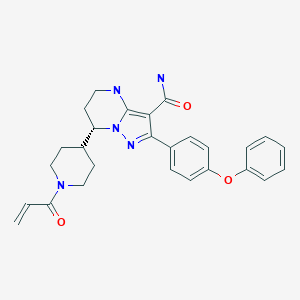
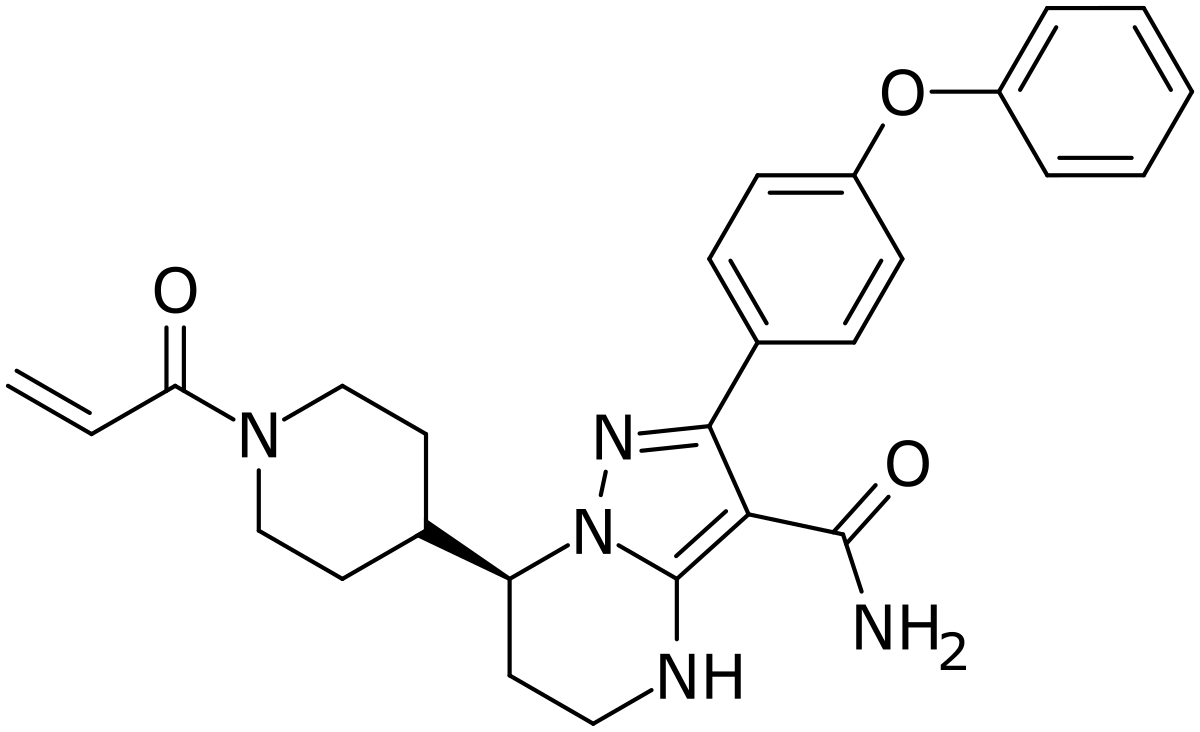
Zanubrutinib, BGB-3111
| Formula |
C27H29N5O3
|
|---|---|
| CAS |
1691249-45-2
|
| Mol weight |
471.5509
|
FDA , 2019/11/14, Brukinsa
ザヌブルチニブ ,
Antineoplastic, Bruton’s tyrosine kinase inhibitor, Mantle cell lymphoma
Zanubrutinib, sold under the brand name Brukinsa, is for the treatment of adult patients with mantle cell lymphoma (MCL) who have received at least one prior therapy.[3]
It was approved for medical use in the United States in November 2019.[4][3][5][6]
Zanubrutinib is classified as a Bruton’s tyrosine kinase (BTK) inhibitor. It is administered orally.
History
Efficacy was evaluated in BGB-3111-206 (NCT03206970), a phase II open-label, multicenter, single-arm trial of 86 patients with mantle cell lymphoma (MCL) who received at least one prior therapy.[5] Zanubrutinib was given orally at 160 mg twice daily until disease progression or unacceptable toxicity.[5] Efficacy was also assessed in BGB-3111-AU-003 (NCT 02343120), a phase I/II, open-label, dose-escalation, global, multicenter, single-arm trial of B‑cell malignancies, including 32 previously treated MCL patients treated with zanubrutinib administered orally at 160 mg twice daily or 320 mg once daily.[5][6]
The primary efficacy outcome measure in both trials was overall response rate (ORR), as assessed by an independent review committee.[5] In trial BGB-3111-206, FDG-PET scans were required and the ORR was 84% (95% CI: 74, 91), with a complete response rate of 59% (95% CI 48, 70) and a median response duration of 19.5 months (95% CI: 16.6, not estimable).[5] In trial BGB-3111-AU-003, FDG-PET scans were not required and the ORR was 84% (95% CI: 67, 95), with a complete response rate of 22% (95% CI: 9, 40) and a median response duration of 18.5 months (95% CI: 12.6, not estimable).[5] Trial 1 was conducted at 13 sites in China, and Trial 2 was conducted at 25 sites in the United States, United Kingdom, Australia, New Zealand, Italy, and South Korea.[6]
The U.S. Food and Drug Administration (FDA) granted zanubrutinib priority review, accelerated approval, breakthrough therapydesignation, and orphan drug designation.[3][5][7]
The FDA approved zanubrutinib in November 2019, and granted the application for Brukinsa to BeiGene USA Inc.[3][5][8]
PAPER
https://www.x-mol.com/paper/5799457
Discovery of Zanubrutinib (BGB-3111), a Novel, Potent, and Selective Covalent Inhibitor of Bruton’s Tyrosine Kinase Journal of Medicinal Chemistry ( IF 6.054 ) Pub Date: 2019-08-19 , DOI: 10.1021 / acs.jmedchem.9b00687
Yunhang Guo, Ye Liu, Nan Hu, Desheng Yu, Changyou Zhou, Gongyin Shi, Bo Zhang, Min Wei, Junhua Liu, Lusong Luo, Zhiyu Tang, Huipeng Song, Yin Guo, Xuesong Liu, Dan Su, Shuo Zhang, Xiaomin Song , Xing Zhou, Yuan Hong, Shuaishuai Chen, Zhenzhen Cheng, Steve Young, Qiang Wei, Haisheng Wang, Qiuwen Wang, Lei Lv, Fan Wang, Haipeng Xu, Hanzi Sun, Haimei Xing, Na Li, Wei Zhang, Zhongbo Wang, Guodong Liu, Zhijian Sun, Dongping Zhou, Wei Li, Libin Liu, Lai Wang, Zhiwei Wang
 |
Bruton’s tyrosine kinase (Btk) belongs to the Tec tyrosine kinase family (Vetrie et al., Nature 361: 226-233, 1993; Bradshaw, Cell Signal. 22: 1175-84, 2010). Btk is primarily expressed in most hematopoietic cells such as B cells, mast cells and macrophages (Smith et al., J. Immunol. 152: 557-565, 1994) and is localized in bone marrow, spleen and lymph node tissue. Btk plays important roles in B-cell receptor (BCR) and FcR signaling pathways, which involve in B-cell development, differentiation (Khan, Immunol. Res. 23: 147, 2001). Btk is activated by upstream Src-family kinases. Once activated, Btk in turn phosphorylates PLC gamma, leading to effects on B-cell function and survival (Humphries et al., J. Biol.Chem. 279: 37651, 2004).
[0003] These signaling pathways must be precisely regulated. Mutations in the gene encoding Btk cause an inherited B-cell specific immunodeficiency disease in humans, known as X-linked agammaglobulinemia (XLA) (Conley et al., Annu. Rev. Immunol. 27: 199-227, 2009). Aberrant BCR-mediated signaling may result in dysregulated B-cell activation leading to a number of autoimmune and inflammatory diseases. Preclinical studies show that Btk deficient mice are resistant to developing collagen- induced arthritis. Moreover, clinical studies of Rituxan, a CD20 antibody to deplete mature B-cells, reveal the key role of B-cells in a number of inflammatory diseases such as rheumatoid arthritis, systemic lupus erythematosus and multiple sclerosis (Gurcan et al, Int. Immunopharmacol. 9: 10-25, 2009). Therefore, Btk inhibitors can be used to treat autoimmune and/or inflammatory diseases.
[0004] In addition, aberrant activation of Btk plays an important role in pathogenesis of B-cell lymphomas indicating that inhibition of Btk is useful in the treatment of hematological malignancies (Davis et al, Nature 463: 88-92, 2010). Preliminary clinical trial results showed that the Btk inhibitor PCI-32765 was effective in treatment of several types of B-cell lymphoma (for example, 54thAmerican Society of Hematology (ASH) annual meeting abstract, Dec. 2012: 686 The Bruton’s Tyrosine Kinase (Btk) Inhibitor, Ibrutinib (PCI- 32765), Has Preferential Activity in the ABC Subtype of Relapsed/Refractory De Novo Diffuse Large B-Cell Lymphoma (DLBCL): Interim Results of a Multic enter, Open-Label, Phase I Study). Because Btk plays a central role as a mediator in multiple signal transduction pathways, inhibitors of Btk are of great interest as anti-inflammatory and/or anti-cancer agents {Mohamed et al., Immunol. Rev. 228: 58-73, 2009; Pan, Drug News perspect 21: 357-362, 200%; Rokosz et al., Expert Opin. Ther. Targets 12: 883-903, 2008; Uckun et al., Anti-cancer Agents Med. Chem. 7: 624-632, 2007; Lou et al, J. Med. Chem. 55(10): 4539-4550, 2012).
[0005] International application WO2014173289A disclosed a series of fused heterocyclic compounds as Btk inhibitors. In particular, WO2014173289A disclosed
(S)-7-(l-acryloylpiperidin-4-yl)-2-(4-phenoxyphenyl)-4,5,6,7-tetra-hydropyrazolo[l,5-a]pyrimi dine-3-carboxamide (hereinafter C
Compound 1
[0006] Compound 1 is a potent, specific and irreversible BTK kinase inhibitor. The data generated in preclinical studies using biochemical, cell based and animal studies suggested that Compound 1 could offer significant benefit in inhibiting tumor growth in B-cell malignancies. As Compound 1 was shown to be more selective than ibrutinib for inhibition of BTK vs. EGFR, FGR, FRK, HER2, HER4, ITK, JAK3, LCK, and TEC, it is expected to give rise to less side-effects than ibrutinib in clinic. In addition, Compound 1 showed significantly less inhibition of rituximab-induced antigen-dependent cell-mediated cytotoxicity (ADCC) than ibrutinib due to weaker ITK inhibition, and therefore may provide better efficacy when combined with rituximab or other ADCC-dependent antibody in treating B-cell malignancies.
[0007] Preclinical safety evaluation has demonstrated that Compound 1 was safer than ibrutinib in terms of the overall tolerance and severe toxicities in both rat and dog single and repeat dose toxicity studies up to 28 days. Additionally, Compound 1 had better bioavailability without accumulation issues observed for ibrutinib. These unique characteristics warrant further evaluation of Compound 1 in clinical studies.
[0008] However, Compound 1 was found to be an amorphous form according to the preparation method for Compound 27 in WO 2014173289A, which was further confirmed by the X-Ray Powder Diffraction pattern of FIG. 7A. The amorphous form was shown to have a low glass transition temperature as shown in FIG. 7B, indicating some difficulties in the drug formulation with the amorphous form, such as low stability and hard to purify. Therefore, it’s necessary to develop a new form of Compound 1 which possesses characteristics such as high melting point and better stability, suitable for drug formulation.
Scheme 1: Preparation of Compound 1 and deuterium-labeled Compound 1
Deuterium-Labeled Compound 1
Step 15: Synthesis of
(S)-7-(l-acryloylpiperidin-4-yl)-2-(4-phenoxyphenyl)-4,5,6,7-tetrahydropyrazolori,5-a1pyrimi dine-3-carboxamide (Compound 1
[0105] Under N2 atmosphere, ACN (12.0 v), water (12.5 v), BG-13 (8.0 Kg, 1.0 eq), and NaHC03 (2.5 eq.) were added to a reactor. The mixture was then cooled to -5-0 °C. To the mixture, the solution of acryloyl chloride (1.1 eq.) in MeCN (0.5 v) was added dropwise and
stirred until the reaction was completed. EA (6.0 v) was then added to the reactor, and stirred. The organic phase was collected. The aqueous layer was further extracted with EA (3.0 v). The organic phases were combined and washed with brine. The organic layer was collected and concentrated.
[0106] The residue was purified by silica gel (2 wt) column, eluted with 3% w/w methanol in DCM (21.0 v). The Compound 1 solution was collected and concentrated under vacuum. The residue was precipitated from EA/MTBE (2.0 v). The cake was collected by centrifugation as the product.
Step 15: Synthesis of (S)-7-(l-acryloylpiperidin-4-yl)-2-(4-phenoxyphenyl)
-4,5,6,7-tetrahydropyrazolori,5-a1pyrimidine-3-carboxamide (Compound 1, alternative method)
[0107] A mixture of CHsCN (10.0 v), purified water (5.0 v), NaOH (1.5 eq.) and BG-13 (1.0 eq.) was stirred to get a clear solution. EtOAc (6.0 v) was then charged to the reaction and separated. The organic phase was collected and washed with 15% brine (3.0 v) twice. The organic phase prepared above was concentrated and the solvent was swapped to CH3CN (residue volume: NMT 5.0 v). CH3CN (7.5 v) and purified water (12.5 v) were charged and cooled to 15-20°C. L-(+)-tartaric acid (0.5 eq) and NaHCCb (2.5 eq.) were charged to the reaction mixture. A solution of acryloyl chloride (1.1 eq.) in CH3CN (0.5 v) was charged drop-wise to the reaction mixture. After the reaction was completed, EtOAc (6.0 v) was charged to the reaction mixture and organic layer was collected. Aqueous phase was further extracted with EA (3.0 v). The organic layers were combined, washed with 15% brine (5.0 v) and concentrated. The solvent was swapped to DCM (volume of residue: 1.5-2.0 v) and purified by silica gel column (silica gel: 100-200 mush, 2.0 w/ w; eluent: 3%> w/ w MeOH in DCM (about 50 v). The collected solution was concentrated and swapped to EtOAc (4.0 v). MTBE (6.4 v) was charged drop-wise to residue at 50°C. The mixture was then cooled to 5°C and the cake was collected centrifugation.
Step 16: Preparation of Crystalline Form A of Compound 1
[0108] The above cake of Compound 1 was dissolved in 7.0 volumes of DCM, and then swapped to solvent EA. After recrystallization from EA/MTBE, the cakes was collected by centrifugation, and was dried under vacuum. This gave 4.44 Kg product (Yield: 70.2%).
[0109] The product was then characterized by X-ray powder diffraction (XRPD) pattern method, which was generated on a PANalytical Empyrean X-ray powder diffractometer with the XRPD parameters as follows: X-Ray wavelength (Cu, ka, Kal (A): 1.540598, Ka2(A): 1.544426; Ka2/Kal intensity ratio: 0.50); X-Ray tube setting (45 Kv, 40mA); divergence slit (automatic); scan mode (Continuous); scan range (°2TH) (3°-40); step size (°2TH) (0.0131); scan speed (°/min) (about 10). The XRPD result found the resultant product as a crystalline shown in FIG. 1.
[0110] The differential scanning calorimetry (DSC) curves shown as in FIG. 2 was generated on a TA Q2000 DSC from TA Instruments. The DSC parameters used includes: temperature (25°C-desired temperature); heating rate (10°C/min) ; method (ramp); sample pan (aluminum, crimped); purge gas (N2). DSC result showed a sharp melting point at 139.4°C (onset temperature).
[0111] The thermo-gravimetric analysis (TGA) curves shown as in FIG. 3 was generated on a TA Q5000 TGA from TA Instruments. The TGA parameters used includes: temperature
(RT-desired temperature); heating rate (10°C/min); method (ramp); sample pan (platinum, open); purge gas (N2). TGA result showed is anhydrous with no weight loss even up to 110 °C.
[0112] The proton nuclear magnetic resonance ^H-NMR) shown as in FIG. 4 was collected on a Bruker 400M NMR Spectrometer in DMSO-de. ¾-NMR (DMSO-de) δ 7.50 (d, J= 8.6 Hz, 2H), 7.46-7.38 (m, 2H), 7.17 (t, J = 7.6 Hz, 1H), 7.08 (d, J= 7.6 Hz, 2H), 7.05 (d, J= 8.8 Hz, 2H), 6.85-6.72 (m, 1H), 6.67 (s, 1H), 6.07 (dd, J= 16.8, 2.2 Hz, 1H), 5.64 (dd, J= 10.4 Hz, 2.2 Hz, 1H), 4.55-4.38 (m, 1H), 4.17-3.94 (m, 2H), 3.33-3.22 (m, 2H), 3.08-2.88 (m, 1H), 2.67-2.51 (m, 1H), 2.36-2.15 (m, 1H), 2.12-1.82 (m, 2H), 1.79-1.65 (m, 1H), 1.63-1.49 (m, 1H), 1.38-1.08 (m, 2H).
[0113] The carbon nuclear magnetic resonance (13C-NMR) shown as in FIG. 5 was collected on a Bruker 400M NMR Spectrometer in DMSO-de. 13C-NMR spectra for Crystalline Form A of Compound 1.
Step 15: Synthesis of (S)-7-(1-acrvlovlpiperidin-4-vl)-2-(4-phenoxvphenyl)-4.5.6.7-tetrahvdropvrazolo[1.5-a1pvrimidine-3-carboxamide (Compound 1)
[0119] Under N2 atmosphere, ACN (12.0 v), water (12.5 v), BG-13 (8.0 Kg, 1.0 eq), and NaHCO3 (2.5 eq.) were added to a reactor. The mixture was then cooled to -5-0 °C. To the mixture, the solution of acryloyl chloride (1.1 eq.) in MeCN (0.5 v) was added dropwise and stirred until the reaction was completed. EA (6.0 v) was then added to the reactor, and stirred. The organic phase was collected. The aqueous layer was further extracted with EA (3.0 v). The organic phases were combined and washed with brine. The organic layer was collected and concentrated.
[0120] The residue was purified by silica gel (2 wt) column, eluted with 3% w/w methanol in DCM (21.0 v). The Compound 1 solution was collected and concentrated under vacuum. The residue was precipitated from EA/MTBE (2.0 v). The cake was collected by centrifugation as the product.
Step 15: Synthesis of (S)-7-(l-acryloylpiperidin-4-yl)-2-(4-phenoxyphenyl) -4.5.6.7-tetrahvdropvrazolori.5-a1pvrimidine-3-carboxamide (Compound 1. alternative method)
[0121] A mixture of CH3CN (10.0 v), purified water (5.0 v), NaOH (1.5 eq.) and BG-13 (1.0 eq.) was stirred to get a clear solution. EtOAc (6.0 v) was then charged to the reaction and separated. The organic phase was collected and washed with 15% brine (3.0 v) twice. The organic phase prepared above was concentrated and the solvent was swapped to CH3CN (residue volume: NMT 5.0 v). CH3CN (7.5 v) and purified water (12.5 v) were charged and cooled to 15-20°C. L-(+)-tartaric acid (0.5 eq) and NaHCO3 (2.5 eq.) were charged to the reaction mixture. A solution of acryloyl chloride (1.1 eq.) in CH3CN (0.5 v) was charged drop-wise to the reaction mixture. After the reaction was completed, EtOAc (6.0 v) was charged to the reaction mixture and organic layer was collected. Aqueous phase was further extracted with EA (3.0 v). The organic layers were combined, washed with 15% brine (5.0 v) and concentrated. The solvent was swapped to DCM (volume of residue: 1.5-2.0 v) and purified by silica gel column (silica gel: 100-200 mush, 2.0 w/ w; eluent: 3% w/ w MeOH in DCM (about 50 v). The collected solution was concentrated and swapped to EtOAc (4.0 v). MTBE (6.4 v) was charged drop-wise to residue at 50°C. The mixture was then cooled to 5°C and the cake was collected centrifugation.
References
- ^ “Zanubrutinib (Brukinsa) Use During Pregnancy”. Drugs.com. 3 January 2020. Retrieved 26 January 2020.
- ^ “Zanubrutinib”. DrugBank. Retrieved 15 November 2019.
- ^ Jump up to:a b c d “FDA approves therapy to treat patients with relapsed and refractory mantle cell lymphoma supported by clinical trial results showing high response rate of tumor shrinkage”. U.S. Food and Drug Administration (FDA) (Press release). 14 November 2019. Retrieved 15 November 2019.
![]() This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ “Brukinsa (zanubrutinib) FDA Approval History”. Drugs.com. 14 November 2019. Archived from the original on 15 November 2019. Retrieved 15 November 2019.
- ^ Jump up to:a b c d e f g h i “FDA grants accelerated approval to zanubrutinib for mantle cell lymphoma”. U.S. Food and Drug Administration (FDA)(Press release). 15 November 2019. Archived from the original on 28 November 2019. Retrieved 27 November 2019.
![]() This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ Jump up to:a b c “Drug Trials Snapshots Brukinsa”. U.S. Food and Drug Administration (FDA). 14 November 2019. Retrieved 26 January 2020.
![]() This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ “Zanubrutinib Orphan Drug Designation and Approval”. U.S. Food and Drug Administration (FDA). 28 November 2019. Archived from the original on 28 November 2019. Retrieved 27 November 2019.
![]() This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ “Drug Approval Package: Brukinsa”. U.S. Food and Drug Administration (FDA). 27 November 2019. Archived from the original on 28 November 2019. Retrieved 27 November 2019.
![]() This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain.
External links
- “Zanubrutinib”. Drug Information Portal. U.S. National Library of Medicine.
 |
|
| Clinical data | |
|---|---|
| Trade names | Brukinsa |
| Other names | BGB-3111 |
| AHFS/Drugs.com | Monograph |
| License data |
|
| Pregnancy category |
|
| Routes of administration |
By mouth |
| Drug class | Bruton’s tyrosine kinase(BTK) inhibitor |
| Legal status | |
| Legal status |
|
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| PubChem SID | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEMBL | |
| Chemical and physical data | |
| Formula | C27H29N5O3 |
| Molar mass | 471.5509 g·mol−1 |
| 3D model (JSmol) | |
/////////////////Zanubrutinib, FDA 2019, ザヌブルチニブ , занубрутиниб , زانوبروتينيب , BGB-3111