17 Dec 2021Vincerx Pharma plans phase II trials for Cancer (IV, Infusion), in the second half of 2022
16 Dec 2021Phase-I clinical trials in Chronic lymphocytic leukaemia (Second-line therapy or greater) in USA (IV)
16 Dec 2021Phase-I clinical trials in Richter’s syndrome (Second-line therapy or greater) (IV) in USA
First-in-human dose escalation study of cyclin-dependent kinase-9 inhibitor VIP152 in patients with advanced malignancies shows early signs of clinical efficacyJennifer R. Diamond, Valentina Boni, Emerson Lim, Grzegorz Nowakowski, Raul Cordoba, Daniel Morillo, Ray Valencia, Isabelle Genvresse, Claudia Merz, Oliver Boix, Melanie M. Frigault, Joy M. Greer, Ahmed M. Hamdy, Xin Huang, Raquel Izumi, Harvey Wong and Victor Moreno DOI: 10.1158/1078-0432.CCR-21-3617
Abstract
Purpose: To report on the first-in-human phase I study of VIP152 (NCT02635672), a potent and highly selective CDK9 inhibitor. Patients and Methods: Adults with solid tumors or aggressive non-Hodgkin lymphoma (NHL) who were refractory to or had exhausted all available therapies received VIP152 monotherapy as a 30-minute intravenous, once weekly infusion, as escalating doses (5, 10, 15, 22.5, or 30 mg in 21-day cycles) until the maximum tolerated dose (MTD) was determined. Results: Thirty-seven patients received {greater than or equal to} 1 VIP152 dose, with 30 mg identified as the MTD based on dose-limiting toxicity of grade 3/4 neutropenia. The most common adverse events were nausea and vomiting (75.7% and 56.8%, respectively), all of grade 1/2 severity. Of the most common events, Grade 3/4 events occurring in > 1 patient were neutropenia (22%), anemia (11%), abdominal pain (8%), increased alkaline phosphatase (8%), and hyponatremia (8%). Day 1 exposure for the MTD exceeded the predicted minimum therapeutic exposure and reproducibly achieved maximal pathway modulation; no accumulation occurred after multiple doses. Seven of 30 patients with solid tumors had stable disease (including 9.5 and 16.8 months in individual patients with pancreatic cancer and salivary gland cancer, respectively), and 2 of 7 patients with high-grade B-cell lymphoma with MYC and BCL2/BCL6 translocations (HGL) achieved durable complete metabolic remission (ongoing at study discontinuation, after 3.7 and 2.3 years of treatment). Conclusion: VIP152 monotherapy, administered intravenously once weekly, demonstrated a favorable safety profile and evidence of clinical benefit in patients with advanced HGL and solid tumors.
CLIP
Preclinical bioconjugation platform designed to overcome limitations of small–molecule and antibody–drug conjugates use to treat cancer
Vincera Pharma, Inc., a biopharmaceutical company aspiring to address the unmet medical needs of patients with cancer through paradigm-shifting therapeutics, today announced the signing of an exclusive license agreement with Bayer AG for the development and commercialization of an early development oncology portfolio. The license will become effective upon the closing of the transaction with LSAC (described below), and Vincera intends to use the funds it will receive upon closing of such transaction to initiate its clinical program.
Under the terms of the license agreement, Vincera will in-license VIP152 (formerly BAY 1251152 & CAS RN.: 1610358-56-9), a clinical-stage, highly selective, positive transcription elongation factor b (PTEFb)/cyclin-dependent kinase 9 (CDK9) inhibitor for the treatment of cancer. Additionally, Vincera will receive assets and license technology for a preclinical bioconjugation platform to address the limitations of small-molecule and antibody-drug conjugates in oncology. The preclinical assets include VIP236, a small molecule drug conjugate (SMDC) targeting advanced and metastatic cancer; as well as VIP943 (formerly BAY-943) and VIP924 (formerly BAY-924), two antibody-drug conjugates (ADC) targeting hematologic tumors; and VIP217, an oral PTEFb/CDK9 inhibitor in discovery. “This license agreement with Bayer creates the foundation of Vincera’s targeted clinical oncology pipeline, with a potentially best-in-class asset, while positioning us for long-term growth across two therapeutic platforms,” said Ahmed Hamdy M.D., Chief Executive Officer of Vincera. “Our lead asset, VIP152, is a small molecule PTEFb/CDK9 inhibitor with very encouraging data from monotherapy Phase 1 studies, including 2 of 7 patients with durable remissions of over 2 years in the very aggressive indication of relapsed/refractory double-hit DLBCL. In addition, preclinical data support our belief that VIP152 is the most selective CDK9 inhibitor in the clinic with on-target depletion of oncogenic MYC and MCL1 mRNA transcripts in patients. These results, combined with the acceptable safety profile seen to date, suggest that VIP152 could be an important new treatment option for patients with MYC- and MCL1-driven malignancies. Importantly, with proof-of-concept clinical data in hand, we are poised to execute on a strategic clinical development plan with the potential for multiple accelerated approvals in the U.S. Expansion of the current Phase 1b study to include these patient populations is expected to begin in 2021.”
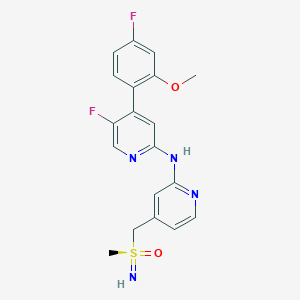
Title: 5-FLUORO-N-(PYRIDIN-2-YL)PYRIDIN-2-AMINE DERIVATIVES CONTAINING A SULFOXIMINE GROUP.
Abstract
The present invention relates to 5-fluoro-N-(pyridin-2-yl)pyridin-2-amine derivatives containing a sulfoximine group of general formula (I) as described and defined herein, and methods for their preparation, their use for the treatment and/or prophylaxis of disorders, in particular of hyper-proliferative disorders and/or virally induced infectious diseases and/or of cardiovascular diseases. The invention further relates to intermediate compounds useful in the preparation of said compounds of general formula (I).
“CDK9 represents a validated target for malignancies such as CLL where other less selective CDK inhibitors have shown clinical activity in high-risk patients,” says Dr. John C. Byrd, Chair of the Scientific Advisory Board of Vincera. “VIP-152 represents an exciting new therapy for this disease, particularly those with prior resistance to ibrutinib and venetoclax where a true unmet need exists for new treatments.”
Dr. Hamdy continued, “In addition to our planned clinical program, we intend to advance, in parallel, the development of our preclinical bioconjugation platform. We believe our next-generation platform has the potential to generate first-in-class and best-in-class opportunities in oncology, improving the specificity of drug targeting and release through a modular platform with innovative warhead design and linker-payload technologies. We are thrilled that the Bayer license will allow us to pursue the commercial potential of this promising oncology portfolio and look forward to providing updates as we execute across our pipeline in the coming quarters.”
In exchange for this license, Vincera will pay Bayer an upfront license fee and development and commercial sales milestone payments. In further consideration of the rights granted, we will also pay an annual royalty on the commercial sale of licensed products in the single- to low-double-digit percentage range on net commercial sales of licensed products.
On September 29, 2020, Vincera announced that it has entered into a merger agreement with LifeSci Acquisition Corp. (“LSAC”), a publicly-traded blank check company targeting biopharma, medical technology, digital health, and healthcare services sectors. Following the completion of the merger, the combined company is expected to have approximately $60 million in cash to fund its preclinical and clinical pipeline. Additional information about the merger and related transactions, including a copy of the merger agreement, are included in a Current Report on Form 8-K filed by LSAC with the SEC on September 29, 2020, and available at www.sec.gov.
About Vincera Pharma, Inc.
Vincera is a recently formed clinical-stage life sciences company focused on leveraging its extensive development and oncology expertise to advance new therapies intended to address unmet medical needs for the treatment of cancer. Vincera’s executive team has assembled a management team of biopharmaceutical experts with extensive experience in building and operating organizations that develop and deliver innovative medicines to patients. Vincera’s current pipeline is derived from an exclusive license agreement with Bayer and includes (i) a clinical-stage and follow-on small molecule drug program and (ii) a preclinical stage bioconjugation/next-generation antibody-drug conjugate platform. The company intends to develop multiple products through clinical proof-of-concept and potentially through Accelerated Approval in the United States. For more information, please visit www.vincerapharma.com.
Preparation of Intermediate 1.12-Chloro-5-fluoro-4-(4-fluoro-2-methoxyphenyl)pyridine
A batch with 2-chloro-5-fluoro-4-iodopyridine (1000 mg; 3.88 mmol; APAC Pharmaceutical, LLC), (4-fluoro-2-methoxyphenyl)boronic acid (660 mg; 3.88 mmol; Aldrich Chemical Company Inc.) and tetrakis(triphenylphosphin)palladium(0) (449 mg; 0.38 mmol) in 1,2-dimethoxyethane (10.0 mL) and 2 M aqueous solution of potassium carbonate (5.8 mL) was degassed using argon. The batch was stirred under an atmosphere of argon for 4 hours at 100° C. After cooling, the batch was diluted with ethyl acetate and THF and washed with a saturated aqueous solution of sodium chloride. The organic phase was filtered using a Whatman filter and concentrated. The residue was purified by column chromatography (hexane to hexane/ethyl acetate 50%) to give the desired product (947 mg; 3.70 mmol).
A batch containing 2-chloro-5-fluoro-4-(4-fluoro-2-methoxyphenyl)pyridine (400 mg; 1.57 mmol), 4-[(methylsulfanyl)methyl]pyridin-2-amine (483 mg; 3.13 mmol; UkrOrgSynthesis Ltd.), (9,9-dimethyl-9H-xanthene-4,5-diyl)bis(diphenylphosphane) (40 mg; 0.07 mmol) and cesium carbonate (765 mg; 2.35 mmol) in dioxane (6.0 mL) was degassed using argon. Tris(dibenzylideneacetone)dipalladium(0) (21 mg; 0.02 mmol) was added under argon and the batch was stirred in a closed pressure tube for 5 hours at 100° C. After cooling, the batch was diluted with an aqueous solution of sodium chloride and extracted with DCM (3×). The combined organic phases were filtered using a Whatman filter and concentrated. The residue was purified by chromatography (hexane to hexane/ethyl acetate 30%) to give the desired product (556 mg; 1.48 mmol).
Under argon, a solution of 2,2,2-trifluoroacetamide (195 mg; 1.73 mmol) in dioxane (0.5 mL) was added dropwise to a solution of sodium tert.-butoxide (111 mg; 1.15 mmol) in dioxane (0.6 mL), so that the temperature of the mixture remained below 10° C. Subsequently, a freshly prepared solution of 1,3-dibromo-5,5-dimethylhydantoin (247 mg; 0.86 mmol) in dioxane (0.6 mL)/THF (1.0 mL) was added dropwise to the stirred mixture, so that the temperature of the mixture remained below 10° C. Then the mixture was stirred for 10 minutes at 10° C. Finally, a solution of 5-fluoro-4-(4-fluoro-2-methoxyphenyl)-N-{4-[(methylsulfanyl)methyl]pyridin-2-yl}pyridin-2-amine (430 mg; 1.15 mmol) in dioxane (1.0 mL) was added dropwise to the stirred mixture, so that the temperature of the mixture remained below 10° C. The mixture was stirred for 60 minutes at 10° C. The batch was diluted with toluene (2.0 mL) under cooling and an aqueous solution of sodium sulfite (145 mg; 1.15 mmol in 2.0 mL water) was added so that the temperature of the mixture remained below 15° C. An aqueous solution of sodium chloride was added and the batch was extracted with ethyl acetate (3×). The combined organic phases were filtered using a Whatman filter and concentrated to give crude 2,2,2-trifluoro-N-{[(2-{[5-fluoro-4-(4-fluoro-2-methoxyphenyl)pyridin-2-yl]amino}pyridin-4-yl)methyl](methyl)-λ 4-sulfanylidene}acetamide, that was used without further purification.
Acetone (6.0 mL) and potassium permanganate (814 mg; 5.15 mmol) were added to the residue and the mixture was stirred at 50° C. for 90 minutes. Additional potassium permanganate (223 mg; 1.42 mmol) was added and stirring was continued at 50° C. for 4 hours. Finally, additional potassium permanganate (305 mg; 1.93 mmol) was added and stirring was continued at 50° C. for 150 minutes. After cooling, the batch was filtered, the residue was washed with acetone and the combined filtrates were concentrated. The residue was dissolved in MeOH (60 mL), potassium carbonate (182 mg; 1.32 mmol) was added and the reaction mixture was stirred for 20 minutes at RT. The batch was diluted with an aqueous solution of sodium chloride and extracted with DCM (3×). The combined organic phases were filtered using a Whatman filter and concentrated. The residue was purified by preparative HPLC to give the desired product (50 mg; 0.12 mmol).
[TABLE-US-00003] System:Waters Autopurificationsystem: Pump 254, Sample Manager 2767, CFO, DAD 2996, SQD 3100Column:XBrigde C18 5 μm 100 × 30 mmSolvent:A = H2O + 0.2% NH3 (32%) B = MeCNGradient:0-8 min 15-50% BFlow:50 mL/minTemperature:RTSolution:132 mg/2 mL DMF/MeOH 1:1Injection:2 × 1 mLDetection:DAD scan range 210-400 nm MS ESI+, ESI−, scan range 160-1000 m/zRetention:3.39-3.88 minMS(ES+):m/z = 404
Alternative Procedure for the Preparation of Intermediate 1.25-Fluoro-4-(4-fluoro-2-methoxyphenyl)-N-{4-[(methylsulfanyl)methyl]pyridin-2-yl}pyridin-2-amine
Preparation of Intermediate 1.3(2-{[5-Fluoro-4-(4-fluoro-2-methoxyphenyl)pyridin-2-yl]amino}pyridin-4-yl)methanol
A batch containing 2-chloro-5-fluoro-4-(4-fluoro-2-methoxyphenyl)pyridine (411 mg; 1.61 mmol), (2-aminopyridin-4-yl)methanol (200 mg; 1.61 mmol; ABCR GmbH & CO. KG), (9,9-dimethyl-9H-xanthene-4,5-diyl)bis(diphenylphosphane) (418 mg; 0.72 mmol) and cesium carbonate (784 mg; 2.41 mmol) in dioxane (8.0 mL) was degassed using argon. Tris(dibenzylideneacetone)dipalladium(0) (147 mg; 0.16 mmol) was added under an atmosphere of argon and the batch was stirred for 29 hours at 100° C. After cooling, additional (2-aminopyridin-4-yl)methanol (100 mg; 0.81 mmol), (9,9-dimethyl-9H-xanthene-4,5-diyl)bis(diphenylphosphane) (118 mg; 0.20 mmol) and tris(dibenzylideneacetone)dipalladium(0) (74 mg; 0.08 mmol) were added and the mixture was stirred for 19 hours at 100° C. After cooling, the batch was diluted with ethyl acetate and washed with an aqueous solution of sodium chloride. The organic phase was filtered using a Whatman filter and concentrated. The residue was purified by chromatography (DCM/EtOH 9:1) to give the desired product (389 mg; 1.13 mmol).
Preparation of End Product (Alternative Preparation of Intermediate 1.2)
Thionyl chloride (0.19 ml; 2.55 mmol) was added dropwise to a stirred solution of (2-{[5-fluoro-4-(4-fluoro-2-methoxyphenyl)pyridin-2-yl]amino}pyridin-4-yl)methanol (350 mg; 1.01 mmol) in DCM (4.0 ml) and NMP (0.4 ml) at 0° C. The mixture was stirred for 7 hours at RT. The batch was diluted with aqueous sodium bicarbonate solution and aqueous sodium chloride solution and extracted with DCM (3×). The combined organic phases were filtered using a Whatman filter and concentrated to give crude N-[4-(chloromethyl)pyridin-2-yl]-5-fluoro-4-(4-fluoro-2-methoxyphenyl)pyridin-2-amine, that was used without further purification in the next step.
The residue was re-dissolved in EtOH (12.0 ml) and the resulting solution was cooled to 0° C. Sodium methanethiolate (158 mg; 2.26 mmol) was added portionwise to the stirred solution at 0° C. The mixture was stirred for 4 hours at RT before it was diluted with DCM and washed with aqueous sodium chloride solution. The organic phase was filtered using a Whatman filter and concentrated. The residue was purified by chromatography (DCM/EtOH 95:5) to give the desired product (301 mg; 0.81 mmol).
Alternative Procedure for the Preparation of Example 1Preparation of Intermediate 1.4(rac)-2,2,2-Trifluoro-N-{[(2-{[5-fluoro-4-(4-fluoro-2-methoxyphenyl)pyridin-2-yl]amino}pyridin-4-yl)methyl](methyl)-λ4-sulfanylidene}acetamide
Under an atmosphere of argon, a solution of 2,2,2-trifluoroacetamide (2.53 g; 22.4 mmol) in THF (10.0 mL) was added dropwise to a solution of sodium tert.-butoxide (1.43 g; 14.9 mmol) in THF (12.0 mL), so that the temperature of the mixture remained below 10° C. Subsequently, a freshly prepared solution of 1,3-dibromo-5,5-dimethylhydantoin (3.20 g; 11.2 mmol) in THF (12.0 mL) was added dropwise to the stirred mixture, so that the temperature of the mixture remained below 10° C. Then the mixture was stirred for 10 minutes at 10° C. Finally, a solution of 5-fluoro-4-(4-fluoro-2-methoxyphenyl)-N-{4-[(methylsulfanyl)methyl]pyridin-2-yl}pyridin-2-amine (5.57 g; 14.9 mmol; Intermediate 1.2) in dioxane (12.0 mL) was added dropwise to the stirred mixture, so that the temperature of the mixture remained below 10° C. The mixture was stirred for 60 minutes at 10° C. The batch was diluted with toluene (40.0 mL) under cooling and an aqueous solution of sodium sulfite (1.88 g; 14.9 mmol in 40.0 mL water) was added so that the temperature of the mixture remained below 15° C. The batch was extracted three times (3×) with ethyl acetate. The combined organic layers were washed with an aqueous solution of sodium chloride, filtered using a Whatman filter and concentrated. The residue was purified by column chromatography on silica gel (DCM to DCM/EtOH 95:5) to give the desired product (4.71 g; 9.72 mmol).
Alternative Preparation of End Product (Example 1)
An aqueous solution of potassium hydroxide (25%) was added dropwise to a stirred solution of 2,2,2-trifluoro-N-{[(2-{[5-fluoro-4-(4-fluoro-2-methoxyphenyl)pyridin-2-yl]amino}pyridin-4-yl)methyl](methyl)-λ 4-sulfanylidene}acetamide (4.64 g; 9.58 mmol) in DMF (350 mL), methanol (100 mL) and water (100 mL) to adjust the pH to 10.5. Oxone® (5.00 g; 8.14 mmol) was added and the mixture was stirred at room temperature for 4.5 hours. During this time, the pH was kept between 10-11, by dropwise addition of an aqueous solution of potassium hydroxide (25%), if necessary. The mixture was filtered and the filter cake was washed with plenty of DCM. The pH of the filtrate was adjusted to 6-7 using an aqueous solution of hydrogen chloride (15%). The filtrate was washed with an aqueous solution of sodium chloride, followed by an aqueous solution of sodium thiosulfate (10%). During evaporation of solvents using a rotary evaporator, a solid substance precipitated from the solution. The precipitated solid was isolated by suction filtration, washed with DCM and diisopropyl ether, and dried to give the desired product (2.61 g; 6.43 mmol).
Enantiomers of 5-Fluoro-4-(4-fluoro-2-methoxyphenyl)-N-{4-[(S-methylsulfonimidoyl)methyl]pyridin-2-yl}pyridin-2-amine
(rac)-5-Fluoro-4-(4-fluoro-2-methoxyphenyl)-N-{4-[(S-methylsulfonimidoyl)methyl]pyridin-2-yl}pyridin-2-amine (3.47 g) was separated into the single enantiomers by preparative chiral HPLC.
Example 4(rac)-5-Fluoro-4-(4-fluoro-2-methoxyphenyl)-N-{6-methyl-4-[(S-methylsulfonimidoyl)methyl]pyridin-2-yl}pyridin-2-amine
Preparation of Intermediate 4.15-Fluoro-4-(4-fluoro-2-methoxyphenyl)pyridin-2-amine
A solution of lithium bis(trimethylsilyl)amide in THF (1M; 20.5 mL; 20.53 mmol; Aldrich Chemical Company Inc.) was added to a mixture of 2-chloro-5-fluoro-4-(4-fluoro-2-methoxyphenyl)pyridine (2.50 g; 9.78 mmol; Intermediate 1.1), tris(dibenzylideneacetone)dipalladium (0) (0.18 g; 0.20 mmol; Aldrich Chemical Company Inc.) and 2-(dicyclohexylphosphino)-2′,4′,6′-triisopropylbiphenyl (0.19 g; 0.39 mmol; Aldrich Chemical Company Inc.) in THF (16.3 mL) under an atmosphere of argon at room temperature. The mixture was stirred at 60° C. for 6 hours. The mixture was cooled to −40° C. and water (10 ml) was added. The mixture was slowly warmed to room temperature under stirring, solid sodium chloride was added and the mixture was extracted with ethyl acetate twice (2×). The combined organic layers were filtered using a Whatman filter and concentrated. The residue was purified by column chromatography on silica gel (hexane to hexane/ethyl acetate 60%) to give the desired product (2.04 g; 8.64 mmol).
Preparation of Intermediate 4.2(2-Chloro-6-methylpyridin-4-yl)methanol
To a stirred solution of 2-chloro-6-methylpyridine-4-carboxylic acid (10.00 g; 55.4 mmol; Maybridge) in THF (100 mL) at 0° C. was added a 1M solution of borane-tetrahydrofuran complex in THF (221.5 mL; 221.5 mmol). The mixture was allowed to react at RT overnight. Then, MeOH (22 mL) was cautiously added to the stirred mixture while cooling with an ice bath. The batch was diluted with ethyl acetate and washed with aqueous sodium hydroxide solution (1N) and saturated aqueous sodium chloride solution. The organic layer was filtered using a Whatman filter and concentrated. The residue was purified by column chromatography on silica gel (DCM/EtOH 95:5) to give the pure product (7.24 g; 45.9 mmol).
Preparation of Intermediate 4.32-Chloro-6-methyl-4-[(methylsulfanyl)methyl]pyridine
To a stirred solution of (2-chloro-6-methylpyridin-4-yl)methanol (7.20 g; 45.7 mmol) in DMF (200 mL) at 0° C. was added dropwise thionyl chloride (8.3 mL; 114.2 mmol). The mixture was allowed to react at 10° C. for 2 hours. Then, the mixture was concentrated to give the crude product 2-chloro-4-(chloromethyl)-6-methylpyridine (17.08 g).
Crude 2-chloro-4-(chloromethyl)-6-methylpyridine (8.04 g).was dissolved in acetone (250 mL) and an aqueous solution of sodium methanethiolate (21%, 18.3 mL, 54.8 mmol; Aldrich Chemical Company Inc.) was added dropwise under stirring. The mixture was stirred at RT for 3 hours before additional aqueous solution of sodium methanethiolate (21%, 15.3 mL, 45.7 mmol; Aldrich Chemical Company Inc.) was added and the mixture was stirred at RT overnight. Finally, additional aqueous solution of sodium methanethiolate (21%, 15.3 mL, 45.7 mmol; Aldrich Chemical Company Inc.) was added and the mixture was stirred at RT for 6 hours. The batch was diluted with ethyl acetate and an aqueous solution of sodium chloride. The mixture was extracted twice with ethyl acetate. The combined organic layers were filtered using a Whatman filter and concentrated. The residue was purified by column chromatography on silica gel (hexane to hexane/ethyl acetate 20%) to give the desired product (7.05 g; 37.6 mmol).
Preparation of Intermediate 4.45-Fluoro-4-(4-fluoro-2-methoxyphenyl)-N-{6-methyl-4-[(methylsulfanyl)methyl]pyridin-2-yl}pyridin-2-amine
A batch containing 5-fluoro-4-(4-fluoro-2-methoxyphenyl)pyridin-2-amine (852 mg; 3.61 mmol), 2-chloro-6-methyl-4-[(methylsulfanyl)methyl]pyridine (677 mg; 3.61 mmol) and cesium carbonate (1410 mg; 4.33 mmol) in dioxane (8.3 mL) was degassed using argon. (9,9-Dimethyl-9H-xanthene-4,5-diyl)bis(diphenylphosphane) (81 mg; 0.14 mmol) and tris(dibenzylideneacetone)dipalladium(0) (69 mg; 0.08 mmol) were added under an atmosphere of argon and the batch was stirred in a closed pressure tube for 3 hours at 100° C. Additional (9,9-dimethyl-9H-xanthene-4,5-diyl)bis(diphenylphosphane) (81 mg; 0.14 mmol) and tris(dibenzylideneacetone)dipalladium(0) (69 mg; 0.08 mmol) were added under an atmosphere of argon and the batch was stirred in the closed pressure tube for additional 20 hours at 100° C.
After cooling, the mixture was diluted with ethyl acetate and washed with an aqueous solution of sodium chloride. The organic layer was filtered using a Whatman filter and concentrated. The residue was purified by column chromatography on silica gel (hexane to hexane/ethyl acetate 50%) to give the desired product (628 mg; 1.62 mmol).
Preparation of Intermediates 4.5 and 4.6(rac)-2,2,2-Trifluoro-N-{[(2-{[5-fluoro-4-(4-fluoro-2-methoxyphenyl)pyridin-2-yl]amino}-6-methylpyridin-4-yl)methyl](methyl)-λ4-sulfanylidene}acetamide and (rac)-N-{[(3-bromo-6-{[5-fluoro-4-(4-fluoro-2-methoxyphenyl)pyridin-2-yl]amino}-2-methylpyridin-4-yl)methyl](methyl)-λ4-sulfanylidene}-2,2,2-trifluoroacetamide
Under an atmosphere of argon, a solution of 2,2,2-trifluoroacetamide (125 mg; 1.11 mmol) in THF (1.0 mL) was added dropwise to a solution of sodium tert.-butoxide (71 mg; 0.74 mmol) in THF (1.0 mL), so that the temperature of the mixture remained below 10° C. Subsequently, a freshly prepared solution of 1,3-dibromo-5,5-dimethylhydantoin (158 mg; 0.55 mmol) in THF (1.0 mL) was added dropwise to the stirred mixture, so that the temperature of the mixture remained below 10° C. Then the mixture was stirred for 10 minutes at 10° C. Finally, a solution of 5-fluoro-4-(4-fluoro-2-methoxyphenyl)-N-{6-methyl-4-[(methylsulfanyl)methyl]pyridin-2-yl}pyridin-2-amine (286 mg; 0.74 mmol) in THF (1.5 mL) was added dropwise to the stirred mixture, so that the temperature of the mixture remained below 10° C. The mixture was stirred for 60 minutes at 10° C. The batch was diluted with toluene (4.0 mL) under cooling and an aqueous solution of sodium sulfite (93 mg; 0.74 mmol in 7.0 mL water) was added so that the temperature of the mixture remained below 15° C. The batch was extracted three times with ethyl acetate. The combined organic layers were washed with an aqueous solution of sodium chloride, filtered using a Whatman filter and concentrated. The residue was purified by column chromatography on silica gel (hexane to hexane/ethyl acetate 100%) to give the desired product 2,2,2-trifluoro-N-{[(2-{[5-fluoro-4-(4-fluoro-2-methoxyphenyl)pyridin-2-yl]amino}-6-methylpyridin-4-yl)methyl](methyl)-λ 4-sulfanylidene}acetamide (134 mg; 0.27 mmol) and the side product N-{[(3-bromo-6-{[5-fluoro-4-(4-fluoro-2-methoxyphenyl)pyridin-2-yl]amino}-2-methylpyridin-4-yl)methyl](methyl)-λ 4-sulfanylidene}-2,2,2-trifluoroacetamide (110 mg; 0.19 mmol).
An aqueous solution of potassium hydroxide (25%) was added dropwise to a stirred solution of 2,2,2-trifluoro-N-{[(2-{[5-fluoro-4-(4-fluoro-2-methoxyphenyl)pyridin-2-yl]amino}-6-methylpyridin-4-yl)methyl](methyl)-λ 4-sulfanylidene}acetamide (126 mg; 0.25 mmol) in methanol (5.0 mL) and water (1.8 mL) to adjust the pH to 10.5. Oxone® (132 mg; 0.22 mmol) was added and the mixture was stirred at room temperature for 4.5 hours. During this time, the pH was kept between 10-11, by dropwise addition of an aqueous solution of potassium hydroxide (25%), if necessary. After 4.5 hours, additional Oxone® (33 mg; 0.05 mmol) was added and the mixture was stirred at room temperature for additional 2.5 hours. The pH was kept between 10-11, by dropwise addition of an aqueous solution of potassium hydroxide (25%), if necessary. The mixture was filtered and the filter cake was washed with plenty of DCM. The filtrate was washed with an aqueous solution of sodium chloride, followed by an aqueous solution of sodium thiosulfate (10%). The organic layer was filtered using a Whatman filter and concentrated. The residue was purified by chromatography (DCM to DCM/ethanol 10%) to give the desired product (38 mg; 0.09 mmol).
Alternative Procedure for the Preparation of Example 4Preparation of Intermediate 4.15-Fluoro-4-(4-fluoro-2-methoxyphenyl)pyridin-2-amine
2-Chloro-5-fluoro-4-(4-fluoro-2-methoxyphenyl)pyridine (20.00 g; 78.23 mmol), tris(dibenzylideneacetone)dipalladium (0) (1.433 g; 1.563 mmol) and 2-(dicyclohexylphosphino)-2′,4′,6′-triisopropylbiphenyl (1.492 g; 3.129 mmol) in anhydrous THF (200 mL) were degassed three times with argon. After 10 minutes of stirring at RT a solution of lithium bis(trimethylsilyl)amide (156.5 mL; 1.0M; THF) was added and the reaction mixture was degassed three more times with argon. The reaction mixture was stirred 2.5 hours at 60° C.
The reaction mixture was cooled to −20° C. Diluted aqueous hydrochloric acid (1.0M) was added so that the pH was adjusted to approximately 5. The reaction mixture was allowed to reach RT and stirred for 10 minutes at this temperature. Then, the pH was adjusted to 10-11 with aqueous sodium hydroxide solution (5.0M). The reaction mixture was diluted with ethyl acetate and washed twice with half saturated sodium chloride solution. The organic layer was dried over magnesium sulfate and concentrated. The residue was purified by column chromatography on silica gel (gradient: hexane to ethyl acetate 100%, with 5% dichloromethane during the first 4 column volumes and afterwards 10% dichloromethane) to give the desired compound (12.04 g; 50.97 mmol).
Preparation of Intermediate 4.32-Chloro-6-methyl-4-[(methylsulfanyl)methyl]pyridine
An aqueous solution of sodium methanethiolate (21%, 13.15 mL, 39.38 mmol) was added dropwise to a stirred solution of 4-(bromomethyl)-2-chloro-6-methylpyridine hydrochloride (4.60 g; 17.90 mmol; Aldlab Chemicals, LLC; for the free base see CAS 1227588-90-0) in acetone (100 mL) while cooling with a water bath at RT. The mixture was stirred at RT over night. EtOAc was added and the layers were separated. The organic layers were washed with saturated aqueous sodium chloride solution, dried over magnesium sulfate and concentrated. The residue was purified by column chromatography on silica gel (gradient: hexane to hexane/EtOAc 8:2) to give the desired product (2.60 g, 13.85 mmol).
Preparation of Intermediate 4.45-Fluoro-4-(4-fluoro-2-methoxyphenyl)-N-{6-methyl-4-[(methylsulfanyl)methyl]pyridin-2-yl}pyridin-2-amine
A batch containing 5-fluoro-4-(4-fluoro-2-methoxyphenyl)pyridin-2-amine (692.2 mg; 2.93 mmol), 2-chloro-6-methyl-4-[(methylsulfanyl)methyl]pyridine (500 mg; 2.66 mmol) and cesium carbonate (1302 mg; 4.00 mmol) in dioxane (15 mL) was degassed with argon. (9,9-Dimethyl-9H-xanthene-4,5-diyl)bis(diphenylphosphane) (67.8 mg; 0.117 mmol) and tris(dibenzylideneacetone)dipalladium(0) (36.6 mg; 0.04 mmol) were added under an atmosphere of argon and the batch was stirred in a closed pressure tube for 10 hours at 100° C.
Five of these batches were combined and diluted with EtOAc. The organic layer was washed twice with saturated aqueous sodium chloride solution, dried over magnesium sulfate and concentrated. The residue was purified by column chromatography on silica gel (gradient: hexane to hexane/EtOAc 1:1) affording the desired product (3.75 g; 9.68 mmol).
Preparation of Intermediate 4.5(rac)-2,2,2-Trifluoro-N-{[(2-{[5-fluoro-4-(4-fluoro-2-methoxyphenyl)pyridin-2-yl]amino}-6-methylpyridin-4-yl)methyl](methyl)-λ4-sulfanylidene}acetamide
Preparation of Intermediate 4.6(rac)-N-{[(3-bromo-6-{[5-fluoro-4-(4-fluoro-2-methoxyphenyl)pyridin-2-yl]amino}-2-methylpyridin-4-yl)methyl](methyl)-λ4-sulfanylidene}-2,2,2-trifluoroacetamide
Under an atmosphere of argon, a solution of 2,2,2-trifluoroacetamide (450.7 mg; 3.99 mmol) in anhydrous THF (2.0 mL) was added dropwise to sodium tert.-butoxide (255.5 mg; 2.60 mmol) in anhydrous THF (3.0 mL), so that the temperature of the mixture remained below 10° C. Subsequently, a freshly prepared solution of 1,3-dibromo-5,5-dimethylhydantoin (456.1 mg; 1.60 mmol) in anhydrous THF (3.0 mL) was added dropwise to the stirred mixture, so that the temperature of the mixture remained below 10° C. Then the mixture was stirred for 10 minutes at 10° C. Finally, a solution of 5-fluoro-4-(4-fluoro-2-methoxyphenyl)-N-{6-methyl-4-[(methylsulfanyl)methyl]pyridin-2-yl}pyridin-2-amine (1030 mg; 2.66 mmol) in anhydrous THF (3.0 mL) was added dropwise to the stirred mixture, so that the temperature of the mixture remained below 10° C. The mixture was stirred 1 hour at 10° C. The batch was diluted with toluene (8.0 mL) under cooling and an aqueous solution of sodium sulfite (335 mg; 2.66 mmol in 15.0 mL water) was added under cooling so that the temperature of the mixture remained below 15° C. After 10 minutes the batch was extracted three times with ethyl acetate. The combined organic phases were washed with saturated aqueous sodium chloride solution, dried over magnesium sulfate and concentrated. The residue was purified by column chromatography on silica gel (gradient: hexane to ethyl acetate 100%) to yield the desired product 2,2,2-trifluoro-N-{[(2-{[5-fluoro-4-(4-fluoro-2-methoxyphenyl)pyridin-2-yl]amino}-6-methylpyridin-4-yl)methyl](methyl)-λ 4-sulfanylidene}acetamide (1202 mg; 2.41 mmol; containing 5,5-dimethylhydantoin) and the side product N-{[(3-bromo-6-{[5-fluoro-4-(4-fluoro-2-methoxyphenyl)pyridin-2-yl]amino}-2-methylpyridin-4-yl)methyl](methyl)-λ 4-sulfanylidene}-2,2,2-trifluoroacetamide (7 mg; 0.012 mmol).
To remove the 5,5-dimethylhydantoin 3.76 g of the product from 4 batches were purified by column chromatography on silica gel (gradient: dichloromethane to dichloromethane/methanol 95:5) to yield the desired product (3.39 g; 6.80 mmol).
3.76 g of racemic 2,2,2-trifluoro-N-{[(2-{[5-fluoro-4-(4-fluoro-2-methoxyphenyl)pyridin-2-yl]amino}-6-methylpyridin-4-yl)methyl](methyl)-λ 4-sulfanylidene}acetamide were separated by chiral HPLC:
[TABLE-US-00005] System:Agilent: Prep 1200, 2xPrep Pump, DLA, MWD, Prep FCColumn:Chiralpak IA 5 μm 250 × 30 mm Nr.: 010Solvent:hexane/ethanol/diethylamine 50:50:0.1 (v/v/v)Flow:45 mL/minTemperature:RTSolution:3760 mg/30.4 mL DCM/MeOHInjection:38 × 0.8 mLDetection:UV 280 nm Fractionsretention time in minpurity in %yieldSpecific optical rotation Intermediate 4.7 5.3-6.8 min95.5%;1520 mg[α]D20 = +113.4° ee: 100%(3.05 mmol)(1.00, DMSO)Intermediate 4.87.2-10.5 min97.1%;1480 mg[α]D20 = −112.1° ee: 98.7%(2.97 mmol)(1.00, DMSO)
Alternative Preparation of End Product (Example 4)(rac)-5-Fluoro-4-(4-fluoro-2-methoxyphenyl)-N-{6-methyl-4-[(S-methylsulfonimidoyl)methyl]pyridin-2-yl}pyridin-2-amine
(rac)-2,2,2-Trifluoro-N-{[(2-{[5-fluoro-4-(4-fluoro-2-methoxyphenyl)pyridin-2-yl]amino}-6-methylpyridin-4-yl)methyl](methyl)-λ 4-sulfanylidene}acetamide (150 mg; 0.301 mmol) was dissolved in methanol (18.0 mL) and water (9.0 mL). At 0-5° C. the pH was adjusted to 9-10 with an aqueous potassium hydroxide solution (15%). At this temperature Oxone® (157.0 mg; 0.256 mmol) was added in several portions and the pH was held at 9-10. The mixture was stirred for 1 hour at 0-5° C. and the pH was held at 9-10.
The reaction mixture was adjusted with 2.0M hydrochloric acid to pH 6-7. Saturated aqueous sodium chloride solution was added and the reaction mixture was extracted three times with dichloromethane. The combined organic phases were washed with an aqueous sodium thiosulfate solution (10%), dried over magnesium sulfate and concentrated. The residue was purified by column chromatography on silica gel (gradient: dichloromethane to dichloromethane/ethanol 9:1) to afford the desired product (100 mg; 0.239 mmol).
An aqueous solution of potassium hydroxide (25%) was added dropwise to a stirred solution of N-{[(3-bromo-6-{[5-fluoro-4-(4-fluoro-2-methoxyphenyl)pyridin-2-yl]amino}-2-methylpyridin-4-yl)methyl](methyl)-λ 4-sulfanylidene}-2,2,2-trifluoroacetamide (161 mg; 0.28 mmol, Intermediate 4.6) in methanol (15.0 mL) and water (5.0 mL) to adjust the pH to 10.5. Oxone® (146 mg; 0.24 mmol) was added and the mixture was stirred at room temperature for 4 hours. During this time, the pH was kept between 10-11, by dropwise addition of an aqueous solution of potassium hydroxide (25%), if necessary. After 4 hours, an additional portion of Oxone® (50 mg; 0.08 mmol) was added and the mixture was stirred at room temperature for additional 2.5 hours. The pH was kept between 10-11, by dropwise addition of an aqueous solution of potassium hydroxide (25%), if necessary. The mixture was filtered and the filter cake was washed with plenty of DCM/MeOH (2:1). The pH of the filtrate was adjusted to pH 6.5 using an aqueous solution of hydrogen chloride (15%), diluted with DCM and washed with an aqueous solution of sodium chloride. The organic layer was finally washed with an aqueous solution of sodium thiosulfate (10%). The organic phase was filtered using a Whatman filter and concentrated. The residue was purified by chromatography (DCM to DCM/ethanol 5%) to give the desired product (44 mg; 0.09 mmol).
Example 6(rac)-5-Fluoro-4-(4-fluoro-2-methoxyphenyl)-N-{6-methoxy-4-[(S-methylsulfonimidoyl)methyl]pyridin-2-yl}pyridin-2-amine
Preparation of Intermediate 6.1:2-Chloro-6-methoxy-4-[(methylsulfanyl)methyl]pyridine
An aqueous solution of sodium methanethiolate (21%, 1.4 mL, 4.2 mmol; Aldrich Chemical Company Inc.) was added dropwise to a stirred solution of 4-(bromomethyl)-2-chloro-6-methoxypyridine (1000 mg; 4.2 mmol, ZereneX Molecular Limited) in acetone (50 mL) while cooling with a water bath at RT. The mixture was stirred at RT for 3 hours. The batch was diluted with ethyl acetate and an aqueous solution of sodium chloride. The mixture was extracted twice (2×) with ethyl acetate. The combined organic layers were filtered using a Whatman filter and concentrated. The residue was purified by column chromatography on silica gel (hexane to hexane/ethyl acetate 10%) to give the desired product (738 mg; 3.6 mmol).
Preparation of Intermediate 6.25-Fluoro-4-(4-fluoro-2-methoxyphenyl)-N-{6-methoxy-4-[(methylsulfanyl)methyl]pyridin-2-yl}pyridin-2-amine
A mixture of 5-fluoro-4-(4-fluoro-2-methoxyphenyl)pyridin-2-amine (1281 mg; 5.4 mmol, Intermediate 4.1), 2-chloro-6-methoxy-4-[(methylsulfanyl)methyl]pyridine (724 mg; 3.6 mmol), chloro(2-dicyclohexylphosphino-2′,4′,6′-tri-iso-propyl-1,1′-biphenyl)[2-(2-aminoethyl)phenyl]palladium(II) methyl-tert-butylether adduct (294 mg; 0.36 mmol; ABCR GmbH & CO. KG) and 2-(dicyclohexylphosphino)-2′,4′,6′-triisopropylbiphenyl (170 mg; 0.36 mmol; Aldrich Chemical Company Inc.) and potassium phosphate (3773 mg; 17.77 mmol) in toluene (84 ml) and NMP (10 mL) was stirred under an atmosphere of argon at 130° C. in a closed vessel for 4 hours. After cooling, the batch was diluted with DCM and washed with aqueous sodium chloride solution. The organic layer was filtered using a Whatman filter and concentrated. The residue was purified by column chromatography on silica gel (hexane to hexane/ethyl acetate 35%) to give the pure product (1212 mg; 3.00 mmol).
Preparation of Intermediate 6.3(rac)-2,2,2-Trifluoro-N-{[(2-{[5-fluoro-4-(4-fluoro-2-methoxyphenyl)pyridin-2-yl]amino}-6-methoxypyridin-4-yl)methyl](methyl)-λ4-sulfanylidene}acetamide
Under an atmosphere of argon, a solution of 2,2,2-trifluoroacetamide (252 mg; 2.23 mmol) in THF (2.0 mL) was added dropwise to a solution of sodium tert.-butoxide (143 mg; 1.49 mmol) in THF (2.0 mL), so that the temperature of the mixture remained below 10° C. Subsequently, a freshly prepared solution of 1,3-dibromo-5,5-dimethylhydantoin (255 mg; 0.89 mmol) in THF (2.0 mL) was added dropwise to the stirred mixture, so that the temperature of the mixture remained below 10° C. Then the mixture was stirred for 10 minutes at 10° C. Finally, a solution of 5-fluoro-4-(4-fluoro-2-methoxyphenyl)-N-{6-methoxy-4-[(methylsulfanyl)methyl]pyridin-2-yl}pyridin-2-amine (600 mg; 1.49 mmol) in THF (3.0 mL) was added dropwise to the stirred mixture, so that the temperature of the mixture remained below 10° C. The mixture was stirred for 3.5 hours at 10° C. The batch was diluted with toluene (8.0 mL) under cooling and an aqueous solution of sodium sulfite (187 mg; 1.49 mmol in 14.0 mL water) was added so that the temperature of the mixture remained below 15° C. The batch was extracted three times with ethyl acetate. The combined organic layers were washed with an aqueous solution of sodium chloride, filtered using a Whatman filter and concentrated. The residue was purified by column chromatography on silica gel (DCM to DCM/ethanol 5%) to give the desired product (37 mg; 0.07 mmol).
An aqueous solution of potassium hydroxide (25%) was added dropwise to a stirred solution of 2,2,2-trifluoro-N-{[(2-{[5-fluoro-4-(4-fluoro-2-methoxyphenyl)pyridin-2-yl]amino}-6-methoxypyridin-4-yl)methyl](methyl)-λ 4-sulfanylidene}acetamide (32 mg; 0.06 mmol) in methanol (1.0 mL) and water (0.6 mL) to adjust the pH to 10.5. Oxone® (32 mg; 0.05 mmol) was added and the mixture was stirred at RT for 2.5 hours. During this time, the pH was kept between 10-11, by dropwise addition of an aqueous solution of potassium hydroxide (25%), if necessary. The mixture was filtered and the filter cake was washed with plenty of DCM. The filtrate was washed with an aqueous solution of sodium chloride, followed by an aqueous solution of sodium thiosulfate (10%). The organic layer was filtered using a Whatman filter and concentrated. The residue was purified by preparative HPLC to give the desired product (9 mg; 0.02 mmol).
[TABLE-US-00006] System:Waters Autopurificationsystem: Pump 2545, Sample Manager 2767, CFO, DAD 2996, ELSD 2424, SQD 3001Column:XBrigde C18 5 μm 100 × 30 mmSolvent:A = H2O + 0.1% HCOOH B = MeCNGradient:0-1 min 1% B, 1-8 min 1-99% B, 8-10 min 99% BFlow:50 mL/minTemperature:RTSolution:Max. 250 mg/max. 2.5 mL DMSO or DMFInjection:1 × 2.5 mLDetection:DAD scan range 210-400 nm MS ESI+, ESI−, scan range 160-1000 m/z
Alternative Procedure for the Preparation of Example 6(rac)-5-Fluoro-4-(4-fluoro-2-methoxyphenyl)-N-{6-methoxy-4-[(S-methylsulfonimidoyl)methyl]pyridin-2-yl}pyridin-2-amine
A freshly prepared 1.5 M solution of sodium ethanolate in ethanol (1.5 mL; 2.25 mmol) was added under an atmosphere of argon to a solution of (rac)-ethyl{[(2-{[5-fluoro-4-(4-fluoro-2-methoxyphenyl)pyridin-2-yl]amino}-6-methoxypyridin-4-yl)methyl](methyl)oxido-λ 6-sulfanylidene}carbamate (290 mg; 0.57 mmol; Example 15) in ethanol (6.3 mL). The batch was stirred at 60° C. for 4 hours. After cooling the batch was diluted with an aqueous solution of sodium chloride and extracted three times with ethyl acetate. The combined organic layers were filtered using a Whatman filter and concentrated to give the desired product (257 mg; 0.0.59 mmol).
Example 7(rac)-N-{6-Chloro-4-[(S-methylsulfonimidoyl)methyl]pyridin-2-yl}-5-fluoro-4-(4-fluoro-2-methoxyphenyl)pyridin-2-amine
Preparation of Intermediate 7.12-Chloro-6-methoxy-4-[(methylsulfanyl)methyl]pyridine
A mixture of 5-fluoro-4-(4-fluoro-2-methoxyphenyl)pyridin-2-amine (2000 mg; 8.47 mmol, Intermediate 4.1), (2,6-dichloropyridin-4-yl)methanol (1507 mg; 8.47 mmol; ABCR GmbH & CO. KG), chloro(2-dicyclohexylphosphino-2′,4′,6′-tri-iso-propyl-1,1′-biphenyl)[2-(2-aminoethyl)phenyl]palladium(II) methyl-tert-butylether adduct (700 mg; 0.85 mmol; ABCR GmbH & CO. KG) and 2-(dicyclohexylphosphino)-2′,4′,6′-triisopropylbiphenyl (404 mg; 0.85 mmol; Aldrich Chemical Company Inc.) and potassium phosphate (8986 mg; 42.33 mmol) in toluene (40 ml) and NMP (4 mL) was stirred under an atmosphere of argon at 110° C. for 135 minutes. After cooling, the batch was diluted with ethyl acetate and washed with aqueous sodium chloride solution. The organic layer was filtered using a Whatman filter and concentrated. The residue was purified by column chromatography on silica gel (hexane to hexane/ethyl acetate 50%) to give the pure product (1350 mg; 3.57 mmol).
Preparation of Intermediate 7.2N-{6-Chloro-4-[(methylsulfanyl)methyl]pyridin-2-yl}-5-fluoro-4-(4-fluoro-2-methoxyphenyl)pyridin-2-amine
To a stirred solution of (2-chloro-6-{[5-fluoro-4-(4-fluoro-2-methoxyphenyl)pyridin-2-yl]amino}pyridin-4-yl)methanol (1.47 g; 3.89 mmol) in DMF (43 mL) at 0° C. was added dropwise thionyl chloride (0.71 mL; 9.73 mmol). The mixture was allowed to react at RT for 2 hours. Then, the mixture was concentrated to give crude N-[6-chloro-4-(chloromethyl)pyridin-2-yl]-5-fluoro-4-(4-fluoro-2-methoxyphenyl)pyridin-2-amine (2.85 g).
Crude N-[6-chloro-4-(chloromethyl)pyridin-2-yl]-5-fluoro-4-(4-fluoro-2-methoxyphenyl)pyridin-2-amine (2.85 g) was dissolved in acetone (87 mL) and an aqueous solution of sodium methanethiolate (21%, 5.2 mL, 15.58 mmol; Aldrich Chemical Company Inc.) was added dropwise under stirring. The mixture was stirred at RT for 6 hours. The mixture was diluted with an aqueous solution of sodium chloride and extracted twice with ethyl acetate. The combined organic layers were filtered using a Whatman filter and concentrated. The residue was purified by column chromatography on silica gel (hexane to hexane/ethyl acetate 20%) to give the desired product (1.24 g; 3.04 mmol).
Preparation of Intermediate 7.3(rac)-N-{[(2-chloro-6-{[5-fluoro-4-(4-fluoro-2-methoxyphenyl)pyridin-2-yl]amino}pyridin-4-yl)methyl](methyl)-λ4-sulfanylidene}-2,2,2-trifluoroacetamide
Under an atmosphere of argon, a solution of 2,2,2-trifluoroacetamide (312 mg; 2.76 mmol) in THF (2.0 mL) was added dropwise to a solution of sodium tert.-butoxide (176 mg; 1.84 mmol) in THF (2.0 mL), so that the temperature of the mixture remained below 10° C. Subsequently, a freshly prepared solution of 1,3-dibromo-5,5-dimethylhydantoin (394 mg; 1.38 mmol) in THF (3.0 mL) was added dropwise to the stirred mixture, so that the temperature of the mixture remained below 10° C. Then the mixture was stirred for 10 minutes at 10° C. Finally, a solution of N-{6-chloro-4-[(methylsulfanyl)methyl]pyridin-2-yl}-5-fluoro-4-(4-fluoro-2-methoxyphenyl)pyridin-2-amine (750 mg; 1.84 mmol) in THF (3.0 mL) was added dropwise to the stirred mixture, so that the temperature of the mixture remained below 5° C. The mixture was stirred for 3 hours at 5° C. The batch was diluted with toluene (5.0 mL) under cooling and an aqueous solution of sodium sulfite (232 mg; 1.84 mmol in 5.0 mL water) was added so that the temperature of the mixture remained below 15° C. The batch was extracted three times with ethyl acetate. The combined organic layers were washed with an aqueous solution of sodium chloride, filtered using a Whatman filter and concentrated. The residue was purified by column chromatography on silica gel (hexane to hexane/ethyl acetate 85%) to give the desired product (363 mg; 0.70 mmol).
An aqueous solution of potassium hydroxide (25%) was added dropwise to a stirred solution of N-{[(2-chloro-6-{[5-fluoro-4-(4-fluoro-2-methoxyphenyl)pyridin-2-yl]amino}pyridin-4-yl)methyl](methyl)-λ 4-sulfanylidene}-2,2,2-trifluoroacetamide (495 mg; 0.95 mmol) in methanol (15.0 mL) and water (6.7 mL) to adjust the pH to 10.5. Oxone® (498 mg; 0.81 mmol) was added and the mixture was stirred at RT for 90 minutes. During this time, the pH was kept between 10-11, by dropwise addition of an aqueous solution of potassium hydroxide (25%), if necessary. The mixture was filtered and the filter cake was washed with plenty of DCM and methanol. The pH of the filtrate was adjusted to 6-7 using an aqueous solution of hydrogen chloride (15%). The filtrate was washed with an aqueous solution of sodium chloride, followed by an aqueous solution of sodium thiosulfate (10%). The organic phase was filtered using a Whatman filter and concentrated. The residue was purified by chromatography (DCM to DCM/ethanol 50%) to give the desired product (118 mg; 0.27 mmol).
A batch with 2-chloro-5-fluoro-4-iodopyridine (1000 mg; 3.88 mmol; APAC Pharmaceutical, LLC), (4-fluoro-2-methoxyphenyl)boronic acid (660 mg; 3.88 mmol; Aldrich Chemical Company Inc.) and tetrakis(triphenylphosphin)palladium(0) (449 mg; 0.38 mmol) in 1,2-dimethoxyethane (10.0 mL) and 2 M aqueous solution of potassium carbonate (5.8 mL) was degassed using argon. The batch was stirred under an atmosphere of argon for 4 hours at 100 °C. After cooling, the batch was diluted with ethyl
acetate and THF and washed with a saturated aqueous solution of sodium chloride. The organic phase was filtered using a Whatman filter and concentrated. The residue was purified by column chromatography (hexane to hexane / ethyl acetate 50%) to give the desired product (947 mg; 3.70 mmol).