeutics Co, LtdTriple Negative Breast CancerJuly 21, 2021Phase 1|Phase 2NCT00063804National Institute of Allergy and Infectious Diseases (NIAID)|AIDS Clinical Trials GroupHIV Infections Phase 1NCT02923531X4 PharmaceuticalsClear Cell Renal Cell CarcinomaDecember 7, 2016Phase 1|Phase 2NCT02680782X4 Pharmaceuticals|CovanceHealthyJanuary 12, 2016Phase 1 |
REF
- [1]. Skerlj RT, et al. Discovery of novel small molecule orally bioavailable C-X-C chemokine receptor 4 antagonists that are potent inhibitors of T-tropic (X4) HIV-1 replication. J Med Chem. 2010 Apr 22;53(8):3376-88. [Content Brief][2]. Chow LN, et al. Impact of a CXCL12/CXCR4 Antagonist in Bleomycin (BLM) Induced Pulmonary Fibrosis and Carbon Tetrachloride (CCl4) Induced Hepatic Fibrosis in Mice. PLoS One. 2016 Mar 21;11(3):e0151765. [Content Brief][3]. Uchida D, et al. Effect of a novel orally bioavailable CXCR4 inhibitor, AMD070, on the metastasis of oral cancer cells. Oncol Rep. 2018 Jul;40(1):303-308. [Content Brief]
/////////////////////////////////////////////////////////////////////////////MAVORIXAFOR, AMD 070, PHASE 2
