
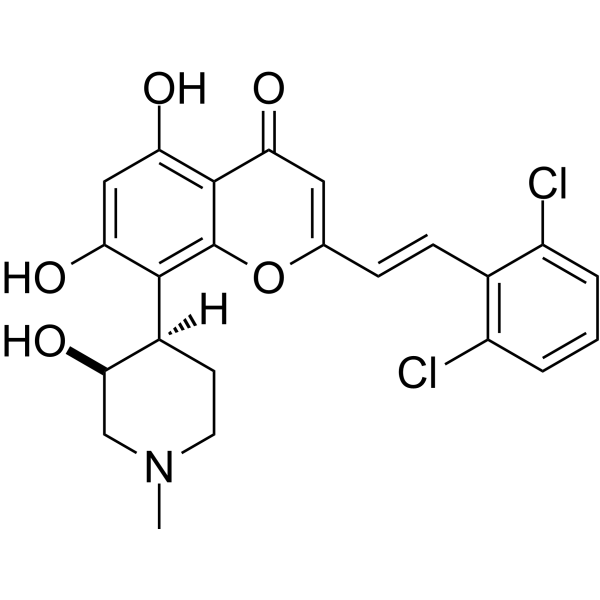

IIIM-290
4H-1-Benzopyran-4-one, 2-[2-(2,6-dichlorophenyl)ethenyl]-5,7-dihydroxy-8-[(3S,4R)-3-hydroxy-1-methyl-4-piperidinyl]-
| Molecular Weight |
462.32 |
|---|---|
| Formula |
C₂₃H₂₁Cl₂NO₅ |
| CAS No. |
2213468-64-3 |
CSIR-IIIM Jammu has filed an IND Application of “IIIM-290” to Drug Controller General of India for conducting Phase I/Phase II clinical trial of its capsule formulation in patients with locally advanced or metastatic pancreatic cancer. This IND candidate has emerged from the eight years of medicinal chemistry/ preclinical efforts of IIIM Jammu in the area of small molecule kinase inhibitors. IIIM-290 (NCE) is an orally bioavailable CDK inhibitor, obtained via semisynthetic modification of a natural product rohitukine. Institute has already secured a patent on this small molecule as well as on its oral capsule formulation.

IIIM-290 is a potent and oral CDK inhibitor with IC50s of 90 and 94 nM for CDK2/A and CDK9/T1.


PAPER
https://pubs.acs.org/doi/pdf/10.1021/acs.jmedchem.7b01765
Discovery and Preclinical Development of IIIM-290, an Orally Active Potent Cyclin-Dependent Kinase Inhibitor
- Sandip B. Bharate
- Vikas Kumar
- Shreyans K. Jain
- Mubashir J. Mintoo
- Santosh K. Guru
- Vijay K. Nuthakki
- Mohit Sharma
- Sonali S. Bharate
- Sumit G. Gandhi
- Dilip M. Mondhe
- Shashi Bhushan
- Ram A. Vishwa
Abstract

Rohitukine (1), a chromone alkaloid isolated from Indian medicinal plant Dysoxylum binectariferum, has inspired the discovery of flavopiridol and riviciclib, both of which are bioavailable only via intravenous route. With the objective to address the oral bioavailability issue of this scaffold, four series of rohitukine derivatives were prepared and screened for Cdk inhibition and cellular antiproliferative activity. The 2,6-dichloro-styryl derivative IIIM-290 (11d) showed strong inhibition of Cdk-9/T1 (IC50 1.9 nM) kinase and Molt-4/MIAPaCa-2 cell growth (GI50 < 1.0 μM) and was found to be highly selective for cancer cells over normal fibroblast cells. It inhibited the cell growth of MIAPaCa-2 cells via caspase-dependent apoptosis. It achieved 71% oral bioavailability with in vivo efficacy in pancreatic, colon, and leukemia xenografts at 50 mg/kg, po. It did not have CYP/efflux-pump liability, was not mutagenic/genotoxic or cardiotoxic, and was metabolically stable. The preclinical data presented herein indicates the potential of 11d for advancement in clinical studies.


Patent
IN201811026240



Patent
InventorRam A. VishwakarmaSandip B. BharateShashi BhushanDilip M. MondheShreyans K. JainSamdarshi MeenaSantosh K. GuruAnup S. PathaniaSuresh KumarAkanksha BehlMubashir J. MintooSonali S. BharatePrashant Joshi Current Assignee Council of Scientific and Industrial Research (CSIR)
https://patents.google.com/patent/US9932327B2/en
The disruption of any internal and external regulation of cellular growth leads to tumorogenesis by uncontrolled proliferation. This loss of control occurs at multiple levels in most of the cancer cases. Cyclin-dependent kinases (CDKs) have been recognized as key regulators of cell cycle progression. Alteration and deregulation of CDK activity have pathogenic link to the cancer. Number of cancers are associated with hyper-activation of CDKs as a result of mutation of the CDK genes or CDK inhibitor genes. Therefore, CDK inhibitors or modulators are of great interest to explore as novel therapeutic agents against cancer (Senderowicz, A. M. Leukemia 2001, 15, 1). Several classes of chemical inhibitors of CDK activity have been described (Zhang, J. et. al. Nat Rev Cancer. 2009, 9, 28) and some of them have reached to clinical pipeline for cancer.
Because CDK inhibitors are ATP competitive ligands; hence earlier they were typically described as purine class of compounds for example dimethylaminopurine, a first substance to be known as a CDK inhibitor (Neant, I. et al. Exp. Cell Res. 1988, 176, 68), olomoucine (Vesely, J. et al. Eur. J. Biochem. 1994, 224, 771) and roscovitine (Meijer, L. et al. Eur. J. Biochem. 1997, 243, 527). The IC50values of these purine class of compounds for CDK1/cyclin B are 120, 7 and 0.2-0.8 μM respectively (Gray, N. et al. Curr. Med. Chem. 1999, 6, 859). Some of the more potent members of this series have been prepared by the Schultz group using combinatorial approaches (Gray, N. S. et al. Science 1998, 281, 533). Number of synthetic flavoalkaloids having potent CDK inhibitory activity has been reviewed recently (Jain, S. K. et al. Mini–Rev. Med. Chem. 2012, 12, 632).
Specific CDKs operate in distinct phases of the cell cycle. CDK complexes with their respective type cyclin partners such as, complex of CDK2 and cyclin A is responsible for the cell’s progression from G1 phase to S phase (Sherr, C. J. Science 1996, 274, 1672). DNA synthesis (S phase) begins with the CDK mediated phosphorylation of Rb (retinoblastoma) protein. Phosphorylated Rb is released from its complex with E2F. The released E2F then promotes the transcription of numerous genes required for the cell to progress through S phase, including thymidylate synthase and dihydrofolate reductase which are required for cell progression (Hatakeyama, M. et. al, Cell Cycle Res. 1995, 1, 9; Zhang, H. S. et. al. Cell 1999, 97, 53). Majority of human cancers have abnormalities in some component of the Rb pathway because of hyper-activation of CDKs resulting from the over-expression of positive cofactors (cyclins/CDKs) or a decrease in negative factors (endogenous CDK inhibitors) or Rb gene mutations (Sausville, E. A. et. al, Pharmacol. Ther. 1999, 82, 285).
The CDK-9 is a member of the Cdc2-like family of kinases. Its cyclin partners are members of the family of cyclin T (T1, T2a and T2b) and cyclin K. The CDK-9/cyclin T complexes appear to be involved in regulating several physiological processes. CDK9/cyclin T1 belongs to the P-TEFb complex, and is responsible for the phosphorylation of carboxyl terminal domain of the RNA Polymerase II, thus promoting general elongation. CDK-9 has also been described as the kinase of the TAK complex, which is homologous to the P-TEFb complex and is involved in HIV replication. CDK9 also appears to be involved in the differentiation program of several cell types, such as muscle cells, monocytes and neurons, suggesting that it may have a function in controlling specific differentiative pathways. In addition, CDK-9 seems to have an anti-apoptotic function in monocytes, that may be related to its control over differentiation of monocytes. This suggests the involvement of CDK-9 in several physiological processes in the cell, the deregulation of which may be related to the genesis of transforming events that may in turn lead to the onset of cancer. In addition, since the complex CDK-9/cyclin T1 is able to bind to the HIV-1 product Tat, the study of the functions of CDK-9/cyclin T may be of interest in understanding the basal mechanisms that regulate HIV replication (Falco, G. D. and Giordano A. Cancer Biol. Therapy 2002, 1, 337).
Rohitukine belongs to a class of chromone alkaloids and it was isolated by chemists at Hoechst India Ltd. in the early 1990’s from Dysoxylum binectariferum Hook. which is phylogenetically related to the Ayurvedic plant, D. malabaricum Bedd., used for rheumatoid arthritis. Rohitukine was isolated as the constituent responsible for anti-inflammatory and immunomodulatory activity (Naik, R. G. et. al. Tetrahedron 1988, 44, 2081; U.S. Pat. No. 4,900,727, 1990). Medicinal chemistry efforts around this nature-derived flavone alkaloid led to discovery of two promising clinical candidates for treatment of cancer viz. flavopiridol of Sanofi-Aventis and P-276-00 of Piramal life sciences. Recently FDA has granted the orphan drug status to flavopiridol for treatment of chronic lymphocytic leukemia (CLL).
The molecular formula of rohitukine is C16H19NO5 and the structure has a molecular weight of 305.32 g/mol. The chemical structure of rohitukine (1) is shown below. The present invention reports new semi-synthetic analogs of rohitukine as promising inhibitors of cyclin-dependent kinases such as CDK-2 and CDK-9.

Synthesis of styryl analog 2-(2,6-dichlorostyryl)-5,7-dihydroxy-8-(3-hydroxy-1-methylpiperidin-4-yl)-4H-chromen-4-one (33)
This compound was synthesized using the procedure as described in example 4. Yellow solid; 1H NMR (DMSO-d6, 400 MHz): δ 7.68 (m, 2H), 7.61 (d, J=16 Hz, 1H), 7.49 (t, J=8 Hz, 1H), 7.14 (d, J=16 Hz, 1H), 6.41 (s, 1H), 5.85 (s, 1H), 4.53 (brs, 1H), 3.10-2.50 (m, 6H of piperidine), 2.65 (s, 3H), 1.62 (m, 1H); 13C NMR (DMSO-d6, 125 MHz): δ 179.68. 171.27, 159.20, 158.02, 154.03, 133.12, 131.49, 129.75, 128.35 (2C), 128.20, 127.90, 108.81, 106.79, 100.88, 100.52, 66.35, 59.82, 54.45, 43.15, 35.79, 22.01, 20.33, ESI-MS: m/z 462.01 [M+H]+; IR (CHCl3): νmax 3400, 2921, 1652, 1577, 1550, 1417, 1380, 1191, 1085 cm−1.
///////////IIIM-290, nda, india, phase 1, dcgi, CSIR, ROHITUKINE
|
OC1=C2C(OC(/C=C/C3=C(Cl)C=CC=C3Cl)=CC2=O)=C([C@]4([H])[C@H](O)CN(C)CC4)C(O)=C1 |