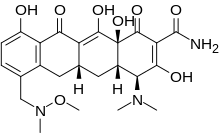
Sarecycline
サレサイクリン
MW 487.5024, MF C24H29N3O8 FREE FORM
Paratek INNOVATOR
FDA 2018/10/1 APPROVED SEYSARA, ALMIRALL, for the oral treatment of inflammatory lesions of non-nodular moderate to severe acne vulgaris in patients 9 years of age and older
P005672,
- P 005672
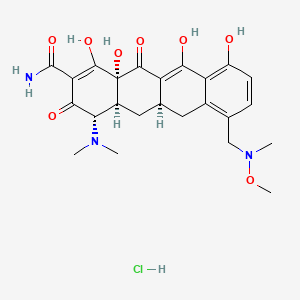
CAS 1035979-44-2 HCl
| Molecular Formula | C24 H29 N3 O8 . Cl H |
| Molecular Weight | 523.963 |
P-005672
PTK-AR-01
SC-1401
WC-3035
Sarecycline (trade name Seysara; development code WC-3035) is a tetracycline-derived antibiotic. In the United States, it was approved by the FDA in October 2018 for the treatment of moderate to severe acne vulgaris.[1]
Paratek Pharmaceuticals, Inc. licensed the US rights to sarecycline for the treatment of acne in the United States to Actavis, a subsidiary of Allergan, while retaining rights in the rest of the world.[2]
Allergan initiated a Phase 3 study in December 2014 evaluating the efficacy and safety of sarecycline tablets 1.5 mg/kg per day taken orally for 12 weeks versus placebo in the treatment of acne vulgaris.[3] Two phase 3 randomized, multi-center, double-blind, placebo-controlled studies evaluating the efficacy and safety of sarecycline in moderate to severe acne reported positive results on 27 March 2017.[4]
SYN
US 2016/0200671


PATENT
WO 2008079363
PATENT
WO 2008079339
PATENT
WO 2012155146
EXAMPLES
[00104] The following examples illustrate the synthesis of the compounds described herein.
Synthesis of (4S,4aS,5aR,12aS)-4-dimethylamino-3,10,12,12a-tetrahydroxy-7-[(methoxy(methyl)amino)-methyl]-l,ll-dioxo-l,4,4a,5,5a,6,ll,12a-octahydro-naphthacene-2-carboxylic acid amide (“the free base”).
[00105] A solution of 7-formylsancycline TFA salt (2.23 g) and N,0-dimethylhydroxylamine hydrochloride (780 mg) in N,N-dimethylacetamide (15 mL) was stirred for 10 minutes at room temperature under argon atmosphere. To this solution was added sodium cyanoborohydride (302 mg). The solution was stirred for 5 minutes and monitored by LC-MS. The reaction mixture was poured into diethyl ether, and the resulting precipitates were collected by filtration under vacuum. The crude product was purified by prep-HPLC using a C18 column (linear gradient 10-40% acetonitrile in 20 mM aqueous triethanolamine, pH 7.4). The prep-HPLC fractions were collected, and the organic solvent (acetonitrile) was evaporated under reduced pressure. The resulting aqueous solution was loaded onto a clean PDVB SPE column, washed with distilled water, then with a 0.1 M sodium acetate solution followed by distilled water. The product was eluted with
acetonitrile. The eluent was concentrated under reduced pressure, 385 mg was obtained as free base.
Synthesis of crystalline mono hydrochloride salt of (4S,4aS,5aR,12aS)-4-dimethylamino-3,10,12,12a-tetrahydroxy-7-[(methoxy(methyl)amino)-methyl]-l,ll-dioxo-l,4,4a,5,5a,6,ll,12a-octahydro-naphthacene-2-carboxylic acid amide (the “Crystalline Mono Hydrochloride Salt”).
[00106] Crude (4S,4aS,5aR,12aS)-4-dimethylamino-3, 10,12,12a-tetrahydroxy-7-[(methoxy(methyl)amino)-methyl]-l ,ll-dioxo-l,4,4a,5,5a,6,l l ,12a-octahydro-naphthacene-2-carboxylic acid amide (lOOg, app. 35% assay) was purified on preparative column chromatography. The desired fractions (8-10 liters) were combined and the pH was adjusted to 7.0-7.5 using ammonium hydroxide. This aqueous solution was extracted 3 times with dichloromethane (4 liters each time). The dichloromethane layers were combined and concentrated under reduced pressure. The residue was suspended in ethanol (800 ml) and 20 ml water was added. The pH was gradually adjusted to pH 1.6-1.3 using 1.25M hydrochloric acid in methanol and the mixture was stirred for 20-60 minutes at which point the free base was completely dissolved. The solution was concentrated under reduced pressure to 200-250 ml and was seeded with (4S,4aS,5aR,12aS)-4-dimethylamino-3,10, 12, 12a-tetrahydroxy-7-[(methoxy(methyl)amino)-methyl]- 1, 11-dioxo-l,4,4a,5,5a,6,l l,12a-octahydro-naphthacene-2-carboxylic acid amide mono HQ crystals (100-200 mg). The stirring was continued for 2-18 hours while the slurry was kept at <5°C. The resulting crystals were filtered, washed with ethanol (50 mL) and dried under reduced pressure to a constant weight. 20g crystalline (4S,4aS,5aR,12aS)-4-dimethylamino-3,10, 12, 12a-tetrahydroxy-7-[(methoxy(methyl)amino)-methyl]- 1, 11-dioxo-l,4,4a,5,5a,6,l l,12a-octahydro-naphthacene-2-carboxylic acid amide mono hydrochloride was isolated in > 90% purity and > 90% assay.
Synthesis of crystalline mono mesylate salt of (4S,4aS,5aR,12aS)-4-dimethylamino-3,10,12,12a-tetrahydroxy-7-[(methoxy(methyl)amino)-methyl]-l,ll-dioxo-l,4,4a,5,5a,6,ll,12a-octahydro-naphthacene-2-carboxylic acid (the “Crystalline Mesylate Salt”).
[00107] (4S,4aS,5aR,12aS)-4-dimethylamino-3, 10,12, 12a-tetrahydroxy-7-[(methoxy(methyl)amino)-methyl]-l,ll-dioxo-l,4,4a,5,5a,6,l l,12a-octahydro-naphthacene-2-carboxylic acid amide free base (74mg) was suspended in ethanol (740μ1) and heated with stirring to 60°C (bath temperature). Methane sulfonic acid (1.1 eq, 167μ1 as 1M solution in THF) was added and most of the solid dissolved. After five minutes, the suspension was cooled to ambient temperature over approximately 1.75 hours (uncontrolled in oil bath). By 53 °C, solid had precipitated which was filtered at ambient temperature under reduced pressure. A further portion of ethanol (200μ1) was added to aid filtration, as the suspension was viscous. The cake was washed with n-hexane (400μ1) and air dried on filter for approximately 30 minutes to yield 59 mg (67% yield) of yellow solid.
Synthesis of crystalline mono sulfate salt of (4S,4aS,5aR,12aS)-4-dimethylamino-3,10,12,12a-tetrahydroxy-7-[(methoxy(methyl)amino)-methyl]-l,ll-dioxo-l,4,4a,5,5a,6,ll,12a-octahydro-naphthacene-2-carboxylic acid (the “Crystalline Sulfate Salt”).
[00108] (4S,4aS,5aR,12aS)-4-dimethylamino-3, 10,12, 12a-tetrahydroxy-7-[(methoxy(methyl)amino)-methyl]-l,l l-dioxo-l,4,4a,5,5a,6,l l,12a-octahydro-naphthacene-2-carboxylic acid amide free base (86mg) was suspended in ethanol (500μ1) and heated with stirring to 63 °C (bath temperature) at which temperature most of the free base had dissolved. Sulfuric acid (1.1 eq, 194μ1 as 1M solution in water) was added and all of the solid dissolved. The solution was cooled to ambient temperature over approximately 1.75 hours (uncontrolled in oil bath) at which temperature no solid had precipitated. Methyl t-butyl ether (MtBE) was added as an antisolvent (4 x 50μ1). Each addition caused a cloud point, but the solid re-dissolved on stirring. The solution was stirred with a stopper for approximately 3 hours after which time solid precipitated. The solid was filtered under reduced pressure and washed with MtBE (3 x 200μ1) and air dried on filter for
approximately 45 minutes to yield 93 mg (90% yield) of yellow solid.
COMPARATIVE EXAMPLE 1
Synthesis of amorphous bis hydrochloride salt of (4S,4aS,5aR,12aS)-4-dimethylamino-3,10,12,12a-tetrahydroxy-7-[(methoxy(methyl)amino)-methyl]-l,ll-dioxo-l,4,4a,5,5a,6,ll,12a-octahydro-naphthacene-2-carboxylic acid amide.
[00109] (4S,4aS,5aR,12aS)-4-dimethylamino-3, 10,12, 12a-tetrahydroxy-7-[(methoxy(methyl)amino)-methyl]-l,l l-dioxo-l,4,4a,5,5a,6,l l,12a-octahydro-naphthacene-2-carboxylic acid amide free base (1 g) was suspended in methanol (50 mL). The freebase was converted to the hydrochloride salt by adding an excess of methanolic HCl followed by under reduced pressure evaporation to give 1.1 g yellow solid: MS (Mz+1 = 488). 1H NMR (300 MHz, CD30D) δ 7.46 (d, 1H, J = 8.6 Hz), 6.81 (d, 1H, J = 8.6 Hz), 4.09 (d, 1H, J = 1.0 Hz), 3.79 (d, 1H, J = 13.1 Hz), 3.73 (d, 1H, J = 13.1 Hz), 3.36 (m, 1H), 3.27 (s, 3H), 3.08-2.95 (8H), 2.61 (s, 3H), 2.38 (t, 1H, J = 14.8), 2.22 (m, 1H), 1.64 (m, 1H). An XRPD pattern is shown in Figure 10 and a TGA and DSC curve overlaid are shown in Figure 11.
COMPARATIVE EXAMPLE 2
Synthesis of amorphous mono hydrochloride salt of (4S,4aS,5aR,12aS)-4- dimethylamino-3,10,12,12a-tetrahydroxy-7-[(methoxy(methyl)amino)-methyl]-l,ll- dioxo-l,4,4a,5,5a,6,ll,12a-octahydro-naphthacene-2-carboxylic acid amide.
[00110] A sample of Crystalline Mono Hydrochloride Salt (2.09 g) was dissolved in water (250 ml, 120 vols), filtered and frozen in a -78°C bath. Water was removed from the solidified sample using a lyophilizer for 110 hours to yield the amorphous mono hydrochloride salt as a fluffy yellow solid, that was confirmed to be amorphous by XRPD analysis .
PATENT
US 20130302442
PATENT
WO 2015153864
PATENT
WO 2018051102
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2003075857
References
- Jump up^ “FDA Approves Sarecycline for Moderate to Severe Acne”. MedScape. October 2, 2018.
- Jump up^ https://www.bloomberg.com/research/stocks/snapshot/snapshot_article.asp?ticker=N4CN:GR&txtsize=s
- Jump up^ “Study to Evaluate Safety & Efficacy of Sarecycline in Treatment of Acne – Full Text View – ClinicalTrials.gov”.
- Jump up^ “Allergan and Paratek Announce Positive Results From Two Phase 3 Trials of Sarecycline for the Treatment of Moderate to Severe Acne”. http://www.globenewswire.com. Retrieved 16 May 2017.
External links
- Sarecycline – Almirall S.A./Paratek Pharmaceuticals, Adis Insight
|
|
| Clinical data | |
|---|---|
| Trade names | Seysara |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| ChemSpider | |
| UNII | |
| Chemical and physical data | |
| Formula | C24H29N3O8 |
| Molar mass | 487.51 g·mol−1 |
| 3D model (JSmol) | |
////////////Sarecycline, Seysara, WC-3035 FDA 2018, サレサイクリン , P-005672 , PTK-AR-01 , SC-1401, WC-3035,