
RADOTINIB
- Molecular FormulaC27H21F3N8O
- Average mass530.504 Da
4-Methyl-N-[3-(4-methyl-1H-imidazol-1-yl)-5-(trifluoromethyl)phenyl]-3-{[4-(2-pyrazinyl)-2-pyrimidinyl]amino}benzamide
4-methyl-N-[3-(4-methylimidazole-l-yl)-5-trifluoromethyl-phenyl] –
3-(4-pyrazine-2-yl-pyrimidine-2-yl amino)benzamide
9242
926037-48-1[RN]
| 926037-48-1 (Radotinib); 926037-85-6 (Radotinib 2HCl); |
Benzamide, 4-methyl-N-[3-(4-methyl-1H-imidazol-1-yl)-5-(trifluoromethyl)phenyl]-3-[[4-(2-pyrazinyl)-2-pyrimidinyl]amino]-
I284LJY110, IY5511
UNII-I284LJY110
радотиниб
رادوتينيب
雷度替尼
MOA:Bcr-Abl tyrosine kinase inhibitor
Indication:Chronic myeloid leukemia (CML )
Company:IL-Yang (Originator)
IY-5511; IY-5511A3001
| Approval Date | Approval Type | Trade Name | Indication | Dosage Form | Strength | Company | Review Classification |
|---|---|---|---|---|---|---|---|
| 2012-01-05 | Marketing approval | Supect | Chronic myeloid leukemia (CML ) | Capsule | 100 mg/200 mg | IL-Yang |
Radotinib dihydrochloride was approved by Korea Food and Drug Administration (KFDA) on January 5, 2012. It was developed and marketed as Supect® by IL-Yang in KR.
Radotinib dihydrochloride is a second-generation tyrosine kinase inhibitor of Bcr-Abl fusion protein and the platelet-derived growth factor receptor (PDGFR). It is indicated for the second-line treatment of patients with Philadelphia chromosome-positive (Ph+) CML that is refractory to Imatinib mesilate.
Supect® is available as capsule for oral use, containing 100 mg or 200 mg of free Radotinib. The recommended dose is 400 mg twice daily.
Radotinib (INN; trade name Supect), and sometimes referred to by its investigational name IY5511, is a drug for the treatment of different types of cancer, most notably Philadelphia chromosome-positive (Ph+) chronic myeloid leukemia (CML)[1] with resistance or intolerance of other Bcr-Abl tyrosine-kinase inhibitors, such as patients resistant or intolerant to imatinib.
Radotinib is being developed by Ilyang Pharmaceutical Co., Ltd of South Korea[2] and is co-marketed by Daewoong Pharmaceutical Co. Ltd, in South Korea.[3] Radotinib completed a multi-national Phase II clinical trial study in 2012[4] and in August 2011, Ilyang initiated a Phase III, multinational, multi-center, open-label, randomized study for first-line indication.[5] Its mechanism of action involves inhibition of the Bcr-Abl tyrosine kinase and of platelet-derived growth factor receptor (PDGFR).[6]
In January 2012, radotinib hydrochloride (marketed as Supect ®) obtained its approval from the KFDA (Korea Food and Drug Administration) for the treatment of patients with Philadelphia chromosomepositive chronic myeloid leukemia (CML) who have become resistant to existing drugs such as Gleevec, Tasigna and Sprycel. Originally developed by IL-YANG pharmaceuticals of South Korea as an orally second-generation tyrosine kinase inhibitor, the drug inhibits both Bcr-Abl fusion protein and the platelet-derived growth factor receptor (PDGFR).
Chemical Synthesis
Because of the structural similarity of radotinib to that of nilotinib (Tasigna ®), the process-scale synthetic route (which is depicted in the scheme) is capable of furnishing both drugs.Claisen condensation of commerical 2-acetylpyrazine (142) with N,N-dimethylformamide dimethylacetal gave rise to the enamino ketone 143 in 81% yield. Under basic conditions, vinylogous amide 143 was coupled with commercial guanidine nitrate 144187 to produce aminopyridine 145. Subsequent condensation with commercial aniline (146) by means of potassium t-butoxide in THF constructed radotinib 147 in 85% yield as the free base, and this material could be converted to the radotinib dihydrochloride (XXII) upon exposure to concentrated hydrochloric acid in chilled acetone. 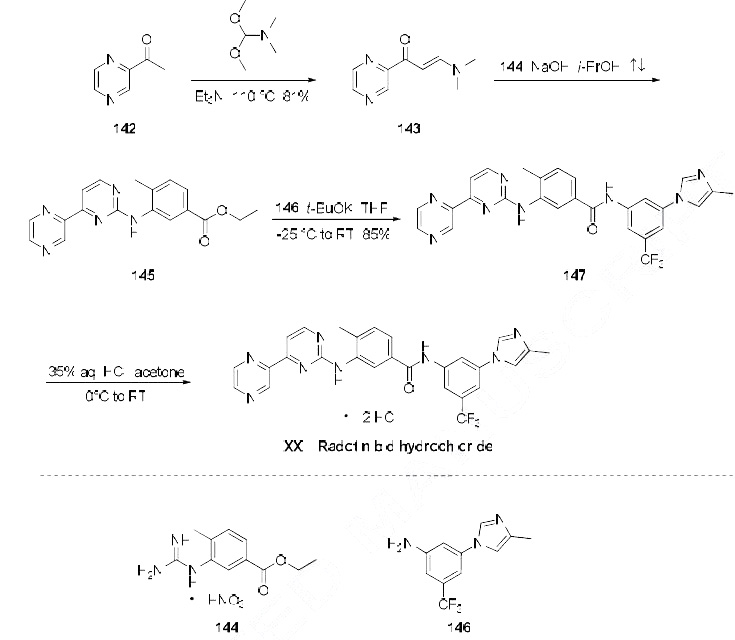
PATENT1.
WO2007018325A1 / US7501424B2.
https://patents.google.com/patent/WO2007018325A1/en
PATENT
WO2010018895A1 / CN101648946A.
https://patents.google.com/patent/WO2010018895A1/en
The compound represented by Formula 1 was disclosed in Korea Patent Registration
No. 10-0674813. A preferred compound according to Formula 1 includes 4-methyl-N- [3-(4-methylimidazole- 1 -yl)-5-trifluoromethyl-phenyl] -3-(4-pyrazine-2-yl -pyrimidine-2-yl amino)benzamide. It has been known that the compound represented by Formula 1 can inhibit at least one kind of tyrosine kinase, for example, c-Abl, Bcr- AbI, and receptor tyrosine kinases (PDGF-R, Flt3, VEGF-R, EGF-R and c-Kit). Accordingly, the compound represented by Formula 1 may be used for treatment of various kinds of cancers in a warm blooded animal, such as lung cancer, stomach cancer, colon cancer, pancreatic cancer, liver cancer, prostate cancer, breast cancer, chronic or acute leukemia, hematological malignancy, brain tumor, bladder cancer, rectal cancer, uterine cervical cancer, lymphoma, etc.
[7] According to a conventional method, the compound represented by Formula 1 is synthesized through hydrolysis of ethyl ester into carboxylic acid and then a reaction with aniline, and herein, diethyl cyano phosphonate is used as a coupling agent (see Reaction Scheme 1).
[8] [Reaction Scheme 1]
NsOIf


( 2 ) { s :

Diethyl cyano phosphate

( 1 )
[10] The above method requires a process of hydrolyzing ethyl ester (2) into carboxylic acid (3). In order to obtain the compound represented by Formula 3 as shown in Reaction Scheme 1, a preparation process and a purifying process require a long time. Also, in the condensation reaction, there have been problems such as high production cost due to a low yield (30 to 40%) of the compound represented by Formula 1. Especially, it is very difficult to treat carboxylic acid (3) after purification and reaction, due to its very low solubility in general organic solvent. Also, diethyl cyano phosphonate used for the condensation reaction is an expensive reagent, and an environmentally harmful and very toxic material, which has LD50 values of 25mg/Kg and 4mg/Kg in mice and rabbits (that is, rodents), respectively. Therefore, there is a requirement for an alternative method of conveniently, consistently, efficiently and rapidly preparing a high-purity compound (represented by Formula 1) with low production cost in high yield, which is not harmful for humans and the environment.


Example 2
[69] Synthesis of 4-methyl-N-[3-(4-methylimidazole-l-yl)-5-trifluoromethyl-phenyl] –
3-(4-pyrazine-2-yl-pyrimidine-2-yl amino)benzamide
[70]
[71] Method A
[72] A pale yellow solid final compound (18.7g, yield 85%) was obtained by reacting
3-(4-methyl-imidazole-l-yl)-5-trifluoromethyl-phenylamine (1Og, 41.46mmol) with 4-methyl -3-(4-pyrazine-2-yl-pyrimidine-2-yl amino)-benzoic acid ethyl ester in a similar manner as described in Method A of Example 1, except that 4-methyl-3-(4-pyrazine-2-yl-pyrimidine-2-yl amino) -benzoic acid ethyl ester (15.3g, 45.60mmol) was used, instead of 4-methyl-3-(4-thiazole-2-yl-pyrimidine-2-yl amino)benzoic acid ethyl ester.
[73] 1H-NMR(DMSOd , δ= 2.21(s,3H), 2.38(s,3H), 7.35(s,lH), 7.39(s,lH), 7.54(s,lH),
7.63(d,lH), 7.75(d,lH), 8.14(d,2H), 8.38(d,2H), 8.54(d,2H), 8.68(s,lH), 9.06(s,lH), 9.45(s, IH), 10.56(s,lH)
[74]
[75] Method B
[76] A pale yellow solid final compound (18.3g, yield 83%) was obtained by reacting
3-(4-methyl-imidazole-l-yl)-5-trifluoromethyl-phenylamine (1Og, 41.46mmol) with 4-methyl -3-(4-pyrazine-2-yl-pyrimidine-2-yl amino)-benzoic acid methyl ester in a similar manner as described in Method A of Example 1, except that 4-methyl-3-(4-pyrazine-2-yl-pyrimidine-2-yl amino) -benzoic acid methyl ester (14.7g, 45.60mmol) was used, instead of 4-methyl-3-(4-thiazole-2-yl-pyrimidine-2-yl amino)benzoic acid ethyl ester.
[77]
[78] Method C
[79] A pale yellow solid final compound (17.2g, yield 78%) was obtained by reacting
3-(4-methyl-imidazole-l-yl)-5-trifluoromethyl-phenylamine (1Og, 41.46mmol) with 4- methyl-3-(4-pyrazine-2-yl-pyrimidine-2-yl amino)benzoic acid methyl ester (14.7g, 45.60mmol) in a similar manner as described in Method A of Example 1, except that sodium tert-butoxide was used, instead of potassium tert-butoxide.
[80]
[81] Method D
[82] A pale yellow solid final compound (16. Ig, yield 73%) was obtained by reacting
3-(4-methyl-imidazole-l-yl)-5-trifluoromethyl-phenylamine (1Og, 41.46mmol) with 4- methyl-3-(4-pyrazine-2-yl-pyrimidine-2-yl amino)benzoic acid phenyl ester in a similar manner as described in Method A of Example 1, except that 4-methyl-3-(4-pyrazine-2-yl-pyrimidine-2-yl amino) -benzoic acid phenyl ester (17.5g, 45.60mmol) was used, instead of 4-methyl-3-(4-thiazole-2-yl-pyrimidine-2-yl amino)benzoic acid ethyl ester.
SYN
https://www.sciencedirect.com/science/article/abs/pii/S0968089614001230


Radotinib hydrochloride (Supect) In January 2012, radotinib hydrochloride (marketed as Supect) obtained its approval from the KFDA (Korea Food and Drug Administration) for the treatment of patients with Philadelphia chromosome-positive chronic myeloid leukemia (CML) who have become resistant to existing drugs such as Gleevec, Tasigna and Sprycel.181 Originally developed by IL-YANG pharmaceuticals of South Korea as an oral second-generation tyrosine kinase inhibitor, the drug inhibits both Bcr-Abl fusion protein and the platelet-derived growth factor receptor (PDGFR).182 Because of the structural similarity of radotinib to that of nilotinib (Tasigna), the processscale synthetic route (which is depicted in Scheme 27) is capable of furnishing both drugs.183–185 Claisen condensation of commerical 2-acetylpyrazine (142) with N,N-dimethylformamide dimethylacetal gave rise to the enamino ketone 143 in 81% yield.186 Under basic conditions, vinylogous amide 143 was coupled with commercial guanidine nitrate 144187 to produce aminopyridine 145. 184 Subsequent condensation with commercial aniline (146) by means of potassium t-butoxide in THF constructed radotinib 147 in 85% yield as the free base, and this material could be converted to the radotinib dihydrochloride (XXII) upon exposure to concentrated hydrochloric acid in chilled acetone.185
181. Droppert, P. In Biotech Strategy Blog: http://biotechstrategyblog.com/2012/01/ radotinib-approved-in-south-korea-for-cml.html/, 2012.
182. Radotinib hydrochloride http://www.cancer.gov/drugdictionary?cdrid= 723999.
183. Davies, S.; Bolos, J.; Serradell, N.; Bayes, M. Drugs Future 2007, 32, 17.
184. Kim, D.-Y.; Cho, D.-J.; Lee, G.-Y.; Kim, H.-Y.; Woo, S.-H.; Kim, Y.-S.; Lee, S.-A.; Han, B.-C. WO Patent 2007/018325 A1, 2007.
185. Kim, D. Y.; Cho, D. J.; Lee, G. Y.; Kim, H. Y.; Woo, S. H. WO Patent 2010/018895 A1, 2010.
///////////////////////////////////////////

AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
References
- ^ Joanne Bronson; Amelia Black; T. G. Murali Dhar; Bruce A. Ellsworth; J. Robert Merritt (2013). “To Market, To Market – 2012”. Radotinib (Anticancer). Annual Reports in Medicinal Chemistry. Vol. 48. pp. 523–524. doi:10.1016/b978-0-12-417150-3.00028-4. ISBN 9780124171503.
- ^ “Il-Yang Pharmaceutical”.
- ^ http://www.dailypharm.com/Users/News/EnglishNews.html?NewsID=3108&nStart=1023&mode=&searchValue=[dead link]
- ^ Kim SH, Menon H, Jootar S, Saikia T, Kwak JY, Sohn SK, Park JS, Jeong SH, Kim HJ, Kim YK, Oh SJ, Kim H, Zang DY, Chung JS, Shin HJ, Do YR, Kim JA, Kim DY, Choi CW, Park S, Park HL, Lee GY, Cho DJ, Shin JS, Kim DW (2014). “Efficacy and safety of radotinib in chronic phase chronic myeloid leukemia patients with resistance or intolerance to BCR-ABL1 tyrosine kinase inhibitors”. Haematologica. 99 (7): 1191–6. doi:10.3324/haematol.2013.096776. PMC 4077080. PMID 24705186.
- ^ https://clinicaltrials.gov/ct2/show/NCT01511289?term=radotinib&rank=1
- ^ “Radotinib hydrochloride”. NCI Drug Dictionary. National Cancer Institute. 2011-02-02.
| Clinical data | |
|---|---|
| Trade names | Supect |
| ATC code | None |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 926037-48-1 |
| PubChem CID | 16063245 |
| ChemSpider | 17222861 |
| UNII | I284LJY110 |
| CompTox Dashboard (EPA) | DTXSID90239069 |
| Chemical and physical data | |
| Formula | C27H21F3N8O |
| Molar mass | 530.515 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI |
Patent
Publication numberPriority datePublication dateAssigneeTitle
WO2003066613A1 *2002-02-072003-08-14Novartis AgN-phenyl-2-pyrimidine-amine derivatives
WO2004005281A1 *2002-07-052004-01-15Novartis AgInhibitors of tyrosine kinases
KR100674813B1 *2005-08-052007-01-29일양약품주식회사N-phenyl-2-pyrimidine-amine derivatives and process for the preparation thereof
Publication numberPriority datePublication dateAssigneeTitle
US9132126B22011-04-192015-09-15Il-Yang Pharm. Co., Ltd.Phenyl-isoxazole derivatives and preparation process thereof
KR20180032784A *2016-09-232018-04-02재단법인 대구경북첨단의료산업진흥재단Novel imidazolyl pyrimidine derivatives, preparation method thereof, and pharmaceutical composition for use in preventing or treating cancer containing the same as an active ingredient
Family To Family Citations
KR101956586B1 *2012-03-272019-03-11일양약품주식회사Pharmaceutical composition and preparation method thereof
////////////////////RADOTINIB, UNII-I284LJY110, радотиниб , رادوتينيب , 雷度替尼 , IY5511, IY 5511, korea 2012, Chronic myeloid leukemia