Image may be NSFW.
Clik here to view.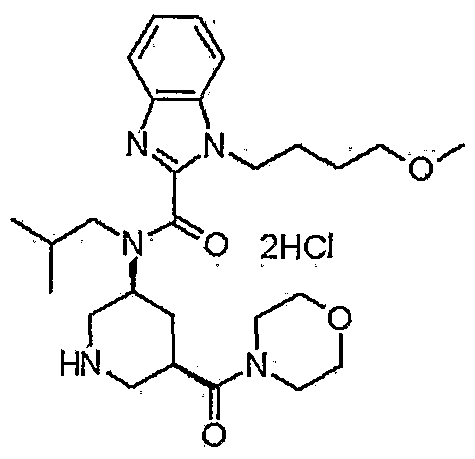
TAK 272
C27 H41 N5 O4 . Cl H, 536.106
Takeda Pharmaceutical Company Limited, INNOVATOR
1-(4-methoxybutyl)-N-(2-methylpropyl)-N-[(3S,5R)-5-(morpholin-4-ylcarbonyl)-piperidin-3-yl]-1H-benzimidazole-2-carboxamide
1- (4-methoxybutyl) -N- (2-methylpropyl) -N- [ (3S, 5R) -5- (morpholin-4-ylcarbonyl) piperidin-3-yl] -lH-benzimidazole-2-carboxamide dihydrochloride
N-Isobutyl-1-(4-methoxybutyl)-N-[5(R)-(morpholin-4-ylcarbonyl)piperidin-3(S)-yl]-1H-benzimidazole-2-carboxamide hydrochloride
1- (4-methoxybutyl) -N- (2- methylpropyl) -N – [(3S, 5R) -5- (morpholin-4-ylcarbonyl) piperidine-3 – yl] -1H- benzimidazole-2-carboxamide hydrochloride,
Image may be NSFW.
Clik here to view.![]()
The compound is used as renin inhibitor for treating diabetic nephropathy and hypertension
Takeda’s TAK-272, was reported to be in phase II in October 2015), an oral renin inhibitor, for treating diabetic nephropathy and hypertension
- 01 Apr 2015Takeda completes a phase I drug-drug interaction trial in Healthy volunteers in Japan (NCT02370615)
- 18 Feb 2015Takeda plans a phase I drug-drug interaction trial in Healthy volunteers in Japan (NCT02370615)
- 13 Feb 2015Takeda plans a phase I pharmacokinetics trial in Renal or Hepatic impairment patients in Japan (NCT02367872)
Image may be NSFW.
Clik here to view.
In the above method, the acid anhydride (BANC) from chiral dicarboxylic acid monoester ((-) – BMPA) were synthesized and then the carboxylic acid after conversion and hydrolysis reaction of the Z amine by the Curtius rearrangement of the carboxylic acid (BAPC) and it was then performs amidation by the condensation reaction with the amine (morpholine), is synthesized heterocyclic amide compound (BMPC). Further, Patent Document 2, the preparation of compounds useful as synthetic intermediates of the above heterocyclic compounds are disclosed.
Image may be NSFW.
Clik here to view.
(Wherein each symbol is as described in Patent Document 2.)
Patent literature
Patent Document 2: International Publication No. 2007/077005
WO2009154300
https://www.google.co.in/patents/WO2009154300A2?cl=en
Reference Example 31 tert-butyl (3S,5R)-3-[{ [1- (4-methoxybutyl) -lH-benzimidazol-2- yl] carbonyl} (2-methylpropyl) amino] -5- (morpholin-4- ylcarbonyl)piperidine-l-carboxylate and 1- (4-methoxybutyl) -N-
(2-methylpropyl) -N- [ (3S, 5R) -5- (morpholin-4- ylcarbonyl)piperidin-3-yl]-lH-benzimidazole-2-carboxamide

tert-Butyl (3S, 5R) -3-{ [ ( {2- [ (4- methoxybutyl) amino] phenyl}amino) (oxo) acetyl] (2- methylpropyl) amino} -5- (morpholin-4-ylcarbonyl) piperidine-1- carboxylate (9.11 g) was dissolved in acetic acid (50 ml), and the mixture was stirred at 8O0C for 15 hr. The reaction mixture was cooled to room temperature and concentrated under reduced pressure, the residue was diluted with aqueous sodium bicarbonate, and the mixture was extracted with ethyl acetate. The extract was washed with saturated brine, and dried over anhydrous magnesium sulfate. The solvent was evaporated under reduced pressure. The residue was subjected to basic silica gel column chromatography, and a fraction eluted with ethyl acetate was concentrated under reduced pressure to give tert- butyl (3S, 5R) -3- [ { [1- (4-methoxybutyl) -lH-benzimidazol-2- yl] carbonyl } (2-methylpropyl) amino] -5- (morpholin-4- ylcarbonyl)piperidine-l-carboxylate (5.85 g) , and a fraction eluted with ethyl acetate-methanol (85:15) was concentrated under reduced pressure to give 1- (4-methoxybutyl) -N- (2- methylpropyl) -N- [ (3S, 5R) -5- (morpholin-4-ylcarbonyl) piperidin- 3-yl] -lH-benzimidazole-2-carboxamide (580 mg) . [0424] tert-butyl (3S,5R)-3-[{ [1- (4-methoxybutyl) -lH-benzimidazol-2- yl] carbonyl} (2-methylpropyl) amino] -5- (morpholin-4- ylcarbonyl ) piperidine-1-carboxylate 1H-NMR (CDCl3) δ 0.63-0.80 (2H, m) , 0.89-1.07 (4H, m) , 1.41- 1.59 (9H, m) , 1.59-1.80 (2H, m) , 1.87-2.23 (4H, m) , 2.30-2.98 (3H, m) , 3.21-3. 46 ( 6H, m) , 3.49-3. 91 (1OH, m) , 3. 95-4 . 47 (5H, m) , 7 . 18-7 . 51 (3H, m) , 7. 56-7 . 84 ( IH, m) .
MS (ESI+, m/e) 600 (M+l )
1- (4-methoxybutyl) -N- (2-methylpropyl) -N- [ (3S, 5R) -5- (morpholin- 4-ylcarbonyl)piperidin-3-yl] -lH-benzimidazole-2-carboxamide BASE
1H-NMR (CDCl3) δ 0.64-0.74 (2H, m) , 0.95-1.07 (4H, m) , 1.43-
1.74 (3H, m) , 1.84-2.41 (4H, m) , 2.48-2.67 (IH, m) , 2.67-3.01
(3H, m), 3.03-3.44 (8H, m) , 3.47-3.78 (9H, m) , 4.06-4.46 (3H, m) , 7.28-7.47 (3H, m) , 7.62-7.81 (IH, m) . MS (ESI+, m/e) 500 (M+l)
Example 10
1- (4-methoxybutyl) -N- (2-methylpropyl) -N- [ (3S, 5R) -5- (morpholin-
4-ylcarbonyl) piperidin-3-yl] -lH-benzimidazole-2-carboxamide dihydrochloride
tert-Butyl (3S,5R)-3-[{ [1- (4-methoxybutyl) -IH- benzimidazol-2-yl] carbonyl} (2-methylpropyl) amino] -5-
(morpholin-4-ylcarbonyl)piperidine-l-carboxylate (5.85 g) was dissolved in methanol (20 ml) , 4M hydrogen chloride-ethyl acetate (20 ml) was added, and the mixture was stirred at room temperature for 15 hr. The reaction mixture was concentrated, and the residue was diluted with aqueous sodium bicarbonate, and the mixture was extracted with ethyl acetate. The extract was washed with saturated brine, and dried over anhydrous magnesium sulfate. The solvent was evaporated under reduced pressure. The residue was subjected to basic silica gel column chromatography, and a fraction eluted with ethyl acetate- methanol (9:1) was concentrated under reduced pressure to give 1- (4-methoxybutyl) -N- (2-methylpropyl) -N- [ (3S, 5R) -5- (morpholin- 4-ylcarbonyl) piperidin-3-yl] -lH-benzimidazole-2-carboxamide (4.40 g) . The obtained 1- (4-methoxybutyl) -N- (2-methylpropyl) – N- [ (3S, 5R) -5- (morpholin-4-ylcarbonyl) piperidin-3-yl] -IH- benzimidazole-2-carboxamide (2.20 g) was dissolved in ethyl acetate (20 ml) , 4M hydrogen chloride-ethyl acetate (5 ml) and methanol (20 ml) were added, and the mixture was stirred at room temperature for 5 min. The reaction mixture was concentrated under reduced pressure to give the object product (2.52 g).
dihydrochloride
1H-NMR (DMSO-d6) δ 0.63-0.76 (2H, m) , 0.85-1.00 (4H, m) , 1.40-
1.60 (2H, m) , 1.68-1.89 (2H, m) , 1.93-2.17 (2H, m) , 2.20-2.44
(2H, m) , 2.81-3.81 (2OH, m) , 4.19-4.39 (3H, m) , 7.23-7.46 (2H, m) , 7.57-7.81 (2H, m) , 8.38-9.77 (2H, m) .
MS (ESI+, m/e) 500 (M+l)
Example 252
1- ( 4-methoxybutyl ) -N- ( 2-methylpropyl ) -N- [ ( 3S 1. 5R) -5- (morpholin- 4-ylcarbonyl ) piperidin-3-yl ] -lH-benzimidazole-2-carboxamide methanesulfonate

l-(4-Methoxybutyl) -N- (2-methylpropyl) -N- [ (3S,5R)-5- (morpholin-4-ylcarbonyl) piperidin-3-yl] -lH-benzimidazole-2- carboxamide (208 mg) was dissolved in ethyl acetate (2 ml) , a solution of methanesulfonic acid (40 μl) in ethyl acetate (1 ml) was added at 75°C, hexane (1 ml) was added, and the mixture was heated under reflux and stood at room temperature overnight. The precipitated crystals were collected by filtration, and dried at 7O0C for 3 hr to give the object product (158 mg) . MS (ESI+, m/e) 500 (M+l) melting point : 144.40C
EXTRAS IF REQD .………….
Example 32
methyl (3R, 5S)-5-[{ [1- (4-methoxybutyl) -lH-benzimidazol-2- yl] carbonyl} (2-methylpropyl) amino] piperidine-3-carboxylate dihydrochloride [0675]

MS (ESI+, m/e) 445 (M+l)
Example 33
(3R, 5S) -5- [ { [1- (4-methoxybutyl) -lH-benzimidazol-2- yljcarbonyl} (2-methylpropyl) amino] piperidine-3-carboxylic acid dihydrochloride

MS (ESI+, m/e) 431 (M+l)
Reference Example 29
{ [ ( 3S , 5R) -1- (tert-butoxycarbonyl ) -5- (morpholin-4- ylcarbonyl ) piperidin-3~yl ] ( 2-itιethylpropyl ) amino } (oxo ) acetic acid

To a solution of tert-butyl (3S,5R)~3-{ [ethoxy (oxo) acetyl] (2-methylpropyl) amino}-5- (morpholin-4- ylcarbonyl) piperidine-1-carboxylate (10.3 g) in ethanol (40 ml) was added 2M aqueous sodium hydroxide solution (22 ml) , and the mixture was stirred at room temperature for 6 hr. The reaction mixture was adjusted to pH 7 with IM hydrochloric acid, and extracted with ethyl acetate. The extract was washed with saturated brine, and dried over anhydrous magnesium sulfate. The solvent was evaporated under reduced pressure to give the object product (10.3 g) .
1H-NMR (CDCl3) δ 0.78-0.99 (6H, m) , 1.37-1.52 (9H, m) , 1.79- 2.16 (3H, m) , 2.38-3.86 (14H, m) , 3.93-4.43 (2H, m) . MS (ESI+, m/e) 442 (M+l)
Reference Example 28
tert-butyl (3S, 5R) -3-{ [ethoxy (oxo) acetyl] (2- methylpropyl ) amino } -5- (morpholin-4-ylcarbonyl) piperidine-1- carboxylate

To a solution of tert-butyl (3S, 5R) -3- [ (2- methylpropyl) amino] -5- (morpholin-4-ylcarbonyl) piperidine-1- carboxylate (9.24 g) and diisopropylethylamine (10.5 ml) in DMA (100 ml) was added dropwise ethyl chloroglyoxylate (3.4 ml) at 0°C. The reaction mixture was stirred at room temperature for 15 hr, and the reaction mixture was concentrated. An aqueous sodium bicarbonate solution was added to the residue, and the mixture was extracted with ethyl acetate. The extract was washed with saturated brine, and dried over anhydrous magnesium sulfate. The solvent was evaporated under reduced pressure. The residue was subjected to silica gel column chromatography, and a fraction eluted with ethyl acetate was concentrated under reduced pressure to give the object product (10.3 g) . 1H-NMR (CDCl3) δ 0.84-1.00 (6H, m) , 1.37 (3H, q) , 1.42-1.53 (9H, m) , 1.80-2.19 (3H, m) , 2.26-2.42 (IH, m) , 2.59-2.96 (IH, in) , 2.97-3.30 (3H, m) , 3.37-3.92 (9H, m) , 4.01-4.26 (2H, m) , 4.26- 4.40 (2H, m) . MS (ESI4-, m/e) 470 (M+l) “
Reference Example 22 tert-butyl (3S, 5R) -3- [ (2-methylpropyl) amino] -5- (morpholin-4- ylcarbonyl)piperidine-l-carboxylate

[0369] tert-Butyl (3S,5R)-3-{ [ (benzyloxy) carbonyl] aminoJ-5- (morpholin-4-ylcarbonyl)piperidine-l-carboxylate (58 g) and palladium (II) hydroxide-carbon (5 g) were suspended in methanol (400 ml) and the mixture was stirred under a hydrogen atmosphere (1 atom) at room temperature for 16 hr. The palladium catalyst was filtered off, and the filtrate was concentrated under reduced pressure. The obtained residue and acetic acid (8.8 ml) were dissolved in methanol (400 ml), 2- methylpropanal (14.0 ml) was added, and the mixture was stirred at room temperature for 1 hr. Sodium triacetoxyborohydride (40.4 g) was added to the reaction mixture, and the mixture was stirred at room temperature for 2 hr. The reaction mixture was concentrated under reduced pressure, and the concentrate was basified with 3.5M aqueous potassium carbonate solution, and the mixture was extracted with ethyl acetate. The extract was washed with saturated brine, and dried over anhydrous magnesium sulfate. The solvent was evaporated under reduced pressure. The residue was subjected to basic silica gel column chromatography, and a fraction eluted with ethyl acetate-hexane (1:5) – ethyl acetate-hexane (1:1) was concentrated under reduced pressure to give the object product (33.3 g) .
1H-NMR (CDCl3) δ: 0.90 (6H, d) , 1.46 (9H, s) , 1.54 (IH, d) , 1.69 (IH, dt), 1.96-2.12 (2H, m) , 2.23-2.37 (IH, m) , 2.47 (3H, d) , 2.66 (IH, d) , 3.61 (IH, br s) , 3.55 (2H, d) , 3.69 (5H, ddd) , 4.01-4.46 (2H, m) .
PATENT
WO2013122260
http://www.google.co.in/patents/WO2013122260A1?cl=en
PATENT
WO 2011158880
http://www.google.co.in/patents/WO2011158880A1?cl=en
Reference Example 1
1- (4-methoxybutyl) -N- (2- methylpropyl) -N – [(3S, 5R) -5- (morpholin-4-ylcarbonyl) piperidin-3-yl] -1H- benzimidazole -2 – carboxamide hydrochloride (A-type crystal)
tert- butyl (3S, 5R) -3 – [{[1- (4- methoxy-butyl) -1H- benzimidazol-2-yl] carbonyl} (2-methylpropyl) amino] -5- (morpholin-4- ylcarbonyl) was suspended dissolved piperidine-1-carboxylate The (300g) in 3N- hydrochloric acid water (1200mL) and Ethyl acetate (60mL), and stirred over 3 h at 25 ~ 35 ℃. After completion of the reaction, it was added ethyl acetate (2400mL) in the same temperature. After the addition, it was added 25% aqueous ammonia (600mL) with cooling. After the addition stirring and extracted the organic layer of 5% aqueous ammonia (600mL) was added and stirred. After stirring, the resulting organic layer it was concentrated until the solvent no longer distilled off. After concentrated, dissolved with ethyl acetate (1500mL), and transferred to solution to the crystallizer vessel, and washed with ethyl acetate (750mL). After washing, it was raised in stirring under 45 ~ 55 ℃. After raising the temperature, at the same temperature 4N- hydrogen chloride – it was dropped ethyl acetate (131.3mL). After dropping, it was to dissolve the precipitate at the same temperature. After dissolution confirmation, it was added heptane (750mL) at 40 ~ 50 ℃, after the addition, then cooled to 25 ~ 35 ℃. After cooling, the addition of A-type crystals of the seed crystals (300mg) which was obtained according to the method described in Example 265 of WO2009 / 154300, and stirred for 30 minutes or more. After stirring, the temperature was raised to 40 ~ 45 ℃, it was dropped heptane (1500mL). After the completion of the dropping, it was stirred at the same temperature. Then gradually cooled to 5 ℃ below, followed by stirring at the same temperature for 1 hour. After stirring, ethyl acetate and filtered crystals – heptane: washed with (1 1,600mL), to obtain a wet crystal. The obtained wet crystals dried under reduced pressure at 50 ℃, 1- (4- methoxybutyl) -N- (2- methylpropyl) -N – [(3S, 5R) -5- (morpholin-4-yl carbonyl) piperidin-3-yl] -1H- obtained a crystalline powder of benzimidazole-2-carboxamide hydrochloride (A-type crystal, 198.82g, 74.1% yield). FINAL PRODUCT
TERT BUTYL DERIVATIVE, N-1
Reference Example 4
tert- butyl (3S, 5R) -3 – [{[1- (4- methoxy-butyl) -1H- benzoimidazol-2-yl] carbonyl} (2-methylpropyl) amino] -5- (morpholin-4- ylcarbonyl) piperidine-1-carboxylate 1)
o- nitro aniline (50.0g, 0.362mol), tetrabutylammonium bromide (58.3g, 0.181mol), potassium bromide (43.1g, 0.362mol) in toluene (500mL ) and it was added. At a temperature of 20 ~ 30 ℃ 1- chloro-4-methoxy-butane (66.6g, 0.543mol) and, I was added to 50w / v% sodium hydroxide solution (145mL, 1.81mol). The reaction was heated to a temperature 85 ~ 95 ℃, and stirred for 6 hours. After cooling to a temperature 20 ~ 30 ℃, the reaction mixture water (250mL), 1N- aqueous hydrochloric acid (250mL × 2), 5w / v% aqueous solution of sodium bicarbonate (250mL), it was washed successively with water (250mL). After concentration under reduced pressure the organic layer to Contents (250mL), was added toluene (100mL), was obtained
N- (4- methoxy-butyl) -2-nitroaniline in toluene (350mL, 100% yield).
1 H-NMR (300MHz, CDCl 3) δ 1.64-1.89 (m, 4H), 3.25-3.39 (m, 2H), 3.35 (s, 3H), 3.44 (t, J = 6.1 Hz, 2H), 6.63 ( ddd, J = 8.5, 6.9, 1.2 Hz, 1H), 6.86 (dd, J = 8.5, 1.2 Hz, 1H), 7.43 (ddd, J = 8.5, 6.9, 1.5 Hz, 1H), 8.07 (br s, 1H ), 8.17 (dd, J = 8.5, 1.5 Hz, 1H).
2) N- (4-methoxy-butyl) -2-10 percent in nitroaniline of toluene solution (350mL) Pd / C (K-type, 50% water-containing product) (10.0g) and toluene (100mL) it was added. Hydrogen pressure of 0.1MPa, it was stirred for 3 hours at a temperature of 20 ~ 30 ℃. A stream of nitrogen, the catalyst was filtered, I was washed with toluene (100mL). After the water in the filtrate was separated off and adding magnesium sulfate (25.0g) at a temperature 20 ~ 30 ℃, and stirred at the same temperature for 30 minutes. Filtered over magnesium sulfate, washed with toluene (100mL), was obtained N- (4- methoxybutyl) -o- toluene solution of phenylenediamine (100% yield).
1 H NMR (500 MHz, CDCl 3) δ1.67-1.78 (m, 4H), 3.12-3.14 (m, 2H), 3.32 (br, 3H), 3.35 (s, 3H), 3.41-3.47 (m, 2H), 6.63-6.69 (m, 2H), 6.69-6.74 (m, 1H), 6.82 (td, J = 7.57, 1.58 Hz, 1H).
3) N- (4- methoxy-butyl) -o- After the toluene solution of phenylenediamine cooled to a temperature 0 ~ 10 ℃, acetic acid (65.2g, 1.09mol) and 2,2,2 trichloroacetimide acid methyl ( 70.3g, 0.398mol) and I were added. After stirring for 30 minutes at a temperature 0 ~ 10 ℃, it was stirred for 3 hours at a temperature of 20 ~ 30 ℃. The reaction was 5w / v% saline (250mL), 2N- aqueous hydrochloric acid / 5w / v% sodium chloride solution: a mixture of (1 1) (250mL × 2), 5w / v% aqueous solution of sodium bicarbonate (250mL), 5w / v It was washed successively with% saline solution (250mL). A stream of nitrogen, was added magnesium sulfate (25.0g) to the organic layer at a temperature 20 ~ 30 ℃, and stirred at the same temperature for 30 minutes. Filtered magnesium sulfate, and washed with toluene (100mL). The filtrate was concentrated under reduced pressure and the amount of contents (150mL). Stir the concentrated solution at a temperature 20 ~ 30 ℃, was allowed to precipitate crystals, was added dropwise heptane (750mL). The crystals bleeding is heated to a temperature 40 ~ 50 ℃, after stirring for 30 min, cooled to a temperature 0 ~ 10 ℃, and the mixture was stirred at the same temperature for 2 hours.The precipitated crystals were collected by filtration, toluene – heptane: was washed with (1 5,150 mL). And dried under reduced pressure at 40 ℃, it was obtained 1- (4-methoxy-butyl) -2-fine brown crystals of trichloromethyl -1H- benzimidazole (96.5g, 82.9% yield from o- nitroaniline).
1 H-NMR (300MHz, CDCl 3) δ: 1.68-1.85 (m, 2H), 1.99-2.17 (m, 2H), 3.37 (s, 3H), 3.48 (t, J = 6.1 Hz, 2H), 4.50 -4.65 (m, 2H), 7.27-7.49 (m, 4H), 7.82-7.93 (m, 1H).
. Anal Calcd for C 13 H 15 Cl 3 N 2 O:. C, 48.55; H, 4.70; N, 8.71; Cl, 33.07 Found: C, 48.30; H, 4.61; N, 8.74; Cl, 33.30.
4) pyridine-3,5-dicarboxylic acid (110g, 0.66mol), it was dropped methanol (660 mL) mixture of concentrated sulfuric acid at a temperature of 50 ℃ or less of (226.0g, 2.30mol). Thereafter, the mixture was stirred and heated to a temperature 55 ~ 65 ℃ 7 hours. The reaction was the temperature 40 ~ 50 ℃, was added water (220mL). And further dropping temperature 40-50 5% aqueous ammonia at ℃ (about 1.10L) was adjusted to pH8.0 ~ 8.5. After stirring at a temperature 40 ~ 50 ℃ 30 minutes and stirred for 1 hour and cooled to a temperature 0 ~ 10 ℃. Was collected by filtration precipitated crystals, methanol – water (1: 3,165mL), and washed successively with water (440mL). To obtain a white crystalline powder pyridine-3,5-dicarboxylic acid dimethyl and dried under reduced pressure at 50 ℃ (105.0g, 82.0% yield).
1 H-NMR (300 MHz, CDCl 3) δ 4.00 (s, 6H), 8.87 (s, 1H), 9.37 (s, 2H).
. Anal Calcd for C 9 H 9 NO 4:. C, 55.39; H, 4.65; N, 7.18; O, 32.79 Found: C, 55.42; H, 4.65; N, 7.16.
5) 1 L autoclave pyridine-3,5-dicarboxylic acid dimethyl (100g, 0.51mol) and was charged with dimethylacetamide (400mL), temperature 30 ℃ below with trifluoroacetic acid (59.2mL, after dropping the 0.77mol), 10% Pd-C (PE-type) the (20.0g) it was added. Hydrogen pressure of 0.5 ~ 0.7MPa, it was stirred for 12 hours at a temperature of 55 ~ 65 ℃. The catalyst was filtered off, it was washed with dimethylacetamide (50mL × 2). Triethylamine and the combined filtrates at a temperature 20 ~ 30 ℃ (77.8g, 0.77mol) was added dropwise, and adjusted to pH9.0 ~ 10.0. Temperature 30 ~ 40 ℃ by di -tert- butyl (134g, 0.614mol) was added dropwise and stirred at the same temperature for 2 hours. After the reaction mixture as a 20 ~ 30 ℃, it was added ethyl acetate (600mL), washed with water (900mL). The aqueous layer it was re-extracted with ethyl acetate (400mL). The combined organic layers 5w / v% citric acid -10w / v% sodium chloride solution (600mL), 3% aqueous sodium bicarbonate (600mL), and washed successively with water (600mL). Contents The organic layer (200mL) until it was concentrated under reduced pressure, methanol (250mL) was added to the concentrated solution, and then concentrated under reduced pressure until Contents (200mL). The addition of methanol (250mL) again concentrate, After concentration under reduced pressure until Contents (200mL), was added methanol (2.40L). The solution in water (18.5g, 1.03mol), cesium carbonate (417g, 1.28mol) was added and stirred for about 24 hours at a temperature 55 ~ 65 ℃. The reaction solution was the temperature 20 ~ 30 ℃, concentrated to Contents (700mL), it was added tetrahydrofuran (500mL). The solution temperature at 15 ~ 35 ℃ 2N- hydrochloric acid solution (1.28L, 2.56mol) was added dropwise and adjusted to pH3.0 ~ 3.5, and the mixture was stirred for 30 minutes at a temperature 20 ~ 30 ℃. Extracted with ethyl acetate (750mL × 2), and the organic layer was washed with 10w / v% aqueous sodium chloride solution (500mL × 3). Contents The organic layer (300mL) until it was concentrated under reduced pressure, to obtain a weight content by adding ethyl acetate (650mL).Heating the concentrate to a temperature of 55 ~ 65 ℃, it was added dropwise heptane (500mL). It cooled to a temperature 20 ~ 30 ℃ and stirred for 1 hour. The precipitated crystals were collected by filtration, ethyl acetate – heptane: was washed with (1 1,120mL). Dried under reduced pressure at 50 ℃ 1- (tert- butoxycarbonyl) to give a white crystalline powder of piperidine-3,5-dicarboxylic acid (113.3g, 80.9% yield).
1 H-NMR (300 MHz, DMSO-d 6) δ 1.40 (s, 9H), 1.44-1.61 (m, 1H), 2.21-2.26 (m, 1H), 2.31-2.41 (m, 2H), 4.10- 4.12 (m, 2H).
. Anal Calcd for C 12 H 19 NO 6:. C, 52.74; H, 7.01; N, 5.13; O, 35.13 Found: C, 52.96; H, 6.99; N, 5.39.
6) Under a nitrogen stream, 1- (tert- butoxycarbonyl) piperidine-3,5-dicarboxylic acid (5.00g, 18.3mmol) was suspended in tetrahydrofuran (10.0mL), trifluoroacetic acid anhydride at a temperature 20 ~ 30 ℃ It was dropping things (3.80mL, 27.5mmol). After the completion of the dropping, it was stirred for 1 hour at a temperature of 20 ~ 30 ℃. It was added dropwise heptane (20.0mL) at a temperature 20 ~ 30 ℃ the reaction solution, and stirred for 3 hours then cooled to a temperature 0 ~ 10 ℃. The precipitated crystals were collected by filtration, and washed with heptane (3.00mL). Dried under reduced pressure at 40 ℃ 2,4- dioxo-3-oxa-7-azabicyclo [3,3,1] white crystalline powder of nonane-7-carboxylic acid tert- butyl was obtained (4.03g, yield 86.1%).
1 H-NMR (300 MHz, CDCl 3) δ 1.43 (s, 9H), 1.93-1.99 (m, 1H), 2.40-2.46 (m, 1H), 3.06-3.11 (m, 4H), 4.50-4.54 ( m, 2H).
. Anal Calcd for C 12 H 17 NO 5:. C, 56.46; H, 6.71; N, 5.49; O, 31.34 Found: C, 56.51; H, 6.63; N, 5.69.
7) Under a nitrogen stream, quinidine (69.9g, 0.215mol) and was charged with tetrahydrofuran (200mL), and cooled to a temperature -5 ~ 5 ℃. At the same temperature 2,4-dioxo-3-oxa-7-azabicyclo [3,3,1] nonane-7-carboxylic acid tert- butyl (50.0g, 0.196mol) was added and washed with tetrahydrofuran (50.0mL) crowded. Temperature -5 ~ 5 methanol at ℃ (9.41g, 0.29 4mol) was added dropwise, and the mixture was stirred for 2 hours at a temperature -5 ~ 5 ℃. Ethyl acetate (350mL) to the reaction mixture, was by adding minute solution 20w / v% citric acid aqueous solution (250mL). The aqueous layer it was re-extracted with ethyl acetate (125mL × 2). The organic layers were combined 20w / v% aqueous solution of citric acid (250mL), I was washed successively with water (250mL × 2). The organic layer it was concentrated under reduced pressure. To the residue ethanol (100mL) was added ethyl acetate (450mL) was heated to a temperature 60 ~ 70 ℃, (R) – was added phenethylamine (23.7g, 0.196mol). Temperature 50-60 for one hour at ℃, 1 hour at a temperature of 20 ~ 30 ℃, it was stirred for 1 hour at a temperature of -5 ~ 5 ℃. The precipitated crystals were collected by filtration, ethanol – ethyl acetate: and washed with (2 9,100mL). And dried under reduced pressure at 50 ℃ (3S, 5R) -1- (tert- butoxycarbonyl) -5- (methoxycarbonyl) piperidin-3 to give a white crystalline powder of the carboxylic acid (1R) -1- phenylethylamine salt It was (55.7g, 69.6% yield).
1 H-NMR (300 MHz, DMSO-d 6) δ 1.42 (s, 9H), 1.43-1.51 (m, 3H), 2.06-2.14 (m, 1H), 2.21-2.26 (m, 1H), 2.39- 2.44 (m, 1H), 2.52-2.53 (m, 1H), 2.57 (br s, 2H), 3.64 (s, 3H), 4.12 (br s, 2H), 4.19-4.26 (m, 1H), 7.30- 7.40 (m, 3H), 7.45-7.48 (m, 2H).
. Anal Calcd for C 21 H 32 N 2 O 6:. C, 61.75; H, 7.90; N, 6.86; O, 23.50 Found: C, 61.54; H, 7.77; N, 6.86.
8) (3S, 5R) -1- (tert- butoxycarbonyl) -5- (methoxycarbonyl) piperidine-3-carboxylic acid (1R) -1- phenylethylamine salt (20.0g, 49.0mmol), methanol (20mL) and it was charged with water (80mL). Temperature 20-30 citric acid at ℃ (11.3g, 58.8mmol) was added dropwise a solution prepared by dissolving in water (20.0mL), and the mixture was stirred 1.5 hours at the same temperature. The precipitated crystals were collected by filtration and washed with water (60mL). And dried under reduced pressure at 50 ℃ (3S, 5R) -1- (tert- butoxycarbonyl) -5- give a white crystalline powder (methoxycarbonyl) piperidine-3-carboxylic acid (13.5g, 96.1% yield ).
1 H-NMR (300 MHz, CDCl 3) δ 1.40 (s, 9H), 1.46-1.59 (m, 1H), 2.22-2.27 (m, 1H), 2.37-2.45 (m, 2H), 2.63-2.73 ( m, 2H), 3.63 (s, 3H), 4.14 (br s, 2H), 12.51 (br s, 1H).
. Anal Calcd for C 13 H 21 NO 6:. C, 54.35; H, 7.37; N, 4.88; O, 33.41 Found: C, 54.14; H, 7.28; N, 4.85.
9) Under a nitrogen stream, (3S, 5R) -1- (tert- butoxycarbonyl) -5- (methoxycarbonyl) piperidine-3-carboxylic acid (30.0g, 104mmol), triethylamine (31.7g, 313mmol) and toluene ( It was charged with 180mL). Diphenylphosphorylazide at a temperature of 15 ~ 35 ℃ (28.7g, 313mmol) I was dropped a toluene (30.0mL) solution. After stirring at a temperature 30 ± 5 ℃ 30 minutes, and the mixture was stirred and heated to a temperature 65 ~ 75 ℃ 30 minutes. Temperature 60 ~ 70 ℃ in the benzyl alcohol (12.4g, 115mmol) it was dropped. To a temperature 80 ~ 90 ℃ was stirred and heated for 3 hours. The reaction mixture was cooled to a temperature 20 ~ 30 ℃, sodium nitrite (7.20g, 104mmol) and after stirring was added a solution prepared by dissolving in water (150mL) 1 hour, the aqueous layer was separated. The organic layer 5w / v% aqueous sodium bicarbonate solution (150mL), 20w / v% aqueous citric acid solution (150mL), washed successively with 5w / v% aqueous sodium chloride solution (150mL), the organic layer was concentrated under reduced pressure. The residue methanol (60.0mL) was added and concentrated under reduced pressure to. The more we went once in the same manner.To the residue was added methanol and the content amount of the (90.0g). Temperature 15 ~ 35 ℃ 2N- aqueous sodium hydroxide (62.6mL, 125mmol) was added and stirred for 1 hour at a temperature 30 ± 5 ℃. Temperature 20 ~ 30 ℃ in methanol (120mL), was added to 20w / v% aqueous citric acid solution (300mL), it was a pH3.0 ~ 3.5. After stirring for 30 minutes at a temperature 50 ~ 60 ℃, cooled to a temperature 20 ~ 30 ℃ and stirred for 1 hour. It was stirred for 1 hour at the temperature 0 ~ 10 ℃. The precipitated crystals were collected by filtration, and washed with water (90.0mL). And dried under reduced pressure at 50 ℃ (3R, 5S) -5 – {[(benzyloxy) carbonyl] amino} -1- (tert- butoxycarbonyl) to yield a white crystalline powder piperidine-3-carboxylic acid (35.0 g, 88.6% yield).
1 H-NMR (300 MHz, DMSO-d 6) δ 1.41 (s, 9H), 2.11 (d, J = 12.4 Hz, 1H), 2.40-2.48 (m, 4H), 2.62 (br s, 1H), 4.08 (t, J = 14.4 Hz, 2H), 5.04 (s, 2H), 7.31-7.41 (m, 5H), 12.53 (br s, 1H).
. Anal Calcd for C 19 H 26 N 2 O 6:. C, 60.30; H, 6.93; N, 7.40; O, 25.37 Found: C, 60.03; H, 6.99; N, 7.41.
10) Under a nitrogen stream, (3R, 5S) -5 – {[(benzyloxy) carbonyl] amino} -1- (tert- butoxycarbonyl) piperidine-3-carboxylic acid (30.0g, 79.3mmol), morpholine (7.60 g, 87.2mmol), 1- hydroxybenzotriazole monohydrate (2.43g, it was charged with 15.9mmol) and dimethylacetamide (90.0mL). Hydrochloride 1-ethyl at a temperature 20 ~ 30 ℃ -3- (3- dimethylaminopropyl) carbodiimide (16.7g, 87.1mmol) after addition and stirred for 1 hour at a temperature 45 ~ 55 ℃. Temperature 45 ~ 55 ℃ with tetrahydrofuran (90.0mL), sequentially dropwise addition of water (210mL), and stirred for 1 hour. After stirring for 1 hour and cooled to a temperature 20 ~ 30 ℃, were collected by filtration the precipitated crystals, tetrahydrofuran – water: washing with (1 3,120mL). And dried under reduced pressure at 50 ℃ tert- butyl piperidine -1- (3S, 5R) -3 – a white crystalline powder of {[(benzyloxy) carbonyl] amino} -5 (morpholin-4-yl-carbonyl) carboxylate It was obtained (32.7g, 92.3% yield).
1 H-NMR (300 MHz, DMSO-d 6) δ 1.41 (s, 9H), 1.49-1.57 (m, 1H), 1.87 (d, J = 12.3 Hz, 1H), 2.43 (br s, 1H), 2.63-2.71 (m, 1H), 2.79-2.83 (m, 1H), 3.37-3.54 (m, 9H), 3.89 (d, J = 11.5 Hz, 1H), 4.06 (br s, 1H), 5.03 (s , 2H), 7.30-7.38 (m, 5H).
. Anal Calcd for C 23 H 33 N 3 O 6:. C, 61.73; H, 7.43; N, 9.39; O, 21.45 Found: C, 61.59; H, 7.50; N, 9.43.
11) tert- Butyl piperidin -1- (3S, 5R) -3 – {[(benzyloxy) carbonyl] amino} -5- (morpholin-4-ylcarbonyl) carboxylate (30.0g, 67.0mmol), isobutyraldehyde (7.25g, 101mmol), it was charged with 10% Pd-C (PE type) (1.50g) and methanol (240mL).Hydrogen pressure of 0.2 ~ 0.3MPa, it was stirred for 4 hours at a temperature of 20 ~ 30 ℃. The catalyst is filtered off and washed with methanol (60.0mL). The filtrate was concentrated under reduced pressure, ethyl acetate was added (60.0mL), and concentrated under reduced pressure again. The residue ethyl acetate was added, followed by the amount of contents (360mL). Temperature 45-55 succinate by heating to ℃ (7.90g, 67.0mmol) was added. After stirring for 1 hour at a temperature 45 ~ 55 ℃, cooled to a temperature 20 ~ 30 ℃, and stirred for 1 hour. The precipitated crystals were collected by filtration, and washed with ethyl acetate (90.0mL). And dried under reduced pressure at 50 ℃ tert- butyl (3S, 5R) -3 – [(2- methyl-propyl) amino] -5- (morpholin-4-yl-carbonyl) piperidine – 1-carboxylate white crystals of alert succinate got sex powder (30.2g, 92.5% yield).
1 H-NMR (300 MHz, D 2 O) δ 1.02 (s, 3H), 1.04 (s, 3H), 1.47 (s, 9H), 1.97-2.09 (m, 2H), 2.26-2.30 (m, 1H ), 2.55 (s, 4H), 2.99 (d, J = 7.0 Hz, 2H), 3.23 (br s, 1H), 3.39-3.45 (m, 2H), 3.53-3.80 (m, 10H), 3.82-3.93 (br s, 1H).
. Anal Calcd for C 23 H 41 N 3 O 8:. C, 56.66; H, 8.48; N, 8.62; O, 26.25 Found: C, 56.48; H, 8.46; N, 8.39.
12) tert- Butyl (3S, 5R) -3 – [(2- methylpropyl) amino] -5- (morpholin-4-ylcarbonyl) piperidine – 1 – carboxylate succinate (30.3g, 62.2mmol), acetonitrile (60.0mL) and, it was charged with water (40.0mL). Then after stirring was added potassium carbonate (34.4g, 0.249mmol) 10 minutes, 1- (4-methoxybutyl) -2-trichloromethyl -1H- benzimidazole (20.0g, 62.2mmol) was added. After stirring for 2 hours at a temperature of 70 ~ 80 ℃, it was added dimethyl sulfoxide (15.0mL), and the mixture was stirred for 6 hours at a temperature 70 ~ 80 ℃. After cooling the reaction mixture to a temperature 20 ~ 30 ℃, water (120mL), it was separated and by adding toluene (240mL). The organic layer 10w / v% sodium chloride solution (100mL), 10w / v% aqueous solution of citric acid (100mL), it was washed sequentially with 10w / v% sodium chloride solution (100mL). The organic layer of activated carbon Shirasagi A a (1.0g) was added, and the mixture was stirred for 30 minutes at a temperature 20 ~ 30 ℃. Activated carbon was filtered, washed with toluene (40.0mL), and concentrated under reduced pressure of the filtrate to 110 mL. By heating to a temperature 35 ~ 45 ℃ was added dropwise heptane (280mL). At a temperature 35 ~ 45 ℃ tert- butyl (3S, 5R) -3 – [{[1- (4- methoxy-butyl) -1H- benzoimidazol-2-yl] carbonyl} (2-methylpropyl) amino] -5 – and the mixture was stirred for 1 hour at (morpholin-4-ylcarbonyl) piperidine-1-carboxylate was added to the same temperature the crystals (10mg) of the acrylate. Heptane (140mL) was stirred and added dropwise to 30 minutes at a temperature 35 ~ 45 ℃. It was cooled to a temperature 20 ~ 30 ℃ and stirred for 2 hours. The precipitated crystals were collected by filtration, toluene – heptane: was washed with (1 5,40.0mL). And dried under reduced pressure at 50 ℃ tert- butyl (3S, 5R) -3 – [{[1- (4- methoxy-butyl) -1H- benzoimidazol-2-yl] carbonyl} (2-methylpropyl) amino] – 5- (morpholin-4-ylcarbonyl) piperidine-1-carboxylate was obtained a pale yellowish crystalline powder of alert (27.7g, 74.2% yield).
1 H-NMR (300 MHz, CDCl 3) δ 0.68-0.80 (m, 3H), 0.96-1.08 (m, 3H), 1.31 (br s, 5H), 1.49 (s, 4H), 1.61-1.71 (m , 2H), 1.71 (br s, 0.5H), 1.92-2.05 (m, 3H), 2.05-2.24 (m, 2H), 2.45 (br s, 1H), 2.60 (br s, 1H), 2.72-2.96 (m, 2H), 3.26-3.35 (m, 3H), 3.35-3.47 (m, 2H), 3.47-3.73 (m, 10H), 4.02-4.26 (m, 2H), 4.26-4.34 (m, 1H) , 4.34-4.47 (m, 0.5H), 7.25-7.29 (m, 1H), 7.29-7.41 (m, 1H), 7.41-7.53 (m, 1H), 7.64 (br s, 0.5H), 7.79 (d , J = 8.2 Hz, 0.5H).
. Anal Calcd for C 32 H 49 N 5 O 6:. C, 64.08; H, 8.23; N, 11.68; O, 16.01 Found: C, 63.82; H, 8.12; N, 11.64.
PATENT
TAKEDA PHARMACEUTICAL COMPANY LIMITED [JP/JP]; 1-1, Doshomachi 4-chome, Chuo-ku, Osaka-shi, Osaka 5410045 (JP)
Provided is a method for producing a synthetic intermediate of a heterocyclic compound having a renin inhibitory activity and effective as a prophylactic or therapeutic drug against diabetic renal disease, hypertension, and the like. A method for producing a compound represented by formula (III-1a), (III-1b), (III-1c), and/or (III-1d) [where the symbols in the formulas are as defined in the description], or a salt thereof, said method characterized in that a compound represented by formula (Ia) or (Ib) [where the symbols in the formulas are as defined in the description] or a salt thereof is reacted with a compound represented by formula (II) [where the symbols in the formula are as defined in the description] or a salt thereof in the presence of an aluminum compound and a chiral amine compound.
Image may be NSFW.
Clik here to view.
In the above method, the acid anhydride (BANC) from chiral dicarboxylic acid monoester ((-) – BMPA) were synthesized and then the carboxylic acid after conversion and hydrolysis reaction of the Z amine by the Curtius rearrangement of the carboxylic acid (BAPC) and it was then performs amidation by the condensation reaction with the amine (morpholine), is synthesized heterocyclic amide compound (BMPC). Further, Patent Document 2, the preparation of compounds useful as synthetic intermediates of the above heterocyclic compounds are disclosed.[Formula 3]
Image may be NSFW.
Clik here to view.
(Wherein each symbol is as described in Patent Document 2.)
Prior art documents
Patent literaturePatent Document 1: Patent No. 4,800,445 Patent
reaction vessel (1R, 3S) – was added to cyclohexane-1,3-dicarboxylic acid (10g) and THF (20mL), 5 It was cooled to ℃. It was added dropwise trifluoroacetic anhydride (8.19mL), and the mixture was stirred for about 1 hour. The reaction mixture was allowed to warm to room temperature, heptane (20mL) was added, up to 5 ℃ was cooled and stirred for about 30 minutes. The precipitate was filtered off, washed with heptane to give the title compound. Yield (6.7g)
reactor in THF (240ml), (3S, 5R) -1- (tert – butoxycarbonyl) -5- (morpholine-4-carbonyl) piperidine-3-carboxylic acid (20.0g), triethylamine (12.2mL) and diphenylphosphoryl azide (15.1mL) They were charged and allowed to react for 1 hour at 60 ℃, cooled to 25 ℃. After cooling the THF (60ml) and sodium trimethyl silanolate (19.7g) to charged 0 ℃ separately reaction vessel, was added dropwise to this was allowed to react before the reaction solution over about 1 hour, 0 at 0 ℃. 5 hours it was allowed to react. 0 slowly added dropwise acetic acid (40mL) at ℃, After stirring for 10 minutes, was added ethanol (60ml) and isobutyraldehyde (5.3mL) at 25 ℃, and stirred for 10 minutes. Then added sodium borohydride (1.88g), and the mixture was stirred for 30 minutes, and further addition of sodium borohydride (1.88g) at 25 ℃, and the mixture was stirred for 30 minutes. After completion of the reaction, water (100mL) was added and stirred for 10 minutes at room temperature. The organic layer was concentrated, then added dropwise slowly toluene (140ml) and 5N aqueous sodium hydroxide solution (120ml), the layers were separated. After washing and addition of aqueous 1N sodium hydroxide (100ml) the organic layer was washed 1N aqueous sodium hydroxide (100ml) was added again organic layer. The aqueous layers were combined and extracted by addition of toluene (100ml). The organic layers were combined, washed with 10w / v% aqueous sodium chloride solution (100ml), and the organic layer was concentrated. It was added ethanol (100ml), after it was concentrated under reduced pressure until about 60ml, warmed to 60 ℃ by the addition of ethyl acetate (40ml). Was added succinic acid (6.9g), After stirring for 30 minutes, it was added dropwise ethyl acetate (200ml) at 60 ℃, and stirred for 30 minutes. After stirring for 1 hour at room temperature, and the mixture was stirred for 1 hour at 0 ℃. The crystals were collected by filtration and washed with a mixture of ethyl acetate / n-heptane (6/1) (60mL). The obtained crystals at an external temperature of 50 ℃ to constant weight and then dried under reduced pressure to give the title compound as almost white crystals. Yield (22.8g)
the reaction vessel in chlorobenzene (7.5mL) and quinine (0.70g ) is added and stirred, it was added dropwise DIBAL1.0M hexane solution (2.16mL). The reaction mixture was cooled to -40 ℃, tert – butyl 2,4-dioxo-3-oxa-7-azabicyclo [3.3.1] was added nonane-7-carboxylic acid ester (0.50g), about 1 hour stirring. Was added chlorobenzene to another reaction vessel (2.5mL) and morpholine (0.17mL), the resulting solution was cooled to -40 ℃ was added dropwise to the previous reaction solution. After completion of the reaction, the mixture was separated with ethyl acetate and 10w / w% aqueous citric acid solution, and the resulting aqueous layer was re-extracted with ethyl acetate. The organic layers were combined, washed with 10w / w% saline, and concentrated to give the title compound. 1 H NMR (500 MHz, DMSO-D 6 ) delta ppm 1.41 (s, 9 H), 1.47 – 1.72 (M, 1 H), 1.89 – 2.10 (M, 1 H), 2.36 – 2.49 (M, 1 H ), 2.55 – 2.83 (m, 3 H), 3.40 – 3.50 (m, 2 H), 3.51 -.. 3.57 (m, 4 H), 3.59 (br s, 2 H), 3.83 – 4.04 (m, 1 H), 4.05 – 4.29 (m, 1 H), 12.52 (s, 1 H) optical purity of 94.3% EE <HPLC analytical conditions> column: CHIRALPAK IC (Co., Ltd. Daicel) column temperature: constant around 15 ℃ Temperature Mobile phase: A solution) 0.02 mol / L KH 2 PO 4 buffer solution (pH3.0): acetonitrile = 70: 30 B solution) 0.02 mol / L KH 2 PO 4 buffer solution (pH3.0): acetonitrile = 50 : 50 gradient program
| WO2010150840A1 | 24 Jun 2010 | 29 Dec 2010 | Dainippon Sumitomo Pharma Co., Ltd. | N-substituted-cyclic amino derivative |
| WO2011158880A1 | 15 Jun 2011 | 22 Dec 2011 | Takeda Pharmaceutical Company Limited | Crystal of amide compound |
| WO2012062687A1 * | 7 Nov 2011 | 18 May 2012 | F. Hoffmann-La Roche Ag | Triazole derivatives and their use for neurological disorders |
| WO2013122260A1 | 14 Feb 2013 | 22 Aug 2013 | Takeda Pharmaceutical Company Limited | Tablet |
| CN103221402B * | 7 Nov 2011 | 17 Jun 2015 | 霍夫曼-拉罗奇有限公司 | 三唑衍生物及其用于神经障碍的用途 |
| US8329691 | 14 Oct 2008 | 11 Dec 2012 | Takeda Pharmaceutical Company Limited | Amide compounds and use of the same |
| US8389511 | 19 Dec 2008 | 5 Mar 2013 | Dainippon Sumitomo Pharma Co., Ltd. | Bicyclic heterocyclic derivative |
| US8658639 | 24 Jun 2010 | 25 Feb 2014 | Dainippon Sumitomo Pharma Co., Ltd | N-substituted-cyclic amino derivative |
| US8742097 | 2 Nov 2011 | 3 Jun 2014 | Hoffmann-La Roche Inc. | Triazole compounds I |
| US9018374 | 15 Jun 2011 | 28 Apr 2015 | Takeda Pharmaceutical Company Limited | Crystal of amide compound |
| US9090601 | 28 Jan 2010 | 28 Jul 2015 | Millennium Pharmaceuticals, Inc. | Thiazole derivatives |
///////////TAK 272, Hypertension
Filed under: Phase2 drugs Tagged: HYPERTENTION, phase 2, SARTAN, TAK 272, TAKEDA Image may be NSFW.
Clik here to view.
 Image may be NSFW.
Image may be NSFW.Clik here to view.
 Image may be NSFW.
Image may be NSFW.Clik here to view.
 Image may be NSFW.
Image may be NSFW.Clik here to view.
 Image may be NSFW.
Image may be NSFW.Clik here to view.
 Image may be NSFW.
Image may be NSFW.Clik here to view.
 Image may be NSFW.
Image may be NSFW.Clik here to view.
 Image may be NSFW.
Image may be NSFW.Clik here to view.
