
Gefapixant
- Molecular FormulaC14H19N5O4S
- Average mass353.397 Da
1015787-98-0[RN]
10642
AF 217
5-[(2,4-Diamino-5-pyrimidinyl)oxy]-4-isopropyl-2-methoxybenzenesulfonamide
5-(2,4-diamino-pyrimidin-5-yloxy)-4-isopropyl-2-methoxy-benzene- sulfonamide
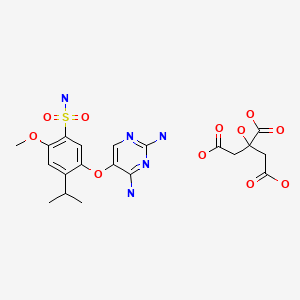
Gefapixant Citrate
| Formula | C14H19N5O4S. C6H8O7 |
|---|---|
| CAS | 2310299-91-1 |
| Mol weight | 545.5203 |
APPROVED JAPAN PMDA 2022/1/20, Lyfnua
ゲーファピキサントクエン酸塩
吉法匹生
| Efficacy | Analgesic, Anti-inflammatory, Antitussive, P2X3 receptor antagonist |
|---|---|
| Comment | Treatment of disorders associated with purinergic receptor activation |
Gefapixant (MK-7264) is a drug which acts as an antagonist of the P2RX3 receptor, and may be useful in the treatment of chronic cough.[1][2][3] It was named in honour of Geoff Burnstock.[4]
Gefapixant is under investigation in clinical trial NCT02397460 (Effect of Gefapixant (AF-219/MK-7264) on Cough Reflex Sensitivity).
PAPER
Organic Process Research & Development (2020), 24(11), 2445-2452.
https://pubs.acs.org/doi/10.1021/acs.oprd.0c00248
A robust, green, and sustainable manufacturing process has been developed for the synthesis of gefapixant citrate, a P2X3 receptor antagonist that is under investigation for the treatment of refractory and unexplained chronic cough. The newly developed commercial process features low process mass intensity (PMI), short synthetic sequence, high overall yield, minimal environmental impact, and significantly reduced API costs. The key innovations are the implementation of a highly efficient two-step methoxyphenol synthesis, an innovative pyrimidine synthesis in flow, a simplified sulfonamide synthesis, and a novel salt metathesis approach to consistently deliver the correct active pharmaceutical ingredient (API) salt form in high purity.

SYN
Organic Process Research & Development (2020), 24(11), 2478-2490.
https://pubs.acs.org/doi/10.1021/acs.oprd.0c00252
Gefapixant citrate (MK-7264) is a P2X3 antagonist for the treatment of chronic cough. The second generation manufacturing route developed for the Step 3A/3B formylation–cyclization reaction to generate the key intermediate diaminopyrimidine (1) (AF-072) required a significant excess of ethyl formate (EF), potassium tert-butoxide (KOt-Bu), and guanidine•HCl (G•HCl) when both steps were run as batch processes. It was imperative to develop an alternative process that required less of each reagent and generated less carbon monoxide byproducts, as the annual production of the final active pharmaceutical ingredient (API) is expected to be over 50 MT. In addition, the second generation process was misaligned with our company’s strategy of having the best science in place at the first regulatory filing. The final flow–batch process described herein, which features a flow-based formylation combined with a batch cyclization, has been performed on a 500 kg scale and now requires 35% less EF (leading to a 70% reduction in waste carbon monoxide), 38% less KOt-Bu, and 50% less G•HCl. These improvements, along with a twofold increase in concentration, have resulted in a 54% reduction in the step process mass intensity (step-PMI) from the second generation two-step batch–batch process (PMI of 17.16) to the flow–batch process (PMI of 7.86), without sacrificing reaction performance.

SYN
H. REN*, K. M. MALONEY* ET AL. (MERCK & CO., INC., RAHWAY USA) Development of a Green and Sustainable Manufacturing Process for Gefapixant Citrate (MK-7264) Part 1: Introduction and Process Overview Org. Process Res. Dev. 2020, 24, 2445–2452, DOI: 10.1021/acs.oprd.0c00248.

SYN
https://pubs.acs.org/doi/abs/10.1021/acs.oprd.0c00247

A scalable two-pot sulfonamidation through the process has been developed for the synthesis of gefapixant citrate, a P2X3 receptor antagonist that is under investigation for the treatment of refractory and unexplained chronic cough. Direct conversion of the diaryl ether precursor to a sulfonyl chloride intermediate using chlorosulfonic acid, followed by treatment with aqueous ammonia hydroxide, provided the desired sulfonamide in high yield. A pH-swing crystallization allowed for the formation of a transient acetonitrile solvate that enables the rejection of two impurities. After drying, the desired anhydrous free base form was isolated in high yield and purity.
SYN
https://www.sciencedirect.com/science/article/abs/pii/S1566070221000898
Gefapixant is the approved generic name for a compound also known as MK-7264, and prior to that AF-219 and RO-4926219. It is the first-in-class clinically developed antagonist for the P2X3 subtype of trimeric ionotropic purinergic receptors, a family of ATP-gated excitatory ion channels, showing nanomolar potency for the human P2X3 homotrimeric channel and essentially no activity at related channels devoid of P2X3 subunits. As the first P2X3 antagonist to have progressed into clinical studies it has now progressed to the point of successful completion of Phase 3 investigations for the treatment of cough, and the NDA application is under review with US FDA for treatment of refractory chronic cough or unexplained chronic cough. The molecule was discovered in the laboratories of Roche Pharmaceuticals in Palo Alto, California, but clinical development then continued with the formation of Afferent Pharmaceuticals for the purpose of identifying the optimal therapeutic indication for this novel mechanism and establishing a clinical plan for development in the optimal patient populations selected. Geoff Burnstock was a close collaborator and advisor to the P2X3 program for close to two decades of discovery and development. Progression of gefapixant through later stage clinical studies has been conducted by the research laboratories of Merck & Co., Inc., Kenilworth, NJ, USA (MRL; following acquisition of Afferent in 2016), who may commercialize the product once authorization has been granted by regulatory authorities.
PATENT
WO 2008040652
https://patents.google.com/patent/WO2008040652A1/en

SCHEME AExample 1: 5-(2,4-Diamino-pyrimidin-5-yloxy)-4-isopropyl-2-methoxy- benzenesulfonamideThe synthetic procedure used in this Example is outlined in Scheme B.


not isolated


SCHEME BStep 1 2-Isopropyl-4-methoxy-phenolTo a cooled solution of l-(2-hydroxy-5-methoxy-phenyl)-ethanone (10.0 kg) in 79.0 kg of THF was gradually added 46.4 kg of 3M solution of MeMgCl in THF at a rate such that the reaction mixture temperature did not exceed 25°C. Following addition of the MeMgCl solution, the reaction mixture was stirred at ambient temperature for 18 hours, at which point HPLC (high pressure liquid chromatography) analysis showed more than 98% conversion of l-(2-hydroxy-5-methoxy-phenyl)-ethanone to 2- (1 -hydroxy- 1- methyl-ethyl)-4-methoxy-phenol (not shown in Scheme D). To the stirred solution was then added 10% palladium on carbon (1.02 kg, 50% water wet) suspended in 3.5 kg of THF. The reaction mixture was cooled and placed under a hydrogen atmosphere at 0.34 atmosphere pressure, and concentrated HCl (19.5 kg) was added while maintaining the reaction temperature at 25°C. The resultant mixture was stirred at ambient temperature for 18 hours, then treated with 44.4 kg water and filtered through a bed of Celite to remove suspended catalyst. The filter cake was rinsed with EtOAc and the combined filtrate was separated. The organic phase was washed with water, then concentrated by distillation to provide an oil. This oil was dissolved in 2-butanone (20.4 kg) and the crude solution was employed directly in the next step. A 161.8 g aliquot of the solution was concentrated under vacuum to provide 49.5 g of 2-isopropyl-4-methoxyphenol as an oil, projecting to 10.4 kg crude contained product in the bulk 2-butanone solution. 1H NMR (DMSO) delta: 1.14 (d, 6H, J = 6.9 Hz), 3.18 (septet, IH, J = 6.9 Hz), 3.65 (s, 3H), 6.56, (dd, IH, J = 8.6 Hz, 3.1 Hz), 6.67 (d, IH, J = 3.1 Hz), 6.69 (d, IH, 8.6 Hz).Step 2 (2-Isopropyl-4-methoxy-phenoxy)-acetonitrileA stirred slurry of toluene-4-sulfonic acid cyanomethyl ester (13.0 kg), potassium carbonate (13.0 kg) and 2-isopropyl-4-methoxyphenol (9.57 kg) in 79.7 kg of 2-butanone was heated to 55-600C for 4 days, then heated to reflux for 18 hours. The resultant slurry was cooled and filtered to remove solids. The filtrate was concentrated under reduced pressure and the residue was redissolved in toluene. The toluene solution was extracted with IN KOH, and the organic phase was concentrated by distillation to give 20.6 g of a 1:1 (by weight) solution of (2-isopropyl-4-methoxy-phenoxy)-acetonitrile in toluene, which was used directly in the next step. A aliquot (96.7 g) of this solution was concentrated to dryness to give 50.9 g of crude (2-isopropyl-4-methoxy-phenoxy)- acetonitrile, projecting to a yield of 10.9 kg in the bulk solution: MS (M+H) = 206; 1H NMR (CDCl3) delta: 1.25 (d, J = 6.9 Hz), 3.31 (septet, IH, J = 6.9 Hz), 3.82 (s, 3H), 4.76 (s, 2H), 6.73 (dd. IH, J = 8.8 Hz, 3.1 Hz), 6.87 (d, IH, J = 3.1 Hz), 6.91 (d, IH, J = 8.8 Hz).Step 3 5-(2-Isopropyl-4-methoxy-phenoxy)-pyrimidine-2,4-diamine An approximately 1:1 (by weight) solution of 10.6 kg of (2-isopropyl-4-methoxy-phen- oxy) -acetonitrile in toluene was concentrated under reduced pressure and the residue was treated with 10.8 kg of tert-butoxybis(dimethylamino)methane (Brederick’s Reagent). The resulting mixture was dissolved in 20.2 kg of DMF and the solution was heated to 1100C for 2 hours, at which point HPLC analysis showed essentially complete conversion to 3,3-bis-dimethylamino-2-(2-isopropyl-4-methoxy-phenoxy)-propionitrile (not isolated, 1H NMR (CDCl3) delta: 1.21 (d, 3H, J = 7.2 Hz), 1.23 (d, 3H, J = 7.1 Hz), 2.46 (s, 6H), 2.48 (s, 6H), 3.43 (d, IH, J = 5.0 Hz), 3.31 (septet, IH, J = 6.9 Hz), 3.79 (s, 3H), 4.93 (d, IH, J = 5.0 Hz), 6.70 (dd, IH, J = 8.8 Hz, 3.0 Hz), 6.82 (d, IH, J = 3.0 Hz), 6.98 (d, IH, J = 8.8 Hz). The DMF solution was cooled and transferred onto 14.7 kg of aniline hydrochloride. The resulting mixture was heated to 1200C for 22 hours, at which point HPLC analysis showed greater than 97% conversion to 2-(2-isopropyl-4-methoxy-phenoxy)-3- phenylamino-acrylonitrile (not isolated, 1H nmr (CDCl3) delta: 1.31 (d, 6H, J = 6.9 Hz), 3.39 (septet, IH, J = 6.9 Hz), 3.82 (s, 3H), 6.61 (d (br), IH, J = 12.7 Hz), 6.73 (dd, IH, J = 8.9 Hz, 3.1 Hz), 6.88 (d, IH, J = 3.0 Hz), 6.93 (m, 2H), 6.97 (d, IH, J = 8.9 Hz), 7.05 (m, IH), 7.17 (d, IH, J = 12.6 Hz), 7.35 (m. 2H)).The mixture was cooled, diluted with 21.5 kg toluene, then with 72.2 L of water. The organic layer was separated, washed with water, and concentrated by distillation. The concentrate was transferred into 23.8 kg DMF, and the DMF solution was transferred onto 6.01 kg of guanidine carbonate. The resulting mixture was heated to 1200C for 3 days, at which point HPLC analysis showed greater than 95% conversion of 2-(2- isopropyl-4-methoxy-phenoxy)-3-phenylamino-acrylonitrile into 5-(2-Isopropyl-4- methoxy-phenoxy)-pyrimidine-2,4-diamine. The reaction mixture was cooled, diluted with 7.8 kg of EtOAc, then reheated to 600C. Water (75.1 L) was added and the resultant mixture was allowed to cool to ambient temperature. The precipitated solid was collected by filtration, rinsed with isopropanol and dried under vacuum at 50 degrees to give 9.62 kg of 5-(2-isopropyl-4-methoxy- phenoxy)-pyrimidine-2,4-diamine: m.p. 170-171 degrees C; MS (M+H) = 275; H nmr (chloroform) delta: 1.25 (d, 6H, J = 6.9 Hz), 3.30 (septet, IH, J = 6.9 Hz), 3.79 (s, 3H), 4.68 (br, 2H), 4.96 (br, 2H), 6.64 (dd, IH, J = 8.9 Hz, 3.0 Hz), 6.73, d, J = 8.9 Hz), 6.85 (d, IH, J = 3 Hz), 7.47 (s, IH).Step 4 5-(2,4-diamino-pyrimidin-5-yloxy)-4-isopropyl-2-methoxy-benzenesulfon- amide, sulfolane solvate Chlorosulfonic acid (13.82 kg) was added to a slurry of 5-(2-isopropyl-4-methoxy-phen- oxy)-pyrimidine-2,4-diamine (10.07 kg) in sulfolane (50.0 kg) at a rate to maintain an internal pot temperature below 65°C. The reaction mixture was aged at 60-650C for 12 hours, at which point HPCL showed that all 5-(2-isopropyl-4-methoxy-phenoxy)- pyrimidine-2,4-diamine starting material had been converted to 5-(2,4-diamino- pyrimidin-5-yloxy)-4-isopropyl-2-methoxy-benzenesulfonic acid. MS (M+H) = 355. Phosphorus oxychloride (3.41 kg) was then added to the reaction mixture at 600C. The reaction mixture was heated to 75°C and aged for 12 hours, at which point HPLC showed that approximately 99% of 5-(2,4-diamino-pyrimidin-5-yloxy)-4-isopropyl-2-methoxy- benzenesulfonic acid had been converted to 5-(2,4-diamino-pyrimidin-5-yloxy)-4-iso- propyl-2-methoxy-benzenesulfonyl chloride. MS (M+H) = 373. The solution of 5-(2,4- diamino-pyrimidin-5-yloxy)-4-isopropyl-2-methoxy-benzenesulfonyl chloride was then cooled to around 2°C).To a cooled (ca. 2°C) solution of ammonia (7N) in MeOH (74.1 kg) was added the cooled sulfolane solution of 5-(2,4-diamino-pyrimidin-5-yloxy)-4-isopropyl-2-methoxy- benzenesulfonyl chloride (a homogeneous syrup) at a rate such that the internal temperature did not exceed 23°C. The resultant slurry was stirred for 18 hours at ambient temperature, then filtered on a coarse porosity frit filter. The collected solids were rinsed with MeOH (15.9 kg), then dried under reduced pressure at 700C to a constant weight of 23.90 kg. HPLC showed 97.5% conversion of 5-(2,4-diamino- pyrimidin-5-yloxy)-4-isopropyl-2-methoxy-benzenesulfonyl chloride to 5-(2,4-diamino- pyrimidin-5-yloxy)-4-isopropyl-2-methoxy-benzenesulfonamide sulfolane solvate. H nmr (DMSOd6) delta: 1.26 (d, 6H, J = 6.9 Hz), 2.07 (sym. m, 8H), 2.99 (sym. m, 8H), 3.41 (septet, IH, J = 6.9 Hz), 3.89 (s, 3H), 6.03 (s (br), 2H), 6.58 (s (br), 2H), 7.00 (s, IH), 7.04 (s (br), 2H), 7.08 (s, IH), 7.35 (s, IH).
Step 5 5-(2,4-diamino-pyrimidin-5-yloxy)-4-isopropyl-2-methoxy-benzene- sulfonamideA slurry of 5-(2,4-diamino-pyrimidin-5-yloxy)-4-isopropyl-2-methoxy-benzenesulfon- amide sulfolane solvate (23.86 kg) in a mixture of ethanol (74.3 kg) and 0.44 N HCl (109.4 kg) was heated to reflux to provide a homogeneous solution of the monohydrochloride salt of 5-(2,4-diamino-pyrimidin-5-yloxy)-4-isopropyl-2-methoxy- benzenesulfonamide. This solution was filterd while hot, then treated with concentrated ammonium hydroxide (3.4 L) to liberate the free base of 5-(2,4-diamino-pyrimidin-5- yloxy)-4-isopropyl-2-methoxy-benzenesulfonamide. The resultant mixture was cooled slowly to 200C and the crystalline product isolated by filtration. The filter cake was washed with water (20.1 kg) and dried under reduced pressure at 700C to a constant weight of 8.17 kg (57.7% yield based on di-solvate of sulfolane).MP = 281-282 0C.1H nmr (DMSOd6) delta: 1.27 (d, 6H, J = 6.9 Hz), 3.41 (septet, IH, J = 6.9 Hz), 3.89 (s, 3H), 5.87 (s (br), 2H), 6.40 (s (br), 2H), 6.98 (s, IH), 7.01 (s (br), 2H), 7.07 (s, IH), 7.36 (s, IH).
PATENT
US 20080207655https://patents.google.com/patent/US20080207655
PATENThttps://patentscope.wipo.int/search/en/detail.jsf?docId=WO2016004358
xample 20
5-(2,4-Diamino-pyrimidin-5-yloxy)-4-isopropyl-2-methoxy-N-methyl-benzenemethylsulfonamide Step 1. 5-(2,4-Diamino-pyrimidin-5-yloxy)-4-isopropyl-2-methoxy- benzenesulfonyl chloride
[211] A mixture of pyrimidine (0.400 g, 1.5 mmol) in 2 ml chlorosulfonic acid was allowed to stir 20 min. The mixture was poured over ice. The precipitate was filtered, washed by cold H2O and dried under vacuum to afford 5-(2,4-diamino-pyrimidin-5-yloxy)-4-isopropyl-2-methoxy-benzenesulfonyl chloride (0.515 g, 95%) as a white solid; [MH]+= 373.
PATENT
WO 2017058645
https://patents.google.com/patent/WO2017058645A1/en
PATENTDisclosed herein is a novel process for preparing Compound A, a phenoxy diaminopyrimidine compound of the following formula, or a pharmaceutically acceptable salt thereof:

Compound A.Also disclosed herein are various salts and solvates of Compound A.
Scheme 1


Step 1. Preparation of 4-Bromo-2-isopropylphenol DABCO Co-crystalStep 1. Preparation of 4-Bromo-2-isopropylphenol DABCO Co-crystalThe following 4-bromo-2-isopropylphenol hemi-DABCO co-crystal is obtained in greater than 99% purity and at about 85-92% yield by the following process:

To a solution of 2-isopropyl phenol (75.0 g, 550 mmol) in acetonitrile (225 mL) was added MSA (0.520 g, 5.41 mmol). The mixture was cooled to -10 °C and NBS (98.01 g, 550 mmol) was added in portions while maintaining the internal temperature below 10 °C. The reaction was aged for 30 min to 1 h and then warmed to 20 °C, diluted with water (450 mL), and extracted with toluene (225 mL). The organic layer was sequentially washed with 9 wt% phosphoric acid (150 mL) and 5 wt% NaCl (150 mL). The organic layers were concentrated to roughly 150 mL and filtered into a clean reactor. The mixture was heated to 30-40 °C and n- heptane (28.5 mL) was added followed by DABCO (30.89 g, 275 mmol). The mixture was seeded (a seed can be synthesized from a previous batch of this procedure preformed without seeding) with 4-bromo-2-isopropylphenol hemi-DABCO co-crystal (75 mg, 0.277 mmol), diluted with 52.5 mL of n-heptane, and stirred for 1 h. The slurry was cooled to 20 °C over 1 h and 370 mL of n-heptane is added over 2 h. The slurry was cooled to 5 °C over 2 h, aged for 2 h, filtered, and washed with n-heptane (2 x 75 mL). The solid was dried at 20-25 °C under vacuum to yield 4-bromo-2-isopropylphenol hemi-DABCO co-crystal (134.8 g, 90 %) as a solid. 1H NMR (400 MHz, DMSO-76) d 7.20 (d, J= 2.5 Hz, 1H), 7.13 (dd, J= 8.5, 2.6 Hz, 2H), 6.73 (d, J = 8.5 Hz, 2H), 3.16 (hept, J= 6.9 Hz, 2H), 2.60 (s, 12H), 1.14 (d, J= 6.9 Hz, 12H).The crystallization of step 1 generates 4-bromo-2-isopropylphenol hemi-DABCO co-crystal, bromophenol mono-DABCO co-crystal, or a mixture of bromophenol hemi-DABCO co-crystal and bromophenol mono-DABCO co-crystal. An XRPD pattern of bromophenol hemi- DABCO co-crystal is shown in Figure 1.
The bromo-phenol mono-DABCO co-crystal can be generated in the following procedure:

bromophenol DABCO co-crystalTo a vial with a stir bar was charged DABCO (1.7 g, 15 mmol), phenol (2.5 g, 15 mmol), and 2 mL of n-heptane. The resulting slurry was stirred at 23 °C overnight. The slurry was then filtered and the resulting wet cake was washed with 2 mL of 5 °C n-heptane. The cake was dried under vacuum with nitrogen sweep to afford 4-bromo-2-isopropylphenol mono- DABCO co-crystal (2.9 g, 70% yield) as a solid. 1H NMR (500 MHz, DMSO-76) d 9.65 (s, 1H), 7.20 (s, 1H), 7.14 (d, J= 8.5 Hz, 1H), 6.74 (d, J= 8.5 Hz, 1H), 3.17 (hept, J= 6.8 Hz, 1H), 2.61(s, 12H), 1.15 (d, 7 = 6.9 Hz, 6H).An XRPD pattern of bromophenol mono-DABCO co-crystal is shown in Figure 2.Step 2a. Preparation of 2-Isopropyl-4-Methoxyphenol
The 2-isopropyl-4-Methoxyphenol shown below is obtained at about 92% yield by the following process:

bromophenol DABCO co-crystal methoxy phenolTo a solution of 4-bromo-2-isopropylphenol hemi-DABCO co-crystal (120 g, 442 mmol) in 25 wt% sodium methoxide in methanol (430 g) was added 60 mL of DMF. The solution was pressure purged with nitrogen, copper (I) bromide (3.23 g, 22.5 mmol) was added to the mixture, and the reaction was heated to reflux for 12-16 h. The reaction is cooled to 0-5 °C and quenched with 6M HC1 until the pH of the solution is less than 5. The slurry is diluted with 492 mL of toluene and 720 mL of water to provide a homogeneous solution with a rag between the layers. The aqueous layer is cut to waste. The organic layer is filtered to remove the rag and washed with 240 mL of water to provide 2-isopropyl-4-methoxylphenol (491 g, 13.3 wt%, 89% assay yield) as a solution in toluene. 1H NMR (500 MHz, DMSO-76) d 8.73 (s, 1H), 6.68 (d, J = 8.6 Hz, 1H), 6.66 (d, 7= 3.0 Hz, 1H), 6.55 (dd, 7= 8.6, 3.1 Hz, 1H), 3.65 (s, 3H), 3.17 (hept, j = 6.9 Hz, 1H), 1.14 (d, 7= 6.9 Hz, 6H).Step 2b. Preparation of 2-Isopropyl-4-Methoxyphenol
Alternatively, the methoxy phenol is obtained by the following process:

To a high-pressure vessel were charged 400 mL of anhydrous toluene, Re2(CO)io (3.16 g, 4.84 mmol) and mequinol (100 g, 806 mmol) at RT. The vessel was then degassed with propylene, and charged with propylene (85.0 g, 2.02 mol). The vessel was sealed and heated to 170 °C. Internal pressure was measured near 250 psi. The reaction was stirred at this condition for 72 h. The vessel was then allowed to cool down to 23 °C. The internal pressure was carefully released to 1 atmospheric pressure, and the toluene solution was assayed as 91% and used directly in the next step or isolated as a solid.Step 2a/2b results in anhydrous 2-isopropyl-4-methoxyphenol form 1. An XRPD pattern of the methoxy phenol form 1 is shown in Figure 3.In another embodiment, the product is isolated as a DMAP co-crystal:

To a vial with a stir bar was charged DMAP (3.67 g, 30.1 mmol), 2.5 ml of toluene, and 2-isopropyl-4-methoxylphenol (5.00 g, 30.1 mmol). The reaction mixture was stirred at RT for 5 min, and a homogeneous solution was formed. The reaction mixture was then cooled to 5 °C. Ten mL of n-heptane was slowly charged over 20 min. The resulting slurry was stirred at 5 °C overnight. The slurry was filtered and the resulting wet cake was washed with 3 mL of 5 °C n-heptane. The cake was dried under vacuum with a nitrogen sweep to provide 2- isopropyl-4-methoxylphenol DMAP co-crystal (7.01 g, 81%) as a solid. 1H NMR (500 MHz, DMSO-76) d 8.78 (s, 1H), 8.10 (d, J= 6.1 Hz, 2H), 6.71 – 6.65 (m, 2H), 6.57 (dd, J= 11.3, 6.0 Hz, 3H), 3.66 (s, 3H), 3.17 (hept, J= 6.8 Hz, 1H), 2.95 (s, 6H), 1.14 (d, J= 6.9 Hz, 6H).The crystallization generates anhydrous 2-isopropyl -4-methoxyphenol DMAP co crystal. An XRPD pattern of the 2-isopropyl-4-methoxyphenol DMAP co-crystal is shown in Figure 4.Step 3a. Preparation of the Cvanoether. 2-(2-isopropyl-4-methoxyphenoxy)acetonitrile
The cyanoether is obtained at about 95 % yield by the following process:

A 12-15 wt% solution of 2-isopropyl-4-methoxylphenol (314.3 g, 12 wt%, 226.8 mmol) was concentrated to greater than 50 wt% 2-isopropyl-4-methoxyphenol in toluene under vacuum at 40-50°C. To the solution was added 189 mL of NMP, and the mixture was cooled to 5 °C. Sodium hydroxide (27.2 g, 50 wt% in water, 340 mmol) and chloroacetonitrile (36 g, 340 mmol) were added sequentially to the mixture while maintaining the internal temperature below 10 °C. The reaction was aged for 2 h and then diluted with 150 mL of toluene and 226 mL of water while maintaining the temperature below 10 °C. The mixture was warmed to 20-25 °C, the layers were separated, and the organic layer was washed with 75 mL of 20 wt% NaCl (aq.). The organic layer was and filtered to provide 2-(2-isopropyl-4-methoxyphenoxy)acetonitrile (56.8 g, 74.6 wt%) as a solution in toluene. The filter was washed with NMP to provide additional 2-(2-isopropyl-4-methoxyphenoxy)acetonitrile (27.1 g, 5.0 wt%) as a solution in NMP. The combined yield was about 94 %. 1H NMR (500 MHz, DMSO-i¾) d 7.05 (d, J= 8.8 Hz, 1H), 6.81 (d, 7= 3.0 Hz, 1H), 6.78 (dd, j= 8.8, 3.1 Hz, 1H), 5.11 (s, 2H), 3.73 (s, 3H), 3.20 (hept, j = 6.9 Hz, 1H), 1.17 (d, 7= 6.9 Hz, 6H).Step 3b. Preparation of the Cvanoether. 2-(2-isopropyl-4-methoxyphenoxy)acetonitrile
Alternatively, the cyanoether shown below is obtained at about 92% yield by the following process:

A solution of 2-isopropyl-4-methoxyphenol in toluene (491 g, 13.3 wt%, 393 mmol) was concentrated and solvent switched to acetonitrile under vacuum at 40-50 °C.Potassium carbonate (164.5 g, 1190 mmol) and tetrabutylammonium hydrogensulfate (1.5 g, 4.42 mmol) were added to a separate vessel, and the vessel was pressure purged with nitrogen gas.The solution of phenol in acetonitrile and chloroacetonitrile was added sequentially to the reaction vessel. The vessel was heated to 40 °C and aged for 4 h. The mixture was allowed to cool to 25 °C, and was diluted with 326 mL water. The layers were separated, and the organic layer was washed with 130 mL of 10 wt% NaCl. A solvent switch to toluene was performed under vacuum, and the organic layer was filtered through two 16D Cuno #5 cartridges. The organic layer was concentrated to provide 2-(2-isopropyl-4-methoxyphenoxy)acetonitrile in toluene (128.2 g, 58 wt%, 92% yield).Step 4 Preparation of the Dia inopyrimidine 5-(2-isopropyl-4-methoxyphenoxy)pyrimidine-2.4-di amineThe diaminopyrimidine is obtained at about 90 % yield by the following process:

A solution of potassium tert-butoxide (44.8 g, 0399 mmol) in NMP (180 mL) was cooled to -10 °C. A solution of 2-(2-isopropyl-4-methoxyphenoxy)acetonitrile, the cyanoether, (59.3 g, 61.4 wt%, 177 mmol) in toluene and ethyl formate (26.3 g, 355 mmol) was charged to the base solution while maintaining the internal temperature between -12 °C and -8 °C. After a 3 h age, guanidine hydrochloride (136 g, 1420 mmol) was added to the mixture and the reaction was heated to 115 °C for 6 h. The mixture was allowed to cool to 90 °C, diluted with 200 mL of water, and aged until the reaction mixture was homogeneous (about 30-45 min). After all solids dissolved, vacuum (400 mm Hg) was applied to the reactor to remove toluene. Vacuum was disconnected and the solution was allowed to cool to 85°C. 5-(2-Isopropyl-4- methoxyphenoxy)pyrimidine-2, 4-diamine seed (49.8 mg) (a seed can be synthesized by a route described in U.S. Patent 7,741,484) was charged, the solution was aged for 2 h, 200 mL of water was added, and the batch was allowed to cool to 20 °C over 6 h. The slurry was aged for 10 h at 20 °C, filtered, washed with 2: 1 water :NMP (3 x 100 mL) and water (3 x 100 mL), and dried under vacuum at 50 °C to provide the title compound (42.2 g, 88%) as a solid. 1H NMR (500 MHz, DMSO-r¾) d 7.23 (s, 1H), 6.83 (d, J= 3.0 Hz, 1H), 6.70 (dd, J= 8.9, 3.0 Hz, 1H), 6.63 (d, j= 8.8 Hz, 1H), 6.32 (s, 2H), 5.75 (s, 2H), 3.71 (s, 3H), 3.28 (hept, j= 6.9 Hz, 1H), 1.20 (d, j = 6.9 Hz, 6H); 13C NMR (126 MHz, DMSO-r¾) d 159.7, 157.2, 155.1, 148.4, 144.2, 139.0, 130.4,116.9, 112.5, 111.3, 55.4, 26.57, 22.83.The crystallization of step 4 generates an anhydrous 5-(2-isopropyl-4- methoxyphenoxy)pyrimidine-2, 4-diamine form 1. An XRPD pattern of the 5-(2-isopropyl-4- methoxyphenoxy)pyrimidine-2, 4-diamine form 1 is shown in Figure 5.In one embodiment, 5-(2-isopropyl-4-methoxyphenoxy)pyrimidine-2, 4-diamineNMP solvate 1 is obtained by adding excess amount of 5-(2-isopropyl-4- methoxyphenoxy)pyrimidine-2, 4-diamine form 1 into NMP in a closed vessel to form a suspension. The suspension is stirred at RT until the completion of form transition. The crystals of 5-(2 -isopropyl -4-methoxyphenoxy)pyrimidine-2, 4-diamine NMP solvate 1 can be collected by filtration and measured immediately by XRPD to prevent desolvation. An XRPD pattern of the 5-(2 -isopropyl -4-methoxyphenoxy)pyrimidine-2, 4-diamine NMP solvate 1 is shown in Figure 6.Step 5. Preparation of Compound A Free BaseCompound A free base is obtained at about 91% yield by a process comprising the steps:

To a suspension of 5-(2 -isopropyl -4-methoxyphenoxy)pyrimidine-2, 4-diamine, the diaminopyrimidine, (47.0 g, 171 mmol) in 141 mL of acetonitrile at -10 °C was added chlorosulfonic acid (63.1 mL, 942 mmol) while maintaining the internal temperature below 25 °C. The solution was aged for 1 h at 25 °C and then heated to 45 °C for 12 h. The solution was allowed to cool to 20 °C and added to a solution of 235 mL ammonium hydroxide and 71 mL of acetonitrile at -10 °C while maintaining the internal temperature below 15 °C. The slurry was aged at l0°C for 1 h, heated to 25 °C, and aged for 1 h. The slurry was diluted with 564 mL of water and 188 mL of 50 wt% sodium hydroxide to provide a homogeneous solution that was heated to 35 °C for 2 h. The solution was allowed to cool to 22 °C and the pH of the solution was adjusted to 12.9 with a 2M solution of citric acid. The solution was seeded with Compound A free base (470 mg, 1.19 mmol) (a seed can be synthesized by a route described in U.S. Patent 7,741,484), aged for 2 h, acidified to pH 10.5-11.3 with a 2M solution of citric acid over 5-10 h, and then aged for 2 h. The slurry was filtered, the resulting cake was washed with 90: 10 water: acetonitrile (2 x 118 mL) and water (2 x 235 mL), and dried at 55 °C under vacuum to provide Compound A free base (50.9 g, 91%) as a solid. 1H NMR (500 MHz, DMSO-i¾) d 7.36 (s, 1H), 7.07 (s, 1H), 7.05 – 6.89 (m, 3H), 6.37 (s, 2H), 5.85 (s, 2H), 3.89 (s, 3H), 3.41 (hept, J = 6.6 Hz, 1H), 1.27 (d, J= 6.8 Hz, 6H).The crystallization of step 5 generates anhydrous Compound A free base form 1. In one embodiment, Compound A free base acetonitrile solvate 1 can be prepared by adding excess amount of Compound A free base form 1 into acetonitrile in a closed vessel to form a suspension. The suspension is stirred at 50 °C until the completion of form transition.The crystals of Compound A free base acetonitrile solvate 1 can be collected by filtration and measured immediately by XRPD to prevent desolvation. An XRPD pattern of Compound A free base acetonitrile solvate 1 is shown in Figure 7.Step 6a. Preparation of Compound A Citrate SaltCompound A citrate salt is obtained by a process comprising the steps:

Compound A free base (30.0 g, 84.9 mmol) and glycolic acid (22.6 g, 297 mmol) were added to methanol (360 mL). The solution was heated to 60 °C, aged for 1 h, and filtered through a 0.6 pm filter into a clean vessel. A solution of citric acid (32.6 g, 170 mmol) in 2- propanol (180 mL) at RT was filtered through a 0.6 pm filter into the methanol solution over 30 min while the temperature of the methanol solution was maintained between 58-62 °C. The solution was seeded with Compound A citrate salt (450 mg, 0.825 mmol) (a seed can be synthesized by a route described in patent application number PCT/US17/66562), aged for 1 h, and diluted with 180 mL of 2-propanol over 3 h while the temperature was maintained between 58-62 °C. The slurry was cooled to 50 °C over 3 h. The slurry was filtered at 50 °C, washed with 1 : 1 methanol :2-propanol (120 mL) and 2-propanol (120 mL) at 50 °C, and dried under vacuum at 35 °C to provide Compound A citrate salt (45.1 g, 97%) as a solid. 1H NMR (400 MHz, DMSO-76) d 10.89 (s, 3H), 7.33 (s, 1H), 7.10 (s, 1H), 7.07 (s, 3H), 7.04 (s, 2H), 6.44 (s, 2H), 3.91 (s, 3H), 3.34 (hept, J= 6.7 Hz, 1H), 2.69 (d, 7= 15.3 Hz, 2H), 2.60 (d, 7= 15.3 Hz, 2H), 1.26 (d, 7= 6.9 Hz, 6H). Step 6b. Alternative preparation of Compound A Citrate SaltAlternatively, Compound A citrate salt is obtained by a process comprising the steps:

To a suspension of Compound A citrate salt (4.5 g, 8.25 mmol) in methanol (72 mL) and 2-propanol (36 mL) at 50 °C were added simultaneously through separate 0.6 pm filters a solution of Compound A free base (30.0 g, 84.9 mmol) and glycolic acid (22.6 g, 297 mmol) in 360 mL of methanol at 50 °C and a solution of citric acid (19.5 g, 101 mmol) in 180 mL of 2- propanol at 25 °C over 8 h while maintaining the seed solution temperature of 60 °C. After the simultaneous addition is complete, citric acid (13.2 g, 68.7 mmol) in 180 mL of 2-propanol was added to the slurry over 8 h while the temperature was maintained at 60 °C. The slurry was allowed to cool to 50 °C and aged for 1 h, filtered at 50 °C, washed with 1 : 1 methanol :2- propanol (2 x 120 mL) and 2-propanol (120 mL), and dried under vacuum at 35 °C to provide Compound A citrate salt (45.1 g, 88%) as a solid.The crystallization of step 6a/6b generates anhydrous Compound A citrate form 1. In another embodiment, Compound A citrate methanol solvate 1 can be prepared via a saturated solution of Compound A citrate form 1 in methanol at 50C. The solution is naturally cooled to ambient temperature or evaporated at ambient temperature until the crystals of Compound A citrate methanol solvate 1 can be acquired. An XRPD pattern of Compound A citrate methanol solvate 1 is shown in Figure 8.
PATENT
https://patents.google.com/patent/CN111635368B/enPreparation of the Compound Gefapixant of example 11Adding compound 7(16g) and dichloromethane (64mL) into a 250mL three-necked bottle, stirring for dissolving, cooling to below 5 ℃ in an ice bath, dropwise adding a mixed solution of chlorosulfonic acid (21.1g) and dichloromethane (16mL) into the reaction solution, and stirring for 1 hour at the temperature of not higher than 5 ℃; then heating to room temperature and continuing stirring for 10 hours, after the reaction is finished, pouring the reaction liquid into ice water, and quickly separating a water layer; the organic layer was washed once with ice water, dried over anhydrous magnesium sulfate and concentrated under reduced pressure to give a crude product. Dissolving the crude product with 30ml of acetonitrile, and cooling to below 5 ℃; 16ml of ammonia water (25-28%) is dripped into the solution, and after the dripping is finished, the solution is heated to room temperature and stirred for 20 hours. After the reaction is completed, concentrating the reaction solution under reduced pressure to remove acetonitrile, and separating out a white solid; and filtering again, and drying the filter cake at 70 ℃ under reduced pressure for 24h to obtain Gefapixant: white powder (19.50g), yield 94.6%, purity: 97.2 percent.Example 12 purification of the Compound GefapixantAdding a compound Gefapixant (20.77g) into a 500mL reaction bottle, adding 0.44N hydrochloric acid (95.4mL), absolute ethyl alcohol (64.4g) and nitrogen protection, heating to 75 ℃, stirring for dissolving, then carrying out heat preservation and reflux for 1 hour, filtering while hot, after filtering, heating the filtrate again to 60 ℃, dropwise adding ammonia water (25-28 percent and 2.96mL), closing and heating after dropwise adding, slowly cooling to room temperature, and gradually precipitating white solids. And continuously cooling the reaction solution to 20 ℃, keeping the temperature and stirring for 4h, filtering, washing a filter cake with 15ml of water, and performing vacuum drying on the obtained wet product at 60 ℃ for 24h to obtain Gefapixant: white powder (6.58g), yield 53.2%, purity: 99.5 percent.1H NMR(400MHz,DMSO)δ7.37(s,1H),7.08(s,1H),7.02(s,2H),7.00(s,1H),6.43(brs,2H),5.89(s,2H),3.90(s,3H),3.42(m,1H),1.28(d,J=8.0Hz,6H);LC-MS:m/z=354.1[M+H]+。
//////////////////////////////////////////

AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
References
- ^ Muccino D, Green S (June 2019). “Update on the clinical development of gefapixant, a P2X3 receptor antagonist for the treatment of refractory chronic cough”. Pulmonary Pharmacology & Therapeutics. 56: 75–78. doi:10.1016/j.pupt.2019.03.006. PMID 30880151.
- ^ Richards D, Gever JR, Ford AP, Fountain SJ (July 2019). “Action of MK-7264 (gefapixant) at human P2X3 and P2X2/3 receptors and in vivo efficacy in models of sensitisation”. British Journal of Pharmacology. 176 (13): 2279–2291. doi:10.1111/bph.14677. PMC 6555852. PMID 30927255.
- ^ Marucci G, Dal Ben D, Buccioni M, Martí Navia A, Spinaci A, Volpini R, Lambertucci C (December 2019). “Update on novel purinergic P2X3 and P2X2/3 receptor antagonists and their potential therapeutic applications”. Expert Opinion on Therapeutic Patents. 29 (12): 943–963. doi:10.1080/13543776.2019.1693542. hdl:11581/435751. PMID 31726893. S2CID 208037373.
- ^ Ford, Anthony P.; Dillon, Michael P.; Kitt, Michael M.; Gever, Joel R. (November 2021). “The discovery and development of gefapixant”. Autonomic Neuroscience. 235: 102859. doi:10.1016/j.autneu.2021.102859.
| Clinical data | |
|---|---|
| ATC code | R05DB29 (WHO) |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 1015787-98-0 |
| PubChem CID | 24764487 |
| DrugBank | DB15097 |
| ChemSpider | 58828660 |
| UNII | 6K6L7E3F1L |
| KEGG | D11349 |
| ChEMBL | ChEMBL3716057 |
| Chemical and physical data | |
| Formula | C14H19N5O4S |
| Molar mass | 353.40 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI |
////////////Gefapixant, Lyfnua, JAPAN 2022, APPROVALS 2022, ゲーファピキサントクエン酸塩 , MK 7264, 吉法匹生 , AF 217
