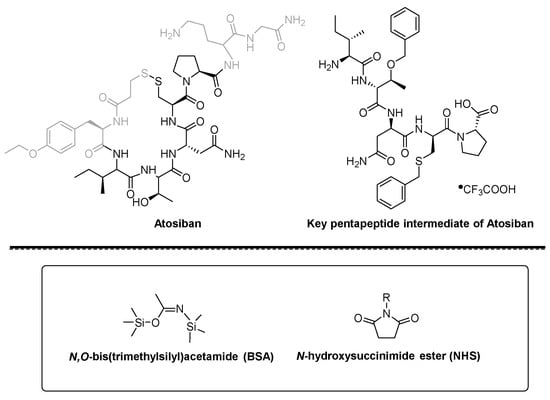
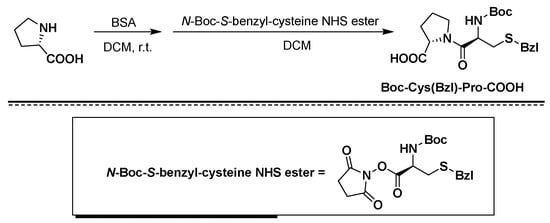
CAS Name: 2-[5-Ethyltetrahydro-5-[tetrahydro-3-methyl-5-[tetrahydro-6-hydroxy-6-(hydroxymethyl)-3,5-dimethyl-2H-pyran-2-yl]-2-furyl]-2-furyl]-9-hydroxy-b-methoxy-a,g,2,8-tetramethyl-1,6-dioxaspiro[4.5]decane-7-butyric acid
Literature References: Polyether antibiotic. Major factor in antibiotic complex isolated from Streptomyces cinnamonensis. Discovery and isolation: Haney, Hoehn, Antimicrob. Agents Chemother.1967, 349. Production: Haney, Hoehn, US3501568 (1970 to Lilly). Structure: Agtarap et al.,J. Am. Chem. Soc.89, 5737 (1967). Crystal structure studies: Lutz et al.,Helv. Chim. Acta53, 1732 (1970); ibid.54, 1103 (1971). Fermentation studies: Stark et al.,Antimicrob. Agents Chemother.1967, 353. Chemistry: Agtarap, Chamberlin, ibid. 359. Stereocontrolled total synthesis: T. Fukuyama et al.,J. Am. Chem. Soc.101, 262 (1979); D. B. Collum et al.,ibid.102, 2117, 2118, 2120 (1980). 13C-NMR study: J. A. Robinson, D. L. Turner, Chem. Commun.1982, 148. Biosynthesis: Day et al.,Antimicrob. Agents Chemother.4, 410 (1973). Review: Stark, “Monensin, A New Biologically Active Compound Produced by a Fermentation Process”, in Fermentation Advances, Pap. Int. Ferment. Symp., 3rd, 1968, D. Perlman, Ed. (Academic Press, New York, 1969) pp 517-540.
Properties: Crystals, mp 103-105° (monohydrate). [a]D +47.7°. pKa 6.6 (in 66% DMF). Very stable under alkaline conditions. Slightly sol in water; more sol in hydrocarbons; very sol in other organic solvents. LD50 of monensin complex in mice, chicks (mg/kg): 43.8 ± 5.2, 284 ± 47 orally (Haney, Hoehn).
Melting point: mp 103-105° (monohydrate)
pKa: pKa 6.6 (in 66% DMF)
Optical Rotation: [a]D +47.7°
Toxicity data: LD50 of monensin complex in mice, chicks (mg/kg): 43.8 ± 5.2, 284 ± 47 orally (Haney, Hoehn)
The structure of monensin was first described by Agtarap et al. in 1967, and was the first polyether antibiotic to have its structure elucidated in this way. The first total synthesis of monensin was reported in 1979 by Kishi et al.[3]
Production / synthesis Monensin is produced in vivo by Streptomyces cinnamonensis as a natural defense against competing bacteria. Monensin presents a formidable challenge to synthetic chemists as it possesses 17 asymmetric centers on a backbone of only 26 carbon atoms. Although its total synthesis has been described (e.g., Kishi et al., 1979), the high complexity of monensin makes an extraction from the bacterium the most economical procedure for its production. The total synthesis has 56 steps and a yield of only 0.26%. The chemical precursors are 2-allyl-1,3-propanediol and 2- (furan-2-yl)acetonitrile. The method used for synthesizing monensin is based on the principle of “absolute asymmetric synthesis”. Molecules are constructed out of prefabricated building blocks in the correct conformation, aiming for higher yields of the desired enantiomer. New stereocenters are also introduced. Using this method, monensin is assembled in two parts, a larger right side and a smaller left one. The penultimate step is connecting the left and the right halves of monensin, which are independently generated, in an Aldol-condensation. The two halves’ keto end groups (C7/ C8) are linked by eliminating a water molecule. The C7 atom is favored over the C1 atom, because it is more reactive. For catalyzing this step, Yoshito Kishi’s group used iPr2NMgBr (Hauser base) and THF to coordinate it at a temperature of − 78°C. Thus, they were able to isolate the molecule in the right conformation at a ratio of 8:1. Due to the low temperature required for a high yield of the correct enantiomer, the reaction is very solw. One of the most difficult steps is the last one: the connection of the spiro center. This is due to a characteristic feature of spiro compounds; they open and close very easily. Therefore, the conditions for forming the right conformation must be optimal in the last step of synthesis. The biosynthesis in a cell culture of Streptomyces cinnamonensis involves a complex medium containing, among other components, glucose, soybean oil, and grit. Cultivation is carried out for a week at a temperature of 30°C and under constant aeration. Product isolation requires filtration, acidification to pH3, extraction with chloroform and purification with activated carbon. In this way, a few grams per liter of monensin are produced and isolated. For crystallization, azeotropic distillation is necessary. In vivo, polyether backbones are assembled by modular polyketide synthases and are modified by two key enzymes, epoxidase and epoxide hydrolase, to generate the product. Precursors of the polyketide pathway are acetate, butyrate and propionate.
A polyether antibiotic, Monensin was the first member of this class of molecules to be structurally characterized.1 The structural features of these polyethers comprise of a terminal carboxylic acid, multiple cyclic ether rings (ex. Tetrahydrofuran and tetrahydropyran), a large amount of stereocenters and (for many of these molecules) one or more spiroketal moieties.2 Monensin was introduced into the market in 1971 and is used to fight coccidial infections in poultry and as an additive in cattle feed.3 Of the 26 carbon atom’s in Monensin’s backbone, 17 are stereogenic and six of those are contiguous. Coupled with a spiroketal moiety, three hydrofuran rings and two hydropyran rings, the molecule was an attractive synthetic target.
1. Agtarap, A.; Chamberlain, J.W.; Pinkerton, M.; Stein-rauf, L. J. Am. Chem. Soc. 1967, 89, 5737 2. Polyether Antibiotics : Naturally Occurring Acid Ionophores. Westley J.W.; Marcel Dekker: New York (1982) Vol. 1-2. 3. Stark, W.M. In Fermentation Advances, Perlman, D., Ed., Academic Press: New York, 1969, 517
Retrosynthetic Analysis of Monensin
//////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
The structure of the sodium (Na+) complex of monensin A.
Monensin A is an ionophore related to the crown ethers with a preference to form complexes with monovalent cations such as: Li+, Na+, K+, Rb+, Ag+, and Tl+.[4][5] Monensin A is able to transport these cations across lipid membranes of cells in an electroneutral (i.e. non-depolarizing) exchange, playing an important role as an Na+/H+antiporter. Recent studies have shown that monensin may transport sodium ion through the membrane in both electrogenic and electroneutral manner.[6] This approach explains ionophoric ability and in consequence antibacterial properties of not only parental monensin, but also its derivatives that do not possess carboxylic groups. It blocks intracellular protein transport, and exhibits antibiotic, antimalarial, and other biological activities.[7] The antibacterial properties of monensin and its derivatives are a result of their ability to transport metal cations through cellular and subcellular membranes.[8]
Uses
Monensin is used extensively in the beef and dairy industries to prevent coccidiosis, increase the production of propionic acid and prevent bloat.[9] Furthermore, monensin, but also its derivatives monensin methyl ester (MME), and particularly monensin decyl ester (MDE) are widely used in ion-selective electrodes.[10][11][12]
In laboratory research, monensin is used extensively to block Golgi transport.[13][14][15]
Toxicity
Monensin has some degree of activity on mammalian cells and thus toxicity is common. This is especially pronounced in horses, where monensin has a median lethal dose 1/100th that of ruminants. Accidental poisoning of equines with monensin is a well-documented occurrence which has resulted in deaths.[16]
^ Kallen, K. J.; Quinn, P.; Allan, D. (1993-02-24). “Monensin inhibits synthesis of plasma membrane sphingomyelin by blocking transport of ceramide through the Golgi: evidence for two sites of sphingomyelin synthesis in BHK cells”. Biochimica et Biophysica Acta (BBA) – Lipids and Lipid Metabolism. 1166 (2–3): 305–308. doi:10.1016/0005-2760(93)90111-l. ISSN0006-3002. PMID8443249.
^ Zhang, G. F.; Driouich, A.; Staehelin, L. A. (December 1996). “Monensin-induced redistribution of enzymes and products from Golgi stacks to swollen vesicles in plant cells”. European Journal of Cell Biology. 71 (4): 332–340. ISSN0171-9335. PMID8980903.
Percent Composition: C 39.85%, H 2.13%, Cl 10.69%, I 38.28%, N 4.22%, O 4.83%
Literature References: Salicylanilide derivative. Prepn: M. A. C. Janssen, V. K. Sipido, BE839481; eidem,US4005218 (1976, 1977 both to Janssen). Effectiveness against Taenia pisiformis in rabbits: R. A. F. Chevis et al.,Vet. Parasitol.7, 333 (1980); against Ancylostoma caninum: J. Guerrero et al.,J. Parasitol.68, 616 (1983); against Fasciola hepatica in sheep: B. E. Stromberg et al.,ibid.70, 446 (1984). Prolonged effect on Haemonchus contortus in sheep: C. A. Hall et al.,Res. Vet. Sci.31, 104 (1981). Acts by uncoupling oxidative phosphorylation: H. Van den Bossche et al.,Arch. Int. Physiol. Biochim.87, 851 (1979); H. J. Kane et al.,Mol. Biochem. Parasitol.1, 347 (1980).
Properties: Crystals from methanol, mp 217.8°.
Melting point: mp 217.8°
Therap-Cat-Vet: Anthelmintic.
N-{5-chloro-4-[(4-chlorophenyl)(cyano)methyl]-2-methylphenyl}-2-hydroxy-3,5-diiodobenzamide is an aromatic amide resulting from the formal condensation of the carboxy group of 3,5-diiodosalicylic acid with the amino group of aniline substituted at positions 2, 4, and 5 by methyl, (4-chlorophenyl)(cyano)methyl, and methyl groups respectively. It is a nitrile, a member of phenols, an organoiodine compound, a monocarboxylic acid amide, an aromatic amide and a member of monochlorobenzenes.
Closantel is a broad-spectrum antiparasitic agent used against
several species and developmental stages of trematodes, nematodes and
arthropods. The anti-trematode activity of closantel is mainly used
against liver fluke. The anti-nematode and anti-arthropod activity is
especially used against those species which feed on blood or plasma.
The drug is widely used in sheep and cattle and can be used
either parenterally (s.c. or i.m.) or orally for both prophylactic and
therapeutic purposes and is available as drench, bolus and injectable
formulations. Closantel has also been combined with mebendazole and
several other benzimidazoles in drench formulations for sheep and with
levamisole in a bolus for cattle (Marsboom et al., 1989).
Closantel has not been evaluated previously by the Joint FAO/WHO
Expert Committee on Food Additives.
Closantel sodium (Closantel Sodium) is a kind of very strong oxidative phosphorylation uncoupler, can suppress the mitochondrial phosphorylation process of polypide, nematode and insect etc. are contacted with blood circulation closely or sucking blood property worm all has and efficiently kills effect, be a kind of broad-spectrum de-worming medicine of efficient, low toxicity, it is huge on market a very large development potentiality.
And 4-chloro-phenyl–(the chloro-4-amino of 2–5-aminomethyl phenyl) cyano group methane is a kind of key intermediate for the synthesis of closantel sodium.But in prior art, the report of the synthetic method of relevant 4-chloro-phenyl–(the chloro-4-amino of 2–5-aminomethyl phenyl) cyano group methane is actually rare, is mainly the iron powder reducing synthetic method.As United States Patent (USP) (US4005218) relates to a kind of with the chloro-α of 4–[the chloro-4-of 2-(hydroxyl imido grpup)-5-methyl-2, the 5-phenylidene] benzyl cyanide (I) is raw material, with excessive iron powder, in ammonium chloride, water and toluene mixing solutions, heating reflux reaction, filter, clean filter cake with a large amount of solvents as tetrahydrofuran (THF) or 4-methyl-2 pentanone, filtrate boils off solvent, then adds the toluene recrystallization to obtain 4-chloro-phenyl–(the chloro-4-amino of 2–5-aminomethyl phenyl) cyano group methane (II).This reaction equation is:
But it is loaded down with trivial details that the shortcoming of the method is operating procedure, the supplementary material consumption is large, and, with producing a large amount of scrap iron powder after iron powder reducing, comparatively thickness, easily comprise product and impurity, and cost recovery is very high, and labour intensity is large, larger to the pollution effect of environment; And product yield and product quality lower.
//////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
Tirzepatide is an agonist of human glucose-dependent insulinotropic polypeptide (GIP) and human glucagon-like peptide-1 (GLP-1) receptors, whose amino acid residues at positions 2 and 13 are 2-methylAla, and the C-terminus is amidated Ser. A 1,20-icosanedioic acid is attached to Lys at position 20 via a linker which consists of a Glu and two 8-amino-3,6-dioxaoctanoic acids. Tirzepatide is a synthetic peptide consisting of 39 amino acid residues.
Tirzepatide, sold under the brand name Mounjaro,[1] is a medication used for the treatment type 2 diabetes.[2][3][4] Tirzepatide is given by injection under the skin.[2] Common side effects may include nausea, vomiting, diarrhea, decreased appetite, constipation, upper abdominal discomfort and abdominal pain.[2]
The large-scale manufacture of complex synthetic peptides is challenging due to many factors such as manufacturing risk (including failed product specifications) as well as processes that are often low in both yield and overall purity. To overcome these liabilities, a hybrid solid-phase peptide synthesis/liquid-phase peptide synthesis (SPPS/LPPS) approach was developed for the synthesis of tirzepatide. Continuous manufacturing and real-time analytical monitoring ensured the production of high-quality material, while nanofiltration provided intermediate purification without difficult precipitations. Implementation of the strategy worked very well, resulting in a robust process with high yields and purity.
Preclinical, phase I, and phase II trials have indicated that tirzepatide exhibits similar adverse effects to other established GLP-1 receptor agonists, such as GLP-1 receptor agonist dulaglutide. These effects occur largely within the gastrointestinal tract.[5] The most frequently observed adverse effects are nausea, diarrhoea and vomiting, which increased in incidence with the dosage amount (i.e. higher likelihood the higher the dose). The number of patients who discontinued taking tirzepatide also increased as dosage increased, with patients taking 15 mg having a 25% discontinuation rate vs 5.1% for 5 mg patients and 11.1% for dulaglutide.[6] To a slightly lesser extent, patients also reported reduced appetite.[5] Other side effects reported were dyspepsia, constipation, abdominal pain, dizziness and hypoglycaemia.[7][8]
Tirzepatide has a greater affinity to GIP receptors than to GLP-1 receptors, and this dual agonist behaviour has been shown to produce greater reductions of hyperglycemia compared to a selective GLP-1 receptor agonist.[3]Signaling studies have shown that this is due to tirzepatide mimicking the actions of natural GIP at the GIP receptor.[13] However, at the GLP-1 receptor, tirzepatide shows bias towards cAMP (a messenger associated with regulation of glycogen, sugar and lipid metabolism) generation, rather than β-arrestin recruitment. This combination of preference towards GIP receptor and distinct signaling properties at GLP-1 suggest this biased agonism increases insulin secretion.[13] Tirzepatide has also been shown to increase levels of adiponectin, an adipokine involved in the regulation of both glucose and lipid metabolism, with a maximum increase of 26% from baseline after 26 weeks, at the 10 mg dosage.[3]
Chemistry
Structure
Tirzepatide is an analog of the human GIP hormone with a C20 fatty-diacid portion attached, used to optimise the uptake and metabolism of the compound.[9] The fatty-diacid section (eicosanedioic acid) is linked via a glutamic acid and two (2-(2-aminoethoxy)ethoxy)acetic acid units to the side chain of the lysine residue. This arrangement allows for a much longer half life, extending the time between doses, because of its high affinity to albumin.[14]
Synthesis
The synthesis of tirzepatide was first disclosed in patents filed by Eli Lilly and Company.[15] This uses standard solid phase peptide synthesis, with an allyloxycarbonylprotecting group on the lysine at position 20 of the linear chain of amino acids, allowing a final set of chemical transformations in which the sidechain amine of that lysine is derivatized with the lipid-containing fragment.
Large-scale manufacturing processes have been reported for this compound.[16]
History
Indiana-based pharmaceutical company Eli Lilly and Company first applied for a patent for a method of glycemic control using tirzepatide in early 2016.[15] The patent was published late that year. After passing phase 3 clinical trials, Lilly applied for FDA approval in October 2021 with a priority review voucher.[17]
Following the completion of the pivotal SURPASS-2 trial no. NCT03987919, the company announced on 28 April that tirzepatide had successfully met their endpoints in obese and overweight patients without diabetes.[18] Alongside results from the SURMOUNT-1 trial no. NCT04184622, they suggest that tirzepatide may potentially be a competitor for existing diabetic medication semaglutide, manufactured by Novo Nordisk.[19][20]
In industry-funded preliminary trials comparing tirzepatide to the existing diabetes medication semaglutide (an injected analogue of the hormone GLP-1), tirzepatide showed minor improvement of reductions (2.01%–2.30% depending on dosage) in glycated hemoglobin tests relative to semaglutide (1.86%).[21] A 10 mg dose has also been shown to be effective in reducing insulin resistance, with a reduction of around 8% from baseline, measured using HOMA2-IR (computed with fasting insulin).[3] Fasting levels of IGF binding proteins like IGFBP1 and IGFBP2 increased following tirzepatide treatment, increasing insulin sensitivity.[3] A meta-analysis published by Dutta et al. showed that over 1-year clinical use, tirzepatide was observed to be superior to dulaglutide, semaglutide, degludec, and insulin glargine with regards to glycemic efficacy and obesity reduction. Tirzepatide is perhaps the most potent agent developed to date to tackle the global problem of “diabesity“.[22]
Society and culture
Names
Tirzepatide is the international nonproprietary name (INN).[23]
^ Dahl D, Onishi Y, Norwood P, Huh R, Bray R, Patel H, Rodríguez Á (February 2022). “Effect of Subcutaneous Tirzepatide vs Placebo Added to Titrated Insulin Glargine on Glycemic Control in Patients With Type 2 Diabetes: The SURPASS-5 Randomized Clinical Trial”. JAMA. 327 (6): 534–545. doi:10.1001/jama.2022.0078. PMID35133415.
^ Jump up to:abUS patent 9474780, Bokvist BK, Coskun T, Cummins RC, Alsina-Fernandez J, “GIP and GLP-1 co-agonist compounds”, issued 2016-10-25, assigned to Eli Lilly and Co
^ Dutta D, Surana V, Singla R, Aggarwal S, Sharma M (November–December 2021). “Efficacy and safety of novel twincretin tirzepatide a dual GIP and GLP-1 receptor agonist in the management of type-2 diabetes: A Cochrane meta-analysis”. Indian Journal of Endocrinology and Metabolism. 25 (6): 475–489. doi:10.4103/ijem.ijem_423_21.
^World Health Organization (2019). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 81”. WHO Drug Information. 33 (1). hdl:10665/330896.
Frías JP (November 2020). “Tirzepatide: a glucose-dependent insulinotropic polypeptide (GIP) and glucagon-like peptide-1 (GLP-1) dual agonist in development for the treatment of type 2 diabetes”. Expert Rev Endocrinol Metab. 15 (6): 379–394. doi:10.1080/17446651.2020.1830759. PMID33030356.
“Tirzepatide”. Drug Information Portal. U.S. National Library of Medicine.
Clinical trial number NCT03954834 for “A Study of Tirzepatide (LY3298176) in Participants With Type 2 Diabetes Not Controlled With Diet and Exercise Alone (SURPASS-1)” at ClinicalTrials.gov
Clinical trial number NCT03987919 for “A Study of Tirzepatide (LY3298176) Versus Semaglutide Once Weekly as Add-on Therapy to Metformin in Participants With Type 2 Diabetes (SURPASS-2)” at ClinicalTrials.gov
Clinical trial number NCT03882970 for “A Study of Tirzepatide (LY3298176) Versus Insulin Degludec in Participants With Type 2 Diabetes (SURPASS-3)” at ClinicalTrials.gov
Clinical trial number NCT03730662 for “A Study of Tirzepatide (LY3298176) Once a Week Versus Insulin Glargine Once a Day in Participants With Type 2 Diabetes and Increased Cardiovascular Risk (SURPASS-4)” at ClinicalTrials.gov
Clinical trial number NCT04039503 for “A Study of Tirzepatide (LY3298176) Versus Placebo in Participants With Type 2 Diabetes Inadequately Controlled on Insulin Glargine With or Without Metformin (SURPASS-5)” at ClinicalTrials.gov
FDA approves Lilly’s Mounjaro (tirzepatide) injection, the first and only GIP and GLP-1 receptor agonist for the treatment of adults with type 2 diabetes
Mounjaro delivered superior A1C reductions versus all comparators in phase 3 SURPASS clinical trials
While not indicated for weight loss, Mounjaro led to significantly greater weight reductions versus comparators in a key secondary endpoint
Mounjaro represents the first new class of diabetes medicines introduced in nearly a decade and is expected to be available in the U.S. in the coming weeks
INDIANAPOLIS, May 13, 2022 /PRNewswire/ — The U.S. Food and Drug Administration (FDA) approved Mounjaro (tirzepatide) injection, Eli Lilly and Company’s (NYSE: LLY) new once-weekly GIP (glucose-dependent insulinotropic polypeptide) and GLP-1 (glucagon-like peptide-1) receptor agonist indicated as an adjunct to diet and exercise to improve glycemic control in adults with type 2 diabetes. Mounjaro has not been studied in patients with a history of pancreatitis and is not indicated for use in patients with type 1 diabetes mellitus.
As the first and only FDA-approved GIP and GLP-1 receptor agonist, Mounjaro is a single molecule that activates the body’s receptors for GIP and GLP-1, which are natural incretin hormones.1
“Mounjaro delivered superior and consistent A1C reductions against all of the comparators throughout the SURPASS program, which was designed to assess Mounjaro’s efficacy and safety in a broad range of adults with type 2 diabetes who could be treated in clinical practice. The approval of Mounjaro is an exciting step forward for people living with type 2 diabetes given the results seen in these clinical trials,” said Juan Pablo Frías, M.D., Medical Director, National Research Institute and Investigator in the SURPASS program.
Mounjaro will be available in six doses (2.5 mg, 5 mg, 7.5 mg, 10 mg, 12.5 mg, 15 mg) and will come in Lilly’s well-established auto-injector pen with a pre-attached, hidden needle that patients do not need to handle or see.
The approval was based on results from the phase 3 SURPASS program, which included active comparators of injectable semaglutide 1 mg, insulin glargine and insulin degludec. Efficacy was evaluated for Mounjaro 5 mg, 10 mg and 15 mg used alone or in combination with commonly prescribed diabetes medications, including metformin, SGLT2 inhibitors, sulfonylureas and insulin glargine. Participants in the SURPASS program achieved average A1C reductions between 1.8% and 2.1% for Mounjaro 5 mg and between 1.7% and 2.4% for both Mounjaro 10 mg and Mounjaro 15 mg. While not indicated for weight loss, mean change in body weight was a key secondary endpoint in all SURPASS studies. Participants treated with Mounjaro lost between 12 lb. (5 mg) and 25 lb. (15 mg) on average.1
Side effects reported in at least 5% of patients treated with Mounjaro include nausea, diarrhea, decreased appetite, vomiting, constipation, indigestion (dyspepsia), and stomach (abdominal) pain. The labeling for Mounjaro contains a Boxed Warning regarding thyroid C-cell tumors. Mounjaro is contraindicated in patients with a personal or family history of medullary thyroid carcinoma or in patients with Multiple Endocrine Neoplasia syndrome type 2.1
“Lilly has a nearly 100-year heritage of advancing care for people living with diabetes – never settling for current outcomes. We’re not satisfied knowing that half of the more than 30 million Americans living with type 2 diabetes are not reaching their target blood glucose levels,” said Mike Mason, president, Lilly Diabetes. “We are thrilled to introduce Mounjaro, which represents the first new class of type 2 diabetes medication introduced in almost a decade and embodies our mission to bring innovative new therapies to the diabetes community.”
Mounjaro is expected to be available in the United States in the coming weeks. Lilly is committed to helping people access the medicines they are prescribed and will work with insurers, health systems and providers to help enable patient access to Mounjaro. Lilly plans to offer a Mounjaro savings card for people who qualify. Patients or healthcare professionals with questions about Mounjaro can visit www.Mounjaro.com or call The Lilly Answers Center at 1-800-LillyRx (1-800-545-5979).
Tirzepatide is also under regulatory review for the treatment of type 2 diabetes in Europe, Japan and several additional markets. A multimedia gallery is available on Lilly.com.
About the SURPASS clinical trial program The SURPASS phase 3 global clinical development program for tirzepatide began in late 2018 and included five global registration trials and two regional trials in Japan. These studies ranged from 40 to 52 weeks and evaluated the efficacy and safety of Mounjaro 5 mg, 10 mg and 15 mg as a monotherapy and as an add-on to various standard-of-care medications for type 2 diabetes. The active comparators in the studies were injectable semaglutide 1 mg, insulin glargine and insulin degludec. Collectively, the five global registration trials consistently demonstrated A1C reductions for participants taking Mounjaro across multiple stages of their type 2 diabetes journeys, from an average around five to 13 years of having diabetes.2-8
SURPASS-1 (NCT03954834) was a 40-week study comparing the efficacy and safety of Mounjaro 5 mg (N=121), 10 mg (N=121) and 15 mg (N=120) as monotherapy to placebo (N=113) in adults with type 2 diabetes inadequately controlled with diet and exercise alone. From a baseline A1C of 7.9%, Mounjaro reduced participants’ A1C by a mean of 1.8%* (5 mg) and 1.7%* (10 mg and 15 mg) compared to 0.1% for placebo. In a key secondary endpoint, from a baseline weight of 189 lb., Mounjaro reduced participants’ weight by a mean of 14 lb.* (5 mg), 15 lb.* (10 mg) and 17 lb.* (15 mg) compared to 2 lb. for placebo.2,3
SURPASS-2 (NCT03987919) was a 40-week study comparing the efficacy and safety of Mounjaro 5 mg (N=470), 10 mg (N=469) and 15 mg (N=469) to injectable semaglutide 1 mg (N=468) in adults with type 2 diabetes inadequately controlled with ≥1500 mg/day metformin alone. From a baseline A1C of 8.3%, Mounjaro reduced participants’ A1C by a mean of 2.0%ꝉ (5 mg), 2.2%* (10 mg) and 2.3%* (15 mg) compared to 1.9% for semaglutide. In a key secondary endpoint, from a baseline weight of 207 lb., Mounjaro reduced participants’ weight by a mean of 17 lb.ꝉ (5 mg), 21 lb.* (10 mg) and 25 lb.* (15 mg) compared to 13 lb. for semaglutide.4,5
SURPASS-3 (NCT03882970) was a 52-week study comparing the efficacy of Mounjaro 5 mg (N=358), 10 mg (N=360) and 15 mg (N=358) to titrated insulin degludec (N=359) in adults with type 2 diabetes treated with metformin with or without an SGLT-2 inhibitor. From a baseline A1C of 8.2%, Mounjaro reduced participants’ A1C by a mean of 1.9%* (5 mg), 2.0%* (10 mg) and 2.1%* (15 mg) compared to 1.3% for insulin degludec. From a baseline weight of 208 lb., Mounjaro reduced participants’ weight by a mean of 15 lb.* (5 mg), 21 lb.* (10 mg) and 25 lb.* (15 mg) compared to an increase of 4 lb. for insulin degludec.6
SURPASS-4 (NCT03730662) was a 104-week study comparing the efficacy and safety of Mounjaro 5 mg (N=328), 10 mg (N=326) and 15 mg (N=337) to insulin glargine (N=998) in adults with type 2 diabetes inadequately controlled with at least one and up to three oral antihyperglycemic medications (metformin, sulfonylureas or SGLT-2 inhibitors), who have increased cardiovascular (CV) risk. The primary endpoint was measured at 52 weeks. From a baseline A1C of 8.5%, Mounjaro reduced participants’ A1C by a mean of 2.1%* (5 mg), 2.3%* (10 mg) and 2.4%* (15 mg) compared to 1.4% for insulin glargine. From a baseline weight of 199 lb., Mounjaro reduced weight by a mean of 14 lb.* (5 mg), 20 lb.* (10 mg) and 23 lb.* (15 mg) compared to an increase of 4 lb. for insulin glargine.7
SURPASS-5 (NCT04039503) was a 40-week study comparing the efficacy and safety of Mounjaro 5 mg (N=116), 10 mg (N=118) and 15 mg (N=118) to placebo (N=119) in adults with inadequately controlled type 2 diabetes already being treated with insulin glargine, with or without metformin. From a baseline A1C of 8.3%, Mounjaro reduced A1C by a mean of 2.1%* (5 mg), 2.4%* (10 mg) and 2.3%* (15 mg) compared to 0.9% for placebo. From a baseline weight of 210 lb., Mounjaro reduced participants’ weight by a mean of 12 lb.* (5 mg), 17 lb.* (10 mg) and 19 lb.* (15 mg) compared to an increase of 4 lb. for placebo.8
*p<0.001 for superiority vs. placebo or active comparator, adjusted for multiplicity ꝉp<0.05 for superiority vs. semaglutide 1 mg, adjusted for multiplicity
About Mounjaro (tirzepatide) injection1 Mounjaro (tirzepatide) injection is FDA-approved as an adjunct to diet and exercise to improve glycemic control in adults with type 2 diabetes mellitus. As the first and only FDA-approved GIP and GLP-1 receptor agonist, Mounjaro is a single molecule that activates the body’s receptors for GIP (glucose-dependent insulinotropic polypeptide) and GLP-1 (glucagon-like peptide-1). Mounjaro will be available in six doses (2.5 mg, 5 mg, 7.5 mg, 10 mg, 12.5 mg, 15 mg) and will come in Lilly’s well-established auto-injector pen with a pre-attached, hidden needle that patients do not need to handle or see.
PURPOSE AND SAFETY SUMMARY WITH WARNINGS Important Facts About MounjaroTM (mown-JAHR-OH). It is also known as tirzepatide.
Mounjaro is an injectable prescription medicine for adults with type 2 diabetes used along with diet and exercise to improve blood sugar (glucose).
It is not known if Mounjaro can be used in people who have had inflammation of the pancreas (pancreatitis). Mounjaro is not for use in people with type 1 diabetes. It is not known if Mounjaro is safe and effective for use in children under 18 years of age.
Warnings Mounjaro may cause tumors in the thyroid, including thyroid cancer. Watch for possible symptoms, such as a lump or swelling in the neck, hoarseness, trouble swallowing, or shortness of breath. If you have a symptom, tell your healthcare provider.
Do not use Mounjaro if you or any of your family have ever had a type of thyroid cancer called medullary thyroid carcinoma (MTC).
Do not use Mounjaro if you have Multiple Endocrine Neoplasia syndrome type 2 (MEN 2).
Do not use Mounjaro if you are allergic to tirzepatide or any of the ingredients in Mounjaro.
Mounjaro may cause serious side effects, including:
Inflammation of the pancreas (pancreatitis). Stop using Mounjaro and call your healthcare provider right away if you have severe pain in your stomach area (abdomen) that will not go away, with or without vomiting. You may feel the pain from your abdomen to your back.
Low blood sugar (hypoglycemia). Your risk for getting low blood sugar may be higher if you use Mounjaro with another medicine that can cause low blood sugar, such as a sulfonylurea or insulin. Signs and symptoms of low blood sugar may include dizziness or light-headedness, sweating, confusion or drowsiness, headache, blurred vision, slurred speech, shakiness, fast heartbeat, anxiety, irritability, or mood changes, hunger, weakness and feeling jittery.
Serious allergic reactions. Stop using Mounjaro and get medical help right away if you have any symptoms of a serious allergic reaction, including swelling of your face, lips, tongue or throat, problems breathing or swallowing, severe rash or itching, fainting or feeling dizzy, and very rapid heartbeat.
Kidney problems (kidney failure). In people who have kidney problems, diarrhea, nausea, and vomiting may cause a loss of fluids (dehydration), which may cause kidney problems to get worse. It is important for you to drink fluids to help reduce your chance of dehydration.
Severe stomach problems. Stomach problems, sometimes severe, have been reported in people who use Mounjaro. Tell your healthcare provider if you have stomach problems that are severe or will not go away.
Changes in vision. Tell your healthcare provider if you have changes in vision during treatment with Mounjaro.
Gallbladder problems. Gallbladder problems have happened in some people who use Mounjaro. Tell your healthcare provider right away if you get symptoms of gallbladder problems, which may include pain in your upper stomach (abdomen), fever, yellowing of skin or eyes (jaundice), and clay-colored stools.
Common side effects The most common side effects of Mounjaro include nausea, diarrhea, decreased appetite, vomiting, constipation, indigestion, and stomach (abdominal) pain. These are not all the possible side effects of Mounjaro. Talk to your healthcare provider about any side effect that bothers you or doesn’t go away.
Tell your healthcare provider if you have any side effects. You can report side effects at 1-800-FDA-1088 or www.fda.gov/medwatch.
Before using
Your healthcare provider should show you how to use Mounjaro before you use it for the first time.
Before you use Mounjaro, talk to your healthcare provider about low blood sugar and how to manage it.
Review these questions with your healthcare provider:
Do you have other medical conditions, including problems with your pancreas or kidneys, or severe problems with your stomach, such as slowed emptying of your stomach (gastroparesis) or problems digesting food?
Do you take other diabetes medicines, such as insulin or sulfonylureas?
Do you have a history of diabetic retinopathy?
Are you pregnant or plan to become pregnant or breastfeeding or plan to breastfeed? It is not known if Mounjaro will harm your unborn baby.
Do you take birth control pills by mouth? These may not work as well while using Mounjaro. Your healthcare provider may recommend another type of birth control when you start Mounjaro or when you increase your dose.
Do you take any other prescription medicines or over-the-counter drugs, vitamins, or herbal supplements?
How to take
Read the Instructions for Use that come with Mounjaro.
Use Mounjaro exactly as your healthcare provider says.
Mounjaro is injected under the skin (subcutaneously) of your stomach (abdomen), thigh, or upper arm.
Use Mounjaro 1 time each week, at any time of the day.
Do not mix insulin and Mounjaro together in the same injection.
If you take too much Mounjaro, call your healthcare provider or seek medical advice promptly.
Learn more For more information, call 1-800-LillyRx (1-800-545-5979) or go to www.mounjaro.com.
This information does not take the place of talking with your healthcare provider. Be sure to talk to your healthcare provider about Mounjaro and how to take it. Your healthcare provider is the best person to help you decide if Mounjaro is right for you.
MounjaroTM and its delivery device base are trademarks owned or licensed by Eli Lilly and Company, its subsidiaries, or affiliates.
About Lilly Lilly unites caring with discovery to create medicines that make life better for people around the world. We’ve been pioneering life-changing discoveries for nearly 150 years, and today our medicines help more than 47 million people across the globe. Harnessing the power of biotechnology, chemistry and genetic medicine, our scientists are urgently advancing new discoveries to solve some of the world’s most significant health challenges, redefining diabetes care, treating obesity and curtailing its most devastating long-term effects, advancing the fight against Alzheimer’s disease, providing solutions to some of the most debilitating immune system disorders, and transforming the most difficult-to-treat cancers into manageable diseases. With each step toward a healthier world, we’re motivated by one thing: making life better for millions more people. That includes delivering innovative clinical trials that reflect the diversity of our world and working to ensure our medicines are accessible and affordable. To learn more, visit Lilly.com and Lilly.com/newsroom or follow us on Facebook, Instagram, Twitter and LinkedIn. P-LLY
This press release contains forward-looking statements (as that term is defined in the Private Securities Litigation Reform Act of 1995) about Mounjaro (tirzepatide 2.5 mg, 5 mg, 7.5 mg, 10 mg, 12.5 mg and 15 mg) injection as a treatment to improve glycemic control in adults with type 2 diabetes, the timeline for supply of Mounjaro to become available, and certain other milestones and ongoing clinical trials of Mounjaro and reflects Lilly’s current beliefs and expectations. However, as with any pharmaceutical product or medical device, there are substantial risks and uncertainties in the process of research, development and commercialization. Among other things, there can be no guarantee that Mounjaro will be commercially successful, that future study results will be consistent with results to date, or that we will meet our anticipated timelines for the commercialization of Mounjaro. For further discussion of these and other risks and uncertainties, see Lilly’s most recent Form 10-K and Form 10-Q filings with the United States Securities and Exchange Commission. Except as required by law, Lilly undertakes no duty to update forward-looking statements to reflect events after the date of this release.
References
Mounjaro. Prescribing Information. Lilly USA, LLC.
Rosenstock, J, et. al. Efficacy and Safety of Once Weekly Tirzepatide, a Dual GIP/GLP-1 Receptor Agonist Versus Placebo as Monotherapy in People with Type 2 Diabetes (SURPASS-1). Abstract 100-OR. Presented virtually at the American Diabetes Association’s 81st Scientific Sessions; June 25-29.
Rosenstock, J, et. al. (2021). Efficacy and safety of a novel dual GIP and GLP-1 receptor agonist tirzepatide in patients with type 2 diabetes (SURPASS-1): a double-blind, randomised, phase 3 trial. Lancet. 2021;398(10295):143-155. doi: 10.1016/S0140-6736(21)01324-6.
Frías JP, Davies MJ, Rosenstock J, et al; for the SURPASS-2 Investigators. Tirzepatide versus semaglutide once weekly in patients with type 2 diabetes. N Engl J Med. 2021;385(6)(suppl):503-515. doi: 10.1056/NEJMoa2107519
Frias, J.P. Efficacy and Safety of Tirzepatide vs. Semaglutide Once Weekly as Add-On Therapy to Metformin in Patients with Type 2 Diabetes. Abstract 84-LB. Presented virtually at the American Diabetes Association’s 81st Scientific Sessions; June 25-29.
Ludvik B, Giorgino F, Jódar E, et al. Once-weekly tirzepatide versus once-daily insulin degludec as add-on to metformin with or without SGLT2 inhibitors in patients with type 2 diabetes (SURPASS-3): a randomised, open-label, parallel-group, phase 3 trial. Lancet. 2021;398(10300):583-598. doi: 10.1016/S0140-6736(21)01443-4
Del Prato S, Kahn SE, Pavo I, et al; for the SURPASS-4 Investigators. Tirzepatide versus insulin glargine in type 2 diabetes and increased cardiovascular risk (SURPASS-4): a randomised, open-label, parallel-group, multicentre, phase 3 trial. Lancet. 2021;398(10313):1811-1824. doi: 10.1016/S0140-6736(21)02188-7
Dahl D, Onishi Y, Norwood P, et al. Effect of subcutaneous tirzepatide vs placebo added to titrated insulin glargine on glycemic control in patients with type 2 diabetes: the SURPASS-5 randomized clinical trial. JAMA. 2022;327(6):534-545. doi:10.1001/jama.2022.0078
Participants taking tirzepatide lost up to 52 lb. (24 kg) in this 72-week phase 3 study
63% of participants taking tirzepatide 15 mg achieved at least 20% body weight reductions as a key secondary endpoint
INDIANAPOLIS, April 28, 2022 /PRNewswire/ — Tirzepatide (5 mg, 10 mg, 15 mg) achieved superior weight loss compared to placebo at 72 weeks of treatment in topline results from Eli Lilly and Company’s (NYSE: LLY) SURMOUNT-1 clinical trial, with participants losing up to 22.5% (52 lb. or 24 kg) of their body weight for the efficacy estimandi. This study enrolled 2,539 participants and was the first phase 3 global registration trial evaluating the efficacy and safety of tirzepatide in adults with obesity, or overweight with at least one comorbidity, who do not have diabetes. Tirzepatide met both co-primary endpoints of superior mean percent change in body weight from baseline and greater percentage of participants achieving body weight reductions of at least 5% compared to placebo for both estimandsii. The study also achieved all key secondary endpoints at 72 weeks.
For the efficacy estimand, participants taking tirzepatide achieved average weight reductions of 16.0% (35 lb. or 16 kg on 5 mg), 21.4% (49 lb. or 22 kg on 10 mg) and 22.5% (52 lb. or 24 kg on 15 mg), compared to placebo (2.4%, 5 lb. or 2 kg). Additionally, 89% (5 mg) and 96% (10 mg and 15 mg) of people taking tirzepatide achieved at least 5% body weight reductions compared to 28% of those taking placebo.
In a key secondary endpoint, 55% (10 mg) and 63% (15 mg) of people taking tirzepatide achieved at least 20% body weight reductions compared to 1.3% of those taking placebo. In an additional secondary endpoint not controlled for type 1 error, 32% of participants taking tirzepatide 5 mg achieved at least 20% body weight reductions. The mean baseline body weight of participants was 231 lb. (105 kg).
“Obesity is a chronic disease that often does not receive the same standard of care as other conditions, despite its impact on physical, psychological and metabolic health, which can include increased risk of hypertension, heart disease, cancer and decreased survival,” said Louis J. Aronne, MD, FACP, DABOM, director of the Comprehensive Weight Control Center and the Sanford I. Weill Professor of Metabolic Research at Weill Cornell Medicine, obesity expert at NewYork-Presbyterian/Weill Cornell Medical Center and Investigator of SURMOUNT-1. “Tirzepatide delivered impressive body weight reductions in SURMOUNT-1, which could represent an important step forward for helping the patient and physician partnership treat this complex disease.”
For the treatment-regimen estimandiii, results showed:
Average body weight reductions: 15.0% (5 mg), 19.5% (10 mg), 20.9% (15 mg), 3.1% (placebo)
Percentage of participants achieving body weight reductions of ≥5%: 85% (5 mg), 89% (10 mg), 91% (15 mg), 35% (placebo)
Percentage of participants achieving body weight reductions of ≥20%: 30% (5 mg, not controlled for type 1 error), 50% (10 mg), 57% (15 mg), 3.1% (placebo)
The overall safety and tolerability profile of tirzepatide was similar to other incretin-based therapies approved for the treatment of obesity. The most commonly reported adverse events were gastrointestinal-related and generally mild to moderate in severity, usually occurring during the dose escalation period. For those treated with tirzepatide (5 mg, 10 mg and 15 mg, respectively), nausea (24.6%, 33.3%, 31.0%), diarrhea (18.7%, 21.2%, 23.0%), vomiting (8.3%, 10.7%, 12.2%) and constipation (16.8%, 17.1%, 11.7%) were more frequently experienced compared to placebo (9.5% [nausea], 7.3% [diarrhea], 1.7% [vomiting], 5.8% [constipation]).
Treatment discontinuation rates due to adverse events were 4.3% (5 mg), 7.1% (10 mg), 6.2% (15 mg) and 2.6% (placebo). The overall treatment discontinuation rates were 14.3% (5 mg), 16.4% (10 mg), 15.1% (15 mg) and 26.4% (placebo).
Participants who had pre-diabetes at study commencement will remain enrolled in SURMOUNT-1 for an additional 104 weeks of treatment following the initial 72-week completion date to evaluate the impact on body weight and the potential differences in progression to type 2 diabetes at three years of treatment with tirzepatide compared to placebo.
“Tirzepatide is the first investigational medicine to deliver more than 20 percent weight loss on average in a phase 3 study, reinforcing our confidence in its potential to help people living with obesity,” said Jeff Emmick, MD, Ph.D., vice president, product development, Lilly. “Obesity is a chronic disease that requires effective treatment options, and Lilly is working relentlessly to support people with obesity and modernize how this disease is approached. We’re proud to research and develop potentially innovative treatments like tirzepatide, which helped nearly two thirds of participants on the highest dose reduce their body weight by at least 20 percent in SURMOUNT-1.”
Tirzepatide is a novel investigational once-weekly GIP (glucose-dependent insulinotropic polypeptide) receptor and GLP-1 (glucagon-like peptide-1) receptor agonist, representing a new class of medicines being studied for the treatment of obesity. Tirzepatide is a single peptide that activates the body’s receptors for GIP and GLP-1, two natural incretin hormones. Obesity is a chronic, progressive disease caused by disruptions in the mechanisms that control body weight, often leading to an increase in food intake and/or a decrease in energy expenditure. These disruptions are multifactorial and can be related to genetic, developmental, behavioral, environmental and social factors. To learn more, visit Lilly.com/obesity.
Lilly will continue to evaluate the SURMOUNT-1 results, which will be presented at an upcoming medical meeting and submitted to a peer-reviewed journal. Additional studies are ongoing for tirzepatide as a potential treatment for obesity or overweight.
About tirzepatide
Tirzepatide is a once-weekly GIP (glucose-dependent insulinotropic polypeptide) receptor and GLP-1 (glucagon-like peptide-1) receptor agonist that integrates the actions of both incretins into a single novel molecule. GIP is a hormone that may complement the effects of GLP-1 receptor agonists. In preclinical models, GIP has been shown to decrease food intake and increase energy expenditure therefore resulting in weight reductions, and when combined with GLP-1 receptor agonism, may result in greater effects on markers of metabolic dysregulation such as body weight, glucose and lipids. Tirzepatide is in phase 3 development for adults with obesity or overweight with weight-related comorbidity and is currently under regulatory review as a treatment for adults with type 2 diabetes. It is also being studied as a potential treatment for non-alcoholic steatohepatitis (NASH) and heart failure with preserved ejection fraction (HFpEF). Studies of tirzepatide in obstructive sleep apnea (OSA) and in morbidity/mortality in obesity are planned as well.
About SURMOUNT-1 and the SURMOUNT clinical trial program
SURMOUNT-1 (NCT04184622) is a multi-center, randomized, double-blind, parallel, placebo-controlled trial comparing the efficacy and safety of tirzepatide 5 mg, 10 mg and 15 mg to placebo as an adjunct to a reduced-calorie diet and increased physical activity in adults without type 2 diabetes who have obesity, or overweight with at least one of the following comorbidities: hypertension, dyslipidemia, obstructive sleep apnea or cardiovascular disease. The trial randomized 2,539 participants across the U.S., Argentina, Brazil, China, India, Japan, Mexico, Russia and Taiwan in a 1:1:1:1 ratio to receive either tirzepatide 5 mg, 10 mg or 15 mg or placebo. The co-primary objectives of the study were to demonstrate that tirzepatide 10 mg and/or 15 mg is superior in percentage of body weight reductions from baseline and percentage of participants achieving ≥5% body weight reduction at 72 weeks compared to placebo. Participants who had pre-diabetes at study commencement will remain enrolled in SURMOUNT-1 for an additional 104 weeks of treatment following the initial 72-week completion date to evaluate the impact on body weight and potential differences in progression to type 2 diabetes at three years of treatment with tirzepatide compared to placebo.
All participants in the tirzepatide treatment arms started the study at a dose of tirzepatide 2.5 mg once-weekly and then increased the dose in a step-wise approach at four-week intervals to their final randomized maintenance dose of 5 mg (via a 2.5 mg step), 10 mg (via steps at 2.5 mg, 5 mg and 7.5 mg) or 15 mg (via steps at 2.5 mg, 5 mg, 7.5 mg, 10 mg and 12.5 mg).
The SURMOUNT phase 3 global clinical development program for tirzepatide began in late 2019 and has enrolled more than 5,000 people with obesity or overweight across six clinical trials, four of which are global studies. Results from SURMOUNT-2, -3, and -4 are anticipated in 2023.
About Lilly
Lilly unites caring with discovery to create medicines that make life better for people around the world. We’ve been pioneering life-changing discoveries for nearly 150 years, and today our medicines help more than 47 million people across the globe. Harnessing the power of biotechnology, chemistry and genetic medicine, our scientists are urgently advancing new discoveries to solve some of the world’s most significant health challenges, redefining diabetes care, treating obesity and curtailing its most devastating long-term effects, advancing the fight against Alzheimer’s disease, providing solutions to some of the most debilitating immune system disorders, and transforming the most difficult-to-treat cancers into manageable diseases. With each step toward a healthier world, we’re motivated by one thing: making life better for millions more people. That includes delivering innovative clinical trials that reflect the diversity of our world and working to ensure our medicines are accessible and affordable. To learn more, visit Lilly.com and Lilly.com/newsroom or follow us on Facebook, Instagram, Twitter and LinkedIn. P-LLY
Tirzepatide results superior A1C and body weight reductions compared to insulin glargine in adults with type 2 diabetes
Newly published data show that participants maintained A1C and weight control up to two years in SURPASS-4, the largest and longest SURPASS trial completed to dateNo increased cardiovascular risk identified with tirzepatide; hazard ratio of 0.74 observed for MACE-4 events
SURPASS-4 is the largest and longest clinical trial completed to date of the phase 3 program studying tirzepatide as a potential treatment for type 2 diabetes. The primary endpoint was measured at 52 weeks, with participants continuing treatment up to 104 weeks or until study completion. The completion of the study was triggered by the accrual of major adverse cardiovascular events (MACE) to assess CV risk. In newly published data from the treatment period after 52 weeks, participants taking tirzepatide maintained A1C and weight control for up to two years.
The overall safety profile of tirzepatide, assessed over the full study period, was consistent with the safety results measured at 52 weeks, with no new findings up to 104 weeks. Gastrointestinal side effects were the most commonly reported adverse events, usually occurring during the escalation period and then decreasing over time.
“We are encouraged by the continued A1C and weight control that participants experienced past the initial 52 week treatment period and up to two years as we continue to explore the potential impact of tirzepatide for the treatment of type 2 diabetes,” said John Doupis, M.D., Ph.D., Director, Diabetes Division and Clinical Research Center, Iatriko Paleou Falirou Medical Center, Athens, Greece and Senior Investigator for SURPASS-4.
Tirzepatide is a novel investigational once-weekly dual glucose-dependent insulinotropic polypeptide (GIP) and glucagon-like peptide-1 (GLP-1) receptor agonist that integrates the actions of both incretins into a single molecule, representing a new class of medicines being studied for the treatment of type 2 diabetes.
SURPASS-4 was an open-label global trial comparing the safety and efficacy of three tirzepatide doses (5 mg, 10 mg and 15 mg) to titrated insulin glargine in 2,002 adults with type 2 diabetes with increased CV risk who were treated with between one and three oral antihyperglycemic medicines (metformin, a sulfonylurea or an SGLT-2 inhibitor). Of the total participants randomized, 1,819 (91%) completed the primary 52-week visit and 1,706 (85%) completed the study on treatment. The median study duration was 85 weeks and 202 participants (10%) completed two years.
Study participants had a mean duration of diabetes of 11.8 years, a baseline A1C of 8.52 percent and a baseline weight of 90.3 kg. More than 85 percent of participants had a history of cardiovascular events. In the insulin glargine arm, the insulin dose was titrated following a treat-to-target algorithm with the goal of fasting blood glucose below 100 mg/dL. The starting dose of insulin glargine was 10 units per day, and the mean dose of insulin glargine at 52 weeks was 43.5 units per day.
About tirzepatide Tirzepatide is a once-weekly dual glucose-dependent insulinotropic polypeptide (GIP) and glucagon-like peptide-1 (GLP-1) receptor agonist that integrates the actions of both incretins into a single novel molecule. GIP is a hormone that may complement the effects of GLP-1. In preclinical models, GIP has been shown to decrease food intake and increase energy expenditure therefore resulting in weight reductions, and when combined with a GLP-1 receptor agonist, may result in greater effects on glucose and body weight. Tirzepatide is in phase 3 development for blood glucose management in adults with type 2 diabetes, for chronic weight management and heart failure with preserved ejection fraction (HFpEF). It is also being studied as a potential treatment for non-alcoholic steatohepatitis (NASH).
About SURPASS-4 and the SURPASS clinical trial program SURPASS-4 (NCT03730662) is a randomized, parallel, open-label trial comparing the efficacy and safety of tirzepatide 5 mg, 10 mg and 15 mg to insulin glargine in adults with type 2 diabetes inadequately controlled with at least one and up to three oral antihyperglycemic medications (metformin, sulfonylureas or SGLT-2 inhibitors), who have increased cardiovascular (CV) risk. The trial randomized 2,002 study participants in a 1:1:1:3 ratio to receive either tirzepatide 5 mg, 10 mg or 15 mg or insulin glargine. Participants were located in the European Union, North America (Canada and the United States), Australia, Israel, Taiwan and Latin America (Brazil, Argentina and Mexico). The primary objective of the study was to demonstrate that tirzepatide (10 mg and/or 15 mg) is non-inferior to insulin glargine for change from baseline A1C at 52 weeks in people with type 2 diabetes and increased CV risk. The primary and key secondary endpoints were measured at 52 weeks, with participants continuing treatment up to 104 weeks or until study completion. The completion of the study was triggered by the accrual of major adverse cardiovascular events (MACE). Study participants enrolled had to have a mean baseline A1C between 7.5 percent and 10.5 percent and a BMI greater than or equal to 25 kg/m2 at baseline. All participants in the tirzepatide treatment arms started the study at a dose of tirzepatide 2.5 mg once-weekly and then increased the dose in a step-wise approach at four-week intervals to their final randomized maintenance dose of 5 mg (via a 2.5 mg step), 10 mg (via steps at 2.5 mg, 5 mg and 7.5 mg) or 15 mg (via steps at 2.5 mg, 5 mg, 7.5 mg, 10 mg and 12.5 mg). All participants in the titrated insulin glargine treatment arm started with a baseline dose of 10 units per day and titrated following a treat-to-target algorithm to reach a fasting blood glucose below 100 mg/dL.
The SURPASS phase 3 global clinical development program for tirzepatide has enrolled more than 20,000 people with type 2 diabetes across 10 clinical trials, five of which are global registration studies. The program began in late 2018, and all five global registration trials have been completed.
About Diabetes
Approximately 34 million Americans2 (just over 1 in 10) and an estimated 463 million adults worldwide3 have diabetes. Type 2 diabetes is the most common type internationally, accounting for an estimated 90 to 95 percent of all diabetes cases in the United States alone2. Diabetes is a chronic disease that occurs when the body does not properly produce or use the hormone insulin.
TIC10 (ONC-201) is a potent, orally active, and stable tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) inducer which acts by inhibiting Akt and ERK, consequently activating Foxo3a and significantly inducing cell surface TRAIL. TIC10 can cross the blood-brain barrier.
ONC-201, also known as TIC10, is a potent, orally active, and stable small molecule that transcriptionally induces TRAIL in a p53-independent manner and crosses the blood-brain barrier. TIC10 induces a sustained up-regulation of TRAIL in tumors and normal cells that may contribute to the demonstrable antitumor activity of TIC10. TIC10 inactivates kinases Akt and extracellular signal-regulated kinase (ERK), leading to the translocation of Foxo3a into the nucleus, where it binds to the TRAIL promoter to up-regulate gene transcription. TIC10 is an efficacious antitumor therapeutic agent that acts on tumor cells and their microenvironment to enhance the concentrations of the endogenous tumor suppressor TRAIL.
Akt/ERK Inhibitor ONC201 is a water soluble, orally bioavailable inhibitor of the serine/threonine protein kinase Akt (protein kinase B) and extracellular signal-regulated kinase (ERK), with potential antineoplastic activity. Upon administration, Akt/ERK inhibitor ONC201 binds to and inhibits the activity of Akt and ERK, which may result in inhibition of the phosphatidylinositol 3-kinase (PI3K)/Akt signal transduction pathway as well as the mitogen-activated protein kinase (MAPK)/ERK-mediated pathway. This may lead to the induction of tumor cell apoptosis mediated by tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL)/TRAIL death receptor type 5 (DR5) signaling in AKT/ERK-overexpressing tumor cells. The PI3K/Akt signaling pathway and MAPK/ERK pathway are upregulated in a variety of tumor cell types and play a key role in tumor cell proliferation, differentiation and survival by inhibiting apoptosis. In addition, ONC201 is able to cross the blood-brain barrier.
Herein, we present a copper-catalyzed tandem reaction of 2-aminoimidazolines and ortho-halo(hetero)aryl carboxylic acids that causes the regioselective formation of angularly fused tricyclic 1,2-dihydroimidazo[1,2-a]quinazolin-5(4H)-one derivatives. The reaction involved in the construction of the core six-membered pyrimidone moiety proceeded via regioselective N-arylation–condensation. The presented protocol been successfully applied to accomplish the total synthesis of TIC10/ONC201, which is an active angular isomer acting as a tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL): a sought after anticancer clinical agent.
ONC201 is the founding member of a novel class of anti-cancer compounds called imipridones that is currently in Phase II clinical trials in multiple advanced cancers. Since the discovery of ONC201 as a p53-independent inducer of TRAIL gene transcription, preclinical studies have determined that ONC201 has anti-proliferative and pro-apoptotic effects against a broad range of tumor cells but not normal cells. The mechanism of action of ONC201 involves engagement of PERK-independent activation of the integrated stress response, leading to tumor upregulation of DR5 and dual Akt/ERK inactivation, and consequent Foxo3a activation leading to upregulation of the death ligand TRAIL. ONC201 is orally active with infrequent dosing in animals models, causes sustained pharmacodynamic effects, and is not genotoxic. The first-in-human clinical trial of ONC201 in advanced aggressive refractory solid tumors confirmed that ONC201 is exceptionally well-tolerated and established the recommended phase II dose of 625 mg administered orally every three weeks defined by drug exposure comparable to efficacious levels in preclinical models. Clinical trials are evaluating the single agent efficacy of ONC201 in multiple solid tumors and hematological malignancies and exploring alternative dosing regimens. In addition, chemical analogs that have shown promise in other oncology indications are in pre-clinical development. In summary, the imipridone family that comprises ONC201 and its chemical analogs represent a new class of anti-cancer therapy with a unique mechanism of action being translated in ongoing clinical trials.
////////////IMIPRIDONE, TIC 10, ONC 201, NSC 350625, OP 10, Fast Track Designation, Orphan Drug Designation, Rare Pediatric Disease Designation, PHASE 3, GLIOMA, CHIMERIX
Arimoclomol maleate is in a phase III clinical trials by Orphazyme for the treatment of Niemann-Pick disease type C (NP-C). It is also in phase II clinical studies for the treatment of amyotrophic lateral sclerosis (ALS).
Arimoclomol (INN; originally codenamed BRX-345, which is a citrate salt formulation of BRX-220) is an experimental drug developed by CytRx Corporation, a biopharmaceutical company based in Los Angeles, California. In 2011 the worldwide rights to arimoclomol were bought by Danish biotech company Orphazyme ApS.[1] The European Medicines Agency (EMA) and U.S. Food & Drug Administration (FDA) granted orphan drug designation to arimoclomol as a potential treatment for Niemann-Pick type C in 2014 and 2015 respectively.[2][3]
Fig. 1 Structures of (±)-bimoclomol (1) and (R)-(+)-arimoclomol (2).
The present disclosure provides an optimized four-step process for preparing an ultra-pure composition comprising arimoclomol citrate, i.e. N-{[(2R)-2-hydroxy-3-piperidin-l-ylpropyl]oxy}pyridine-3-carboximidoyl chloride 1-oxide citrate. The optimized process comprises a plurality of optimized sub-steps, each contributing to an overall improved process, providing the ultra-pure composition comprising arimoclomol citrate. The ultra-pure composition comprising arimoclomol citrate meets the medicines agencies’ high regulatory requirements. An overview of the four-steps process is outlined below:
Step 1: Overview of process for preparing ORZY-01
Step 2: Overview of process for preparing ORZY-03
Step 4: Overview of process for preparing BRX-345 (ORZY-05)
The previously reported two-step synthesis of ORZY-01 as shown below includes a 2 hour reflux in step 1A, followed by purification of intermediate compound (V) to increase the batch quality.
(R,Z)-3-(N’-(2-hydroxy-3-(piperidin-1-yl)propoxy)carboximidoyl chloride)pyridine-1-oxide1 – (R)-(+)-Arimoclomol – 2 A solution of (R,Z)-3-(N’-(2-hydroxy-3-(piperidin-1-yl)propoxy)carbamimidoyl)pyridine-1-oxide 12 (205 mg, 0.70 mmol) in conc. hydrochloric acid (1.1 mL, 13.9 mmol) and water (3 mL) was cooled to -5 °C for 15 minutes. Sodium nitrite (63 mg, 0.91 mmol) in water (0.5 mL) was then added dropwise to the reaction mixture and the reaction was stirred at -5 °C for 2.5 hours. The reaction mixture was made alkaline with NaOH (7 M, 3 mL). An additional 10 mL of water was added followed by DCM (30 mL) containing EtOAc (5 mL) and the organics were dried over MgSO4, filtered and concentrated in vacuo. The residue was purified by FCC on Biotage Isolera using Biotage SNAP 10 g Si cartridge eluting with gradient elution 0-30% MeOH:DCM both containing 0.1% Et3N to afford the title compound (160 mg, 73% yield) as a colourless semi-solid. Analytical data was consistent with literature values. See ESI section SFC traces for specific enantiomeric ratios of 2 synthesised under the various methodologies quoted in the text. Optical rotation was not determined as it was determined in the ultimate product of this 2·citrate and comparative run times on SFC. 1H NMR (600 MHz, CDCl3) δ: 8.63 (t, J = 1.4 Hz, 1H), 8.16 (ddd, J = 6.4, 1.6, 0.9 Hz, 1H), 7.66 – 7.62 (m, 1H), 7.25 (dd, J = 8.0, 6.6 Hz, 1H), 4.26 (qd, J = 11.3, 5.2 Hz, 2H), 4.07 (dd, J = 9.2, 4.7 Hz, 1H), 2.62 (s, 2H), 2.47 – 2.31 (m, 4H), 1.65 – 1.51 (m, 4H), 1.42 (s, 2H); 13C NMR (151 MHz, CDCl3) δ: 140.3, 137.7, 133.1, 132.5, 125.7, 123.9, 78.7, 64.9, 60.9, 54.8, 25.8, 24.0.
(R)-(+)- Arimoclomol citrate – 2·citrate (R,Z)-3-(N’-(2-hydroxy-3-(piperidin-1-yl)propoxy)carboximidoyl chloride)pyridine-1-oxide (159 mg, 0.51 mmol) was dissolved in acetone (3 mL) and citric acid (97 mg, 0.51 mmol) was added. The reaction mixture was left to stir at room temperature for 18 hours. After this time the mixture was sonicated and the precipitate was filtered, rinsed with cold acetone (1 mL) and dried under vacuum to afford the title compound (165 mg, 64% yield) as a white amorphous solid. Analytical data was consistent with literature values. m.p. 161-162 °C, Acetone (lit. 163-165 °C, EtOH); [α]D 20 +8.0 (c=1, H2O); IR νmax (neat): 3423, 3228, 2949, 2868, 1722, 1589, 1483, 1433, 1307, 1128, 972, 829 cm-1; 1H NMR (600 MHz, d6-DMSO) δ: 8.54 (t, J = 1.5 Hz, 1H), 8.39 – 8.35 (m, 1H), 7.72 – 7.68 (m, 1H), 7.55 (dd, J = 8.0, 6.5 Hz, 1H), 4.28 (ddd, J = 17.6, 13.3, 7.4 Hz, 3H), 3.35 (br. s, 2H), 3.13 – 2.74 (m, 6H), 2.59 (d, J = 15.2 Hz, 2H), 2.56 – 2.51 (m, 2H), 1.77 – 1.61 (m, 4H), 1.48 (s, 2H); 13C NMR (151 MHz, d6-DMSO) δ: 176.6, 171.3, 140.5, 136.4, 132.7, 131.5, 126.8, 123.3, 77.8, 71.4, 63.8, 58.7, 53.1, 44.0, 30.7, 23.0, 21.9; HRMS (m/z TOF MS ES+) for C14H20ClN3O3 [M+H]+ calc. 314.1271, observed 314.1263; SFC er purity R:S >99:1
Procedure for the conversion of (R)-(+)-Bimoclomol 1 into (R)-(+)-Arimoclomol 2 To a solution of (R)-(+)-bimoclomol (61 mg, 0.21 mmol) in acetone (2 mL) was added benzenesulfonic acid (33 mg, 0.21 mmol). The reaction mixture was stirred at room temperature for 1.5 hours. The reaction mixture was concentrated in vacuo. Separately to a suspension of hydrogen peroxide-urea adduct (39 mg, 0.41 mmol) in acetonitrile (6 mL) at -5°C (ice-salt bath) was added trifluoroacetic anhydride (58 μL, 0.41 mmol) dropwise. A suspension of (R)-(+)-bimoclomol, 1, benzenesulfonic acid salt, as made above, in acetonitrile (3 mL) was then added dropwise to this solution. The reaction mixture was stirred for 18 hours, whilst slowly warming to room temperature. Aqueous Na2S2O5 solution (0.5 M, 1 mL) was added and the reaction mixture stirred for 1 hour. The reaction mixture was made alkaline with NaOH (7 M) and extracted with DCM (2 x 30 mL). The combined organics were washed with brine, dried over MgSO4, filtered and concentrated in vacuo. The residue was purified by FCC on a Biotage Isolera using Biotage SNAP 10g Si cartridge eluting with gradient elution 0-35% MeOH in DCM to afford the title compound (35 mg, 55% yield) as a colourless semi-solid. Analytical data of the products was consistent with literature and/or previous samples synthesised above.
Arimoclomol is believed to function by stimulating a normal cellular protein repair pathway through the activation of molecular chaperones. Since damaged proteins, called aggregates, are thought to play a role in many diseases, CytRx believes that arimoclomol could treat a broad range of diseases.
Arimoclomol has been shown to extend life in an animal model of ALS[11] and was well tolerated in healthy human volunteers in a Phase I study. CytRx is currently conducting a Phase II clinical trial.[12]
Arimoclomol also has been shown to be an effective treatment in an animal model of Spinal Bulbar Muscular Atrophy (SBMA, also known as Kennedy’s Disease).[13]
Arimoclomol was discovered by Hungarian researchers, as a drug candidate to treat insulin resistance[14][15] and diabetic complications such as retinopathy, neuropathy and nephropathy. Later, the compound, along with other small molecules, was screened for further development by Hungarian firm Biorex, which was sold to CytRx Corporation, who developed it toward a different direction from 2003.
^ Kieran D, Kalmar B, Dick JR, Riddoch-Contreras J, Burnstock G, Greensmith L (April 2004). “Treatment with arimoclomol, a coinducer of heat shock proteins, delays disease progression in ALS mice”. Nat. Med. 10 (4): 402–5. doi:10.1038/nm1021. PMID15034571. S2CID2311751.
^ Kalmar B, Greensmith L, Malcangio M, McMahon SB, Csermely P, Burnstock G (December 2003). “The effect of treatment with BRX-220, a co-inducer of heat shock proteins, on sensory fibers of the rat following peripheral nerve injury”. Exp. Neurol. 184 (2): 636–47. doi:10.1016/S0014-4886(03)00343-1. PMID14769355. S2CID5316222.
^ Rakonczay Z, Iványi B, Varga I, et al. (June 2002). “Nontoxic heat shock protein coinducer BRX-220 protects against acute pancreatitis in rats”. Free Radic. Biol. Med. 32 (12): 1283–92. doi:10.1016/S0891-5849(02)00833-X. PMID12057766.
^ Kalmar B, Burnstock G, Vrbová G, Urbanics R, Csermely P, Greensmith L (July 2002). “Upregulation of heat shock proteins rescues motoneurones from axotomy-induced cell death in neonatal rats”. Exp. Neurol. 176 (1): 87–97. doi:10.1006/exnr.2002.7945. PMID12093085. S2CID16071543.
Ulcerative colitis (UC) is a disease characterized by chronic inflammation of the rectal and colonic mucosa, affecting the innermost lining in the first stage. The disease is recurrent, with both active and inactive stages that differ in pathology, symptoms and treatment. The underlying cause of UC is not understood, nor is it known what triggers the disease to recur between its inactive and active forms (Irvine, EJ (2008) Inflamm Bowel Dis 14(4): 554-565). Symptoms of active UC include progressive loose stools with blood and increased frequency of bowel movements. Active mucosal inflammation is diagnosed by endoscopy.
The stools contain pus, mucous and blood and are often associated with abdominal cramping with urgency to evacuate (tenesmi). Diarrhoea may have an insidious onset or, more rarely, start quite suddenly. In severe cases the symptoms may include fever and general malaise. In severe stages, deep inflammation of the bowel wall may develop with abdominal tenderness, tachycardia, fever and risk of bowel perforation. Furthermore, patients with UC may suffer extra intestinal manifestations such as arthralgia and arthritis, erythema nodosum, pyoderma gangrenosum and inflammation in the eyes. In the case of remission or inactive UC, patients are usually free of bowel symptoms.
The extent of inflamed and damaged mucosa differs among patients with UC. UC that affects only the rectum is termed ulcerative proctitis. The condition is referred to as distal or left sided colitis when inflammatory changes are present in the left side of the colon up to the splenic flexure. In extensive UC the transverse colon is also affected, and pancolitis designates a disease involving the entire colon.
Active mucosal inflammation is diagnosed by endoscopy and is characterized by a loss of vascular patterning, oedema, petechia, spontaneous bleeding and fibrinous exudates. The endoscopic picture is that of continuous inflammation, starting in the rectum and extending proximally to a variable extent into the colon. Biopsies obtained at endoscopy and subjected to histological examination help to diagnose the condition. Infectious causes, including Clostridium difficile, camphylobacter, Salmonella and Shigella, may mimic UC and can be excluded by stool cultures.
The medical management of UC is divided into treatment of active disease and maintenance of remission.
The treatment of patients with active UC aims to reduce inflammation and promote colon healing and mucosal recovery. In milder cases the disease may be controlled with conventional drugs including sulphasalazine, 5 -aminosalicylic acid (5-ASA) (Sutherland, L., F. Martin, S. Greer, M. Robinson, N. Greenberger, F. Saibil, T Martin, J. Sparr, E. Prokipchuk and L. Borgn (1987) Gastroenterology 92: 1894-1898) and glucocorticosteroids (GCS) (Domenech, E., M. Manosa and E. Cabre (2014). Dig Dis 32( 4): 320-327).
GCS are generally used to treat disease flare-ups and are not recommended for maintenance of remission since there are significant side effects in long-term use, and the possible development of steroid dependent disease. Glucocorticoid drugs act non-selectively, so in the long run they may impair many healthy anabolic processes. As a result, maintenance treatment with systemic GCS is not advised (Prantera, C. and S.
For patients who become refractory to GCS and suffer from severe or moderately severe attacks of UC, the addition of immunomodulatory agents such as cyclosporine, 6-mercaptopurine and azathioprine may be used. However, immunomodulators are slow-
acting and the induction of remission in these patients is often temporary (Khan, KJ, MC Dubinsky, AC Ford, TA Ullman, NJ Talley and P. Moayyedi (2011) Am J Gastroenterol 106(4): 630-642).
Further treatment options for UC include biologic agents (Fausel, R. and A. Afzali (2015) Ther Clin Risk Manag 11: 63-73). The three TNF-α inhibitors currently approved for the treatment of moderate to severe UC are infliximab, adalimumab, and golimumab. All three carry potential risks associated with their use, and should be avoided in certain patients, eg those with uncontrolled infections, advanced heart failure, neurologic conditions and in patients with a history of malignancy, due to a potential risk of accelerating the growth of a tumor. Other potential adverse effects of TNF-α inhibitor therapy include neutropenia, hepatotoxicity, serum sickness, leukocytoclastic vasculitis, rash including psoriasiform rash, induction of autoimmunity, and injection or infusion site reactions, including anaphylaxis, convulsions, and hypotension.
All three TNF-α inhibitor agents and their related biosimilar/derivative counterparts may be used to induce and maintain clinical response and remission in patients with UC.
Combination therapy with azathioprine is also used for inducing remission.
However, more than 50% of patients receiving TNF-α inhibitor agents fail to respond to induction dosing, or lose response to the TNF-α inhibitor agents over time (Fausel, R. and A. Afzali (2015) Ther Clin Risk Manag 11 : 63-73).
Vedolizumab, an a4b7 integrin inhibitor, was recently approved for the treatment of UC. In the GEMINI 1 trial, vedolizumab was found to be more effective than placebo for inducing and maintaining clinical response, clinical remission, and mucosal healing (Feagan, BG, P. Rutgeerts, BE Sands, S. Hanauer, JF Colombel, WJ Sandbom, G. Van Assche, J. Axler, HJ Kim, S. Danese, I. Fox, C. Milch, S. Sankoh, T. Wyant, J. Xu, A. Parikh and GS Group (2013) “Vedolizumab as induction and maintenance therapy for ulcerative colitis.” N Engl J Med 369(8): 699-710.).
Ulcerative colitis patients, who are chronically active and refractory to known treatments pose a serious medical challenge and often the only remaining course of action is
colectomy. A total colectomy is a potentially curative option in severe UC, but is a life-changing operation that entails risks as complications, such as pouch failure, pouchitis, pelvic sepsis, infertility in women, and nocturnal faecal soiling, may follow. Therefore, surgery is usually reserved for patients with severe refractory disease, surgical or other emergencies, or patients with colorectal dysplasia or cancer.
An emerging third line treatment for UC is cobitolimod (Kappaproct/DIMS0150), a modified single strand deoxyribonucleic acid (DNA)-based synthetic oligonucleotide of 19 bases in length. Cobitolimod has the sequence 5′- G*G*A*ACAGTTCGTCCAT*G*G*C-3′ (SEQ ID NO:1), wherein the CG dinucleotide is unmethylated.
Cobitolimod functions as an immunomodulatory agent by targeting the Toll-like receptor 9 (TLR9) present in immune cells. These immune cells (ie, B-cells and plasmacytoid dendritic cell (pDCs) reside in high abundance in mucosal surfaces, such as colonic and nasal mucosa. The immune system is the key mediator of the changes of UC. The mucosa of the colon and rectum of patients with UC is chronically inflamed and contains active immune cells. Cobitolimod may be topically administered in the region of inflammation, which places the drug in close contact with a high number of intended target cells, ensuring that the drug will reach an area rich in TLR9 expressing cells.The activation of these cells by cobitolimod induces various cytokines,
The clinical efficacy of cobitolimod has been demonstrated in the “COLLECT” (CSUC-01/10 ) clinical trial, which involved the administration to patients of 30 mg doses of cobitolimod, at 4 week intervals and also in the “CONDUCT” (CSUC- 01/16 ) clinical trial, which involved testing different dosage regimes. The details of the “COLLECT” trial were published in Journal of Crohn’s and Colitis (Atreya et al. J Crohn’s Colitis, 2016 May 20) and are summarized in Reference Example 1. The details of the “CONDUCT” clinical trial were published in The Lancet Gastroenterology and Hepatology (Atreya et al 2020. Lancet Gastroenterol Hepatol. 2020 Dec;5(12): 1063-1075) and are summarized in Reference Example 2. Overall, data on cobitolimod support a positive benefit-risk
assessment for patients with chronic UC which is in an active phase (occasionally referred to herein as “chronic active UC”). Cobitolimod is safe and well tolerated and has been shown to be effective to induce clinical response and remission in patients with chronic UC which is in an active phase, as well as symptomatic and endoscopic remission in patients with treatment refractory, moderate to severe chronic UC which is in an active phase. Despite the clinical trial results obtained this far, there still remains a need for additional effective dosages of cobitolimod which exhibit both good efficacy and safety.
In the COLLECT study, which involved administration of a relatively low (30mg) dose of cobitolimod, topical administration of cobitolimod was performed using a spray catheter device, administered during an endoscopy. This is an invasive medical procedure which is necessarily carried out by a medical professional. Further, before the topical administration of the cobitolimod to the patients, the colon of each patient was cleaned to remove faecal matter. That was done to enable the cobitolimod to reach the intestinal epithelial cells within the colon and to enable the endoscopist to view the colonic mucosa. Thus, it is well known in the art that oligonucleotides such as cobitolimod bind to organic matter such as faeces.
As noted above, patients suffering from chronic ulcerative colitis, who are in an active disease state and refractory to known treatments pose a serious medical challenge and often the only remaining course of action is colectomy. For this reason, patients will tolerate medical intervention which requires both colonic cleaning to remove faecal matter and topical administration via spray catheter, despite the inconvenience and discomfort involved in such invasive procedures. However, it would be therapeutically desirable to provide a topical treatment for ulcerative colitis patients which does not require colonic cleaning to remove faecal matter and which, preferably, can be self-administered by the patient.
PATENTS
WO2001074344
WO2005080568
WO2007004977
WO2007004979
WO2007050034
EP2596806
WO2018206722
WO2018206713
WO2018206711
WO2020099585
WO2021037764
//////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
InDex Pharmaceuticals Holding AB (publ) announced that the company has entered an agreement for services with global clinical research organisation (CRO) Parexel Biotech for the phase III study CONCLUDE. The study will evaluate the efficacy and safety of the drug candidate cobitolimod for the treatment of moderate to severe left-sided ulcerative colitis.
“We are excited to advance cobitolimod into phase III, which is the final stage of development before applying for market approval. After the successful collaboration in our recent phase IIb study CONDUCT, we are very pleased to collaborate once again with Parexel Biotech as our clinical development partner”, says Peter Zerhouni, CEO of InDex Pharmaceuticals. “Parexel Biotech is a leading global CRO with considerable experience managing phase III studies in inflammatory bowel disease, which will ensure an efficient execution of the study.”
CONCLUDE is a randomised, double-blind, placebo-controlled, global phase III study to evaluate cobitolimod as a novel treatment for patients with moderate to severe left-sided ulcerative colitis. The induction study will include approximately 400 patients, and the primary endpoint will be clinical remission at week 6. Patients responding to cobitolimod in the induction study will be eligible to continue in a one-year maintenance study, where they will be treated with either cobitolimod or a placebo. Apart from the dosing 250 mg x 2, which was the highest dose and the one that showed the best efficacy in the phase IIb study CONDUCT, the phase III study will also evaluate a higher dose, 500 mg x 2, in an adaptive study design. This higher dose has the potential to provide even better efficacy than what was observed in the phase IIb study.
“We are pleased to partner with InDex Pharmaceuticals on phase III clinical trial CONCLUDE to evaluate a potential new therapy for patients with moderate to severe ulcerative colitis,” said Jim Anthony, Senior Vice President and Global Head, Parexel Biotech. “Our collaboration with InDex Pharmaceuticals demonstrates our commitment to designing innovative solutions that draw from our global clinical experience and therapeutic expertise to fulfil unmet medical needs on behalf of patients worldwide.”
///////////COBITOLIMOD, WHO 10066, IDX 0150, DIMS 0150, Kappaproct
Difluprednate is a topical corticosteroid used for the symptomatic treatment of inflammation and pain associated with ocular surgery.
Difluprednate is a corticosteroid, It is chemically a butyrate ester of 6(alpha),9(alpha)-difluoro prednisolone acetate. Accordingly, difluprednate is sometimes abbreviated DFBA, for difluoroprednisolone butyrate acetate.
Difluprednate is a topical corticosteroid indicated for the treatment of infammation and pain associated with ocular surgery. It is a butyrate ester of 6(α), 9(α)-difluoro prednisolone acetate. Difluprednate is abbreviated DFBA, or difluoroprednisolone butyrate acetate. It is indicated for treatment of endogenous anterior uveiti.
Approval
On June 24, 2008, the US Food and Drug Administration (FDA) approved difluprednate for the treatment of post-operative ocular inflammation and pain.[1] It is marketed by Alcon under the tradename Durezol.
WO/2022/118271DIFLUPREDNATE FOR REDUCING THE ADVERSE EFFECTS OF OCULAR INFLAMMATION
SYN 1
Synthetic Reference
Process for preparation of Difluprednate from sterol fermentation product; Ding, Kai; Xu, Feifei; Assignee Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, Peop. Rep. China; East China University of Science and Technology; 2014; Patent Information; Aug 06, 2014; CN; 103965277; A
SYN 2
Synthetic Reference
Preparation method of Difluprednate; Tian, Yuan; Zhou, Shengan; Guo, Bin; Xu, Zhiguo; Assignee Guangzhou Renheng Pharmaceutical Technology Co., Ltd., Peop. Rep. China 2017; Patent Information; May 10, 2017; CN; 106632561; A
SYN3
Synthetic Reference
Shailesh, Singh; Bharat, Suthar; Jain, Ashish; Gaikwad, Vinod; Kulkarni, Kuldip. Process for preparing difluprednate. Assignee Ajanta Pharma Ltd., India. IN 2013MU02535. (2015).
SYN4
Synthetic Reference
Sun, Hongbin; Chen, Bo. Method for preparation of Difluprednate. Assignee China Pharmaceutical University, Peop. Rep. China. CN 103509075. (2014).
Embodiment 1:4, pregnant steroid-17 α of 9 (11)-diene, 21-dihydroxyl-3,20-diketone-21-acetic ester (formula III compound)
10g hydrocortisone-21 acetic ester (formula II compound) is joined in 250mL eggplant type bottle, add 50mL N, dinethylformamide and 8.8mL pyridine, slowly heat up and make material dissolution complete, slowly cooling afterwards, slowly be added dropwise to 4.4mL methylsulfonyl chloride, add rear solution to be yellow completely.Be warming up to 85 ℃ of stirrings, the reaction solution thick one-tenth that can slowly become sticky is faint yellow, adds slightly some DMFs and makes reaction solution dilution, can normally stir, and keeps this thermotonus one hour, and reaction solution slowly becomes grey black during this period.TLC follows the tracks of (sherwood oil: ethyl acetate=1: 1) show that reaction finishes.Stop heating, treat that the backward reaction solution of slow cooling adds 200mL methyl alcohol, stir 1min, reaction flask is placed in to crystallization under ice-water bath.Suction filtration after 1h, makes water and methanol wash filter cake, crude product productive rate 100%.With methyl alcohol-methylene dichloride mixed solvent system recrystallization, obtain sterling, M.P.231-235 ℃, productive rate 90%. 1H-NMR(300MHz,CDCl 3):δ(ppm)5.75(1H,s,4-H),5.55(1H,s,11-H),5.07(1H,d,J=5Hz,21-H),4.84(1H,d,J=5Hz,21-H),2.15(3H,s,H-21-OAc),1.31(3H,s,19-CH 3),0.65(3H,s,18-CH 3),0.66-2.90(m,17H,backbone).
By 9.4g4, pregnant steroid-17 α of 9 (11)-diene, 21-dihydroxyl-3,20-diketone-21-acetic ester (formula III compound) and 10g4-Dimethylamino pyridine add in 1000mL eggplant-shape bottle, add again 50mL diethylene glycol dimethyl ether and 260mL methylene dichloride, heated and stirred makes dissolution of solid, slowly adds 32mL butyryl oxide slightly after cooling, is warming up to 80 ℃ of return stirrings.After 23h, TLC follows the tracks of, and raw material primitive reaction is complete, stops heating and stirs.Vacuum concentration is removed methylene dichloride.After being down to room temperature, add frozen water in reaction flask, white solid standing to be separated out.Suction filtration, saturated sodium bicarbonate aqueous solution washing leaching cake, dries under infrared lamp, obtain 4,9 (11)-diene-17 α, 21-dihydroxyl-3,20-ketone-21-acetic ester 17 iophenoxic acid esters (formula IV compound) sterling 10.65g, M.P220-224 ℃, productive rate 95.9%. 1H-NMR(500MHz,CDCl 3):δ(ppm)5.75(1H,s,4-H),5.54(1H,m,11-H),4.87(1H,d,J=4.8Hz,O=C-CH 2-O,21-H),4.64-4.91(2H,ABq,J=16.6Hz,21-H),2.75(2H,m,2-H),0.70(3H,s,18-CH 3),0.95(3H,t,J=4.4Hz),1.34(3H,s,18-CH 3),1.66(2H,m,-CH 2CH 3),2.17(3H,s,O=C-CH 3),2.32(2H,t,J=4.3Hz,O=C-CH 2),? 13C-NMR(75MHz,CDCl 3):δ(ppm)199.1,198.9,173.4,170.4,169.1,144.1,124.1,118.5,94.5,66.9,48.2,46.3,40.9,37.5,36.4,34.2,33.8,32.7,32.2,32.1,30.6,26.2,24.5,20.5,18.3,13.7,13.6;ESI-MS?m/z:457.2[M+H +],479.2[M+Na +];HRMS?for?C 27H 36O 6+Na +?calcd?479.2410,found479.2402.
10g4, pregnant steroid-17 α of 9 (11)-diene, 21-dihydroxyl-3,20-diketone-21-acetic ester 17 iophenoxic acid esters add in 250mL eggplant type bottle, then add 80mL methylvinyl acetate, slowly drip while stirring the 1mL vitriol oil.Be warming up to 80 ℃ of stirring reactions, solution is thin out yellow clarification slowly.(sherwood oil: ethyl acetate=3: 1), raw material reaction is complete produces new point to TLC after 30min.Stop heating, wait to be cooled to 50 ℃, add 1mL triethylamine, be stirred to and be down to room temperature.Add water in reaction solution, ethyl acetate aqueous layer extracted three times, saturated common salt water washing organic phase twice, anhydrous sodium sulfate drying.After 30min, steam organic solvent and obtain brown color oily matter.Column chromatography is purified and is obtained 3,5,9 (11) pregnant steroid-3 of triolefin, 17 α, 21 trihydroxy–3,20-diketone-3,21-diacetate esters 17 iophenoxic acid esters, productive rate 90%. 1H-NMR(300MHz,CDCl 3):δ(ppm)5.74(1H,s,4-H),5.53(1H,s,11-H),5.45(1H,s,6-H),4.64-4.91(2H,ABq,J=16.6Hz,21-H),2.17(3H,s,-COCH 3),1.17(3H,s,19-CH 3),0.96(3H,t,J=7.5Hz),0.70(3H,s,18-CH 3).
14g4, 9 (11)-diene-6 α-fluoro-17 α, 21-dihydroxyl-3, 20-diketone-21-acetic ester 17 iophenoxic acid esters (formula VII) and 9 (11)-diene-6 β-fluoro-17 α, 21-dihydroxyl-3, the mixture of 20-diketone-21-acetic ester 17 iophenoxic acid esters (formula VI) adds in dry three-necked bottle, add while stirring 400mL acetum, under room temperature, slowly pass into anhydrous hydrogen chloride gas (98% vitriol oil is added dropwise in 37% concentrated hydrochloric acid solution and makes) until saturated, be stirred to raw material and be dissolved into yellow solution completely, continue to stir 2h, TLC monitoring reacts completely, stop stirring, in reaction solution, add the aqueous solution, after separating out solid, suction filtration, saturated sodium bicarbonate aqueous solution washing, dry, be weighed as 13g, productive rate is 93%. 1H?NMR(300MHz,CDCl 3):δ(ppm)6.10(s,1H),5.61(s,1H),5.41-5.16(m,1H),4.64-4.91(2H,ABq,J=16.6Hz,21-H),2.82(dd,J=28.3,15.7Hz,3H),2.50(s,2H),2.32(t,J=7.4Hz,2H),2.17(s,3H),1.96(s,5H),1.66(d,J=7.4Hz,2H),1.46(s,2H),1.33(s,3H),0.96(s,3H),0.71(s,3H).
13g 6 α-fluoro-4; 9; (11)-diene-pregnant steroid-3,20-22 ketone-17-butyric ester-20-acetic ester is dissolved in and fills 300mL1, in the eggplant type bottle of 4 dioxane; add while stirring 40mL 0.46mol/L high chloro acid solution; under room temperature, stir after several minutes, add 14g N-succinimide in reaction system, under nitrogen protection, stir; raw material dissolves gradually, and it is faint yellow that reaction solution is.(the sherwood oil: ethyl acetate=12: 5) monitoring, raw material primitive reaction is complete, adds 10%Na of TLC after 2h 2sO 3unnecessary N-succinimide is fallen in aqueous solution cancellation, and checks (it is blue that test paper no longer becomes) with starch-kalium iodide test paper.Add water in reaction flask, ethyl acetate extraction three times, twice of saturated common salt water washing organic phase, anhydrous sodium sulfate drying organic phase, after 30min, be spin-dried for organic phase, obtain faint yellow oily matter, column chromatography purification (sherwood oil: ethyl acetate=12: 1) obtain white solid 6 α-fluoro-9 α-bromo-11 beta-hydroxies-4-alkene-pregnant steroid-3, the about 14g of 20-diketone-17-butyric ester-20-acetic ester, productive rate is 89%. 1H-NMR(300MHz,CDCl 3):δ(ppm)5.93(1H,d,J=4.5,4-H),5.06(1H,m,6-H),4.64-4.91(2H,ABq,J=16.6Hz,21-H),2.17(3H,s,-COCH 3),1.84(3H,s,18-CH 3),0.96(3H,t,J=7.5Hz),1.02(3H,s,19-CH 3),4.72(1H,s,11-H);ESI-MS?m/z:593.3,595.3[M+Na +].
100mg 6 α-fluoro-9 β, 11 beta epoxides-4-alkene-pregnant steroid-3,20-diketone-17-butyric ester-20-acetic ester drops in the Plastic Bottle of tetrafluoroethylene, adds 2mL methylene dichloride to dissolve, and stirs at-20 ℃.1mL Olah reagent with under 1mL methylene dichloride low temperature, mix after, be slowly added dropwise in reaction system, maintain low temperature and stir 2 hours, TLC monitoring reaction finishes.Reaction flask shifts out low-temp reaction groove, is slowly added dropwise to the 1mol/L NaOH aqueous solution by excessive HF cancellation, is adjusted to pH7~8.Add chloroform in reaction system, extraction, organic layer is used respectively aqueous hydrochloric acid and the saturated common salt water washing of 3mol/L, anhydrous sodium sulfate drying, after standing 30min, steams except organic solvent, column chromatography is further purified and is obtained white solid powder 6 α, 9 α-fluoro-11 beta-hydroxies-4-alkene-pregnant steroid-3,20-diketone-17-butyric ester-20-acetic ester, productive rate 90%. 1H-NMR(300MHz,CDCl 3):δ(ppm)?6.11(1H,d,J=4.5Hz,4-H),5.27(1H,m,6-H),4.64-4.91(2H,ABq,J=16.6Hz,21-H),2.17(3H,s,-COCH 3),4.40(1H,d,J=4.5Hz,11-H),1.02(3H,s,18-CH 3),0.96(3H,t,J=7.5Hz),1.52(3H,s,19-CH 3);ESI-MS?m/z:533.3[M+Na +]
40mg 6 α, 9 α-fluoro-11 beta-hydroxies-4-alkene-pregnant steroid-3,20-diketone-17-butyric ester-20-acetic ester is dissolved in 3mL dioxane, adds 28mgDDQ, and 100 ℃ of return stirrings heat up.TLC monitoring reaction (sherwood oil: ethyl acetate=12: 8) after 13h, generate the larger product of polarity, steam except organic solvent dioxane, obtain brown color oily matter, add a small amount of methylene dichloride lysate, suction filtration, elimination solid residue, filtrate is washed with sodium bicarbonate aqueous solution after adding a small amount of methylene dichloride again, steams except organic phase rear pillar Chromatographic purification, obtain white solid powder 6 α, 9 α-fluoro-11 beta-hydroxies-Isosorbide-5-Nitrae-diene-pregnant steroid-3,20-diketone-17-butyric ester-20-acetic ester, be title molecule difluprednate, productive rate 70%. 1h-NMR (300MHz, CDCl 3): δ (ppm) 7.20 (1H, d, J=4.5Hz, 1-H), 6.43 (1H, s, 4-H), 6.38 (1H, d, J=6Hz, 2-H), 5.36 (1H, m, 6-H), 4.64-4.91 (2H, ABq, J=16.6Hz, 21-H), 4.43 (1H, d, J=4.5Hz, 11-H), 2.27 (2H, m ,-CH 2-CH 3), 2.17 (3H, s, O=C-CH 3), 1.55 (3H, s, 19-CH 3), 1.02 (3H, s, 18-CH 3), 0.93 (3H, t, J=4.5Hz, 0=C-CH 2cH 2cH 3); ESI-MS m/z:509.3[M+H +]; HRMS for C 27h 35o 7f 2+ H +calcd 509.2351, found 509.2356.M.P.188-190 ℃ (literature value M.P.190-194 ℃); [α] d22=+30.1 ° of (literature values [α] d22=+31.7 °).
Claims (6)
Hide Dependent
1. a method of preparing difluprednate, as following reaction formula:
Specifically comprise the following steps:
(1) by hydrocortisone-21-acetic ester (formula II compound):
Carry out dehydration reaction, generate formula III compound:
(2) formula III compound is carried out to butyric acid esterification, obtains formula IV compound:
(3) formula IV compound is carried out to the reaction of enolization esterifying reagent, obtains formula V compound:
(4) formula V compound is reacted with fluoro reagent and obtains formula VI and formula VII compound:
(5) by formula VI compound, through configuration reversal, reaction obtains formula VII compound;
(6) formula VII compound is reacted with N-bromo-succinimide and water, obtains formula VIII compound:
(7) formula VIII compound epoxidation under alkaline condition is obtained to formula IX compound:
(8) formula IX compound is reacted with fluorination reagent and obtains formula X compound:
(9) dehydrogenation of formula X compound oxidation is obtained to formula I compound (difluprednate).
2. method as claimed in claim 1, is characterized in that, in step (2), formula III compound is obtained to formula IV compound through fourth esterification, and the fourth esterifying reagent adopting is butyryl oxide or butyryl chloride; The alkaline catalysts adopting is pyridine, triethylamine or DMAP; The solvent adopting is methylene dichloride, diethylene glycol dimethyl ether, 1, the mixture of the optional solvents in 2-ethylene dichloride, dioxane, trichloromethane, DMF, methyl-sulphoxide, N,N-dimethylacetamide or above-mentioned solvent.
3. method as claimed in claim 1, is characterized in that, in step (3), formula IV compound is obtained to formula V compound through enolization esterification, and the enolization esterifying reagent adopting is diacetyl oxide, Acetyl Chloride 98Min., methylvinyl acetate or vinyl-acetic ester; The catalyzer adopting is the vitriol oil or tosic acid; The solvent adopting is the mixture of the optional solvents in methylene dichloride, chloroform, toluene, methylvinyl acetate, vinyl-acetic ester or above-mentioned solvent.
4. method as claimed in claim 1, is characterized in that, in step (4), formula V compound is obtained to formula VI compound and formula VII compound through fluoridizing, and the fluoro reagent adopting is Selectfluor or Accufluor; The solvent adopting is the mixture of the optional solvents in methylene dichloride, chloroform, toluene, acetonitrile or above-mentioned solvent.
5. method as claimed in claim 1, it is characterized in that, in step (8), formula IX compound is obtained to formula X compound through fluoridizing open loop, the fluorination reagent adopting is aqueous hydrogen fluoride solution, hydrogen fluoride pyridine solution (Olah reagent) or hydrogen fluoride triethylamine solution; The solvent adopting is methylene dichloride, chloroform, 1, the mixture of the optional solvents in 2-ethylene dichloride, tetrahydrofuran (THF), toluene or above-mentioned solvent; Range of reaction temperature is-50~50 ℃.
6. a key intermediate compound for synthetic difluprednate, shown in IV compound:
Difluprednate ophthalmic emulsion 0.05% is also being studied in other ocular inflammatory diseases, including a phase 3 study evaluating difluprednate for the treatment of anterior uveitis[2][3]
Vutrisiran Vutrisiran Sodium is a sodium salt of an siRNA derivative targeting transthyretin (TTR) covalently linked to a triantennary GalNAc3 complex at the 3’ end of the sense strand. The siRNA moiety is composed of a duplex oligonucleotide of sense strand consisting of chemically modified 21 nucleotide residues and antisense strand consisting of chemically modified 23 nucleotide residues each.
Vutrisiran is a double-stranded small interfering ribonucleic acid (siRNA) that targets wild-type and mutant transthyretin (TTR) messenger RNA (mRNA).7 This siRNA therapeutic is indicated for the treatment of neuropathies associated with hereditary transthyretin-mediated amyloidosis (ATTR), a condition caused by mutations in the TTR gene.2 More than 130 TTR mutations have been identified so far,3 but the most common one is the replacement of valine with methionine at position 30 (Val30Met).2 The Val30Met variant is the most prevalent among hereditary ATTR patients with polyneuropathy, especially in Portugal, France, Sweden, and Japan.2
TTR mutations lead to the formation of misfolded TTR proteins, which form amyloid fibrils that deposit in different types of tissues. By targeting TTR mRNA, vutrisiran reduces the serum levels of TTR.6,7 Vutrisiran is commercially available as a conjugate of N-acetylgalactosamine (GalNAc), a residue that enables the delivery of siRNA to hepatocytes.5,7 This delivery platform gives vutrisiran high potency and metabolic stability, and allows for subcutaneous injections to take place once every three months.8 Another siRNA indicated for the treatment of polyneuropathy associated with hereditary ATTR is patisiran.2 Vutrisiran was approved by the FDA in June 2022.
Schematic illustrations of the working mechanisms of miRNA (a) and siRNA (b)
Structures of chemical modifications and analogs used for siRNA and ASO decoration. According to the modification site in the nucleotide acid, these structures can be divided into three classes: phosphonate modification, ribose modification and base modification, which are marked in red, purple and blue, respectively. R = H or OH, for RNA or DNA, respectively. (S)-cEt-BNA (S)-constrained ethyl bicyclic nucleic acid, PMO phosphorodiamidate morpholino oligomer
Representative designs for the chemical modification of siRNA. The sequences and modification details for ONPATTRO®, QPI-1007, GIVLAARI and inclisiran are included. The representative siRNA modification patterns developed by Alnylam (STC, ESC, advanced ESC and ESC+) and arrowhead (AD1-3 and AD5) are shown. Dicerna developed four GalNAc moieties that can be positioned at the unpaired G–A–A–A nucleotides of the DsiRNA structure. 2′-OMe 2′-methoxy, 2′-F 2′-fluoro, GNA glycol nucleic acid, UNA unlocked nucleic acid, SS sense strand, AS antisense strand
siRNA delivery platforms that have been evaluated preclinically and clinically. Varieties of lipids or lipidoids, siRNA conjugates, peptides, polymers, exosomes, dendrimers, etc. have been explored and employed for siRNA therapeutic development by biotech companies or institutes. The chemical structures of the key component(s) of the discussed delivery platforms, including Dlin-DMA, Dlin-MC3-DMA, C12-200, cKK-E12, GalNAc–siRNA conjugates, MLP-based DPC2.0 (EX-1), PNP, PEI, PLGA-based LODER, PTMS, GDDC4, PAsp(DET), cyclodextrin-based RONDEL and dendrimer generation 3 are shown. DLin-DMA (1,2-dilinoleyloxy-3-dimethylaminopropane), DLin-MC3-DMA (6Z,9Z,28Z,31Z)-heptatriaconta-6,9,28,31-tetraen-19-yl-4-(dimethylamino) butanoate, DPC Dynamic PolyConjugates, MLP membrane-lytic peptide, CDM carboxylated dimethyl maleic acid, PEG polyethylene glycol, NAG N-acetylgalactosamine, PNP polypeptide nanoparticle, PEI poly(ethyleneimine), LODER LOcal Drug EluteR, PLGA poly(lactic-co-glycolic) acid, PTMS PEG-PTTMA-P(GMA-S-DMA) poly(ethylene glycol)-co-poly[(2,4,6-trimethoxybenzylidene-1,1,1-tris(hydroxymethyl))] ethane methacrylate-co-poly(dimethylamino glycidyl methacrylate), GDDC4 PG-P(DPAx-co-DMAEMAy)-PCB, where PG is guanidinated poly(aminoethyl methacrylate) PCB is poly(carboxybetaine) and P(DPAx-co-DMAEMAy) is poly(dimethylaminoethyl methacrylate-co-diisopropylethyl methacrylate), PEG-PAsp(DET) polyethylene glycol-b-poly(N′-(N-(2-aminoethyl)-2-aminoethyl) aspartamide), PBAVE polymer composed of butyl and amino vinyl ether, RONDEL RNAi/oligonucleotide nanoparticle delivery
Vutrisiran SodiumVutrisiran Sodium is a sodium salt of an siRNA derivative targeting transthyretin (TTR) covalently linked to a triantennary GalNAc3 complex at the 3’ end of the sense strand. The siRNA moiety is composed of a duplex oligonucleotide of sense strand consisting of chemically modified 21 nucleotide residues and antisense strand consisting of chemically modified 23 nucleotide residues each.C530H672F9N171Na43O323P43S6 : 17289.77 [1867157-35-4 , Vutrisiran]
REF
Nucleic Acids Research (2019), 47(7), 3306-3320.
Drug Metabolism & Disposition (2019), 47(10), 1183-1201.
The present invention relates to pharmaceutical compositions and methods of treatment comprising administering to a patient in need thereof a combination of a benzoxazole derivative transthyretin stabilizer or a pharmaceutically acceptable salt or prodrug thereof and an additional therapeutic agent for the treatment of transthyretin amyloidosis. Particularly, the present invention relates to pharmaceutical compositions and methods of treatment comprising administering to a patient in need thereof 2-(3,5-dichlorophenyl)-1,3-benzoxazole-6-carboxylic acid or a pharmaceutically acceptable salt or prodrug thereof and one or more additional therapeutic agent for the treatment of transthyretin amyloidosis.
The present invention relates to pharmaceutical compositions and methods of treatment comprising administering to a patient in need thereof a combination of a benzoxazole derivative transthyretin stabilizer or a pharmaceutically acceptable salt or prodrug thereof and one or more additional therapeutic agent. Particularly, the present invention relates to pharmaceutical compositions and methods of treatment comprising administering to a patient in need thereof 2-(3,5-dichlorophenyl)-1,3-benzoxazole-6-carboxylic acid or a pharmaceutically acceptable salt or prodrug thereof and one or more additional therapeutic agent. The compositions and methods of the invention are useful in stabilizing transthyretin, inhibiting transthyretin misfolding, proteolysis, and treating amyloid diseases associated thereto.
Transthyretin (TTR) is a 55 kDa homotetrameric protein present in serum and cerebral spinal fluid and which functions as a transporter of L-thyroxine (T4) and holo-retinol binding protein (RBP). TTR has been found to be an amyloidogenic protein that, under certain conditions, can be transformed into fibrils and other aggregates which can lead to disease pathology such as polyneuropathy or cardiomyopathy in humans.
US Patent Nos. 7,214,695; 7,214,696; 7,560,488; 8, 168.683; and 8,653,119 each of which is incorporated herein by reference, discloses benzoxazole derivatives which act as transthyretin stabilizers and are of the formula
or a pharmaceutically acceptable salt thereof; wherein Ar is 3,5-difluorophenyl, 2,6-difluorophenyl, 3,5-dichlorophenyl, 2,6-dichlorophenyl, 2-(trifluoromethyl)phenyl or 3-(trifluoromethyl)phenyl. Particularly, 2-(3,5-dichlorophenyl)-1,3-benzoxazole-6-carboxylic acid (tafamidis) of the formula
is disclosed therein. Tafamidis is an orally active transthyretin stabilizer that inhibits tetramer dissociation and proteolysis that has been approved in certain jurisdictions for the treatment of transthyretin polyneuropathy (TTR-PN) and is currently in development for the treatment of transthyretin cardiomyopathy (TTR-CM). US Patent No. 9,249, 112, also incorporated herein by reference, discloses polymorphic forms of the meglumine salt of 2-(3,5-dichlorophenyl)-1,3-benzoxazole-6-carboxylic acid (tafamidis meglumine). US Patent No. 9,770,441 discloses polymorphic forms of the free acid of 2-(3,5-dichlorophenyl)-1,3-benzoxazole-6-carboxylic acid (tafamidis), and is also incorporated by reference herein.
Summary of the Invention
The present invention provides pharmaceutical compositions and methods comprising the compound 2-(3,5-dichlorophenyl)-1,3-benzoxazole-6-carboxylic acid or a pharmaceutically acceptable salt or prodrug thereof, and one or more additional therapeutic agent. Particular embodiments of this invention are pharmaceutical compositions and methods comprising 2-(3,5-dichlorophenyl)-1,3-benzoxazole-6-carboxylic acid or a pharmaceutically acceptable salt or prodrug thereof, and one or more additional therapeutic agents selected from the group consisting of agents that lower plasma levels of TTR such as an antisense therapy, TTR gene editing therapy, transcriptional modulators, translational modulators, TTR protein degraders and antibodies that bind and reduce TTR levels; amyloid reduction therapies such as anti amyloid antibodies (either TTR selective or general), stimulators of amyloid clearance, fibril disruptors and therapies that inhibit amyloid nucleation; other TTR stabilizers; and TTR modulators such as therapeutics which inhibit TTR cleavage. Particularly, the present invention provides pharmaceutical compositions and methods comprising tafamidis or tafamidis meglumine salt with one or more additional therapeutic agents. More particularly, the present invention provides pharmaceutical compositions and the present invention provides pharmaceutical compositions and methods comprising tafamidis or tafamidis meglumine salt with one or more additional therapeutic agents. More particularly, the present invention provides pharmaceutical compositions and the present invention provides pharmaceutical compositions and methods comprising tafamidis or tafamidis meglumine salt with one or more additional therapeutic agents. More particularly, the present invention provides pharmaceutical compositions and
methods comprising a polymorphic form of tafamidis free acid or a polymorphic form of tafamidis meglumine salt with one or more additional therapeutic agents.
The present invention also provides a method of treating or preventing transthyretin amyloidosis in a patient, the method comprising administering to a patient in need thereof a therapeutically or prophylactically effective amount of 2-(3,5-dichlorophenyl)-1,3-benzoxazole- 6-carboxylic acid or a pharmaceutically acceptable salt or prodrug thereof, and one or more additional therapeutic agents.
A particular embodiment of the present method of treatment is the method comprising a pharmaceutical composition comprising 2-(3,5-dichlorophenyl)-1,3-benzoxazole-6-carboxylic acid or a pharmaceutically acceptable salt or prodrug thereof, and one or more additional therapeutic agent are administered orally. Additional embodiments of this invention are methods of treatment as described above wherein the 2-(3,5-dichlorophenyl)-1,3-benzoxazole-6-carboxylic acid or a pharmaceutically acceptable salt or prodrug thereof, and one or more additional therapeutic agent are administered parenterally (intravenously or subcutaneously). Further embodiments of this invention are methods of treatment wherein the 2-(3,5-dichlorophenyl)-1, 3-benzoxazole-6-carboxylic acid or a pharmaceutically acceptable salt or prodrug thereof is administered orally and the one or more additional therapeutic agent is administered either orally or parenterally. Another embodiment of the present invention is wherein a pharmaceutical composition comprising 2-(3,5-dichlorophenyl)-1,3-benzoxazole-6-carboxylic acid or a pharmaceutically acceptable salt or prodrug thereof in combination with one or more additional therapeutic agent is administered parenterally and then 2-(3,5-dichlorophenyl)-1,3-benzoxazole-6-carboxylic acid or a pharmaceutically acceptable salt or prodrug thereof is administered orally. A particular method of treatment is a method of treating TTR amyloidosis such as TTR polyneuropathy or TTR Another embodiment of the present invention is wherein a pharmaceutical composition comprising 2-(3,5-dichlorophenyl)-1,3-benzoxazole-6-carboxylic acid or a pharmaceutically acceptable salt or prodrug thereof in combination with one or more additional therapeutic agent is administered parenterally and then 2-(3,5-dichlorophenyl)-1,3-benzoxazole-6-carboxylic acid or a pharmaceutically acceptable salt or prodrug thereof is administered orally. A particular method of treatment is a method of treating TTR amyloidosis such as TTR polyneuropathy or TTR Another embodiment of the present invention is wherein a pharmaceutical composition comprising 2-(3,5-dichlorophenyl)-1,3-benzoxazole-6-carboxylic acid or a pharmaceutically acceptable salt or prodrug thereof in combination with one or more additional therapeutic agent is administered parenterally and then 2-(3,5-dichlorophenyl)-1,3-benzoxazole-6-carboxylic acid or a pharmaceutically acceptable salt or prodrug thereof is administered orally. A particular method of treatment is a method of treating TTR amyloidosis such as TTR polyneuropathy or TTR 5-dichlorophenyl)-1,3-benzoxazole-6-carboxylic acid or a pharmaceutically acceptable salt or prodrug thereof is administered orally. A particular method of treatment is a method of treating TTR amyloidosis such as TTR polyneuropathy or TTR 5-dichlorophenyl)-1,3-benzoxazole-6-carboxylic acid or a pharmaceutically acceptable salt or prodrug thereof is administered orally. A particular method of treatment is a method of treating TTR amyloidosis such as TTR polyneuropathy or TTR
cardiomyopathy, the method comprising administering to a patient in need thereof a therapeutically effective amount of 2-(3,5-dichlorophenyl)-1,3-benzoxazole-6-carboxylic acid or a pharmaceutically acceptable salt or prodrug thereof in combination with one or more additional therapeutic agents.
Exemplary RNAi agents that reduce the expression of TTR include patisiran and vutrisiran.
The ter s “antisense polynucleotide agent”, “antisense oligonucleotide”, “antisense compound”, and “antisense agent” as used interchangeably herein, refer to an agent comprising a single-stranded oligonucleotide that specifically binds to the target nucleic acid molecules via hydrogen bonding (e.g., Watson-Crick, Hoogsteen, or reversed Hoogsteen hydrogen bonding) and inhibits the expression of the targeted nucleic acid by an antisense mechanism of action, e.g., by RNase H. In some embodiments, an antisense agent is a nucleic acid therapeutic that acts by reducing the expression of a target gene, thereby reducing the expression of the polypeptide encoded by the target gene. Exemplary antisense agents that reduce the expression of TTR include inotersen and Ionis 682884/ ION-TTR-LRx (see, e.g., WO2014179627 which is incorporated by reference in its entirety). Further antisense agents that reduce the expression of TTR are provided, for example in WO2011139917 and WO2014179627, each of which is incorporated by reference in its entirety.
Tissues targeted by siRNA and miRNA therapeutics currently being investigated at the clinical stage. The corresponding therapeutic names are shown beside the tissues
Alnylam announces 3-month extension of review period for new drug application for vutrisiran to treat ATTR amyloidosis.
Alnylam Pharmaceuticals, Inc., a RNAi therapeutics company, announced that the FDA has extended the review timeline of the New Drug Application (NDA) for vutrisiran, an investigational RNAi therapeutic in development for the treatment of transthyretin-mediated (ATTR) amyloidosis, to allow for the review of newly added information related to the new secondary packaging and labelling facility.
Alnylam recently learned that the original third-party secondary packaging and labelling facility the Company planned to use for the vutrisiran launch was recently inspected and the inspection requires classification for the FDA to take action on the vutrisiran NDA. The inspection observations were not directly related to vutrisiran. In order to minimize delays to approval, Alnylam has identified a new facility to pack and label vutrisiran and submitted an amendment to the NDA for review by the FDA. The updated Prescription Drug User Fee Act (PDUFA) goal date to allow for this review is July 14, 2022. No additional clinical data have been requested by the FDA.
////////////Vutrisiran sodium, APPROVALS 2022, FDA 2022, FDA APPROVED, AMVUTTRA, 2022/6/13, ブトリシランナトリウム , ALN 65492, Votrisiran, siRNA
Originator University of California at San Francisco; University of Michigan; University of Washington
Developer University of California at San Francisco; University of Michigan; University of Washington; ViewPoint Therapeutics
Class Small molecules; Sterols
Mechanism of Action Amyloid inhibitors; Protein aggregation inhibitors; Protein folding inhibitors
28 Jun 2020 No recent reports of development identified for research development in Presbyopia in USA (Ophthalmic, Drops)
28 Dec 2019 No recent reports of development identified for preclinical development in Cataracts in USA (Ophthalmic, Drops)
01 Apr 2016 ViewPoint Therapeutics receives SBIR grant from National Eye Institute for VP1 001 development in Cataracts (ViewPoint Therapeutics website)
We previously identified an oxysterol, VP1-001 (also known as compound 29), that partially restores the transparency of lenses with cataracts. To understand the mechanism of VP1-001, we tested the ability of its enantiomer, ent-VP1-001, to bind and stabilize αB-crystallin (cryAB) in vitro and to produce a similar therapeutic effect in cryAB(R120G) mutant and aged wild-type mice with cataracts. VP1-001 and ent-VP1-001 have identical physicochemical properties. These experiments are designed to critically evaluate whether stereoselective binding to cryAB is required for activity.
Investigators are exploring chemical compounds to restore transparency to the crystalline lens.
Surgery is an effective but costly means of managing cataracts, and, like all surgical interventions, it carries risks. Moreover, a substantial number of people worldwide, particularly in developing countries, lack access to cataract surgery. The World Health Organization estimates that 65.2 million people globally are blind or visually impaired from cataracts.1 The development of an eye drop that restores transparency and flexibility to the crystalline lens would therefore be a game-changer as a less expensive, noninvasive option for treating a leading cause of blindness.
RESEARCH RESULTS
The crystalline lens is composed of epithelial and fiber cells. One of the major lens proteins in the fiber cells is alpha-crystallin, a chaperone protein thought to maintain homeostasis of the crystalline lens, thereby preserving its transparency and flexibility. As a person ages, alpha-crystallin proteins become prone to misfolding, causing them to clump together and form insoluble high-molecular-weight protein aggregates, which can lead to cataract formation.
Compound 29 (full name, 5-cholesten-3beta,25-diol), also known as VP1-001 (ViewPoint Therapeutics) and as 25-hydroxy-cholesterol, is an oxysterol, a derivative of cholesterol. Usha Andley, PhD, FARVO, is an investigator conducting research on this chemical compound’s use as a treatment for cataracts.2,3 Another oxysterol being investigated for this purpose is lanosterol. In a recent study, neither oxysterol was effective at reducing opacities of in vitro cultured lenses treated with various reagents to induce opacification in vitro.4 In an interview with ME, however, Dr. Andley stated that VP1-001 appears to be more effective than lanosterol at reducing lens opacity. VP1-001 differs from lanosterol in terms of solubility, she said, allowing VP1-001 to penetrate the eye better.
The goal of the research being conducted by Dr. Andley and her colleagues, she said, is twofold:
No. 1: To ensure that the compound is not toxic to the cornea; and
No. 2: To show that the compound is capable of reducing the tendency of alpha-crystallins to aggregate and perhaps reverse their aggregation.
In proof-of-concept studies, VP1-001 was incorporated into an eye drop formulation of 8% cyclodextrin. Dr. Andley and colleagues administered the drops three times per week in a mouse model for 2 to 4 weeks. According to Dr. Andley, the compound seemed to increase the stability of the alpha-crystallin protein so that it increased the soluble fraction of proteins from mouse and human lens cataracts. The compound also increased the solubility of two other lens crystallins, beta- and gamma-crystallin, in the mouse lens, and it seemed to reduce the abundance of high-molecular-weight aggregates in the lens.2,3
ViewPoint Therapeutics is currently developing this technology for use in humans. According to Dr. Andley, who is working with the company, ViewPoint Therapeutics is using different model systems for in vitro and protein-binding studies in an attempt to improve on earlier results with the chemical compound. Their research has identified new compounds, nonsterol ligands for alpha-crystallin, that exhibited in vitro activity and efficacy similar to or better than those of VP1-001 in mouse models of cataracts.5 The discovery of these nonsterols supports the hypothesis that pharmacologic chaperones targeting alpha-crystallin can prevent or reverse cataracts, Dr. Andley said.
CHALLENGES AND FUTURE DIRECTIONS
In studies to date, Dr. Andley and her colleagues have treated mice in one eye with the VP1-001 formulation, and the contralateral untreated eyes have served as controls. They then compared the two eyes after the conclusion of treatment (Figure). She is looking forward to conducting masked and randomized studies that include baseline measurements of lens opacity. According to Dr. Andley, this research will begin this year. Animal testing, however, can advance understanding only so far. Human testing of safety and efficacy will be a major step forward in the research on VP1-001 and newer variants.
Figure | Representative slit-lamp images from aged wild-type mouse lenses show the extent of opacity treated with vehicle (left) or drug (right). Mice were treated topically with the drug in one eye and vehicle in the contralateral eye three times per week for 2 weeks. Slit-lamp examinations were performed on conscious, live mice. Mouse 1 and 2 were treated with VP1-001.
A major challenge in the development of a pharmacologic treatment for cataract is how to determine if a chemical compound can be maintained in the lens at a sufficient concentration and for an adequate duration to achieve the desired outcome. A second challenge relates to detecting change. Dr. Andley and her colleagues are seeking a more quantitative method by which to assess the extent of lenticular opacity before and after treatment. They have a modified Lens Opacities Classification System, she said, but it is less objective than methods such as Scheimpflug photography. A goal, then, is to develop a more standardized, objective way of measuring results.
In addition to age-related cataract, Dr. Andley suggested, a topical agent could be advantageous for the treatment of congenital cataracts that form because of a mutation in alpha A crystallin or alpha B crystallin. Retaining the crystalline lens instead of extracting it would allow pediatric eyes to develop normally, she noted.
The idea of an eye drop formulation to treat cataracts may seem like science fiction, but Dr. Andley expects it to become a more realistic possibility within the next few years. The utility of such a chemical compound could extend beyond cataracts to the treatment of presbyopia. The hypothesis, she said, is that softening the crystalline lens would improve accommodative amplitude.
2. Makley LN, McMenimen KA, DeVree BT, et al. Pharmacological chaperone for α-crystallin partially restores transparency in cataract models. Science. 2015;350(6261):674-677.
3. Molnar KS, Dunyak BM, Su B, et al. Mechanism of action of VP1-001 in cryAB(R120G)-associated and age-related cataracts. Invest Ophthalmol Vis Sci. 2019;60(10):3320-3331.
4. Daszynski DM, Santhoshkumar P, Phadte AS, et al. Failure of oxysterols such as lanosterol to restore lens clarity from cataracts. Sci Rep. 2019;9(1):8459.
5. Dunyak B, Su B, Molnar K, et al. Discovery of non-sterol aB-crystallin ligands as potential cataract therapeutics. Invest Ophthalmol Vis Sci. 2019;60(9):5691.
Pimitespib (TAS-116) is an oral bioavailable, ATP-competitive, highly specific HSP90α/HSP90β inhibitor (Kis of 34.7 nM and 21.3 nM, respectively) without inhibiting other HSP90 family proteins such as GRP94. Pimitespib demonstrates less ocular toxicity.
Formula
C25H26N8O
CAS
1260533-36-5
Mol weight
454.5269
JAPAN APPROVED 2022/6/20, ピミテスピブ
Jeselhy
Taiho. originator
Pimitespib is a specific inhibitor of heat shock protein 90 (Hsp90) subtypes alpha and beta, with potential antineoplastic and chemo/radiosensitizing activities. Upon oral administration, pimitespib specifically binds to and inhibits the activity of Hsp90 alpha and beta; this results in the proteasomal degradation of oncogenic client proteins, which inhibits client protein dependent-signaling, induces apoptosis, and inhibits the proliferation of cells overexpressing HSP90alpha/beta. Hsp90, a family of molecular chaperone proteins that are upregulated in a variety of tumor cells, plays a key role in the conformational maturation, stability, and function of “client” proteins within the cell,; many of which are involved in signal transduction, cell cycle regulation and apoptosis, including kinases, cell-cycle regulators, transcription factors and hormone receptors. As TAS-116 selectively inhibits cytosolic HSP90alpha and beta only and does not inhibit HSP90 paralogs, such as endoplasmic reticulum GRP94 or mitochondrial TRAP1, this agent may have less off-target toxicity as compared to non-selective HSP90 inhibitors.
3-Ethyl-4-fluorobenzonitrile is an important intermediate for the preparation of a variety of new drugs under development, such as TAS-116, a Phase II clinical drug of Taiho Pharmaceuticals for the treatment of gastrointestinal stromal tumors.
Patent WO2005105760 discloses its preparation method. In the method, tetrakis(triphenylphosphine) palladium is used as a catalyst, and 3-bromo-4-fluorobenzonitrile is coupled with tetraethyl tin in a solvent hexamethylphosphoramide for a heating reaction for 15 hours to obtain 3 -Ethyl-4-fluorobenzonitrile. The method uses highly toxic tetraethyl tin, which brings great harm to operators and the environment, and is difficult to carry out industrial production. Meanwhile, the product 3-ethyl-4-fluorobenzonitrile obtained by the preparation method is an oily substance, which is purified by column chromatography with complicated operation, which is unfavorable for industrial production, and the specific purity of the product is not described.
Therefore, looking for a new method for preparing 3-ethyl-4-fluorobenzonitrile with cheap and easy-to-obtain raw materials, safe and simple operation, high product purity and low cost suitable for industrial production, which will speed up the research process of related new drugs under development. , it is of great significance to reduce the production cost of related new drugs.
Example 1 3-Bromo-4-fluorobenzonitrile
3-Bromo-4-fluorobenzaldehyde (250g, 1.23mol) was dissolved in acetonitrile (1.5L), then hydroxylamine sulfonic acid (67g, 1.48mol) was added, and the reaction was refluxed for 4h. TLC showed that the conversion of the raw materials was complete, and the reaction solution was concentrated. To a small volume, add water (2L) and stir for 30min, cool to 5-10°C and continue stirring for 10min, filter, dissolve the filter cake with methyl tert-butyl ether (1.2L), wash twice with 500ml of water, saturated with 200ml Washed with sodium bicarbonate solution, dried over anhydrous sodium sulfate, filtered, the filtrate was adsorbed with activated carbon (10g), filtered, concentrated under reduced pressure to remove the solvent, added n-heptane (250ml), cooled and stirred in an ice-salt bath for 1h, filtered, reduced Press drying to give 3-bromo-4-fluorobenzonitrile (217 g, 88% yield). 1 H NMR (CDCl 3 ,400MHz):δ7.91(m,1H),7.63(m,1H),7.24(m,1H)。
Example 2 3-Bromo-4-fluorobenzonitrile
Add tetrahydrofuran (100ml) to a 250ml reaction flask, add 3-bromo-4-fluorobenzaldehyde (10g, 49.2mmol) and ammonia (40ml) under stirring, add elemental iodine (25g, 98.5mmol) in batches under cooling to 5°C ), then raised to ambient temperature and reacted for 2 to 3 hours, the reaction was completed, the reaction solution was poured into a 10% aqueous solution of sodium sulfite (200g), extracted twice with methyl tert-butyl ether (100ml), dried over anhydrous sodium sulfate , concentrated under reduced pressure to remove the solvent, added n-heptane (20 ml), cooled to 0-10 °C and stirred for 1 h, filtered, and dried under reduced pressure to obtain 3-bromo-4-fluorobenzonitrile (9.6 g, yield: 97.5 %). The NMR spectrum of this compound was determined and was identical to the product of Example 1.
Example 3 3-ethyl-4-fluorobenzonitrile
3-Bromo-4-fluorobenzonitrile (200 g, 1 mol) and [1,1-bis(diphenylphosphino)ferrocene]palladium(II) chloride dichloromethane complex (4.08 g, 5mmol) was dissolved in THF (1.2L), 1.0M/L diethylzinc n-hexane solution (600mL, 0.6mol) was added at 40-50°C, and the temperature was raised to 50-60°C for 4-5h. TLC showed The raw materials reacted completely. After the reaction solution was cooled to room temperature, it was added to 5% dilute hydrochloric acid (1 L), the layers were separated, the organic layer was washed twice with 500 ml of water, and then concentrated under reduced pressure to remove the solvent. Then n-hexane (600mL) and activated carbon (20g) were added, refluxed for 0.5h, cooled to room temperature, filtered, then added activated carbon (10g) to the filtrate, refluxed for 0.5h, cooled to room temperature, filtered, and cooled to -50°C to -60°C and filtered, and the filter cake was dried under reduced pressure at 10-20°C to obtain 3-ethyl-4-fluorobenzonitrile (112 g, yield: 75%) as an off-white solid, melting point 23.1-27.4°C. 1 H NMR (CDCl 3 , 400MHz): δ 7.50 (m, 2H), 7.09 (m, 1H), 2.69 (q, J=7.6Hz, 2H), 1.24 (t, 3H, J=7.6Hz), HPLC purity 99.6%.
Acylation of 2-fluoro-4-iodopyridine with isobutyric anhydride in presence of BuLi and DIEA in THF at -78 °C gives 1-(2-fluoro-4-iodo-3-pyridinyl)-2-methylpropan-1-one ,
This upon cyclization using hydrazine hydrate at 65 °C gives 4-iodo-3-isopropylpyrazolo[3,4-b]pyridine.
N-Protection of intermediate with PMB-Cl in the presence of base NaH in solvent DMF at 0 °C affords 4-iodo-3-isopropyl-1-(4-methoxybenzyl)pyrazolo[3,4-b]pyridine,
This is coupled with 4-(4-imidazolyl)-1-methylpyrazole in the presence of Cu2O, 4,7-dimethoxy-1,10-phenanthroline, Cs2CO3 and PEG-diamine in solvent NMP or DMSO at 130 °C to furnish 4-[4-(4-pyrazolyl)-imidazol-1-yl]pyrazolo[3,4-b]pyridine derivative .
N-Deprotection of PMB-protected pyrazolo[3,4-b]pyridine derivative by using TFA and anisole gives free pyrazolo[3,4-b]pyridine ,
This on condensation with 3-ethyl-4-fluorobenzonitrile in the presence of Cs2CO3 in DMF at 95 °C yields 4-(pyrazolo[3,4-b]pyridin-1-yl)benzonitrile .
Finally, partial hydrolysis of nitrile by means of aqueous NaOH and H2O2 in DMSO/EtOH gives the Pimitespib TAS-116 .
The molecular chaperone heat shock protein 90 (HSP90) is a promising target for cancer therapy, as it assists in the stabilization of cancer-related proteins, promoting cancer cell growth, and survival. A novel series of HSP90 inhibitors were discovered by structure–activity relationship (SAR)-based optimization of an initial hit compound 11a having a 4-(4-(quinolin-3-yl)-1H-indol-1-yl)benzamide structure. The pyrazolo[3,4-b]pyridine derivative, 16e (TAS-116), is a selective inhibitor of HSP90α and HSP90β among the HSP90 family proteins and exhibits oral availability in mice. The X-ray cocrystal structure of the 16e analogue 16d demonstrated a unique binding mode at the N-terminal ATP binding site. Oral administration of 16e demonstrated potent antitumor effects in an NCI-H1975 xenograft mouse model without significant body weight loss.
Compound 1 in the present invention is 3-ethyl-4- {3-isopropyl-4- (4- (1-methyl-1H-pyrazol-4-yl) -1H-imidazole-1-yl) -1H-. Pyrazolo [3,4-b] pyridin-1-yl} benzamide (formula below). Compound 1 is known to have HSP90 inhibitory activity and exhibit excellent antitumor activity. Compound 1 can be synthesized based on the production methods described in Patent Documents 1 and 2.
[0013]
[hua 1]
Patent Document 1: International Publication No. 2012/093708 Patent Document 2: International Publication No. 2011/004610
Comparative Example 1 3-Ethyl-4- {3-isopropyl-4- (4- (1-methyl-1H-pyrazole-4-yl) -1H-imidazole-1-yl) -1H-pyrazolo [3, 4-b] Pyridine-1-yl} Synthesis of type I crystals of benzamide 3-Ethyl-4 obtained according to the production method described in International Publication No. 2012/093708 and International Publication No. 2011/004610. -{3-Isopropyl-4- (4- (1-methyl-1H-pyrazole-4-yl) -1H-imidazole-1-yl) -1H-pyrazolo [3,4-b] pyridin-1- A white solid (3.58 g) of yl} benzamide was added to ethanol (7.84 mL) and stirred at room temperature for 2 hours. After sampling, it was washed with ethanol (7.84 mL) and dried under reduced pressure at 70 to 80 ° C. for 20 hours to obtain type I crystals (yield: 2.40 g, yield: 61.2%, purity: 98.21%). rice field. Further, as shown in FIG. 1, the type I crystal has a diffraction angle (2θ) of 8.1 °, 10.9 °, 12.1 °, 14.0 °, and 14.9 in the powder X-ray diffraction spectrum. °, 16.2 °, 17.7 °, 20.2 °, 21.0 °, 21.5 °, 22.6 °, 24.3 °, 25.4 ° 26.4 °, 27.0 ° , 28.3 °, 30.2 °, 30.9 °, 31.5 °, 32.7 °, 34.7 °, 35.4 ° and 36.6 ° showed characteristic peaks.
Synthesis of Test Compound The following synthesis example compounds (Synthesis Examples 1 to 3) were synthesized according to the method described in WO2011 / 004610.
28 Mar 2022No recent reports of development identified for phase-I development in Peripheral-T-cell-lymphoma in China (IV, Injection)
26 Jan 2022ZIOPHARM Oncology is now called Alaunos Therapeutics
11 Dec 2021Safety and efficacy data from a phase II trial in Peripheral T-cell lymphoma presented at the 63rd American Society of Hematology Annual Meeting and Exposition (ASH-2021)
Darinaparsin is a small-molecule organic arsenical with potential antineoplastic activity. Although the exact mechanism of action is unclear, darinaparsin, a highly toxic metabolic intermediate of inorganic arsenicals (iAs) that occurs in vivo, appears to generate volatile cytotoxic arsenic compounds when glutathione (GSH) concentrations are low. The arsenic compounds generated from darinaparsin disrupt mitochondrial bioenergetics, producing reactive oxygen species (ROS) and inducing ROS-mediated tumor cell apoptosis; in addition, this agent or its byproducts may initiate cell death by interrupting the G2/M phase of the cell cycle and may exhibit antiangiogenic effects. Compared to inorganic arsenic compounds such as arsenic trioxide (As2O3), darinaparsin appears to exhibit a wide therapeutic window.
Darinaparsin, also know as ZIO-101 and SP-02, is a small-molecule organic arsenical with potential antineoplastic activity. Although the exact mechanism of action is unclear, darinaparsin, a highly toxic metabolic intermediate of inorganic arsenicals (iAs) that occurs in vivo, appears to generate volatile cytotoxic arsenic compounds when glutathione (GSH) concentrations are low. The arsenic compounds generated from darinaparsin disrupt mitochondrial bioenergetics, producing reactive oxygen species (ROS) and inducing ROS-mediated tumor cell apoptosis; in addition, this agent or its byproducts may initiate cell death by interrupting the G2/M phase of the cell cycle and may exhibit antiangiogenic effects.
Darinaparsin is an organic arsenical composed of dimethylated arsenic linked to glutathione, and is being investigated for antitumor properties in vitro and in vivo. While other arsenicals, including arsenic trioxide, have been used clinically, none have shown significant activity in malignancies outside of acute promyelocytic leukemia. Darinaparsin has significant activity in a broad spectrum of hematologic and solid tumors in preclinical models. Here, we review the literature describing the signaling pathways and mechanisms of action of darinaparsin and compare them to mechanisms of cell death induced by arsenic trioxide. Darinaparsin has overlapping, but distinct, signaling mechanisms. We also review the current results of clinical trials with darinaparsin (both intravenous and oral formulations) that demonstrate significant antitumor activity.
[0071] Sterile water (15.5 L) and ethyl alcohol (200 proof, 15.5 L) were charged in a reaction flask prior to the addition of L-glutathione (3.10 kg). While being stirred, the reaction mixture was cooled to 0-5 °C prior to the addition of triethylamine (1.71 L). Stirring was continued until most of the solids were dissolved and the solution was filtered. After filtration, the reaction mixture was cooled to 0-5 °C prior to the addition of chlorodimethylarsine (1.89 kg) over 115 minutes while maintaining the temperature at 0-5 °C. Stirring continued at 0-5 °C for 4 hours before acetone (30.6 L) was added over 54 minutes while maintaining the temperature at 0-5 °C. The suspension was stored at 0-5°C overnight prior to filtration. The solid was collected in a filter funnel, washed successively with ethyl alcohol (200 proof, 13.5 L) and acetone (13.5 L) and dried in suction for 23 minutes. A second similar run was performed and the collected solids from both runs were combined. Ethyl alcohol (200 proof, 124 L) and the combined solids (11.08 kg) were charged in a vessel. The slurry was stirred at ambient temperature for 2 hours before filtration, washing successively with ethyl alcohol (200 proof, 27 L) and acetone (27 L) and dried in suction for 60 minutes. The resulting solid was transferred to drying trays and dried in a vacuum oven at ambient temperature for 66 hours to provide darinaparsin as a solid with the differential scanning calorimetry (DSC) thermogram of Figure 1, with an extrapolated onset temperature at about 191.36° C and a peak temperature at about 195.65° C.
PATENT
WO 2010021928
Step 1
Dimethylchloroarsine. Dimethylarsinic acid, (CH3)2As(O)OH was supplied by the Luxembourg Chemical Co., Tel Aviv, Israel. The product was accompanied by a statement of its purity and was supplied as 99.7% pure. The dimethylarsinic acid was dissolved in water-hydrochloric acid to pH 3. A stream of sulfur dioxide was passed through this solution for about one hour. Dimethylchloroarsine separated as a heavy, colorless oil. The two liquid phases, water/(CH3)2AsCl were separated using a separatory funnel. The chlorodimethylarsine was extracted into diethylether and the ether solution was dried over anhydrous sodium sulfate. The dried solution was transferred to a distillation flask which was heated slowly to evaporate the ether. The remaining liquid, dimethylchloroarsine was purified by distillation. The fraction boiling at 106-109°C was collected. The product, a colorless oil. 1H NMR resonance at 1.65 ppm.
Step 2
SGLU-1: Glutathione (14.0 g, 45.6 mmol) was stirred rapidly in glyme while dimethylchoroarsine (6.5 g, 45.6 mmol) was added dropwise. Pyridine (6.9 g, 91.2 mmol) was then added to the slurry and the mixture was subsequently heated to reflux. The heat was removed immediately and the mixture stirred at room temperature for 4 h. Isolation of the resultant insoluble solid and recrystallization from ethanol afforded 4 as the pyridine hydrochloride complex (75% yield). mp 115-118°C; NMR (D20) δ1.35 (s, 6H), 1.9-4.1 (m’s, 10H), 7.8-9.0 (m, 5H); mass spectrum (m/e) 140, 125, 110, 105, 79, 52, 45, 36.
PATENT
WO 2009075870
Step 1
Example 1. Preparation of Dimethylchloroarsine (DMCA). A 3-neck round-bottom flask (500 mL) equipped with mechanical stirrer, inlet for nitrogen, thermometer, and an ice bath was charged with cacodylic acid (33 g, 0.23 mol) and cone. hydrochloric acid (67 mL). In a separate flask, a solution of SnCl2·2H2O (54 g, 0.239 mol) in cone. hydrochloric acid (10 mL) was prepared. The SnCl2·2 H2O solution was added to the cacodylic acid in HCl solution under nitrogen while maintaining the temperature between 5 °C and 10 °C. After the addition was complete, the ice bath was removed and the reaction mixture was stirred at ambient temperature for 1 h. The reaction mixture was transferred to a separatory funnel and the upper layer (organic) collected. The bottom layer was extracted with dichloromethane (DCM) (2 × 25 mL). The combined organic extract was washed with 1 N HCl (2 × 10 mL) and water (2 × 20 mL). The organic extract was dried over MgSO4 and DCM was removed by rotary evaporation (bath temperature 80 °C, under nitrogen, atmospheric pressure). The residue was further distilled under nitrogen. Two tractions of DMCA were collected. The first fraction contained some DCM and the second fraction was of suitable quality (8.5 g, 26% yield). The GC analysis confirmed the identity and purity of the product.
Step 2
Example 3. Preparation of S-Dimethylarsinoglutathione (SGLU-1). In a 3 L three-neck flask equipped with a mechanic stirrer, dropping funnel and thermometer under an inert atmosphere was prepared a suspension of glutathione (114.5 g, 0.37 mol) in a 1:1 (v/v) mixture of water/ethanol (1140 mL) and cooled to below 5 °C. The mixture was treated slowly (over 15 min) with triethylamine (63.6 mL, 0.46 mol) while maintaining the temperature below 20 °C. The mixture was cooled to 4 °C and stirred for 15 min and then the traces of undissolved material removed by filtration. The filtrate was transferred in a clean 3 L three-neck flask equipped with a mechanic stirrer, dropping funnel, nitrogen inlet, and thermometer and DMCA (70 g, 0.49 mol) (lot # 543-07-01-44) was added slowly while maintaining the temperature at 3-4°C. The reaction mixture was stirred at 1-4°C for 4 h, and acetone (1.2 L) was added over a period of 1 h. The mixture was stirred for 90 min between 2 and 3°C and the resulting solid was isolated by filtration. The product was washed with ethanol (2 × 250 mL) and acetone (2 × 250 mL) and the wet solids were suspended in ethanol 200 Proof (2000 mL). The product was isolated by filtration, washed with ethanol (2 × 250 mL) and acetone (2 × 250 mL) and dried in vacuum for 2 days at RT to give 115 g (75%) of SGLU-1, HPLC purity > 99.5% (in process testing).
PATENT
WO 2007027344
Example 2 Preparation of S-Dimethylarsinoglutathione A 5 L, three necked round bottom flask was equipped with a mechanical stirrer assembly, thermometer, addition funnel, nitrogen inlet, and a drying tube was placed in a cooling bath. A polyethylene crock was charged with glutathione-reduced (200 g) and deionized water (2 L) and stirred under a nitrogen atmosphere to dissolve all solids. The mixture was filtered to remove any insoluble material and the filtrate was transferred to the 5 L flask. While stirring, ethanol, 200 proof (2 L) was added and the clear solution was cooled to 0-5° C. using an ice/methanol bath. Pyridine (120 g) was added followed by a dropwise addition of Me2AsCl (120 g) over a minimum of 1 hour. The reaction mixture was stirred at 0-5° C. for a minimum of 2 hours prior to removal of the cooling bath and allowing the mixture to warm to room temperature under a nitrogen atmosphere with stirring. The reaction mixture was stirred overnight (>15 hrs) at room temperature under a nitrogen atmosphere at which time a white solid may precipitate. The reaction mixture was concentrated to a slurry (liquid and solid) at 35-45° C. using oil pump vacuum to provide a white solid residue. As much water as possible is removed, followed by two coevaporations with ethanol to azeotrope the last traces of water. The white solid residue was slurried in ethanol, 200 pf. (5 L) under a nitrogen atmosphere at room temperature overnight. The white solid was filtered and washed with ethanol, 200 pf. (2×500 mL) followed by acetone, ACS (2×500 mL). The resulting solid was transferred to drying trays and vacuum oven dried overnight at 25-35° C. using oil pump vacuum to provide pyridinium hydrochloride-free S-dimethylarsinoglutathione as a white solid. melting point of 189-190° C.
PATENT
WO 20060128682
Step 1
Dimethylchloroarsine. Dimethylarsinic acid, (CH3)2As(O)OH was supplied by the Luxembourg Chemical Co., Tel Aviv, Israel. The product was accompanied by a statement of its purity and was supplied as 99.7% pure. The dimethylarsinic acid was dissolved in water-hydrochloric acid to pH 3. A stream of sulfur dioxide was passed through this solution for about one hour. Dimethylchloroarsine separated as a heavy, colorless oil. The two liquid phases, water/(CH3)2AsCl were separated using a separatory funnel. The chlorodimethylarsine was extracted into diethylether and the ether solution was dried over anhydrous sodium sulfate. The dried solution was transferred to a distillation flask which was heated slowly to evaporate the ether. The remaining liquid, dimethylchloroarsine was purified by distillation. The fraction boiling at 106-109° C. was collected. The product, a colorless oil. 1H NMR resonance at 1.65 ppm.
Step 2
Pyridine Hydrochloride Free Synthesis of S-Dimethylarsinoglutathione (GLU) Dimethylarsinoglutathione is made using an adapted of Chen (Chen, G. C., et al. Carbohydrate Res. (1976) 50: 53-62) the contents of which are hereby incorporated by reference in their entirety. Briefly, dithiobis(dimethylarsinoglutamine) is dissolved in dichloromethane under nitrogen. Tetramethyldiarsine is added dropwise to the solution and the reaction is stirred overnight at room temperature under nitrogen and then exposed to air for 1 h. The mixture is then evaporated to dryness and the residue is washed with water and dried to give a crude solid that is recrystallized from methanol to give S-dimethylarsinoglutathione.
//////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
Solasia Pharma K.K. (TSE: 4597, Headquarters: Tokyo, Japan, President & CEO: Yoshihiro Arai, hereinafter “Solasia”) today announced submission of a New Drug Application (NDA) for its new anti-cancer drug darinaparsin (generic name, development code: SP-02) as a treatment for relapsed or refractory peripheral T-cell lymphoma to the Ministry of Health, Labour and Welfare (MHLW). Based on positive results of R&D on darinaparsin, centered primarily on the results of the Asian Multinational Phase 2 Study (study results released in June 2020), Solasia filed an NDA for the drug with the regulatory authority in Japan ahead of anywhere else in the world.
Solasia expects to obtain regulatory approval in 2022 and to also launch in the same year. If approved and launched, darinaparsin would be the third drug Solasia successfully developed and brought to market since its founding and is expected to contribute to the treatment of PTCL.
Mr. Yoshihiro Arai, President and CEO of Solasia, commented as follows: “No standard treatment has been established for relapsed or refractory PTCL as of yet. I firmly believe that darinaparsin, with its novel mechanism of action that differs from those of already approved drugs, will contribute to patients and healthcare providers at clinical sites as a new treatment option for relapsed or refractory PTCL. Since founding, Solasia has conducted R&D on five pipeline drugs. Of the five, we have successfully developed and brought to market two drugs, i.e., began providing them to patients, and today, we submitted an NDA for our first anti-cancer drug. Under our mission to provide patients with ‘Better Medicine for a Brighter Tomorrow’, we will continue aiming to contribute to patients’ treatment and enhanced quality of life. ”
About darinaparsin (SP-02) Darinaparsin, an organoarsenic compound with anticancer activity, is a novel mitochondrial-targeted agent being developed for the treatment of various hematologic and solid tumors. The proposed mechanism of action of the drug involves the disruption of mitochondrial function, increased production of reactive oxygen species, and modulation of intracellular signal transduction pathways. Darinaparsin is believed to exert anticancer effect by inducing cell cycle arrest and apoptosis. Darinaparsin has been granted orphan drug designation in the US and EU. For more information, please visit at https://solasia.co.jp/en/pipeline/sp-02.html
About Asian Multinational Phase 2 Study The Asian Multinational Phase 2 Study was a multinational, multicenter, single-arm, open-label, non-randomized study to evaluate the efficacy and safety of darinaparsin monotherapy in patients with relapsed or refractory PTCL conducted in Japan, Korea, Taiwan, and Hong Kong. (CT.gov Identifier: NCT02653976). Solasia plans to present the results of the study at an international academic conference to be held in the near future.
Atosiban, sold under the brand name Tractocile among others, is an inhibitor of the hormones oxytocin and vasopressin. It is used as an intravenousmedication as a labour repressant (tocolytic) to halt premature labor. It was developed by Ferring Pharmaceuticals in Sweden and first reported in the literature in 1985.[5] Originally marketed by Ferring Pharmaceuticals, it is licensed in proprietary and generic forms for the delay of imminent preterm birth in pregnant adult women.
Atosiban is an inhibitor of the hormones oxytocin and vasopressin. It is used intravenously to halt premature labor. Although initial studies suggested it could be used as a nasal spray and hence would not require hospital admission, it is not used in that form. Atobisan was developed by the Swedish company Ferring Pharmaceuticals. It was first reported in the literature in 1985. Atosiban is licensed in proprietary and generic forms for the delay of imminent pre-term birth in pregnant adult women.
Medical uses
Atosiban is used to delay birth in adult women who are 24 to 33 weeks pregnant, when they show signs that they may give birth pre-term (prematurely).[4] These signs include regular contractions lasting at least 30 seconds at a rate of at least four every 30 minutes,[4] and dilation of the cervix (the neck of the womb) of 1 to 3 cm and an effacement (a measure of the thinness of the cervix) of 50% or more.[4] In addition, the baby must have a normal heart rate.[4]
Pharmacology
Mechanism of action
Atosiban is a nonapeptide, desamino-oxytocin analogue, and a competitive vasopressin/oxytocin receptor antagonist (VOTra). Atosiban inhibits the oxytocin-mediated release of inositol trisphosphate from the myometrial cell membrane. As a result, reduced release of intracellular, stored calcium from the sarcoplasmic reticulum of myometrial cells and reduced influx of Ca2+ from the extracellular space through voltage-gated channels occur. In addition, atosiban suppresses oxytocin-mediated release of PGE and PGF from the decidua.[6]
In human preterm labour, atosiban, at the recommended dosage, antagonises uterine contractions and induces uterine quiescence. The onset of uterus relaxation following atosiban is rapid, uterine contractions being significantly reduced within 10 minutes to achieve stable uterine quiescence.
Other uses
Atosiban use after assisted reproduction
Atosiban is useful in improving the pregnancy outcome of in vitro fertilization-embryo transfer (IVF-ET) in patients with repeated implantation failure.[7] The pregnancy rate improved from zero to 43.7%.[8]
First- and second-trimester bleeding was more prevalent in ART than in spontaneous pregnancies. From 2004 to 2010, 33 first-trimester pregnancies with vaginal bleeding after ART with evident uterine contractions, when using atosiban and/or ritodrine, no preterm delivery occurred before 30 weeks.[9]
In a 2010 meta-analysis,[10] nifedipine is superior to β2 adrenergic receptor agonists and magnesium sulfate for tocolysis in women with preterm labor (20–36 weeks), but it has been assigned to pregnancy category C by the U.S. Food and Drug Administration, so is not recommended before 20 weeks, or in the first trimester.[9] A report from 2011 supports the use of atosiban, even at very early pregnancy, to decrease the frequency of uterine contractions to enhance success of pregnancy.[7]
Pharmacovigilance
Following the launch of atosiban in 2000, the calculated cumulative patient exposure to atosiban (January 2000 to December 2005) is estimated as 156,468 treatment cycles. To date, routine monitoring of drug safety has revealed no major safety issues.[11]
Regulatory affairs
Atosiban was approved in the European Union in January 2000 and launched in the European Union in April 2000.[12][4] As of June 2007, atosiban was approved in 67 countries, excluding the United States and Japan.[12] It was understood that Ferring did not expect to seek approval for atosiban in the US or Japan, focusing instead on development of new compounds for use in Spontaneous Preterm Labor (SPTL).[12] The fact that atosiban only had a short duration before it was out of patent that the parent drug company decided not to pursue licensing in the US.[13]
Systematic reviews
In a systematic review of atosiban for tocolysis in preterm labour, six clinical studies — two compared atosiban to placebo and four atosiban to a β agonist — showed a significant increase in the proportion of women undelivered by 48 hours in women receiving atosiban compared to placebo. When compared with β agonists, atosiban increased the proportion of women undelivered by 48 hours and was safer compared to β agonists. Therefore, oxytocin antagonists appear to be effective and safe for tocolysis in preterm labour.[14]
A 2014 systematic review by the Cochrane Collaboration showed that while atosiban had fewer side effects than alternative drugs (such as ritodrine), other beta blockers, and calcium channel antagonists, it was no better than placebo in the major outcomes i.e. pregnancy prolongation or neonatal outcomes. The finding of an increase in infant deaths in one placebo-controlled trial warrants caution. Further research is recommended.[15]
PATENT
WO 2021207870
Atosiban (Atosiban) is an oxytocin and vasopressin V1A combined receptor antagonist, which can be used as a competitive antagonist of cyclic peptide oxytocin receptors in the uterus, decidua and fetal membrane. Atosiban is a disulfide-bonded cyclic polypeptide composed of 9 amino acids. It is a modified oxytocin molecule at positions 1, 2, 4 and 8. The N-terminal of the peptide is 3-mercaptopropionic acid (thiol and [ Cys] 6 thiol forms a disulfide bond), the C-terminal is in the form of an amide, and the second amino acid at the N-terminal is ethylated [D-Tyr(Et)] 2 . Atosiban is generally present in medicines in the form of acetate salt, commonly known as atosiban acetate. Its chemical formula is C 45 H 71 N 11 O 14 S 2 , its molecular weight is 994.19, and its structural formula is as follows:
[0003]
[0004]
In the prior art, atosiban is usually synthesized by a solid-phase peptide synthesis (SPPS) method, an amino resin is used as a starting carrier resin, and protected amino acids are sequentially connected, and the obtained atosiban is oxidized and then cleaved to obtain atosiban. However, the above-mentioned existing process has high cost, generates a large amount of solvent waste, and is not easy to monitor during the cyclization process. In addition, the above-mentioned prior art has deficiencies in the overall yield of crude peptides. Moreover, due to the existence of D-Tyr(Et) in the structure of atosiban, Fmoc-D-Tyr(Et) easily undergoes a racemization reaction during the peptide attachment process, resulting in [Tyr(Et) 2 ]-A The impurity of tosiban, which is similar in polarity to atosiban itself, is difficult to completely remove through purification, thus affecting the quality of atosiban.
[table 0001]
Amino acid name
alphabetic symbols
Glycine
Gly
Ornithine
Orn
Proline
Pro
cysteine
Cys
Asparagine
Asn
Threonine
Thr
Isoleucine
Ile
D-tyrosine (oxyethyl)
D-Tyr(ET)
Table 3 List of intermediates and Fmoc protected amino acids
According to the most preferred embodiment of the present invention, the method of the present invention comprises the following steps:
[0046]
The first step: Fmoc-Gly Rink resin can be directly purchased, which reduces the first step of synthesis and improves the synthesis efficiency;
[0047]
The second step: preparing a deprotection solution: the deprotection solution is a mixture of piperidine/N,N-dimethylformamide, preferably piperidine/N,N-dimethylformamide in a volume ratio of 1/4.
[0048]
The third step: preparation of Fmoc-Orn(Boc)-Gly Rink resin: deprotect the Fmoc-Gly Rink resin obtained in the first step, wash with DMF, add Fmoc-Orn(Boc)-OH in DMF solution, Condensation reaction is carried out under the condition of peptide coupling condensing agent to obtain Fmoc-Orn(Boc)-Gly Rink resin;
[0049]
The fourth step: preparation of Fmoc-Pro-Orn(Boc)-Gly Rink resin: the peptide resin obtained in the fourth step is deprotected and washed, and then reacted with Fmoc-Pro-OH under the condition of a peptide coupling agent to obtain Fmoc-Pro-Orn(Boc)-Gly Rink resin;
[0050]
The fifth step: preparation of Fmoc-Cys(Trt)-Pro-Orn(Boc)-Gly Rink resin. The peptide resin obtained in the fifth step is deprotected and washed, and then reacted with Fmoc-Cys(Trt)-OH under the condition of peptide coupling agent to obtain Fmoc-Cys(Trt)-Pro-Orn(Boc)-Gly Rink resin;
[0051]
The sixth step: preparation of Fmoc-Asn-Cys(Trt)-Pro-Orn(Boc)-Gly Rink resin. The peptide resin obtained in the sixth step is deprotected and washed, and then reacted with Fmoc-Asn-OH under the condition of peptide coupling agent to obtain Fmoc-Asn-Cys(Trt)-Pro-Orn(Boc)-Gly Rink resin ;
[0052]
The seventh step: preparation of Fmoc-Thr(tBu)-Asn-Cys(Trt)-Pro-Orn(Boc)-Gly Rink resin. The peptide resin obtained in the seventh step was deprotected and washed, and then reacted with Fmoc-Thr(tBu)-OH under the condition of a peptide coupling agent. Obtain Fmoc-Thr(tBu)-Asn-Cys(Trt)-Pro-Orn(Boc)-Gly Rink resin;
[0053]
The eighth step: preparation of Fmoc-Ile-Thr(tBu)-Asn-Cys(Trt)-Pro-Orn(Boc)-Gly Rink resin. The peptide resin obtained in the eighth step is deprotected and washed, and then reacted with Fmoc-Ile-OH under the condition of a peptide coupling agent to obtain Fmoc-Ile-Thr(tBu)-Asn-Cys(Trt)-Pro-Orn (Boc)-Gly Rink resin;
[0054]
The ninth step: preparation of Fmoc-D-Tyr(RT)-Ile-Thr(tBu)-Asn-Cys(Trt)-Pro-Orn(Boc)-Gly Rink resin. The peptide resin obtained in the ninth step is deprotected and washed, and then reacted with Fmoc-D-Tyr(ET)-OH under the condition of a peptide coupling agent to obtain Fmoc-D-Tyr(RT)-Ile-Thr(tBu )-Asn-Cys(Trt)-Pro-Orn(Boc)-Gly Rink resin;
[0055]
The tenth step: preparation of Mpa(Trt)-D-Tyr(ET)-Ile-Thr(tBu)-Asn-Cys(Trt)-Pro-Orn(Boc)-Gly Rink resin. The peptide resin obtained in the tenth step is deprotected and washed, and then reacted with Mpa(Trt) under the condition of a peptide coupling agent to obtain Mpa(Trt)-D-Tyr(ET)-Ile-Thr(tBu)-Asn -Cys(Trt)-Pro-Orn(Boc)-Gly Rink resin;
[0056]
The eleventh step: Mpa(Trt)-D-Tyr(ET)-Ile-Thr(tBu)-Asn-Cys(Trt)-Pro-Orn(Boc)-Gly Rink resin in TFA/TIS/EDT/H2O =90/54/10/5 TFA, cleaved for 3 hours, and filtered to obtain crude peptide solution;
[0057]
The twelfth step: sedimentation and washing of the crude peptide solution with methyl tert-butyl ether, centrifugation at 2000 rpm, and vacuum drying to obtain a pale yellow solid powder of atosiban linear crude peptide;
[0058]
The thirteenth step: prepare three solutions for atosiban cyclization: solution A-sodium acetate buffered aqueous solution, solution B-aqueous solution of linear peptide atosiban crude peptide acetic acid, solution C: 30%-60% hydrogen peroxide solution ;
[0059]
The fourteenth step: Mix the above three solutions of A, B, and C at 15-25 ° C, and stir for 1-3 hours after mixing, so that the Mpa at the 1st position and the Cys at the 6th position form a disulfide bond to obtain Cyclized atosiban crude peptide.
[0060]
Step fifteen: Purify crude atosiban by preparative high performance liquid chromatography with a water/acetonitrile gradient from 100% water to 100% acetonitrile in 20 minutes.
[0061]
The sixteenth step: freeze-dry the purified atosiban solution at -50 to -70° C. for 18-48 hours with a freeze dryer.
[0062]
The purity of atosiban obtained by the method of the invention is more than 99.5%, and the total product yield is 55%-65%.
[0063]
The advantage of the method for preparing atosiban of the present invention is:
[0064]
The traditional SPPS synthesis of atosiban usually produces a large amount of waste with high disposal costs. This process adopts high-temperature SPPS process and selects different condensing agent combinations, which is faster than the conventional SPPS process, the product purity can reach more than 99.9%, the purity is better than that of the conventional atosiban process, the impurity content is low, and the product quality is high. The total yield can reach 55%-65%.
Detailed ways
[0065]
The invention will now be described with reference to specific embodiments. It must be understood that these examples are merely illustrative of the invention and are not intended to limit the scope of the invention. Unless otherwise stated, percentages and parts are by weight. Unless otherwise specified, experimental materials and reagents used in the following examples were obtained from commercial sources.
[0066]
Example 1:
[0067]
Using Rink-Fmoc-Gly resin (40 g, substitution amount 0.61 mmol/g) as the starting material, the stepwise Fmoc-SPPS (solid phase peptide synthesis) method was used to synthesize the peptide. Fmoc deprotection was performed with piperidine in DMF (1:4 v/v). Subsequently, other amino acids in the sequence are connected in the following order, and the coupling reagents are N,N-diisopropylcarbodiimide, 2-(7-benzotriazole)-N,N,N’,N ‘-Tetramethylurea hexafluorophosphate mixed in a volume ratio of 1:1, Fmoc-Orn(Boc)-OH, Fmoc-Pro-OH, Fmoc-Cys(Trt)-OH, Fmoc-Asn-OH, Fmoc-Thr(tBu)-OH, Fmoc-Ile-OH, Fmoc-D-Tyr(ET)-OH, Mpa(Trt). Coupling and deprotection of amino acids were carried out at 90°C for 2-3 min and monitored with the Kaiser test. The peptide was cleaved with the lysing solution of TFA for 3 hours, precipitated and washed twice with methyl tert-butyl ether, and after centrifugal drying, the atosiban linear crude peptide was cyclized by the method of liquid phase synthesis, and the volume ratio was 1: 2:2 A solution-acetic acid-sodium acetate buffer aqueous solution (concentration is 30g/L), B solution-linear peptide atosiban crude peptide acetic acid aqueous solution and C solution: 60% hydrogen peroxide solution.
[0068]
The crude peptide yield was 85%. Crude atosiban was purified by preparative high performance liquid chromatography with a water/acetonitrile gradient from 100% water to 100% acetonitrile in 20 minutes. The purified atosiban solution is freeze-dried at -50 to -70° C. for 18 hours with a freeze dryer, the obtained atosiban has a purity of more than 99.5%, and the total product yield is 56%.
[0069]
Example 2:
[0070]
Using Rink-Fmoc-Gly resin (40 g, substitution amount 0.36 mmol/g) as the starting material, the stepwise Fmoc-SPPS (solid phase peptide synthesis) method was used to synthesize the peptide. Fmoc deprotection was performed with piperidine in DMF (1:4 v/v). Subsequently, the other amino acids in the sequence are connected in the following order, and the coupling reagents are N,N-tert-dicyclohexylcarbodiimide, 1-hydroxybenzotriazole and Oxyma, which are mixed in a volume ratio of 1:1:1 , Fmoc-Orn(Boc)-OH, Fmoc-Pro-OH, Fmoc-Cys(Trt)-OH, Fmoc-Asn-OH, Fmoc-Thr(tBu)-OH, Fmoc-Ile-OH, Fmoc-D- Tyr(ET)-OH, Mpa(Trt). Coupling and deprotection of amino acids were carried out at 90°C for 2-3 min and monitored with the Kaiser test. The peptide was cleaved with the lysing solution of TFA for 3 hours, precipitated with methyl tert-butyl ether and washed twice, and after centrifugal drying, the atosiban linear crude peptide was cyclized by the method of liquid phase synthesis, and the volume ratio was 1: 3:2 solution A-formic acid-sodium formate buffer aqueous solution (concentration 25g/L), solution B-linear peptide atosiban crude peptide formic acid aqueous solution and solution C: 30% hydrogen peroxide solution, and oxygen was introduced.
[0071]
The crude peptide yield was 83%. Crude atosiban was purified by preparative high performance liquid chromatography with a water/acetonitrile gradient from 100% water to 100% acetonitrile in 20 minutes. The purified atosiban solution is freeze-dried at -50 to -70° C. for 18 hours with a freeze dryer, the obtained atosiban has a purity greater than 99.5%, and the total product yield is 57%.
[0072]
Example 3:
[0073]
Using Rink-Fmoc-Gly resin (40 g, substitution amount 0.36 mmol/g) as the starting material, the stepwise Fmoc-SPPS (solid phase peptide synthesis) method was used to synthesize the peptide. Fmoc deprotection was performed with piperidine in DMF (1:4 v/v). Subsequently, other amino acids in the sequence were connected in the following order, and the coupling reagents were N,N-diisopropylethylamine, 2-(7-benzotriazole)-N,N,N’,N’- Two kinds of tetramethylurea hexafluorophosphate mixed in a 1:1 volume ratio, Fmoc-Orn(Boc)-OH, Fmoc-Pro-OH, Fmoc-Cys(Trt)-OH, Fmoc-Asn-OH, Fmoc- Thr(tBu)-OH, Fmoc-Ile-OH, Fmoc-D-Tyr(ET)-OH, Mpa(Trt). Coupling and deprotection of amino acids were carried out at 75°C for 2-3 min and monitored with the Kaiser test. The peptide was cleaved with the lysing solution of TFA for 3 hours, precipitated and washed twice with methyl tert-butyl ether, and after centrifugal drying, the atosiban linear crude peptide was cyclized by the method of liquid phase synthesis, and the volume ratio was 1: 2:3 solution A-sodium phosphate buffered aqueous solution (concentration 15g/L), solution B-linear peptide atosiban crude peptide phosphoric acid aqueous solution and solution C: DMSO aqueous solution (volume 1:1).
[0074]
The crude peptide yield was 80%. Crude atosiban was purified by preparative high performance liquid chromatography with a water/acetonitrile gradient from 100% water to 100% acetonitrile in 20 minutes. The purified atosiban solution is freeze-dried at -50 to -70 DEG C for 28 hours with a freeze dryer, the obtained atosiban has a purity of more than 99.5%, and the total product yield is 55%.
[0075]
Example 4:
[0076]
Using Rink-Fmoc-Gly resin (40 g, substitution amount 0.36 mmol/g) as the starting material, the stepwise Fmoc-SPPS (solid phase peptide synthesis) method was used to synthesize the peptide. Fmoc deprotection was performed with piperidine in DMF (1:3 by volume). Subsequently, the other amino acids in the sequence were connected in the following order, and the coupling reagents were selected from 2-oxime ethyl cyanoacetate, N,N-diisopropylcarbodiimide, and 1-hydroxybenzotriazole in a volume ratio of 1. :1:1 mix, Fmoc-Orn(Boc)-OH, Fmoc-Pro-OH, Fmoc-Cys(Trt)-OH, Fmoc-Asn-OH, Fmoc-Thr(tBu)-OH, Fmoc-Ile-OH , Fmoc-D-Tyr(ET)-OH, Mpa(Trt). Coupling and deprotection of amino acids were carried out at 80°C for 2-3 min and monitored with the Kaiser test. The peptide was cleaved with the lysing solution of TFA for 3 hours, precipitated with methyl tert-butyl ether and washed twice, and after centrifugal drying, the atosiban linear crude peptide was cyclized by the method of liquid phase synthesis, and the volume ratio was 1: 3:4 solution of A-trifluoroacetic acid-aqueous ammonia solution (concentration of 45 g/L), solution B-aqueous solution of linear peptide atosiban crude peptide trifluoroacetic acid and solution C: saturated aqueous iodine solution.
[0077]
The crude peptide yield was 78%. Crude atosiban was purified by preparative high performance liquid chromatography with a water/acetonitrile gradient from 100% water to 100% acetonitrile in 20 minutes. The purified atosiban solution is freeze-dried at -50 to -70° C. for 38 hours with a freeze dryer, the obtained atosiban has a purity of more than 99.5%, and the total product yield is 52%.
Atosiban Acetate Injection was first listed in Austria on March 23, 2000 under the trade name: Atosiban, a new type of anti-prematurity drug developed by Ferring GmbH, which is an oxytocin The analog is a competitive antagonist of oxytocin receptors in the uterus, decidua, and fetal membranes. It is a first-line drug recommended by the European Medical Association; it can inhibit the binding of oxytocin and oxytocin receptors, thereby directly inhibiting the effect of oxytocin. In the uterus, it can inhibit uterine contraction; it can also inhibit the hydrolysis of phosphatidylinositol.
Atosiban is a cyclic nonapeptide whose molecular formula is C 43 H 67 N 11 O 12 S 2 ; molecular weight is 994.19; CAS registration number is 90779-69-4; its peptide sequence is as follows:
In the Chinese patents with announcement numbers CN101314613B and CN101696236B, the solid-phase synthesis of atosiban uses Rink Amide AM Resin resin solid-phase coupling stepwise to obtain Mpa(Trt)-D-Tyr(Et)-Ile-Thr(tBu)- Asn(Trt)-Cys(Trt)-Pro-Orn(Boc)-Gly-Resin is directly oxidized in solid phase to generate disulfide bonds, and then cleaved to obtain atosiban. The Rink Amide AM Resin resin used in the prior art needs to be cracked under a strong acid environment, which is not conducive to product stability and has a greater operational risk; Mpr and Cys both have sulfhydryl groups, and the sulfhydryl groups have the ability to capture tBu to generate double tBu impurities, When the peptide resin after solid-phase oxidation is cleaved to remove the protective group and resin, due to the presence of tBu or tBu source Boc protective group, it requires high capture agent, which is not conducive to product quality control and reduces product yield.
The Chinese patent with publication number CN105408344B discloses a method for synthesizing atosiban starting from Fmoc-Orn-Gly-NH2, wherein Fmoc-Orn-Gly-NH2 is connected to trityl through the side chain of ornithine On the base resin, impurities can be effectively controlled. However, using dipeptide and trityl-type resin for coupling, the resin attached to the Orn side chain of the dipeptide increases the steric hindrance of the subsequent Pro coupling and prolongs the coupling time, which is easy to cause missing peptide impurities.
Example 1. Synthesis of Fmoc-Pro-Orn-Gly-NH 2 tripeptide
[0027]
Fmoc-Pro-OH (134.94 g, 400 mmol) and N-hydroxysuccinimide (46.00 g, 400 mmol) were weighed into 1600 ml of tetrahydrofuran, and stirred at room temperature. The temperature was controlled at about 5°C, and a solution of DCC (90.72g, 440mmol) in tetrahydrofuran (320ml) was slowly added and stirred at room temperature for 2.5h, filtered, concentrated and added to petroleum ether for recrystallization to precipitate a solid, washed and dried, and the obtained activated ester was The solid was dissolved in 400 ml of tetrahydrofuran, and H-Orn(Boc)-NH 2 (92.92 g, 400 mmol) was dissolved in 300 ml of tetrahydrofuran and slowly added dropwise to the above solution. After dropping, the reaction was continued at room temperature. Concentrate to dryness under reduced pressure, add N-hydroxysuccinimide (46.00 g, 400 mmol) and 1600 ml of tetrahydrofuran to dissolve, and stir at room temperature. The temperature was controlled at about 5°C, and a solution of DCC (90.72g, 440mmol) in tetrahydrofuran (320ml) was slowly added and stirred at room temperature for 2.5h, filtered, concentrated and added to petroleum ether for recrystallization to precipitate a solid, washed and dried, and the obtained activated ester was The solid was dissolved in 400 ml of tetrahydrofuran, and H-Gly-NH 2 (29.64 g, 400 mmol) was dissolved in 300 ml of tetrahydrofuran and slowly added dropwise to the above solution, and the reaction was continued at room temperature after dropping, and the monitoring of the raw materials was completed. The reaction was filtered, and the filtrate was concentrated under reduced pressure. Dry, add 1000 mL of 5% TFA/DCM solution to the reaction solution, continue to react for 1 h, and concentrate to dryness to obtain a yellow oil, which is recrystallized from isopropanol to obtain 171.56 g of white solid with a yield of 69%.
[0028]
Example 2. Synthesis of Fmoc-Pro-Orn (trityl resin)-Gly-NH 2 peptide resin with a degree of substitution of 0.42 mmol/g
[0029]
Trityl resin (37.5 g, 30 mmol, substitution degree: 0.80 mmol/g) was weighed into a solid-phase reaction synthesis column. 400 mL of dry DMF was added to swell for 30 min, and the DMF was removed. The resin was washed with 3*400 mL of dry DMF, and the DMF was removed. Fmoc-Pro-Orn-Gly-NH 2 (37.30 g, 60 mmol) prepared in Example 1 , DIEA (11.63 g, 90 mmol) were added, 100 mL of dry DMF was added to dissolve and clarified, added to the resin to react for 2 h, and methanol (9.61 mmol) was added. g, 300 mmol) reacted for 20 min, sucked dry, washed the resin with 3*400 mL of CH 2 Cl 2 , and removed CH 2 Cl 2 . The resin was taken out and dried under vacuum at 25-35° C. to obtain 52.14 g of Fmoc-Pro-Orn (trityl resin)-Gly-NH 2 resin with a measured substitution degree of 0.42 mmol/g.
[0030]
Example 3. Synthesis of Fmoc-Pro-Orn(2-CTC Resin)-Gly-NH 2 peptide resin with a degree of substitution of 0.50 mmol/g
[0031]
2-CTC Resin resin (30.0 g, 30 mmol, substitution degree: 1.00 mmol/g) was weighed into a solid-phase reaction synthesis column. 400 mL of dry DMF was added to swell for 30 min, and the DMF was removed. The resin was washed with 3*400 mL of dry DMF, and the DMF was removed. Fmoc-Pro-Orn-Gly-NH 2 (37.30 g, 60 mmol) prepared in Example 1 , DIEA (11.63 g, 90 mmol) were added, 100 mL of dry DMF was added to dissolve and clarified, added to the resin to react for 2 h, and methanol (9.61 mmol) was added. g, 300 mmol) reacted for 20 min, sucked dry, washed the resin with 3*400 mL of CH 2 Cl 2 , and removed CH 2 Cl 2 . The resin was taken out and dried under vacuum at 25-35° C. to obtain 43.80 g of Fmoc-Pro-Orn(2-CTC Resin)-Gly-NH 2 resin with a measured substitution degree of 0.50 mmol/g.
[0032]
Example 4. Synthesis of Fmoc-Pro-Orn (4-methyl-trityl resin)-Gly-NH 2 peptide resin with a degree of substitution of 0.50 mmol/g
[0033]
4-methyl-trityl resin (33.33 g, 30 mmol, substitution degree: 0.90 mmol/g) was weighed into a solid-phase reaction synthesis column. 400 mL of dry DMF was added to swell for 30 min, and the DMF was removed. The resin was washed with 3*400 mL of dry DMF, and the DMF was removed. Fmoc-Pro-Orn-Gly-NH 2 (37.30 g, 60 mmol) prepared in Example 1 , DIEA (11.63 g, 90 mmol) were added, 100 mL of dry DMF was added to dissolve and clarified, added to the resin to react for 2 h, and methanol (9.61 mmol) was added. g, 300 mmol) reacted for 20 min, sucked dry, washed the resin with 3*400 mL of CH 2 Cl 2 , and removed CH 2 Cl 2 . The resin was taken out and dried under vacuum at 25-35° C. to obtain 43.89 g of Fmoc-Pro-Orn (4-methyl-trityl resin)-Gly-NH 2 resin with a measured substitution degree of 0.50 mmol/g.
[0034]
Example 5. Synthesis of Fmoc-Pro-Orn (4-methoxy-trityl resin)-Gly-NH 2 peptide resin with a degree of substitution of 0.50 mmol/g
[0035]
4-Methoxy-trityl resin (30.0 g, 30 mmol, substitution degree: 1.00 mmol/g) was weighed into a solid-phase reaction synthesis column. 400 mL of dry DMF was added to swell for 30 min, and the DMF was removed. The resin was washed with 3*400 mL of dry DMF, and the DMF was removed. Fmoc-Pro-Orn-Gly-NH 2 (37.30 g, 60 mmol) prepared in Example 1 , DIEA (11.63 g, 90 mmol) were added, 100 mL of dry DMF was added to dissolve and clarified, added to the resin to react for 2 h, and methanol (9.61 mmol) was added. g, 300 mmol) reacted for 20 min, sucked dry, washed the resin with 3*400 mL of CH 2 Cl 2 , and removed CH 2 Cl 2 . The resin was taken out and dried under vacuum at 25-35° C. to obtain 43.69 g of Fmoc-Pro-Orn (4-methoxy-trityl resin)-Gly-NH 2 resin with a measured substitution degree of 0.50 mmol/g.
[0036]
Example 6. Synthesis of Atosiban Linear Peptide Resin 1
[0037]
Fmoc-Pro-Orn (trityl resin)-Gly-NH 2 (35.71 g) prepared in Example 2 was weighed into a solid-phase reaction synthesis column. 400 mL of DMF was added to swell for 30 min, and the DMF was removed. The resin was washed with 3*200 mL of dry DMF, and the DMF was removed. 200 mL of DBLK solution (20% piperidine/DMF solution, V/V) was added and deprotected twice, the first time was 5 min and the second time was 15 min. After deprotection, the resin was washed with 200 mL of DMF each time, and washed 6 times. After the fourth washing, a little resin was taken with a glass rod. The ninhydrin test was positive, indicating that Fmoc had been removed.
[0038]
Weigh 17.57g Fmoc-Cys(Trt)-OH and 4.86g HOBt, add 100mL DMF to dissolve, after complete dissolution, cool the solution to below 5°C, then add 5.68g DIC (pre-cooled to <0°C), Activated in the solution for about 3 to 5 minutes, the activated solution was added to the reaction column under control, and reacted at 20 to 35 °C for 2 to 3 hours. The ninhydrin test was negative. The reaction solution was removed, and 200 mL of DMF was added to wash the resin. 6 times. After washing, the washing liquid was removed to obtain Fmoc-Cys(Trt)-Pro-Orn (trityl resin)-Gly-NH 2 .
[0039]
Repeat the step of receiving the peptide and remove the Fmoc protective group. According to the amino acid sequence of atosiban, Fmoc-Cys(Trt)-Pro-Orn (trityl resin)-Gly-NH 2 was coupled to Fmoc- Asn-OH, Fmoc-Thr-OH, Fmoc-Ile-OH, Fmoc-D-Tyr(Et)-OH, Mpa(Trt)-OH give Mpa(Trt)-D-Tyr(Et)-Ile-Thr- Asn-Cys(Trt)-Pro-Orn (trityl resin)-Gly- NH2 . After washing with DMF, the washing solution was removed. The resin was washed with 200 ml of DCM each time, 4 times, 5 min/time, the DCM was removed, and the resin was vacuum-dried at room temperature (20-35° C.) until it was quicksand. The peptide resin was 48.72g after drying, and the resin weight gain was 89.0%.
[0040]
Example 7. Synthesis of atosiban linear peptide resin 2
[0041]
Fmoc-Pro-Orn(2-CTC Resin)-Gly-NH 2 (30.00 g) prepared in Example 3 was weighed into a solid-phase reaction synthesis column. 400 mL of DMF was added to swell for 30 min, and the DMF was removed. The resin was washed with 3*200 mL of dry DMF, and the DMF was removed. 200 mL of DBLK solution (20% piperidine/DMF solution, V/V) was added and deprotected twice, the first time was 5 min and the second time was 15 min. After deprotection, the resin was washed with 200 mL of DMF each time, and washed 6 times. After the fourth washing, a little resin was taken with a glass rod. The ninhydrin test was positive, indicating that Fmoc had been removed.
[0042]
Weigh 17.57g Fmoc-Cys(Trt)-OH and 13.65g HBTU, add 100mL DMF to dissolve, after complete dissolution, cool the solution to below 5°C, then add 5.82g DIEA (pre-cooled to <0°C), put Activated in the solution for about 3 to 5 minutes, the activated solution was added to the reaction column under control, and reacted at 20 to 35 °C for 2 to 3 hours. The ninhydrin test was negative. The reaction solution was removed, and 200 mL of DMF was added to wash the resin. 6 times. After washing, the washing solution was removed to obtain Fmoc-Cys(Trt)-Pro-Orn(2-CTC Resin)-Gly-NH 2 .
[0043]
Fmoc-D-Tyr(Et)-OH (86.30 g, 200 mmol) and N-hydroxysuccinimide (23.00 g, 200 mmol) were weighed into 800 ml of tetrahydrofuran, and stirred at room temperature. The temperature was controlled at about 5°C, and a solution of DCC (45.36g, 220mmol) in tetrahydrofuran (160ml) was slowly added and stirred at room temperature for 2.5h, filtered, concentrated and added to petroleum ether for recrystallization to precipitate a solid, washed and dried, and the obtained activated ester was The solid was dissolved in 200 ml of tetrahydrofuran, and H-Ile-OH (26.24 g, 200 mmol) was dissolved in 150 ml of tetrahydrofuran and slowly added dropwise to the above solution. After dropping, the reaction was continued at room temperature. The monitoring of the raw materials was completed. After filtration, the solution was concentrated under reduced pressure. , the concentrated solution was added to petroleum ether to separate out the solid, the solid was washed and then dried, recrystallized and dried with isopropanol to obtain 75.60 g of Fmoc-D-Tyr(Et)-Ile-OH with a yield of 75%.
[0044]
Repeat the step of receiving the peptide and removing the Fmoc protective group. According to the amino acid sequence of atosiban, sequentially couple Fmoc-Asn on Fmoc-Cys(Trt)-Pro-Orn(2-CTC Resin)-Gly-NH 2 -OH, Fmoc-Thr-OH, Fmoc-D-Tyr(Et)-Ile-OH, Mpa(Trt)-OH to give Mpa(Trt)-D-Tyr(Et)-Ile-Thr-Asn-Cys(Trt )-Pro-Orn( 2 -CTC Resin)-Gly-NH2 . After washing with DMF, the washing solution was removed. The resin was washed with 200 ml of DCM each time, 4 times, 5 min/time, the DCM was removed, and the resin was vacuum-dried at room temperature (20-35° C.) until it was quicksand. The peptide resin was 42.77g after drying, and the resin weight gain rate was 87.4%.
[0045]
Example 8. Synthesis of atosiban linear peptide resin 3
[0046]
Fmoc-Pro-Orn (4-methyl-trityl resin)-Gly-NH 2 (30.00 g) prepared in Example 4 was weighed into a solid-phase reaction synthesis column. 400 mL of DMF was added to swell for 30 min, and the DMF was removed. The resin was washed with 3*200 mL of dry DMF, and the DMF was removed. 200 mL of DBLK solution (20% piperidine/DMF solution, V/V) was added and deprotected twice, the first time was 5 min and the second time was 15 min. After deprotection, the resin was washed with 200 mL of DMF each time, and washed 6 times. After the fourth washing, a little resin was taken with a glass rod, and the ninhydrin test was positive, indicating that Fmoc had been removed.
[0047]
Weigh 17.57g Fmoc-Cys(Trt)-OH, 13.65g HBTU and 4.05g HOBt, add 100mL DMF to dissolve, after complete dissolution, cool the solution to below 5°C, then add 5.82g DIEA (pre-cooled to <0 ℃), activate in the solution for about 3-5min, add the activated solution to the reaction column, react at 20-35 ℃ for 2-3h, the ninhydrin test is negative, remove the reaction solution, add 200mL of DMF The resin was washed 6 times. After washing, the washing liquid was removed to obtain Fmoc-Cys(Trt)-Pro-Orn(4-methyl-trityl resin)-Gly-NH 2 .
[0048]
Mpa(Trt)-OH (69.69 g, 200 mmol) and N-hydroxysuccinimide (23.00 g, 200 mmol) were weighed into 800 ml of tetrahydrofuran, and stirred at room temperature. The temperature was controlled at about 5°C, and a solution of DCC (45.36g, 220mmol) in tetrahydrofuran (160ml) was slowly added and stirred at room temperature for 2.5h, filtered, concentrated and added to petroleum ether for recrystallization to precipitate a solid, washed and dried, and the obtained activated ester was The solid was dissolved in 200 ml of tetrahydrofuran, and HD-Tyr(Et)-OH (41.85 g, 200 mmol) was dissolved in 150 ml of tetrahydrofuran and slowly added dropwise to the above solution. After dropping, the reaction was continued at room temperature. Concentrate under reduced pressure, add the concentrated solution to petroleum ether to precipitate a solid, wash the solid and then dry it, recrystallize and dry with isopropanol to obtain Mpa(Trt)-D-Tyr(Et)-OH 77.98g, yield 72%.
[0049]
Repeat the step of receiving the peptide and removing the Fmoc protective group, according to the amino acid sequence of atosiban, on Fmoc-Cys(Trt)-Pro-Orn (4-methyl-trityl resin)-Gly- NH 2 Fmoc-Asn-OH, Fmoc-Thr-OH, Fmoc-Ile-OH, Mpa(Trt)-D-Tyr(Et)-OH were sequentially coupled to obtain Mpa(Trt)-D-Tyr(Et)-Ile-Thr -Asn-Cys(Trt)-Pro-Orn(4-methyl-trityl resin)-Gly- NH2 . After washing with DMF, the washing solution was removed. The resin was washed with 200 ml of DCM each time, 4 times, 5 min/time, the DCM was removed, and the resin was vacuum-dried at room temperature (20-35° C.) until it was quicksand. The peptide resin was 42.91g after drying, and the resin weight gain rate was 88.3%.
[0050]
Example 9. Synthesis of atosiban linear peptide resin 4
[0051]
Fmoc-Pro-Orn (4-methoxy-trityl resin)-Gly-NH 2 (30.00 g) prepared in Example 5 was weighed into a solid-phase reaction synthesis column. 400 mL of DMF was added to swell for 30 min, and the DMF was removed. The resin was washed with 3*200 mL of dry DMF, and the DMF was removed. 200 mL of DBLK solution (20% piperidine/DMF solution, V/V) was added and deprotected twice, the first time was 5 min and the second time was 15 min. After deprotection, the resin was washed with 200 mL of DMF each time, and washed 6 times. After the fourth washing, a little resin was taken with a glass rod. The ninhydrin test was positive, indicating that Fmoc had been removed.
[0052]
Fmoc-Asn-OH (70.87 g, 200 mmol) and N-hydroxysuccinimide (23.00 g, 200 mmol) were weighed into 800 ml of tetrahydrofuran, and stirred at room temperature. The temperature was controlled at about 5°C, and a solution of DCC (45.36g, 220mmol) in tetrahydrofuran (160ml) was slowly added and stirred at room temperature for 2.5h, filtered, concentrated and added to petroleum ether for recrystallization to precipitate a solid, washed and dried, and the obtained activated ester was The solid was dissolved in 200 ml of tetrahydrofuran, and H-Cys(Trt)-OH (79.96 g, 200 mmol) was dissolved in 150 ml of tetrahydrofuran and slowly added dropwise to the above solution. After dropping, the reaction was continued at room temperature. Concentrate under reduced pressure, add the concentrated solution to petroleum ether to precipitate a solid, wash the solid and then dry, recrystallize and dry with isopropanol to obtain Fmoc-Asn-Cys(Trt)-OH 102.17g, yield 73%.
[0053]
Weigh 20.99g Fmoc-Asn-Cys(Trt)-OH and 13.65g HCTU, add 100mL DMF to dissolve, after complete dissolution, cool the solution to below 5°C, then add 5.82g DIEA (pre-cool to <0°C) , activate in the solution for about 3-5min, add the activated solution to the reaction column, react at 20-35°C for 2-3h, the ninhydrin test is negative, remove the reaction solution, add 200mL of DMF to wash the resin , wash 6 times. After washing, the washing liquid was removed to obtain Fmoc-Asn-Cys(Trt)-Pro-Orn(4-methoxy-trityl resin)-Gly-NH 2 .
[0054]
Repeat the step of receiving the peptide and removing the Fmoc protective group. According to the amino acid sequence of atosiban, in Fmoc-Asn-Cys(Trt)-Pro-Orn(4-methoxy-trityl resin)-Gly- Fmoc-Thr-OH, Fmoc-Ile-OH, Fmoc-D-Tyr(Et)-OH, Mpa(Trt)-OH were sequentially coupled on NH 2 to obtain Mpa(Trt)-D-Tyr(Et)-Ile- Thr-Asn-Cys(Trt)-Pro-Orn(4-methoxy-trityl resin)-Gly- NH2 . After washing with DMF, the washing solution was removed. The resin was washed with 200 ml of DCM each time, 4 times, 5 min/time, the DCM was removed, and the resin was vacuum-dried at room temperature (20-35° C.) until it was quicksand. The peptide resin was 42.28g after drying, and the resin weight gain was 84.0%.
[0055]
Example 10. Synthesis of atosiban crude peptide 1
[0056]
Configure 487.2ml of TFA/DCM=2/98 (V/V) lysis solution, cool to 5-10°C, add 48.72g of peptide resin prepared in Example 6 into the lysis solution, at room temperature (20-35°C) React for 5h, filter, wash the peptide resin twice with acetonitrile, 50ml/time, combine into the filtrate, spin the filtrate to dry, obtain a solid after drying, wash with isopropyl ether, filter, and dry under reduced pressure at 20-35°C to constant weight To obtain 14.77g of atosiban linear peptide, dissolve 14.30g of atosiban linear peptide in 0.75L of glacial acetic acid, add 6.75L of water to dilute, add 0.1M/L iodine ethanol solution dropwise until the solution changes color, react at room temperature for 1.0h, That is, the crude atosiban peptide is obtained, and its HPLC spectrum is shown in Figure 1.
[0057]
Example 11. Synthesis of atosiban crude peptide 2
[0058]
Configure TFA/DCM=5/95 (V/V) lysate 448.6ml, cool to 5~10℃, add 42.77g of peptide resin prepared in Example 7 into the lysate, at room temperature (20~35℃) React for 3h, filter, wash the peptide resin twice with acetonitrile, 50ml/time, combine into the filtrate, spin the filtrate, dry to obtain a solid, wash with isopropyl ether, filter, and dry under reduced pressure at 20-35°C to constant weight To obtain 14.21g of atosiban linear peptide, dissolve 14.21g of atosiban linear peptide in 1.5L of glacial acetic acid, add 6L of water to dilute, add 0.1M/L iodoethanol solution dropwise until the solution changes color, react at room temperature for 1.0h, that is The crude atosiban peptide was obtained, and its HPLC chromatogram was similar to that in Figure 1.
[0059]
Example 12. Synthesis of atosiban crude peptide 3
[0060]
Configure 450.5ml of TFA/DCM=20/80(V/V) lysis solution, cool to 5~10℃, add 45.05g of peptide resin prepared in Example 8 into the lysis solution, at room temperature (20~35℃) React for 2h, filter, wash the peptide resin twice with acetonitrile, 50ml/time, combine into the filtrate, spin the filtrate to dry, obtain a solid after drying, wash with isopropyl ether, filter, and dry under reduced pressure at 20-35°C to constant weight To obtain 14.63g of atosiban linear peptide, dissolve 14.63g of atosiban linear peptide in 1.5L of glacial acetic acid, add 6L of water to dilute, add 10% hydrogen peroxide solution, and react at room temperature for 1.0h to obtain atosiban Crude peptide, its HPLC chromatogram is similar to Figure 1.
[0061]
Example 13. Synthesis of atosiban crude peptide 4
[0062]
Configure TFA/DCM=1/99 (V/V) lysate 442.7ml, cool to 5~10℃, add 44.27g of peptide resin prepared in Example 9 into the lysate, at room temperature (20~35℃) React for 5h, filter, wash the peptide resin twice with acetonitrile, 50ml/time, combine into the filtrate, spin the filtrate to dry, obtain a solid after drying, wash with isopropyl ether, filter, and dry under reduced pressure at 20-35°C to constant weight To obtain 14.13 g of atosiban linear peptide, dissolve 14.13 g of atosiban linear peptide in 1.5 L of glacial acetic acid, add 6 L of water to dilute, add 30% hydrogen peroxide solution, and react at room temperature for 1.0 h to obtain atosiban Crude peptide, its HPLC chromatogram is similar to Figure 1.
[0063]
Example 14. Purification of atosiban crude peptide 1
[0064]
The atosiban crude peptide prepared in Example 10 was dissolved in 15% acetonitrile aqueous solution and filtered, purified by preparative reverse-phase HPLC (C18 column), transferred to salt, collected more than 99% of the fraction, concentrated and lyophilized to obtain 10.12g , the yield is 64%, the purity is 99%, and the HPLC spectrum of the obtained atosiban peptide is shown in Figure 2.
[0065]
Example 15. Purification of atosiban crude peptide 2
[0066]
The crude atosiban peptide obtained in Example 11 was dissolved in a 15% acetonitrile aqueous solution and filtered, purified by preparative reverse-phase HPLC (C18 column), transferred to salt, collected more than 99% of the fraction, concentrated and lyophilized to obtain 9.80 g , the yield is 62%, the purity is 99%, and the obtained atosiban peptide HPLC spectrum is similar to Figure 2.
[0067]
Example 16. Purification of atosiban crude peptide 3
[0068]
The crude atosiban peptide obtained in Example 12 was dissolved in a 15% acetonitrile aqueous solution and filtered, purified by preparative reverse-phase HPLC (C18 column), transferred to salt, collected more than 99% of the fraction, concentrated and lyophilized to obtain 10.28g , the yield is 65%, the purity is 99%, and the HPLC spectrum of the obtained atosiban peptide is similar to that in Figure 2.
[0069]
Example 17. Purification of atosiban crude peptide 4
[0070]
The crude atosiban peptide obtained in Example 13 was dissolved in 15% acetonitrile aqueous solution and filtered, purified by preparative reverse-phase HPLC (C18 column), transferred to salt, collected more than 99% of the fraction, concentrated and lyophilized to obtain 10.27g , the yield is 65%, the purity is 99%, and the HPLC spectrum of the obtained atosiban peptide is similar to that in Figure 2.
Atosiban is a nonapeptide which contains three non-natural amino acids: D-Tyr(Et), Mpa and Orn, and a pair of disulfide bonds looped between Mpa and Cys, the structural formula is: c[Mpa-D-Tyr(Et)-Ile-Thr-Asn-Cys]-Pro-Orn-Gly-NH2.
By means of competing for oxytocin receptor with oxytocin, Atosiban can inhibit the combination between oxytocin and oxytocin receptor, and directly prevent the oxytocin from acting on uterus, and then inhibit the uterine contraction; as another hand, atosiban can also inhibit the hydrolysis of phosphatidylinositol and then block the generation of messenger and activity of Ca2+, with the decreasing of activity from oxytocin, the contraction of uterine is indirectly inhabited.
At present, there are many reports about synthesis process method in China and abroad A report in China shows that the inventor found a simple process by adopting solid phase oxidation, resulting in a low purity crude product, with low yield and low application value. The aforementioned reports about atosiban synthesis process reveal that most of them adopt the method using Boc solid phase synthetic and cleaving peptide with liquid ammonia, then oxidating with liquid phase oxidation, and purifying. Those respective processes result in “the three wastes” and are too complex for industrial production. See U.S. Pat. No. 4,504,469.
Example 1Preparing the Linear Atosiban Peptide Resin
(i) 6.25 g of Rink Amide resin (substitutability=0.8 mmol/g) is put into a reaction bottle, DMF is added into the bottle and washed twice, then swelled for 30 min with DMF. Fmoc protecting group of Rink Amide resin is removed with 30-40 ml of 20% DBLK, washed for 4 times with DMF, then washed twice with DCM after removal, the product is detected by ninhydrin detecting method, the resin is reddish-brown.
(ii) 4.46 g of Fmoc-Gly-OH and 2.43 g of HOBt dissolved in a suitable amount of DMF, which had been pre-activated with 3.05 ml DIC; the mixture is, added to the reaction bottle, and reacted for 2 h, the resin is negative by ninhydrin detecting method, after the reaction, the product is washed for 4 times with DMF, then washed twice with DCM, if the resin is positive, repeating the above condensation reaction until negative.
(iii) Fmoc-Orn(Boc)-OH, Fmoc-Pro-OH, Fmoc-Cys(Trt)-OH, Fmoc-Asn(Trt)-OH, Fmoc-Thr(tBu)-OH, Fmoc-Ile-OH, Fmoc-D-Tyr(ET)-OH and Mpa(Trt)-OH are coupled orderly.
Example 2Cleaving the Linear Atosiban Peptide Resin
5.15 g of linear atosiban is prepared by washing the linear atosiban peptide resin obtained from Example 1 for 3 times with 30 ml of methanol, adding the dry resin obtained to 150 ml of mixed solution with a volume ratio of TFA:H2O=95:5, reacting for 2 hours at 25° C. and filtering, washing the resin for 3 times with few trifluoroacetic acid, combining the filtrate and pouring into 1500 ml glacial ether, making rest for 2 hours, centrifugally separating the linear atosiban, washing for 3 times, and drying in a vacuum drier, MS: 995.3, HPLC: 91.5%, content: 65.5%, synthesis yield: 68%.
Example 3Oxidizing the Linear Atosiban
2.85 g of atosiban acetate is prepared by dissolving the linear atosiban obtained from Example 2 in 250 ml of 5% acetonitrile aqueous solution, adjusting the pH value to 8 to 9 with 30% ammonia water, adding 0.60 g of H2O2, reacting for 10 min at 25° C., monitoring with HPLC (HPLC: 75.6%), filtering after reaction, purifying filtrate by preparative RP-HPLC (column C18 or C8), transferring salt, and freeze-drying, MS: 994.5, HPLC: 99.4%.
Example 4Oxidizing the Linear Atosiban
3.01 g of atosiban acetate is prepared by dissolving the linear atosiban obtained from Example 2 in 250 ml of 10% acetonitrile aqueous solution, adjusting the pH value to 8 to 9 with 30% ammonia water, adding 0.85 g of H2O2, reacting for 30 min at 25° C., monitoring with HPLC (HPLC: 89.5%), filtering after reaction, purifying filtrate by preparative RP-HPLC (column C18 or C8), transferring salt, and freeze-drying, MS: 994.5, HPLC: 99.6%.
Example 5Oxidizing the Linear Atosiban
2.95 g of atosiban acetate is prepared by dissolving the linear atosiban obtained from Example 2 in 250 ml of 10% acetonitrile aqueous solution, adjusting the pH value to 8 to 9 with 30% ammonia water, adding 0.85 g of H2O2, reacting for 60 min at 25° C., monitoring with HPLC (HPLC: 83.5%), filtering after reaction, purifying filtrate by preparative RP-HPLC (column C18 or C8), transferring salt, and freeze-drying, MS: 994.5, HPLC: 99.4%.
The above is the further detailed description of the invention in conjunction with specific preferred examples, but it should not be considered that the specific examples of the invention are only limited to the these descriptions. For one of ordinary skill in the art, many deductions and replacements can be made without departing from the inventive concept. Such deductions and replacements should fall within the scope of protection of the invention.
A 2013 retrospective study comparing the efficacy and safety of atosiban and nifedipine in the suppression of preterm labour concluded that atosiban and nifedipine are effective in delaying delivery for seven days or more in women presenting with preterm labour.[16] A total of 68.3% of women in the atosiban group remained undelivered at seven days or more, compared with 64.7% in the nifedipine group.[16] They have the same efficacy and associated minor side effects.[16] However, flushing, palpitation, and hypotension were significantly higher in the nifedipine group.[16]
A 2012 clinical trial compared tocolytic efficacy and tolerability of atosiban with that of nifedipine.[17] Forty-eight (68.6%) women allocated to atosiban and 39 (52%) to nifedipine did not deliver and did not require an alternate agent at 48 hours, respectively (p=.03).[17] Atosiban has fewer failures within 48 hours.[17] Nifedipine may be associated with a longer postponement of delivery.[17]
A 2009 randomised controlled study demonstrated for the first time the direct effects of atosiban on fetal movement, heart rate, and blood flow.[18] Tocolysis with either atosiban or nifedipine combined with betamethasone administration had no direct fetal adverse effects.[18]
Atosiban vs. ritodrine
Multicentre, controlled trial of atosiban vs. ritodrine in 128 women shows a significantly better tocolytic efficacy after 7 days in the atosiban group than in the ritodrine group (60.3 versus 34.9%), but not at 48 hours (68.3 versus 58.7%). Maternal adverse events were reported less frequently in the atosiban group (7.9 vs 70.8%), resulting in fewer early drug terminations due to adverse events (0 versus 20%). Therefore, atosiban is superior to ritodrine in the treatment of preterm labour.[19]
Brand names
In India it is marketed under the brand name Tosiban by Zuventus healthcare ltd.
^ Akerlund M, Carlsson AM, Melin P, Trojnar J (1985). “The effect on the human uterus of two newly developed competitive inhibitors of oxytocin and vasopressin”. Acta Obstet Gynecol Scand. 64 (6): 499–504. doi:10.3109/00016348509156728. PMID4061066. S2CID25799128.
^ Lamont, Ronald F; Kam, KY Ronald (March 2008). “Atosiban as a tocolytic for the treatment of spontaneous preterm labor”. Expert Review of Obstetrics & Gynecology. 3 (2): 163–174. doi:10.1586/17474108.3.2.163. ISSN1747-4108.
^ Jump up to:abc Lamont, Ronald F.; Kam, KY Ronald (2008). “Atosiban as a tocolytic for the treatment of spontaneous preterm labor”. Expert Review of Obstetrics & Gynecology. 3 (2): 163–174. doi:10.1586/17474108.3.2.163.
^ Lamont CD, Jørgensen JS, Lamont RF (September 2016). “The safety of tocolytics used for the inhibition of preterm labour”. Expert Opinion on Drug Safety. 15 (9): 1163–73. doi:10.1080/14740338.2016.1187128. PMID27159501. S2CID4937942. It was for this reason and the fact that Tractocile (atosiban) only had a short duration before it was out of patent that the parent drug company decided not to pursue licensing in the USA.
^ Coomarasamy, A; Knox, EM; Gee, H; Khan, KS (November 2002). “Oxytocin antagonists for tocolysis in preterm labour — a systematic review”. Med Sci Monit. 8 (11): RA268–73. PMID12444392.
^ Jump up to:ab de Heus R, Mulder EJ, Derks JB, Visser GH (June 2009). “The effects of the tocolytics atosiban and nifedipine on fetal movements, heart rate and blood flow”. J Matern Fetal Neonatal Med. 22 (6): 485–90. doi:10.1080/14767050802702349. PMID19479644. S2CID35810758.
^ Shim JY, Park YW, YoonBH, Cho YK, Yang JH, Lee Y, Kim A. “Multicentre, parallelgroup, randomised, single-blind study of the safety and efficacy of atosibanversus ritodrine in the treatment of acute preterm labour in Korean women. BJOG 2006Nov;113(11):1228-34.
External links
“Atosiban”. Drug Information Portal. U.S. National Library of Medicine.
01 Jun 2022Takeda Pharmaceuticals completes a phase I clinical trials in Respiratory disorder (In adults) in Netherlands (IV) (ISRCTN63027076)
02 Apr 2022Efficacy and safety data from phase a Ib trial in Hypersomnia presented at the 74th Annual Meeting of the American Academy of Neurology 2022 (AAN-2022)
10 Mar 2022Phase-I clinical trials in Sleep apnoea syndrome in Australia (IV) (NCT05180890)
TAK-925, a potent, selective, and brain-penetrant orexin 2 receptor (OX2R) agonist, [methyl (2R,3S)-3-((methylsulfonyl)amino)-2-(((cis-4-phenylcyclohexyl)oxy)methyl)piperidine-1-carboxylate, 16], was identified through the optimization of compound 2, which was discovered by a high throughput screening (HTS) campaign. Subcutaneous administration of compound 16 produced wake-promoting effects in mice during the sleep phase. Compound 16 (TAK-925) is being developed for the treatment of narcolepsy and other related disorders.
^ Evans, R., Hazel, J., Faessel, H., Wu, J., Hang, Y., Alexander, R., … & Hartman, D. (2019). Results of a phase 1, 4-period crossover, placebo-controlled, randomized, single dose study to evaluate the safety, tolerability, pharmacokinetics, and pharmacodynamics of TAK-925, a novel orexin 2 receptor agonist, in sleep-deprived healthy adults, utilizing modafinil as an active comparator. Sleep Medicine, 64, S106. https://scholar.google.com/scholar?cluster=10933819770107034612
^ Evans R, Tanaka S, Tanaka S, Touno S, Shimizu K, Sakui S, et al. (December 2019). “A Phase 1 single ascending dose study of a novel orexin 2 receptor agonist, TAK-925, in healthy volunteers (HV) and subjects with narcolepsy type 1 (NT1) to assess safety, tolerability, pharmacokinetics, and pharmacodynamic outcomes”. Sleep Medicine. 64: S105–S106. doi:10.1016/j.sleep.2019.11.290.
Tegafur/gimeracil/oteracil, sold under the brand names Teysuno and TS-1,[3][4] is a fixed-dose combination medication used for the treatment of advanced gastric cancer when used in combination with cisplatin,[3] and also for the treatment of head and neck cancer, colorectal cancer, non–small-cell lung, breast, pancreatic, and biliary tract cancers.[5]: 213
The most common severe side effects when used in combination with cisplatin include neutropenia (low levels of neutrophils, a type of white blood cell), anaemia (low red blood cell counts) and fatigue (tiredness).[3]
Tegafur/gimeracil/oteracil (Teysuno) was approved for medical use in the European Union in March 2011.[3] It has not been approved by the U.S. Food and Drug Administration (FDA).[5]: 213
Medical uses
In the European Union tegafur/gimeracil/oteracil is indicated in adults for the treatment of advanced gastric cancer when given in combination with cisplatin.[3]
Contraindications
In the European Union, tegafur/gimeracil/oteracil must not be used in the following groups:
people receiving another fluoropyrimidine (a group of anticancer medicines that includes tegafur/gimeracil/oteracil) or who have had severe and unexpected reactions to fluoropyrimidine therapy;[3]
people known to have no DPD enzyme activity, as well as people who, within the previous four weeks, have been treated with a medicine that blocks this enzyme;[3]
Tegafur is the actual chemotherapeutic agent. It is a prodrug of the active substance fluorouracil (5-FU).[3] Tegafur, is a cytotoxic medicine (a medicine that kills rapidly dividing cells, such as cancer cells) that belongs to the ‘anti-metabolites’ group. Tegafur is converted to the medicine fluorouracil in the body, but more is converted in tumor cells than in normal tissues.[3] Fluorouracil is very similar to pyrimidine.[3] Pyrimidine is part of the genetic material of cells (DNA and RNA).[3] In the body, fluorouracil takes the place of pyrimidine and interferes with the enzymes involved in making new DNA.[3] As a result, it prevents the growth of tumor cells and eventually kills them.[3]
Within the medication, the molar ratio of the three components (tegafur:gimeracil:oteracil) is 1:1:0.4.[7]
The maximum tolerated dose differed between Asian and Caucasian populations (80 mg/m2 and 25 mg/m2 respectively), perhaps due to differences in CYP2A6genotype.[5]: 213
^ Jump up to:abc DeVita, DeVita; Lawrence, TS; Rosenberg, SA (2015). DeVita, Hellman, and Rosenberg’s Cancer: Principles and Practice of Oncology (10th ed.). LWW. ISBN978-1451192940.
^ Jump up to:ab A. Klement (22 July 2013). “Dreier-Kombination gegen Magenkrebs: Teysuno”. Österreichische Apothekerzeitung (in German) (15/2013): 23.
^ Peters GJ, Noordhuis P, Van Kuilenburg AB et al. (2003). “Pharmacokinetics of S-1, an oral formulation of ftorafur, oxonic acid and 5-chloro-2,4-dihydroxypyridine (molar ratio 1:0.4:1) in patients with solid tumors”. Cancer Chemother. Pharmacol. 52 (1): 1–12. doi:10.1007/s00280-003-0617-9. PMID12739060. S2CID10858817.
“Tegafur”. Drug Information Portal. U.S. National Library of Medicine.
“Gimeracil”. Drug Information Portal. U.S. National Library of Medicine.
“Oteracil”. Drug Information Portal. U.S. National Library of Medicine.
Gimeracil is an adjunct to antineoplastic therapy, used to increase the concentration and effect of the main active componets within chemotherapy regimens. Approved by the European Medicines Agency (EMA) in March 2011, Gimeracil is available in combination with Oteracil and Tegafur within the commercially available product “Teysuno”. The main active ingredient in Teysuno is Tegafur, a pro-drug of Fluorouracil (5-FU), which is a cytotoxic anti-metabolite drug that acts on rapidly dividing cancer cells. By mimicking a class of compounds called “pyrimidines” that are essential components of RNA and DNA, 5-FU is able to insert itself into strands of DNA and RNA, thereby halting the replication process necessary for continued cancer growth.
Gimeracil’s main role within Teysuno is to prevent the breakdown of Fluorouracil (5-FU), which helps to maintin high enough concentrations for sustained effect against cancer cells 2. It functions by reversibly and selectively blocking the enzyme dihydropyrimidine dehydrogenase (DPD), which is involved in the degradation of 5-FU 1. This allows higher concentrations of 5-FU to be achieved with a lower dose of tegafur, thereby also reducing toxic side effects.
/////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
aReagents and conditions: (a) CH3C(OCH3)3, MeOH, then (CH3)2NHCH(OCH3)2, reflux, 92%; (b) aq AcOH, 130 °C, 2 h, 95%; (c) SO2Cl2, HOAc, 50 °C, 0.5 h, 91%; (d) 40% H2SO4, 130 °C, 4 h, 91%; (e) SO2Cl2, HOAc, 50 °C, 45 min, 86%; (f) 75% H2 SO4, 140 °C, 3 h, then NaOH, then pH 4–4.5, 89%
In 1953, Kolder and Hertog reported a synthesis of the TS-1 additive gimeracil 20, which was completed in seven steps using 4-nitropyridine N-oxide as starting material.222 Later, Yano et al. reported an alternative gram-scale synthesis (Scheme 15).223 The one-pot, three component condensation of malononitrile 111, 1,1,1-trimethoxyethane, and 1,1-dimethyoxytrimethylamine generated the dicyano intermediate 112, which was into 2(1H)-pyridinone 113.224 Selective chlorination of 113 was followed by acid-mediated demethylation, hydrolysis, and decarboxylation, to afford gimeracil 20. Interestingly, Xu et al. found that treatment of intermediate 113 with sulfuryl chloride resulted in dichloro 115 formation, which could still be converted to gimeracil 20 by treatment with sulfuric acid.225
(222) Kolder CR; den Hertog HJ Synthesis and reactivity of 5-chloro-2,4-dihydroxypyridine. Rec. Trav. Chim 1953, 72, 285–295. [Google Scholar]
(223) Yano S; Ohno T; Ogawa K Convenient and practical synthesis of 5-chloro-4-hydroxy-2(1H)-pyridinone. Heterocycles 1993, 36, 145–148. [Google Scholar]
(224) Mittelbach M; Kastner G; Junek H Synthesen mit Nitrilen, 71. Mitt. Zur Synthese von 4-Hydroxynicotinsaure aus Butadiendicarbonitrilen. Arch. Pharm 1985, 318 (6), 481–486. [Google Scholar]
(225) Xu Y; Mao D; Zhang F CN Patent 1915976, 2007.
NEW DRUG APPROVALS
THIS MAY NOT RUN WITHOUT SUBSCRIPTION HELP. AVOID CLOSURE OF THIS BLOG
4,6-Dihydroxy-1,3,5-triazine-2-carboxylic acid potassium salt
KOX
NSC 28841
Oxonate
Oxonate, potassium
CDSCO APPROVED,01.02.2022
Gimeracil bulk & Oteracil potassium bulk and Tegafur 15mg/20mg, Gimeracil 4.35mg/5.8mg and Oteracil 11.8mg/15.8mg capsules
indicated in adults for the treatment of advanced gastric cancer when given in combination with cisplatin.
Oteracil Potassium is the potassium salt of oxonate, an enzyme inhibitor that modulates 5- fluorouracil (5-FU) toxicity. Potassium oxonate inhibits orotate phosphoribosyltransferase, which catalyzes the conversion of 5-FU to its active or phosphorylated form, FUMP. Upon oral administration, Oxonate is selectively distributed to the intracellular sites of tissues lining the small intestines, producing localized inhibitory effects within the gastrointestinal tract. As a result, 5-FU associated gastrointestinal toxic effects are reduced and the incidence of diarrhea or mucositis is decreased in 5-FU related therapy.
Oteracil is an adjunct to antineoplastic therapy, used to reduce the toxic side effects associated with chemotherapy. Approved by the European Medicines Agency (EMA) in March 2011, Oteracil is available in combination with Gimeracil and Tegafur within the commercially available product “Teysuno”. The main active ingredient in Teysuno is Tegafur, a pro-drug of Fluorouracil (5-FU), which is a cytotoxic anti-metabolite drug that acts on rapidly dividing cancer cells. By mimicking a class of compounds called “pyrimidines” that are essential components of RNA and DNA, 5-FU is able to insert itself into strands of DNA and RNA, thereby halting the replication process necessary for continued cancer growth.
Oteracil’s main role within Teysuno is to reduce the activity of 5-FU within normal gastrointestinal mucosa, and therefore reduce’s gastrointestinal toxicity 1. It functions by blocking the enzyme orotate phosphoribosyltransferase (OPRT), which is involved in the production of 5-FU.
/////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
Poje et al. reported a two-step, gram-scale preparation of the TS-1 additive oteracil 21 (Scheme 16).226 Iodine-mediated-oxidation of uric acid 116 produced dehydroallantoin 117 as the major product, and subsequent treatment with potassium hydroxide resulted in the rearranged product oteracil 21.227
Synthesis of Oteracil 21a
aReagents and conditions: (a) LiOH, I2, H2O, 5 °C, 5 min, then AcOH, 75%; (b) aq KOH, 20 min, rt, 82%.
(226) Poje M; Sokolić-Maravić L The mechanism for the conversion of uric acid into allantoin and dehydro-allantoin: A new look at an old problem. Tetrahedron 1986, 42 (2), 747–751. [Google Scholar]
(227) Sugi M; Igi M EP Patent 0957096, 1999.
EP0957096A1 *1998-05-111999-11-17SUMIKA FINE CHEMICALS Co., Ltd.Method for producing potassium oxonate
CN101475539A *2009-02-112009-07-08鲁南制药集团股份有限公司Refining method for preparing high-purity oteracil potassium
CN102250025A *2011-05-182011-11-23深圳万乐药业有限公司Preparation method suitable for industrially producing oteracil potassium
CN102746244A *2012-07-272012-10-24南京正大天晴制药有限公司Refining method of oteracil potassium
//////////OTERACIL POTTASIUM, KOX, NSC 28841, Oxonate, Oxonate potassium, INDIA 2022, APPROVALS 2022, CANCER
Percent Composition: C 41.07%, H 4.21%, F 14.44%, N 15.97%, O 24.32%
Literature References: Prepn: L. W. Hertel, GB2136425; idem,US4808614 (1984, 1989 both to Lilly); L. W. Hertel et al.,J. Org. Chem.53, 2406 (1988); T. S. Chou et al.,Synthesis1992, 565. Antitumor activity: L. W. Hertel et al.,Cancer Res.50, 4417 (1990). Mode of action study: V. W. T. Ruiz et al.,Biochem. Pharmacol.46, 762 (1993). Clinical pharmacokinetics and toxicity: J. L. Abbruzzese et al.,J. Clin. Oncol.9, 491 (1991). Review of clinical studies: B. Lund et al.,Cancer Treat. Rev.19, 45-55 (1993).
Properties: Crystals from water, pH 8.5. [a]365 +425.36°; [a]D +71.51° (c = 0.96 in methanol). uv max (ethanol): 234, 268 (e 7810, 8560). LD10 i.v. in rats: 200 mg/m2 (Abbruzzese).
Optical Rotation: [a]365 +425.36°; [a]D +71.51°
Absorption maximum: uv max (ethanol): 234, 268 (e 7810, 8560)
Toxicity data: LD10 i.v. in rats: 200 mg/m2 (Abbruzzese)
Derivative Type: Hydrochloride
CAS Registry Number: 122111-03-9
Molecular Formula: C9H11F2N3O4.HCl
Molecular Weight: 299.66
Percent Composition: C 36.07%, H 4.04%, F 12.68%, N 14.02%, O 21.36%, Cl 11.83%
Properties: Crystals from water-acetone, mp 287-292° (dec). [a]D +48°; [a]365 +257.9° (c = 1.0 in deuterated water). uv max (water): 232, 268 nm (e 7960, 9360).
Gemcitabine is a nucleoside metabolic inhibitor used as adjunct therapy in the treatment of certain types of ovarian cancer, non-small cell lung carcinoma, metastatic breast cancer, and as a single agent for pancreatic cancer.
Gemcitabine hydrochloride was first approved in ZA on Jan 10, 1995, then approved by the U.S. Food and Drug Administration (FDA) on May 15, 1996, and approved by Pharmaceuticals and Medicals Devices Agency of Japan (PMDA) on Aug 31, 2001. It was developed and marketed as Gemzar® by Eli Lilly.
Gemcitabine hydrochloride is a nucleoside metabolic inhibitor. It kills cells undergoing DNA synthesis and blocks the progression of cells through the G1/S-phase boundary. It is indicated for the treatment of advanced ovarian cancer that has relapsed at least 6 months after completion of platinum-based therapy, in combination with paclitaxel, for first-line treatment of metastatic breast cancer after failure of prior anthracycline-containing adjuvant chemotherapy, unless anthracyclines were clinically contraindicated, and it is also indicated in combination with cisplatin for the treatment of non-small cell lung cancer, and treated as a single agent for the treatment of pancreatic cancer.
Gemzar® is available as injection of lyophilized powder for intravenous use, containing 200 mg or 1000 mg of free Gemcitabine per vial. The recommended initial dosage is 1000 mg/m2 over 30 minutes on days 1 and 8 of each 21 day cycle for ovarian cancer, 1250 mg/m2 over 30 minutes on days 1 and 8 of each 21 day cycle for breast cancer, 1000 mg/m2 over 30 minutes on days 1, 8, and 15 of each 28 day cycle or 1250 mg/m2 over 30 minutes on days 1 and 8 of each 21 day cycle for non-small cell lung cancer, and 1000 mg/m2 over 30 minutes once weekly for the first 7 weeks, then one week rest, then once weekly for 3 weeks of each 28 day cycle for pancreatic cancer.
Gemcitabine was patented in 1983 and was approved for medical use in 1995.[6] Generic versions were introduced in Europe in 2009 and in the US in 2010.[7][8] It is on the WHO Model List of Essential Medicines.[9]
Taking gemcitabine can also affect fertility in men and women, sex life, and menstruation. Women taking gemcitabine should not become pregnant, and pregnant and breastfeeding women should not take it.[15]
As of 2014, drug interactions had not been studied.[11][10]
SYN
. Hertel, L. W.; Kroin, J. S.; Misner, J. W.; Tustin, J. M. J. Org. Chem. 1988, 53, 2406– 2409.
NEXT
a) Noe, C. R.; Jasic, M.; Kollmann, H.; Saadat, K. WO009147, 2007.; b) Noe, C. R.; Jasic, M.; Kollmann, H.; Saadat, K. US0249119, 2008. Note: no stereochemistry was indica
NExT
15. Hanzawa, Y.; Inazawa, K.; Kon, A.; Aoki, H.; Kobayashi, Y. Tetrahedron Lett. 1987, 28, 659–662. 16. Wirth, D. D. EP0727432, 1996
Synthesis Reference
John A. Weigel, “Process for making gemcitabine hydrochloride.” U.S. Patent US6001994, issued May, 1995.US6001994Route 1
Reference:1. J. Org. Chem.1988, 53, 2406-2409.
2. US4808614A.Route 2
Reference:1. CN102417533A.Route 3
Reference:1. Nucleosides, Nucleotides and Nucleic Acids2010, 29, 113-122.Route 4
Reference:1. CN102617677A.Route 5
Reference:1. CN103012527A.
SYN
U.S. Patent No. 4,808,614 (the ‘614 patent) describes a process for synthetically producing gemcitabine, which process is generally illustrated in Scheme Scheme 1
5
SYN
U.S. Patent No. 4,965,374 (the ‘374 patent) describes a process for producing gemcitabine from an intermediate 3,5-dibenzoyl ribo protected lactone of the formula:
11 where the desired erythro isomer can be isolated in a crystalline form from a mixture of erythro and threo isomers. The process described in the ‘374 patent is generally outlined in Scheme 2.
Scheme 2
mixture of α and β anomers
SYN
U.S. Patent No. 5,521,294 (the ‘294 patent) describes l-alkylsulfonyl-2,2- difluoro-3 -carbamoyl ribose intermediates and intermediate nucleosides derived therefrom. The compounds are reportedly useful in the preparation of 2′-deoxy-2′,2’- difluoro-β-cytidine and other β-anomer nucleosides. The ‘294 patent teaches, inter alia, that the 3-hydroxy carbamoyl group on the difluororibose intermediate may enhance formation of the desired β-anomer nucleoside derivative. The ‘294 patent describes converting the lactone 4 to the dibenzoyl mesylate 13, followed by deprotection at the 3 position to obtain the 5-monobenzoyl mesylate intermediate 15, which is reacted with various isocyanates to obtain the compounds of formula 16. The next steps involve coupling and deprotection using methods similar to those described in previous patents. The process and the intermediates 15 and 16 are illustrated by scheme 3 below: Scheme 3
[0045] This example demonstrates the preparation of 2-deoxy-2,2-difluoro-D- ribofuranose-3,5-dicinnamate-l-p-toluenesulfonate.
[0046] Crude 2-deoxy-2,2-difluoro-D-riboufuranose-3,5-dicinnamate (2.5g, 6 mmol) was dissolved in dichloromethane (20 ml) in a round flask, and diethylamine (0.7g, 9.6 mmol) was added followed by p-toluenesulfonyl chloride (1.32 g, 6.92 mmol), which was added drop wise while cooling to 0-50C. The mixture was stirred for 1 hour, and washed with IN HCl (15 ml), concentrated solution OfNaHCO3 (15 ml), and dried over MgSO4. The solvent was distilled off under reduced pressure to obtain crude 2-deoxy-2,2-difluoro-D-ribofuranose-3,5-dicinnamate-l-p- toluenesulfonate as light oil. Yield: 3.22 g, (5.6 mmol), 93%.
EXAMPLE 2
[0047] This example demonstrates the preparation of 3′,5′-dicinnamoyl-2′-deoxy- 2′,2′-difluorocytidine.
[0048] Dry 1 ,2-dichloroethane (800 ml) was added to N,O-bis(trimethylsilyl)- cytosine (136 g, 487 mmol) under nitrogen blanket to produce a clear solution, followed by adding trimethylsilyl triflate (Me3SiOTf), (100 ml, 122.8 g, 520 mmol) and stirred for 30 minutes. A solution of 2-deoxy-2,2-difluoro-D-ribofuranose-3,5- dicinnamate-1-p-toluenesulfonate (128 g, 224 mmol) in 1 ,2-dichloroethane (400 ml) was added drop wise, and the mixture was refluxed overnight. After cooling, the solvent was distilled off to obtain crude 3,5-dicinnamoyl-N4-trimethylsilyl-2′-deoxy- 2′,2′-difluorocytidine as a light yellow solid. The residue was dissolved in ethyl acetate (1600 ml) and washed 3 times with water (3X400 ml). The ethyl acetate phase was mixed with concentrated solution OfNaHCO3 (800 ml) for about 5 minutes, and then the mixture was set aside for about 20 minutes without stirring. The thus formed solid, which was precipitated in the inter-phase of the two layers, was filtered off and washed with 60 ml of ethyl acetate. The solid was dried under reduced pressure to obtain 116.7 g (223 mmol, 99.5%) of the crude 3′,5′-dicinnamoyl- 2′-deoxy-2′,2′- difluorocytidine containing 73.3 % of the β-anomer and 11.8 % of the α-anomer.
EXAMPLE 3
[0049] This example demonstrates the preparation of 3′,5′-dicinnamoyl-2′-deoxy- 2′,2′-difluorocytidine.
[0050] Dry 1,2-dichloroethane (1.5 L) was added to bis(trimethylsilyl)cytosine (417 g, 1.49 mol) under nitrogen blanket to produce a clear solution followed by adding trimethylsilyl triflate (Me3SiOTf), (300 ml, 368.4 g, 1.56 mol) and stirred for 30 minutes. A solution of 2-deoxy-2,2-difluoro-D-ribofuranose-3,5-dicinnamate-l-p- toluenesulfonate (384 g, 673 mmol) in 1,2-dichloroethane (1.2 L) was added drop wise, and the mixture was refluxed overnight. After cooling, the solvent was distilled off to obtain crude 3,5-dicinnamoyl-N4-trimethylsilyl-2l-deoxy-2′,2′-difluorocytidine as a light yellow solid. The residue was dissolved in ethyl acetate (2.4 L) and washed 3 times with water (3X1.2 L). The ethyl acetate phase was mixed with concentrated solution OfNaHCO3 (1.34 L) for about 20 minutes. The thus formed solid, which was precipitated in the inter-phase of the two layers, was filtered off and washed with 180 ml of ethyl acetate. The solid was dried under reduced pressure to obtain 346.5 g (0.66 mol, 99.9% yield) of the crude 3l,5l-dicinnamoyl-2′-deoxy-2′,2′-difluorocytidine containing 43 % of the β-anomer and 52 % of the α-anomer.
EXAMPLE 4
[0051] This example demonstrates the preparation of gemcitabine hydrochloride. [0052] To a solution of ammonia-methanol (15.8 %, 4.57 L), the crude 3,5- dicirmamoyl-2′-deoxy-2′,2′-difluorocytidine of example 3 was added (346.5 g, 0.66 mol), and stirred at ambient temperature for 6 hours. The mixture was concentrated to afford a light yellow solid (306 g). Purified water (3 L) was added to the solid, followed by addition of ethyl acetate (1.8 L), and stirring was maintained for about 10 minutes. The aqueous layer was separated and the organic layer was extracted with water (1.05 L). The aqueous layers were combined and water was removed by evaporation under reduced pressure to obtain an oil (154.7 g). Water was added (660 ml) and the mixture was heated to 50-550C to dissolve the solid. The mixture was cooled to 5-1O0C during about one hour and mixed for about 16 hours at that temperature. The thus formed solid was filtered and dried to afford 46.75 g (0.177 mol), containing 98 % of the β-anomer and 1.3 % of the α-anomer. 0.5N HCl (936 ml) was added followed by addition of dichloromethane (300 ml) with stirring. The water phase was separated and the aqueous phase was washed with dichloromethane (300 ml). After filtration, the aqueous phase was concentrated to dryness under reduced pressure to obtain gemcitabine hydrochloride as a solid (46.9 g). The solid was dissolved in water (187 ml) at ambient temperature and the mixture was heated to 500C to afford a clear solution and cooled to ambient temperature. Acetone (1.4 L) was added and stirring was maintained for about one hour. Then, the precipitate was collected by filtration and washed twice with acetone (2X30 ml) and dried at 450C under vacuum to obtain 39.2 g of gemcitabine hydrochloride, containing 99.9% of the β-anomer
EXAMPLE 5
[0053] This example demonstrates the preparation of gemcitabine hydrochloride. [0054] To a solution of ammonia-methanol (about 15.8 %, 1.35 L), the crude 3′,5′- dicinnamoyl-2′-deoxy-2′,2′-difluorocytidine prepared as described in example 2 was added (96 g, 183.4 mmol), and stirred at ambient temperature for 4 hours. The mixture was concentrated to afford a light yellow solid (80.5 g). Purified water (1 L) was added to the solid, followed by addition of ethyl acetate (600 ml), and stirring was maintained for about 10 minutes. The aqueous layer was separated and the organic layer was extracted with water (350 ml). The aqueous layers were combined and water was removed by evaporation under reduced pressure to obtain an oil (46.4 g). Water was added (220 ml) and the mixture was heated to 50-550C to dissolve the solid. The mixture was cooled to 0-50C during about one hour and mixed for about 16 hours at that temperature. The thus formed solid was filtered and dried to afford 11.1 g of gemcitabine free base. 0.5N HCl (240 ml) was added followed by addition of dichloromethane (100 ml) with stirring. The water phase was separated and the aqueous phase was washed with dichloromethane (300 ml). After filtration, the aqueous phase was concentrated to dryness under reduced pressure to obtain gemcitabine hydrochloride as a solid (12.0 g). The solid was dissolved in water (48 ml) at ambient temperature and the mixture was heated to 5O0C to afford a clear solution and cooled to ambient temperature. Acetone (360 ml) was added and stirring was maintained for about one hour. Then, the precipitate was collected by filtration and washed twice with acetone (2X30 ml) and dried at 450C under vacuum to obtain 9.9 g of gemcitabine hydrochloride, containing 99.6% of the β-anomer.
EXAMPLE 6
[0055] This example demonstrates the slurrying procedure of the 3 ‘,5′- dicinnamoyl-2′-deoxy-2’,2l-difluorocytidine in different solvents. [0056] 1 g of the crude 3′,5′-dicinnamoyl-2′-deoxy-2l,2′-difluorocytidine, containing 73.7 % of the β-anomer and 17.5 % of the α-anomer, was placed in flask and 10 ml of a solvent was added and the mixture was mixed at ambient temperature for one hour. Then, the solid was obtained by filtration, washed with 5 ml of the solvent and dried. The liquid obtained after filtering the solid and the liquid obtained after washing the solid were combined (hereinafter the mother liquor). The ratio between the β-anomer and the α-anomer in the solid and in the mother liquor was determined by HPLC and the results are summarized in Table 1.
Table 1
/////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
Gemcitabine is a chemotherapy drug that works by killing any cells that are dividing.[10] Cancer cells divide rapidly and so are targeted at higher rates by gemcitabine, but many essential cells also divide rapidly, including cells in skin, the scalp, the stomach lining, and bone marrow, resulting in adverse effects.[16]: 265
More than 10% of users develop adverse effects, including difficulty breathing, low white and red blood cells counts, low platelet counts, vomiting and nausea, elevated transaminases, rashes and itchy skin, hair loss, blood and protein in urine, flu-like symptoms, and edema.[10][15]
Common adverse effects (occurring in 1–10% of users) include fever, loss of appetite, headache, difficulty sleeping, tiredness, cough, runny nose, diarrhea, mouth and lip sores, sweating, back pain, and muscle pain.[10]
Thrombotic thrombocytopenic purpura (TTP) is a rare but serious side effect that been associated with particular chemotherapy medications including gemcitabine. TTP is a blood disorder and can lead to microangipathic hemolytic anemia (MAHA), neurologic abnormalities, fever, and renal disease.[18]
Pharmacology
Gemcitabine is hydrophilic and must be transported into cells via molecular transporters for nucleosides (the most common transporters for gemcitabine are SLC29A1 SLC28A1, and SLC28A3).[19][20] After entering the cell, gemcitabine is first modified by attaching a phosphate to it, and so it becomes gemcitabine monophosphate (dFdCMP).[19][20] This is the rate-determining step that is catalyzed by the enzymedeoxycytidine kinase (DCK).[19][20] Two more phosphates are added by other enzymes. After the attachment of the three phosphates gemcitabine is finally pharmacologically active as gemcitabine triphosphate (dFdCTP).[19][21]
When gemcitabine is incorporated into DNA it allows a native, or normal, nucleoside base to be added next to it. This leads to “masked chain termination” because gemcitabine is a “faulty” base, but due to its neighboring native nucleoside it eludes the cell’s normal repair system (base-excision repair). Thus, incorporation of gemcitabine into the cell’s DNA creates an irreparable error that leads to inhibition of further DNA synthesis, and thereby leading to cell death.[2][19][20]
The form of gemcitabine with two phosphates attached (dFdCDP) also has activity; it inhibits the enzyme ribonucleotide reductase (RNR), which is needed to create new DNA nucleotides. The lack of nucleotides drives the cell to uptake more of the components it needs to make nucleotides from outside the cell, which also increases uptake of gemcitabine.[2][19][20][22]
The synthesis described and pictured below is the original synthesis done in the Eli Lilly Company labs. Synthesis begins with enantiopure D-glyceraldehyde (R)-2 as the starting material which can made from D-mannitol in 2–7 steps. Then fluorine is introduced by a “building block” approach using ethyl bromodifluroacetate. Then, Reformatsky reaction under standard conditions will yield a 3:1 anti/syn diastereomeric mixture, with one major product. Separation of the diastereomers is carried out via HPLC, thus yielding the anti-3 gemcitabine in a 65% yield.[23][24] At least two other full synthesis methods have also been developed by different groups.[24]
Illustration of the original synthesis process used and published by Hertel et al. in 1988 of Lilly laboratories.
History[
Gemcitabine was first synthesized in Larry Hertel’s lab at Eli Lilly and Company during the early 1980s. It was intended as an antiviral drug, but preclinical testing showed that it killed leukemia cells in vitro.[25]
During the early 1990s gemcitabine was studied in clinical trials. The pancreatic cancer trials found that gemcitabine increased one-year survival time significantly, and it was approved in the UK in 1995[10] and approved by the FDA in 1996 for pancreatic cancers.[4] In 1998, gemcitabine received FDA approval for treating non-small cell lung cancer and in 2004, it was approved for metastatic breast cancer.[4]
European labels were harmonized by the EMA in 2008.[26]
By 2008, Lilly’s worldwide sales of gemcitabine were about $1.7 billion; at that time its US patents were set to expire in 2013 and its European patents in 2009.[27] The first generic launched in Europe in 2009,[7] and patent challenges were mounted in the US which led to invalidation of a key Lilly patent on its method to make the drug.[28][29] Generic companies started selling the drug in the US in 2010 when the patent on the chemical itself expired.[29][8] Patent litigation in China made headlines there and was resolved in 2010.[30]
Because it is clinically valuable and is only useful when delivered intravenously, methods to reformulate it so that it can be given by mouth have been a subject of research.[31][32][33]
Research into pharmacogenomics and pharmacogenetics has been ongoing. As of 2014, it was not clear whether or not genetic tests could be useful in guiding dosing and which people respond best to gemcitabine.[19] However, it appears that variation in the expression of proteins (SLC29A1, SLC29A2, SLC28A1, and SLC28A3) used for transport of gemcitabine into the cell lead to variations in its potency. Similarly, the genes that express proteins that lead to its inactivation (deoxycytidine deaminase, cytidine deaminase, and NT5C) and that express its other intracellular targets (RRM1, RRM2, and RRM2B) lead to variations in response to the drug.[19] Research has also been ongoing to understand how mutations in pancreatic cancers themselves determine response to gemcitabine.[34]
It has been studied as a treatment for Kaposi sarcoma, a common cancer in people with AIDS which is uncommon in the developed world but not uncommon in the developing world.[35]
^World Health Organization (2019). World Health Organization model list of essential medicines: 21st list 2019. Geneva: World Health Organization. hdl:10665/325771. WHO/MVP/EMP/IAU/2019.06. License: CC BY-NC-SA 3.0 IGO.
^ Zhang XW, Ma YX, Sun Y, Cao YB, Li Q, Xu CA (June 2017). “Gemcitabine in Combination with a Second Cytotoxic Agent in the First-Line Treatment of Locally Advanced or Metastatic Pancreatic Cancer: a Systematic Review and Meta-Analysis”. Targeted Oncology. 12 (3): 309–321. doi:10.1007/s11523-017-0486-5. PMID28353074. S2CID3833614.
^ Jain A, Kwong LN, Javle M (November 2016). “Genomic Profiling of Biliary Tract Cancers and Implications for Clinical Practice”. Current Treatment Options in Oncology. 17 (11): 58. doi:10.1007/s11864-016-0432-2. PMID27658789. S2CID25477593.
^ Jump up to:ab Macmillan Cancer Support. “Gemcitabine”. Macmillan Cancer Support. Archived from the original on 25 March 2017. Retrieved 6 May 2017.
^ Fatima, M., Iqbal Ahmed, M. M., Batool, F., Riaz, A., Ali, M., Munch-Petersen, B., & Mutahir, Z. (2019). Recombinant deoxyribonucleoside kinase from Drosophila melanogaster can improve gemcitabine based combined gene/chemotherapy for targeting cancer cells. Bosnian Journal of Basic Medical Sciences, 19(4), 342-349. https://doi.org/10.17305/bjbms.2019.4136
^ Dyawanapelly S, Kumar A, Chourasia MK (2017). “Lessons Learned from Gemcitabine: Impact of Therapeutic Carrier Systems and Gemcitabine’s Drug Conjugates on Cancer Therapy”. Critical Reviews in Therapeutic Drug Carrier Systems. 34 (1): 63–96. doi:10.1615/CritRevTherDrugCarrierSyst.2017017912. PMID28322141.
^ Birhanu G, Javar HA, Seyedjafari E, Zandi-Karimi A (April 2017). “Nanotechnology for delivery of gemcitabine to treat pancreatic cancer”. Biomedicine & Pharmacotherapy. 88: 635–643. doi:10.1016/j.biopha.2017.01.071. PMID28142120.
^ Dubey RD, Saneja A, Gupta PK, Gupta PN (October 2016). “Recent advances in drug delivery strategies for improved therapeutic efficacy of gemcitabine”. European Journal of Pharmaceutical Sciences. 93: 147–62. doi:10.1016/j.ejps.2016.08.021. PMID27531553.
US4526988A *1983-03-101985-07-02Eli Lilly And CompanyDifluoro antivirals and intermediate therefor
US4751221A *1985-10-181988-06-14Sloan-Kettering Institute For Cancer Research2-fluoro-arabinofuranosyl purine nucleosides
US5223608A *1987-08-281993-06-29Eli Lilly And CompanyProcess for and intermediates of 2′,2′-difluoronucleosides
US4965374A *1987-08-281990-10-23Eli Lilly And CompanyProcess for and intermediates of 2′,2′-difluoronucleosides
US5256798A *1992-06-221993-10-26Eli Lilly And CompanyProcess for preparing alpha-anomer enriched 2-deoxy-2,2-difluoro-D-ribofuranosyl sulfonates
US5371210A *1992-06-221994-12-06Eli Lilly And CompanyStereoselective fusion glycosylation process for preparing 2′-deoxy-2′,2′-difluoronucleosides and 2′-deoxy-2′-fluoronucleosides
US5256797A *1992-06-221993-10-26Eli Lilly And CompanyProcess for separating 2-deoxy-2,2-difluoro-D-ribofuranosyl alkylsulfonate anomers
US5480992A *1993-09-161996-01-02Eli Lilly And CompanyAnomeric fluororibosyl amines
US5521294A *1995-01-181996-05-28Eli Lilly And Company2,2-difluoro-3-carbamoyl ribose sulfonate compounds and process for the preparation of beta nucleosides
US5559222A *1995-02-031996-09-24Eli Lilly And CompanyPreparation of 1-(2′-deoxy-2′,2′-difluoro-D-ribo-pentofuranosyl)-cytosine from 2-deoxy-2,2-difluoro-β-D-ribo-pentopyranose
US5602262A *1995-02-031997-02-11Eli Lilly And CompanyProcess for the preparation of 2-deoxy-2,2-difluoro-β-D-ribo-pentopyranose
US5633367A *1995-03-241997-05-27Eli Lilly And CompanyProcess for the preparation of a 2-substituted 3,3-difluorofuran
GB9514268D0 *1995-07-131995-09-13Hoffmann La RochePyrimidine nucleoside
US5756775A *1995-12-131998-05-26Eli Lilly And CompanyProcess to make α,α-difluoro-β-hydroxyl thiol esters
CA2641719A1 *2006-02-072007-08-16Chemagis Ltd.Process for preparing gemcitabine and associated intermediates
US20070249823A1 *2006-04-202007-10-25Chemagis Ltd.Process for preparing gemcitabine and associated intermediates
WO2010029574A22010-03-18An improved process for the preparation of gemcitabine and its intermediates using novel protecting groups and ion exchange resins
KR20080090950A2008-10-09Process for preparing gemcitabine and associated intermediates
ClassAmines; Antineoplastics; Azetidines; Fluorinated hydrocarbons; Isoquinolines; Pyrazolones; Pyridines; Small molecules
Mechanism of ActionSelective estrogen receptor degraders
Phase IIIBreast cancer
13 Jun 2022AstraZeneca initiates a phase I drug-drug interaction trial of AZD 9833 Healthy postmenopausal female volunteers, in USA (NCT05438303)
10 Jun 2022AstraZeneca and Quotient Sciences complete the phase I QSC205863 trial in Breast cancer (In volunteers) in United Kingdom (PO, Liquid) (NCT05364255)
03 Jun 2022Safety, efficacy and pharmacokinetics data from the phase I SERENA 1 trial for Breast cancer presented at the 58th Annual Meeting of the American Society of Clinical Oncology (ASCO-2022)
Mechanism:selective estrogen receptor degrader
Area under investigation:estrogen receptor +ve breast cancer
Date commenced phase:Q1 2019
Estimated Filing Acceptance:
CountryDateUS: EU: Japan: China:
AZD9833 is an orally available selective estrogen receptor degrader (SERD), with potential antineoplastic activity. Upon administration, SERD AZD9833 binds to the estrogen receptor (ER) and induces a conformational change that results in the degradation of the receptor. This prevents ER-mediated signaling and inhibits the growth and survival of ER-expressing cancer cells
Camizestrant is an orally available selective estrogen receptor degrader (SERD), with potential antineoplastic activity. Upon administration, camizestrant binds to the estrogen receptor (ER) and induces a conformational change that results in the degradation of the receptor. This prevents ER-mediated signaling and inhibits the growth and survival of ER-expressing cancer cells
Discovery of AZD9833, a Potent and Orally Bioavailable Selective Estrogen Receptor Degrader and Antagonist J. Med. Chem. 2020, 63, 14530–14559, DOI: 10.1021/acs.jmedchem.0c01163.
SYN
doi: 10.1021/acs.jmedchem.0c01163.
aReagents and Conditions: (a) n-BuLi, THF, −78 oC to 0 oC, 1 h, then 4 N HCl/dioxane, RT, 1 h, 60%; (b) alkyl triflate, DIPEA, 1,4-dioxane, 90 oC, 63-74% or isobutyrylaldehyde, Na(OAc)3BH, THF, 0 oC, 56%; (c) benzophenone imine, Pd2dba3, Rac-BINAP, NaOtBu, toluene, 90 oC, then 1 N aq. HCl, 71-85%; (d) nBuLi, THF, −78 oC to 0 oC, 1 h, then 4 N HCl/dioxane, RT, 4 h; e) NH2OH, NH2OH.HCl, EtOH, reflux. 84% over 2 steps; (f) alkyl triflate, DIPEA, 1,4-dioxane, 90 oC, 44-100% or 1-fluorocyclopropane-1- carboxylic acid, HATU, Et3N, DMF, RT, 61%, then BH3.THF, THF, 65 oC, 82%.
AZD9833 is selective oestrogen receptor degrader (SERD). It works by breaking down the site where oestrogen attaches to the cancer cell. This can help stop or slow the growth of hormone receptor breast cancer. Researchers think that AZD9833 with palbociclib might work better than anastrozole and palbociclib.
AZD9833 + palbociclib
The patients will receive AZD9833 (75 mg, PO, once daily) + palbociclib (PO, once daily, 125 mg for 21 consecutive days followed by 7 days off treatment) + anastrozole placebo (1 mg, PO, once daily)
Camizestrant (AZD-9833) is a potent and orally active estrogen receptor (ER) antagonist. Camizestrant is used for the study of ER+ HER2-advanced breast cancer[1].
Camizestrant is extracted from patent US20180111931A1, example 17[1].MCE has not independently confirmed the accuracy of these methods. They are for reference only.
In Vivo
Camizestrant (oral administration; 0.2-50 mg/kg; 20 days) exhibits anti-tumour efficacy as a dose-dependent manner in human parental MCF7 mice xenograft[1]. Camizestrant (oral administration; 0.8-40 mg/kg; 30 days) decreases tumor growth as a dose-dependent manner. It gives almost complete tumour growth inhibition at the doses >10 mg/kg in mice[1]. MCE has not independently confirmed the accuracy of these methods. They are for reference only.Animal Model:Human ESR1 mutant breast cancer patient derived xenograft with CTC174 cells in female NSG mice[1]Dosage:0.8 mg/kg, 3 mg/kg, 10 mg/kg, 20 mg/kg, 40 mg/kgAdministration:Oral administration; 30 days; once dailyResult:Inhibited tumor growth in a dose-dependent manner.
Clinical Trial
NCT NumberSponsorConditionStart DatePhaseNCT04711252AstraZenecaER-Positive HER2-Negative Breast CancerJanuary 28, 2021Phase 3NCT04964934AstraZenecaER-Positive HER2-Negative Breast CancerJune 30, 2021Phase 3NCT04214288AstraZenecaAdvanced ER-Positive HER2-Negative Breast CancerApril 22, 2020Phase 2NCT04588298AstraZenecaHER2-negative Breast CancerNovember 2, 2020Phase 2NCT04541433AstraZenecaER&addition; HER2- Advanced Breast CancerSeptember 29, 2020Phase 1NCT03616587AstraZenecaER&addition; HER2- Advanced Breast CancerOctober 11, 2018Phase 1NCT04546347AstraZeneca|Quotient SciencesHealthy VolunteersSeptember 17, 2020Phase 1NCT04818632AstraZenecaER&addition;, HER2-, Metastatic Breast CancerOctober 11, 2021Phase 1
////////////Camizestrant, AZD 9833, AZ 14066724, UNII-JUP57A8EPZ, WHO 11592, PHASE 2, ASTRA ZENECA, CANCER
DR ANTHONY MELVIN CRASTO is Chief Advisor Industry Advisory Board Amity Univ. Noida, INDIA
Journey begins with online meet. 26th aug 2022 Service to education is service to humanity Brochure for First Industry Advisory Board meeting at Amity Institute of Pharmacy is attached. Date 26th Aug 2022 it as hybrid-Online/offline.
Icenticaftor (QBW251) is an orally active CFTR channel potentiator, with EC50s of 79 nM and 497 nM for F508del and G551D CFTR, respectively. Icenticaftor can be used for chronic obstructive pulmonary disease (COPD) and cystic fibrosis research.
Cystic fibrosis (CF) is the most prevalent life-threatening Mendelian disorder in Caucasian populations. CF arises from mutations of the gene for the cystic fibrosis transmembrane conductance regulator (CFTR) protein. The CFTR ion channel orchestrates gating of chloride and bicarbonate ions across epithelial cell membranes in various tissues, including the lung, pancreas, intestine, reproductive tract, and sweat glands. While CF is a systemic disorder, the primary mortality derives from reduced CFTR activity in the airways. Subsequent acidification3 and dehydration leads to accumulation of a viscous mucus layer, occluding the airways and trapping bacteria, leading to infections, reduced lung function, and ultimately, respiratory failure. The most common CFTR mutation, F508del (Class II, found in 90% of CF patients), impairs folding of the CFTR protein (a Class II trafficking defect), resulting in a reduced amount of channel present at the plasma membrane. With the G551D mutation (class III), theamount of protein at the membrane is unaffected, but its open probability (Po) is reduced, also resulting in a reduced channel gating. Thus, to address the underlying causes of CF, two distinct CFTR modulators are required: correctors to increase CFTR levels at the plasma membrane and potentiators to enable effective opening of the channel
Chronic obstructive pulmonary disease (COPD) is anticipated to shortly become the third leading cause of death globally. COPD is characterized by persistent airflow obstruction with cigarette smoke exposure recognized as the primary risk factor. Airflow limitation is associated with all COPD patients; however, the disease is heterogeneous, with variable phenotypes ranging from chronic bronchitis (CB) to emphysema. Small airway disease exhibits increased numbers of goblet cells and mucus plugging with associated smooth muscle hyperplasia, airway fibrosis, and increased inflammation. Excess mucus secretion is believed to play an important role in COPD pathogenesis and is associated with progression of the disease.
Cystic fibrosis (CF) is a fatal genetic disease caused by mutations in the gene encoding the Cystic Fibrosis Transmembrane Conductance Regulator (CFTR), a protein kinase A activated epithelial anion channel involved in salt and fluid transport in multiple organs, including the lung. Most CF mutations either reduce the number of CFTR channels at the cell surface (e.g. synthesis or processing mutations) or impair channel function (e.g. gating or conductance mutations) or both.
PCT publication No. WO 2011/113894 describes compounds which restore or enhance the function of mutant and/or wild type CFTR for the treatment of cystic fibrosis, primary ciliary dyskinesia, chronic bronchitis, chronic obstructive pulmonary disease, asthma and other CFTR related diseases. The compounds described therein include (S)-3-amino-6-methoxy-N-(3,3,3-trifluoro-2-hydroxy-2-methylpropyl)-5-(trifluoromethyl)picolinamide (Example 5 of WO 2011/113894).
The synthesis described in WO 2011/113894 to make (S)-3-amino-6-methoxy-N-(3,3,3-trifluoro-2-hydroxy-2-methylpropyl)-5-(trifluoromethyl)picolinamide is long, uses expensive starting materials and toxic reagents. Schemes 1 and 2 outline a synthesis from WO 2011/113894 used to make(S)-3-amino-6-methoxy-N-(3,3,3-trifluoro-2-hydroxy-2-methylpropyl)-5-(trifluoromethyl)picolinamide.
In Scheme 1, the intermediate ethyl 3-amino-5-(trifluoromethyl)picolinate (B4) is made via a Buchwald-Hartwig coupling reaction which requires the use of an expensive starting material (B1) and an expensive palladium catalyst which has to be controlled in the final product. Also, the conversion of B4 to B5 requires the use of NBS, a mutagenic reagent which has to be controlled in the API.
Moreover, the conversion of B5 to B8 is accomplished through the addition of 2,5-hexanedione, a well-known neurotoxin, as shown in Scheme 2. Transformation of the pyrrole in B8 to the amine B9 uses hydroxylamine which is a mutagenic and thermally unstable compound that is dangerous to use in large quantities. The overall process described in WO 2011/113894 requires many protecting group manipulations that lead to a low atom economy and afford a lot of waste. Thus there is a need for an improved synthetic process for making (S)-3-amino-6-methoxy-N-(3,3,3-trifluoro-2-hydroxy-2-methylpropyl)-5-(trifluoromethyl)picolinamide.
5-bromo-2-methoxy-3-(trifluoromethyl)pyridine (III) (1.4 kg, 5.47 mol), tetramethyl ethylene diamine (TMEDA) (1.75 kg, 15 mol) and tetrahydrofuran (THF) (10kg) were charged to a dry and inert reactor. At -25°C a solution of 2,2,6,6-tetramethyl-piperidinylmagnesium chloride lithiumchloride complex, 1 M in THF/toluene (TMPMgCl.LiCl)(14.5 kg, 15 mol) was slowly added. After stirring the reaction mixture for 30 min., CO2 gas was carefully bubbled into the reactor so that the temperature of the exothermic reaction did not exceed -20°C. The reaction mixture was then quenched onto a mixture of t-butyl methyl ether (TBME) and 5% aq. H2SO4 (50 kg). The biphasic mixture was separated and the organic phase was extracted with 2M NaOH solution. The aqueous phase was acidified to pH 1-2 with 5% aq. H2SO4 and extracted with TBME. After a distillative solvent change to cyclohexane the product was crystallized from cyclohexane to yield 1.1 kg 3-bromo-6-methoxy-5-(trifluoromethyl)picolinic acid (65% yield).
HRMS: [M-H]- expected C8H4BrF3NO3, 297.9405; found C8H4BrF3NO3, 297.9337
Example 2: Methyl 3-bromo-6-methoxy-5-(trifluoromethyl)picolinate
5-bromo-2-methoxy-3-(trifluoromethyl)pyridine (III) (5.0 g, 19.53 mmol) was added to a 100 ml reactor followed by toluene (20 ml) and dimethylcarbonate (17.59 g, 195.30 mmol). To the stirred solution at 20 °C was slowly added 2,2,6, 6-tetramethyl-piperidinylmagnesium chloride lithium chloride complex as a 1 M solution in THF/toluene (27.34 ml, 27.34 mmol) within 45 minutes. A sample was taken and diluted in acetic acid for HPLC analysis in order to confirm full conversion of II to the methylester. Within the same vessel 5% aq. H2SO4 (36 ml) was slowly added to the reaction mixture until a pH below 2 was obtained (caution, exothermic). The biphasic mixture was separated and the lower aqueous phase back-extracted with toluene (10 ml).
In order to isolate the methylester the organic phases were combined and concentrated by rotary evaporation to yield a residue which was chromatographed on reverse-phase silica to yield the final product: methyl 3-bromo-6-methoxy-5-(trifluoromethyl)picolinate as a yellow solid, 5.3 g, 86 % yield. The solid was optionally recrystallized from methanol and water to further increase purity.
HRMS: MH+ expected C9H8BrF3NO3, 313.9561 ; found C9H8BrF3NO3, 313.9634
HPLC Conditions:
HPLC: Column : Agilent Zorbax SB-C18 (150 mm x 3.0 mm, particle size 3.5 urn)
Eluent A : Water / TFA = 1000/1 (v/v)
Eluent B: Acetonitrile / TFA = 1000/1 (v/v)
Wavelength : 230 nm
Flow-rate : 0.8 ml/min
Gradient: eluent B: 45% to 90% over 9 mins
Retention time 3-bromo-6-methoxy-5-(trifluoromethyl)picolinate: 5.80 min
Alternative synthesis for 3-bromo-6-methoxy-5-(trifluoromethyl)picolinic acid:
Isolation of Example 1
In order to proceed to Example 1 without the isolation of VII, the work-up continues from the combined toluene phases post-H2SO4 quench as follows:
To the combined organic phases was slowly added 50% aq. sodium hydroxide (30 ml) until a pH of above 10 was obtained. The reaction mixture was heated to 35 °C and after 15 mins addition of water (30 ml) followed by 30 mins further stirring preceded sample-taking to ensure full hydrolysis of the methylester to Example 1 by HPLC. Water was added (130 ml), followed by TBME (60 ml) and the phases separated. To the aqueous phase was cautiously added concentrated H2SO4 (30 g) until a pH of below 2.5 was obtained (caution, exothermic and release of CO2 causes foaming). TBME (100 ml) was added and the phases separated. The organic phase contained the C2, and could be evaporated to dryness by rotary evaporation to confirm the yield, 5.4 g C2, 92 % yield.
HRMS: M-H- expected C8H4BrF3NO3, 297.9405; found C8H4BrF3NO3, 297.9333
For HPLC method details see above. Retention time C2: 2.94 min
Alternative synthesis for ethyl 3-bromo-6-methoxy-5-(trifluoromethyl)picolinate:
5-bromo-2-methoxy-3-(trifluoromethyl)pyridine (III) (0.5 g, 1.95 mmol) was added to a reactor followed by THF (2 ml) and the solution cooled to 0 °C. To the mixture was added 2,2,6,6-tetramethyl-piperidinylmagnesium chloride lithium chloride complex as a 1 M solution in THF/toluene (4.88 ml, 3.91 mmol), and the mixture was left to stir for 15 minutes at 0 °C. An aliquot of the solution (50 ul) was then added to a reactor containing diethylcarbonate (20 ul, 19.5 mmol). A second aliquot (50 ul) was taken of the metallated II and added to a reactor containing ethyl chloroformate (14 ul, 19.5 mmol). After 2 minutes both reactors were quenched with a 1 :1 mixture of acetonitrile/HCl (1 M). The reaction with diethylcarbonate gave 56 A% of ethyl 3-bromo-6-methoxy-5-(trifluoromethyl)picolinate and the reaction with ethyl chloroformate gave 68 A% of ethyl 3-bromo-6-methoxy-5-(trifluoromethyl)picolinate product according to the HPLC method described above.
Example 3: Synthesis of (S)-3-amino-6-methoxy-N-(3,3,3-trifluoro-2-hydroxy-2-methylpropyl)-5-(trifluoromethyl)picolinamide
Step 1: 3-bromo-6-methoxy-5-(trifluoromethyl)picolinic acid (1.3 kg, 4.33 mol) and
copper(II)sulfate pentahydrate (0.108 kg, 0.433 mol) were charged into an inert autoclave
followed by aqueous ammonia 25% (12 kg). The mixture was stirred and heated up to 100 °C, whereby a pressure of 7 bar resulted. The solution was stirred for 2 hr and then cooled down to
5 °C. Sulfuric acid (8 M) was dosed upon cooling, so that a temperature range of 5 °C to 30 °C was held until a pH of about 5 was reached. Isopropylacetate was added and the pH was
further adjusted to 1-2. The phases were separated and the organic phase was azeotropically dried by partial distillation. n-Heptane was added and the mixture stirred for 15 hr at 20 °C
during which the product crystallized out. After filtration and drying 3-amino-6-methoxy-5-(trifluoromethyl)picolinic acid was obtained as a yellow solid (0.92 kg, 90%).
13C NMR (101 MHz, DMSO-d6): δ ppm 53.59, 116.76 m, 123.27, 126.36-117.40 m, 128.04, 142.56, 148.65, 167.62
Step 2: 3-amino-6-methoxy-5-(trifluoromethyl) picolinic acid (20 g, 84.7 mmol) and HATU (38.6 g, 101.6 mmol) were charged to a reactor followed by a solution of (S)-3-amino-1 ,1 ,1-trifluoro-2- methylpropan-2-ol in isopropylacetate (7 %, 188 g, 93 mmol). The solution was stirred at room temperature, diisopropyl ethyl amine (21.9 g, 169 mmol) was added and stirring was continued for at least 16h at 25 °C. Water (250 ml) was then added dropwise within 15 min. keeping the temperature below 25 °C. The water phase was separated and the organic phase was extracted with 5% aqueous HCl , 5% potassium carbonate solution, and water. The organic layer was concentrated to about 60% solution. At 50 °C n-heptane (41 g) was added and the solution was cooled by a linear ramp to 5 °C while adding more n-heptane (131 g). The precipitate was filtered off and dried at 50 °C resulting in a yellow to beige product (S)-3-amino-6-methoxy-N- (3,3,3-trifluoro-2-hydroxy-2-methylpropyl)-5-(trifluoromethyl)picolinamide (21.1 g, 69 % yield).
13C NMR (101 MHz, DMSO-d6): δ ppm 18.92, 42.15, 53.52, 72.40, 115.5-116.5 m, 118-126 m, 122-130.7 m, 124.82, 128.3 m, 140.95, 148.49, 166.27
Example 4: Telescoped process for the synthesis of the HCl salt of 3-amino-6-methoxy- 5-(trifluoromethyl)picolinic acid (V)
1 Equivalent* of (III) and 6 equivalents of dimethyl carbonate (DMC) were dissolved in 3.5 parts** of toluene at room temperature. To this solution 1.5 equivalent of TMPMgCl.LiCl solution in THF was added at 15-25°C within ca. 1 h. Tert butyl methyl ether (MTBE, 5.9 parts) was added and the mixture was quenched in 7.3 parts of 10% sulfuric acid at 25-40°C. The water phase was discarded and to the organic phase 6.2 parts of 30% sodium hydroxide solution were added. The mixture was stirred well at 40°C for 1-2h. After the successful conversion of (VIII) to (IV), 2.5 parts of water were added to dissolve the partially precipitated sodium carbonate. The water phase was discarded and the organic phase was cooled to 20°C and extracted with 4.8 parts of 25% aqueous ammonia. The aqueous phase was transferred in an autoclave and 0.0979 parts (10mol%) of copper sulfate pentahydrate were added. The autoclave was well inertized by a pressure method and heated up to 100°C, while the pressure raises up to ca. 8 bar absolute pressure. After the successful conversion of (IV) to (V), the green solution was added to a mixture of 3.7 parts of MTBE and 6.8 parts of 50% sulfuric acid resulting in a biphasic solution of pH 1-2. The water phase was separated and the organic phase washed two times with 2.5 parts of water each. The organic phase was dried by distillation at JT 50°C/400mbar while 3.7 parts of MTBE were added/replaced. To the dried organic solution 0.41 parts of HCl gas was dosed at 0-5°C under or over solvent level. The suspension was stirred for ca.1 h, then filtered off and washed with 48 parts of TBME. The product was dried at 40°C/20 mbar for ca. 12h. (yield from (III): 72%, slightly beige solid).
*equivalents are based on the molar amount of the starting material (III) = 1 equivalent
13C NMR (101 MHz, DMSO-d6): δ ppm 53.59, 116.76 m, 123.27, 126.36-117.40 m, 128.04, 142.56, 148.65, 167.62
Example 5: Alternative synthesis of (S)-3-amino-6-methoxy-N-(3,3,3-trifluoro-2-hydroxy-2-methylpropyl)-5-(trifluoromethyl)picolinamide
Step 1 : (VIII) (1.0 g), (S)-3-amino-1 ,1 ,1-trifluoro-2-methylpropan-2-ol as mandellic acid salt (1.128 g, 1.2 eq.) and 2,3,4,6, 7, 8-hexahydro-1H-pyrimido[1,2-a]pyrimidine (TBD, 0.588 g, 1.3 eq.) were added to a pre-dried flask as solids. To this was added the anhydrous THF (10 ml) and the cloudy solution heated to 55 °C. Sampling and analytical determination of purity at 2.5 hrs confirmed 88 A% product upon which water (10 ml) was added and the phases separated. The organic phase was distilled to a concentrated mixture upon which toluene (20 ml) was added. The organic layer was extracted with 10% aq. citric acid (10 ml) followed by three consecutive extractions with 1 M aq. NaOH. The organic phase was then dried with magnesium sulfate and evaporated to dryness to give 1.196 g of (S)-3-bromo-6-methoxy-N-(3,3,3-trifluoro- 2-hydroxy-2-methylpropyl)-5-(trifluoromethyl)picolinamide (IX) as a white solid (95 A%, 88% yield).
HRMS: MH+ expected C12H12BrF6N2O3, 424.9857; found C12H12BrF6N2O3, 424.9931
HPLC (method described above): retention time = 4.94 min
Step 2: IX (79 mg, 0.186 mmol) was combined with copper(II)sulfate pentahydrate (4.6 mg, 0.019 mmol), methanol (0.6 ml) and 23% aqueous ammonium hydroxide solution (559 ul) within a glass microwave vial. The headspace was inertized with nitrogen, then the vial sealed and placed in the microwave unit for heating to 105 °C for 7.5 hrs. Isopropylacetate (5 ml) was added to the deep green reaction mixture and a solvent-switch brought about by rotary evaporation. To the mixture now in water and isopropyl acetate was added 8M H2SO4 (5 ml), the phases mixed and then left to separate. The aqueous phase was further extracted with isopropylacetate and the combined organic phases washed with aq. NaCl (5 ml). The organic phase was dried over MgSO4 and evaporated to yield of a yellow residue, 66 mg.
A portion of the residue (16 mg) was re-dissolved in heptane / ethyl acetate and submitted for combiflash purification (n-heptane / ethyl acetate gradient, elution at 20% ethyl acetate) providing (S)-3-amino-6-methoxy-N-(3,3,3-trifluoro-2-hydroxy-2-methylpropyl)-5-(trifluoromethyl)picolinamide (VII) as a residue on evaporation in 91 A% purity containing trace residual solvents (17 mg, corrected to 13 mg by 1H NMR, 80 % yield back-calculated).
Examples 4, 5 and 6: 3-Amino-6-methoxy-5-trifluoromethyl-pyridine-2-carboxylic acid (3,3,3-trifluoro-2-hydroxy-2-methyl-propyl)-amide and its enantiomers
Example 4: 3-Amino-6-methoxy-5-trifluoromethyl-pyridine-2-carboxylic acid (3,3,3-trifluoro-2-hydroxy-2-methyl-propyl)-amide,
was prepared according to the following procedure:
A solution comprising 3-amino-6-methoxy-5-trifluoromethyl-pyridine-2-carboxylic acid (Intermediate D)(4 g, 16.94 mmol) and 3-amino-1,1,1-trifluoro-2-methylpropan-2-ol hydrochloride (Intermediate R) (3.04 g, 16.94 mmol) in NMP (188 ml) was treated with HATU (7.73 g, 20.33 mmol) followed by dropwise addition (2 ml portions) of DIPEA (8.88 ml, 50.8 mmol) over 1 hour. After stirring for a further hour, the reaction mixture was poured into water (450 ml) and EtOAc (450 ml). The aqueous phase was acidified with 5M HCl (50 ml) and the layers were separated. The organic portion was washed with 2M NaOH (200 ml), water (4×200 ml), brine (2×100 ml), dried over MgSO 4, filtered and concentrated in vacuo to afford a brown solid. Purification of the solid by chromatography on silica (220 g pre-packed silica cartridge) eluting with 0-50% EtOAc in iso-hexane afforded the racemate, 3-amino-6-methoxy-5-trifluoromethyl-pyridine-2-carboxylic acid (3,3,3-trifluoro-2-hydroxy-2-methyl-propyl)-amide (Ex. 4) as a yellow solid;
Chiral separation of the racemate by Supercritical Fluid Chromatography was carried out using the following conditions to afford the compounds listed hereinafter:
Mobile Phase: 12% 2-propanol+0.1% DEA/50% CO 2
Column: Chiralcel OD-H, 250×10 mm id, 5 μm (2 columns linked in series)
Detection: UV @ 220 nm
Flow rate: 10 ml/min
Sample concentration: 3.5 g in 30 ml EtOH
Injection volume: 100 μl
Examples 5 and 6 are Entantiomers
Example 5: First eluted peak Rt=7.30 minutes. 3-Amino-6-methoxy-5-trifluoromethyl-pyridin e-2-carboxylic acid ((S)-3,3,3-trifluoro-2-hydroxy-2-methyl-propyl)-amide (“Compound A”):
Optical rotation [α] 21D at 589 nm −20.83° (c=0.513, MeOH).
The stereochemistry of this compound was confirmed by X-ray crystallography.
Example 6: Second eluted peak Rt=8.29 minutes. 3-Amino-6-methoxy-5-trifluoromethyl-pyridine-2-carboxylic acid ((R)-3,3,3-trifluoro-2-hydroxy-2-methyl-propyl)-amide
LC-MS Rt=1.15 mins [M+H]+ 362.4 (Method 2 minLC_v003).
Alternatively, Example 5 may be prepared according to the following method: To a solution of 3-amino-6-methoxy-5-trifluoromethyl-pyridine-2-carboxylic acid (Intermediate D) (10 g, 42.3 mmol) and (S)-3-amino-1,1,1-trifluoro-2-methylpropan-2-ol hydrochloride (Intermediate RA)(7.60 g, 42.3 mmol) in NMP (400 ml) was added HATU (19.3 g, 50.8 mmol) followed by dropwise addition of DIPEA (22.19 ml, 127 mmol) over ˜1 hr. After stirring at room temperature for 30 min, the mixture was added to EtOAc (2 L), washed with 1M NaOH (2×1 L), water (1 L), brine (1 L), dried (MgSO 4) and evaporated under reduced pressure to give the crude product as a dark brown oil. Purification by chromatography on silica eluting with a gradient of 1 to-25% of EtOAc in iso-hexane afforded a yellow oil. Recrystallisation of the oil from iso-hexane/DCM afforded 3-amino-6-methoxy-5-trifluoromethyl-pyridine-2-carboxylic acid ((S)-3,3,3-trifluoro-2-hydroxy-2-methyl-propyl)-amide as a crystalline solid;
Mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) ion channel are established as the primary causative factor in the devastating lung disease cystic fibrosis (CF). More recently, cigarette smoke exposure has been shown to be associated with dysfunctional airway epithelial ion transport, suggesting a role for CFTR in the pathogenesis of chronic obstructive pulmonary disease (COPD). Here, the identification and characterization of a high throughput screening hit 6 as a potentiator of mutant human F508del and wild-type CFTR channels is reported. The design, synthesis, and biological evaluation of compounds 7–33 to establish structure–activity relationships of the scaffold are described, leading to the identification of clinical development compound icenticaftor (QBW251) 33, which has subsequently progressed to deliver two positive clinical proofs of concept in patients with CF and COPD and is now being further developed as a novel therapeutic approach for COPD patients.
a Reagents and conditions: (i) aq NaOH, THF, RT, 97%; (ii) aq Me2NH or MeNH2, THF, RT, 56−92%; (iii) 41, HATU, Et3N, NMP, RT, 52− 78%; (iv) NH2OH·HCl, Et3N, EtOH−water, reflux, then chiral HPLC, 34−36%; (v) aq NaOH, MeOH, 60°C, 97%; (vi) cat H2SO4, MeOH, reflux, 75%; (vii) TMSCl, KI, MeCN, reflux, 54%; (viii) EtOH, DEAD, Ph3P, dioxane, RT, 61%; (ix) aq NaOH, THF, reflux, 26%; (x) (S)-41, HATU, DIPEA, DMF, RT, 89%; (xi) NH2OH·HCl, Et3N, EtOH−water, reflux, 37−53%; (xii) (S)-41, HATU, DIPEA, NMP, RT, 59%.
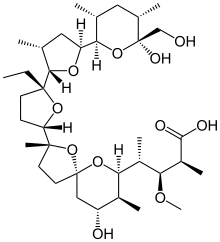
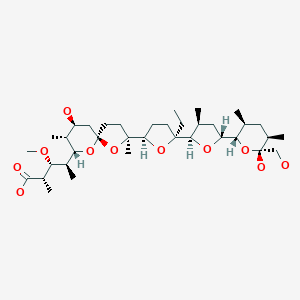









 verify (what is ?)Infobox references
verify (what is ?)Infobox references

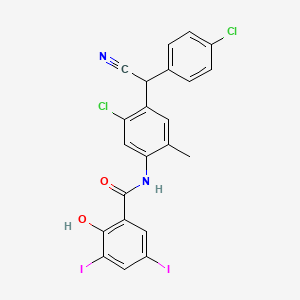




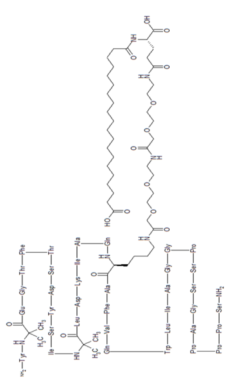







 (tirzepatide) injection, the first and only GIP and GLP-1 receptor agonist for the treatment of adults with type 2 diabetes
(tirzepatide) injection, the first and only GIP and GLP-1 receptor agonist for the treatment of adults with type 2 diabetes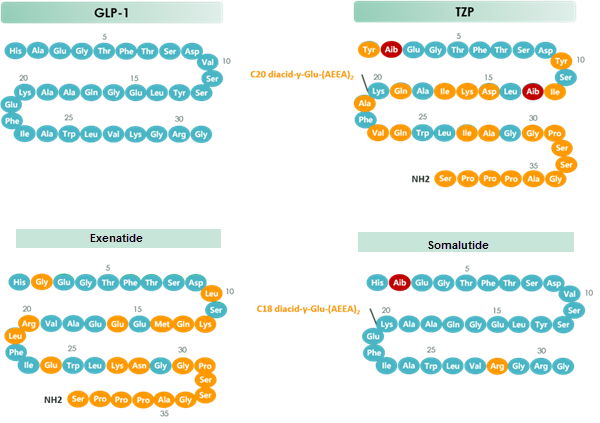

![7-Benzyl-4-(2-methylbenzyl)-1,2,6,7,8,9-hexahydroimidazo[1,2-A]pyrido[3,4-E]pyrimidin-5(4H)-one.png](http://pubchem.ncbi.nlm.nih.gov/image/imgsrv.fcgi?cid=73777259&t=l)
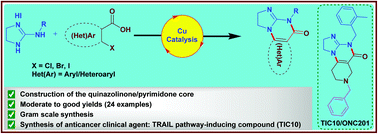












![[thin space (1/6-em)]](http://www.rsc.org/images/entities/char_2009.gif) :
:




















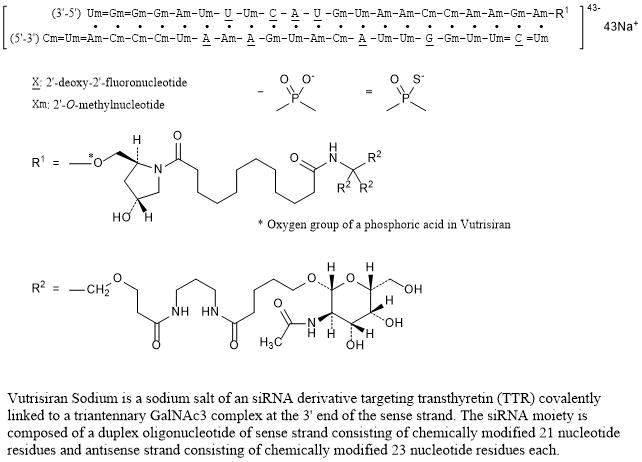
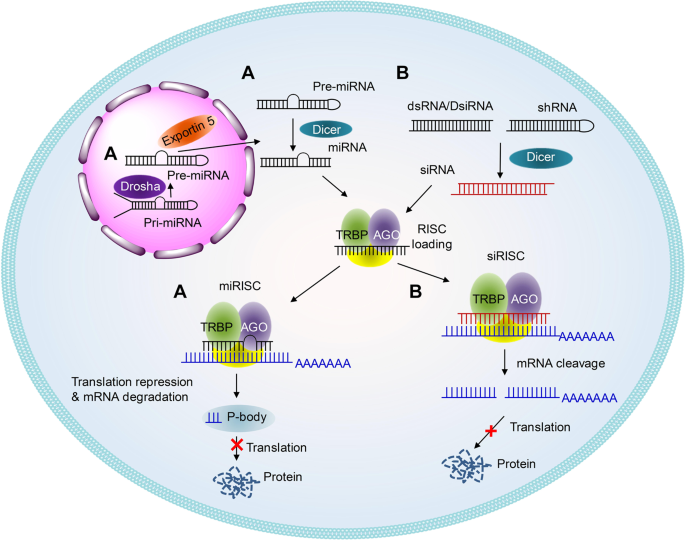
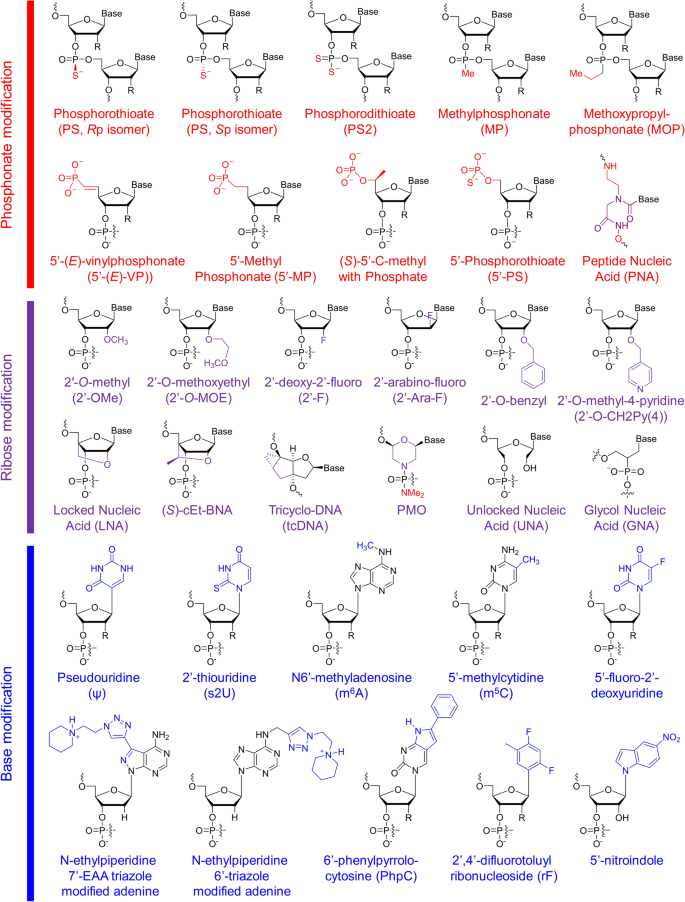
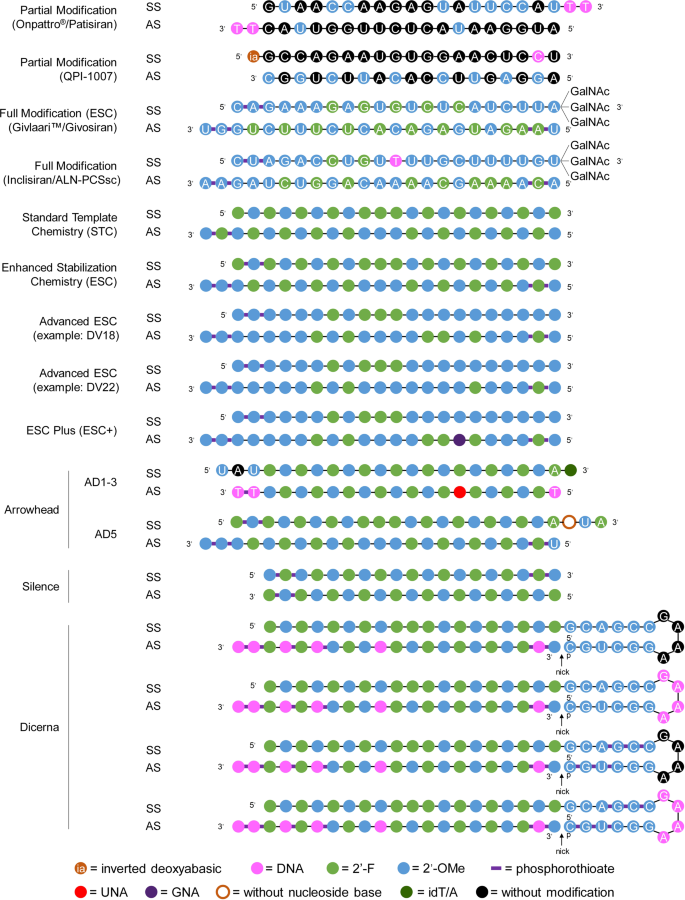
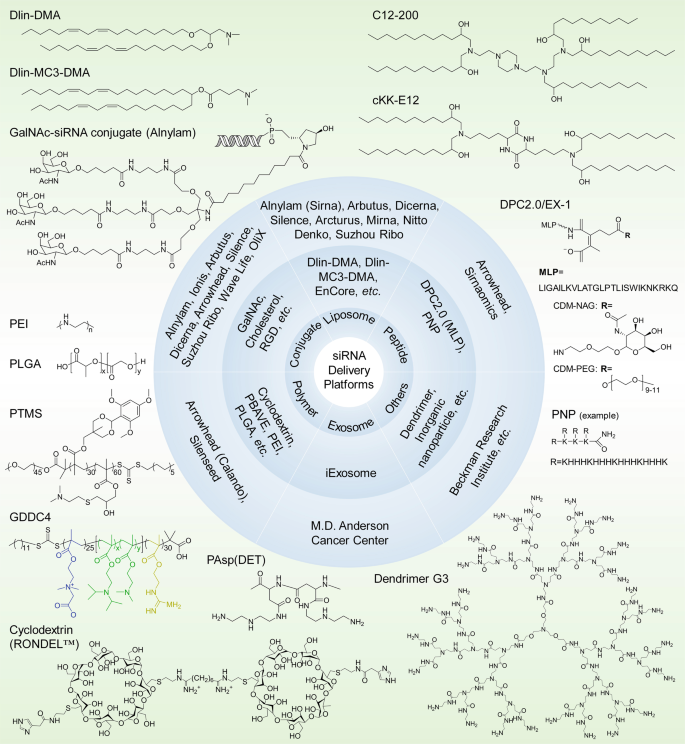

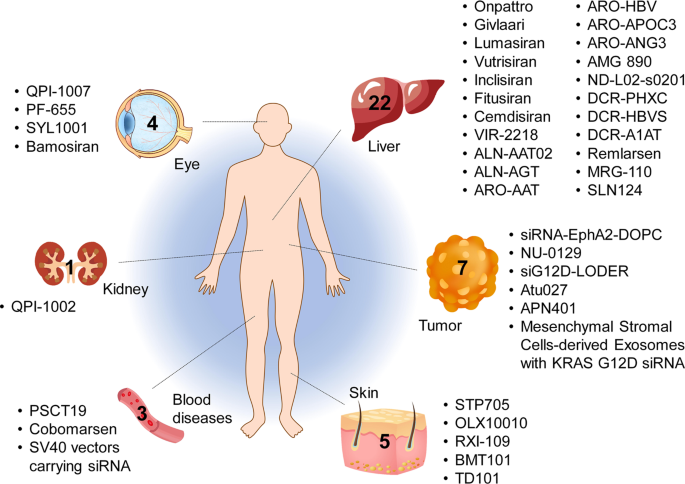







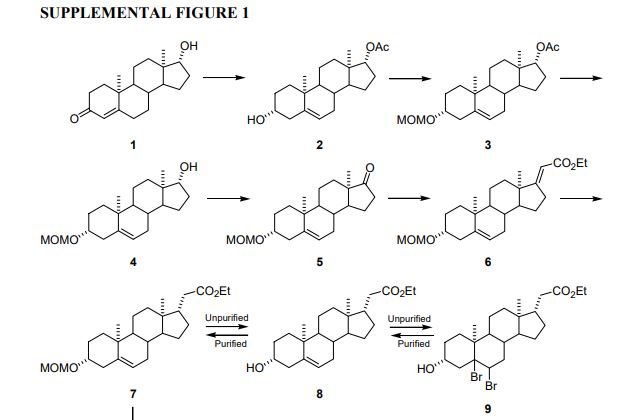







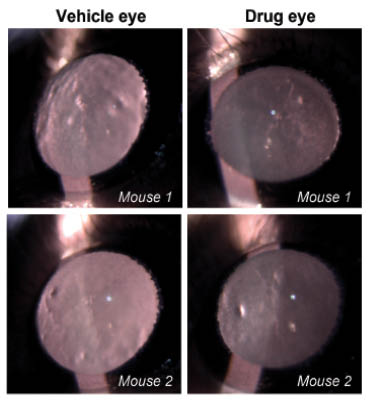
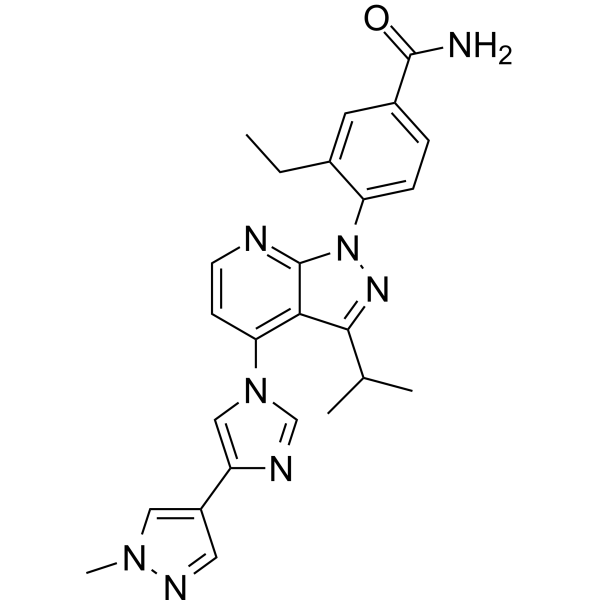
![Benzamide, 3-ethyl-4-[3-(1-methylethyl)-4-[4-(1-methyl-1H-pyrazol-4-yl)-1H-imidazol-1-yl]-1H-pyrazolo[3,4-b]pyridin-1-yl]-.png](http://pubchem.ncbi.nlm.nih.gov/image/imgsrv.fcgi?cid=67501411&t=l)