


Synthesis
Pramipexole can be synthesized from a cyclohexanone derivative by the following route:

Pramipexole synthesis:[14]
Research
Pramipexole has been evaluated for the treatment of cluster headache and to counteract problems with sexual dysfunction experienced by some users of selective serotonin reuptake inhibitor (SSRI) antidepressants.[15] Pramipexole has shown effects on pilot studies in a placebo-controlled proof of concept study in bipolar disorder.[8][16][17] It is also being investigated for the treatment of clinical depression and fibromyalgia.[18][19][20]
Paper
http://pubs.acs.org/doi/abs/10.1021/op1000989

Pramipexole is a dopamine D2 subfamily receptor agonist that is used for the treatment of Parkinson’s disease. We report here on the successful application of the Fukuyama alkylation protocol to the development of a novel and scalable process for synthesis of pramipexole and its pharmaceutically acceptable salts. The synthesis consists of converting the crucial intermediate (S)-2,6-diamino-4,5,6,7-tetrahydrobenzothiazole to (6S)-N-(2-amino-4,5,6,7-tetrahydrobenzothiazole-6-yl)-2-nitrobenzenesulfonamide, which is in turn monoalkylated to (6S)-N-(2-amino-4,5,6,7-tetrahydrobenzothiazole-6-yl)-2-nitro-N-propylbenzenesulfonamide. Deprotection of the latter yields pramipexole base, which is finally converted to a crude pramipexole dihydrochloride monohydrate with a yield of over 50% over four steps. The process allows for the telescoping of the final three steps, has high conversion rates of intermediates, offers ease of purification, and preserves high optical purity throughout all of the stages.
pramipexole dihydrochloride monohydrate 13 (315 g) with a yield of 70% (calculated from 12) and an HPLC purity of 94.4%. 1H NMR (300 MHz, DMSO-d6) δ 0.89 (t, J = 7.5 Hz, 3H), 1.62−1.75 (m, J = 7.6 Hz, 2H), 1.87−2.00 (m, 1H), 2.24−2.28 (m, 1H), 2.55−2.67 (m, 2H), 2.71−2.79 (m, 1H), 2.86−2.89 (m, 2H), 2.99−3.06 (m, 1H), 3.47 (m, 1H), 9.50 (m, 2H). 13C NMR (300 MHz, DMSO-d6) 11.1, 19.1, 20.9, 23.5, 24.8, 46.0, 52.3, 111.0, 132.9, 168.7. FT-IR (cm−1): 3150−3450 (NH2 stretching), 2700−3000 (C−H stretching), 1600−1650 (C═N stretching), 1550−1600 (heteroaromatic ring skeleton).

POSTER
A NEW, EFFICIENT AND ECONOMIC METHOD FOR PREPARATION OF PRAMIPEXOLE. |
| Roman Balicki , Agnieszka Ciesielska , Michał Odrowąż-Sypniewski |
| Pharmaceutical Research Institute (IF), Rydygiera 8, Warszawa 01-793, Poland |
| Abstract |
|
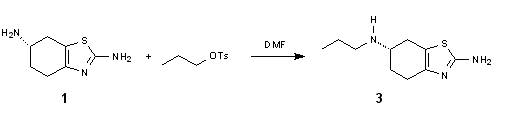
Pramipexole is a novel nonergot dopamine agonist which has high selectivity for interacting with dopamine D2 receptors. It is effective in early Parkinson,s disease as monotherapy and as adjunctive therapy with L-dopa in advanced stages of the disease. Known, two-steps method for preparation of pramipexole (3) is based on acylation reaction of diamine 1 with propionic anhydride. The obtained amide 2 is subsequently reduced using borane to give final product 3 with 65% yield. Now, we present novel, more economic and safe procedure for obtaining pramipexole. Our one-step method requires only alkylation of 1 using n-propyl tosylate. Dangerous reduction with borane is eliminated and the final compound is obtained with similar yield as in a previous method.
|
Patent
http://www.google.com/patents/US7741490
Pramipexole, of formula (A)

is a dopaminergic agonist, known from U.S. Pat. No. 4,843,086, used in the treatment of Parkinson’s disease in the form of dihydrochloride monohydrate.
US 2002/0103240 discloses inter alia a method for the resolution or the enrichment of (R,S)-2-amino-6-propylamino-4,5,6,7-tetrahydrobenzothiazole in the single (R) or (S) enantiomers, in particular in the (S) enantiomer. The same application illustrates in detail the synthetic routes known for the preparation of pramipexole, in particular those described in U.S. Pat. No. 4,886,812, EP 186087, EP 207696 and J. Med. Chem. 30. 494 (1987). From what reported it is evident that the synthetic pathways up to now available make use of synthetic steps that do not fulfill the requirements for the production of pramipexole on the industrial scale. Therefore there is the need for an improved process, which is simpler, easier to carry out and meets the requirements for the industrial production of pramipexole.
Example 13 Intermediate (VIII) Ra=H; Pramipexole Free Base
A 2 liter reactor under nitrogen is loaded with 53.3 g of, 33.0 g of (S) N-(6-propionylamino-4,5,6,7-tetrahydro-benzothiazol-2-yl)-amine, 95% sodium borohydride and 260 ml of tetrahydrofuran (THF). A solution of 98.7 g of iodine in 160 ml of THF is dropped therein in about 3 hours, keeping the temperature at approx. 20-25° C. The reaction mixture is kept under stirring for further 2 hours at about 20-25° C. The reaction mixture is poured into a solution of 60.0 g of 37% HCl in 260 ml of water. The mixture is heated to 50-55° C. and left under stirring for an hour. The complete cleavage of the boran-complexes is checked by HPLC. The mixture is added with 250 g of 50% aqueous NaOH, keeping the temperature at about 20-25° C. After that, 315 ml of toluene are added and the mixture is heated to about 30-35° C. Stirring is interrupted and the two phases are separated. The organic phase are washed, concentrated to a residue and dissolved in 420 ml of ethyl acetate.
The solution is concentrated under vacuum at a temperature below 50° C. to about 150 ml volume. The resulting suspension is refluxed, then cooled to about 10-15° C., stirred for further 1-2 hours, then filtered with suction and the precipitate is washed twice with 30 ml of ethyl acetate. The product is dried under vacuum at 40° C. 32 g of (S)-2-amino-6-propylamino-4,5,6,7-tetrahydrobenzothiazole are obtained.
PATENT
https://www.google.sc/patents/WO2008097203A1?cl=en
Example 1
Synthesis of N-(2-amino-4,5, 6, 7-tetrahydrobenzo[d]thiazole-6-yl)-2- n itrobenzenesulfonam ide

o-Nitrobenzenesulfonyl chloride (8.865 g, 40 mmol) was dissolved in 100 ml of THF and during stirring cooled to -100C (ice + salt). Then, first 3 equiv. of triethylamine (Et3N) (120 mmol, 16.8 ml) and then also 1.1 equiv. of 4,5,6,7-tetrahydrobenzo[J]thiazol-2,6-diamine (7.605 g, 45 mmol) were added. The formed suspension was, during stirring, gradually heated to room temperature and was left standing until the reaction was completed. In the course of the reaction, in addition to a soluble product, also in THF non-soluble Et3NH+Cl“ was formed, which was, at the end of the reaction, filtered off by suction and the reaction mixture was evaporated to dryness on a rotatory evaporator. The residue was poured over with H2O (300 ml), whereby on the bottom of a round-bottom flask an orange viscous liquid was obtained. After rubbing with the glass stick a yellow precipitate (N-(2- amino-4,5,6,7-tetrahydrobenzo[^thiazole-6-yl)-2-nitrobenzenesulfonamide) was formed. The precipitate was filtered off and washed with 100 ml of cold ethylether and dried. The yield of the reaction was 95%.
Example 2
Synthesis of N-(2-amino-4,5,6, 7-tetrahydrobenzo[d]thiazole-6-yl)-2-nitro-N- propylbenzenesulfonamide

Process A:
N-(2-amino-4,5,6,7-tetrahydrobenzo[rf]thiazole-6-yl)-2-nitrobenzenesulfonamide (1.770 g, 5 mmol) and K2CO3 (2.856 g, 20 mmol) were suspended in acetonitrile (40 ml) and was, during stirring, heated to 600C. Then propylbromide (1.65 ml, 18 mmol) was added and the reaction was left to run over night (the course of the reaction was followed by the use of a suitable method).
After the reaction was completed, the present precipitate was filtered off by suction. The reaction mixture was evaporated to dryness and the residue was dissolved in dichloromethane (150 ml). Organic phase was washed with IM NaOH (3 x 50 ml), saturated NaCl solution (2 x 50 ml) and dried with Na2SO4. After evaporation of dichloromethane an orange oily product N-(2-amino-4, 5,6,7- tetrahydrobenzo[^thiazole-6-yl)-2-nitro-Ν-propylbenzenesulfonamide was obtained.
Process B:
N-(2-amino-4,5,6,7-tetrahydrobenzo[rf]thiazole-6-yl)-2-nitrobenzenesulfonamide (1.770 g, 5 mmol), Cs2CO3 (3.909 g, 12 mmol) and KI (0.415 g, 2.5 mmol) was suspended in acetonitrile (40 ml) and heated to 600C. Then propyl bromide (0.9 ml, 10 mmol) was added and the course of the reaction was followed by the use of a suitable method.
The process of the isolation was the same as in the process A.
Example 3
Synthesis of lf-propyl-4,5,6, 7-tetrahydrobenzo[d]thiazole-2,6-diamine

Process A:
K2CO3 (2.073 g, 15 mmol) was suspended in 20 ml of DMF (stored above molecular sieves), thioglycolic acid (SHCH2COOH, 0.6 ml, 7.5 mmol) was added and stirred for 30 minutes. Then N-(2-amino-4,5,6,7-tetrahydrobenzo[d]thiazole-6-yl)-2-nitro-N- propylbenzenesulfonamide ( 0,99 g, 2,5 mmol), dissolved in 20 ml of DMF was added and the reaction was left to run over night. After the reaction was completed, DMF was evaporated, the residue was poured over with H2O (100 ml) and IM NaOH (200 ml). The aqueous phase was then washed with dichloromethane (3 x 80 ml) and the combined organic fractions were dried with z Na2SO4. After the evaporation of the solvent an oily residue of orange-red colour (presence of DMF is possible) was obtained.
Process B:
LiOH (0.5 g, 20 mmol) was suspended in 20 ml of DMF (stored above molecular sieves), thioglycolic acid (SHCH2COOH, 0.6 ml, 7.5 mmol) was added and stirred for 30 minutes. Then N-(2-amino-4,5,6,7-tetrahidrobenzo[cT|thiazole-6-yl)-2-nitro-N- propylbenzenesulfonamide (0.99 g, 2.5 mmol), dissolved in 20 ml of DMF was added and the reaction was left to run over night (the solution was coloured in orange-red).
After the reaction was completed, DMF was evaporated off, the residue was poured over with H2O (100 ml) and IM NaOH (200 ml). The aqueous phase was then washed with dichloromethane (3 x 80 ml) and the combined organic fractions were dried with Na2SO4. After the evaporation of the solvent an oily residue of an orange- red colour was obtained.
Example 4
Pramipexole dihydrochloride monohydrate
(S)-(-)-2-Amino-6-(N-propylamino)-4,5,6,7-tetrahydrobenzothiazole (9.15 g, 43.28 mmol) in 500 ml round-bottom flask was dissolved in 30 ml of ethanol and water (0.78 g, 43.33 mmol) was added. A solution was cooled in an ice bath to 00C and gaseous HCl^)1 was blown through whereby a white precipitate fell out. The round- bottomed flask was sealed and it was stirred over night at room temperature. The next day the precipitate was filtered off by suction and washed with a small amount of anhydrous ethanol. The precipitate was transferred into 100 ml round-bottom flask and anhydrous ethanol (50 ml) was added. The suspension was heated to 45°C and ethanol was evaporated on a rotatory evaporator. The process was repeated for another two times in order to drive out all of the excessive HCl(g). The product was recrystallized from methanol: a salt was dissolved in methanol (70 ml) at 450C, approximately 40 ml of methanol was evaporated and 20 ml of ethanol were added. It was cooled to room temperature and the resulting precipitate was filtered by suction, washed with some cooled anhydrous ethanol and dried in vacuum over P2O5 and NaOH. Yield: 11.631 g (89.01 %)
Example •§
Synthesis of N-(2-amino-4, 5, 6, 7-tetrahydrobenzo[d]thiazole-6-yl)-2- nitrobenzenesulfonamide

2-Nitrobenzenesulfonyl chloride (390 g, 1.76 mol) was dissolved in 4 1 of THF. The solution was cooled to approximately -100C. Triethylamine (Et3N) (740 g, 7.313 mol) and (6S)-4,5,6,7-tetrahydrobenzotiazole-2,6-diamine (327 g, 1.932 mol) were added. The suspension was heated during mixing to approximately 25°C and allowed to react at this temperature for approximately 1 hour.
Precipitated triethylammonium chloride (Et3NH+Cl“) was filtered off. The filtrate was concentrated to about 1/3 of the volume and water (2 1) was added. Again, approximately 1/2 of the solvent was distilled off. Water (2 1) was added, the mixture was cooled to about 25°C and mixed for about 1 hour. The precipitated product ((6S)- N-(2-amino-4,5,6,7-tetrahydrobenzotiazole-6-yl)-2-nitrobenzenesulfonamide) was separated by filtration or centrifuging.
Example g
Synthesis of (S)-(-)-2-Amino-6-(N-propylamino)-4,5,6, 7-tetrahidrobenzo[d]thiazole dihydrochloride hydrate

Potassium carbonate (1890 g, 13.675 mol), (6S)-N-(2-amino-4,5,6,7- tetrahydrobenzotiazole-6-yl)-2-nitrobenzenesulfonamide (590 g, 1.665 mol) and propyl bromide (1.09 1, 12 mol) were suspended in 4.1 1 of acetonitrile. The mixture was heated during stirring to approximately 600C and mixed at this temperature for about 12 hours. The mixture was cooled to about 25°C. Potassium bromide was removed by filtration. The solution was concentrated to about 1/4 of the volume (not exceeding 600C) and cooled to the room temperature. Methylene chloride (2 1) and 1 M NaOH water solution (2.43 1) were added and the mixture was mixed for about 30 minutes. Phases were separated and water phase was washed again with methylene chloride (1.46 1). Organic phases were collected and concentrated to about 1/10 of the volume. 0.83 1 of ethanol was added and the solution was concentrated to 1/10 of the volume. 3.35 1 of ethanol was added and ethanolic solution of (6S)-N-(2-amino- 4,5 ,6,7-tetrahydrobenzotiazole-6-yl)-2-nitro-Ν-propylbenzenesulfonamide was stored for further reaction.
Ethanol (2.35 1) and of LiOH (288 g, 12 mol) were put into the reactor and the suspension was cooled to 0 – 5°C. During about 30 minutes thioglycolic acid (SHCH2COOH) (720 g, 7.816 mol) was added (the temperature must not exceed 25°C). The suspension was heated to about 25°C and mixed for about 45 minutes. Ethanolic solution of (6S)-N-(2-amino-4,5,6,7-tetrahydrobenzotiazole-6-yl)-2-nitro-N- propylbenzenesulfonamide was added. The air in the reactor was replaced by nitrogen. The mixture was heated to about 500C and mixed at this temperature for about 4 hours. The mixture was cooled to about 25°C and filtrated. The filtrate was concentrated at 400C to about 1/4 of the volume and cooled to the ambient temperature. Methylene chloride (4.23 1) and of IM aqueous NaOH solution solution (2.53 1) were added and the mixture was mixed for about 30 minutes. Phases were separated and water phase was washed again with 4.23 1 of methylene chloride. Organic phases containing pramipexole were collected and concentrated to about 1/4 of the volume. 5 1 of ethanol was added.
To the ethanolic solution of pramipexole water was added (27.6 ml, 1.53 mol) and solution was cooled to about -100C. Gaseous HCl(g) was introduced into the solution (200 g). The temperature of the solution and later the suspension must not exceed 250C during addition of gaseous HCl(g) . After the addition the suspension was heated to about 4O0C and concentrated to 2/3 of the volume. 2.65 1 of ethanol was added and the suspension was concentrated to 1/2 of the volume. Again 3.5 1 of ethanol was added and the suspension was concentrated to 1/2 of the volume. The solution was cooled to about -15°C and the product was separated by filtration. The product was dried at 25°C and finally at 400C on air.
PATENT
http://www.google.com/patents/WO2008041240A1?cl=en
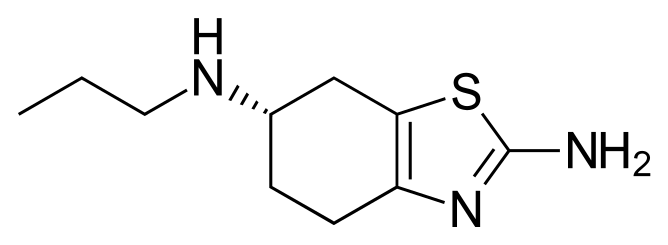
(S)-2-amino-6-propylaminio-4,5,6,7- tetrahydrobenzothiazole of formula (I), which is more commonly known as Pramipexole. Pramipexole is the commercial product marketed, in a form of a dihydrochloride salt in a peroral formulation, under several brand names e.g. Mirapexin[TM].
The compound of formula (I) has one symmetric carbon and they may exist either as a single enantiomer or in a mixed or racemic form. The pharmacological activity of compounds of formula (I) is generally connected only or mainly with one isomer thereof. Accordingly, the dopaminergic activity of the (S) isomer is twice as high as that of the (R) enantiomer.
A general process for the preparation of Pramipexole dihydrochloride has been described in US 4886812, EP 186087 and EP 207696. The process comprises the protection of amino function of 4-aminocyclohexanol to give the intermediate compound wherein, Rl is acyl or alkoxycarbonyl and R2 is hydrogen or Rl and R2 together form an amino protective group such as pthalimido group which on oxidation with an oxidising agent, followed by halogenation (preferably bromination) of protected ketone to give alpha halogenatedketone which on reaction with thiourea, followed by deprotection yielded the racemic 2,6-diaminotetrahydrobenzothiazole. Reductive alkylation of diaminotetrahydrobenzothiazole with n-propanal furnished the racemic pramipexole. Although the (S) isomer of pramipexole is mentioned therein, it is not clear at what stage the chiral resolution has been carried out. The general process steps are indicated in Scheme- Ia below.

H2N
Racemate Resolution

n-Propyl Bromide –


Scheme-la
Another process for preparing optically pure pramipexole dihydrochloride was disclosed in J. Med. Chem. 1987, 30, 494-498, wherein, racemic 2,6-diamino-4,5,6,7- tetrahydrobenzo- thiazole was resolved, using L (+) tartaric acid to give optically pure (S)-2,6-diamino-4,5,6,7-tetrahydrobenzothiazole, which was converted to optically pure pramipexole by reacting (S)- 2,6-diamino-4,5,6,7-tetrahydro benzothiazole with propionic anhydride in THF and followed by reduction with borane THF complex . The reaction steps are shown in Scheme-lb as under:

(VIII) (II)
(CH3CH2CO)2O

2HCl Scheme- Ib
However, the variants of the above general process prepare only racemate.
Thus, the synthesis of pramipexole by the above process yields R,S(±)-2~amino-6- propylamino-4,5,6,7-tetrahydrobenzothiazole. The above-mentioned acknowledge that the produced racemic compound may be resolved into single enantiomers by classical methods such as chromatography on a chiral phase or fractional crystallization of a salt with an optically active acid. However, even though the S(-)-enantiomer of pramipexole was disclosed and characterized therein, no information is provided how it was prepared; i.e. whether it was prepared by a resolution of racemic pramipexole of form some optically active precursor. Further, no information is provided on how to produce the S(-)- enantiomer of pramipexole.
WO 2006/003677 Al discloses the improved process the preparation of biologically active tetrahydrobenzothiazole derivative. The patent application discloses the process that has tried to solve the problems of prior art. However, much improvement over the prior art process has still been achieved. Moreover, the process discloses the formation of 2,6-diamino-4,5,6,7-tetrahydrobenzothiazole via an isolated bromo intermediate, which on reaction with thiourea gets converted to tetrahydrobenzothiazole. The isolation of bromo intermediate can also be avoided. The halogenation of the protected cyclohexanone derivative is performed in presence of Lewis acid catalysts like AICI3, ZnCl2 or SnCl2 etc. which will give aluminous waste though increase the yield during the halognation reaction. Moreover, the overall steps of the reaction will increase by performing isolation and work up for bromo intermediate.
US 6,727,637 B2 discloses the monobasic acid addition salts and the mixed salts of 2-amino-6-propylamino-4,5,6,7-tetrahydrobenzothiazole wherein the monobasic acid includes hydrochloric, hydrobromic, hydroiodic, nitric, benzoic, acetic, methane sulfonic, ethane sulfonic, trifluromethane sulfonic, benzene sulfonic, and p- toluene sulfonic acids. Further the patent US ‘637 B2 discloses the formation of mixed salts like of 2-amino-6-propylamino-4,5,6,7-tetrahydrobenzothiazole monohydrochloride monotartrate, of 2-amino-6-propylamino -4,5,6,7- tetrahydrobenzothiazole monohydrobromide monotartrate or of 2-amino-6- propylamino-4,5,6,7-tetrahydrobenzothiazole. monomethane sulfonate dibenzoyl-D- tartrate. The process as disclosed in US ‘637 B2 converts the racemic pramipexole into monohydrochloride salt of pramipexole which is then resolved with a optically active auxilliary acid to give mixed salt like of 2-amino-6-propylamino-4,5,6,7-tetrahydro- benzothiazole monohydrochloride monotartrate which is then converted to (S)- pramipexole free base and then to the desired pharmaceutically active ingredient (S)- pramipexole dihydrochloride.
US 6,770,761 B2 also discloses the process for preparation of 2-amino-6(alkyl)- amino-4,5,6,7-tetrahydrobenzothiazoles which includes the bromination of 1,4- cyclohexadione by bromine in an alcoholic solvent, followed by treatment of the reaction mixture with thiourea or N-acylthiourea and isolation of compound (A), that is further treated with an amine R1-NH2 or a chiral amine to get an imine intermediate and reducing it by reaction with said reducing agent or by hydrogenation, to yield the compound of formula (B)


(A) (B) Polymorphism is the occurrence of different crystalline forms of a single compound and it is a property of some compounds and complexes. Thus, polymorphs are distinct solids sharing the same molecular formula, yet each polymorph may have distinct physical properties. Therefore, a single compound may give rise to a variety of polymorphic forms where each form has different and distinct physical properties, such as different solubility profiles, different melting point temperatures and/or different x- ray diffraction peaks. Since the solubility of each polymorph may vary, identifying the existence of pharmaceutical polymorphs is essential for providing pharmaceuticals with predicable solubility profiles. It is desirable to investigate all solid-state forms of a drug, including all polymorphic forms, and to determine the stability^ dissolution and flow properties of each polymorphic form. Polymorphic forms of a compound can be distinguished in a laboratory by X-ray diffraction spectroscopy and by other methods such as, infrared spectrometry. For a general review of polymorphs and the pharmaceutical applications of polymorphs see G. M. Wall, Pharm Manuf. 3, 33 (1986); J. K. Haleblian and W. McCrone, J. Pharm. ScL, 58, 911 (1969); and J. K. Haleblian, J. Pharm. ScL, 64, 1269 (1975), all of which are incorporated herein by reference.

Example-1: Preparation of 2-amino-6-phthaIimido-4,5,6,7- tetrahydrobenzothiazole A) Preparation of chromic acid:
0.278 kg of chromium trioxide was added in 0.428 L of water at 150C to 35°C. The reaction mixture was cooled to 50C to 1O0C. 0.198 L of sulfuric acid Was added slowly within 25 to 30 minutes. 1.0 L of water was added to get the clear solution. B) Preparation of 2-amino-6-phthalimido-4,5,6,7-tetrahydrobenzothiazole via 4- (phthalimido)-cyclohexanone
1.0 Kg of 4-(phthalimido)-cyclohexanol was added in 20.0 L of acetone at 250C to 350C. The reaction mixture was cooled to 50C to 100C and treated with chromic acid solution. 0.2 L of isopropanol was added and stirred for 30 min. The reaction mixture was filtered and washed with acetone (1.0 L). The filtrate was treated with 0.4 kg sodium bicarbonate at 250C to 350C and stirred for 1 h. The reaction mass was again filtered, washed with acetone (1.0 L). Excess of acetone was distilled under vacuum. The residue was treated with 0.5 L ethanol followed by distillation of ethanol under vacuum. The reaction mass was cooled and treated with 3.36 L ethanol at 450C to 250C while gradual cooling. The reaction mixture was further cooled to 150C to 2O0C and treated with 0.22 L of bromine and 0.43 Kg of thiourea under stirring for 1 h. The reaction mixture was heated to reflux at 750C to 780C for 6 hrs. The reaction mixture was cooled and stirred for 1 hr at 50C to 1O0C. The product was isolated by centrifuge, washing with ethanol 0.66 L and drying under vacuum at 5O0C t0 550C. (yield: 0.70 Kg).
ExampIe-2: Preparation of 2, 6-diamino-4,5,6,7-tetrahydrobenzothiazole
1.595 kg of 2-amino-6-phthalimido-4,5,6,7-tetrahydrobenzothiazole was treated with 40% aqueous solution of monomethylamine at 250C to 350C. The reaction mass was allowed to stir for 5-10 minutes and heated at 45°C to 5O0C for 1 – 1.5 hr. The reaction mixture was cooled gradually to 50C to 1O0C and maintained for 30 minutes. The product thus obtained was filtered, washed with chilled water and dried at 5O0C to 550C to obtained racemic 2,6-diamino-4,5,6,7-tetrahydrobenzothiazole. (Yield: 0.522 kg)
Example-3: Preparation of 2, 6-diamino-4,5,6,7-tetrahydrobenzothiazoIe tartrate salt
1.0 Kg of 2, 6-diamino-4,5,6,7-tetrahydrobenzothiazole was added in 9.5 L of water and heated at 750C to 850C. 0.888 Kg of L-(+)-tartaric acid was added to the reaction mixture and maintained for 30 minutes. The reaction mixture was fine filtered at high temperature and washed with 0.5 L of water. The filtrate was gradually cooled to 250C to 300C and maintained for 16 hours. The product was centrifuge and washed with 1 L water. The wet cake was treated with 6.0 L water and heated at 8O0C to 9O0C with addition of excess water to ensure clear solution. The reaction mass was fine filtered at high temperature and washed with 0.5 L water. The filtrate thus obtained was gradually cooled to 5°C to 1O0C and maintained for 2 hrs. The product was centrifuge and washed with 1 L chilled water. The wet cake was treated with 6.0 L water and heated at 8O0C to 9O0C with addition of excess water to ensure clear solution. The reaction mass was gradually cooled to 950C to 25°C and maintained for 2 hrs. The product was centrifuge, washed with 1 L chilled water, dried at 5O0C to 550C and cooled to 250C to 350C. (Yield: 0.70 Kg). ExampIe-4: Preparation of (S)-2,6-diamino-4,5,6,7-tetrahydrobenzothiazole
1.0 Kg of 2, 6-diamino-4,5,6,7-tetrahydrobenzothiazole tartrate salt was treated with 1.5 L of water and stirred for 15 minutes at 25°C to 35°C. 0.245 Kg of sodium hydroxide solution in 0.612 L of water was added to adjust the pH 11.0 to 12.0 within 35 to 40 minutes and stirred for 1 hr. The product was centrifuge, washed with 1.0 L water and dried at 500C to 550C. The product was cooled to 20°C-40°C to obtain (S)- 2,6-diamino-4,5,6,7-tetrahydrobenzothiazole. (Yield: 0.37 Kg). Example-5: Preparation of Pramipexole crude
To the solution of 1.0 Kg of (S)-2,6-diamino-4,5,6,7-tetrahydrobenzothiazole and 0.1225 Kg of potassium carbonate in 10.0 L isopropanol was added 0.540 L n- propyl bromide. The reaction mixture was stirred for 15 minutes and heated to reflux on a water bath up to 8O0C and was maintained for 5 hours. 0.3236 L of n-propyl bromide was further added in two portions at 8O0C to 82°C and maintained for 5 hours. The isopropanol was removed completely by distillation under vacuum at 550C to 750C. 7.5 L of process water was added into the reaction mass and stirred for 30 minutes. The reaction mixture was cooled to 250C to 350C. 40% sodium hydroxide solution (0.108 Kg in 0.27 L water) was added to adjust the constant pH 10.0 to 10.5 followed by treatment with 5.0 L methylene dichloride twice and separating the organic layer. The organic layer was treated with 5.0 L of process water and stirred for 30 minutes. The separated organic layer was subjected to distillation to remove methylene dichloride under vacuum. 5.0 L of isopropanol was added at 4O0C to 450C and heated up to 6O0C to 650C. Acidic isopropanol 0.440L was added to adjust the pH 7.0 to 7.5 and stirred for 1 hour. The reaction mass was cooled to 250C to 35°C. The product was obtained by centrifuge, washing with 0.5 L of isopropanol and drying at 5O0C to 550C followed by cooling. (Yield: 1.0 Kg)
ExampIe-6: Preparation of Pramipexole dihydrochloride monohydrate
1.0 Kg of crude Pramipexole was added in 5.0 L of ethanol and heated to reflux using water bath at 800C. The reaction mixture was maintained for 1 hour and cooled to 250C to 35°C and stirred for 1 hour. The product was centrifuge and washed with 0.5 L ethanol. The wet cake thus obtained was further treated with 5.0 L of ethanol and heated to reflux using water bath at 8O0C. The reaction mixture was maintained for 1 hour and cooled to 250C to 350C and stirred for 1 hour. The product was centrifuge and washed with 0.5 L ethanol. The wet cake was treated with 5.0 L isopropanol and heated to 6O0C to 65°C using water bath. Acidic isopropanol 0.35 L was added to adjust the pH 1.7 to 2.3 and maintained for 1 hour. The product was centrifuge and washed with 1 L of isopropanol and dried in hot air oven at 5O0C to 550C to give Pramipexole dihydrochloride pure, which is converted to Pramipexole dihydrochloride monohydrate upon cooling the dried material under airflow. (Purity: 99.5% by HPLC and having known individual impurities less than 0.1% and total impurities less than 1.0%.) Example-7.: Preparation of Pramipexole dihydrochloride monohydrate
1.0 Kg of crude Pramipexole was added in 5.0 L of ethanol and heated to reflux using water bath at 8O0C. The reaction mixture was maintained for 1 hour and cooled to 250C to 350C and stirred for 1 hour. The product was centrifuge and washed with 0.5 L ethanol. The wet cake thus obtained was further treated with 5.0 L of ethanol and heated to reflux using water bath at 800C. The reaction mixture was maintained for 1 hour and cooled to 250C to 350C and stirred for 1 hour. The product was centrifuge and washed with 0.5 L ethanol. The wet cake was treated with 5.0 L isopropanol and heated to 600C to 650C using water bath. Isopropanolic HCl (0.35 L) containing water was added to adjust the pH 1.7 to 2.3 and maintained for 1 hour. The product was centrifuge and washed with 1 L of isopropanol and dried at 4O0C to 5O0C to give Pramipexole dihydrochloride monohydrate
PATENT
New patent, WO 2015155704, An improved process for the preparation of pramipexole dihydrochloride monohydrate
WO 2015155704, An improved process for the preparation of pramipexole dihydrochloride monohydrate
PIRAMAL ENTERPRISES LIMITED [IN/IN]; Piramal Tower, Ganpatrao Kadam Marg Lower Parel Mumbai 400013 (IN)
|




References
- “Once-daily MIRAPEX ER now approved by FDA for both early and advanced Parkinson’s disease”. Boehringer Ingelheim Pharmaceuticals, Inc. Retrieved 19 December 2013.
- National Prescribing Service (2009). “Pramipexole for Parkinson’s Disease”. Medicines Update. Available athttp://www.nps.org.au/consumers/publications/medicine_update/issues/Pramipexole_for_Parkinsons_disease
- Kvernmo T, Härtter S, Burger E (August 2006). “A review of the receptor-binding and pharmacokinetic properties of dopamine agonists”. Clinical Therapeutics 28 (8): 1065–78.doi:10.1016/j.clinthera.2006.08.004. PMID 16982285.
- Newman-Tancredi A, Cussac D, Audinot V, et al. (November 2002). “Differential actions of antiparkinson agents at multiple classes of monoaminergic receptor. II. Agonist and antagonist properties at subtypes of dopamine D(2)-like receptor and alpha(1)/alpha(2)-adrenoceptor”. The Journal of Pharmacology and Experimental Therapeutics 303 (2): 805–14.doi:10.1124/jpet.102.039875. PMID 12388667.
- Millan MJ, Maiofiss L, Cussac D, Audinot V, Boutin JA, Newman-Tancredi A (November 2002). “Differential actions of antiparkinson agents at multiple classes of monoaminergic receptor. I. A multivariate analysis of the binding profiles of 14 drugs at 21 native and cloned human receptor subtypes”. The Journal of Pharmacology and Experimental Therapeutics 303 (2): 791–804. doi:10.1124/jpet.102.039867. PMID 12388666.
- Weber, M; Chang W; Breier M; Ko D; Swerdlow NR (December 2008). “Heritable strain differences in sensitivity to the startle gating-disruptive effects of D2 but not D3 receptor stimulation”. Behav Pharmacol 19 (8): 786–795. doi:10.1097/FBP.0b013e32831c3b2b. PMC 3255557. PMID 19020413.
- Chang, W; Weber M; Breier MR; Saint Marie RL; Hines SR; Swerdlow NR (February 2012). “Stereochemical and neuroanatomical selectivity of pramipexole effects on sensorimotor gating in rats”. Brain Res 1437: 69–76. doi:10.1016/j.brainres.2011.12.007. PMID 22227455.
- Zarate CA, Payne JL, Singh J, et al. (July 2004). “Pramipexole for bipolar II depression: a placebo-controlled proof of concept study”. Biol. Psychiatry 56 (1): 54–60.doi:10.1016/j.biopsych.2004.03.013. PMID 15219473.
- Corrigan MH, Denahan AQ, Wright CE, Ragual RJ, Evans DL (2000): Comparison of pramipexole, fluoxetine, and placebo in patients with major depression. Depress Anxiety 11:58 –65.
- Jump up^ “MedlinePlus Drug Information: Pramipexole (Systemic)”. United States National Library of Medicine. Archived from the original on 2006-09-26. Retrieved 2006-09-27.
- “FDA Prescribing Information: Mirapex (pramipexole dihydrochloride)” (PDF). Food and Drug Administration (United States). Retrieved 2008-12-31.
- Wolters ECh, van der Werf YD, van den Heuvel OA (September 2008). “Parkinson’s disease-related disorders in the impulsive-compulsive spectrum”. J. Neurol. 255 Suppl 5: 48–56.doi:10.1007/s00415-008-5010-5. PMID 18787882.
- Jump up^ Bostwick JM, Hecksel KA, Stevens SR, Bower JH, Ahlskog JE (April 2009). “Frequency of new-onset pathologic compulsive gambling or hypersexuality after drug treatment of idiopathic Parkinson disease”. Mayo Clin. Proc. 84 (4): 310–6. doi:10.4065/84.4.310. PMC 2665974. PMID 19339647.
- Jump up^ Schneider, C. S.; Mierau, J. (1987). “Dopamine autoreceptor agonists: Resolution and pharmacological activity of 2,6-diaminotetrahydrobenzothiazole and an aminothiazole analog of apomorphine”. Journal of Medicinal Chemistry 30 (3): 494–8. doi:10.1021/jm00386a009. PMID 3820220.
- Jump up^ DeBattista C, Solvason HB, Breen JA, Schatzberg AF. (2000). “Pramipexole augmentation of a selective serotonin reuptake inhibitor in the treatment of depression.”. J Clin Psychopharmacol. 20 (2): 274–275. doi:10.1097/00004714-200004000-00029. PMID 10770475.
- Jump up^ Goldberg JF, Burdick KE, Endick CJ (March 2004). “Preliminary, randomized, double-blind, placebo-controlled trial of pramipexole added to mood stabilizers for treatment resistant bipolar depression.”. American Journal of Psychiatry 161 (3): 161:564–566. doi:10.1176/appi.ajp.161.3.564. PMID 14992985.
- Jump up^ Guy M. Goodwina, A. Martinez-Aranb, David C. Glahn c, Eduard Vieta b (November 2008). “Cognitive impairment in bipolar disorder: Neurodevelopment or neurodegeneration? An ECNP expert meeting report”. European Neuropsychopharmacology 18 (11): 787–793. doi:10.1016/j.euroneuro.2008.07.005. PMID 18725178.
- Lattanzi L, Dell’Osso L, Cassano P, Pini S, Rucci P, Houck PR, Gemignani A, Battistini G, Bassi A, Abelli M, Cassano GB. (2002). “Pramipexole in treatment-resistant depression: a 16-week naturalistic study.”. Bipolar Disord. 4 (5): 307–314. doi:10.1034/j.1399-5618.2002.01171.x. PMID 12479663.
- Cassano P, Lattanzi L, Soldani F, Navari S, Battistini G, Gemignani A, Cassano GB. (2004). “Pramipexole in treatment-resistant depression: an extended follow-up.”. Depression and Anxiety20 (3): 131–138. doi:10.1002/da.20038. PMID 15549689.
- Holman AJ, Myers RR. (2005). “A randomized, double-blind, placebo-controlled trial of pramipexole, a dopamine agonist, in patients with fibromyalgia receiving concomitant medications.”.Arthritis Rheum. 52 (8): 2495–2505. doi:10.1002/art.21191. PMID 16052595.
External links
- Mirapex.com – Manufacturer’s website
| WO2006003677A1 * | Apr 25, 2005 | Jan 12, 2006 | Alembic Ltd | Improved process for the preparation of biologically active tetrahydrobenzthiazole derivative |
| EP0186087A1 * | Dec 16, 1985 | Jul 2, 1986 | Dr. Karl Thomae GmbH | Tetrahydro-benzothiazoles, their production and their use as intermediates or drugs |
| EP0207696A1 * | Jun 20, 1986 | Jan 7, 1987 | Eli Lilly And Company | Dialkylaminotetrahydrobenzothiazoles and oxazoles |
| EP1731514A1 * | Jun 2, 2005 | Dec 13, 2006 | Sandoz AG | Process for the preparation of Pramipexole |
| BOEHRINGER INGELHEIM: “Mirapex“[Online] 2006, pages 4-31, XP002444888 Retrieved from the Internet: URL:http://www.fda.gov/medwaTCH/safety/200 6/Nov_PIs/Mirapex_PI.pdf> | ||
| 2 | * | SCHNEIDER C S ET AL: “Dopamine autoreceptor agonists: resolution and pharmacological activity of 2,6-diaminotetrahydrobenzothiazole and aminothiazole analogue of apomorphine” JOURNAL OF MEDICINAL CHEMISTRY, AMERICAN CHEMICAL SOCIETY. WASHINGTON, US, vol. 30, no. 3, March 1987 (1987-03), pages 494-498, XP002186199 ISSN: 0022-2623 cited in the application |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| EP2137171A2 * | Mar 14, 2008 | Dec 30, 2009 | Knopp Neurosciences, Inc. | Synthesis of chirally purified substituted benzothiazole diamines |
 |
|
|
|
| Systematic (IUPAC) name | |
|---|---|
|
(S)-N 6-propyl-4,5,6,7-tetrahydro-1,3-benzothiazole-2,6-diamine
|
|
| Clinical data | |
| Trade names | Mirapex, Mirapexin, Sifrol |
| AHFS/Drugs.com | monograph |
| MedlinePlus | a697029 |
| Pregnancy category |
|
| Legal status |
|
| Routes of administration |
Oral |
| Pharmacokinetic data | |
| Bioavailability | >90% |
| Protein binding | 15% |
| Biological half-life | 8–12 hours |
| Excretion | Urine (90%), Feces(2%) |
| Identifiers | |
| CAS Registry Number | 104632-26-0  |
| ATC code | N04BC05 |
| PubChem | CID: 119570 |
| IUPHAR/BPS | 953 |
| DrugBank | DB00413 |
| ChemSpider | 106770 |
| UNII | 83619PEU5T |
| KEGG | D05575 |
| ChEBI | CHEBI:8356 |
| ChEMBL | CHEMBL301265 |
| Chemical data | |
| Formula | C10H17N3S |
| Molecular mass | 211.324 g/mol |
Filed under: Uncategorized Tagged: Pramipexole







