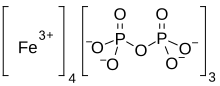
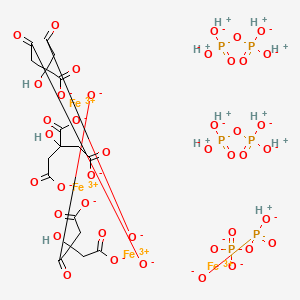

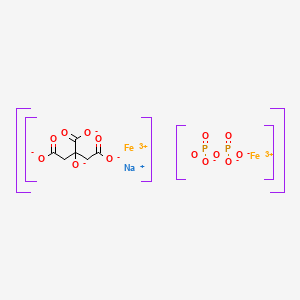

Ferric pyrophosphate citrate
1802359-96-1
tetrairon(3+) bis((phosphonooxy)phosphonic acid) tris(2-hydroxypropane-1,2,3-tricarboxylate) (hydrogen phosphonooxy)phosphonate
Iron(3+) diphosphate (4:3)
Proper name: ferric pyrophosphate citrate Chemical names: Iron (3+) cation; 2-oxidopropane-1,2,3-tricarboxylate; diphosphate 1,2,3-propanetricarboxylic acid, 2-hydroxy-, iron (3+), diphosphate Molecular formula: [Fe4 3+(C6H5O7)3(P2O7)3] Molecular mass: 1313
Physicochemical properties: TRIFERIC AVNU (ferric pyrophosphate citrate) contains no asymmetric centers. Ferric pyrophosphate citrate is a yellow to green amorphous powder. The drug substance does not melt, or change state, below 300 °C. Thermal decomposition was observed at 263 ± 3ºC. Ferric pyrophosphate citrate is freely soluble in water (>100 g/L). Ferric pyrophosphate citrate is completely insoluble in most organic solvents (MeOH, Acetone, THF, DMF, DMSO). A 5% solution in water exhibits a solution pH of about 6. … https://pdf.hres.ca/dpd_pm/00060816.PDF
- Ferric pyrophosphate citrate
- FPC
- SFP
- Tetraferric nonahydrogen citrate pyrophosphate
- Triferic
Active Moieties
| NAME | KIND | UNII | CAS | INCHI KEY |
|---|---|---|---|---|
| Ferric cation | ionic | 91O4LML611 | 20074-52-6 | VTLYFUHAOXGGBS-UHFFFAOYSA-N |
CANADA

Summary Basis of Decision – Triferic AVNU – Health Canada
Date SBD issued:2021-07-29
The following information relates to the new drug submission for Triferic AVNU.
Iron (supplied as ferric pyrophosphate citrate)
Drug Identification Number (DIN):
DIN 02515334 – 1.5 mg/mL iron (supplied as ferric pyrophosphate citrate), solution, intravenous administration
Rockwell Medical Inc.
New Drug Submission Control Number: 239850
On April 22, 2021, Health Canada issued a Notice of Compliance to Rockwell Medical Inc. for the drug product Triferic AVNU.
The market authorization was based on quality (chemistry and manufacturing), non-clinical (pharmacology and toxicology), and clinical (pharmacology, safety, and efficacy) information submitted. Based on Health Canada’s review, the benefit-harm-uncertainty profile of Triferic AVNU is favourable for the replacement of iron to maintain hemoglobin in adult patients with hemodialysis-dependent chronic kidney disease (CKD-HD). Triferic AVNU is not intended for use in patients receiving peritoneal dialysis and has not been studied in patients receiving home hemodialysis.
Triferic AVNU, an iron preparation, was authorized for the replacement of iron to maintain hemoglobin in adult patients with hemodialysis-dependent chronic kidney disease (CKD-HD). Triferic AVNU is not intended for use in patients receiving peritoneal dialysis and has not been studied in patients receiving home hemodialysis.
Triferic AVNU is not authorized for use in pediatric patients (<18 years of age), as its safety and effectiveness have not been established in this population. No overall differences in efficacy or safety were observed in geriatric patients (≥65 years of age) compared to younger patients in clinical trials.
Triferic AVNU is contraindicated for patients who are hypersensitive to this drug or to any ingredient in the formulation, or component of the container.
Triferic AVNU was approved for use under the conditions stated in its Product Monograph taking into consideration the potential risks associated with the administration of this drug product.
Triferic AVNU (1.5 mg/mL iron [supplied as ferric pyrophosphate citrate]) is presented as a solution. In addition to the medicinal ingredient, the solution contains water for injection.
For more information, refer to the Clinical, Non-clinical, and Quality (Chemistry and Manufacturing) Basis for Decision sections.
Additional information may be found in the Triferic AVNU Product Monograph, approved by Health Canada and available through the Drug Product Database.
Health Canada considers that the benefit-harm-uncertainty profile of Triferic AVNU is favourable for the replacement of iron to maintain hemoglobin in adult patients with hemodialysis-dependent chronic kidney disease (CKD-HD). Triferic AVNU is not intended for use in patients receiving peritoneal dialysis and has not been studied in patients receiving home hemodialysis.
Chronic kidney disease (CKD) is a worldwide public health concern. One of the most common comorbidities of CKD-HD patients is anemia, which may be due to low body iron stores (as a result of blood loss during dialysis) and impaired utilization of iron. Consequently, there is an ongoing need to replenish body iron in CKD-HD patients.
Iron deficiency anemia in CKD-HD patients is generally treated using parenteral (intravenous) iron administration used in conjunction with erythropoiesis stimulating agents (ESAs). Intravenous administration is preferred, as oral iron is not well absorbed and gastrointestinal intolerance is common. At the time of authorization of Triferic AVNU, there were four other intravenous iron products marketed in Canada: Dexiron, an iron dextran (≥1,000 mg/dose); Ferrlecit (sodium ferric gluconate; 125 mg/dose); Venofer (iron sucrose; 200 mg/dose); and the more recently approved Monoferric (Iron Isomaltoside 1,000; up to 500 mg/bolus injection and up to 1,500 mg/infusion). Each of these intravenous iron products are indicated for the treatment of iron deficiency anemia and are associated with safety concerns for hypersensitivity reactions. Serious hypersensitivity reactions have been reported, including life threatening and fatal anaphylactic/anaphylactoid reactions.
Triferic AVNU is an iron replacement product delivered via intravenous infusion into the blood lines pre- and post-dialyzer in CKD-HD patients at each hemodialysis treatment. It is a preservative-free sterile solution containing 1.5 mg elemental iron/mL in water for injection.
Triferic AVNU has been shown to be efficacious in maintaining hemoglobin (Hb) during the treatment period in CKD-HD patients. The market authorization was primarily based on the results of two pivotal, randomized, placebo-controlled, single blind, Phase III clinical studies (Studies SFP-4 and SFP-5). Both studies were identical in design and enrolled a combined total of 599 adult patients with CKD-HD who were iron-replete. Patients were randomized to receive either Triferic AVNU added to bicarbonate concentrate with a final concentration of 110 μg of iron/L in dialysate or placebo (standard dialysate) administered 3 to 4 times per week during hemodialysis. All patients were to remain randomized in their treatment group until pre-specified Hb or ferritin criteria were met, indicating the need for a change in anemia management, or until they had completed 48 weeks of treatment. After randomization, patients’ ESA product, doses, or route of administration were not to be changed and oral or intravenous iron administration were not allowed.
The primary efficacy endpoint (mean change in Hb level from baseline to the end-of-treatment period) was met in both pivotal studies. In Study SFP-4, the mean Hb decreased 0.04 g/dL in the Triferic AVNU group compared to 0.39 g/dL in the placebo group. In Study SFP-5, the mean Hb decreased 0.09 g/dL in the Triferic AVNU group compared to 0.45 g/dL in the placebo group. In both studies, the treatment difference in mean hemoglobin change was 0.36 g/dL (p = 0.011) between the Triferic AVNU and the placebo groups. This value was statistically significant for both studies. The treatment difference of 0.35 g/dL was also statistically significant (p = 0.010) for both studies in the analysis using the intent-to-treat population. A high proportion of patients did not complete the planned 48 weeks of study treatment mainly due to protocol-mandated changes in anemia management (ESA dose changes). However, the proportion was similar for both arms and the analysis of Hb change in this subgroup was consistent with that of the primary efficacy analysis. Secondary endpoints which included changes in reticulocyte Hb content, serum ferritin, and pre-dialysis serum iron panel to the end of treatment, were consistent with the primary efficacy results.
The safety of Triferic AVNU was evaluated in seven controlled and uncontrolled Phase II/III studies, which included the two pivotal studies. In total, 1,411 CKD-HD patients were exposed to Triferic AVNU in the clinical program. In the pivotal studies, 78% of patients in the Triferic AVNU group and 75% of patients in the placebo group had at least one treatment-emergent adverse event (TEAE). The most common TEAEs in the Triferic AVNU group (which were higher than the placebo group) were procedural hypotension (21.6%), muscle spasms (9.6%), headache (9.2%), pain in extremity (6.8%), edema peripheral (6.8%) and dyspnoea (5.8%). Serious TEAEs were reported at similar rates for the two groups at 27.7% for the Triferic AVNU group and 27.4% for the placebo group. The most common serious TEAEs occurring in the Triferic AVNU group (which were higher than the placebo group) were cardiac arrest (1.7%), arteriovenous fistula thrombosis (1.7%), and pulmonary edema (1.4%). Few patients discontinued study treatment due to TEAEs (4.5% in the Triferic AVNU group and 2.4% in the placebo group).
In the overall clinical program, there were two cases (0.1%) of hypersensitivity reactions related to treatment out of the 1,411 patients treated with Triferic AVNU. There were no cases of serious hypersensitivity reaction and no cases of anaphylaxis related to Triferic AVNU treatment. A Serious Warnings and Precautions box describing a warning for hypersensitivity reaction has been included in the Product Monograph for Triferic AVNU.
A Risk Management Plan (RMP) for Triferic AVNU was submitted by Rockwell Medical Inc. to Health Canada. The RMP is designed to describe known and potential safety issues, to present the monitoring scheme and when needed, to describe measures that will be put in place to minimize risks associated with the product. In the RMP, the sponsor included ‘hypersensitivity reactions’ as an important identified risk; ‘systemic/serious infections’ as an important potential risk; and ‘use in pregnant and breastfeeding women’, ‘use in children’ and ‘concomitant use with other intravenous iron product’ as missing information. Labelling for these safety concerns has been included in the Product Monograph and the sponsor has committed to systemically review clinical and post-marketing safety data as part of routine pharmacovigilance activities. Upon review, the RMP was considered to be acceptable.
The submitted inner and outer labels, package insert and Patient Medication Information section of the Triferic AVNU Product Monograph meet the necessary regulatory labelling, plain language and design element requirements.
A review of the submitted brand name assessment, including testing for look-alike sound-alike attributes, was conducted and the proposed name Triferic AVNU was accepted.
Overall, the therapeutic benefits of Triferic AVNU therapy seen in the pivotal studies are positive and are considered to outweigh the potential risks. Triferic AVNU has an acceptable safety profile based on the non-clinical data and clinical studies. The identified safety issues can be managed through labelling and adequate monitoring. Appropriate warnings and precautions are in place in the Triferic AVNU Product Monograph to address the identified safety concerns.
This New Drug Submission complies with the requirements of sections C.08.002 and C.08.005.1 and therefore Health Canada has granted the Notice of Compliance pursuant to section C.08.004 of the Food and Drug Regulations. For more information, refer to the Clinical, Non-clinical, and Quality (Chemistry and Manufacturing) Basis for Decision sections.
- See MedEffect Canada for the latest advisories, warnings and recalls for marketed products.
- See the Notice of Compliance (NOC) Database for a listing of the authorization dates for all drugs that have been issued an NOC since 1994.
- See the Drug Product Database (DPD) for the most recent Product Monograph. The DPD contains product-specific information on drugs that have been approved for use in Canada.
- See the Notice of Compliance with Conditions (NOC/c)-related documents for the latest fact sheets and notices for products which were issued an NOC under the Notice of Compliance with Conditions (NOC/c) Guidance Document, if applicable. Clicking on a product name links to (as applicable) the Fact Sheet, Qualifying Notice, and Dear Health Care Professional Letter.
- See the Patent Register for patents associated with medicinal ingredients, if applicable.
- See the Register of Innovative Drugs for a list of drugs that are eligible for data protection under C.08.004.1 of the Food and Drug Regulations, if applicable.
The Chemistry and Manufacturing information submitted for Triferic AVNU has demonstrated that the drug substance and drug product can be consistently manufactured to meet the approved specifications. Proper development and validation studies were conducted, and adequate controls are in place for the commercial processes. Changes to the manufacturing process and formulation made throughout the pharmaceutical development are considered acceptable upon review. Based on the stability data submitted, the proposed shelf life of 36 months is acceptable when the drug product is stored protected from light in the aluminum pouch at room temperature (15 ºC to 30 ºC).
Proposed limits of drug-related impurities are considered adequately qualified (i.e. within International Council for Harmonisation [ICH] limits and/or qualified from toxicological studies).
All sites involved in production are compliant with Good Manufacturing Practices.
None of the excipients used in the formulation of Triferic AVNU are of human or animal origin. All non-medicinal ingredients (described earlier) found in the drug product are acceptable for use in drugs according to the Food and Drug Regulations.
DIN:
02515334
Product Monograph/Veterinary Labelling:
Date: 2021-04-21 Product monograph/Veterinary Labelling (PDF version ~ 175K)
Company:
ROCKWELL MEDICAL INC
30142 S Wixom Rd
Wixom
Michigan
United States 48393
Class:
Human
Dosage form(s):
Solution
Route(s) of administration:
Intravenous
Number of active ingredient(s):
1
Schedule(s):
Prescription
Biosimilar Biologic Drug:
No
American Hospital Formulary Service (AHFS):See footnote3
20:04.04 IRON PREPARATIONS
Anatomical Therapeutic Chemical (ATC):See footnote4
B03AC IRON, PARENTERAL PREPARATIONS
Active ingredient group (AIG) number:See footnote5
0108536041
| Active ingredient(s) | Strength |
|---|---|
| IRON (FERRIC PYROPHOSPHATE CITRATE) | 1.5 MG / ML |
RXLIST
TRIFERIC®
(ferric pyrophosphate citrate) Solution, for Hemodialysis Use
TRIFERIC®
(ferric pyrophosphate citrate) powder packet for hemodialysis use
DESCRIPTION
Triferic (ferric pyrophosphate citrate) solution, an iron replacement product, is a mixed-ligand iron complex in which iron (III) is bound to pyrophosphate and citrate. It has a molecular formula of Fe4(C6H4O7)3(H2P2O7)2(P2O7) and a relative molecular weight of approximately 1313 daltons. Ferric pyrophosphate citrate has the following structure:
|
Triferic Solution
Triferic (ferric pyrophosphate citrate) solution–is a clear, slightly yellow-green color sterile solution containing 27.2 mg of elemental iron (III) per 5 mL (5.44 mg iron (III) per mL) filled in a 5 mL or 272 mg of elemental iron (III) per 50 mL (5.44 mg iron (III) per mL) filled in a 50 Ml low density polyethylene (LDPE) ampule. Each Triferic ampule contains iron (7.5-9.0% w/w), citrate (15-22% w/w), pyrophosphate (15-22% w/w), phosphate (< 2% w/w), sodium (18-25% w/w) and sulfate (20-35%). One Triferic 5 mL ampule is added to 2.5 gallons (9.46 L) of bicarbonate concentrate. One Triferic 50 mL ampule is added to 25 gallons (94.6 L) of master bicarbonate mix.
Triferic Powder Packets
Triferic (ferric pyrophosphate citrate) powder is a slightly yellow-green powder, packaged in single use paper, polyethylene and aluminum foil packets, each containing 272.0 mg of elemental iron (III). Each Triferic packet contains iron (7.5-9.0% w/w), citrate (15-22% w/w), pyrophosphate (15-22% w/w), phosphate (< 2% w/w), sodium (18-25% w/w) and sulfate (20- 35%). One Triferic powder packet is added to 25 (94.6 L) gallons of master bicarbonate mix.
Ferric pyrophosphate citrate (FPC), a novel iron-replacement agent, was approved by the US Food and Drug Administration in January 2015 for use in adult patients receiving chronic hemodialysis (HD). This iron product is administered to patients on HD via the dialysate.
Ferric pyrophosphate citrate is a soluble iron replacement product. Free iron presents several side effects as it can catalyze free radical formation and lipid peroxidation as well as the presence of interactions of iron in plasma. The ferric ion is strongly complexed by pyrophosphate and citrate.1 FPC is categorized in Japan as a second class OTC drug.6 This category is given to drugs with ingredients that in rare cases may cause health problems requiring hospitalization or worst.7 It is also FDA approved since 2015.Label
Iron(III) pyrophosphate is an inorganic chemical compound with the formula Fe4(P2O7)3.
Synthesis
Anhydrous iron(III) pyrophosphate can be prepared by heating the mixture of iron(III) metaphosphate and iron(III) phosphate under oxygen with the stoichiometric ratio 1:3. The reactants can be prepared by reacting iron(III) nitrate nonahydrate with phosphoric acid.[2]
It can be also prepared via the following reaction:[3]3 Na4P2O7(aq) + 4 FeCl3(aq) → Fe4(P2O7)3(s) + 12 NaCl(aq)
References
- ^ W.M.Haynes. CRC Handbook of Chemistry and Physics (97th edition). New York: CRC Press, 2016. pp 4-68
- ^ Elbouaanani, L.K; Malaman, B; Gérardin, R; Ijjaali, M (2002). “Crystal Structure Refinement and Magnetic Properties of Fe4(P2O7)3 Studied by Neutron Diffraction and Mössbauer Techniques”. Journal of Solid State Chemistry. Elsevier BV. 163 (2): 412–420. doi:10.1006/jssc.2001.9415. ISSN 0022-4596.
- ^ Rossi L, Velikov KP, Philipse AP (May 2014). “Colloidal iron(III) pyrophosphate particles”. Food Chem. 151: 243–7. doi:10.1016/j.foodchem.2013.11.050. PMID 24423528.
- Gupta A, Amin NB, Besarab A, Vogel SE, Divine GW, Yee J, Anandan JV: Dialysate iron therapy: infusion of soluble ferric pyrophosphate via the dialysate during hemodialysis. Kidney Int. 1999 May;55(5):1891-8. doi: 10.1046/j.1523-1755.1999.00436.x. [Article]
- Naigamwalla DZ, Webb JA, Giger U: Iron deficiency anemia. Can Vet J. 2012 Mar;53(3):250-6. [Article]
- Fidler MC, Walczyk T, Davidsson L, Zeder C, Sakaguchi N, Juneja LR, Hurrell RF: A micronised, dispersible ferric pyrophosphate with high relative bioavailability in man. Br J Nutr. 2004 Jan;91(1):107-12. [Article]
- Pratt RD, Swinkels DW, Ikizler TA, Gupta A: Pharmacokinetics of Ferric Pyrophosphate Citrate, a Novel Iron Salt, Administered Intravenously to Healthy Volunteers. J Clin Pharmacol. 2017 Mar;57(3):312-320. doi: 10.1002/jcph.819. Epub 2016 Oct 3. [Article]
- Underwood E. (1977). Trace elements in human and animal nutrition (4th ed.). Academic press.
- KEGG [Link]
- Nippon [Link]
- FDA Reports [Link]
| Names | |
|---|---|
| Other namesFerric pyrophosphate | |
| Identifiers | |
| CAS Number | 10058-44-3 (anhydrous) 10049-18-0 (nonahydrate) |
| 3D model (JSmol) | Interactive image |
| ChEBI | CHEBI:132767 |
| ChemSpider | 23258 |
| DrugBank | DB09147 |
| ECHA InfoCard | 100.030.160 |
| EC Number | 233-190-0 |
| PubChem CID | 24877 |
| UNII | QK8899250F 1ZJR117WBQ (nonahydrate) |
| CompTox Dashboard (EPA) | DTXSID6047600 |
| showInChI | |
| showSMILES | |
| Properties | |
| Chemical formula | Fe4(P2O7)3 |
| Molar mass | 745.224 (anhydrate) 907.348 (nonahydrate) |
| Appearance | yellow solid (nonahydrate)[1] |
| Solubility in water | insoluble |
| Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa). | |
| Infobox references |
CLIP
https://link.springer.com/article/10.1007/s10534-018-0151-1
Iron deficiency is a significant health problem across the world. While many patients benefit from oral iron supplements, some, including those on hemodialysis require intravenous iron therapy to maintain adequate iron levels. Until recently, all iron compounds suitable for parenteral administration were colloidal iron–carbohydrate conjugates that require uptake and processing by macrophages. These compounds are associated with variable risk of anaphylaxis, oxidative stress, and inflammation, depending on their physicochemical characteristics. Ferric pyrophosphate citrate (FPC) is a novel iron compound that was approved for parenteral administration by US Food and Drug Administration in 2015. Here we report the physicochemical characteristics of FPC. FPC is a noncolloidal, highly water soluble, complex iron salt that does not contain a carbohydrate moiety. X-ray absorption spectroscopy data indicate that FPC consists of iron (III) complexed with one pyrophosphate and two citrate molecules in the solid state. This structure is preserved in solution and stable for several months, rendering it suitable for pharmaceutical applications in solid or solution state.
Iron deficiency with or without associated anemia represents a significant health problem worldwide. While many patients can restore iron levels with the use of oral iron supplements, oral supplementation is not suitable in some patients, including those undergoing chronic hemodialysis for chronic kidney disease (CKD) (Fudin et al. 1998; Macdougall et al. 1996; Markowitz et al. 1997). The limitations of oral iron replacement in patients undergoing hemodialysis likely arise from excessive ongoing losses and insufficient absorption, thus intravenous (IV) iron has become the primary route of administration in such patients (Shah et al. 2016). Multiple IV iron formulations are available, including iron dextran, iron sucrose, sodium ferric gluconate, iron carboxymaltose, ferrumoxytol, and iron isomaltoside (Macdougall et al. 1996). All such formulations are iron–carbohydrate macromolecular complexes, and the majority consist of an iron oxide core surrounded by a carbohydrate moiety (Macdougall et al. 1996; Markowitz et al. 1997).
Intravenous iron products have been used extensively for over 30 years for the treatment of iron-deficiency anemia and to maintain iron balance in hemodialysis patients since these patients have obligatory excessive losses. While these agents are generally well tolerated, they have been associated with risk of anaphylaxis (Wang et al. 2015). Compared to oral iron agents, there may be an increased risk of cardiovascular complications and infections in nondialysis patients with CKD (Macdougall et al. 1996). Additionally, higher mortality rates have been reported with use of high-dose IV iron in hemodialysis patients (Bailie et al. 2015).
Iron possesses oxidizing properties that may cause injury to cells and tissues (Koskenkorva-Frank et al. 2013; Vaziri 2013). Iron loading in general is associated with endocrinological, gastrointestinal, infectious, neoplastic, neurodegenerative, obstetric, ophthalmic, orthopedic, pulmonary, and vascular complications. In addition, excessive or misplaced tissue iron also can contribute to aging and mortality (Weinberg 2010). Normally, the body is able to protect tissues from the damaging effects of iron by regulating iron absorption in the intestine and sequestering iron with iron-binding proteins. However, the concentrations of iron introduced into the bloodstream with IV iron therapy can be as much as 100 times more than that absorbed normally through the intestine. Combined with the fact that IV iron is administered over a period of minutes compared to the slow, regulated absorption in the gut, it is possible that the increased iron load may damage cells and tissues.
A novel parenteral iron formulation, ferric pyrophosphate citrate (FPC), potentially offers a more physiologic delivery of iron. Unlike previous forms of IV iron, FPC contains no carbohydrate shell. Soluble ferric pyrophosphate-citrate complexes, generally referred to as soluble ferric pyrophosphate (SFP) were first described in the mid-1800s by Robiquet and Chapman (Chapman 1862; Robiquet 1857). This class of food-grade iron salts has been available for over 100 years as oral iron supplements and for fortification of food. In the late-1990s, Gupta et al. demonstrated that food-grade SFP could be administered to hemodialysis patients via the dialysate (Gupta et al. 1999). However, the commercially available compounds are poorly characterized and not suitable for further development as a parenteral iron supplement. Therefore, a pharmaceutical-grade SFP was developed. This product had a higher solubility than food-grade SFP and was granted a new USAN name—FPC. In 2015 FPC was approved by the US Food and Drug Administration (FDA) for parenteral delivery by hemodialysis to replace iron losses and thereby maintain hemoglobin levels in hemodialysis-dependent patients with CKD (Rockwell Medical Inc 2018). FPC is currently marketed under the trade name Triferic® (Rockwell Medical Inc., Wixom, Michigan, USA). FPC is the first carbohydrate-free, noncolloidal, water-soluble iron salt suitable for parenteral administration.
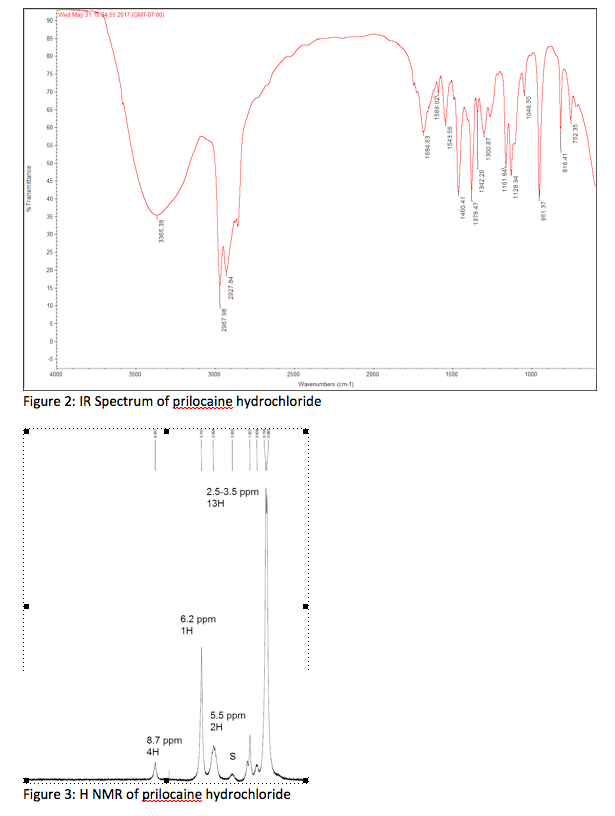
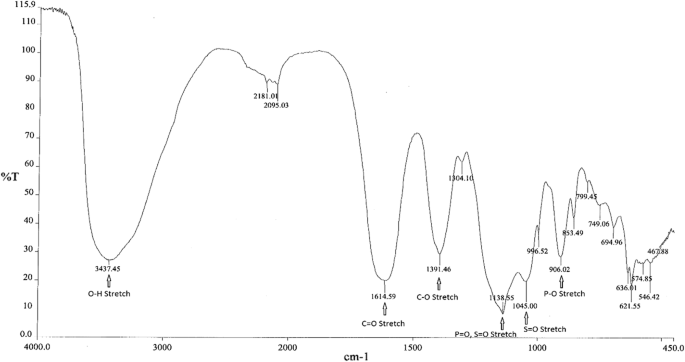
Infrared spectroscopy
Infrared (IR) spectroscopy was used to determine the main functional groups present in FPC. Figure 1 shows a representative IR spectrum of FPC. Peak assignments and positions for FPC as well as for sodium citrate, sodium pyrophosphate, and ferric sulfate, which were used to confirm the peak assignments, are shown in Table 1.
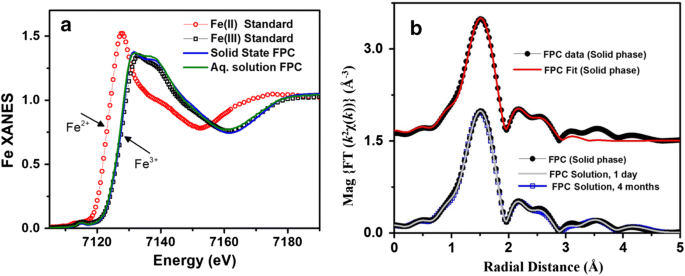
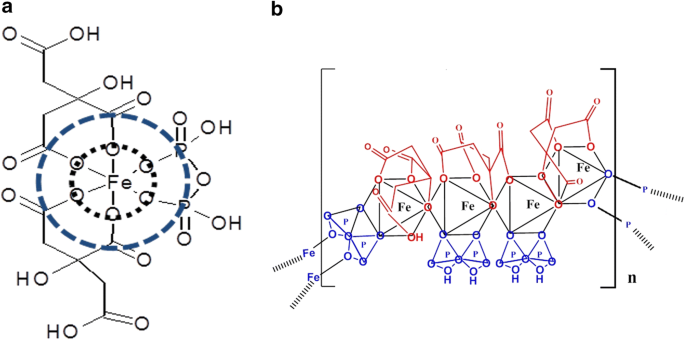
X-ray spectra of solid and aqueous iron standards and FPC. a XANES spectra of iron (II) and iron (III) standards as well as FPC in the solid and solution phases show that FPC consists exclusively of iron (III) and that the solid-phase structure is maintained in solution. b EXFAS modeling of FPC in the solid phase (top) and in solution (bottom) at Day 1 and Month 4
Chemical composition of ferric pyrophosphate citrate
From: Physicochemical characterization of ferric pyrophosphate citrate
| Ion | Percentage |
|---|---|
| Iron | 8 |
| Citrate | 19 |
| Pyrophosphate | 18 |
| Phosphate | < 1 |
| Sulfate | 25–28 |
PATENT
https://patents.google.com/patent/WO2017040937A1/enProperties of Conventional SFP

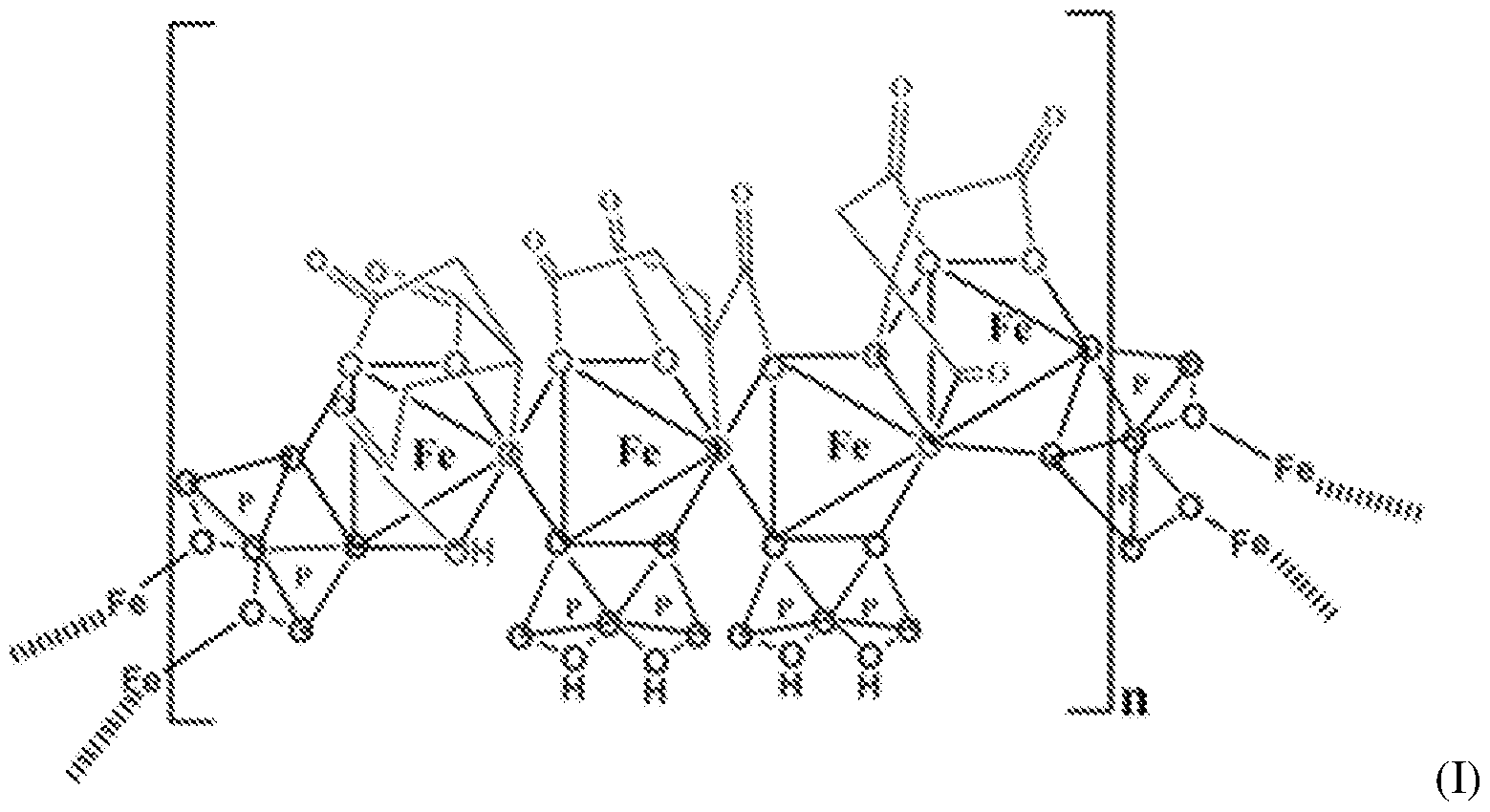
Another example of SFP is the composition is the chelate composition described in US Patent Nos. 7,816,404 and 8,178,709. The SFP may be a ferric pyrophosphate citrate (FPC) comprising a mixed-ligand iron compound comprising iron chelated with citrate andpyrophosphate, optionally FPC has the following formula: Fe4(C6H407)3(H2P207)2(P207) (relative MW 1313 daltons), e.g., structure (I):
[0036] An exemplary SFP according to the present disclosure is known to have the properties described in Table 3.Table 3 – Properties of SFP according to the present disclosure


////////////ferric pyrophosphate citrate, Triferic AVNU, , Ferric pyrophosphate citrate, FPC, SFP, Tetraferric nonahydrogen citrate pyrophosphate, Triferic, FDA 2015, APPROVALS 2021, CANADA 2021, hemodialysis-dependent chronic kidney disease
[Fe+3].[Fe+3].[Fe+3].[Fe+3].OP(O)(=O)OP(O)(O)=O.OP(O)(=O)OP(O)(O)=O.OP([O-])(=O)OP([O-])([O-])=O.OC(CC([O-])=O)(CC([O-])=O)C([O-])=O.OC(CC([O-])=O)(CC([O-])=O)C([O-])=O.OC(CC([O-])=O)(CC([O-])=O)C([O-])=O












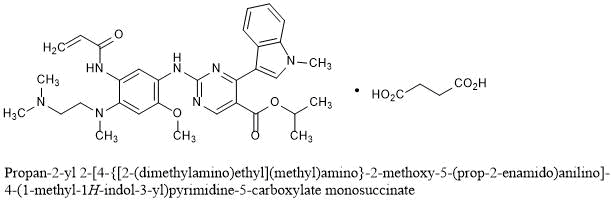
 C4H6O4 : 703.78
C4H6O4 : 703.78 (mobocertinib) Approved by U.S. FDA as the First Oral Therapy Specifically Designed for Patients with EGFR Exon20 Insertion+ NSCLC……..
(mobocertinib) Approved by U.S. FDA as the First Oral Therapy Specifically Designed for Patients with EGFR Exon20 Insertion+ NSCLC…….. 



































.jpg)
.jpg)
.jpg)
.jpg)
.jpg)
.jpg)
.jpg)
.jpg)
.jpg)
.jpg)



































.jpg)








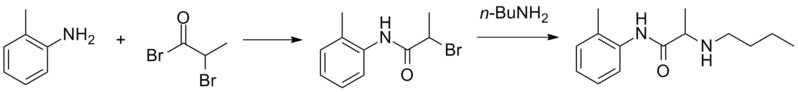
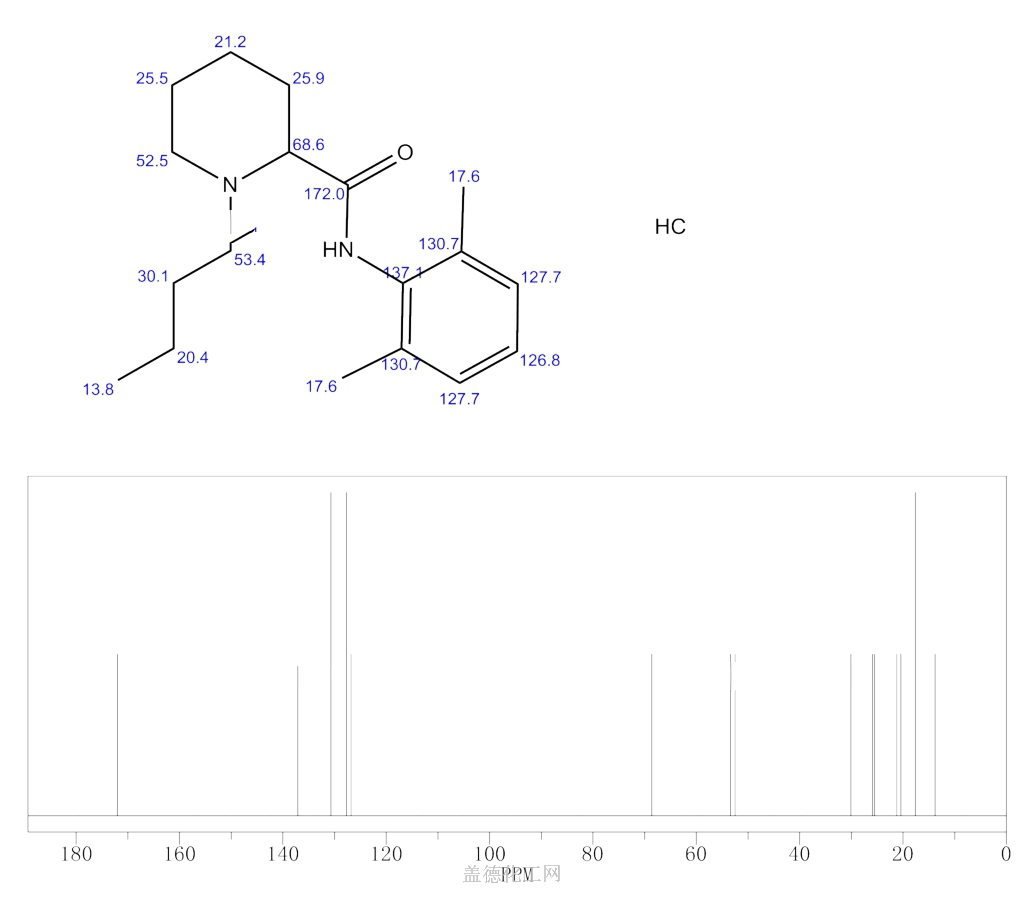
![1 H-nuclear magnetic resonance ( 1 H-NMR) spectra of prilocaine solution after sterilization with the assignment of the prilocaine hydrogens. [Prilocaine] = 5 mM, 20°C, 500 MHz.](http://www.researchgate.net/profile/Francisco-Groppo/publication/23458262/figure/fig3/AS:394557704425486@1471081295276/1-H-nuclear-magnetic-resonance-1-H-NMR-spectra-of-prilocaine-solution-after.png) 1 H-nuclear magnetic resonance ( 1 H-NMR) spectra of prilocaine solution after sterilization with the assignment of the prilocaine hydrogens. [Prilocaine] = 5 mM, 20°C, 500 MHz.
1 H-nuclear magnetic resonance ( 1 H-NMR) spectra of prilocaine solution after sterilization with the assignment of the prilocaine hydrogens. [Prilocaine] = 5 mM, 20°C, 500 MHz.