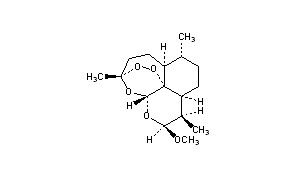
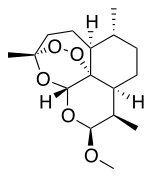
ARTEMETHER
- Molecular FormulaC16H26O5
- Average mass298.375 Da
(3R,5aS,6R,8aS,9R,10S,12R,12aR)-10-methoxy-3,6,9-trimethyldecahydro-3,12-epoxy[1,2]dioxepino[4,3-i]isochromene
(4S,5R,8S,9R,10S,12R,13R)-10-Methoxy-1,5,9-trimethyl-11,14,15,16-tetraoxatetracyclo[10.3.1.04,13.08,13]hexadecane[
3,12-Epoxy-12H-pyrano[4,3-j]-1,2-benzodioxepin, decahydro-10-methoxy-3,6,9-trimethyl-, (3R,5aS,6R,8aS,9R,10S,12R,12aR)-
3,12-Epoxy-12H-pyrano[4,3-j]-1,2-benzodioxepin, decahydro-10-methoxy-3,6,9-trimethyl-, (5aS,6R,8aS,9R,10S,12R,12aR)-
71963-77-4[RN]
dihydroartemisinin methyl ether
Dihydroqinghaosu Methyl Ether
KD4165000
PALUTHER
- SM 224
- SM-224
- 3,12-Epoxy-12H-pyrano[4,3-j]-1,2-benzodioxepin, decahydro-10-methoxy-3,6,9-trimethyl-, [3R-(3α,5aβ,6β,8aβ,9α,10α,12β,12aR*)]-
- (3R,5aS,6R,8aS,9R,10S,12R,12aR)-Decahydro-10-methoxy-3,6,9-trimethyl-3,12-epoxy-12H-pyrano[4,3-j]-1,2-benzodioxepin
- (+)-Artemether
Artemether
CAS Registry Number: 71963-77-4
CAS Name: (3R,5aS,6R,8aS,9R,10S,12R,12aR)-Decahydro-10-methoxy-3,6,9-trimethyl-3,12-epoxy-12H-pyrano[4,3-j]-1,2-benzodioxepin
Additional Names: dihydroartemisinin methyl ether; dihydroqinghaosu methyl ether; o-methyldihydroartemisinin
Manufacturers’ Codes: SM-224
Trademarks: Paluther (RPR)
Molecular Formula: C16H26O5, Molecular Weight: 298.37,
Percent Composition: C 64.41%, H 8.78%, O 26.81%
Literature References: Derivative of artemisinin, q.v. Prepn: Y. Li et al.,K’o Hsueh T’ung Pao24, 667 (1979), C.A.91, 211376u (1979); eidem,Acta Pharm. Sin.16, 429 (1981). Absolute configuration: X.-D. Luo et al.,Helv. Chim. Acta67, 1515 (1984). NMR spectral study: F. S. El-Feraly et al.,Spectrosc. Lett.18, 843 (1985). Inhibition of protein synthesis: H. M. Gu et al.,Biochem. Pharmacol.32, 2463 (1983). Antimalarial activity: S. Thaithong, G. H. Beale, Bull. WHO63, 617 (1985). Series of articles on chemistry, pharmacology and antimalarial efficacy: China Cooperative Research Group on Qinghaosu, J. Tradit. Chin. Med.2, 3-50 (1982). Toxicity data: eidem,ibid. 31. Clinical trial in cerebral malaria in children: M. B. van Hensbroek et al.,N. Engl. J. Med.335, 69 (1996). Review: R. N. Price, Expert Opin. Invest. Drugs9, 1815-1827 (2000).
Properties: Crystals, mp 86-88°. [a]D19.5 +171° (c = 2.59 in CHCl3). LD50 i.m. in mice: 263 mg/kg (China Cooperative Research Group on Qinghaosu).
Melting point: mp 86-88°
Optical Rotation: [a]D19.5 +171° (c = 2.59 in CHCl3)
Toxicity data: LD50 i.m. in mice: 263 mg/kg (China Cooperative Research Group on Qinghaosu)
Therap-Cat: Antimalarial.
Keywords: Antimalarial.
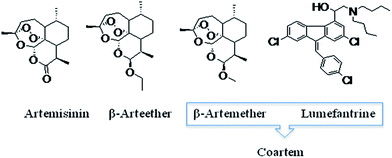
Artemether is an antimalarial agent used in combination with lumefantrine for the treatment of acute uncomplicated malaria caused by Plasmodium falciparum.
Artemether is an antimalarial agent used to treat acute uncomplicated malaria. It is administered in combination with lumefantrine for improved efficacy. This combination therapy exerts its effects against the erythrocytic stages of Plasmodium spp. and may be used to treat infections caused by P. falciparum and unidentified Plasmodium species, including infections acquired in chloroquine-resistant areas.
Artemether is a natural product which effectively kills both malarial parasites P. falciparum and P. vivax. Artemether is usually used in combination with Lumefantrine for the treatment of malaria. Arthemether also kills trematodes of the species Schistosoma, providing protection against schistosomiasis. Sesquiterpene lactones like artemether, artesunate, and artemisinin have potential applications in certain types of cancer and inflammatory conditions.
Artemether is a medication used for the treatment of malaria.[2] The injectable form is specifically used for severe malaria rather than quinine.[2] In adults, it may not be as effective as artesunate.[2] It is given by injection in a muscle.[2] It is also available by mouth in combination with lumefantrine, known as artemether/lumefantrine.[3][4]
Artemether causes relatively few side effects.[5] An irregular heartbeat may rarely occur.[5] While there is evidence that use during pregnancy may be harmful in animals, there is no evidence of concern in humans.[5] The World Health Organization (WHO) therefore recommends its use during pregnancy.[5] It is in the artemisinin class of medication.[5]
Artemether has been studied since at least 1981, and been in medical use since 1987.[6] It is on the World Health Organization’s List of Essential Medicines.[7]
Synthesis Reference
Haynes RK, Vonwiller SC: Extraction of artemisinin and artemisinic acid: preparation of artemether and new analogues. Trans R Soc Trop Med Hyg. 1994 Jun;88 Suppl 1:S23-6. Pubmed.
REF
ChemMedChem (2007), 2, (10), 1448-1463
PAT
| Malaria is a serious parasitic disease caused by Plasmodium parasites in the human body. Plasmodium falciparum, Plasmodium vivax, Plasmodium ovale, Plasmodium malaria and Plasmodium knowlesi are the parasites that live in humans, of which P. vivax and P. falciparum are the most common. |
| Traditional anti-malarial drugs mainly include quinine, chloroquine, primaquine, and pyrimethamine. In 1972, the antimalarial active ingredient artemisinin extracted from the Compositae plant Artemisia annuaL by Chinese scientists is the most popular antimalarial effect after chloroquine, pyrimethamine, primary amine and sulfonamide. Drugs, especially for the treatment of cerebral malaria and anti-chloroquine malaria. |
| At present, a large number of artemisinin derivatives have been synthesized and screened for antimalarial activity. Artemether is a compound with excellent curative effect. In addition to the advantages of artemisinin’s quick effect and low toxicity, its solubility in oil is also higher than that of artemisinin. Artemisinin is large, which is especially beneficial for the preparation of preparations. Since artemether has two products, α and β epimers, and the antimalarial activity of artemether is mainly isomer β, so the industrial automation and intelligent production of β-artemether and the improvement of the process are realized. , reducing the impurities produced by the reaction, improving the quality of the product, and improving the purity of the product are the problems that need to be solved in today’s scientific research. |
| Patent CN104557965B discloses a preparation process of β-artemether, which mainly includes adding dihydroartemisinin and etherification reagent to alcohol to form a reaction system, and then adding acid to the reaction system for reaction. Water or non-alkaline aqueous solution is added to the reaction system to crystallize, namely β-artemether. The preparation process claims to effectively inhibit the production of isomer α-artemether in the reaction, and can make the etherification reaction proceed mildly, with simple post-treatment and high purity; although the purity of the product has been improved, the yield and Purity needs to be further improved. |
| Patent CN102731523B discloses a method for preparing β-artemether, which mainly includes the reaction of artemisinin under the action of a reducing agent to generate dihydroartemisinin, and the reaction of dihydroartemisinin with p-toluenesulfonic acid to generate β-artemisinin. The crude artemether is crystallized with methanol, ethanol, ethylene glycol or isopropanol, filtered, washed and dried. The method for preparing B-artemether of the invention has mild conditions, is environmentally friendly, is suitable for industrial production, and has a product yield of over 90 percent and a purity of 99.2 percent. The crystallization step of the invention adopts organic reagents, which adversely affects the quality control of subsequent products. |
| Patent CN103180325B discloses a method for preparing β-artemether, which uses dihydroartemisinin as a raw material and undergoes etherification reaction with trimethyl orthoformate in organic solvents including esters and alkanes to obtain β-artemether. The method of the invention is easy to control in process operation, high in yield, low in cost and high in product quality, and is suitable for industrial production. The method requires vacuum distillation, the obtained crude product needs to be redissolved with methanol, decolorized with activated carbon, etc., new impurities are easily introduced, the operation is not simple enough, and the efficiency is low. |
| Patent CN107793428A discloses a preparation method of artemether, hydrogenating artemisinin to obtain dihydroartemisinin, adding trimethyl orthoformate, reacting with boron trifluoride ether solution, slowly adding saturated sodium bicarbonate solution dropwise, The system was adjusted to neutrality, the liquids were separated, the aqueous phase was extracted with dichloromethane, the organic phases were combined, washed with saturated brine, dried over anhydrous sodium sulfate, and the solvent was removed under reduced pressure to obtain a solid; the obtained solid was dissolved in methanol, and an appropriate amount of activated carbon was added to obtain a solid. Reflux and decolorize, filter, add pure water dropwise to the filtrate, crystallize, wash with water, and dry to obtain artemether. However, this method requires steps such as extraction with an organic reagent dichloromethane and decolorization with activated carbon, which is cumbersome to handle. |
| Therefore, the following problems generally exist in the process of preparing β-artemether at present: |
| (1) when preparing β-artemether, the reaction time is longer, the impurities are large, and the purity and yield of the product are not high enough; |
| (2) The use of organic reagents in the subsequent purification process has a certain impact on the quality control of the product; |
| (3) The batch production equipment is adopted, the subsequent process steps are many, the degree of industrialization is low, the production efficiency is low, and it does not meet the requirements of GMP. |
| Example 1 |
| This embodiment includes the following steps: |
| (1) at room temperature, add methanol 2400L in the 3000L stirred tank (1), then add 600kg of dihydroartemisinin through the solid feed pump, and circulate and disperse evenly; |
| (2) add etherification agent trimethyl orthoformate and acid catalyst acetyl chloride through three-way automatic feeding mixing reactor again, the volume ratio is 500:100:3, the mixing reactor control temperature is 5 ℃, and the flow rate of control feeding is 5L /min; |
| (3) in the continuous flow pipeline, enter the second mixer and add 5% sodium bicarbonate solution to neutralize, and the adding speed is 1.0L/min, and is filtered through the fine filter; |
| (4) Then directly enter the 2000L crystallization reaction kettle 11 with 300L of water added in advance and keep the temperature at 10°C. At the same time, purified water was added to the reaction kettle at a rate of 12L/min, and the crystallization was continued for 1.5h; the jacket of the crystallization kettle was fed with -10°C chilled water for 30min, and the temperature of the system was controlled to 5°C. |
| (5) centrifugal washing, obtaining crude artemether 704.5kg, drying to obtain artemether fine product 608.6kg, β-artemether purity 99.83%, α-artemether impurity 0.12%, and other single impurities less than 0.1%, The content is 99.8%, the mass yield is 96.1%, and the molar yield is 91.42%. |
| Example 2 |
| This embodiment includes the following steps: |
| (1) at room temperature, 2400L of methanol was pumped into the 3000L reactor 1, and then 800kg of dihydroartemisinin was added by the solid feed pump, and the circulation was uniformly dispersed; |
| (2) add etherifying agent dimethyl phosphate and acid catalyst boron trifluoride ether through the three-way automatic feeding mixing reactor again, the volume ratio is 500:105:3.5, the mixing reactor control temperature is 3 ℃, and the control feeding flow rate is 3L/min; |
| (3) in the continuous flow pipeline, enter the second mixer and add 3% sodium bicarbonate solution to neutralize, and the speed of addition is 1.8L/min, through the fine filter; |
| (4) Directly enter the 2000L crystallization reaction kettles 11 and 12 with 300L of water added in advance and the temperature kept at 10°C. At the same time, purified water was added to the reaction kettle at 9 L/min, and the crystallization was continued for 2.5 hours; the jacket of the crystallization kettle was fed with -10 °C chilled water for 30 minutes, and the temperature of the system was controlled to 10 °C |
| (5) centrifugal washing, obtain crude artemether 939.3kg, oven dry to obtain artemether fine product 809.7kg, β-artemether purity 99.81%, α-artemether impurity 0.11%, other single impurities are less than 0.1%, The content is 99.8%, the mass yield is 96.2%, and the molar yield is 91.6%. |
| Example 3 |
| This embodiment includes the following steps: |
| (1) 2400L of methanol was pumped into the 3000L reactor F1 at room temperature, and then 400kg of dihydroartemisinin was added through the solid feed pump, and the circulation was uniformly dispersed; |
| (2) Add etherification agent dimethyl phosphate and acid catalyst trimethylchlorosilane through the three-way automatic feeding mixing reactor, the volume ratio is 500:95:2.5, the mixing reactor is controlled at a temperature of 8 °C, and the feeding liquid is controlled to be added. The flow rate is 7L/min, and the reaction time is; |
| (3) in the continuous flow pipeline, enter the second mixer and add 8% sodium bicarbonate solution for neutralization, and the rate of addition is 0.6L/min, passing through the fine filter; |
| (4) Directly enter into the 2000L crystallization reactor J2 with 300L water added in advance and keeping the temperature at 10°C. At the same time, purified water was added to the reaction kettle at 15 L/min, and the crystallization was continued for 1 hour; the jacket of the crystallization kettle was fed with -10 °C chilled water for 30 minutes, and the temperature of the system was controlled to 0 °C |
| (5) centrifugal washing, obtain crude artemether 939.3kg, oven dry to obtain artemether fine product 809.7kg, β-artemether purity 99.81%, α-artemether impurity 0.11%, other single impurities are less than 0.1%, The content is 99.8%, the mass yield is 95.5%, and the molar yield is 90.9%. |
| Comparative Example 1 |
| The difference between this embodiment and Example 1 is that hydrochloric acid is used instead of the acidic catalyst. Finally, 633.6kg of crude artemether was obtained, and 550.3kg of fine artemether was obtained by drying. The purity of β-artemether was 94.20%, and the impurities of α-artemether were 3.66%. %, and the molar yield was 80.6%. |
| Comparative Example 2 |
| The difference between this embodiment and Example 1 is that the step of adding water in advance in the crystallization kettle is removed. Finally, 645.1kg of crude artemether was obtained, and 562.2kg of fine artemether was obtained by drying. The purity of β-artemether was 99.68%, the impurity of α-artemether was 0.22%, and the average of single and impurity was less than 0.1%. The mass yield was 88.7%. %, and the molar yield was 84.4%. |
| In Comparative Example 2, the step of adding water in advance in the crystallization was removed, the purity of β-artemether was 99.68%, and the yield was 88.7%. The yield dropped by 7.6%. |
| The above detailed description is a specific description of one of the feasible embodiments of the present invention, and this embodiment is not intended to limit the patent scope of the present invention. Any equivalent implementation or modification that does not depart from the present invention shall be included in the present invention. within the scope of the technical solution. |
SYN1
Synthetic Reference
Continuous synthesis of artemisinin-derived medicines; Gilmore, Kerry; Kopetzki, Daniel; Lee, Ju Weon; Horvath, Zoltan; McQuade, D. Tyler; Seidel-Morgenstern, Andreas; Seeberger, Peter H. Chemical Communications (Cambridge, United Kingdom); Volume 50; Issue 84; Pages 12652-12655; Journal; 2014

SYN2
Synthetic Reference
An Improved Manufacturing Process for the Antimalaria Drug Coartem. Part I; Boehm, Matthias; Fuenfschilling, Peter C.; Krieger, Matthias; Kuesters, Ernst; Struber, Fritz; Organic Process Research & Development; Volume 11; Issue 3; Pages 336-340; Journal; 2007

SYN3
Synthetic Reference
Some transition metal complexes bearing artemisinin derivatives and (N-N-O) tridentate chromium (III) complexes ligated by 2-benzolmidazo-yl-6-acetyl-pyridines for catalytic behaviour towards ethylene; Obaleye, Joshua Ayoola; Amolegbe, Saliu Alao; Adewuyi, Sheriff; Sun, Wenhua; Oshodi, Margaret Damilola; Journal of Chemistry and Chemical Engineering; Volume 4; Issue 12; Pages 23-32; Journal; 2010

SYN4
Synthetic Reference
Method and apparatus for the synthesis of dihydroartemisinin and artemisinin derivatives; Kopetzki, Daniel; McQuade, David Tyler; Seeberger, Peter H.; Gilmore, Kerry; Assignee Max-Planck-Gesellschaft zur Foerderung der Wissenschaften e.V., Germany; 2015; Patent Information; Jan 21, 2015; EP 2826779 A1

PAPER
https://pubs.rsc.org/en/content/articlehtml/2014/ra/c4ra05531d
An efficient one pot green synthesis of β-artemether/arteether from artemisinin has been developed using a sodium borohydride-cellulose sulfuric acid (CellSA) catalyst system. The green methodology is high yielding and the catalyst has good recyclability.
Experimental section
Representative procedure for catalyst preparation
Preparation of cellulose sulfuric acid.To a magnetically stirred mixture of 5.00 g of cellulose (DEAE for column chromatography, Merck) in 20 ml of n-hexane, 1.0 g of chlorosulfonic acid (9 mmol) was added dropwise at 0 °C over 2 h. HCl gas was removed from the reaction vessel immediately. After the addition was complete, the mixture was stirred for 2 h. Then, the mixture was filtered, washed with 30 ml of acetonitrile, and dried at room temperature to obtain 5.47 g cellulose sulfuric acid as a white powder.17
General procedure for the arteether from artemisinin in one-pot
To a solution of artemisinin (200 mg, 0.71 mmol) in ethanol (15 ml) and trimethyl orthoacetate (0.5 ml) was added NaBH4 (67 mg, 1.77 mmol, 2.5 equ.) and cellulose sulfuric acid (0.015 g). Reaction mixture was carried out at −5 to 0 °C for 60 min, and then stirred at room temperature for 1.5 h. Then we added a solution of sodium bicarbonate to quenched the reaction. The slurry was stirred in an below 20 °C for 1 h and allowed to settle for 30 min. Solid crude arteether was collected by filtration, and the cake was washed with of ethanol. The reaction mass was heated to 40 ± 5 °C in water. The reaction mass was seeded with pure β-arteether. Then it was filtered, washed with chilled 50% solution of ethanol in water and dried.
General procedure for the artemether from artemisinin in one-pot
Artemisinin (200 mg, 0.71 mmol) in methanol (15 ml) and trimethylorthoformate (0.5 ml), cellulose sulfuric acid (0.015 g), was carried out at −5 to 0 °C for 60 min, and then stirred at room temperature for 1.5 h. The reaction was monitored by TLC and HPLC to check completion of the reaction. The cellulose sulfuric acid was removed by filtration, the filtrate was concentrated. Then we added a solution of sodium bicarbonate to terminate the reaction. Then, follow above recrystallization method.
Preparation of cellulose sulfuric acid. To a magnetically stirred mixture of 5.00 g of cellulose (DEAE for column chromatography, Merck) in 20 ml of n-hexane, 1.0 g of chlorosulfonic acid (9 mmol) was added dropwise at 0 0 C over 2 h. HCl gas was removed from the reaction vessel immediately. After the addition was complete, the mixture was stirred for 2 h. Then, the mixture was filtered, washed with 30 ml of acetonitrile, and dried at room temperature to obtain 5.47 g cellulose sulfuric acid as a white powder. K General procedure for the arteether from artemisinin in one-pot. To a solution of artemisinin (200 mg, 0.71 mmol) in ethanol (15 mL) and trimethyl orthoacetate (0.5 mL) was added NaBH4 (67 mg, 1.77 mmol, 2.5 equ.) and cellulose sulfuric acid (0.015 g). Reaction mixture was was carried out at -5 to 0°C for 60 min, and then stirred at room temperature for 1.5 h. Then we added a solution of sodium bicarbonate to quenched the reaction. The slurry was stirred in an below 20 0 C for 1 h and allowed to settle for 30 min. Solid crude arteether was collected by filtration, and the cake was washed with of ethanol. The reaction mass was heated to 40± 5 0 C in water. The reaction mass was seeded with pure β–arteether. Then it was filtered, washed with chilled 50% solution of ethanol in water and dried. General procedure for the artemether from artemisinin in one-pot. Artemisinin (200 mg, 0.71 mmol) in methanol (15 ml) and trimethylorthoformate (0.5 ml), cellulose sulfuric acid (0.015 g), was carried out at -5 to 0°C for 60 min, and then stirred at room temperature for 1.5 h. The reaction was monitored by TLC and HPLC to check completion of the reaction. The cellulose sulfuric acid was removed by filtration, the filtrate was concentrated. Then we added a solution of sodium bicarbonate to terminate the reaction. Then, follow above recrystallization method.


PATENT
https://patents.google.com/patent/US6683193B2/en
Approximately, out of the 4 billion people suffering from malaria, 1-3 million, mostly children die every year worldwide. The rapidly spreading multidrug resistant parasite to standard quinoline based antimalarial drugs such as chloroquine and mefloquine based antimalarial complicate chemotherapy treatment of malaria patients.
Artemether is a methyl ether derivative of dihydroartemisinin. Dihydroartemisinin is derived from arternisinin, a novel sesquiterpene endoperoxide isolated from the plant Artemisia annua. Artemisinin and its derivative artemether, arteether, artelinate and artesunate a novel class of antimalarials derived from Artemisia annua are now proving their promising activity and being used for the treatment; of uncomplicated severe complicated/cerebral and multi drug resistant malaria.
Artemether, developed in France and China has undergone extensive preclinical, animal, toxicological studies as well as clinical studies. Artemether is more potential as compared to artemisinin and an antimalarial drug especially for treating multi drug resistant and complicated strains of Plasmodium falciparum.
Artemether shows rapid shizonticidal action with quicker parasite clearance rate, short half life less side effect and low recrudence rate. Brossi, et al (Brossi, A; Venugopalan, B, Domingueg, G L; Yeh, H. J. C; Flippend-Anderson, J. L.; Buchs, P; Luo, X. D.; Milhous,W and peters, W; J. Med. Chem. 31, 646-649, 1988) reported the preparation of arteether, the ethyl ether derivative of dihydroartemisinin in two steps: First artemisinin was reduced with an excess of sodium borohydride in methanol at 0 to −5 degree C. in 3 hours to dihydroartemisinin in 79% yield. In the second step arteether is prepared by dissolving the dihydroartemisinin in the solvent mixture of benzene and ethanol at 45 degree C. followed by addition of BF3 etherate and refluxing the reaction mixture at 70 degree C. for one hour. After completion of the reaction it was worked up, dried over anhydrous sodium sulphate with removal of the solvent dichloromethane. The reaction yielded arteether along with some impurities. Column chromatography of the reaction mixture over silica gel, 1:20 ratio yielded pure alpha and beta arteether in nearly qualitative yield.
EL-Feraly etal. (E L Feraly, F. S; Al-Yahya M A; Orabi, K. Y; Mc-Phail D R and Me Phail A. T. J.Nat.Prod. 55, 878-883 1992) reported the preparation of arteether by a process in which anhydrodihydroartemisinin, prepared from artemisinin was dissolved in absolute alcohol. The reaction mixture was stirred in the presence of p-toluene sulphonic acid used as a catalyst. On workup it yielded a mixture of beta arteether and C-11 epimer in the ratio of 3:1. In this process only beta arteether, is obtained and separation of C-11 epimer is difficult and preparation of anhydrodihydroartemisinin is a tedious process. The reaction took 22 hours to complete. The lewis acid catalyst used in this reaction is required in large amount (60 mg. acid catalyst by 100 mg. anhydrodihydroartemisinin).
In another method Bhakuni etal (Bhakuni, R. S.; Jain D. C and Sharma R. P. Indian. J. Chemistry, 34B, 529-30, 1995) arteether, artemether and other ether derivatives were prepared from dihydroartemisinin in different alcohol and benzene in the presence of chlorotrimethylsilane catalyst in 2-4 hours at room temperature. After workup of the reaction mixture and removal of the solvent, the impure reaction products were purified over silica gel column to obtained the pure mixture of alpha, beta ethers.
Another method is reported by Lin et al. (Lin, A. J. and Miller, R. E, J.Med Chero. 38,764-770, 1995) In this method the new ether derivatives were prepared by dissolving dihydroarternisinin in anhydrous ether and appropriate alcohol followed by BF3-etherate. The reaction mixture was stirred at room temperature for 24 hours. The yield of the purified products ranged from 40-90%. Purification was achieved by the use of silica gel chromatography.
Another method described by Jain et al (Jain D. C, Bhakuni R. S, Saxena S, kumar, S and Vishwakarma, R. A.) the preparation of arteether from artemisinin comprises: Reduction of artemisinin into dihydroartemisinin. Isolation of dihydroartemisinin. Acylation of dihydroartemisinin by dissolving it in alcohol and adding trialkylorthoformate in the reaction mixture, which produce ethers in quantitative yield in 10 hours at 40 degree C.
The above mentioned methods carry some disadvantages being less cost effective and more time consuming as compared to the present invention. Moreover, benzene, a carcinogenic solvent, used in the previous methods is not acceptable according to the health standard. Further, all the above methods require at least two separate steps to convert artemisinin into ethers i.e. reduction of the artemisinin into dihydroartemisinin in the first pot followed by isolation of dihydroartemisinin and then comes the second step of conversion of dihydroartemisinin into different ethers in the second pot. However, the present invention provide an efficient method for conversion of artemisinin into artemether
EXAMPLE 1
Artemisinin (3 g.) was dissolved in dry methanol (40 ml) at room temperature. It was cooled to −5 degree C. Now sodium borohydride (700 mg) was added slowly for 30 minutes and the reaction mixture was stirred for about 1.5 hours. The reaction was monitored by TLC to check completion of the reduction step. Now cation exchange resin (8 g) was added slowly at cooling temperature and the reaction mixture was further stirred at room temperature for about 2 hours. Cooled water was added to the reaction mixture and the resin was filtered.
The filtrate was neutralized with 5% sodium bicarbonate solution followed by extracting with dichloromethane (3×50 ml). The dichloromethane extract was dried over anhydrous sodium sulphate and evaporation of the solvent yielded 3.21 g, of artemether along with some impurities. The impure artemether was purified over silica gel column (1:5 ratio) in hexane:ethyl acetate (96:4) furnished pure alpha and beta artemether 2.43 g (81% w/w). Small portion of artemether was separated by prep TLC into alpha and beta isomers and characterized by the analysis of their IR, Mass and 1H NMR data.
EXAMPLE 2
The experiment was carried out following example 1 except in place of solid acid catalyst in the second reaction. Liquid acid catalyst chlorotrimethylsilane was added at cooling temperature for methylation reaction. The overall yield of pure alpha, beta artemether after column chromatography was 2.46 gm (82% w/w).
EXAMPLE 3
Artemisinin (100 g.) was dissolved in dry methanol (3 ml). Added sodium borohydride (30 mg.) at −5° C. The reaction mixture was stirred for 2 hours. After completion of the reaction, trifluroacetic acid (0.5 ml) was added and the reaction mixture was stirred for 5 hours. The methylation was incompleted and after workup the artemether was purified by prep TLC to yield 46 mg (46%) pure alpha, beta artemether.
EXAMPLE 4
The experiment was carried following example 1 except before column chromatography, the beta isomer (40%) was recrystallized in hexane from impure artemether and remaining mother liquor was purified over silica gel column in 1:5 ratio to yield alpha and beta artemether in 80% w/w.

PAPER
https://www.sciencedirect.com/science/article/abs/pii/S0920586114003307
The earlier developed flow protocol for stoichiometric reduction of an important biologically derived pharmaceutical precursor, artemisinin, to dihydroartemisinin was extended to a sequential reaction to produce one of the final APIs, artemether. A highly active heterogeneous catalyst was found for the etherification reaction. The use of QuadraSil catalyst allows to eliminate one step of reaction workup. A comparative Life Cycle Assessment of both reactions has shown advantages of the flow process over the optimized literature batch protocols. Results of LCA highlight the significance of solvents in pharmaceuticals manufacture and the advantage of flow technology, enabling small solvent inventories to be used.
Graphical abstract

PAPER
http://chem.vander-lingen.nl/articles/Target:_Artemether/id/126/itemid/663
In a previous episode chemical company Sanofi was granted exclusive access to certain yeast cells that produce a precursor to anti-malarial drug artemisinin. One of the charities making this all possible is the Bill and Melinda Gates Foundation. Another charity that has apparently entered into the drug business is the Clinton Health Access Initiative. Bill together with Rodger Stringham and David Teager report on an improved process for the conversion of artemisinin to artemether in Organic Process Research & Development (DOI).
Does the Clinton Health Access Initiative have a pilot-plant facility or even an organic lab? Unless it is all cramped in suite 400 on Dorchester Avenue in Boston, the article is not very explicit. The acknowledgements mention Mangalam Drugs and Organics.
Case at hand: artemether has the carbonyl group replaced by a methoxy group in a two-step reduction – methylation. So far so good. The point is that principal supplier Novartis reports up to 68% overall yields but that many Indian and Chinese suppliers working with the procedure generously supplied by same Novartis, report considerably lower figures (58-62%). But Why? And how can the process be improved?
Any organic chemist knows reported yields in the literature should be considered with caution. Chemists tend to be over-optimistic / self-delusionional but this scenario was not considered. No bottlenecks were encountered in step 1, the reduction with sodium borohydride. Only the beta form was isolated due to its poor solubility in the quench. Drying the product without heat prevented formation of one byproduct. Moving on to step two, the methylation with HCl in methanol was more troublesome. The byproducts lurking around the corner are the anomer and the elimination product. Co-solvent (co-reagent?) trimethyl orthoformate made all the difference. The critical element in the workup was first adding more methanol before adding the base quench otherwise you end up with a nasty gum. The new record yield for the improved synthesis is 72%.
But what have all these suppliers been doing wrong with the existing Novartis procedure? The answer to that question, remains unclear. The Novartis yield for step two with co-solvent methylacetate (not the formate) was confirmed so no surprise there.
///////////////////////////////////////////

AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
Medical uses
Artemether is an antimalarial drug for uncomplicated malaria caused by P. falciparum (and chloroquine-resistant P. falciparum) or chloroquine-resistant P. vivax parasites.[8] Artemether can also be used to treat severe malaria.[2]
The World Health Organization (WHO) recommends the treatment of uncomplicated P. falciparum with artemisinin-based combination therapy.[9] Given in combination with lumefantrine, it may be followed by a 14-day regimen of primaquine to prevent relapse of P. vivax or P. ovale malarial parasites and provide a complete cure.[10]
Artemether can also be used in treating and preventing trematode infections of schistosomiasis when used in combination with praziquantel.[11]
Artemether is rated category C by the FDA based on animal studies where artemisinin derivatives have shown an association with fetal loss and deformity. Some studies, however, do not show evidence of harm.[12][13]
Side effects
Possible side effects include cardiac effects such as bradycardia and QT interval prolongation.[14] Also, possible central nervous system toxicity has been shown in animal studies.[15][16]
Drug interactions
Plasma artemether level was found to be lower when the combination product was used with lopinavir/ritonavir.[16] There is also decreased drug exposure associated with concurrent use with efavirenz or nevirapine.[17][18]
Artemether/lumefantrine should not be used with drugs that inhibit CYP3A4.[19]
Hormonal contraceptives may not be as efficacious when used with artemether/lumefantrine.[19]
Pharmacology
Mechanism of action
Artemether is an artemisinin derivative and the mechanism of action for artemisinins is.[medical citation needed]
Artemether interact with ferriprotoporphyrin IX (heme) or ferrous ions in the acidic parasite food vacuole, and generates cytotoxic radical species
The accepted mode of action of the peroxide containing drug involve its interaction with heme (byproduct of hemoglobin degradation), derived from proteolysis of haemoglobin. This interaction results in the formation of toxic oxygen and carbon centered radicals.
One of the proposed mechanisms is that through inhibiting anti-oxidant and metabolic enzymes, artemisinin derivatives inflict oxidative and metabolic stress on the cell. Some pathways affected may concern glutathione and glucose metabolism. As a consequence, lesions and reduced growth of the parasite may result.[20]
Another possible mechanism of action suggests that arteristinin drugs exert their cidal action through inhibiting PfATP6. Since PfATP6 is an enzyme regulating cellular calcium concentration, its malfunctioning will lead to intracellular calcium accumulation, which in turns causes cell death.[21]
Pharmacokinetics
Absorption of artemether is improved 2- to 3-fold with food. It is highly bound to protein (95.4%). Peak concentrations of artemether are seen 2 hours after administration.[4]
Artemether is metabolized in the human body to the active metabolite, dihydroartemisinin, primarily by hepatic enzymes CYP3A4/5.[4] Both the parent drug and active metabolite are eliminated with a half-life of about 2 hours.[4]
Chemistry
Artemether is a methyl ether derivative of artemisinin, which is a peroxide-containing lactone isolated from the antimalarial plant Artemisia annua. It is also known as dihydroartemisinin methyl ether, but its correct chemical nomenclature is (+)-(3-alpha,5a-beta,6-beta,8a-beta, 9-alpha,12-beta,12aR)-decahydro-10-methoxy-3,6,9-trimethyl-3,12-epoxy-12H-pyrano(4,3-j)-1,2-benzodioxepin. It is a relatively lipophilic and unstable drug,[22] which acts by creating reactive free radicals in addition to affecting the membrane transport system of the plasmodium organism.[14]
References
- ^ “Artemether – Drugs.com”. http://www.drugs.com. Archived from the original on 20 December 2016. Retrieved 7 December 2016.
- ^ Jump up to:a b c d e f Esu, Ekpereonne B.; Effa, Emmanuel E.; Opie, Oko N.; Meremikwu, Martin M. (18 June 2019). “Artemether for severe malaria”. The Cochrane Database of Systematic Reviews. 6: CD010678. doi:10.1002/14651858.CD010678.pub3. ISSN 1469-493X. PMC 6580442. PMID 31210357.
- ^ “Artemether and Lumefantrine”. The American Society of Health-System Pharmacists. Archived from the original on 20 December 2016. Retrieved 28 November 2016.
- ^ Jump up to:a b c d “Coartem- artemether and lumefantrine tablet”. DailyMed. 5 August 2019. Retrieved 26 April 2020.
- ^ Jump up to:a b c d e Kovacs, SD; Rijken, MJ; Stergachis, A (February 2015). “Treating severe malaria in pregnancy: a review of the evidence”. Drug Safety. 38 (2): 165–81. doi:10.1007/s40264-014-0261-9. PMC 4328128. PMID 25556421.
- ^ Rao, Yi; Zhang, Daqing; Li, Runhong (2016). Tu Youyou and the Discovery of Artemisinin: 2015 Nobel Laureate in Physiology or Medicine. World Scientific. p. 162. ISBN 9789813109919. Archived from the original on 2017-09-10.
- ^ World Health Organization model list of essential medicines: 21st list 2019. Geneva: World Health Organization. 2019. hdl:10665/325771. WHO/MVP/EMP/IAU/2019.06. License: CC BY-NC-SA 3.0 IGO.
- ^ Makanga, Michael; Krudsood, Srivicha (2009-10-12). “The clinical efficacy of artemether/lumefantrine (Coartem)”. Malaria Journal. 8 (Suppl 1): S5. doi:10.1186/1475-2875-8-S1-S5. ISSN 1475-2875. PMC 2760240. PMID 19818172.
- ^ Treatment of Uncomplicated Plasmodium falciparum Malaria. World Health Organization. 2015-01-01. Archived from the original on 2017-09-10.
- ^ Treatment Of Uncomplicated Malaria Caused By P. vivax, P. ovale, P. malariae or P. knowlesi. World Health Organization. 2015-01-01. Archived from the original on 2017-09-10.
- ^ Pérez del Villar, Luis; Burguillo, Francisco J.; López-Abán, Julio; Muro, Antonio (2012-01-01). “Systematic review and meta-analysis of artemisinin based therapies for the treatment and prevention of schistosomiasis”. PLOS ONE. 7 (9): e45867. Bibcode:2012PLoSO…745867P. doi:10.1371/journal.pone.0045867. ISSN 1932-6203. PMC 3448694. PMID 23029285.
- ^ Dellicour S, Hall S, Chandramohan D, Greenwood B (2007). “The safety of artemisinins during pregnancy: a pressing question”. Malaria Journal. 6: 15. doi:10.1186/1475-2875-6-15. PMC 1802871. PMID 17300719.
- ^ Piola P, Nabasumba C, Turyakira E, et al. (2010). “Efficacy and safety of artemether—lumefantrine compared with quinine in pregnant women with uncomplicated Plasmodium falciparum malaria: an open-label, randomised, non-inferiority trial”. Lancet Infect Dis. 10 (11): 762–769. doi:10.1016/S1473-3099(10)70202-4. hdl:10144/116337. PMID 20932805.
- ^ Jump up to:a b “Artemether”. http://www.antimicrobe.org. Archived from the original on 2017-02-23. Retrieved 2016-11-09.
- ^ “WHO Model Prescribing Information: Drugs Used in Parasitic Diseases – Second Edition: Protozoa: Malaria: Artemether”. apps.who.int. Archived from the original on 2016-11-10. Retrieved 2016-11-09.
- ^ Jump up to:a b Askling, Helena H.; Bruneel, Fabrice; Burchard, Gerd; Castelli, Francesco; Chiodini, Peter L.; Grobusch, Martin P.; Lopez-Vélez, Rogelio; Paul, Margaret; Petersen, Eskild (2012-01-01). “Management of imported malaria in Europe”. Malaria Journal. 11: 328. doi:10.1186/1475-2875-11-328. ISSN 1475-2875. PMC 3489857. PMID 22985344.
- ^ van Geertruyden, J.-P. (2014). “Interactions between malaria and human immunodeficiency virus anno 2014”. Clinical Microbiology and Infection. 20 (4): 278–285. doi:10.1111/1469-0691.12597. PMC 4368411. PMID 24528518.
- ^ Kiang, Tony K. L.; Wilby, Kyle J.; Ensom, Mary H. H. (2013-10-26). “Clinical Pharmacokinetic Drug Interactions Associated with Artemisinin Derivatives and HIV-Antivirals”. Clinical Pharmacokinetics. 53 (2): 141–153. doi:10.1007/s40262-013-0110-5. ISSN 0312-5963. PMID 24158666. S2CID 1281113.
- ^ Jump up to:a b Stover, Kayla R.; King, S. Travis; Robinson, Jessica (2012-04-01). “Artemether-Lumefantrine: An Option for Malaria”. Annals of Pharmacotherapy. 46 (4): 567–577. doi:10.1345/aph.1Q539. ISSN 1060-0280. PMID 22496476. S2CID 7678606.
- ^ Saeed, ME; Krishna, S; Greten, HJ; Kremsner, PG; Efferth, T (August 2016). “Antischistosomal activity of artemisinin derivatives in vivo and in patients”. Pharmacological Research. 110: 216–26. doi:10.1016/j.phrs.2016.02.017. PMID 26902577.
- ^ Guo, Zongru (2016-03-01). “Artemisinin anti-malarial drugs in China”. Acta Pharmaceutica Sinica B. 6 (2): 115–124. doi:10.1016/j.apsb.2016.01.008. PMC 4788711. PMID 27006895.
- ^ De Spiegeleer, B.M.J.; D’Hondt, M.; Vangheluwe, E.; Vandercruyssen, K.; De Spiegeleer, B.G.I.; Jansen, H.; Koijen, I.; Van Gompel, J. (2012). “Relative response factor determination of artemether degradants with a dry heat stress approach”. Journal of Pharmaceutical and Biomedical Analysis. 70: 111–116. doi:10.1016/j.jpba.2012.06.002. hdl:1854/LU-2938963. PMID 22770733.
| Clinical data | |
|---|---|
| Trade names | Many[1] |
| AHFS/Drugs.com | International Drug Names |
| Routes of administration | Intramuscular[2] |
| ATC code | P01BE02 (WHO) |
| Legal status | |
| Legal status | UK: POM (Prescription only) |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 71963-77-4 |
| PubChem CID | 68911 |
| DrugBank | DB06697 |
| ChemSpider | 62138 |
| UNII | C7D6T3H22J |
| KEGG | D02483 |
| ChEBI | CHEBI:195280 |
| ChEMBL | ChEMBL1237051 |
| PDB ligand | D8Z (PDBe, RCSB PDB) |
| CompTox Dashboard (EPA) | DTXSID7040651 |
| ECHA InfoCard | 100.189.847 |
| Chemical and physical data | |
| Formula | C16H26O5 |
| Molar mass | 298.379 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| Melting point | 86 to 88 °C (187 to 190 °F) |
| showSMILES | |
| showInChI | |
| (what is this?) (verify) |
///////////ARTEMETHER, ANTIMALARIAL, SM 224, SM-224
[H][C@@]12CC[C@@H](C)[C@]3([H])CC[C@@]4(C)OO[C@@]13[C@]([H])(O[C@H](OC)[C@@H]2C)O4
