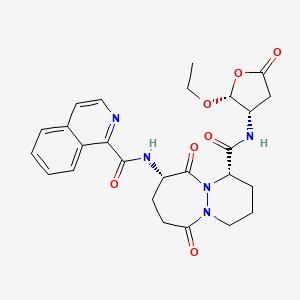
Pralnacasan
VX 740
cas 192755-52-5
(4S,7S)-N-[(2R,3S)-2-ethoxy-5-oxooxolan-3-yl]-7-(isoquinoline-1-carbonylamino)-6,10-dioxo-2,3,4,7,8,9-hexahydro-1H-pyridazino[1,2-a]diazepine-4-carboxamide
N-[(4S,7S)-4-{[(2R,3S)-2-ethoxy-5-oxooxolan-3-yl]carbamoyl}-6,10-dioxo-octahydro-1H-pyridazino[1,2-a][1,2]diazepin-7-yl]isoquinoline-1-carboxamide
(1S,9S)-N-((2R,3S)-2-Ethoxy-5-oxotetrahydrofuran-3-yl)-9-((isoquinolin-1-ylcarbonyl)amino)-6,10-dioxooctahydro-6-H-pyridazino(1,2-a)(1,2)diazepine-1-carboxamide
6H-Pyridazino(1,2-a)(1,2)diazepine-1-carboxamide, N-((2R,3S)-2-ethoxytetrahydro-5-oxo-3-furanyl)octahydro-9-((1-isoquinolinylcarbonyl)amino)-6,10-dioxo-, (1S,9S)-
- HMR 3480
- HMR3480
- HMR3480/VX-740
- Pralnacasan
- UNII-N986NI319S
- VX 470
- VX-740
C26H29N5O7, 523.543
NSAID, ICE inhibitor & metastasis inhibitor.пралнаказан [Russian] [INN]برالناكاسان [Arabic] [INN]普那卡生 [Chinese] [INN]

Pralnacasan is an orally bioavailable pro-drug of a potent, non-peptide inhibitor of interleukin-1beta converting enzyme (ICE).Pralnacasan is a potent, non-peptide inhibitor of interleukin-1beta converting enzyme (ICE, aka Caspase-1). It was originally discovered by Vertex Pharmaceuticals and licensed for development to Aventis Pharma. In 2003 Aventis and Vertex Pharmaceuticals agreed to voluntarily discontinue development based on results from a 9-month animal toxicity trial that showed liver abnormalities due to chronic high doses of pralnacasan. Pralnacasan has also been investigated for the treatment of Partial Epilepsy; advancing to Phase II clinical trials.Pralnacasan is a potent, non-peptide inhibitor of interleukin-1beta converting enzyme (ICE). Pralnacasan is an oral, anti-cytokine drug candidate licensed for development by Aventis Pharma from Vertex Pharmaceuticals. In November 2003, Aventis and Vertex Pharmaceuticals announced that they had voluntarily suspended the phase II clinical trials of pralnacasan due to results from an animal toxicity study that demonstrated liver abnormalities after a nine-month exposure to pralnacasan at high doses. While no similar liver toxicity has been seen to date in human trials, the companies will evaluate the animal toxicity results before proceeding with the phase II clinical program.Pralnacasan inhibits interleukin-1beta converting enzyme (ICE), an enzyme that regulates the production of IL-1 and IFN gamma – intercellular mediators that initiate and sustain the process of inflammation. Inhibiting ICE may be an effective strategy for curtailing damaging inflammatory processes common to a number of acute and chronic conditions, such as rheumatoid arthritis (RA) and osteoarthritis.
PAPERhttps://pubs.rsc.org/en/content/articlelanding/2017/ob/c7ob01403a/unauth
IDrugs (2003), 6(2), 154-158.
Chemistry (Weinheim an der Bergstrasse, Germany) (2017), 23(2), 360-369PAPER
Bioorganic & Medicinal Chemistry Letters (2006), 16(16), 4233-4236.https://www.sciencedirect.com/science/article/abs/pii/S0960894X06006184?
Abstract
Novel 1-(2-acylhydrazinocarbonyl)cycloalkyl carboxamides were designed as peptidomimetic inhibitors of interleukin-1β converting enzyme (ICE). A short synthesis was developed and moderately potent ICE inhibitors were identified (IC50 values <100 nM). Most of the synthesized examples were selective for ICE versus the related cysteine proteases caspase-3 and caspase-8, although several dual-acting inhibitors of ICE and caspase-8 were identified. Several of the more potent ICE inhibitors were also shown to inhibit IL-1β production in a whole cell assay (IC50 < 500 nM).
Graphical abstract
Novel 1-(2-acylhydrazinocarbonyl)cycloalkyl carboxamides were designed and synthesized as selective peptidomimetic inhibitors of interleukin-1β converting enzyme (ICE IC50 values <100 nM).

PAPEROrganic letters (2014), 16(13), 3488-91.https://pubs.acs.org/doi/10.1021/ol501425b
Abstract

Peptides containing N2-acyl piperazic or 1,6-dehydropiperazic acids can be formed efficiently via a novel multicomponent reaction of 1,4,5,6-tetrahydropyridazines, isocyanides, and carboxylic acids. Remarkably, the reaction’s induced intramolecularity can enable the regiospecific formation of products with N2-acyl piperazic acid, which counters the intrinsic and troublesome propensity for piperazic acids to react at N1 in acylations. The utility of the methodology is demonstrated in the synthesis of the bicyclic core of the interleukin-1β converting enzyme inhibitor, Pralnacasan.
PatentWO 9722619WO 9903852WO 9952935
PATENTWO 2000042061https://patents.google.com/patent/WO2000042061A1/enThe invention particularly relates to the process as defined above in which the compound of formula (I) is 9- (1, 3-dihydro-1,3, dioxo-2H-isoindol-2-yl) -3 ,, 7, 8, 9, 10-hexahydro-6, 10-dioxo-6H-pyridazino- [1,2- a] [1, 2] ethyl diazepine-1-carboxylate:

The invention particularly relates to the process as defined above in which the compound of formula (Iopt) is- (lS-cis) -9- (1, 3-dihydro-l, 3-dioxo-2H-isoindol-2-yl) – 3,4,7,8,9, 10-hexahydro-β, 10-dioxo -6H-pyridazino- [1,2- a] [1, 2] ethyl diazeρine-1-carboxylate:

The compounds of formula (I) can be generally used for the synthesis of medicaments as indicated in patent EP 94095. The compounds of formulas (II) and (III) and (F) are known and can be prepared according to the experimental method described below.The invention also relates to the application of the process as defined above as an intermediate step for the preparation of a compound of formula (V)

via the compound of formula (Iopt) as defined above, characterized in that this process comprises the steps of the process for the preparation of the compounds of formula (Iopt) from the compounds of formula (II) as defined above.The subject of the invention is also the application as defined above, characterized in that the compound of formula (Iopt) is (lS-cis) -9- (1, 3-dihydro-l, 3-dioxo -2H- isoindol-2-yl) -3,4,7,8,9, 10-hexahydro-6, 10-dioxo-6H- pyridazino- [1,2-a] [1, 2] diazepine-1- ethyl carboxylate

The subject of the invention is also the application of the process as defined above as an intermediate step in the overall process for preparing the compounds of formula (I) and (Iopt) as defined above. Finally, the subject of the invention is, as intermediate compound, the compound of formula (IA) as defined above.Preparation 1 Preparation of bis (phenylmethyl) 1,2-hydrazinecarboxylate1.5 liters of methanol and 25 g of 80% hydrazine monohydrate are placed under nitrogen. Cooled to 0 ° C and then introduced 75 g of benzyl chloroformate and a solution of 93 g of sodium carbonate in 1100 ml of demineralized water. Maintaining the reaction mixture for 1 hour at 0 ° C, drained and washed by displacement with a mixture of 100 ml of methanol and 100 ml of water, then washed by displacement with 500 ml of water at 0 C °. Dried and obtained 107.6 g of the desired product. Preparation 2Preparation of N-phthaloyl-L-glutamic anhydride D (+) 2-tetrahydro-2,6,6-dioxo-2H-pyran-3-yl-1H-isoindole-1,3 (2H) – dione (R)Stage a: N-phthaloyl-L-glutamic acid2- (1, 3-dihydro-1,3, dioxo-2H-isoindole-2-yl) acid – pentanedioic (2S)To a solution of 14.4 g of sodium carbonate in 180 ml of water is added 10 g of L-glutamic acid then 16 g of N-carbethoxyphthalimide (nefkens reagent, commercial). The mixture is stirred at ambient temperature for 2 hours and then extracted with ethyl acetate. The organic phase is evaporated under reduced pressure until a dry extract is obtained and 2.74 g of crude product is obtained. Washing is carried out with sodium bicarbonate, then after return to the acid and extraction with ethyl acetate, 370 mg of expected product and H 2 N-C0 2 Et are isolated. Furthermore, the aqueous phase is brought to pH = 2 with 36% hydrochloric acid at a temperature below 5 ° C and then extracted with ethyl acetate, washed with a saturated chloride solution. sodium, dry, filter and concentrate under reduced pressure until 22.7 g of expected product is obtained in the form of an oil.Mass spectrum (MH) “ = 276 “ Infrared (Nujol):1775 cm “1 (m), 1720 cm ” 1 (F, complex): CO 1611 cm “1 : Aromatic Stage b:To the product obtained in stage a), 160 ml of tetrahydrofuran are added and 18.6 g of DCC (1, 3-Dicyclohexyl-carbodiimide) dissolved in 55 ml of tetrahydrofuran are added dropwise over 30 minutes. Stirred for 1 hour at 15-17 ° C, then filtered, rinsed with tetrahydrofuran, evaporated under reduced pressure until a dry extract is obtained which is taken up in isopropyl ether. After 30 minutes of stirring, the filter is washed and dried. 14.98 g of expected product are obtained. α D = -52.63 λ H NMR (DMSO) 2.12 (m, 1H); 2.61 (m, 1H); 2.98 (dm, 1H); 3.16 (ddd, 1H); 5.48 (dd, 1H); 7.82 (m,> 4H)Example 1: (IS-cis) -9- (1, 3-dihydro-1,3, dioxo-2H-isoindol-2-yl) -3,4,7,8,9,10-hexahydro-6,10 -dioxo-6H-pyridazino- [1,2- a] [1,2] diazepine-1-ethyl carboxylate. Stage a: Preparation of 2,5-dibromopentanoic acid 39 ml of bromine are added to a mixture of 106 g of 5-bromopentanoic acid and 1 ml of phosphorus tribromide. The reaction mixture is brought to 70-80 ° C for 16 h 30. The reaction medium is brought to 100 ° C for 15 minutes and allowed to return to room temperature. 147 g of sought product is obtained.Stage b: Preparation of ethyl 2,5-dibromopentanoate24.37 g of oxalyl chloride are added to a mixture containing 50 g of the acid prepared in the preceding stage, 15 drops of dimethylformamide and 300 ml of dichloromethane. The reaction mixture is kept under stirring at at room temperature, until the reaction is complete. The reaction mixture is cooled to 10 ° C and 50 ml of ethyl alcohol are added. Stirred for 30 minutes at 10 ° C, allowed to return to room temperature and stirred for 3 hours at room temperature. It is brought to dryness and the desired product is obtained. Stage c: CyclizationPreparation of (S) -tetrahydro-1,2,3-pyridazinetricarboxylate of 3-ethyl-1,2-bis (phenylmethyl) and (R) -tetrahydro-1,2,3-pyridazinetricarboxylate of 1,2 -bis (phenylmethyl). A suspension of 12.1 g of ethyl 2,5-dibromopentanoate (stage b) in 50 ml of diglyme is introduced at 20-25 ° C. in a suspension containing 10.42 g of 1,2-hydrazine carboxylate of bis (phenylmethyl) (preparation 1), 65 ml of diglyme and 8.26 g of potassium carbonate. The suspension obtained is heated to 90 ° C. and stirring is continued for 48 hours. Cooled to 20 ° C, poured into a solution containing 50 ml of 2N hydrochloric acid and 150 ml of a mixture of water and ice. Extraction is carried out with ethyl acetate, washing with water and drying. It is filtered, rinsed with ethyl acetate and dried. Finally, the crude product is purified by chromatography on silica, eluting with a heptane / ethyl acetate mixture 40/20 and 10.71 g of sought product is obtained. Stage d: Acylation and hydrogenolysisPreparation of α, (IS) – [3-oxo-3- (tetrahydro-3-ethoxycarbonyl-1 (2H) -pyridazinyl) propyl] -1,3-dihydro-1,3-dioxo-2H-isoindole acid -2-aceticThe mixture consisting of 15g of tetrahydro-1,2,3-pyridazinetricarboxylate of 3-ethyl-1,2-bis (phenylmethyl) is placed under hydrogen pressure (1.3 bar) for 24 hours. R + S mixture as prepared in stage c 150 ml of tetrahydrofuran, 2.5 g of palladium on carbon (10%) and 9.08 g of phthaloylglutamic acid anhydride as prepared according to preparation 2. After filtration, we evaporated under reduced pressure until a dry extract is obtained which is taken up in 100 ml of ethyl acetate and 150 ml of a saturated solution of sodium bicarbonate. It is extracted 3 times and the bicarbonate solution is acidified to pH = 3 with 36% hydrochloric acid. It is extracted 3 times with dichloromethane and washed with water. 13.16 g of crude product are obtained, which product is purified by chromatography on silica, eluting with a toluene / ethyl acetate / acetic acid 20/80 / 1.5 mixture to obtain 12.7 g of the expected product.NMR (250MHz, CDC1 3 ): 1.24 (d, 3H, OCH 2 CH 3 ); 4.12 (q, 2H, OCH 2 CH 3 ); 4.36-4.40 (m, 1H, Hl in alpha or beta position); 4.69-4.92 (m, 1H, H9 in the alpha position); 7.70 – 7.86 H aromatic. Stage el: cyclization with POCl 3– (lS-cis) -9- (1, 3-dihydro-l, 3-dioxo-2H-isoindol-2-yl) – 3,4,7,8,9, 10-hexahydro-6, 10-dioxo -6H-pyridazino- [1,2- a] [1, 2] ethyl diazepine-1-carboxylate. – (lR-trans) -9- (1, 3-dihydro-1,3, dioxo-2H-isoindol-2-yl) – 3,4,7,8, 9, 10-hexahydro-6,10-dioxo -6H-pyridazino- [1,2-a] [1,2] diazepine-1-ethyl carboxylate.To a solution of 20 ml of dichloroethane heated beforehand to 75 ° C., the following solutions A and B are added over 3 hours: A: 417 mg of the ester prepared in stage d in 4 ml of dichloroethane to which 1 ml of a solution of 1.2 ml of 2,6-lutidine in 5 ml of dichloroethane. B: 1 ml of a solution of 1.9 ml of P0Cl 3 in 10 ml of dichloroethane, then the mixture is stirred for 1 hour at this temperature. Cool to 10 ° C., add demineralized water, extract with dichloromethane and evaporate under reduced pressure to obtain a crude product (0.415 g) which is purified by chromatography on silica eluting with the heptane / dichloromethane mixture. / ethyl acetate 1/1/1. 161.8 mg of the SS diastereoisomer, 126.7 mg of the SR diastereoisomer and 5.8 mg of the SS + SR mixture are isolated. Stage e2: cyclization with POBr 3– (lS-cis) -9- (1, 3-dihydro-l, 3-dioxo-2H-isoindol-2-yl) – 3,4,7, 8, 9, 10-hexahydro-6, 10-dioxo -6H-pyridazino- [1, 2- a] [1, 2] ethyl diazepine-1-carboxylate. – (lR-trans) -9- (1, 3-dihydro-l, 3-dioxo-2H-isoindol-2-yl) – 3,4,7, 8, 9, 10-hexahydro-6, 10-dioxo -6H-pyridazino- [1, 2- a] [1, 2] ethyl diazepine-1-carboxylate.To a solution of 20 ml of dichloroethane heated beforehand to 80 ° C., the following solutions A and B are added over 3 hours:A: 417 mg of the ester prepared in stage d in 4 ml of dichloroethane to which 1 ml of a solution of 2.4 ml of 2,6-lutidine in 10 ml of dichloroethane was added. B: 1 ml of a solution of 5.85 g of POBr 3 in 10 ml of dichloroethane, then the mixture is stirred for 1 hour at this temperature. Cool to 10 ° C, add demineralized water, extract with dichloromethane and evaporate under reduced pressure to obtain a crude product (0.419 g) which is purified by chromatography on silica eluting with the heptane / dichloromethane / mixture 1/1/1 ethyl acetate. 163 mg of the SS diastereoisomer, 143 mg of the SR diastereoisomer and 6.2 mg of the SS + SR mixture are isolated.Stage f: deracemization / epimerization – (lS-cis) -9- (1, 3-dihydro-l, 3-dioxo-2H-isoindol-2-yl) – 3,4,7,8, 9, 10-hexahydro -6,10-dioxo-6H-pyridazino- [1, 2- a] [1, 2] ethyl diazepine-1-carboxylate.Is introduced at a temperature of -45 / -48 ° C in one hour 30 minutes, a solution containing 0.029 g of potassium terbutylate and 0.3 ml of dimethylformamide in a mixture containing 0.194 g of the mixture SS + SR prepared in stage d , 1.5 ml of dimethylformamide and 0.75 ml of terbutanol. The mixture is kept stirring for 1 hour and, after cooling to -50 ° C., 0.4 g of powdered ammonium chloride is introduced. Stirred 10 minutes at -45 ° C, add 1 ml of ammonium chloride at 20 ° C and stirred again 10 minutes. 2 ml of water are added after 5 minutes demineralized. Extracted with ethyl acetate, washed with demineralized water, decanted, concentrated and dried. 0.166 g of expected SS diastereoisomer is obtained. ” D = -75.3 ° (1% in methanol) NMR (250MHz, CDC1 3 ): 1.73 (m, 3H, H-2alpha H-3alpha H-3beta; 1.24 (d, 3H, OCH 2 CH 3 ); 2.38 (m, 3H, H2beta, H7alpha, H8 alpha); 2.92 (m, 1H, H4alpha); 3.39 – 3.44 (m, 1H, H8beta); 3.62 (m, 1H, H7beta); 4.23 (m, 2H, OCH 2 CH 3 ); 4.66-4.71 (m, 1H, H4 in beta position); 5.26-5.41 (m, 2H, Hl and H9 in the alpha position); 7.72 – 7.88 H aromatics.
PATENT
WO 2000010979https://patents.google.com/patent/WO2000010979A1/en

formula II, said compound has the structure:
In the synthesis of these inhibitors, the terminal carbon of Ri adjacent the -COOH moiety contains a protecting substituent. Preferably that protecting
substituent is
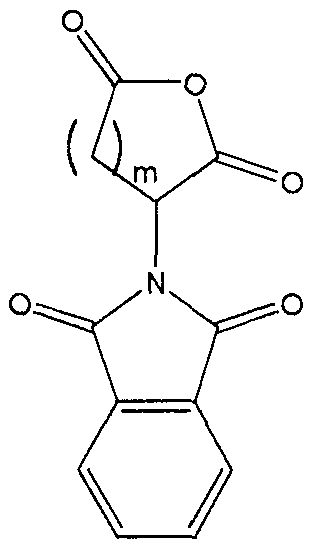
The synthesis steps from compound H to the inhibitors set forth above involve removal of the protecting substituent on Rx; coupling of the R5-NH- or R5′-NH- moiety in its place; hydrolysis of the R2 group;N .(CJ2)m.—Tand coupling of the amine ( (Ch,2)a Rs or -NH-Z)in its place. The removal of the protecting substituent on Ri is typically carried out with hydrazine. The subsequent coupling of the R5-NH- or R5′-NH- moiety is achieved with standard coupling reagents, such as EDC, DCC or acid chloride . Depending upon the nature of R2, its hydrolysis may be achieved with an acid (when R2 is t-butyl), a hydroxide (when R2 is any other alkyl, alkenyl or alkynyl or Ar) or hydrogenolysis (when R2 is an Ar-substituted alkyl, alkenyl or alkynyl) . This produces the corresponding acid from the ester.The acid is then coupled to the amine with standard coupling reagents, such as EDC, DCC or acid chloride .In order that this invention be more fully understood, the following examples are set forth. These examples are for the purpose of illustration only and are not to be construed as limiting the scope of the invention in any way. EXAMPLE 1Synthesis of a 7,6 Scaffold for a Caspase InhibitorA.

Compound A’ was dissolved m 5 equivalents of S0C12 and then heated to 80°C for 1 hour. The solution was then cooled to 50°C and 2 equivalents of bromine were added. The solution was incubated at 50°C for an additional 12 hours until the red color disappeared. We then cooled the solution to 10°C and added 4 volumes of water. The solution was then re-heated to 50°C for another hour. We then separated the organic and aqueous layer, washed the organic layer consecutively with water, Na2S0 and then brme, removing the aqueous layer after each washing. The final organic layer was then isolated, dried over Na2S0 and concentrated to produce compound B’ as an amber oil.B.

Compound B’ was treated with 1 equivalent of tert-butanol and 0.1 equivalents of 4- (dimethylammo) – pyπdme a solution of and the resulting solution cooled to 7°C. We then added a solution of 1 equivalent of DCC m toluene while maintaining reaction temperature at less than 22°C. The cooling bath was removed and the reaction was stirred at ambient temperature under a nitrogen atmosphere for 16 hours. The reaction mixture was then diluted with hexane and cooled to 9°C . The resulting solids were removed by filtration. The filtrate was washed consecutively with 0. IN HC1, water, and then sodium bicarbonate. The filtrate was then dried over sodium sulfate and concentrated in vacuo to afford compound C as a yellow oil.C.

Compound D’ was combined with 1.2 equivalents of compound C and dissolved in DMF at ambient temperature under nitrogen atmosphere. We then added granular sodium sulfate, 2.5 equivalents of LiOH monohydrate, and then 0.1 equivalents Bu4NI to the resulting solution. The reaction temperature was maintained at between 20°C and 30°C and allowed to stir for 16 hours. The reaction mixture was then diluted with ethyl acetate and water and the layers separated. The organic layer was washed with water and then brine, dried over sodium sulfate and concentrated in vacuo to produce an amber oil. This oil was then dissolved in 5 volumes of ethanol at ambient temperature. We then added 2.5 volumes of water. The resulting mixture was allowed to stir until a white solid formed (approximately 5 hours) . The crystallized product was isolated via filtration then dried in vacuo to afford compound E’ as a white solid.D.

We dissolved compound E’ in THF. We then added, at ambient temperature under a nitrogen atmosphere, 0.02 equivalents of triethylamine and 0.01 equivalents of Pd(OAc)2. A solution of 2.5 equivalents of triethylsilane (Et3SiH) in THF was then added and the resulting black solution was allowed to stir for 16 hours to complete the reaction. We then added a saturated, aqueous solution of sodium bicarbonate followed by a solution of compound F’ in THF. After 30 minutes, the layers were separated and the aqueous layer acidified to pH 4.5 with aqueous citric acid. The product in the aqueous layer was then extracted into ethyl acetate. The organic layer was isolated, washed with brine, dried over sodium sulfate and concentrated in vacuo to produce a white foam. This crude product was then recrystallized from MTBE to afford compound G’ as a white powder. E.
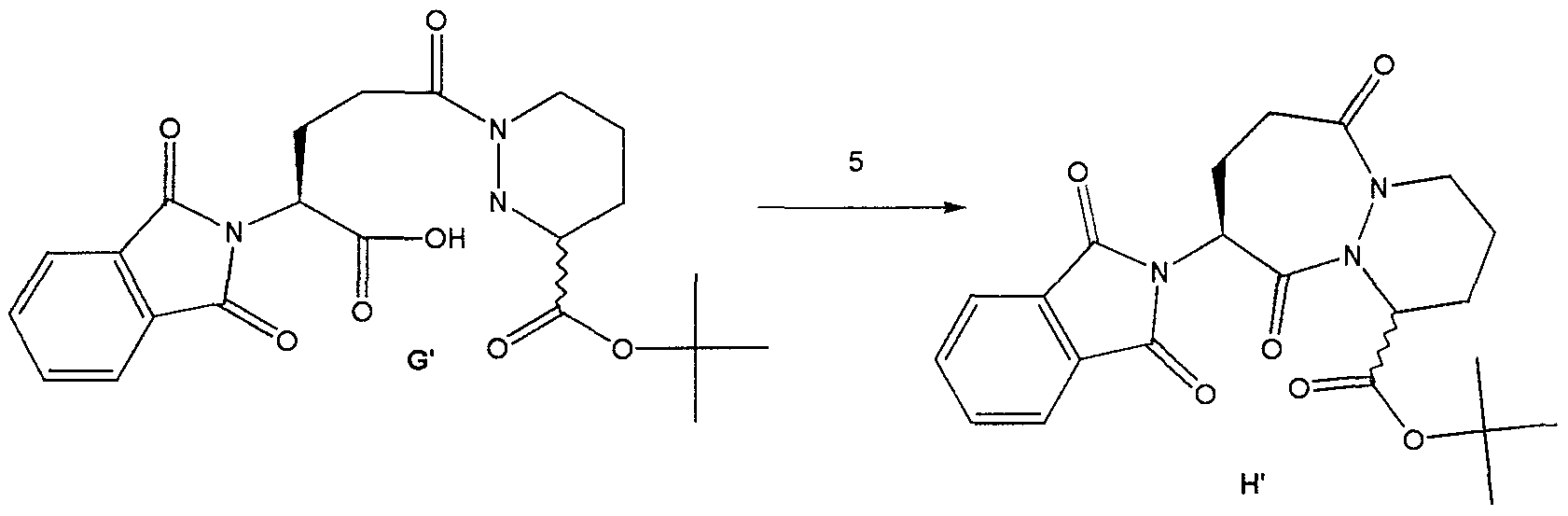
Method #1:To a suspension of compound G’ and 0.1 equivalents of DMF m dichloroethane, at 70°C we added 5 equivalents of 2, 6-lutιdme simultaneously with 2.5 equivalents of S0C12 over a period of 3 hours. The reaction was then diluted with toluene and washed consecutively with NaHC03 and br e. The solution was then dried over Na2S04 and concentrated in vacuo to afford compound H’ as a yellow solid.Method #2:To a suspension of compound G’ m dichloroethane, at 70°C, we added 4 equivalents of 2,6- lutid e followed by 2 equivalents of methanesulfonyl chloride. The resulting solution was stirred at 70°C for 12 hours. The reaction was then diluted with toluene and washed consecutively with NaHC03 and brme. The solution was then dried over Na2S04 and concentrated in vacuo to afford compound H’ as a white solid. Method #2 produced a significantly higher yield of H’ as compared to Method #1. EXAMPLE 2 Use of Intermediate H’ to Produce an Inhibitor of ICE A.

t-Butyl-9-amino-6 , 10-dioxo-l ,2,3,4,7,8,9, 10-octahydro-6- H-pyridazino [1 ,2-a] [1 ,2] diazepine-1-carboxylate (GB2,128,984) To a suspension of H’ (107 g, 0.25 mol) in ethanol (900 iriL) was added hydrazine (27 L, 0.55 mol) and the resulting mixture was allowed to stir at ambient temperature. After 4 hours, the reaction was concentrated in vacuo and the resulting white solid was suspended in acetic acid (IL of 2N) and allowed to stir at ambient temperature for 16 hours. The resulting white solid was filtered off and washed with water. The filtrate was made basic by the addition of solid sodium carbonate and the product extracted with dichloromethane. The organic layer was washed with brine, dried over magnesium sulfate and concentrated in vacuo to afford 79 mg of compound I’ as a yellow viscous oil.B.

t-Butyl-9- (isoquinolin-1-oylamino) -6, 10-dioxo- 1,2,3,4,7,8,9, 10-octahydro-6-H-pyridazino [ 1 , 2-a] [1,2] diazepine-1-carboxylate To a solution of the amine I’ (79 g, 0.265 mol) and isoquinolin-1-carboxylic acid (56g, 0.32 mol) in dichloromethane : DMF (400mL: 400mL) was added hydroxybenztriazole (54 g, 0.4 mol) and l-(3- dimethylaminopropyl) -3-ethylcarbodiimide hydrochloride (74 g, 0.39 mol) and the resulting mixture was allowed to stir at ambient temperature for 16 hours. The reaction mixture was poured into water and extracted with ethyl acetate. The ethyl acetate layer was washed with 0.5N sodium bisulfate, water, sodium bicarbonate, brine, dried over sodium sulfate and concentrated in vacuo to afford 122 g of compound J’ as an orange solid-foam.C.

9- (isoquinolin-1-oylamino) -6, 10-dioxo-l ,2 ,3 ,4 , 7 , 8 , 9 , 10- octahydro-6-H-pyridazino [1 ,2-a] [1,2] diazepine-1- carboxylate A solution of the ester J’ (122 g) in dichloromethane and trifluoroacetic acid (200 mL) was allowed to stir at ambient temperature for 16 hours. The reaction mixture was concentrated to a black oil which was then triturated with acetonitrile and ether to afford 98 g of compound K’ as a pale yellow solid. D .

K'[IS, 9S (2RS, 3S) ] N-(2-benzyloxγ-5-oxotetrahydrofuran-3- yl) -6 , 10-dιoxo-9- (ιsoquιnolιn-1-oγlamιno) -1,2,3,4,7,8,9, 10-octahydro-6-H-pyrιdazιno [ 1 , 2-a] [1,2] dιazepιne-l-carboxamιde To a solution of (3S, 2RS) 3- allyloxycarbonylammo-2- (4-chlorobenzyl) oxy-5- oxotetrahydrofuran [Bioorg. & Med. Chem. Lett., 2, pp. 615-618 (1992)] (4.4 g, 15.1 mmol) in dichloromethane was added N, N-dimethylbarbituric acid (5.9g, 3.8 mmol) then tetrakispalladium ( 0) tπphenyl phosphme (1.7 g, 1.5 mmol) and the resulting mixture was allowed to stir at ambient temperature for 15 minutes. To the resulting mixture was added the acid, compound K’ (5.0 g, 12.6 mmol), hydroxybenztπazole (2.0 g, 14.8 mmol) then and 1- (3-dιmethylammopropyl) -3-ethylcarbodιιmιde hydrochloride (2.7g, 14 mmol) and the reaction was allowed to stir for 3 hours at ambient temperature. The reaction mixture was then poured into water and extracted with ethyl acetate. The organics were washed with 0.5M sodium bisulfate, water, sodium bicarbonate, br e, dried over magnesium sulfate and concentrated m vacuo to afford 2.6 g of the crude product as a yellow foam. The crude material was purified by column chromatography (Sι02, dichloromethane : acetone 9:1 – 3:1) to afford 1.2 g of the compound L’ . Compound L’ and related compounds that may be synthesized using the method of this invention as an intermediate step are described in WO 97/22619, the disclosure of which is herein incorporated by reference. Those related compounds may be synthesized from the product of the method of this invention, H or H’ , through modifications of the procedure set forth in Example 2. Such modifications are well known in the art.
PATENTWO 2001083458https://patents.google.com/patent/WO2001083458A2/enScheme IV

C 2 5,> R’==OH (S)-VI-a *•* 6 6., R R”==<CI

Example 1

(S) -t-butyl- bis- (1,2-benzyloxycarbonyl) – hexahydropyridazine-3-carboxylate (>90% ee) : To a solution of bis-Cbz hydrazine and (R) -t-butyl-2, 5- dimesylvalerate (from the diol prepared by the method of Schmidt et al., Synthesis, p. 223 (1996)) in DMF was added Na2S04 then TBAF (2.5 equivalents). The resulting reaction mixture was allowed to stir at room temperature for 24 hrs. The reaction was then diluted with ethyl acetate. The organic layer was washed sequentially with 10% citric acid and brine, dried over anhydrous Na2S04 and concentrated in vacuo to afford the title compound. The optical purity of the title compound was greater than 90% ee as determined by HPLC using a ChiralPak® AD column and eluting with ethanol at 0.7 ml per minute.Example 2

(S) -t-butyl-bis- (1 ,2-benzyloxycarbonyl) – hexahydropyridazine-3-carboxylate (40% ee) : To a solution of bis-Cbz hydrazine and (R) -t-butyl-2, 5-dimesylvalerate(96.5% ee) in DMF was added Na2S04 then K2C03 (5 equivalents) and TBAI (0.1 equivalents). The resulting reaction mixture was heated at 80°C for 24 hrs. The reaction was allowed to cool and diluted with ethyl acetate. The organic layer was washed sequentially with 10% citric acid and brine, dried over anhydrous Na2S04 and concentrated in vacuo to afford the title compound as a 70:30 mixture of the S:R enantiomers (40% ee, as determined by HPLC using a ChiralPak® AD column, eluting with ethanol at 0.7 ml/min) .Example 3

Racemic t-butyl- bis- (1 ,2-benzyloxycarbonyl) – hexahydropyridazine-3-carboxylate: To a solution of bis- Cbz hydrazine and (R) -t-butyl-2, 5-dimesylvalerate (96.5% ee) in THF was added NaH (2 equivalents) . The resulting reaction mixture was stirred at room temperature. The reaction was quenched then diluted with ethyl acetate. The organic layer was washed sequentially with 10% citric acid and brine, dried over anhydrous Na2S04 and concentrated in vacuo to afford the title compound as a racemic mixture.Example 4 A. Deprotection and salt formation

Hexahydro-pyridazine-3-carboxylic acid tert-butyl ester , L-tartaric acid salt (B) : Compound A was combined with 10% Pd/C (10% w/w) in tetrahydrofuran. The resulting suspension was stirred at 60 °C under a hydrogen atmosphere until deprotection complete. The catalyst was removed via filtration, to the filtrate was added L- tartaric acid (1 equivalent) and the resulting solution concentrated in vacuo.B. Enantiomeric Enrichment
Hθ

The concentrate (B) was taken up in n-butanol(10 volumes), heated to reflux, then allowed to slowly cool to ambient temperature while stirring. The resulting solids were collected via filtration to afford(S) -piperazic acid, t-butyl ester as the tartrate salt (C) in 33% yield.C. Chiral AnalysisCompound (C) was suspended in water and DCM and cooled. We then added NaOH to basify the aqueous layer. The layers were then separated and to the organic layer we added two equivalents of benzyl chloroformate andNaOH. After stirring for 1 hour, the layers were again separated and the organic layer was washed with water.The organic layer was then dried over MgS04 and then concentrated in vacuo to produce the bis-Cbz piperazic acid, t-butyl ester for chiral HPLC analysis. The bis-Cbz piperazic acid, t-butyl ester was applied to a Chiralpak AD HPLC column (Chiral Technologies, Exton, PA) and eluted with ethanol at 0.8 ml/minute. Fractions from the column were quantitate by absorption at 210 nm. The results demonstrated that (S)- piperazic acid, t-butyl ester accounted for 94.5% of the piperazic acid, t-butyl ester present in the preparation.
Example 5 Conversion of Intermediate IV to Intermediate Vl-a Cbzy


IV’ C02t-Bu yi-a C02t-Bu Tetrahydro-pyridazine-l,3-dicarboxylic acid 1-benzyl ester 3-tert-butyl ester (Vl-a) : Compound IV (1 mmol) is combined with toluene and sodium hydroxide (aqueous, 2M, 3 equivalents) and the resulting mixture cooled to 1 °C. A solution of benzylchloroformate (1.05 equivalents) in toluene is added while maintaining the reaction pH at 10 or higher by the addition of sodium hydroxide, as needed. After stirring an additional 1 hour, allow the mixture to warm to room temperature then extract with ethyl acetate. The organic layer is washed with brine, dried over sodium sulfate and concentrated to afford Vl-a.Example 6 Conversion of Intermediate X to an Inhibitor of ICE
A. Phthalimide removal to form IX-b

X IX-b t-Butyl-9-amino-6 , 10-dioxo-l ,2,3,4,7,8,9, 10-octa ydro-6-H-pyridazino[l,2-a] [1,2] diazepine-1-carboxylate (GB 2,128,984): To a suspension of X (107 g, 0.25 mol) in ethanol (900 mL) was added hydrazine (27 mL, 0.55 mol) and the resulting mixture was allowed to stir at ambient temperature. After 4 hours, the reaction was concentrated in va cuo and the resulting white solid was suspended in acetic acid (1L of 2N) and allowed to stir at ambient temperature for 16 hours. The resulting white solid was filtered off and washed with water. The filtrate was made basic by the addition of solid sodium carbonate and the product extracted with dichloromethane. The organic layer was washed with brine, dried over magnesium sulfate and concentrated in va cuo to afford 79g of compound IX-b as a yellow viscous oil.B. Formation of compound XII

IX-b XII t-Butyl-9- (isoquinolin-1-oylamino) -6 , 10-dioxo- 1,2,3,4,7,8,9, 10-octahydro-6-H-pyridazino [1 , 2-a] [1,2] diazepine-1-carboxylate (XII) : To a solution of IX-b (79 g, 0.265 mol) and isoquinolin-1-carboxylic acid (56g, 0.32 mol) in dichloromethane and DMF (400mL: 00mL) was added hydroxybenzotriazole (54 g, 0.4 mol) and l-(3- dimethylaminopropyl) -3-ethylcarbodiimide hydrochloride (74 g, 0.39 mol) and the resulting mixture was allowed to stir at ambient temperature for 16 hours. The reaction mixture was poured into water and extracted with ethyl acetate. The ethyl acetate layer was washed with 0.5N sodium bisulfate, water, sodium bicarbonate, brine, dried over sodium sulfate and concentrated in vacuo to afford 122 g of compound XII as an orange solid-foam.t-Butyl ester hydrolysis to form compound XIII

XIII 9- (isoquinolin-1-oylamino) -6 , 10-dioxo-l ,2,3,4,7,8,9, 10- octahydro-6-H-pyridazino [1 , 2-a] [1 , 2] diazepine-1- carboxylate (XIII) : A solution of the ester XII (from step B) (122 g) in dichloromethane and trifluoroacetic acid (200 mL) was allowed to stir at ambient temperature for 16 hours. The reaction mixture was concentrated to a black oil which was then triturated with acetonitrile and ether to afford 98 g of compound XIII as a pale yellow solid.D. Formation of compound 4-b

[1S, 9S (2RS,3S) ]N- (2-benzyloxy-5-oxotetrahydrofuran-3- yl) -6,10-dioxo-9- (isoquinolin-1-oylamino) – 1,2,3,4,7,8,9, 10-octahydro-6-H-pyridazino [1 , 2-a] [1,2] diazepine-1-carboxamide (4-b) : To a solution of (3S, 2RS) 3-allyloxycarbonylamino-2-benzyloxy-5-oxotetrahydrofuran [Bioorq. & Med. Chem. Lett., 2, pp. 615-618 (1992)] (4.4 g, 15.1 mmol) in dichloromethane was added N,N- dimethylbarbituric acid (5.9g, 3.8 mmol) then tetrakispalladium(O) triphenyl phosphine (1.7 g, 1.5 mmol) and the resulting mixture was allowed to stir at ambient temperature for 15 minutes. To the resulting mixture was added the acid, compound XIII (from step C) (5.0 g, 12.6 mmol), hydroxybenzotriazole (2.0 g, 14.8 mmol), then 1- (3-dimethylaminopropyl) -3-ethylcarbodiimide hydrochloride (2.7g, 14 mmol) and the reaction was allowed to stir for 3 hours at ambient temperature. The reaction mixture was then poured into water and extracted with ethyl acetate. The organics were washed with 0.5M sodium bisulfate, water, sodium bicarbonate, brine, dried over magnesium sulfate and concentrated in vacuo to afford 2.6 g of the crude product as a yellow foam. The crude material was purified by column chromatography (Si02, dichloromethane: acetone 9:1 – 3:1) to afford 1.2 g of the compound 4-b. Compounds of formulae VII and VIII, and related compounds, that may be synthesized using the method of this invention as an intermediate step are described in WO 97/22619 and United States Patent 6,204,261 the disclosure of which is herein incorporated by reference. Those related compounds may be synthesized from the product of the method of this invention, I, IV, or V, through modifications of the procedure set forth in Examples 4 through 6. Such modifications are well known in the art.PATENTUS 6559304https://patents.google.com/patent/US6559304B1PATENTWO 2008074816https://patents.google.com/patent/WO2008074816A1/en
Patent
Publication numberPriority datePublication dateAssigneeTitleEP0094095A2 *1982-05-121983-11-16F. Hoffmann-La Roche AgBicyclic carboxylic acids and their alkyl and aralkyl estersUS4692438A *1984-08-241987-09-08Hoffmann-La Roche Inc.Pyridazo-diazepines, diazocines, and -triazepines having anti-hypertensive activityWO1993023403A1 *1992-05-151993-11-25Merrell Dow Pharmaceuticals Inc.NOVEL MERCAPTOACETYLAMIDO PYRIDAZO[1,2]PYRIDAZINE, PYRAZOLO[1,2]PYRIDAZINE, PYRIDAZO[1,2-a][1,2]DIAZEPINE AND PYRAZOLO[1,2-a][1,2]DIAZEPINE DERIVATIVES USEFUL AS INHIBITORS OF ENKEPHALINASE AND ACEWO1994011353A1 *1992-11-121994-05-26University College LondonProcess for the preparation of (3r)- and (3s)-piperazic acid derivativesWO1995035308A1 *1994-06-171995-12-28Vertex Pharmaceuticals IncorporatedINHIBITORS OF INTERLEUKIN-1β CONVERTING ENZYMEFamily To Family CitationsUS6204261B11995-12-202001-03-20Vertex Pharmaceuticals IncorporatedInhibitors of interleukin-1β Converting enzyme inhibitorsFR2777888B11998-04-272004-07-16Hoechst Marion Roussel IncNOVEL DERIVATIVES OF ACID (3,4,7,8,9,10-HEXAHYDRO-6,10- DIOXO-6H-PYRIDAZINO [1,2-A] [1,2] DIAZEPINE-1-CARBOXYLIC, THEIR PROCESS OF PREPARATION AND THEIR APPLICATION TO THE PREPARATION OF MEDICINESFR2777889B11998-04-272004-07-09Hoechst Marion Roussel IncNOVEL DERIVATIVES OF OCTAHYDRO-6,10-DIOXO-6H- PYRIDAZINO [1,2-A] [1,2] DIAZEPINE-1-CARBOXYLIC, THEIR PREPARATION PROCESS AND THEIR APPLICATION TO THE PREPARATION OF THERAPEUTICALLY ACTIVE COMPOUNDS
////////////////Pralnacasan, VX 740, VX 470, HMR 3480, пралнаказан , برالناكاسان , 普那卡生 ,
CCOC1C(CC(=O)O1)NC(=O)C2CCCN3N2C(=O)C(CCC3=O)NC(=O)C4=NC=CC5=CC=CC=C54
