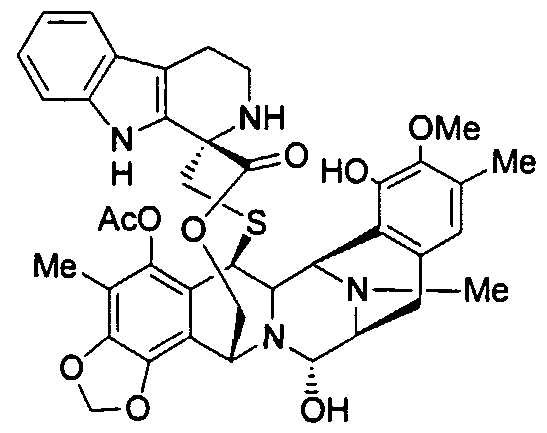
Berotralstat, also known as BCX-7353, is a kallikrein inhibitor. BCX7353 is a synthetic, once-daily, small molecule drug that can be taken as an oral capsule to treat HAE attacks and for prophylaxis.
Hereditary angioedema (HAE) is rare disorder caused by a SERPING1 gene mutation that triggers severe swelling of the skin and upper airway. Treatment options for HAE with deficient and dysfunctional C1-inhibitor are expanding to include small-molecule drugs that inhibit protein interactions in the kallikrein-kinin system
Serine proteases make up the largest and most extensively studied group of proteolytic enzymes. Their critical roles in physiological processes extend over such diverse areas as blood coagulation, fibrinolysis, complement activation, reproduction, digestion, and the release of physiologically active peptides. Many of these vital processes begin with cleavage of a single peptide bond or a few peptide bonds in precursor protein or peptides. Sequential limited proteolytic reactions or cascades are involved in blood clotting, fibrinolysis, and complement activation. The biological signals to start these cascades can be controlled and amplified as well. Similarly, controlled proteolysis can shut down or inactivate proteins or peptides through single bond cleavages.
Kallikreins are a subgroup of serine proteases. In humans, plasma kallikrein (KLKB1) has no known homologue, while tissue kallikrein-related peptidases (KLKs) encode a family of fifteen closely related serine proteases. Plasma kallikrein participates in a number of pathways relating to the intrinsic pathway of coagulation, inflammation, and the complement system.
Coagulation is the process by which blood forms clots, for example to stop bleeding. The physiology of coagulation is somewhat complex insofar as it includes two separate initial pathways, which converge into a final common pathway leading to clot formation. In the final common pathway, prothrombin is converted into thrombin, which in turn converts fibrinogen into fibrin, the latter being the principal building block of cross- linked fibrin polymers which form a hemostatic plug. Of the two initial pathways upstream of the final common pathway, one is known as the contact activation or intrinsic pathway, and the other is known as the tissue factor or extrinsic pathway.
The intrinsic pathway begins with formation of a primary complex on collagen by high-molecular- weight kininogen (HMWK), prekallikrein, and FXII (Factor XII; Hageman factor). Prekallikrein is converted to kallikrein, and FXII is activated to become FXIIa. FXIIa then converts Factor XI (FXI) into FXIa, and FXIa in turn activates Factor IX (FIX), which with its co-factor F Villa form the“tenase” complex, which activates Factor X (FX) to FXa. It is FXa which is responsible for the conversion of prothrombin into thrombin within the final common pathway.
Prekallikrein, the inactive precursor of plasma kallikrein, is synthesized in the liver and circulates in the plasma bound to FDVTWK or as a free zymogen. Prekallikrein is cleaved by activated factor XII(FXIIa) to release activated plasma kallikrein (PK). Activated plasma kallikrein displays endopeptidase activity towards peptide bonds after arginine (preferred) and lysine. PK then generates additional FXIIa in a feedback loop which in turn activates factor XI (FXI) to FXIa to connect to the common pathway. Although the initial activation of the intrinsic pathway is through a small amount of FXIIa activating a small amount of PK, it is the subsequent feedback activation of FXII by PK that controls the extent of activation of the intrinsic pathway and hence downstream coagulation. Hathaway, W. E., et al. (1965) Blood 26:521-32.
Activated plasma kallikrein also cleaves HMWK to release the potent vasodilator peptide bradykinin. It is also able to cleave a number of inactive precursor proteins to generate active products, such as plasmin (from plasminogen) and urokinase (from prourokinase). Plasmin, a regulator of coagulation, proteolytically cleaves fibrin into fibrin degradation products that inhibit excessive fibrin formation.
Patients who have suffered acute myocardial infarction (MI) show clinical evidence of being in a hypercoagulable (clot-promoting) state. This hypercoagulability is
paradoxically additionally aggravated in those receiving fibrinolytic therapy. Increased generation of thrombin, as measured by thrombin-antithrombin III (TAT) levels, is observed in patients undergoing such treatment compared to the already high levels observed in those receiving heparin alone. Hoffmeister, H. M. et al. (1998) Circulation 98:2527-33. The increase in thrombin has been proposed to result from plasmin-mediated activation of the intrinsic pathway by direct activation of FXII by plasmin.
Not only does the fibrinolysis-induced hypercoagulability lead to increased rates of reocclusion, but it is also probably responsible, at least in part, for failure to achieve complete fibrinolysis of the clot (thrombus), a major shortcoming of fibrinolytic therapy (Keeley, E. C. et al. (2003) Lancet 361 : 13-20). Another problem in fibrinolytic therapy is the accompanying elevated risk of intracranial hemorrhage. Menon, V. et al. (2004) (Chest l26:549S-575S; Fibrinolytic Therapy Trialists’ Collaborative Group (1994) Lancet 343 :311-22. Hence, an adjunctive anti -coagulant therapy that does not increase the risk of bleeding, but inhibits the formation of new thrombin, would be greatly beneficial. Plasma kallikrein inhibitors also have therapeutic potential for treating hereditary angioedema (HAE). HAE is is a serious and potentially life-threatening rare genetic illness, caused by mutations in the Cl -esterase inhibitor (C1INH) gene, located on chromosome 1 lq. HAE is inherited as an autosomal dominant condition, although one quarter of diagnosed cases arise from a new mutation. HAE has been classed as an orphan disease in Europe, with an estimated prevalence of 1 in 50,000. Individuals with HAE experience recurrent acute attacks of painful subcutaneous or submucosal edema of the face, larynx, gastrointestinal tract, limbs or genitalia which, if untreated, may last up to 5 days. Attacks vary in frequency, severity and location and can be life-threatening. Laryngeal attacks, with the potential for asphyxiation, pose the greatest risk. Abdominal attacks are especially painful, and often result in exploratory procedures or unnecessary surgery. Facial and peripheral attacks are disfiguring and debilitating.
HAE has a number of subtypes. HAE type I is defined by CllNH gene mutations which produce low levels of Cl -inhibitor, whereas HAE type II is defined by mutations which produce normal levels of ineffective Cl protein. HAE type III has separate pathogenesis, being caused by mutations in the F12 gene which codes for the serine protease known as Factor XII. Diagnostic criteria for distinguishing the subtypes of HAE, and distinguishing HAE from other angioedemas, can be found in Ann Allergy Asthma Immunol 2008; l00(Suppl2): S30-S40 and J Allergy Clin Immunol 2004; 114: 629-37, incorporated herein by reference.
Current treatments for HAE fall into two main types. Older non-specific treatments including androgens and antifibrinolytics are associated with significant side effects, particularly in females. Newer treatments are based on an understanding of the molecular pathology of the disease, namely that CllNH is the most important inhibitor of kallikrein in human plasma and that CllNH deficiency leads to unopposed activation of the kallikrein- bradykinin cascade, with bradykinin the most important mediator of the locally increased vascular permeability that is the hallmark of an attack. All of the currently available targeted therapies are administered by intravenous or subcutaneous injection. There is currently no specific targeted oral chronic therapy for HAE.
Therefore, a need exists to develop inhibitors of PK that can tip the balance of fibrinolysis/thrombosis at the occluding thrombus toward dissolution, thereby promoting reperfusion and also attenuating the hypercoagulable state, thus preventing thrombus from reforming and reoccluding the vessel. In particular, the creation of plasma kallikrein inhibitors that are specific and capable of being formulated for in vivo use could lead to a new class of therapeutics. Thus, what is needed are improved compositions and methods for preparing and formulating plasma kallikrein inhibitors.
For example, in patients with angioedema conditions, small polypeptide PK inhibitor DX-88 (ecallantide) alleviates edema in patients with hereditary angioedema (HAE). Williams, A. et al. (2003) Transfus. Apher. Sci. 29:255-8; Schneider, L. et al.
(2007) J Allergy Clin Immunol. 120:416-22; and Levy, J. H. et al. (2006) Expert Opin. Invest. Drugs 15: 1077-90. A bradykinin B2 receptor antagonist, Icatibant, is also effective in treating HAE. Bork, K. et al. (2007) J. Allergy Clin. Immunol. 119:1497-1503. Because plasma kallikrein generates bradykinin, inhibition of plasma kallikrein is expected to inhibit bradykinin production.
For example, in coagulation resulting from fibrinolytic treatment (e.g., treatment with tissue plasminogen activator or streptokinase), higher levels of plasma kallikrein are found in patients undergoing fibrinolysis. Hoffmeister, H. M. et al. (1998) J. Cardiovasc. Pharmacol. 31 :764-72. Plasmin-mediated activation of the intrinsic pathway has been shown to occur in plasma and blood and was markedly attenuated in plasma from individuals deficient in any of the intrinsic pathway components. Ewald, G. A. et al. (1995) Circulation 91 :28-36. Individuals who have had an acute MI were found to have elevated levels of activated plasma kallikrein and thrombin. Hoffmeister, H. M., et al. (1998) Circulation 98:2527-33.
DX-88 reduced brain edema, infarct volume, and neurological deficits in an animal model of ischemic stroke. Storini, C. et al. (2006) J Pharm. Exp. Ther. 318:849-854. Cl- inhibitor reduced infarct size in a mouse model of middle cerebral artery occlusion
(MCAO). De Simoni, M. G. et al. (2004) Am. J. Pathol. 164: 1857-1863; and Akita, N. et al. (2003) Neurosurgery 52:395-400). B2 receptor antagonists were found to reduce the infarct volume, brain swelling, and neutrophil accumulation and were neuroprotective in an MCAO animal model. Zausinger, S. et al. (2003 ) Acta Neurochir. Suppl. 86:205-7;
Lumenta, D. B. et al. (2006) Brain Res. 1069:227-34; Ding-Zhou, L. et al. (2003) Br. J Pharmacol. 139: 1539-47.
Regarding blood loss during cardiopulmonary bypass (CPB), it has been found that the kallikrein-kinin (i.e., contact) system is activated during CABG. Wachtfogel, Y. T. (1989) Blood 73:468. Activation of the contact system during CPB results in up to a 20- fold increase in plasma bradykinin. Cugno, M. et al. (2006) Chest 120:1776-82; and Campbell, D. J. et al. (2001 ) Am. J. Physiol. Reg. Integr. Comp. Physiol. 281 : 1059-70.
Plasma kallikrein inhibitors P8720 and PKSI-527 have also been found to reduce joint swelling in rat models of arthritis. De La Cadena, R. A. et al. (1995) FASEB J. 9:446- 52; Fujimori, Y. (1993) Agents Action 39:42-8. It has also been found that inflammation in animal models of arthritis was accompanied by activation of the contact system. Blais, C. Jr. et al. (1997) Arthritis Rheum. 40: 1327-33.
Additionally, plasma kallikrein inhibitor P8720 has been found to reduce inflammation in an acute and chronic rat model of inflammatory bowel disease (IBD). Stadnicki, A. et al. (1998) FASEB J. 12:325-33; Stadnicki, A. et al. (1996) Dig. Dis. Sci.
41 :9l2-20; and De La Cadena, R. A., et al. (1995) FASEB J. 9:446-52. The contact system is activated during acute and chronic intestinal inflammation. Sartor, R. B. et al. (1996) Gastroenterology 110: 1467-81. It has been found that B2 receptor antagonist, an antibody to high molecular weight kininogen, or reduction in levels of kininogen reduced clinicopathology in animal models of IBD. Ibid !; Arai, Y. et al. (1999) Dig. Dis. Sci.
44:845-51; and Keith, J. C. et al. (2005) Arthritis Res. Therapy 7 :R769-76.
H-D-Pro-Phe-Arg-chloromethylketone (CMK), an inhibitor of PK and FXII and a physiological inhibitor (Cl -inhibitor), has been found to reduce vascular permeability in multiple organs and reduce lesions in lipopolysaccharide (LPS)- or bacterial-induced sepsis in animals. Liu, D. et al. (2005) Blood 105:2350-5; Persson, K. et al. (2000) J. Exp. Med. 192: 1415-24. Clinical improvement was observed in sepsis patients treated with Cl- inhibitor. Zeerleder, S. et al. (2003) Clin. Diagnost. Lab. Immunol. 10:529-35; Caliezi, C., et al. (2002) Crit. Care Med. 30:1722-8; and Marx, G. et al. (1999) Intensive Care Med.
25: 1017-20. Fatal cases of septicemia are found to have a higher degree of contact activation. Martinez-Brotons, F. et al. (1987) Thromb. Haemost. 58:709-713; and Kalter, E. S. et al. (1985) J. Infect. Dis. 151 : 1019-27.
It has also been found that prePK levels are higher in diabetics, especially those with proliferative retinopathy, and correlate with fructosamine levels. Gao, B.-B., et al. (2007) Nature Med. 13: 181-8; and Kedzierska, K. et al. (2005) Archives Med. Res. 36:539- 43. PrePK is also found to be highest in those with a sensorimotor neuropathy. Christie,
M. et al. (1984) Thromb. Haemostas. (Stuttgart) 52:221-3. PrePK levels are elevated in diabetics and are associated with increased blood pressure. PrePK levels independently correlate with the albumin excretion rate and are elevated in diabetics with
macroalbuminuria, suggesting prePK may be a marker for progressive nephropathy. Jaffa, A. A. et al. (2003) Diabetes 52: 1215-21. Bl receptor antagonists have been found to decrease plasma leakage in rats treated with streptozotocin. Lawson, S. R. et al. (2005)
Eur. J. Pharmacol. 514:69-78. Bl receptor antagonists can also prevent streptozotocin- treated mice from developing hyperglycemia and renal dysfunction. Zuccollo, A. et al. (1996) Can. J. Physiol. Pharmacol. 74:586-9.
Example 1 : Synthetic protocol for racemic compound 54e
Reproduced from WO 2015/134998 and U.S. Patent Application Publication No. 2017/0073314 A1 (both incorporated by reference)
Preparation of 1 -(3-(aminomethyl)phenyl)-N-(5-((3-cyanophenyl)(cyclopropyl- methylamino)methyl)-2-fluorophenyl)-3-(trifluoromethyl)-lH-pyrazole-5-carboxamide
(54e)
Step-l : Preparation of 3-((3-amino-4-fluorophenyl)(hydroxy)methyl)benzonitrile (54b)
To a solution of 3-formylbenzonitrile (54a) (29 g, 217 mmol) in tetrahydrofuran (200 mL) cooled to 0 °C was added freshly prepared Grignard reagent (52c) (245 mL, 221 mmol, ~ 0.9 M in THF) stirred at 0 °C for 1 h, and room temperature for 18 h. The reaction mixture was quenched with 1 N HC1 (aq. 440 mL), stirred for 3 h, neutralized with NaOH (2 N, aq.) to pH = ~ 8. The reaction mixture was extracted with ethyl acetate (600, 300 mL). The combined extracts were washed with brine (120 mL), dried over MgS04, filtered and concentrated in vacuum. The crude product was purified by flash column
chromatography [silica gel, eluting with hexanes/ethyl acetate (1 :0 to 1 : 1) to give 3-((3- amino-4-fluorophenyl)(hydroxy)methyl)benzonitrile (54b) (36.28 g) as a brown gum which was used as such for next step; MS (ES+) 265.3 (M+23).
Step-2: Preparation of tert-butyl 3-(5-(5-((3-cyanophenyl)(hydroxy)methyl)-2- fluorophenylcarbamoyl)-3-(trifluoromethyl)-lH-pyrazol-l-yl)benzylcarbamate (54c)
To a solution of 3-((3-amino-4-fluorophenyl)(hydroxy)methyl)benzonitrile (54b) (24.682 g, 102 mmol) in DMF (480 mL) was added l-(3-((tert- butoxycarbonylamino)methyl)phenyl)-3-(trifluoromethyl)-lH-pyrazole-5-carboxylic acid (lOd) (35.0 g, 91 mmol), N-ethyl-N-isopropylpropan-2-amine (132 mL, 758 mmol), bromotripyrrolidin-l-ylphosphonium hexafluorophosphate(V) (PyBrOP, 42.8 g, 91 mmol) and stirred at room temperature for 19 h. The reaction mixture was diluted with ethyl acetate (1000 mL), washed with water (500, 400 mL), brine (400 mL), dried over MgS04, filtered and concentrated in vacuum. The crude product was purified by flash column chromatography [silica gel, eluting with hexanes/ethyl acetate (1 :0 to 1 : 1)] to afford tert- butyl 3-(5-(5-((3-cyanophenyl)(hydroxy)methyl)-2-fluorophenylcarbamoyl)-3- (trifluoromethyl)-lH-pyrazol-l-yl)benzylcarbamate (54c) (4.583 g, 5% for two steps) as a yellow solid; ¾ NMR (300 MHz, DMSO-i¾) d 10.57 (s, 1H), 7.81 (t, J= 1.7 Hz, 1H), 7.73 – 7.66 (m, 2H), 7.64 – 7.19 (m, 10H), 6.25 (d, J= 4.0 Hz, 1H), 5.78 (d, J= 4.0 Hz, 1H), 4.19 (d, J= 6.1 Hz, 2H), 1.37 (s, 9H); 19F NMR (282 MHz, DMSO-i¾) d -60.81 , -123.09; MS (ES+) 632.3 (M+23).
Step-3: Preparation of tert-butyl 3-(5-(5-((3- cyanophenyl)(cyclopropylmethylamino)methyl)-2-fluorophenylcarbamoyl)-3- (trifluoromethyl)-lH-pyrazol-l-yl)benzylcarbamate (54d)
To a solution of tert-butyl 3-(5-(5-((3-cyanophenyl)(hydroxy)methyl)-2- fluorophenylcarbamoyl)-3-(trifluoromethyl)-lH-pyrazol-l-yl)benzylcarbamate (54c) (1.333 g, 2.187 mmol) in dichloromethane (40 mL) at 0°C was added thionyl chloride (0.340 mL, 4.59 mmol) and warmed to room temperature over 2 h. The reaction mixture was quenched with triethyl amine (2.0 mL, 14.35 mmol) stirred at room temperature for 1 h. It was then treated with cyclopropylmethanamine (4.30 mL, 48.0 mmol), concentrated to remove most of dichloromethane followed by addition of acetonitrile (30 mL), stirring at 70 °C for 14 h, and concentration in vacuum to dryness. The residue was treated with chlorofrom (200 mL), washed with water (100 mL), dried over MgS04 followed by filtration and
concentration. The crude product was purified by flash column chromatography [silica gel eluting with hexanes/ethyl acetate (1 :0 to 2: 1)] to afford tert-butyl 3-(5-(5-((3- cyanophenyl)(cyclopropylmethylamino)methyl)-2-fluorophenylcarbamoyl)-3- (trifluoromethyl)-lH-pyrazol-l-yl)benzylcarbamate (54d) (184 mg, 13%) as colorless gum; ¾ NMR (300 MHz, DMSO-ά) d 10.56 (s, 1H), 7.89 (t, J= 1.7 Hz, 1H), 7.77 – 7.71 (m, 1H), 7.70 – 7.30 (m, 10H), 7.22 (dd, J= 10.3, 8.5 Hz, 1H), 4.93 (s, 1H), 4.19 (d, J= 6.2 Hz, 2H), 2.26 (d, J= 6.6 Hz, 2H), 1.37 (s, 9H), 1.00 – 0.80 (m, 1H), 0.45 – 0.28 (m, 2H), 0.12 – -0.01 (m, 2H); 19F NMR (282 MHz, DMSO-i¾) d -60.80 , -123.20; MS (ES+) 663.4 (M+l). Step-4: Preparation of l-(3-(aminomethyl)phenyl)-N-(5-((3-cyanophenyl)(cyclopropyl- methylamino)methyl)-2-fluorophenyl)-3-(trifluoromethyl)-lH-pyrazole-5-carboxamide (54e)
To a solution of tert-butyl 3-(5-(5-((3- cyanophenyl)(cyclopropylmethylamino)methyl)-2-fluorophenylcarbamoyl)-3- (trifluoromethyl)-lH-pyrazol-l-yl)benzylcarbamate (54d) (161 mg, 0.243 mmol) in 1,4- Dioxane (18 mL) was added hydrogen chloride (2.60 mL, 10.40 mmol, 4 M in l,4-dioxane) and stirred at room temperature for 16 h. the reaction mixture was treated with hexanes, decanted, washed with hexanes, and decanted again. The insoluble crude product was purified by flash column chromatography [silica gel, eluting with chloroform/CMA80 (1 :0 to 2:1)] to afford l-(3-(aminomethyl)phenyl)-N-(5-((3-cyanophenyl)(cyclopropyl- methylamino)methyl)-2-fluorophenyl)-3-(trifluoromethyl)-lH-pyrazole-5-carboxamide (54e). The pure product was dissolved in methanol (10 mL) and added 4 N HC1 (aq. 0.14 mL) followed by concentration in vacuum to dryness to give HC1 salt of l-(3- (aminomethyl)phenyl)-N-(5-((3-cyanophenyl)(cyclopropyl-methylamino)methyl)-2- fluorophenyl)-3-(trifluoromethyl)-lH-pyrazole-5-carboxamide (54e) (74 mg, 48%) white solid; ¾ NMR (300 MHz, DMSO- d, D20 ex NMR) d 8.13 (t, J = 1.7 Hz, 1H), 7.98 – 7.84 (m, 3H), 7.73 – 7.64 (m, 3H), 7.63 – 7.48 (m, 4H), 7.44 (dd, J = 10.2, 8.6 Hz, 1H),
Example 2: Separation of enantiomers of racemic compound 54e
Reproduced from WO 2015/134998 and U.S. Patent Application Publication No. 2017/0073314 A1 (both incorporated by reference)
Compound I (free base) Separation of (+)-l-(3-(aminomethyl)phenyl)-N-(5-((3-cyanophenyl)(cyclopropyl- methylamino)methyl)-2-fluorophenyl)-3-(trifluoromethyl)-lFl-pyrazole-5-carboxamide (Compound I), and (-)-l-(3-(aminomethyl)phenyl)-N-(5-((3-cyanophenyl)(cyclopropyl- methylamino)methyl)-2-fluorophenyl)-3-(trifluoromethyl)-lFl-pyrazole-5-carboxamide ((-
)-enantiomer)
Isomers of Racemic l-(3-(aminomethyl)phenyl)-N-(5-((3- cyanophenyl)(cyclopropyl-methylamino)methyl)-2-fluorophenyl)-3-(trifluoromethyl)-lF[- pyrazole-5-carboxamide (54e) (0.4 g) were separated by using preparative SFC method using the following conditions to furnish:
Preparative SFC Method used:
Column 20mm x 25.0 cm ChromegaChiral CCS from
Regis Technologies (Morton Grove, IL)
CO2 Co-solvent (Solvent B) Methanol: Isopropanol (1 : 1) with 1%
Isopropylamine
Isocratic Method 20 % Co-solvent at 80 mL/min
System Pressure 200 bar
Column Temperature 25 °C
Sample Diluent Methanol: Isopropanol
Chiral Purity of peaks was determined by following Analytical SFC Method:
Column 4.6 x 100 mm ChiralPak AS from Chiral
Technologies (West Chester, PA)
CO2 Co-solvent (Solvent B) Methanol: Isopropanol (1 : 1) with 0.1%
Isopropylamine
Isocratic Method 5-65 % Co-solvent Gradient at 4 mL/min System Pressure 100 bar
Column Temperature 25 °C
Sample Diluent Methanol
Peak-l (Compound I) 2.1 min 144 mg >95% ee (UV 254)
98.6 % purity (UV 254)
Peak-2 ((-)-enantiomer) 2.4 min 172 mg 95.5 % ee (UV 254)
96.5 % purity (UV 254) 1. Peak-l assigned as (+)-l-(3-(aminomethyl)phenyl)-N-(5-((3- cyanophenyl)(cyclopropyl-methylamino)methyl)-2-fluorophenyl)-3- (trifluoromethyl)-lH-pyrazole-5-carboxamide (Compound I) (144 mg, >95%ee) free base as white solid; Optical rotation: [O]D = (+) 6.83 [CH3OH, 1.2]; ‘H NMR (300 MHz, DMSO-£¾) d 10.53 (s, 1H, D2O exchangeable), 7.88 (t, J= 1.7 Hz, 1H), 7.77 – 7.71 (m, 1H), 7.67 (dt, 7= 7.7, 1.4 Hz, 1H), 7.63 (dd, J= 7.5, 2.1 Hz, 1H), 7.56 (s, 1H), 7.54 – 7.47 (m, 2H), 7.47 – 7.38 (m, 2H), 7.34 (ddt, J= 8.6, 5.9, 2.8 Hz, 2H), 7.22 (dd, J= 10.3, 8.5 Hz, 1H), 4.93 (s, 1H), 3.77 (s, 2H), 2.31 – 2.21 (m, 2H), 0.97 – 0.80 (m, 1H), 0.42 – 0.33 (m, 2H), 0.10 – -0.02 (m, 2H); 19F NMR (282 MHz, DMSO-Ts) d -60.73 , -123.20; MS (ES+) 563.3 (M+l), 561.3 (M-l). To a solution of free base mixture of (+)-l-(3-(aminomethyl)phenyl)-N-(5-((3- cyanophenyl)(cyclopropyl-methylamino)methyl)-2-fluorophenyl)-3- (trifluoromethyl)-lH-pyrazole-5-carboxamide (Compound I) (120 mg) in methanol (15 mL) was added hydrogen chloride (0.969 mL, 1.938 mmol), stirred at room temperature for 10 min, evaporated to dryness to afford (+)-l-(3- (aminomethyl)phenyl)-N-(5-((3-cyanophenyl)(cyclopropyl-methylamino)methyl)-2- fluorophenyl)-3-(trifluoromethyl)-lH-pyrazole-5-carboxamide (Compound I) (100 mg) hydrochloride salt as white solid; ¾ NMR (300 MHz, DMSO-Ts) d 10.84 (s, 1H, D2O exchangeable), 10.44 (s, 2H, D2O exchangeable), 8.44 (s, 3H, D2O exchangeable), 8.30 (s, 1H, D2O exchangeable), 8.09 (d, J= 7.9 Hz, 1H), 7.99 (d, J = 6.8 Hz, 1H), 7.91 – 7.83 (m, 1H), 7.80 – 7.50 (m, 7H), 7.42 (dd, J= 10.3, 8.6 Hz, 1H), 5.78 (d, J= 6.9 Hz, 1H), 4.13 (d, J= 5.7 Hz, 2H), 2.88 – 2.62 (m, 2H), 1.42 – 0.99 (m, 1H), 0.73 – 0.46 (m, 2H), 0.32 (d, J= 4.4 Hz, 2H); 19F NMR (282 MHz, DMSO-i¾) d -60.81 , -119.99; MS (ES+): MS (ES+) 563.3 (M+l), MS (ES-) 561.3 (M-l), 597.3 (M+Cl); Analysis calculated for C30H26F4N6O 2HC1 l.75H20: C, 54.02; H, 4.76; Cl, 10.63; N, 12.60; Found: C, 54.12; H, 4.83; Cl, 10.10; N, 11.97. Peak-2 assigned as (-)-l-(3-(aminomethyl)phenyl)-N-(5-((3-cyanophenyl)(cyclopropyl- methylamino)methyl)-2-fluorophenyl)-3-(trifluoromethyl)-lH-pyrazole-5-carboxamide ((-)-enantiomer) (172 mg, 95.5 % ee) as free base was repurified by flash column chromatography (silica gel 12 g, eluting 0-30% MeOH in chloroform for 15 min) to afford (-)-l-(3-(aminomethyl)phenyl)-N-(5-((3-cyanophenyl)(cyclopropyl- methylamino)methyl)-2-fluorophenyl)-3-(trifluoromethyl)-lH-pyrazole-5-carboxamide ((-)-enantiomer) free base as an off-white solid; Optical rotation: [O]D = (-) 5.44
2H), 7.34 (ddq, J= 8.7, 6.1, 3.5, 2.8 Hz, 2H), 7.22 (dd, J= 10.3, 8.5 Hz, 1H), 4.93 (s, 1H), 3.78 (s, 2H), 2.25 (d, J= 6.9 Hz, 2H), 0.90 (ddd, J= 9.8, 8.0, 5.2 Hz, 1H), 0.47 – 0.29 (m, 2H), 0.04 (dd, J= 5.0, 1.5 Hz, 2H); 19F NMR (282 MHz, DMSO-i¾) d -60.73 , -123.19; MS (ES+) 563.3 (M+l), MS (ES-), 561.3 (M-l). To a solution of free base of (-)-l-(3-(aminomethyl)phenyl)-N-(5-((3-cyanophenyl)(cyclopropyl- methylamino)methyl)-2-fluorophenyl)-3-(trifluoromethyl)-lH-pyrazole-5-carboxamide ((-)-enantiomer) (0.124 g, 0.220 mmol) in methanol (15 mL) was added hydrogen chloride (1.102 mL, 2.204 mmol), stirred at room temperature for 10 min, evaporated to dryness to afford (-)-l-(3-(aminomethyl)phenyl)-N-(5-((3-cyanophenyl)(cyclopropyl- methylamino)methyl)-2-fluorophenyl)-3-(trifluoromethyl)-lH-pyrazole-5-carboxamide ((-)-enantiomer) (0.121 g) hydrochloride salt as an off-white solid; Ή NMEE ¾ NMR (300 MHz, DMSO-i¾) d 10.82 (s, 1H, D20 exchangeable), 10.36 (s, 2H, D2O exchangeable), 8.38 (s, 3H, D2O exchangeable), 8.27 (s, 1H), 8.06 (d, J= 7.9 Hz, 1H), 7.98 (d, J= 6.7 Hz, 1H), 7.87 (d, J= 7.7 Hz, 1H), 7.78 – 7.49 (m, 7H), 7.48 – 7.37 (m, 1H), 5.78 (s, 1H), 4.13 (d, j= 5.7 Hz, 2H), 2.72 (s, 2H), 1.14 (s, 1H), 0.56 (d, j= 7.7 Hz, 2H), 0.31 (d, J= 5.0 Hz, 2H); 19F NMR (282 MHz, DMSO-i¾) d -60.82 , -120.03; MS (ES+): MS (ES+) 563.3 (M+l), MS (ES-), 561.3 (M-l), 597.2 (M+Cl); Analysis calculated for C30H26F4N6O.2HCI. I .75H2O: C, 54.02; H, 4.76; Cl, 10.63; N, 12.60; Found: C, 54.12; H, 4.83; Cl, 10.10; N, 11.97.
Example 3 : Preparation of a Seed Crystal of Compound I*2
A solution of Compound I ( see Example 2) in methyl tert-butyl ether (MTBE) (1 equiv) is added to a solution of HC1 (aq) (2 equiv) in methanol (cold), followed by heating to about 30°C, and keeping it at about 30°C for not longer than 5 hours while stirring at about 115 rpm. Compound I bis(HCl) is collected by filtration and dried. The crystalline material obtained can be used as a seed for the crystallization protocol described in
Example 4. Example 4: Large-Scale Synthetic & Crystallization Protocol for Compound I*2(HC1 )
Compound I (free base) Compound I bis(HCI)
37% Aqueous hydrochloric acid (38.1 kg, 32.3 L, 2.14 equiv.) was charged to a clean and empty crystallization vessel, methanol (228.9 kg, 39.5 equiv.) was added, and the contents were cooled to -7 ± 3°C. A solution of Compound I free base (approx. 101.8 kg; 180.9 moles) in MTBE (approx. 1,300 L) was filtered through a polish filter into the crystallization vessel at temperature -5 ± 5°C. After rinse with MTBE, pre-weighed Compound I»2(HCl) seed crystals (1.39 kg, 0.012 equiv.; Example 3) were charged to the crystallization vessel via the manhole. The vessel content was heated to 30-33°C, and the agitation speed was set to 25-50 rpm. After confirmed crystallization, the slurry was agitated for another three to four hours. The product slurry was transferred to centrifuge and isolated by centrifugation. The product was washed with MTBE (585 L). After dry spinning the wet product, Compound I*2(HC1), it was discharged from the centrifuge, and the product was dried at < 40°C under vacuum in a cone drier. Product Compound I»2(HCl) yield: 100 kg; 157.4 mol; approx. 85%.
‘H NMR (300 MHz, DMSO-c/i,) data is shown in the following table:
19F NMR (282 MHz, DMSO- is) data is shown in the following table:
Compound I has two basic sites. The conjugate acid of the primary amine was calculated to have a pKa value of 8.89, and the conjugate acid of the secondary amine was calculated to have a pKa value of 7.86.
The XRPD pattern of Compound I»2(HCl) is shown in Fig. 1. Compound I»2(HCl) has characteristic peaks in its XRPD pattern at values of two theta (°2Q) of 5.28, 8.96, 14.27, 16.18, 19.79, 21.16, 22.01, 23.31, 24.64, and 30.31. TG-IR analysis indicated two, distinct weight loss regions: the first was completed by 125 °C while the second began at approximately 208 °C. IR analysis of the off gasses from this experiment detected only trace amounts of water at the initial weight loss while HC1 gas was detected at the 208°C event. No other solvents were detected in the sample. Thus, it was determined that Compound I*2(HC1) initially loses water when heated and, when heated to above 200°C, the salt begins to break apart and HC1 gas is evolved. The IR signal for all these events is very weak indicating that they are occurring over a range and not at a specified temperature. An exemplary TG-IR spectrum is shown in Fig. 2.
REFERENCES
1: Sohtome Y, Sodeoka M. Development of Chaetocin and S-Adenosylmethionine Analogues as Tools for Studying Protein Methylation. Chem Rec. 2018 Dec;18(12):1660-1671. doi: 10.1002/tcr.201800118. Epub 2018 Oct 16. Review. PubMed PMID: 30324709.
2: Bensussen A, Torres-Sosa C, Gonzalez RA, Díaz J. Dynamics of the Gene Regulatory Network of HIV-1 and the Role of Viral Non-coding RNAs on Latency Reversion. Front Physiol. 2018 Sep 28;9:1364. doi: 10.3389/fphys.2018.01364. eCollection 2018. PubMed PMID: 30323768; PubMed Central PMCID: PMC6172855.
Tirabrutinib (Velexbru®) is an orally administered, small molecule, Bruton’s tyrosine kinase (BTK) inhibitor being developed by Ono Pharmaceutical and its licensee Gilead Sciences for the treatment of autoimmune disorders and haematological malignancies. Tirabrutinib irreversibly and covalently binds to BTK in B cells and inhibits aberrant B cell receptor signalling in B cell-related cancers and autoimmune diseases. In March 2020, oral tirabrutinib was approved in Japan for the treatment of recurrent or refractory primary central nervous system lymphoma. Tirabrutinib is also under regulatory review in Japan for the treatment of Waldenström’s macroglobulinemia and lymphoplasmacytic lymphoma. Clinical development is underway in the USA, Europe and Japan for autoimmune disorders, chronic lymphocytic leukaemia, B cell lymphoma, Sjogren’s syndrome, pemphigus and rheumatoid arthritis. This article summarizes the milestones in the development of tirabrutinib leading to the first approval of tirabrutinib for the treatment of recurrent or refractory primary central nervous system lymphoma in Japan.
The preparation of the compound 3- (1- {3- [5- (1-Methyl-piperidin-4-ylmethoxy) -pyrimidin-2-yl] -benzyl} -6-oxo-1,6-dihydro-pyridazin-3 -yl) -benzonitrile (“A257”) takes place analogously to the following scheme
40.1 17.7 g (67.8 mmol) triphenyl are added to a suspension of 13.0 g (56.5 mmol) 3- (5-hydroxypyrimidin-2-yl) -benzoic acid methyl ester and 13.4 g (62.1 mmol) N-Boc-piperidinemethanol in 115 ml THF -phosphine and cooled to 5 ° C. To the suspension kept at this temperature, 13.3 ml (67.8 mmol) of diisopropylazodicarboxylate are added dropwise with stirring within 45 minutes. The reaction mixture is stirred for 1 hour at room temperature. Then a further 22.2 g (84.7 mmol) triphenylphosphine and 16.6 ml (84.7 mmol)
Diisopropyl azodicarboxylate added. The reaction mixture turns 18
Stirred for hours at room temperature and concentrated in vacuo. The resulting solid is filtered off with suction, washed with diethyl ether and chromatographed on a silica gel column with dichloromethane / methanol as the mobile phase: 4- [2- (3-methoxycarbonyl-phenyl) -pyrimidin-5-yloxymethyl] -piperidine-1-carboxylic acid tert .-butyl ester as lemon yellow crystals; 166 ° C .; ESI 428.
40.2 To a suspension of 1.71 g (3.99 mmol) of 4- [2- (3-methoxycarbonyl-phenyl) -pyrimidin-5-yloxymethyl] -piperidine-1-carboxylic acid tert-butyl ester in 20 ml of THF are added under nitrogen 25 ml (25 mmol) of a 1 M solution of diisobutylaluminum hydride in THF were added dropwise. The reaction mixture is stirred at room temperature for 1 hour, and 1 ml of a saturated sodium sulfate solution is added. The resulting precipitate is filtered off with suction and washed with THF and hot 2-propanol. The filtrate is evaporated and recrystallized from tert-butyl methyl ether: {3- [5- (1-Methyl-piperidin-4-ylmethoxy) -pyrimidin-2-yl] -phenyl} -methanol as beige crystals; Mp 175 ° C; ESI 314.
40.3 To a solution of 313 mg (1.00 mmol) {3- [5- (1-methyl-piperidin-4-ylmethoxy) -pyrimidin-2-yl] -phenyl} -methanol in 2 ml THF are successively added 264 mg (1.30 mmol) 3- (6-oxo-1, 6-dihydro-pyridazin-3-yl) benzonitrile and 397 mg (1.5 mmol) triphenylphosphine are added. The reaction mixture is cooled in an ice bath and 294 μl (1.5 mmol) of diisopropylazodicarboxylate are added dropwise with stirring. The
The reaction mixture is stirred for 18 hours at room temperature and evaporated. The residue is chromatographed on a silica gel column using dichloromethane / methanol. The product-containing fractions are combined, evaporated, the residue digested with tert-butyl methyl ether, filtered off with suction and dried in vacuo: 3- (1- {3- [5- (1-methylpiperidin-4-ylmethoxy) pyrimidine) -2-yl] -benzyl} -6-oxo-1,6-dihydro-pyridazin-3-yl) -benzonitrile as colorless crystals; M.p. 177 ° C; ESI 493; 1 H-NMR (de-DMSO): δ [ppm] = 1.33 (m, 2H), 1.75 (m, 3H), 1.89 (m, 2H), 2.17 (S, 3H), 2.80 (m, 2H), 4.05 (d, J = 6.1 Hz 1 2H), 5.45 (s, 2H) 1 7.16 (d, J = 10 Hz, 1 H), 7.49 (m, 2H), 7.73 (t, J = 7.8 Hz, 1H ), 7.93 (d, J = 7.8 Hz, 1H) 1 8.17 (d, J = 10 Hz, 1H), 8.24 (m, 2H), 8.38 (m, 2H), 8.64 (s, 2H).
The hemisulfate, citrate, tartrate, sulfate, succinate and hydrochloride are obtained from “A257” by salt formation.
Scheme 1. Reagents and conditions: a) PdCl2(PPh3)2, Na2CO3, ethanol/toluene/water, 90 °C, 8 h; b) SOCl2, CHCl3, reflux; c) SeO2, dioxane:H2O = 10:1, reflux, 12 h; d) NaOH, −30 °C; e) NaH, DMF/THF, 0 °C—room temperature, 12 h; f) dry ethanol, reflux; g) NaOH, DMF/H2O, 60 °C, 8 h, N2.
Scheme 2. Reagents and conditions: a) N,N-diisopropylethylamine, dry CH2Cl2, 0 °C—room temperature, 6 h; b) PdCl2(PPh3)2, Na2CO3, ethanol/toluene/water, 90 °C, 8 h; c) 10% aq. HCl, MeOH, reflux; d) K2CO3, dry DMF, 80 °C, 12 h; e) NaOH, DMF/H2O, 60 °C, 8 h, N2; f) PPh3, DIAD, THF, 0 °C—room temperature; g) SOCl2, CHCl3, reflux; h) 35% formaldehyde, NaBH4, MeOH.
Scheme 3. Reagents and conditions: a) PdCl2(PPh3)2, Na2CO3, ethanol/toluene/water, 90 °C, 8 h; b) NaBH4, MeOH, 0 °C—room temperature, 1 h; c) SOCl2, CHCl3, reflux; d) K2CO3, dry DMF, 80 °C, 12 h; e) 31a–31b: NaOH, DMF/H2O, 60 °C, 8 h, N2; f) 31c–31g: NaH, dry DMF, 0 °C—room temperature, 5 h.
Scheme 4. Reagents and conditions: a) K2CO3, dry DMF, 80 °C, 12 h; b) PdCl2(PPh3)2, Na2CO3, DME/DMF/water, 89 °C, 12 h; c) NaOH, DMF/H2O, 60 °C, 8 h, N2.
Scheme 5. Reagents and conditions: a) K2CO3, dry DMF, 80 °C, 12 h; b) PdCl2(PPh3)2, Na2CO3, DME/DMF/water, 89 °C, 12 h; c) NaOH, DMF/H2O, 60 °C, 8 h, N2.
With the development of atomic science, radiation therapy such as cobalt hexahydrate, linear accelerator, and electron beam has become one of the main methods of cancer treatment. However, traditional photon or electron therapy is limited by the physical conditions of the radiation itself. While killing the tumor cells, it also causes damage to a large number of normal tissues on the beam path. In addition, due to the sensitivity of tumor cells to radiation, traditional radiation therapy For the more radiation-resistant malignant tumors (such as: glioblastoma multiforme, melanoma), the treatment effect is often poor.
In order to reduce the radiation damage of normal tissues around the tumor, the concept of target treatment in chemotherapy has been applied to radiation therapy; and for tumor cells with high radiation resistance, it is currently actively developing with high relative biological effects (relative Biological effectiveness, RBE) radiation sources, such as proton therapy, heavy particle therapy, neutron capture therapy. Among them, neutron capture therapy combines the above two concepts, such as boron neutron capture therapy, by the specific agglomeration of boron-containing drugs in tumor cells, combined with precise neutron beam regulation, providing better radiation than traditional radiation. Cancer treatment options.
Boron Neutron Capture Therapy (BNCT) is a high-capture cross-section of thermal neutrons using boron-containing ( 10 B) drugs, with 10 B(n,α) 7 Li neutron capture and nuclear splitting reactions. Two heavy charged particles of 4 He and 7 Li are produced. The average energy of the two charged particles is about 2.33 MeV, which has high linear energy transfer (LET) and short range characteristics. The linear energy transfer and range of α particles are 150 keV/μm and 8 μm, respectively, while the 7 Li heavy particles are For 175 keV/μm, 5 μm, the total range of the two particles is equivalent to a cell size, so the radiation damage caused to the organism can be limited to the cell level, when the boron-containing drug is selectively aggregated in the tumor cells, with appropriate The sub-radiation source can achieve the purpose of locally killing tumor cells without causing too much damage to normal tissues.
Since the effectiveness of boron neutron capture therapy depends on the concentration of boron-containing drugs in the tumor cell position and the number of thermal neutrons, it is also called binary cancer therapy; thus, in addition to the development of neutron sources, The development of boron-containing drugs plays an important role in the study of boron neutron capture therapy.
4-( 10 B)dihydroxyboryl-L-phenylalanine (4-( 10 B)borono-L-phenylalanine, L- 10 BPA) is currently known to be able to utilize boron neutron capture therapy (boron neutron capture therapy) , BNCT) An important boron-containing drug for the treatment of cancer.
Therefore, various synthetic methods of L-BPA have been developed. As shown in the following formula (A), the prior art L-BPA synthesis method includes two methods of forming a bond (a) and a bond (b):
Among them, the method for synthesizing L-BPA by forming the bond (a) is to try to introduce a substituent containing a dihydroxylboryl group or a borono group into the skeleton of the phenylalanine, thereby the pair of the amide substituent. The position forms a carbon-boron bond to produce L-BPA.
J. Org. Chem. 1998, 63, 8019 discloses a method for the cross-coupling reaction of (S)-4-iodophenylalanine with a diboron compound by palladium-catalyzed amine end treatment. Amine-protected (S)-4-iodophenylalanine (eg (S)-N-tert-butoxycarbonyl-4-iodophenylalanine ((S)-N-Boc-4-) Iodophenylalanine)) is prepared by cross-coupling with a diboron compound such as bis(pinacolato diboron) to give (S)-N-tert-butoxycarbonyl-4-pentanoylboryl phenylalanine The amine-terminated (S)-4-boranyl ester phenylalanine of the acid ((S)-N-Boc-4-pinacolatoborono phenylalanine); afterwards, the protecting group on the amine end and the boronic end are removed. The above substituents complete the preparation of L-BPA.
However, since the selected 10 B-doped divaleryl diboron is not a commercially available compound, this method requires additional pretreatment of the preparation of the borating agent, resulting in a high process complexity and a long time consuming process. It is impossible to prepare a high yield of L-BPA. In addition, the carboxylic acid group of the protected (S)-4-iodophenylalanine at the amine end needs to be protected by a substituent to form a benzyl ester group to increase the process yield to 88%; however, The preparation of L-BPA in this manner also requires an additional step of deprotecting the carboxylic acid group, which in turn increases the process complexity of L-BPA.
Accordingly, the method provided in this document not only involves pre-treatment of the preparation of the borating agent, but also requires a large amount of process time and synthesis steps to complete the steps of protecting and deprotecting the carboxylic acid group, and is not advantageous as an industry. The main method of synthesizing L-BPA.
On the other hand, a method for synthesizing L-BPA by forming a bond (b) is a coupling reaction of an amino acid with a boron-containing benzyl fragment or a boron-containing benzaldehyde fragment. To synthesize L-BPA. Biosci. Biotech. Biochem. 1996, 60, 683 discloses an enantioselective synthesis of L-BPA which gives the hands of a cyclic ethers of boronic acid and L-proline The chiral derivatives from L-valine are subjected to a coupling reaction to produce L-BPA. However, this method requires the formation of a cyclic ether compound of boric acid from 4-boronobenzylbromide, followed by a coupling reaction with a chiral derivative of L-proline, and in the latter stage. The amino acid undergoes an undesired racemization in the synthesis step, so that the method requires an enzymatic resolution step to reduce the yield to obtain L-BPA having a certain optical purity.
Accordingly, the method provided in the literature still includes the steps of pretreatment of the preparation of the borating agent and post-treatment of the enzymatic resolution, so that the process involved in the method is complicated and takes a long time, and cannot be obtained. High yield of L-BPA.
In addition, L- 10 BPA (4-( 10 B)borono-L-phenylalanine, 4-( 10 B)dihydroxyboryl-L-phenylalanine) containing 10 boron is currently known to accumulate in tumor cells. The key factor is to use the thermal neutron beam to irradiate the boron element accumulated in the tumor cells to kill the tumor cells by capturing the high-energy particles generated by the reaction, thereby achieving the purpose of treating cancer. Therefore, 10 boron can promote the treatment of L- 10 BPA by boron neutron capture treatment.
However, the boron element present in nature contains about 19.9% of 10 boron and about 80.1% of 11 boron. Therefore, many researchers are still actively developing methods that can be applied to the synthesis of L-BPA, especially for the synthesis of 10- boron-rich L-BPA.
J.Org.Chem.1998,63,8019 additionally provides a method of synthesizing 10 boronated agents, since the method involves multiple steps, it is easy to greatly reduce the boron content of 1010 boron enriched material in the manufacturing process. Therefore, the method provided in this document is not suitable for the synthesis of 10- boron-rich L-BPA.
Another example is the Biosci.Biotech.Biochem.1996,60,683, before the enzymatic resolution step is not performed, the method provided by the articles could not be obtained with a certain L-BPA optical purity; 10 and the method for preparing boronated agents when also relates to multi-step, resulting in conversion of boron-rich material 10 occurs during the manufacturing process. Therefore, the method provided in this document is also not suitable for the synthesis of 10- boron-rich L-BPA.
Furthermore, Bull. Chem. Soc. Jpn. 2000, 73, 231 discloses the use of palladium to catalyze 4-iodo-L-phenylalanine with 4,4,5,5-tetramethyl-1,3,2 A method in which a dioxonium pentoxide (common name: pinacolborane) is subjected to a coupling reaction. However, this document does not mention how to prepare articles 10 boron enriched L-BPA using this method, and 4,4,5,5-tetramethyl-1,3,2-dioxaborolane not a commercial 10 The compounds available in the literature are not suitable for the synthesis of 10- boron-rich L-BPA.
In addition, Synlett. 1996, 167 discloses a method for coupling a iodophenylborate with a zinc derivative of L-serine zinc derivatives, which involves first preparing phenyl iodoborate. The ester and the preparation of a zinc derivative of L-type serine acid, etc., result in a lower yield of the produced L-BPA. In addition, since the 10- boron-rich triiodide 10 boron and 1,3-diphenylpropane-1,3-diol selected for this method are not commercially available compounds, the methods provided in this document are also provided. Still not suitable for the synthesis of 10- boron-rich L-BPA.
SYN
Repub. Korean Kongkae Taeho Kongbo, 2018060319,
PAPER
Research and Development in Neutron Capture Therapy, Proceedings of the International Congress on Neutron Capture Therapy, 10th, Essen, Germany, Sept. 8-13, 2002 (2002), 1-8.
PAPER
European Journal of Pharmaceutical Sciences (2003), 18(2), 155-163
Before preparing (S)-N-tert-butoxycarbonyl-4-dihydroxyborylphenylalanine from (S)-N-tert-butoxycarbonyl-4-iodophenylalanine, it is necessary to reveal Process for preparing (S)-N-tert-butoxycarbonyl-4-iodophenylalanine by using (S)-4-iodophenylalanine as a starting material and a process for preparing 10 tributyl borate with 10 boric acid.
1. Preparation of (S)-N-tert-butoxycarbonyl-4-iodophenylalanine from (S)-4-iodophenylalanine
Please refer to the following reaction formula I, which is (S)-4-iodophenylalanine in a solvent of 1,4-dioxane (1,4-dioxane) and water (H 2 O) with hydrogen peroxide. Sodium (NaOH) and di-tert-butyl dicarbonate (Boc 2 O) are reacted to obtain a chemical reaction formula of (S)-N-tert-butoxycarbonyl-4-iodophenylalanine.
In the preparation process, two reaction vessels were selected for the reaction.
The specific operation process is as follows:
1. Set up a reaction using a 3L three-neck bottle.
2. (S)-4-iodo-L-phenylalanine (200.00 g, 687.10 mmol, 1.00 eq) was added to the reaction system.
3. Add 1,4-dioxane (1.00 L) and water (1.00 L) to the reaction system, respectively.
4. Sodium hydroxide (68.71 g, 1.72 mol, 2.50 eq) was added to the reaction system, the solution gradually became clear, and the temperature rose slightly to 19 °C.
5. When the system is cooled to 0-10 ° C, di-tert-butyl dicarbonate (254.93 g, 1.17 mol, 268.35 mL, 1.70 eq) is added to the reaction system, and the temperature of the reaction system is naturally raised to 10 to 30 ° C and Stir at room temperature (about 30 ° C) for 8 hours.
6. The reaction was detected using high performance liquid chromatography (HPLC) until the starting of the reaction.
7. The temperature of the control system is less than 40 ° C, and the 1,4-dioxane in the reaction solution is concentrated.
8. The reaction system was lowered to room temperature (about 25 ° C), 100 mL of water was added, and the pH was adjusted to 1.8-2 with hydrochloric acid (2M (ie, molarity, M)).
9. Extract three times with ethyl acetate (2 L).
10. Combine the organic phases and wash twice with saturated brine (1 L).
11. The organic phase was dried over sodium sulfate (200 g).
12. Continue drying in an oven (40-45 ° C) to give (S)-N-tert-butoxycarbonyl-4-iodo-L-phenylalanine (250.00 g, 626.28 mmol, HPLC analysis, yield 93.00 %, purity 98%).
The prepared (S) -N- tert-butoxycarbonyl-4-iodo-phenylalanine was -L- Hydrogen 1 nuclear magnetic resonance spectrum analysis (1 HNMR) as follows:
Second, tributyl borate 10 was prepared from boronic acid 10
See the following reaction formulas II, 10 as boric acid (H 2 SO 4) is reacted with sulfuric acid in a solvent (butan-1-ol), and toluene (Toluene) in n-butanol, to obtain 10 tributyl borate (10 The chemical reaction formula of B(OBu) 3 ).
The specific operation process is as follows:
1. Set up a reaction device R1 using a 3L three-necked bottle, and configure a water separator on the device.
2. 10 boric acid (150.00 g, 2.46 mol, 1.00 eq) was added to the reaction R1 at room temperature (about 25 ° C).
3. Add n-butanol (1.00 L) to the reaction R1 at room temperature (about 25 ° C) and stir, and most of the boric acid cannot be dissolved.
4. Toluene (1.00 L) was added to the reaction R1 at room temperature (about 25 ° C) and stirred.
5. Concentrated sulfuric acid (4.82 g, 49.16 mmol, 2.62 mL, 0.02 eq) was added dropwise to the reaction at room temperature (about 25 ° C), at which time a large amount of solid remained undissolved.
6. The reaction system was heated to 130 ° C, and the water was continuously removed, stirred for 3.5 hours, and water (about 140 g) was formed in the water separator. The solids were all dissolved, and the solution changed from colorless to brown. .
8. Distill off most of the toluene at atmospheric pressure.
9. After most of the toluene is distilled off, the temperature of the system is lowered to 20 to 30 ° C, and the reaction liquids of the two reactions are combined, and the apparatus is changed for distillation.
10. Oil bath external temperature 108-110 ° C pump distillation under reduced pressure, Kelvin thermometer 45 ° C, distilled n-butanol.
11. Oil bath external temperature 108-110 ° C oil pump distillation under reduced pressure, the residual butanol was distilled off.
12. Oil bath external temperature 118-120 ° C oil pump vacuum distillation, Kelvin thermometer 55 ° C, began to produce products.
13. The temperature is raised to 135-140 ° C oil pump vacuum distillation, the product is completely distilled.
14. The product is obtained as a colorless liquid 10 tributyl borate (830.00g, 3.62mol, yield 73.58%).
The results of the 1 H NMR analysis of the obtained tributyl 10 borate were as follows:
Three, -N- tert-butoxycarbonyl-4-iodo-phenylalanine was prepared (S) of (S) -N- tert-butoxycarbonyl-4-hydroxy-10-yl -L- phenylalanine boron
Please refer to the following reaction formula III, which is (S)-N-tert-butoxycarbonyl-4-iodophenylalanine with tributyl 10 borate, t-butyl magnesium chloride (t-BuMgCl) and bis (2-A) yl aminoethyl) ether (BDMAEE) reaction, to produce (S) -N- tert-butoxycarbonyl group -4- (10 B) dihydroxyboryl -L- phenylalanine chemical reaction.
In the preparation process, two reaction vessels were selected for the reaction.
The specific operation process is as follows:
1. Set up a reaction using a 3L three-neck bottle.
2. Tributyl 10 borate (187.60 g, 87.98 mmol, 3.20 eq) was placed in the reaction system at room temperature (about 22 ° C).
3. Sodium hydride (20.45 g, 511.24 mmol, purity 60%, 2.00 eq) was added to the reaction system at room temperature (about 22 ° C). The reaction solution was a suspension and stirred at room temperature (about 22 ° C). 5 minutes.
4. Bis(2-methylaminoethyl)ether (327.73 g, 2.04 mol, 8.00 eq) was added to the reaction at room temperature (about 22 ° C).
5. N-tert-Butoxycarbonyl-4-iodo-L-phenylalanine (100.00 g, 255.62 mmol, 1.00 eq) was added to the reaction system at room temperature (about 22 ° C), and a large amount of solid was not dissolved.
6. Lower the temperature of the reaction system to 0-5 ° C, add t-butyl magnesium chloride (1.7 M, 1.20 L, 2.04 mol, 8.00 eq) to the reaction, control the temperature between 0-10 ° C, the dropping time is about It is 1.5 hours.
7. After the completion of the charging, the temperature of the reaction system was naturally raised to room temperature (20 to 30 ° C) and stirred at this temperature for 12 hours.
8. Using high performance liquid chromatography (HPLC) to detect about 9.00% of the remaining material.
9. When the temperature of the reaction system was lowered to -5 to 0 ° C, it was quenched by dropwise addition of 500 mL of water.
10. Lower the temperature of the system to 0-5 ° C, add methyl tert-butyl ether (500 mL) to the reaction system and adjust the pH to 2.9-3.1 (using a pH meter) with 37% HCl (about 500 mL). Exothermic, the temperature of the control system is between 0-15 °C.
11. The aqueous phase obtained by liquid separation was extracted once with methyl tert-butyl ether (500 mL), and the obtained organic phases were combined to give an organic phase of about 1.1 L.
12. Slowly add a sodium hydroxide aqueous solution (1 M, 400 mL) to the obtained organic phase, exotherm during the dropwise addition, and control the system temperature between 0-15 °C.
13. After the completion of the dropwise addition, the pH of the system was about 10, and the pH was adjusted to between 12.10 and 12.6 with an aqueous sodium hydroxide solution (4M). (measured with a pH meter)
14. Dispensing.
15. The aqueous phase 1 obtained after liquid separation was extracted once with n-butanol (500 ml) to obtain aqueous phase 2.
16. Combine the aqueous phase 2 of the two reaction vessels.
17. Adjust the pH of the aqueous phase to 2.9-3.1 with 37% HCl, stir for about 40 minutes, and precipitate a large amount of solid.
18. Filtration gave a white solid which was washed once with dichloromethane (50 mL).
19. At 25 ° C, the precipitated solid was slurried with dichloromethane (150 mL) and stirred for 10 min.
20. A white solid was filtered to give (S) -N- tert-butoxycarbonyl group -4- (10 B) dihydroxyboryl -L- phenylalanine (75.00g, 240.82mmol, by HPLC analysis, a yield of 47.11% , purity 99%).
The prepared (S) -N- tert-butoxycarbonyl group -4- (10 B) results dihydroxyboryl -L- phenylalanine 1 HNMR was as follows:
Preparation of L- 10 BPA from (S)-N-tert-Butoxycarbonyl-4-dihydroxyboryl-L-phenylalanine
See the following reaction scheme IV, which is (S) -N- tert-butoxycarbonyl group -4- (10 B) of amine end dihydroxyboryl -L- phenylalanine deprotection of the chemical reaction, to obtain L- 10 BPA.
The specific operation process is as follows:
1. Set up a reaction using a 1L three-neck bottle.
2. room temperature (20-30 deg.] C) to (S) -N- tert-butoxycarbonyl group -4- (10 B) dihydroxyboryl -L- phenylalanine (67.00g, 217.31mmol, 1.00eq) was added the reaction In the system.
3. room temperature (20-30 deg.] C) water (23.75mL) and acetone (Acetone, 420.00mL) were added dropwise to the reaction flask, stirred (S) -N- tert-butoxycarbonyl group -4- (10 B) dihydroxy Boronyl-L-phenylalanine.
4. Concentrated hydrochloric acid (23.93 g, 656.28 mmol, 23.46 mL, 3.02 eq) was added dropwise to the reaction system at room temperature (20-30 ° C). After the addition was completed, the reaction system was heated to 55-60 ° C and stirred for 4.5 hours.
5. HPLC detection until the reaction of the starting material is completed.
6. The temperature is controlled below 40 ° C, and the acetone in the reaction system is concentrated.
7. Lower the concentrated system to below 15 °C, adjust the pH of the system to about 1.5 with sodium hydroxide solution (4M) (pH meter detection), stir for 40 minutes and continue to adjust the pH of the system to 6.15 using sodium hydroxide solution (4M). ~6.25, a large amount of white solid precipitated, which was filtered to give a white solid, and rinsed with acetone (200mL).
8. Obtained as a white solid L- 10 BPA (36.00 g, 171.17 mmol, HPLC, yield 78.77%, purity 99%).
The analytical results obtained by the L- 10 BPA 1 HNMR are as follows:
Preparation of (S)-N-tert-butoxycarbonyl-4-dihydroxyboryl-L-phenylalanine from (S)-N-tert-butoxycarbonyl-4-iodophenylalanine
Please refer to the following reaction formula VII, which is a reaction of (S)-N-tert-butoxycarbonyl-4-iodophenylalanine with tributyl borate and t-butylmagnesium chloride (t-BuMgCl) to obtain (S The chemical reaction formula of -N-tert-butoxycarbonyl-4-dihydroxyboryl-L-phenylalanine.
The specific operation process is as follows:
1. Construct a reaction unit with a 250 mL three-neck bottle.
2. Tributyl borate (17.65 g, 76.68 mmol, 3.00 eq) was placed in a 250 mL reaction flask at 20-30 °C.
3. Sodium hydride (1.02 g, 25.56 mmol, 1.00 eq) was added to a 250 mL reaction vial at 20-30 °C.
4. (S)-N-tert-Butoxycarbonyl-4-iodo-L-phenylalanine (10.00 g, 25.56 mmol, 1.00 eq) was added to a 250 mL reaction vial at 20-30 °C.
5. Reduce the temperature of the reaction system to 0 ° C under nitrogen atmosphere, slowly add t-butyl magnesium chloride (1.7 M in THF, 120 mL, 8.00 eq) to the reaction, the dropping time is about 30 minutes, and the control temperature is 0. Between °C and 10 °C.
Stir at 20.20 ~ 30 ° C for 20 hours.
7. HPLC detection of the basic reaction of the raw materials, leaving only about 0.7% of the raw materials.
8. At a temperature of 0 ° C, 5 mL of water was added dropwise to the reaction to quench it. After complete quenching, stirring was continued for 10 minutes.
9. Cool down to 0 ° C, add methyl tert-butyl ether (50 mL) to the reaction and adjust the pH to 3 with 37% HCl (about 50 mL) (detected with a pH meter), adjust the pH during the process to exotherm, control the temperature at 0 Between °C and 15 °C.
12. The aqueous phase obtained by liquid separation was extracted once with methyl t-butyl ether (50 mL) and the organic phases were combined.
12. Add NaOH solution (1M, 55mL) to the obtained organic phase to adjust the pH to between 12.10-12.6. The process is exothermic and the temperature is controlled between 0 °C and 15 °C.
13. Liquid separation, the obtained aqueous phase was extracted once with n-butanol (50 mL), and most of the impurities were extracted and removed.
14. The aqueous phase obtained by liquid separation was adjusted to pH 3 with 37% HCl and stirred for about 30 minutes to precipitate a white solid.
15. Filtration gave a white solid which was washed once with dichloromethane (50 mL).
16. The precipitated solid was slurried with 25 mL of dichloromethane at 25 ° C and stirred for 10 minutes.
Please continue to refer to Reaction Scheme VII. The specific operation process is as follows:
1. Construct a reaction unit with a 250 mL three-neck bottle.
2. Tributyl borate (8.82 g, 38.34 mmol, 3.00 eq) was added to a 250 mL reaction vial at 20-30 °C.
3. Sodium hydride (511.25 mg, 12.78 mmol, 1.00 eq) was added to a 250 mL reaction vial at 20-30 °C.
4. (S)-N-tert-Butoxycarbonyl-4-iodo-L-phenylalanine (5.00 g, 12.78 mmol, 1.00 eq) was added to a 250 mL reaction vial at 20-30 °C.
5. The temperature of the reaction system was lowered to 0 ° C under nitrogen atmosphere, and t-butyl magnesium chloride (1.7 M in THF, 60 mL, 8.00 eq) was added dropwise to the reaction, the dropwise addition time was about 30 minutes, and the control temperature was 0 ° C. -10 ° C between.
Stir at 6.20 ~ 30 ° C for 22 hours.
7. HPLC detection of the raw material reaction is completed.
8. At a temperature of 0 ° C, 2.5 mL of water was added dropwise to the reaction to quench it. After complete quenching, stirring was continued for 10 minutes.
9. Cool down to 0 ° C, add methyl tert-butyl ether (25 mL) to the reaction and adjust the pH to 3 with 37% HCl (about 25 mL) (detected with a pH meter), adjust the pH during the process to exotherm, control the temperature at 0 Between °C and 15 °C.
12. The aqueous phase obtained by liquid separation was extracted once with methyl t-butyl ether (25 mL) and the organic phases were combined.
12. Add NaOH solution (1M, 30mL) to the obtained organic phase to adjust the pH to between 12.10-12.6. The process is exothermic and the temperature is controlled between 0 °C and 15 °C.
13. Liquid separation, the obtained aqueous phase was extracted once with n-butanol (25 ml), and most of the impurities were extracted and removed.
14. The aqueous phase obtained by liquid separation was adjusted to pH 3 with 37% HCl and stirred for about 30 minutes to precipitate a white solid.
15. Filtration gave a white solid which was washed once with dichloromethane (25 mL).
16. The precipitated solid was slurried with 15 mL of dichloromethane at 25 ° C and stirred for 10 minutes.
17. Filtration gave (S)-N-tert-butoxycarbonyl-4-dihydroxyboryl-L-phenylalanine (3.4 g, obtained by HPLC, yield: 85.26%, purity 98%).
Bis(2-methylaminoethyl)ether is a complexing agent for Mg, which can reduce the occurrence of side reactions in the reaction. The reactions of Examples 6 and 7 were carried out without adding bis(2-methylaminoethyl)ether. The analysis showed that the iodine impurity in the reaction of Example 6 was about 17%, and the iodine impurity in the reaction of Example 7 was observed. About 28%. Therefore, it has been proved from the side that the addition of bis(2-methylaminoethyl)ether can protect the reaction from reducing iodine.
The BPA or 10 BPA obtained in the above examples were analyzed by chiral HPLC, and the ratio of the L-enantiomer to the D-enantiomer was 100:0.
The boron-containing drug L-BPA for neutron capture therapy disclosed in the present invention is not limited to the contents described in the above examples. The above-mentioned embodiments are only examples for convenience of description, and the scope of the claims should be determined by the claims.
The most common side effects include neutropenia (low levels of neutrophils, a type of white blood cell), infusion reactions, pneumonia (infection of the lungs), upper respiratory tract infection (such as nose and throat infections), diarrhoea and bronchitis (inflammation of the airways in the lungs).[3]
In the European Union it is indicated, in combination with pomalidomide and dexamethasone, for the treatment of adults with relapsed and refractory multiple myeloma (MM) who have received at least two prior therapies including lenalidomide and a proteasome inhibitor (PI) and have demonstrated disease progression on the last therapy.[3]
Researchers started a Phase I study with isatuximab in combination with pomalidomide and dexamethasone for the treatment of patients with multiple myeloma (MM). The results during the Phase I trial showed that 26 out of the 45 patients discontinued the treatment due to progression of the disease. The patients had already been heavily pretreated. The latter lead to a manageable safety profile where the dose of isatuximab in combination with pomalidomide and dexamethasone would be capped to the maximum of 10 mg/kg weekly every two weeks for future studies.[12]
Based on the remarkable findings during the Phase I trial, a Phase II trial was launched where researchers investigated isatuximab as a single agent in patients with MM. The heavily pretreated patients reacted well to the single administration of isatuximab during Phase II of the trial.[13]
A Phase III combination trial for plasma cell myeloma is comparing pomalidomide and dexamethasone with and without isatuximab is in progress with an estimated completion date of 2021.[medical citation needed]
Additionally, two Phase III trials were added in 2017. The first trial highlights whether there is an added value in the combination of isatuximab with bortezomib, lenalidomide and dexamethasone. The latter will be tested in patients with newly diagnosed MM who are not qualified for a transplant (IMROZ trial). The second trial evaluates the combinations of isatuximab with carfilzomib and dexamethasone compared to carfilzomib with dexamethasone. The second trial was designed for patients who were previously treated with one to three prior lines (IKEMA). There is currently[when?] no treatment for MM, however promising improvements have been made and the study is still ongoing.[14][15]
In March 2020, it was approved for medical use in the United States.[8][9][10]
The U.S. Food and Drug Administration (FDA) approved isatuximab-irfc in March 2020, based on evidence from a clinical trial (NCT02990338) of 307 subjects with previously treated multiple myeloma.[10] The trial was conducted at 102 sites in Europe, North America, Asia, Australia and New Zealand.[10]
The trial evaluated the efficacy and side effects of isatuximab-irfc in subjects with previously treated multiple myeloma.[10] Subjects were randomly assigned to receive either isatuximab-irfc (in combination with pomalidomide and low-dose dexamethasone) or active comparator (pomalidomide and low-dose dexamethasone).[10] Treatment was administered in both groups in 28-day cycles until disease progression or unacceptable toxicity.[10] Both subjects and health care providers knew which treatment was given.[10] The trial measured the time patients lived without the cancer growing (progression-free survival or PFS).[10]
It was approved for medical use in the European Union in May 2020.[3]
Structure and reactivity
The structure of isatuximab consists of two identical immunoglobulin kappa light chains and also two equal immunoglobulin gamma heavy chains. Chemically, isatuximab is similar to the structure and reactivity of daratumumab, hence both drugs show the same CD38 targeting. However, isatuximab shows a more potent inhibition of its ectozyme function. The latter gives potential for some non-cross reactivity. Isatuximab shows action of an allosteric antagonist with the inhibition of the CD38 enzymatic activity. Additionally, isatuximab shows potential where it can induce apoptosis without cross linking.[16] Lastly, Isatuximab reveals direct killing activity when a larger increase in apoptosis is detected in CD38 expressing cancer cells. Furthermore, isatuximab demonstrated a dose dependent inhibition of CD38 enzymatic activity. However, daratumumab with the same experimental conditions shows a more limited inhibition without a dose response.[17]
Reactions
Isatuximab binds uniquely to an epitope on the CD38 receptor and is the only CD38 antibody which can start apoptosis directly.[18] Isatuximab binds to a different CD38 epitopeamino-acid sequence than does the anti-CD38 monoclonal antibody daratumumab.[19] The binding with the CD38 receptor is mainly via the gamma heavy chains and are more potent than other CD38 antibodies such as daratumumab which can inhibit the enzymatic activity of CD38. Moreover, isatuximab inhibits the hydrolase activity of CD38.[medical citation needed]
The antibodies show signs of improving antitumor immunity by eliminating regulatory T cells, B cells and myeloid-derived suppressor cells. The difference in binding between isatuximab and daratumumab is in the recognition of the different amino acid groups. Isatuximab identifies 23 amino acids of CD38 to the contrary with daratumumab who has 27. The residue of Glu233 has a flexible sidechain and faces the N-terminal of Asp1 residue in the isatuximab light chain. The latter light chain of isatuximab is also flexible which makes the interaction between CD38/Glu233 and the Asp1 weaker than the other interactions between CD38 and isatuximab. The caspase-dependent apoptotic pathway and the lysosomal mediated cell death pathway in MM cells is induced by isatuximab. The MM cell death follows the downstream reactions of the lysosomal activation. The latter also activates the production of reactive oxygen species.[20]
Available forms
Isatuximab or isatuximab-irfc is available as a drug in an intravenous infusion form. Injection doses are 100 mg/5 mL (20 mg/mL) solution in single-dose vial or 500 mg/25 mL (20 mg/mL) solution in single-dose vial.[4]
Mechanism of action
Cancer of the blood that is distinguished by an overproduction of malignant plasma cells in the bone marrow is called multiple myeloma. The myeloma cells are marked with uniformed overexpression of CD38 surface glycoproteins. Although these proteins are also expressed on other myeloid and lymphoid cells, the extent is relatively minor compared to myeloma cells. The fact that CD38 glycoproteins carry out various important cellular functions, and that they are plentiful on the surface of myeloma cells, has made them an appealing target for multiple myeloma treatment.[21] CD38 was first described as an activation marker, but later the molecule displayed functions in adhesion to endothelial CD31 proteins, e.g. as an aiding component of the synapse complex, as well as an ectoenzyme implicated in the metabolism of extracellular NAD+ and cytoplasmic NADP. The tumour cells can evade the immune system, possibly due to adenosine, an immunosuppressive molecule that arises as a product of the ectoenzymatic activity of CD38.[22]
Isatuximab-irfc is an IgG1-derived monoclonal antibody that selectively binds to the CD38 that exists on the exterior of hematopoietic and multiple myeloma cells (as well as other tumor cells). This drug induces apoptosis of tumor cells and activates immune effector mechanisms such as complement dependent cytotoxicity (CDC), antibody-dependent cellular phagocytosis (ADCP), and antibody-dependent cell-mediated cytotoxicity (ADCC). Isatuximab-irfc is able to stimulate natural killer (NK) cells in the absence of CD38-positive target tumor cells and blocks CD38-positive T-regulatory cells.[4] Furthermore, the NADase activity of CD38 is adjusted by isatuximab, similarly to other CD38 antibodies. Contrarily to daratumumab however, isatuximab can incite apoptosis directly without cross-linking, and in its binding epitope.[23] According to the FDA, isatuximab-irfc alone has reduced ADCC and direct tumor cell killing activity in vitro in comparison to when it is combined with pomalidomide. As well as increased anti-tumor activity as opposed to isatuximab-irfc or pomalidomide only in a human multiple myeloma xenograft model.[4]
Metabolism and toxicity
Metabolism
Isatuximab-irfc is likely to be metabolized through catabolic pathways into smaller peptides. When isatuximab is at a constant state it is expected that the ≥99% elimination will occur approximately two months after the last dose was administered. The clearance percentage diminished when the dosages were increased over time, as well as when multiple doses were administered. However, the elimination of isatuximab-irfc did not differ when applied as a single agent or as a combination therapy.[4]
Toxicity
A dose-limiting toxicity (DLT) has characterized been characterized as the development of any of the following: grade ≥ 3 non-hematologic toxicity; grade 4 neutropenia or grade 4 thrombocytopenia lasting more than 5 days; grade ≥ 2 allergic reactions or hypersensitivity (i.e., infusion reactions); or any other toxicity considered to be dose-limiting by the investigators or sponsor. Grade ≤ 2 infusion reactions were excluded from the DLT definition, because, with suitable care, patients that suffered a grade 2 infusion reaction prior to completion of the infusion were able to finalize isatuximab administration.[23]
There is no recommended reduced dose of isatuximab-irfc. In the eventuality of hematological toxicity it may be necessary to delay administration so that the blood count may be recovered.[4] Although there is no counteracting agent for isatuximab, clinical experience with overdoses is seemingly nonexistent as well. Overdose symptoms will probably be in line with the side effects attached to isatuximab. Therefore, infusion reactions, gastrointestinal disturbances and an elevated risk of infections may occur. It is necessary to carefully monitor the patient in case of an overdose and to employ clinically indicated symptomatic and supportive procedures.[21]
No studies have been conducted with isatuximab concerning carcinogenicity, genotoxicity or fertility.[4]
Pregnancy
When given to pregnant women isatuximab-irfc can cause fetal injury, due to the mechanism of action. It can precipitate depletion of immune cells as well as decreased bone density in the fetus. Pregnant women are therefore notified of the potential risks to a fetus, and women that are able to reproduce are advised to use effective contraceptives during treatment and at least five months subsequent to the last dose of isatuximab-irfc.
Furthermore, it is not recommended to combine isatuximab-irfc with pomalidomide in women that are carrying a child, because pomalidomide may cause birth defects and death of the unborn child.[4]
Indications
Isatuximab is indicated as a CD38-directed cytolytic antibody. By inhibiting the enzymatic activity of CD38.
The binding of isatuximab to CD38 on multiple myeloma (MM) cells leads to a trigger to several mechanisms leading to direct apoptosis of target cancer cells. The triggered pathways are the caspase-dependent apoptotic and the lysosome-mediated cell death pathway in MM cells.[24]
It is used in a combination with dexamethasone and pomalidomide. The drug is thus to treat patients with multiple myeloma. Restrictions for the use of isatuximab is that the patients have to be adults who have at least received two previous treatments with lenalidomide and a proteasome inhibitor.[4]
Isatuximab is currently[when?] also being tested in a Phase II trial as a monotherapy against refractory/recurrent systemic light-chain amyloidosis.[24]
Efficacy and side effects
Efficacy
A Phase III study of patients with refractory and relapsed MM, who were resistant to lenalidomide and a proteasome inhibitor, and could not have received daratumumab, another anti-CD38 monoclonal antibody was published in 2019 (ICARIA-MM). The addition of isatuximab to pomalidomide and dexamethasone improved progression free survival to 11.5 months compared to 6.5 months, with an overall response rate of 63%.[25]
Side effects
Adverse reactions to isatuximab-irfc may include neutropenia, infusion-related reactions and/or secondary primary malignancies.[4] Of these three the most commonly occurring ones are the infusion-related reactions.[24] Examples of the most frequent symptoms of infusion-related reactions are dyspnea, cough, chills, and nausea, while the severest signs and symptoms included hypertension and dyspnea.[4]
Effects on animals
The activity of isatuximab has been researched in mouse tumor models. It has been proven that isatuximab leads to antitumor activity in MM cells. Furthermore, the combination of isatuximab and pomalidomide will lead to an extra enhanced antitumor activity in MM cells. Thus, pomalidomide in vivo and in vitro leads to an increase of the activity of isatuximab.[24]
Animal studies in reproduction toxicity have not yet been carried out. So, the risks of birth defects and miscarriage risks are unknown.[4]
^ Orlowski RZ, Goldschmidt H, Cavo M, Martin TG, Paux G, Oprea C, Facon T (20 May 2018). “Phase III (IMROZ) study design: Isatuximab plus bortezomib (V), lenalidomide (R), and dexamethasone (d) vs VRd in transplant-ineligible patients (pts) with newly diagnosed multiple myeloma (NDMM)”. Journal of Clinical Oncology. 36 (15_suppl): TPS8055. doi:10.1200/JCO.2018.36.15_suppl.TPS8055.
“Isatuximab”. Drug Information Portal. U.S. National Library of Medicine.
Clinical trial number NCT02990338 for “Multinational Clinical Study Comparing Isatuximab, Pomalidomide, and Dexamethasone to Pomalidomide and Dexamethasone in Refractory or Relapsed and Refractory Multiple Myeloma Patients (ICARIA-MM)” at ClinicalTrials.gov
Molecular Formula: C13H16N2O2Molecular Weight: 232.28Percent Composition: C 67.22%, H 6.94%, N 12.06%, O 13.78%
Literature References: A hormone of the pineal gland, also produced by extra-pineal tissues, that lightens skin color in amphibians by reversing the darkening effect of MSH, q.v. Melatonin has been postulated as the mediator of photic-induced antigonadotrophic activity in photoperiodic mammals and has also been shown to be involved in thermoregulation in some ectotherms and in affecting locomotor activity rhythms in sparrows. Isoln from the pineal glands of beef cattle: Lerner et al.,J. Am. Chem. Soc.80, 2587 (1958); Wurtman et al.,Science141, 277 (1963). Structure: Lerner et al.,J. Am. Chem. Soc.81, 6084 (1959). Crystal and molecular structure: A. Wakahara, Chem. Lett.1972, 1139. Synthesis from 5-methoxyindole as starting material by two different routes: Szmuszkovicz et al.,J. Org. Chem.25, 857 (1960). Biochemical role of melatonin: Chem. Eng. News45, 40 (May 1, 1967). Pharmacological studies: Barchas et al.,Nature214, 919 (1967). Identification of antigonadal action sites in mouse brain: J. D. Glass, G. R. Lynch, Science214, 821 (1981). Binding studies in human hypothalamus: S. M. Reppert et al.,Science242, 78 (1988). Efficacy in control of estrus in red deer: G. W. Asher, Anim. Reprod. Sci.22, 145 (1990). Reviews: M. K. Vaughn, Int. J. Rev. Physiol.24, 41-95 (1981); D. C.Klein et al.,Life Sci.28, 1975-1986 (1981). Book: Advan. Biosci.vol. 29, N. Birau, W. Schlott, Eds. (Pergamon Press, New York, 1981) 420 pp. Review of etiological role in clinical disease: A. Miles, D. Philbrick, Crit. Rev. Clin. Lab. Sci.25, 231-253 (1987); in psychiatric disorders: eidem,Biol. Psychiatry23, 405-425 (1988).Properties: Pale yellow leaflets from benzene, mp 116-118°. uv max: 223, 278 nm (e 27550, 6300).Melting point: mp 116-118°Absorption maximum: uv max: 223, 278 nm (e 27550, 6300)Therap-Cat-Vet: Control of estrus.
Melatonin is a hormone primarily released by the pineal gland that regulates the sleep–wake cycle.[3][4] As a dietary supplement, it is often used for the short-term treatment of insomnia, such as from jet lag or shift work, and is typically taken by mouth.[5][6][7] Evidence of its benefit for this use, however, is not strong.[8] A 2017 review found that sleep onset occurred six minutes faster with use, but found no change in total time asleep.[6] The melatonin receptor agonist medication ramelteon may work as well as melatonin supplements,[6] at greater cost but with different adverse effects, for some sleep conditions.[9]
Melatonin was discovered in 1958.[3] It is sold over the counter in Canada and the United States;[10][13] in the United Kingdom, it is a prescription-only medication.[7] It is not approved by the US Food and Drug Administration (FDA) for any medical use.[10] In Australia and the European Union, it is indicated for difficulty sleeping in people over the age of 54.[20][11] In the European Union, it is indicated for the treatment of insomnia in children and adolescents.[12] It was approved for medical use in the European Union in 2007.[11]
Chemical Synthesis of Melatonin The methods for the chemical synthesis of melatonin are generally not so complicated and do not involve more than three steps of conversion. Three synthesis reactions of melatonin from primary literatures are shown below;
Reaction 1
In 1958 melatonin was first isolated and characterised by A.B.Lerner. It was know as one of a substituted 5-hydroxyindole derivative in the pineal gland that could lighten pigment cells. It had not been know to exist in biological tissue although it had been isolated as a urinary excretion product in rats after administration of 5-hydroxytryptamine. Melatonin or N-acetyl-5-methoxytryptamine (40 mg) was prepared by reducing 100 mg of 5-methoxyindole-3-acetonitrile with 160 mg of sodium and 2 ml of ethanol. Then the product was acetylated with 4 ml of both glacial acetic acid and acetic anhydride at 100 oC for 1 minute. Purification was achieved by countercerrent distribution and silicic acid chromatography.
Reaction 2
5-Methoxytryptamine hydrochloride (1g, 4.75 mmole) was dissolved in pyridine (10 ml) and acetic anhydride (10 ml) and kept overnight at 20 oC. The solution was poured onto iced, neutralised with dilute hydrochloric acid and extracted with chloroform (2×25 ml). The combined extracts were washed with water, dried in MgSO4 and evaporated to afford a liquid of N,N diacetyltryptamine derivative. The liquid was then poured into water (50 ml) and extracted with chlroform (2×25 ml). The combined organic layers were washed with water (25 ml), dried in MgSO4 and evaporated to dryness. The residual solid crystallised from benzene to afford melatonin 819 mg, 80% yield.
Reaction 3
The more reactive indoles (1a-1d) were alkylated at the 3 position by reaction with nitroethene generated in situ by thermolysis of nitroethyl acetate. The nitroethyl acetate used for this purpose was prepared by acetylation of nitroethanol with acetic anhydride using NaOAc as a catalyst. These conditions constitute a substantial improvement of the overal yield of the reation. Reduction of the nitroethylated indoles (2a-d) by hydrogenation over PtO2, followed by acetylation fo the resluting tryptamines with acetic anhydride-pyridine completed the synthesis of melatonin and its derivatives (4a-d).
Biological Synthesis and Metabolism of Melatonin
The biosynthesis of melatonin (Fig.1) is initiated by the uptake of the essential amino acid tryptophan into pineal parenchymal cells. Tryptophan is the least abundant of essential amino acids in normal diets. It is converted to another amino acid, 5-hydroxytryptophan, through the action of the enzyme tryptopahn hydroxylase and then to 5-hydroxytryptamine (serotonin) by the enzyme aromatic amino acid decarboxylase. Serotonin concentrations are higher in the pineal than in any other organ or in any brain region. They exhibit a striking diurnal rhythm remaining at a maximum level during the daylight hours and falling by more than 80% soon after the onset of darkness as the serotonin is converted to melatonin, 5-hydroxytryptophol and other methoxyindoles. Serotonin’s conversion to melatonin involves two enzymes that are characteristic of the pineal : SNAT (serotonin-N-acetyltransferase) which converts the serotonin to N-acetylserotonin, and HIOMT (hydroxyindole-O-methyltrasferase) which trasfers a methyl group from S-adenosylmethionine to the 5-hydroxyl of the N-acetylserotonin. The activities of both enzymes rise soon after the onset of darkness because of the enhanced release of norepinephrine from sympathetic neurons terminating on the pineal parenchymal cells. Another portion of the serotonin liberated from pineal cells after the onset of darkness is deaminated by the enzyme monoamine oxidase (MAO) and then either oxidized to form 5-hydroxyindole acetic acid or reduced to form 5-hydroxytryptophol (Fig.1). Both of these compounds are also substrates for HIOMT and can thus be converted in the pineal to 5-methoxyindole acetic acid 5-methoxytryptophol (Fig.1). The level of this latter indole, like that of melatonin, rises markedly in the pineal with the onset of darkness. Since 5-methoxytryptophol synthesis does not require the acetylation of serotonin, the nocturnal increase in pineal SNAT activity cannot be the trigger that causes pineal methoxyindole levels to rise. More likely, a single unexplained process- the intraparenchymal release of stored pineal serotonin, which then becomes accessible to both SNAT and MAO. This process ultimately controls the rates at which all three major pineal methoxyindoles are synthesized and generates the nocturnal increases in pineal melatonin and 5-methoxytryptophol. The proportion of available serotonin acetylated at any particular time of day or night depends on the relative activities of pineal SNAT and MAO at that time. The rates of methylation of all three 5-hydroxyindoles formed from pinela serotonin depends on HIOMT activity.Fig.1 Biosynthesis of pineal methoxyindoles from serotonin
Serotonin may be either acetylated to form N-acetylserotonin through the action of the enzyme serotonin-N-acetyltransferase (SNAT), or oxidatively deaminated by monoamine oxidase (MAO) to yield an unstable aldehyde. This compound is then either oxidized to 5-hydroxyindole acetic acid by the enzyme aldehyde dehydrogenase (ADH), or reduced to from 5-hydroxytryptophol by aldehyde reductase (AR). Each of these 5-hydroxyindole derivatives of serotonin is a substrate for hydroxyindole-O-methyltrasferase (HIMOT). The enzymatic trasfer of a methyl group from S-adenosylmethionine to these hydroxyindoles yields melatonin (5-hydroxy-N-acetyltryptamine), 5-methoxyindole acetic acid and 5-methoxytryptophol respectively. Pineal serotonin is synthesized from the essential amino acid tryptophan by 5-hydroxylation folloed by decarboxylation. The first step in ths enzymic sequence is catalysed by tryptophan hydroxylase. The second step is catalysed by aromatic L-amino acid decarboxylase.
Medical uses
In the European Union it is indicated for the treatment of insomnia in children and adolescents aged 2–18 with autism spectrum disorder (ASD) and / or Smith–Magenis syndrome, where sleep hygiene measures have been insufficient[12] and for monotherapy for the short-term treatment of primary insomnia characterized by poor quality of sleep in people who are aged 55 or over.[11]
Sleep disorders
Positions on the benefits of melatonin for insomnia are mixed.[8] An Agency for Healthcare Research and Quality (AHRQ) review from 2015 stated that evidence of benefit in the general population was unclear.[8] A review from 2017, found a modest effect on time until onset of sleep.[3] Another review from 2017 put this decrease at six minutes to sleep onset but found no difference in total sleep time.[6] Melatonin may also be useful in delayed sleep phase syndrome.[3] Melatonin appears to work as well as ramelteon but costs less.[6]
A 2020 Cochrane review found no evidence that melatonin helped sleep problems in people with moderate to severe dementia due to Alzheimer’s disease.[26] A 2019 review found that while melatonin may improve sleep in minimal cognitive impairment, after the onset of Alzheimer’s it has little to no effect.[27] Melatonin may, however, help with sundowning.[28]
Jet lag and shift work
Melatonin is known to reduce jet lag, especially in eastward travel. If the time it is taken is not correct, however, it can instead delay adaption.[29]
Melatonin appears to have limited use against the sleep problems of people who work shift work.[30] Tentative evidence suggests that it increases the length of time people are able to sleep.[30]
Adverse effects
Melatonin appears to cause very few side effects as tested in the short term, up to three months, at low doses.[clarification needed] Two systematic reviews found no adverse effects of exogenous melatonin in several clinical trials and comparative trials found the adverse effects headaches, dizziness, nausea, and drowsiness were reported about equally for both melatonin and placebo.[31][32] Prolonged-release melatonin is safe with long-term use of up to 12 months.[33] Although not recommended for long term use beyond this, low-dose melatonin is generally safer, and a better alternative, than many prescription and over the counter sleep aids if a sleeping medication must be used for an extended period of time. Low-doses of melatonin are usually sufficient to produce a hypnotic effect in most people. Higher doses do not appear to result in a stronger effect, but instead appear to cause drowsiness for a longer period of time.[34]
In those taking warfarin, some evidence suggests there may exist a potentiating drug interaction, increasing the anticoagulant effect of warfarin and the risk of bleeding.[41]
Functions
When eyes receive light from the sun, the pineal gland’s production of melatonin is inhibited and the hormones produced keep the human awake. When the eyes do not receive light, melatonin is produced in the pineal gland and the human becomes tired.
Circadian rhythm
In animals, melatonin plays an important role in the regulation of sleep–wake cycles.[42] Human infants’ melatonin levels become regular in about the third month after birth, with the highest levels measured between midnight and 8:00 am.[43] Human melatonin production decreases as a person ages.[44] Also, as children become teenagers, the nightly schedule of melatonin release is delayed, leading to later sleeping and waking times.[45]
Antioxidant
Melatonin was first reported as a potent antioxidant and free radical scavenger in 1993.[46] In vitro, melatonin acts as a direct scavenger of oxygen radicals and reactive nitrogen species including OH•, O2−•, and NO•.[47][48] In plants, melatonin works with other antioxidants to improve the overall effectiveness of each antioxidant.[48] Melatonin has been proven to be twice as active as vitamin E, believed to be the most effective lipophilic antioxidant.[49] Via signal transduction through melatonin receptors, melatonin promotes the expression of antioxidant enzymes such as superoxide dismutase, glutathione peroxidase, glutathione reductase, and catalase.[50][51]
Melatonin occurs at high concentrations within mitochondrial fluid which greatly exceed the plasma concentration of melatonin.[52][53][54] Due to its capacity for free radical scavenging, indirect effects on the expression of antioxidant enzymes, and its significant concentrations within mitochondria, a number of authors have indicated that melatonin has an important physiological function as a mitochondrial antioxidant.[50][52][53][54][55]
The melatonin metabolites produced via the reaction of melatonin with reactive oxygen species or reactive nitrogen species also react with and reduce free radicals.[51][55] Melatonin metabolites generated from redox reactions include cyclic 3-hydroxymelatonin, N1-acetyl-N2-formyl-5-methoxykynuramine (AFMK), and N1-acetyl-5-methoxykynuramine (AMK).[51][55]
Immune system
While it is known that melatonin interacts with the immune system,[56][57] the details of those interactions are unclear. An antiinflammatory effect seems to be the most relevant. There have been few trials designed to judge the effectiveness of melatonin in disease treatment. Most existing data are based on small, incomplete trials. Any positive immunological effect is thought to be the result of melatonin acting on high-affinity receptors (MT1 and MT2) expressed in immunocompetent cells. In preclinical studies, melatonin may enhance cytokine production,[58] and by doing this, counteract acquired immunodeficiences. Some studies also suggest that melatonin might be useful fighting infectious disease[59] including viral, such as HIV, and bacterial infections, and potentially in the treatment of cancer.
In bacteria, protists, fungi, and plants, melatonin is synthesized indirectly with tryptophan as an intermediate product of the shikimate pathway. In these cells, synthesis starts with D-erythrose 4-phosphate and phosphoenolpyruvate, and in photosynthetic cells with carbon dioxide. The rest of the synthesising reactions are similar, but with slight variations in the last two enzymes.[62][63]
It has been hypothesized that melatonin is made in the mitochondria and chloroplasts.[64]
Mechanism
Mechanism of melatonin biosynthesis
In order to hydroxylate L-tryptophan, the cofactor tetrahydrobiopterin (THB) must first react with oxygen and the active site iron of tryptophan hydroxylase. This mechanism is not well understood, but two mechanisms have been proposed:
1. A slow transfer of one electron from the THB to O2 could produce a superoxide which could recombine with the THB radical to give 4a-peroxypterin. 4a-peroxypterin could then react with the active site iron (II) to form an iron-peroxypterin intermediate or directly transfer an oxygen atom to the iron.
2. O2 could react with the active site iron (II) first, producing iron (III) superoxide which could then react with the THB to form an iron-peroxypterin intermediate.
Iron (IV) oxide from the iron-peroxypterin intermediate is selectively attacked by a double bond to give a carbocation at the C5 position of the indole ring. A 1,2-shift of the hydrogen and then a loss of one of the two hydrogen atoms on C5 reestablishes aromaticity to furnish 5-hydroxy-L-tryptophan.[65]
A decarboxylase with cofactor pyridoxal phosphate (PLP) removes CO2 from 5-hydroxy-L-tryptophan to produce 5-hydroxytryptamine.[66] PLP forms an imine with the amino acid derivative. The amine on the pyridine is protonated and acts as an electron sink, enabling the breaking of the C-C bond and releasing CO2. Protonation of the amine from tryptophan restores the aromaticity of the pyridine ring and then imine is hydrolyzed to produce 5-hydroxytryptamine and PLP.[67]
It has been proposed that histidine residue His122 of serotonin N-acetyl transferase is the catalytic residue that deprotonates the primary amine of 5-hydroxytryptamine, which allows the lone pair on the amine to attack acetyl-CoA, forming a tetrahedral intermediate. The thiol from coenzyme A serves as a good leaving group when attacked by a general base to give N-acetylserotonin.[68]
N-acetylserotonin is methylated at the hydroxyl position by S-adenosyl methionine (SAM) to produce S-adenosyl homocysteine (SAH) and melatonin.[67][69]
Regulation
In vertebrates, melatonin secretion is regulated by activation of the beta-1 adrenergic receptor by norepinephrine.[70] Norepinephrine elevates the intracellular cAMP concentration via beta-adrenergic receptors and activates the cAMP-dependent protein kinase A (PKA). PKA phosphorylates the penultimate enzyme, the arylalkylamine N-acetyltransferase (AANAT). On exposure to (day)light, noradrenergic stimulation stops and the protein is immediately destroyed by proteasomalproteolysis.[71] Production of melatonin is again started in the evening at the point called the dim-light melatonin onset.
Blue light, principally around 460–480 nm, suppresses melatonin biosynthesis,[72] proportional to the light intensity and length of exposure. Until recent history, humans in temperate climates were exposed to few hours of (blue) daylight in the winter; their fires gave predominantly yellow light.[citation needed] The incandescent light bulb widely used in the 20th century produced relatively little blue light.[73] Light containing only wavelengths greater than 530 nm does not suppress melatonin in bright-light conditions.[74] Wearing glasses that block blue light in the hours before bedtime may decrease melatonin loss. Use of blue-blocking goggles the last hours before bedtime has also been advised for people who need to adjust to an earlier bedtime, as melatonin promotes sleepiness.[75]
When used several hours before sleep according to the phase response curve for melatonin in humans, small amounts (0.3 mg[77]) of melatonin shift the circadian clock earlier, thus promoting earlier sleep onset and morning awakening.[78] Melatonin is rapidly absorbed and distributed, reaching peak plasma concentrations after 60 minutes of administration, and is then eliminated.[61] Melatonin has a half life of 35–50 minutes.[79] In humans, 90% of orally administered exogenous melatonin is cleared in a single passage through the liver, a small amount is excreted in urine, and a small amount is found in saliva.[5] The bioavalibility of melatonin is between 10 and 50%.[61]
Melatonin is metabolized in the liver by cytochrome P450 enzyme CYP1A2 to 6-hydroxymelatonin. Metabolites are conjugated with sulfuric acid or glucuronic acid for excretion in the urine. 5% of melatonin is excreted in the urine as the unchanged drug.[61]
Some of the metabolites formed via the reaction of melatonin with a free radical include cyclic 3-hydroxymelatonin, N1-acetyl-N2-formyl-5-methoxykynuramine (AFMK), and N1-acetyl-5-methoxykynuramine (AMK).[51][55]
In 1958, dermatology professor Aaron B. Lerner and colleagues at Yale University, in the hope that a substance from the pineal might be useful in treating skin diseases, isolated the hormone from bovine pineal gland extracts and named it melatonin.[85] In the mid-70s Lynch et al. demonstrated that the production of melatonin exhibits a circadian rhythm in human pineal glands.[86]
The discovery that melatonin is an antioxidant was made in 1993.[87] The first patent for its use as a low-dose sleep aid was granted to Richard Wurtman at MIT in 1995.[88] Around the same time, the hormone got a lot of press as a possible treatment for many illnesses.[89]The New England Journal of Medicine editorialized in 2000: “With these recent careful and precise observations in blind persons, the true potential of melatonin is becoming evident, and the importance of the timing of treatment is becoming clear.”[90]
It was approved for medical use in the European Union in 2007.[11]
Other animals
In vertebrates, melatonin is produced in darkness, thus usually at night, by the pineal gland, a small endocrine gland[91] located in the center of the brain but outside the blood–brain barrier. Light/dark information reaches the suprachiasmatic nuclei from retinal photosensitive ganglion cells of the eyes[92][93] rather than the melatonin signal (as was once postulated). Known as “the hormone of darkness”, the onset of melatonin at dusk promotes activity in nocturnal (night-active) animals and sleep in diurnal ones including humans.
Many animals use the variation in duration of melatonin production each day as a seasonal clock.[94] In animals including humans,[95] the profile of melatonin synthesis and secretion is affected by the variable duration of night in summer as compared to winter. The change in duration of secretion thus serves as a biological signal for the organization of daylength-dependent (photoperiodic) seasonal functions such as reproduction, behavior, coat growth, and camouflage coloring in seasonal animals.[95] In seasonal breeders that do not have long gestation periods and that mate during longer daylight hours, the melatonin signal controls the seasonal variation in their sexual physiology, and similar physiological effects can be induced by exogenous melatonin in animals including mynah birds[96] and hamsters.[97] Melatonin can suppress libido by inhibiting secretion of luteinizing hormone and follicle-stimulating hormone from the anterior pituitary gland, especially in mammals that have a breeding season when daylight hours are long. The reproduction of long-day breeders is repressed by melatonin and the reproduction of short-day breeders is stimulated by melatonin.
During the night, melatonin regulates leptin, lowering its levels.
Cetaceans have lost all the genes for melatonin synthesis as well as those for melatonin receptors.[98] This is thought to be related to their unihemispheric sleep pattern (one brain hemisphere at a time). Similar trends have been found in sirenians.[98]
Plants
Until its identification in plants in 1987, melatonin was for decades thought to be primarily an animal neurohormone. When melatonin was identified in coffee extracts in the 1970s, it was believed to be a byproduct of the extraction process. Subsequently, however, melatonin has been found in all plants that have been investigated. It is present in all the different parts of plants, including leaves, stems, roots, fruits, and seeds, in varying proportions.[19][99] Melatonin concentrations differ not only among plant species, but also between varieties of the same species depending on the agronomic growing conditions, varying from picograms to several micrograms per gram.[63][100] Notably high melatonin concentrations have been measured in popular beverages such as coffee, tea, wine, and beer, and crops including corn, rice, wheat, barley, and oats.[19] In some common foods and beverages, including coffee[19] and walnuts,[101] the concentration of melatonin has been estimated or measured to be sufficiently high to raise the blood level of melatonin above daytime baseline values.
Although a role for melatonin as a plant hormone has not been clearly established, its involvement in processes such as growth and photosynthesis is well established. Only limited evidence of endogenous circadian rhythms in melatonin levels has been demonstrated in some plant species and no membrane-bound receptors analogous to those known in animals have been described. Rather, melatonin performs important roles in plants as a growth regulator, as well as environmental stress protector. It is synthesized in plants when they are exposed to both biological stresses, for example, fungal infection, and nonbiological stresses such as extremes of temperature, toxins, increased soil salinity, drought, etc.[63][102][103]
Occurrence
Dietary supplement
Melatonin is categorized by the US Food and Drug Administration (FDA) as a dietary supplement, and is sold over-the-counter in both the US and Canada.[5] FDA regulations applying to medications are not applicable to melatonin,[15] though the FDA has found false claims that it cures cancer.[104] As melatonin may cause harm in combination with certain medications or in the case of certain disorders, a doctor or pharmacist should be consulted before making a decision to take melatonin.[29] In many countries, melatonin is recognized as a neurohormone and it cannot be sold over-the-counter.[105]
Food products
Naturally-occurring melatonin has been reported in foods including tart cherries to about 0.17–13.46 ng/g,[106] bananas and grapes, rice and cereals, herbs, plums,[107] olive oil, wine[108] and beer. When birds ingest melatonin-rich plant feed, such as rice, the melatonin binds to melatonin receptors in their brains.[109] When humans consume foods rich in melatonin, such as banana, pineapple, and orange, the blood levels of melatonin increase significantly.[110]
Beverages and snacks containing melatonin were being sold in grocery stores, convenience stores, and clubs in May 2011.[111] The FDA considered whether these food products could continue to be sold with the label “dietary supplements”. On 13 January 2010, it issued a Warning Letter to Innovative Beverage, creators of several beverages marketed as drinks, stating that melatonin, while legal as a dietary supplement, was not approved as a food additive.[112] A different company selling a melatonin-containing beverage received a warning letter in 2015.[113]
Commercial availability
Immediate-release melatonin is not tightly regulated in countries where it is available as an over-the-counter medication. It is available in doses from less than half a milligram to 5 mg or more. Immediate-release formulations cause blood levels of melatonin to reach their peak in about an hour. The hormone may be administered orally, as capsules, gummies, tablets, or liquids. It is also available for use sublingually, or as transdermal patches.[medical citation needed]
Formerly, melatonin was derived from animal pineal tissue, such as bovine. It is now synthetic, which limits the risk of contamination or the means of transmitting infectious material.[15][114]
Melatonin is the most popular over-the-counter sleep remedy in the US, resulting in sales in excess of US$400 million during 2017.[115]
Research
A bottle of melatonin tablets. Melatonin is available in timed-release and in liquid forms.
Various uses and effects of melatonin have been studied. A 2015 review of studies of melatonin in tinnitus found the quality of evidence low, but not entirely without promise.[116]
Headaches
Tentative evidence shows melatonin may help reduce some types of headaches including cluster and hypnic headaches.[117][118]
Cancer
A 2013 review by the National Cancer Institutes found evidence for use to be inconclusive.[119] A 2005 review of unblinded clinical trials found a reduced rate of death, but that blinded and independently conducted randomized controlled trials are needed.[120]
Protection from radiation
Both animal[121] and human[122][123][124] studies have shown melatonin to protect against radiation-induced cellular damage. Melatonin and its metabolites protect organisms from oxidative stress by scavenging reactive oxygen species which are generated during exposure.[125] Nearly 70% of biological damage caused by ionizing radiation is estimated to be attributable to the creation of free radicals, especially the hydroxyl radical that attacks DNA, proteins, and cellular membranes. Melatonin has been described as a broadly protective, readily available, and orally self-administered antioxidant that is without known, major side effects.[126]
Epilepsy
A 2016 review found no beneficial role of melatonin in reducing seizure frequency or improving quality of life in people with epilepsy.[127]
Secondary dysmenorrhoea
A 2016 review suggested no strong evidence of melatonin compared to placebo for dysmenorrhoea secondary to endometriosis.[128]
Delirium
A 2016 review suggested no clear evidence of melatonin to reduce the incidence of delirium.[129]
Gastroesophageal reflux disease
A 2011 review said melatonin is effective in relieving epigastric pain and heartburn.[130]
Psychiatry
Melatonin might improve sleep in people with autism.[131] Children with autism have abnormal melatonin pathways and below-average physiological levels of melatonin.[132][133] Melatonin supplementation has been shown to improve sleep duration, sleep onset latency, and night-time awakenings.[132][134][135] However, many studies on melatonin and autism rely on self-reported levels of improvement and more rigorous research is needed.
While the packaging of melatonin often warns against use in people under 18 years of age, studies suggest that melatonin is an efficacious and safe treatment for insomnia in people with ADHD, including children. However, larger and longer studies are needed to establish long-term safety and optimal dosing.[136]
Melatonin in comparison to placebo is effective for reducing preoperative anxiety in adults when given as premedication. It may be just as effective as standard treatment with midazolam in reducing preoperative anxiety. Melatonin may also reduce postoperative anxiety (measured 6 hours after surgery) when compared to placebo.[137]
Some supplemental melatonin users report an increase in vivid dreaming. Extremely high doses of melatonin increased REM sleep time and dream activity in people both with and without narcolepsy.[138] Some evidence supports an antidepressant effect.[139]
^ Jump up to:abc Buscemi N, Vandermeer B, Pandya R, Hooton N, Tjosvold L, Hartling L, et al. (November 2004). “Melatonin for treatment of sleep disorders” (PDF). Evidence Report/Technology Assessment No. 108. (Prepared by the University of Alberta Evidence-based Practice Center, Under Contract No. 290-02-0023.) AHRQ Publication No. 05-E002-2. Rockville, MD: Agency for Healthcare Research and Quality. Agency for Healthcare Research and Quality (AHRQ), US Department of Health and Human Services (108): 1–7. doi:10.1037/e439412005-001. PMC4781368. PMID15635761. Retrieved 5 June2013.
^ Jump up to:abcdef Matheson E, Hainer BL (July 2017). “Insomnia: Pharmacologic Therapy”. American Family Physician. 96 (1): 29–35. PMID28671376.
^ Jump up to:abc Brasure M, MacDonald R, Fuchs E, Olson CM, Carlyle M, Diem S, et al. (2015). “Management of Insomnia Disorder[Internet]”. AHRQ Comparative Effectiveness Reviews. 15 (16): EHC027–EF. PMID26844312. Evidence for benzodiazepine hypnotics, melatonin agonists in the general adult population, and most pharmacologic interventions in older adults was generally insufficient
^ Adams, Katie S. (2014). “Melatonin agonists in the management of sleep disorders: A focus on ramelteon and tasimelteon”. Mental Health Clinician. 4 (2): 59–64. doi:10.9740/mhc.n190087. Retrieved 25 October 2020. However, the clinical relevance of this objective and therefore the author’s conclusion that these results support the potential use of ramelteon in circadian rhythm sleep disorders is questionable. … It is unclear whether Takeda Pharmaceuticals will pursue FDA indications for ramelteon for circadian rhythm disorders given these results.
^ Boutin JA, Audinot V, Ferry G, Delagrange P (August 2005). “Molecular tools to study melatonin pathways and actions”. Trends in Pharmacological Sciences. 26 (8): 412–9. doi:10.1016/j.tips.2005.06.006. PMID15992934.
^ Hardeland R (July 2005). “Antioxidative protection by melatonin: multiplicity of mechanisms from radical detoxification to radical avoidance”. Endocrine. 27 (2): 119–30. doi:10.1385/ENDO:27:2:119. PMID16217125.
^“Australian Public Assessment Report for Melatonin” (PDF). Australian Government Department of Health and Ageing Therapeutic Goods Administration. January 2011. pp. 2, 4. Retrieved 9 January 2019. Monotherapy for the short term treatment of primary insomnia characterised by poor quality of sleep in patients who are aged 55 or over.
^ Gao C, Scullin MK, Bliwise DL (2019). “Mild Cognitive Impairment and Dementia”. In Savard J, Ouellet MC (eds.). Handbook of Sleep Disorders in Medical Conditions. Academic Press. pp. 253–276. doi:10.1016/b978-0-12-813014-8.00011-1. ISBN978-0-12-813014-8.
^ Lyseng-Williamson KA (November 2012). “Melatonin prolonged release: in the treatment of insomnia in patients aged ≥55 years”. Drugs & Aging. 29 (11): 911–23. doi:10.1007/s40266-012-0018-z. PMID23044640. S2CID1403262.
^ Database of Abstracts of Reviews of Effects (DARE): Quality-assessed Reviews [Internet]. York (UK): Centre for Reviews and Dissemination (UK); 1995. Optimal dosages for melatonin supplementation therapy in older adults: a systematic review of current literature. 2014.
^ Morera AL, Henry M, de La Varga M (2001). “[Safety in melatonin use]” [Safety in melatonin use]. Actas Espanolas de Psiquiatria (in Spanish). 29 (5): 334–7. PMID11602091.
^ Ardura J, Gutierrez R, Andres J, Agapito T (2003). “Emergence and evolution of the circadian rhythm of melatonin in children”. Hormone Research. 59 (2): 66–72. doi:10.1159/000068571. PMID12589109. S2CID41937922.
^ Poeggeler B, Saarela S, Reiter RJ, Tan DX, Chen LD, Manchester LC, Barlow-Walden LR (November 1994). “Melatonin—a highly potent endogenous radical scavenger and electron donor: new aspects of the oxidation chemistry of this indole accessed in vitro”. Annals of the New York Academy of Sciences. 738 (1): 419–20. Bibcode:1994NYASA.738..419P. doi:10.1111/j.1749-6632.1994.tb21831.x. PMID7832450. S2CID36383425.
^ Pieri C, Marra M, Moroni F, Recchioni R, Marcheselli F (1994). “Melatonin: a peroxyl radical scavenger more effective than vitamin E”. Life Sciences. 55 (15): PL271-6. doi:10.1016/0024-3205(94)00666-0. PMID7934611.
^ Jump up to:abcdefgh Jockers R, Delagrange P, Dubocovich ML, Markus RP, Renault N, Tosini G, et al. (September 2016). “Update on melatonin receptors: IUPHAR Review 20”. British Journal of Pharmacology. 173 (18): 2702–25. doi:10.1111/bph.13536. PMC4995287. PMID27314810. Hence, one melatonin molecule and its associated metabolites could scavenge a large number of reactive species, and thus, the overall antioxidant capacity of melatonin is believed to be greater than that of other well‐known antioxidants, such as vitamin C and vitamin E, under in vitro or in vivo conditions (Gitto et al., 2001; Sharma and Haldar, 2006; Ortiz et al., 2013).
^ Jump up to:abc Reiter RJ, Rosales-Corral S, Tan DX, Jou MJ, Galano A, Xu B (November 2017). “Melatonin as a mitochondria-targeted antioxidant: one of evolution’s best ideas”. Cellular and Molecular Life Sciences. 74 (21): 3863–3881. doi:10.1007/s00018-017-2609-7. PMID28864909. S2CID23820389. melatonin is specifically targeted to the mitochondria where it seems to function as an apex antioxidant … The measurement of the subcellular distribution of melatonin has shown that the concentration of this indole in the mitochondria greatly exceeds that in the blood.
^ Jump up to:abc Reiter RJ, Mayo JC, Tan DX, Sainz RM, Alatorre-Jimenez M, Qin L (October 2016). “Melatonin as an antioxidant: under promises but over delivers”. Journal of Pineal Research. 61 (3): 253–78. doi:10.1111/jpi.12360. PMID27500468. S2CID35435683. There is credible evidence to suggest that melatonin should be classified as a mitochondria-targeted antioxidant.
^ Jump up to:abc Manchester LC, Coto-Montes A, Boga JA, Andersen LP, Zhou Z, Galano A, et al. (November 2015). “Melatonin: an ancient molecule that makes oxygen metabolically tolerable”. Journal of Pineal Research. 59 (4): 403–19. doi:10.1111/jpi.12267. PMID26272235. S2CID24373303. While originally thought to be produced exclusively in and secreted from the vertebrate pineal gland [53], it is now known that the indole is present in many, perhaps all, vertebrate organs [54] and in organs of all plants that have been investigated [48, 55, 56]. That melatonin is not relegated solely to the pineal gland is also emphasized by the reports that it is present in invertebrates [57–59], which lack a pineal gland and some of which consist of only a single cell.
^ Carrillo-Vico A, Guerrero JM, Lardone PJ, Reiter RJ (July 2005). “A review of the multiple actions of melatonin on the immune system”. Endocrine. 27 (2): 189–200. doi:10.1385/ENDO:27:2:189. PMID16217132. S2CID21133107.
^ Arushanian EB, Beĭer EV (2002). “[Immunotropic properties of pineal melatonin]”. Eksperimental’naia i Klinicheskaia Farmakologiia (in Russian). 65 (5): 73–80. PMID12596522.
^ Carrillo-Vico A, Reiter RJ, Lardone PJ, Herrera JL, Fernández-Montesinos R, Guerrero JM, Pozo D (May 2006). “The modulatory role of melatonin on immune responsiveness”. Current Opinion in Investigational Drugs. 7 (5): 423–31. PMID16729718.
^ Tan DX, Manchester LC, Liu X, Rosales-Corral SA, Acuna-Castroviejo D, Reiter RJ (March 2013). “Mitochondria and chloroplasts as the original sites of melatonin synthesis: a hypothesis related to melatonin’s primary function and evolution in eukaryotes”. Journal of Pineal Research. 54 (2): 127–38. doi:10.1111/jpi.12026. PMID23137057. S2CID206140413.
^ Sumi-Ichinose C, Ichinose H, Takahashi E, Hori T, Nagatsu T (March 1992). “Molecular cloning of genomic DNA and chromosomal assignment of the gene for human aromatic L-amino acid decarboxylase, the enzyme for catecholamine and serotonin biosynthesis”. Biochemistry. 31 (8): 2229–38. doi:10.1021/bi00123a004. PMID1540578.
^ Hickman AB, Klein DC, Dyda F (January 1999). “Melatonin biosynthesis: the structure of serotonin N-acetyltransferase at 2.5 A resolution suggests a catalytic mechanism”. Molecular Cell. 3 (1): 23–32. doi:10.1016/S1097-2765(00)80171-9. PMID10024876.
^ Lerner AB, Case JD, Takahashi Y (July 1960). “Isolation of melatonin and 5-methoxyindole-3-acetic acid from bovine pineal glands”. The Journal of Biological Chemistry. 235: 1992–7. PMID14415935.
^ Poeggeler B, Reiter RJ, Tan DX, Chen LD, Manchester LC (May 1993). “Melatonin, hydroxyl radical-mediated oxidative damage, and aging: a hypothesis”. Journal of Pineal Research. 14 (4): 151–68. doi:10.1111/j.1600-079X.1993.tb00498.x. PMID8102180. S2CID23460208.
^US patent 5449683, Wurtman RJ, “Methods of inducing sleep using melatonin”, issued 12 September 1995, assigned to Massachusetts Institute of Technology
^ Arendt J (August 2005). “Melatonin: characteristics, concerns, and prospects”. Journal of Biological Rhythms. 20 (4): 291–303. doi:10.1177/0748730405277492. PMID16077149. S2CID19011222. There is very little evidence in the short term for toxicity or undesirable effects in humans. The extensive promotion of the miraculous powers of melatonin in the recent past did a disservice to acceptance of its genuine benefits.
^ Reiter RJ (May 1991). “Pineal melatonin: cell biology of its synthesis and of its physiological interactions”. Endocrine Reviews. 12 (2): 151–80. doi:10.1210/edrv-12-2-151. PMID1649044. S2CID3219721.
^ Richardson GS (2005). “The human circadian system in normal and disordered sleep”. The Journal of Clinical Psychiatry. 66 Suppl 9: 3–9, quiz 42–3. PMID16336035.
^ Perreau-Lenz S, Pévet P, Buijs RM, Kalsbeek A (January 2004). “The biological clock: the bodyguard of temporal homeostasis”. Chronobiology International. 21 (1): 1–25. doi:10.1081/CBI-120027984. PMID15129821. S2CID42725506.
^ Jump up to:ab Arendt J, Skene DJ (February 2005). “Melatonin as a chronobiotic”. Sleep Medicine Reviews. 9 (1): 25–39. doi:10.1016/j.smrv.2004.05.002. PMID15649736. Exogenous melatonin has acute sleepiness-inducing and temperature-lowering effects during ‘biological daytime’, and when suitably timed (it is most effective around dusk and dawn), it will shift the phase of the human circadian clock (sleep, endogenous melatonin, core body temperature, cortisol) to earlier (advance phase shift) or later (delay phase shift) times.
^ Chaturvedi CM (1984). “Effect of Melatonin on the Adrenl and Gonad of the Common Mynah Acridtheres tristis”. Australian Journal of Zoology. 32 (6): 803–09. doi:10.1071/ZO9840803.
^ Chen HJ (July 1981). “Spontaneous and melatonin-induced testicular regression in male golden hamsters: augmented sensitivity of the old male to melatonin inhibition”. Neuroendocrinology. 33 (1): 43–6. doi:10.1159/000123198. PMID7254478.
^ Paredes SD, Korkmaz A, Manchester LC, Tan DX, Reiter RJ (1 January 2009). “Phytomelatonin: a review”. Journal of Experimental Botany. 60 (1): 57–69. doi:10.1093/jxb/ern284. PMID19033551. S2CID15738948.
^ Bonnefont-Rousselot D, Collin F (November 2010). “Melatonin: action as antioxidant and potential applications in human disease and aging”. Toxicology. 278 (1): 55–67. doi:10.1016/j.tox.2010.04.008. PMID20417677.
^ Reiter RJ, Manchester LC, Tan DX (September 2005). “Melatonin in walnuts: influence on levels of melatonin and total antioxidant capacity of blood”. Nutrition. 21 (9): 920–4. doi:10.1016/j.nut.2005.02.005. PMID15979282.
^ Burkhardt S, Tan DX, Manchester LC, Hardeland R, Reiter RJ (October 2001). “Detection and quantification of the antioxidant melatonin in Montmorency and Balaton tart cherries (Prunus cerasus)”. Journal of Agricultural and Food Chemistry. 49 (10): 4898–902. doi:10.1021/jf010321. PMID11600041.
^ Lamont KT, Somers S, Lacerda L, Opie LH, Lecour S (May 2011). “Is red wine a SAFE sip away from cardioprotection? Mechanisms involved in resveratrol- and melatonin-induced cardioprotection”. Journal of Pineal Research. 50 (4): 374–80. doi:10.1111/j.1600-079X.2010.00853.x. PMID21342247. S2CID8034935.
^ Hattori A, Migitaka H, Iigo M, Itoh M, Yamamoto K, Ohtani-Kaneko R, et al. (March 1995). “Identification of melatonin in plants and its effects on plasma melatonin levels and binding to melatonin receptors in vertebrates”. Biochemistry and Molecular Biology International. 35(3): 627–34. PMID7773197.
^ Sae-Teaw M, Johns J, Johns NP, Subongkot S (August 2013). “Serum melatonin levels and antioxidant capacities after consumption of pineapple, orange, or banana by healthy male volunteers”. Journal of Pineal Research. 55 (1): 58–64. doi:10.1111/jpi.12025. PMID23137025. S2CID979886.
^ Rodriguez RR (13 January 2010). “Warning Letter”. Inspections, Compliance, Enforcement, and Criminal Investigations. U.S. Food and Drug Administration. Archived from the original on 12 January 2017.
^Bebida Beverage Company U.S. Food and Drug Administration 4 March 2015 (Accessed 8 December 2017)
^ Miroddi M, Bruno R, Galletti F, Calapai F, Navarra M, Gangemi S, Calapai G (March 2015). “Clinical pharmacology of melatonin in the treatment of tinnitus: a review”. European Journal of Clinical Pharmacology. 71 (3): 263–70. doi:10.1007/s00228-015-1805-3. PMID25597877. S2CID16466238.
^ Peres MF, Masruha MR, Zukerman E, Moreira-Filho CA, Cavalheiro EA (April 2006). “Potential therapeutic use of melatonin in migraine and other headache disorders”. Expert Opinion on Investigational Drugs. 15 (4): 367–75. doi:10.1517/13543784.15.4.367. PMID16548786. S2CID28114683.
^ Mills E, Wu P, Seely D, Guyatt G (November 2005). “Melatonin in the treatment of cancer: a systematic review of randomized controlled trials and meta-analysis”. Journal of Pineal Research. 39 (4): 360–6. doi:10.1111/j.1600-079X.2005.00258.x. PMID16207291. S2CID22225091.
^ Meltz ML, Reiter RJ, Herman TS, Kumar KS (March 1999). “Melatonin and protection from whole-body irradiation: survival studies in mice”. Mutation Research. 425 (1): 21–7. doi:10.1016/S0027-5107(98)00246-2. PMID10082913.
^ Reiter RJ, Herman TS, Meltz ML (December 1996). “Melatonin and radioprotection from genetic damage: in vivo/in vitro studies with human volunteers”. Mutation Research. 371 (3–4): 221–8. doi:10.1016/S0165-1218(96)90110-X. PMID9008723.
^ Reiter RJ, Herman TS, Meltz ML (February 1998). “Melatonin reduces gamma radiation-induced primary DNA damage in human blood lymphocytes”. Mutation Research. 397 (2): 203–8. doi:10.1016/S0027-5107(97)00211-X. PMID9541644.
^ Tan DX, Manchester LC, Terron MP, Flores LJ, Reiter RJ (January 2007). “One molecule, many derivatives: a never-ending interaction of melatonin with reactive oxygen and nitrogen species?”. Journal of Pineal Research. 42 (1): 28–42. doi:10.1111/j.1600-079X.2006.00407.x. PMID17198536. S2CID40005308.
^ Giannotti F, Cortesi F, Cerquiglini A, Bernabei P (August 2006). “An open-label study of controlled-release melatonin in treatment of sleep disorders in children with autism”. Journal of Autism and Developmental Disorders. 36 (6): 741–52. doi:10.1007/s10803-006-0116-z. PMID16897403. S2CID19724241.
^ Bendz LM, Scates AC (January 2010). “Melatonin treatment for insomnia in pediatric patients with attention-deficit/hyperactivity disorder”. The Annals of Pharmacotherapy. 44(1): 185–91. doi:10.1345/aph.1M365. PMID20028959. S2CID207263711.
FDA 11/25/2020, Imcivree, To treat obesity and the control of hunger associated with pro-opiomelanocortin deficiency, a rare disorder that causes severe obesity that begins at an early age Drug Trials Snapshot, 10MG/ML, SOLUTION;SUBCUTANEOUS, Orphan
DESCRIPTION
IMCIVREE contains setmelanotide acetate, a melanocortin 4 (MC4) receptor agonist. Setmelanotide is an 8 amino acid cyclic peptideanalog of endogenous melanocortin peptide α-MSH (alpha-melanocyte stimulating hormone).
The chemical name for setmelanotide acetate is acetyl-L-arginyl-L-cysteinyl-D-alanyl-Lhistidinyl-D-phenylalanyl-L-arginyl-L-tryptophanyl-L-cysteinamide cyclic (2→8)-disulfide acetate. Its molecular formula is C49H68N18O9S2 (anhydrous, free-base), and molecular mass is 1117.3 Daltons (anhydrous, free-base).
The chemical structure of setmelanotide is:
IMCIVREE injection is a sterile clear to slightly opalescent, colorless to slightly yellow solution. Each 1 mL of IMCIVREE contains 10 mg of setmelanotide provided as setmelanotide acetate, which is a salt with 2 to 4 molar equivalents of acetate, and the following inactive ingredients: 100 mg N-(carbonyl-methoxypolyethylene glycol 2000)-1,2-distearoyl-glycero-3phosphoethanolamine sodium salt, 8 mg carboxymethylcellulose sodium (average MWt 90,500), 11 mg mannitol, 5 mg phenol, 10 mg benzyl alcohol, 1 mg edetate disodium dihydrate, and Water for Injection. The pH of IMCIVREE is 5 to 6.
Setmelanotide is a peptide drug and investigational anti-obesity medication which acts as a selective agonist of the MC4 receptor. Setmelanotide binds to and activates MC4 receptors in the paraventricular nucleus (PVN) of the hypothalamus and in the lateral hypothalamic area (LHA), areas involved in the regulation of appetite, and this action is thought to underlie its appetite suppressant effects. Setmelanotide increases resting energy expenditure in both obese animals and humans. Setmelanotide has been reported to possess the following activity profile (cAMP, EC50): MC4 (0.27 nM) > MC3 (5.3 nM) ≈ MC1 (5.8 nM) > MC5 (1600 nM) ≟ MC2 (>1000 nM).
Setmelanotide, sold under the brand name Imcivree, is a medication for the treatment of obesity.[1]
The most common side effects include injection site reactions, skin hyperpigmentation (skin patches that are darker than surrounding skin), headache and gastrointestinal side effects (such as nausea, diarrhea, and abdominal pain), among others.[1] Spontaneous penile erections in males and adverse sexual reactions in females have occurred with treatment.[1] Depression and suicidal ideation have also occurred with setmelanotide.[1]
SYN
WO 2011060355
Medical uses
Setmelanotide is indicated for chronic weight management (weight loss and weight maintenance for at least one year) in people six years and older with obesity due to three rare genetic conditions: pro-opiomelanocortin (POMC) deficiency, proprotein subtilisin/kexin type 1 (PCSK1) deficiency, and leptin receptor (LEPR) deficiency confirmed by genetic testing demonstrating variants in POMC, PCSK1, or LEPR genes considered pathogenic (causing disease), likely pathogenic, or of uncertain significance.[1] Setmelanotide is the first FDA-approved treatment for these genetic conditions.[1]
Setmelanotide is not approved for obesity due to suspected POMC, PCSK1, or LEPR deficiency with variants classified as benign (not causing disease) or likely benign or other types of obesity, including obesity associated with other genetic syndromes and general (polygenic) obesity.[1]
Setmelanotide has been reported to possess the following activity profile (cAMP, EC50): MC4 (0.27 nM) > MC3 (5.3 nM) ≈ MC1 (5.8 nM) > MC5 (1600 nM) ≟ MC2 (>1000 nM).[5] (19.6-fold selectivity for MC4 over MC3, the second target of highest activity.)
History
Setmelanotide was evaluated in two one-year studies.[1] The first study enrolled participants with obesity and confirmed or suspected POMC or PCSK1 deficiency while the second study enrolled participants with obesity and confirmed or suspected LEPR deficiency; all participants were six years or older.[1] The effectiveness of setmelanotide was determined by the number of participants who lost more than ten percent of their body weight after a year of treatment.[1]
The effectiveness of setmelanotide was assessed in 21 participants, ten in the first study and eleven in the second.[1] In the first study, 80 percent of participants with POMC or PCSK1 deficiency lost ten percent or more of their body weight.[1] In the second study, 46 percent of participants with LEPR deficiency lost ten percent or more of their body weight.[1]
The study also assessed the maximal (greatest) hunger in sixteen participants over the previous 24 hours using an eleven-point scale in participants twelve years and older.[1] In both studies, some, but not all, of participants’ weekly average maximal hunger scores decreased substantially from their scores at the beginning of the study.[1] The degree of change was highly variable among participants.[1]
The U.S. Food and Drug Administration (FDA) granted the application for setmelanotide orphan disease designation, breakthrough therapy designation, and priority review.[1] The FDA granted the approval of Imcivree to Rhythm Pharmaceutical, Inc.[1]
Synthesis of Example 1i.e., Ac-Arg-cyclo(Cys-D-Ala-His-D-Phe-Arg-Trp-Cys)-NH2
The title peptide having the above structure was assembled using Fmoc chemistry on an Apex peptide synthesizer (Aapptec; Louisville, Ky., USA). 220 mg of 0.91 mmol/g (0.20 mmoles) Rink Amide MBHA resin (Polymer Laboratories; Amherst, Mass., USA) was placed in a reaction well and pre-swollen in 3.0 mL of DMF prior to synthesis. For cycle 1, the resin was treated with two 3-mL portions of 25% piperidine in DMF for 5 and 10 minutes respectively, followed by 4 washes of 3-mL DMF—each wash consisting of adding 3 mL of solvent, mixing for 1 minute, and emptying for 1 minute. Amino acids stocks were prepared in NMP as 0.45M solutions containing 0.45M HOBT. HBTU was prepared as a 0.45M solution in NMP and DIPEA was prepared as a 2.73M solution in NMP. To the resin, 2 mL of the first amino acid (0 9 mmoles, Fmoc-Cys(Trt)-OH) (Novabiochem; San Diego, Calif., USA) was added along with 2 mL (0.9 mmoles) of HBTU and 1.5 mL (4.1 mmoles) of DIPEA. After one hour of constant mixing, the coupling reagents were drained from the resin and the coupling step was repeated. Following amino acid acylation, the resin was washed with two 3-mL aliquots of DMF for 1 minute. The process of assembling the peptide (deblock/wash/acylate/wash) was repeated for cycles 2-9 identical to that as described for cycle 1. The following amino acids were used: cycle 2) Fmoc-Trp(Boc)-OH (Genzyme; Cambridge, Mass., USA); cycle 3) Fmoc-Arg(Pbf)-OH (Novabiochem); cycle 4) Fmoc-DPhe-OH (Genzyme); cycle 5) Fmoc-His(Trt)-OH (Novabiochem); cycle 6) Fmoc-D-Ala-OH (Genzyme); cycle 7) Fmoc-Cys(Trt)-OH, (Novabiochem); and cycle 8) Fmoc-Arg(Pbf)-OH (Genzyme). The N-terminal Fmoc was removed with 25% piperidine in DMF as described above, followed by four 3-mL DMF washes for 1 minute. Acetylation of the N-terminus was performed by adding 0.5 mL of 3M DIPEA in NMP to the resin along with 1.45 mL of 0.45M acetic anhydride in NMP. The resin was mixed for 30 minutes and acetylation was repeated. The resin was washed with 3 mL of DMF for a total of 5 times followed with 5 washes with 5 mL of DCM each.
To cleave and deprotect the peptide, 5mL of the following reagent was added to the resin: 2% TIS/5% water/5% (w/v) DTT/88% TFA. The solution was allowed to mix for 3.5 hours. The filtrate was collected into 40 mL of cold anhydrous ethyl ether. The precipitate was pelleted for 10 minutes at 3500 rpm in a refrigerated centrifuge. The ether was decanted and the peptide was re-suspended in fresh ether. The ether workup was performed three times. Following the last ether wash, the peptide was allowed to air dry to remove residual ether.
The peptide was dissolved in 10% acetonitrile and analyzed by mass spectrometry and reverse-phase HPLC employing a 30×4.6 cm C18 column (Vydac; Hesperia, Calif., USA) with a gradient of 2-60% acetonitrile (0.1% TFA) over 30 minutes. This analysis identified a product with ˜53% purity. Mass analysis employing electrospray ionization identified a main product containing a mass of 1118.4 corresponding to the desired linear product. The crude product (˜100 mg) was diluted to a concentration of 2 mg/mL in 5% acetic acid. To this solution, 0.5M iodine/methanol was added dropwise with vigorous stirring until a pale yellow color was achieved. The solution was vigorously stirred for another 10 minutes. Excess iodine was then quenched by adding 1.0M sodium thiosulfate under continuous mixing until the mixture was rendered colorless. The peptide was re-examined by mass spectrometry analysis and HPLC. Mass spectrometry analysis identified a main species with a mass of 1116.4 which indicated successful oxidation to form the cyclic peptide. The peptide solution was purified on a preparative HPLC equipped with a C18 column using a similar elution gradient. The purified product was re-analyzed by HPLC for purity (>95%) and mass spectrometry (1116.9 which is in agreement with the expected mass of 1117.3) and subsequently lyophilized. Following lyophilization, 28 mg of purified product was obtained representing a 24% yield.
The other exemplified peptides were synthesized substantially according to the procedure described for the above-described synthetic process. Physical data for select exemplified peptides are given in Table 1.
The acetate salt of Example 1 (200 mg, 0.18 mmole) was dissolved in 10 mL of water. Sodium pamoate (155 mg, 0.36 mmole) was dissolved in 10 mL of water. The two solutions were combined and mixed well. The precipitates were collected by centrifugation at 3000 rpm for 20 minutes, washed for three times with water, and dried by lyophilization.
^ Jump up to:abc Lee EC, Carpino PA (2015). “Melanocortin-4 receptor modulators for the treatment of obesity: a patent analysis (2008-2014)”. Pharmaceutical Patent Analyst. 4 (2): 95–107. doi:10.4155/ppa.15.1. PMID25853469.
^ Jackson VM, Price DA, Carpino PA (August 2014). “Investigational drugs in Phase II clinical trials for the treatment of obesity: implications for future development of novel therapies”. Expert Opinion on Investigational Drugs. 23 (8): 1055–66. doi:10.1517/13543784.2014.918952. PMID25000213. S2CID23198484.
The peptide sequence is Ac-Arg-Cys(1)-D-Ala-His-D-Phe-Arg-Trp-Cys(1)-NH2. It is being researched by Rhythm Pharmaceuticals for the treatment of obesity and diabetes. In addition, Rhythm Pharmaceuticals is conducting trials of setmelanotide for the treatment of Prader–Willi syndrome (PWS), a genetic disorder which includes MC4 receptor deficiency and associated symptoms such as excessive appetite and obesity. As of December 2014, the drug is in phase II clinical trials for obesity and PWS.
1: Lee EC, Carpino PA. Melanocortin-4 receptor modulators for the treatment of obesity: a patent analysis (2008-2014). Pharm Pat Anal. 2015;4(2):95-107. doi: 10.4155/ppa.15.1. PubMed PMID: 25853469.
2: Chen KY, Muniyappa R, Abel BS, Mullins KP, Staker P, Brychta RJ, Zhao X, Ring M, Psota TL, Cone RD, Panaro BL, Gottesdiener KM, Van der Ploeg LH, Reitman ML, Skarulis MC. RM-493, a melanocortin-4 receptor (MC4R) agonist, increases resting energy expenditure in obese individuals. J Clin Endocrinol Metab. 2015 Apr;100(4):1639-45. doi: 10.1210/jc.2014-4024. Epub 2015 Feb 12. PubMed PMID: 25675384; PubMed Central PMCID: PMC4399297.
3: Clemmensen C, Finan B, Fischer K, Tom RZ, Legutko B, Sehrer L, Heine D, Grassl N, Meyer CW, Henderson B, Hofmann SM, Tschöp MH, Van der Ploeg LH, Müller TD. Dual melanocortin-4 receptor and GLP-1 receptor agonism amplifies metabolic benefits in diet-induced obese mice. EMBO Mol Med. 2015 Feb 4;7(3):288-98. doi: 10.15252/emmm.201404508. PubMed PMID: 25652173; PubMed Central PMCID: PMC4364946.
4: Jackson VM, Price DA, Carpino PA. Investigational drugs in Phase II clinical trials for the treatment of obesity: implications for future development of novel therapies. Expert Opin Investig Drugs. 2014 Aug;23(8):1055-66. doi: 10.1517/13543784.2014.918952. Epub 2014 Jul 7. Review. PubMed PMID: 25000213.
5: Kievit P, Halem H, Marks DL, Dong JZ, Glavas MM, Sinnayah P, Pranger L, Cowley MA, Grove KL, Culler MD. Chronic treatment with a melanocortin-4 receptor agonist causes weight loss, reduces insulin resistance, and improves cardiovascular function in diet-induced obese rhesus macaques. Diabetes. 2013 Feb;62(2):490-7. doi: 10.2337/db12-0598. Epub 2012 Oct 9. PubMed PMID: 23048186; PubMed Central PMCID: PMC3554387.
6: Kumar KG, Sutton GM, Dong JZ, Roubert P, Plas P, Halem HA, Culler MD, Yang H, Dixit VD, Butler AA. Analysis of the therapeutic functions of novel melanocortin receptor agonists in MC3R- and MC4R-deficient C57BL/6J mice. Peptides. 2009 Oct;30(10):1892-900. doi: 10.1016/j.peptides.2009.07.012. Epub 2009 Jul 29. PubMed PMID: 19646498; PubMed Central PMCID: PMC2755620.
External links
“Setmelanotide”. Drug Information Portal. U.S. National Library of Medicine.
RNA, (Gm-sp-Am-sp-Cm-Um-Um-Um-(2′-deoxy-2′-fluoro)C-Am-(2′-deoxy-2′-fluoro)U-(2′-deoxy-2′-fluoro)C-(2′-deoxy-2′-fluoro)C-Um-Gm-Gm-Am-Am-Am-Um-Am-Um-Am), 3′-[[(2S,4R)-1-[29-[[2-(acetylamino)-2-deoxy-β-D-galactopyranosyl]oxy]-14,14-bis[[3-[[3-[[5-[[2-(acetylamino)-2-deoxy-β-D-galactopyranosyl]oxy]-1-oxopentyl]amino]propyl]amino]-3-oxopropoxy]methyl]-1,12,19,25-tetraoxo-16-oxa-13,20,24-triazanonacos-1-yl]-4-hydroxy-2-pyrrolidinyl]methyl hydrogen phosphate], complex with RNA (Um-sp-(2′-deoxy-2′-fluoro)A-sp-Um-Am-Um-(2′-deoxy-2′-fluoro)U-Um-(2′-deoxy-2′-fluoro)C-(2′-deoxy-2′-fluoro)C-Am-Gm-Gm-Am-(2′-deoxy-2′-fluoro)U-Gm-(2′-deoxy-2′-fluoro)A-Am-Am-Gm-Um-Cm-sp-Cm-sp-Am) (1:1)
Nucleic Acid Sequence
Sequence Length: 44, 23, 2115 a 8 c 7 g 14 umultistranded (2); modified
OXLUMO is supplied as a sterile, preservative-free, clear, colorless-to-yellow solution for subcutaneous administration containing the equivalent of 94.5 mg of lumasiran (provided as lumasiran sodium) in 0.5 Ml of water for injection and sodium hydroxide and/or phosphoric acid to adjust the pH to ~7.0.
Lumasiran An investigational RNAi Therapeutic for Primary Hyperoxaluria Type 1 (PH1)
Overview • Lumasiran (ALN-GO1) is an investigational, subcutaneously administered (under the skin) RNA interference (RNAi) therapeutic targeting glycolate oxidase (GO) in development for the treatment of primary hyperoxaluria type 1 (PH1).
• PH1 is a rare, life-threatening disease that can cause serious damage to kidneys and progressively to other organs.1
• PH1 is characterized by the pathologic overproduction of oxalate by the liver. Oxalate is an end product of metabolism that, when in excess, is toxic and accumulates in the kidneys forming calcium oxalate crystals.1,2
• Symptoms of PH1 are often associated with recurrent kidney stones and include flank pain, urinary tract infections, painful urination, and blood in the urine.2,3
• Currently, the only curative treatment is a liver transplant, to correct the metabolic defect, combined with a kidney transplant, to replace the terminally damaged kidneys.1,3 Clinical Development
• The safety and efficacy of lumasiran are being evaluated in a randomized, double-blind, placebo-controlled, global, multicenter Phase 3 study of approximately 30 PH1 patients, called ILLUMINATE-A (NCT03681184).
• The primary endpoint is percent change in 24-hour urinary oxalate excretion from baseline to Month 6.
• Key secondary and exploratory endpoints in ILLUMINATE-A will evaluate additional measures of urinary oxalate, estimated glomerular filtration rate (eGFR), safety, and tolerability.
Regulatory Designations • Breakthrough Therapy Designation by the U.S. Food and Drug Administration (FDA) • Priority Medicines (PRIME) Designation from the European Medicines Agency (EMA) • Orphan Drug Designations in both the U.S. and the European Union
PSMA-11, also known as HBED-CC-PSMA or Psma-hbed-CC, is used to make gallium Ga 68-labeled PSMA-11, which has potential use as a tracer for PSMA-expressing tumors during positron emission tomography (PET). Upon intravenous administration of gallium Ga 68-labeled PSMA-11, the Glu-urea-Lys(Ahx) moiety targets and binds to PSMA-expressing tumor cells. Upon internalization, PSMA-expressing tumor cells can be detected during PET imaging. PSMA, a tumor-associated antigen and type II transmembrane protein, is expressed on the membrane of prostatic epithelial cells and overexpressed on prostate tumor cells
Name: PSMA-11 CAS#: 1366302-52-4 Chemical Formula: C44H62N6O17 Exact Mass: 946.4171
The Food and Drug Administration (FDA) has approved Gallium 68 PSMA-11 (Ga 68 PSMA-11), the first drug for positron emission tomography (PET) imaging of prostate-specific membrane antigen (PSMA) positive lesions in men with prostate cancer.
Ga 68 PSMA-11, a radioactive diagnostic agent, is indicated for patients with suspected prostate cancer metastasis who are potentially curable by surgery or radiation therapy. It is also indicated for patients with suspected prostate cancer recurrence based on elevated serum prostate-specific antigen (PSA) levels.
The approval was based on efficacy and safety data from 2 prospective clinical trials (Trial 1 and 2) with a total of 960 men with prostate cancer who each received 1 injection of Ga 68 PSMA-11. Trial 1 included 325 patients with biopsy-proven prostate cancer who underwent PET/CT or PET/MRI scans performed with Ga 68 PSMA-11. Results from the study showed that positive readings in the pelvic lymph nodes on Ga 68 PSMA-11 PET were associated with a clinically important rate of metastatic cancer confirmed by surgical pathology in those who proceeded to surgery.
In Trial 2, 635 patients with rising serum PSA levels after prostate surgery or radiotherapy received a single Ga 68 PSMA-11 PET/CT scan or PET/MR scan. Findings demonstrated that 74% of patients had at least 1 positive lesion detected by Ga 68 PSMA-11 PET, and local recurrence or metastasis of prostate cancer was confirmed in 91% of cases.
This is the first drug approved for PET imaging of prostate-specific membrane antigen positive lesions in men with prostate cancer.
REF
REFERENCES
1: Meißner S, Janssen JC, Prasad V, Brenner W, Diederichs G, Hamm B, Hofheinz F, Makowski MR. Potential of asphericity as a novel diagnostic parameter in the evaluation of patients with (68)Ga-PSMA-HBED-CC PET-positive prostate cancer lesions. EJNMMI Res. 2017 Oct 23;7(1):85. doi: 10.1186/s13550-017-0333-9. PubMed PMID: 29058157; PubMed Central PMCID: PMC5651532.
2: Verburg FA, Pfister D, Drude NI, Mottaghy FM, Behrendt F. PSA levels, PSA doubling time, Gleason score and prior therapy cannot predict measured uptake of [(68)Ga]PSMA-HBED-CC lesion uptake in recurrent/metastatic prostate cancer. Nuklearmedizin. 2017 Oct 18;56(6). doi: 10.3413/Nukmed-0917-17-07. [Epub ahead of print] PubMed PMID: 29044297.
3: Amor-Coarasa A, Kelly JM, Gruca M, Nikolopoulou A, Vallabhajosula S, Babich JW. Continuation of comprehensive quality control of the itG (68)Ge/(68)Ga generator and production of (68)Ga-DOTATOC and (68)Ga-PSMA-HBED-CC for clinical research studies. Nucl Med Biol. 2017 Oct;53:37-39. doi: 10.1016/j.nucmedbio.2017.07.006. Epub 2017 Jul 14. PubMed PMID: 28803001.
4: Janssen JC, Woythal N, Meißner S, Prasad V, Brenner W, Diederichs G, Hamm B, Makowski MR. [(68)Ga]PSMA-HBED-CC Uptake in Osteolytic, Osteoblastic, and Bone Marrow Metastases of Prostate Cancer Patients. Mol Imaging Biol. 2017 Dec;19(6):933-943. doi: 10.1007/s11307-017-1101-y. PubMed PMID: 28707038.
5: Damle NA, Tripathi M, Chakraborty PS, Sahoo MK, Bal C, Aggarwal S, Arora G, Kumar P, Kumar R, Gupta R. Unusual Uptake of Prostate Specific Tracer (68)Ga-PSMA-HBED-CC in a Benign Thyroid Nodule. Nucl Med Mol Imaging. 2016 Dec;50(4):344-347. Epub 2016 Mar 22. PubMed PMID: 27994690; PubMed Central PMCID: PMC5135692.
6: Behrendt F, Krohn T, Mottaghy F, Verburg FA. [(68)Ga]PSMA-HBED-CC PET/CT to differentiate between diffuse bone metastases of prostate cancer and osteopoikilosis. Nuklearmedizin. 2016 Dec 6;55(6):N64-N65. PubMed PMID: 27922151.
7: Krohn T, Birmes A, Winz OH, Drude NI, Mottaghy FM, Behrendt FF, Verburg FA. The reconstruction algorithm used for [(68)Ga]PSMA-HBED-CC PET/CT reconstruction significantly influences the number of detected lymph node metastases and coeliac ganglia. Eur J Nucl Med Mol Imaging. 2017 Apr;44(4):662-669. doi: 10.1007/s00259-016-3571-6. Epub 2016 Nov 29. PubMed PMID: 27900518.
8: Berliner C, Tienken M, Frenzel T, Kobayashi Y, Helberg A, Kirchner U, Klutmann S, Beyersdorff D, Budäus L, Wester HJ, Mester J, Bannas P. Detection rate of PET/CT in patients with biochemical relapse of prostate cancer using [(68)Ga]PSMA I&T and comparison with published data of [(68)Ga]PSMA HBED-CC. Eur J Nucl Med Mol Imaging. 2017 Apr;44(4):670-677. doi: 10.1007/s00259-016-3572-5. Epub 2016 Nov 28. PubMed PMID: 27896369.
9: Sathekge M, Lengana T, Modiselle M, Vorster M, Zeevaart J, Maes A, Ebenhan T, Van de Wiele C. (68)Ga-PSMA-HBED-CC PET imaging in breast carcinoma patients. Eur J Nucl Med Mol Imaging. 2017 Apr;44(4):689-694. doi: 10.1007/s00259-016-3563-6. Epub 2016 Nov 8. PubMed PMID: 27822700; PubMed Central PMCID: PMC5323468.
10: Rauscher I, Maurer T, Beer AJ, Graner FP, Haller B, Weirich G, Doherty A, Gschwend JE, Schwaiger M, Eiber M. Value of 68Ga-PSMA HBED-CC PET for the Assessment of Lymph Node Metastases in Prostate Cancer Patients with Biochemical Recurrence: Comparison with Histopathology After Salvage Lymphadenectomy. J Nucl Med. 2016 Nov;57(11):1713-1719. Epub 2016 Jun 3. PubMed PMID: 27261524.
13: Noto B, Vrachimis A, Schäfers M, Stegger L, Rahbar K. Subacute Stroke Mimicking Cerebral Metastasis in 68Ga-PSMA-HBED-CC PET/CT. Clin Nucl Med. 2016 Oct;41(10):e449-51. doi: 10.1097/RLU.0000000000001291. PubMed PMID: 27355852.
14: Pfob CH, Ziegler S, Graner FP, Köhner M, Schachoff S, Blechert B, Wester HJ, Scheidhauer K, Schwaiger M, Maurer T, Eiber M. Biodistribution and radiation dosimetry of (68)Ga-PSMA HBED CC-a PSMA specific probe for PET imaging of prostate cancer. Eur J Nucl Med Mol Imaging. 2016 Oct;43(11):1962-70. doi: 10.1007/s00259-016-3424-3. Epub 2016 May 20. PubMed PMID: 27207281.
15: Amor-Coarasa A, Schoendorf M, Meckel M, Vallabhajosula S, Babich JW. Comprehensive Quality Control of the ITG 68Ge/68Ga Generator and Synthesis of 68Ga-DOTATOC and 68Ga-PSMA-HBED-CC for Clinical Imaging. J Nucl Med. 2016 Sep;57(9):1402-5. doi: 10.2967/jnumed.115.171249. Epub 2016 Apr 21. PubMed PMID: 27103024.
16: Prasad V, Steffen IG, Diederichs G, Makowski MR, Wust P, Brenner W. Biodistribution of [(68)Ga]PSMA-HBED-CC in Patients with Prostate Cancer: Characterization of Uptake in Normal Organs and Tumour Lesions. Mol Imaging Biol. 2016 Jun;18(3):428-36. doi: 10.1007/s11307-016-0945-x. PubMed PMID: 27038316.
17: Pfister D, Porres D, Heidenreich A, Heidegger I, Knuechel R, Steib F, Behrendt FF, Verburg FA. Detection of recurrent prostate cancer lesions before salvage lymphadenectomy is more accurate with (68)Ga-PSMA-HBED-CC than with (18)F-Fluoroethylcholine PET/CT. Eur J Nucl Med Mol Imaging. 2016 Jul;43(8):1410-7. doi: 10.1007/s00259-016-3366-9. Epub 2016 Mar 19. PubMed PMID: 26993315.
18: Kanthan GL, Coyle L, Kneebone A, Schembri GP, Hsiao E. Follicular Lymphoma Showing Avid Uptake on 68Ga PSMA-HBED-CC PET/CT. Clin Nucl Med. 2016 Jun;41(6):500-1. doi: 10.1097/RLU.0000000000001169. PubMed PMID: 26914565.
19: Kanthan GL, Hsiao E, Kneebone A, Eade T, Schembri GP. Desmoid Tumor Showing Intense Uptake on 68Ga PSMA-HBED-CC PET/CT. Clin Nucl Med. 2016 Jun;41(6):508-9. doi: 10.1097/RLU.0000000000001192. PubMed PMID: 26909712.
20: Eiber M, Weirich G, Holzapfel K, Souvatzoglou M, Haller B, Rauscher I, Beer AJ, Wester HJ, Gschwend J, Schwaiger M, Maurer T. Simultaneous (68)Ga-PSMA HBED-CC PET/MRI Improves the Localization of Primary Prostate Cancer. Eur Urol. 2016 Nov;70(5):829-836. doi: 10.1016/j.eururo.2015.12.053. Epub 2016 Jan 18. PubMed PMID: 26795686.
//////////Gallium 68 PSMA-11, FDA 2020, 2020 APPROVALS, RADIO ACTIVE
Lonafarnib, sold under the brand name Zokinvy, is a medication used to reduce the risk of death due to Hutchinson-Gilford progeria syndrome and for the treatment of certain processing-deficient progeroidlaminopathies in people one year of age and older.[1][2]
The most common side effects included nausea vomiting, diarrhea, infection, decreased appetite and fatigue.[1]
Lonafarnib is contraindicated for co-administration with strong or moderate CYP3A inhibitors and inducers, as well as midazolam and certain cholesterol-lowering medications.[1]
History
Lonafarnib, a farnesyltransferase inhibitor, is an oral medication that helps prevent the buildup of defective progerin or progerin-like protein.[1] The effectiveness of lonafarnib for the treatment of Hutchinson-Gilford progeria syndrome was demonstrated in 62 patients from two single-arm trials (Trial 1/NCT00425607 and Trial 2/NCT00916747) that were compared to matched, untreated patients from a separate natural history study.[1][2] Compared to untreated patients, the lifespan of Hutchinson-Gilford progeria syndrome patients treated with lonafarnib increased by an average of three months through the first three years of treatment and by an average of 2.5 years through the maximum follow-up time of 11 years.[1] Lonafarnib’s approval for the treatment of certain processing-deficient progeroid laminopathies that are very rare took into account similarities in the underlying genetic mechanism of disease and other available data.[1] The participants were from 34 countries around the world, including the United States.[2]
Lonafarnib is a farnesyltransferase inhibitor (FTI) that has been investigated in a human clinical trial as a treatment for progeria, which is an extremely rare genetic disorder in which symptoms resembling aspects of aging are manifested at a very early age.[3][4]
Lonafarnib is a synthetic tricyclichalogenatedcarboxamide with antineoplastic properties.[5] As such, it is used primarily for cancer treatment. For those with progeria, research has shown that the drug reduces the prevalence of stroke and transient ischemic attack, and the prevalence and frequency of headaches while taking the medication.[6] A phase II clinical trial was completed in 2012, which showed that a cocktail of drugs that included lonafarnib and two other drugs met clinical efficacy endpoints that improved the height and diminished the rigidity of the bones of progeria patients.
SYN
EP 1019392; EP 1380581; JP 1999501671; WO 9723478
Introduction of a bromine atom at the 10-position of the benzocycloheptapyridine (I) was achieved by the following sequence. Nitration of (I) using NaNO3-H2SO4 afforded a mixture of nitro compounds (II) and (III), from which the major 9-nitro isomer (III) was separated by silica gel chromatography. Reduction of the nitro group of (III) with iron filings and CaCl2 in refluxing aqueous ethanol gave amine (IV), which was brominated at position 10 with Br2 in AcOH. The brominated aniline (VI) was then deaminated by diazotization, followed by reduction of the resulting diazonium salt with hypophosphorous acid to give trihalo compound (VI). Hydrolysis of carbamate group of (VI) in boiling concentrated HCl afforded piperidine (VII). Subsequent reduction of the C-11 double bond of (VII) was carried out using DIBAL-H in refluxing toluene to afford the corresponding racemic piperidine. Separation of enantiomers was achieved by HPLC on a ChiralPak AD column or by chemical resolution using N-acetyl-L-phenylalanine as the resolving agent. The appropriate R-(+) enantiomer (VIII) was coupled with N-Boc-piperidylacetic acid (IX) in the presence of EDC and HOBt to yield protected amide (X). Hydrolysis of the Boc protecting group was performed with trifluoroacetic acid, and the resulting piperidine (XI) was finally treated with trimethylsilyl isocyanate to give the desired carboxamide (3-5).
SYN2
EP 1091954; JP 2002519419; WO 0001689
J Org Chem 2000,65(18),5451
The starting product is the benzocyclohetapyridine (VII), already reported as intermediate (VII) in the synthesis of 25468001a. Compound (VII) is resolved into its atropaisomers by digestion with Toyobo LIP-300 enzyme in the presence of trifluroethyl isobutyrate (XII) to give a mixture of unreacted (-)-(XIII) and acylated compound (+)-(XIV) that are separated by acid extraction. The undesired atropaisomer (-)-(XIII) can be recovered by thermal razemization in diethyleneglycol dibutyl ether at 210 C and new enzymatic separation. The acid hydrolysis of the separated amide (+)-(XIV) produces the desired atropaisomer (+)-(XIII), which is reduced to the (R)-(+)-(VIII), intermediate already reported with no. (VIII) in the synthesis of 25468001a. (6,7)
SYN 3
1) By carboxylation of 8-chloro-6,11-dihydro-11-(4-piperidylidene)-5H-benzo[5,6]cyctohepta[1,2-b]pyridine (I) with ethyl chloroformate (II) in refluxing benzene.
SYN 4
2) By reaction of 8-chloro-6,11-dihydro-5H-benzo[5,6]cyclohepta[1,2-b]pyridin-11-one (III) with the Grignard reagent (IV) to give the tertiary carbinol (V), which is dehydrated with 85% H2SO4 affording 8-chloro-11-piperidinylidene derivative (VI). Finally, cornpound (VI) is treated with ethyl chloroformate (II) in toluene.
SYN 5
J Med Chem 1997,40(26),4290
The nitration of loratadine (I) (1) by means of tetrabutylammonium nitrate and trifluoroacetic anhydride (TFAA) in dichloromethane gives the 3-nitro derivative (II), which is reduced with iron filings and CaCl2 in refluxing ethanol/water to yield the 3-amino derivative (III). Treatment of compound (III) with NaNO2, HBr and Br2 provides 4-(3-bromo-8-chloro-5,6-dihydro-1H-benzo[5,6]-cyclohepta[1,2-b]pyridin-11-ylidene)piperidine-1-carboxylic acid ethyl ester (IV) (see scheme 25468001a, intermediate (I).(2)
Benzocycloheptapyridine (I) was nitrated with NaNO3 and H2SO4 to afford (II) as the major isomer. Reduction of (III) with iron and CaCl2 gave amine (III), which was brominated to provide (IV). Removal of the amino group of (IV) was accomplished by diazotization, followed by reduction with hypophosphorous acid to give (V). Then, hydrolysis of the carbamate group of (V) in refluxing hydrochloric acid furnished piperidine (VI). Subsequent coupling of (VI) with pyridineacetic acid N-oxide (VII) using EDC and HOBt yielded the corresponding amide. Finally, separation of the target (+)-atropoisomer was achieved by chiral chromatography.
^ Liu G, Marrinan CH, Taylor SA, Black S, Basso AD, Kirschmeier P, et al. (September 2007). “Enhancement of the antitumor activity of tamoxifen and anastrozole by the farnesyltransferase inhibitor lonafarnib (SCH66336)”. Anti-Cancer Drugs. 18 (8): 923–31. doi:10.1097/CAD.0b013e3280c1416e (inactive 2020-09-10). PMID17667598.
Mechanism of ActionAngiogenesis inhibitors; Src-Family kinase inhibitors; Tubulin polymerisation inhibitors
PreregistrationActinic keratosis
Phase IIPsoriasis
Phase I/IISolid tumours
Phase IPhotodamage
PreclinicalSkin cancer
09 Mar 2020FDA assigns PDUFA action date of 30/12/2020 for tirbanibulin for Actinic keratosis
09 Mar 2020US FDA accepts NDA for tirbanibulin for Actinic keratosis for review
02 Mar 2020European Medicines Agency accepts Marketing Authorization Application for tirbanibulin for Actinic keratosis for review
KX-01 is a dual inhibitor of Src kinase and tubulin polymerization. KX01 promotes the induction of p53, G2/M arrest of proliferating cell populations and subsequent apoptosis via the stimulation of Caspase-3 and PARP cleavage. The drug was developed by Kinex Pharmaceuticals and reached phase II of clinical trials for the treatment of Castration-Resistant Prostate Cancer and Actinic Keratosis. KX-01 demonstrated good in vitro pofile against different cancer cell lines with IC50 in nanomolar range.
Tirbanibulin (Mesylate) (KX2-391 (Mesylate)) is an inhibitor of Src that targets the peptide substrate site of Src, with GI50 of 9-60 nM in cancer cell lines.
Tirbanibulin (KX2-391) is a Src inhibitor that is directed to the Src substrate pocket. Tirbanibulin (KX2-391) shows steep dose-response curves against Huh7 (GI50=9 nM), PLC/PRF/5 (GI50=13 nM), Hep3B (GI50=26 nM), and HepG2 (GI50=60 nM), four hepatic cell cancer (HCC) cell lines[1]. Tirbanibulin (KX2-391) is found to inhibit certain leukemia cells that are resistant to current commercially available drugs, such as those derived from chronic leukemia cells with the T3151 mutation. Tirbanibulin (KX2-391) is evaluated in engineered Src driven cell growth assays inNIH3T3/c-Src527F and SYF/c-Src527F cells and exhibits GI50 with 23 nM and 39 nM, respectively[2].
Orally administered Tirbanibulin (KX2-391) is shown to inhibit primary tumor growth and to suppress metastasis, in pre-clinical animal models of cancer[2].
[1]. Lau GM, et al. Expression of Src and FAK in hepatocellular carcinoma and the effect of Src inhibitors on hepatocellular carcinoma in vitro. Dig Dis Sci, 2009, 54(7), 1465-1474. [2]. Fallah-Tafti A, et al. Thiazolyl N-benzyl-substituted acetamide derivatives: synthesis, Src kinase inhibitory and anticancer activities. Eur J Med Chem, 2011, 46(10), 4853-4858.
Approval allows Almirall to move forward with the topical ointment for individuals with AK on the face or scalp.
The US Food and Drug Administration (FDA) has approved tirbanibulin (Klisyri) as a topical treatment for actinic keratosis (AK).
The approval, awarded to Almirall, S.A., will allow the novel, topical first-in-class microtubule inhibitor for treatment of the disease on the face or scalp, representing a significant breakthrough in treatment of AK because of its short treatment protocol of once daily application for 5 days.
Actinic keratosis represents the second most common diagnosis in dermatology in the US, with a reported prevalence between 11-25%.
“Early diagnosis and treatment of actinic keratosis (AK) is critical, since those who already have an AK are likely to develop more actinic keratoses (plural) in the future,” said Deborah S. Sarnoff, MD, President of the Skin Cancer Foundation, said in a statement. “Patients with AK are at higher risk for skin cancer, since AKs can progress into squamous cell carcinoma (SCC), a common and sometimes invasive form of skin cancer.”
The approval is based on recent data from a large phase 3 clinical study, as well as 2 randomized, double-blind, vehicle-controlled phase 3 studies evaluating the efficacy and safety of tirbanibulin ointment 1% in adults with AK on the face or scalp.
“These studies enrolled a total of 702 patients across 62 sites in the United States, providing robust data,” Andrew Blauvelt, MD, MBA, President of Oregon Medical Research Center, and one of the lead investigators of the studies, said in a statement. “Tirbanibulin achieved a significantly higher number of patients with complete (100%) clearance of AK lesions in the treated area compared to vehicle (44% vs. 5% in study 1 and 54% vs. 13% in study 2), as well as reaching the secondary endpoint of partial (≥75%) clearance of lesions.”
[0374]A 1 L single-necked round-bottomed flask was charged with 7 (61.4 g, 0.172 mol), benzyl amine (55.6 g, 0.519 mol, 3 eq), and anhydrous anisole (300 g) and then stirred at reflux until reaction was essentially complete (23 h, 165° C. oil bath temperature; internal temperature was 147° C.) and then allowed to cool to near room temperature. A portion (1 mL) of the reaction mixture was diluted with toluene (1 mL) resulting in the complete crystallization of that portion. This seed was then added to the reaction mixture and allowed to stand until the whole reaction mixture had crystallized to a single block. Toluene (150 mL) was added and the mixture swirled to break up the solid. Heptane/toluene (1:1, 100 mL) was added and the solid mixture broken up further. Finally, heptane (50 mL, then 25 mL) was added and the mixture broken up even further, allowing to stand an additional 30 min before filtering the solid. Filtration of the solid, washing with 2:1 toluene/heptane (300 mL), 1:2 toluene/heptane (300 mL), and then heptane (2×300 mL), and then drying (air, then high vac) gave 60.16 g (yield of 81%) of title product as a white solid (>98.9% AUC). Another 2.5 g of less pure (97.4%) material was obtained from the mother liquors.
[0376]To a stirred suspension of KX2-391 (free base, 60.00 g) in absolute EtOH (600 mL) was added 170 mL of 2.5 M HCl (in ethanol), 25 mL EtOH being added to wash down the sides of the flask. The resulting homogeneous solution was stirred at room temperature (20 min) and then evaporated to near dryness (to frothing). After chasing with EtOH (2×150 mL), the residue was taken up again in EtOH (150 mL) and then was followed by the slow addition of heptane until the mixture appeared saturated (33 mL required for cloudiness to remain). After sitting overnight, two layers had formed. After adding additional heptane (250 mL) crystallization still could not be induced and so the reaction mixture was concentrated to a volume of ˜200 mL at which time the mixture was homogeneous. This thick homogeneous solution was added dropwise to very rapidly stirred (mechanical) EtOAc (2 L). After the addition was complete, a 25 mL EtOH rinse of the original flask and addition funnel was added to the rapidly stirred mixture. The rapid stirring was continued for another ˜1 h and then the mixture was filtered and the solid (partly gummy) was washed with EtOAc (300 mL) and then heptane. As soon as the heptane wash began, the solid got much gummier. The fritted Buchner funnel and its contents were covered (paper towel/rubber band) and immediately placed in the vacuum oven. After overnight vacuum at ˜45° C., the vacuum was released under nitrogen, and the Buchner funnel containing the product (foamy solid) was immediately placed in a zip-lock back and then, under nitrogen (glove bag), transferred to a bottle and the foamy solid broken up (spatula) to a powder. A second night under high vacuum (˜45° C.) resulted in only 1.3 g of additional weight loss. Constant weight was essentially attained with the third night of high vacuum (˜45° C.) where only 0.2 g of weight was lost. The final weight of material was 68.05 g (yield of 97%), containing 0.29 eq (4.8% w/w) of EtOAc, 0.035 eq (0.3% w/w) EtOH, and 0.03 eq (0.6% w/w) heptane. The purity was 99.6%.
[0379]Calculated (%): C, 60.03; H, 6.54; N, 7.65; Cl, 12.91
[0380]Observed (%):C, 59.85/59.97; H, 6.54/6.47; N, 7.67/7.67; Cl, 13.10/13.24
[0381]Calculated FW: 534.63 (does not take into account the 0.8 H2O which probably arose during handling of this very hygroscopic powder, since 1H NMR shows no evidence for H2O).
[0382]The ethyl chloride level in this material was measured and found to be 98 ppm. The sample was also analyzed and found to contain 5,800 ppm of heptane.
[0383]Analysis of another portion of this sample yielded the following results: 99.6% AUC, 1640 ppm ethanol, 41,480 ppm ethyl acetate, 5600 ppm heptane, no anisole detected, and 120 ppm ethyl chloride.
[0384]A procedure for recrystallizing the salt was also developed using the above dried salt. This procedure would work just was well on the highly pure crude salt (containing residual EtOH) obtained from concentrating the HCl salt-forming reaction mixture:
[0385]The salt (575 mg) was dissolved in twice the mass of absolute EtOH (1.157 g) and then heated under nitrogen. To this hot solution (stirred) was added 1.6 g of 25% EtOH (in EtOAc) followed by the addition of EtOAc (0.25 mL) resulting in a cloudiness that remained. The cloudy hot solution was allowed to cool to room temperature during which time crystallization occurred. After crystallization was complete (2 h), the crystalline solid was filtered, washed with anhydrous EtOAc (˜40 mL), and vacuum dried to give 424 mg of the dihydrochloride salt of KX2-391 as a free-flowing solid (tiny beads, 99.8% AUC) containing only 0.05 eq (0.45% w/w) of EtOH and 0.015 eq (0.26% w/w) of EtOAc. Slightly better recovery (460 mg from 586 mg) was attained using isopropanol/EtOAc but the level of solvent entrapment was higher [0.085 eq (1.0% w/w) of isopropanol and 0.023 eq (0.4% w/w) of EtOAc].
Preparation of 2-(5-(4-(2-morpholinoethoxy)phenyl)pyridin-2-yl)-N-benzylacetamide (Compound (I) free base).
[ 000242 ] A l L single-necked round-bottomed flask was charged with 7 (61.4 g, 0.172 mol), benzyl amine (55.6 g, 0.519 mol, 3 eq), and anhydrous anisole (300 g) and then stirred at reflux until reaction was essentially complete (23 h, 165 0C oil bath temperature; internal temperature was 147 0C) and then allowed to cool to near room temperature. A portion (1 mL) of the reaction mixture was diluted with toluene (1 mL) resulting in the complete crystallization of that portion. This seed was then added to the reaction mixture and allowed to stand until the whole reaction mixture had crystallized to a single block. Toluene (150 mL) was added and the mixture swirled to break up the solid. Heptane/toluene (1:1, 100 mL) was added and the solid mixture broken up further. Finally, heptane (50 mL, then 25 mL) was added and the mixture broken up even further, allowing to stand an additional 30 min before filtering the solid. Filtration of the solid, washing with 2:1 toluene/heptane (300 mL), 1:2 toluene/heptane (300 mL), and then heptane (2 x 300 mL), and then drying (air, then high vac) gave 60.16 g (yield of 81%) of title product as a white solid (>98.9% AUC). Another 2.5 g of less pure (97.4%) material was obtained from the mother liquors.
Preparation of 4-(2-(4-(6-(2-(benzylamino)-2-oxoethyl)pyridinium-3-yl)phenoxy)ethyl)- morpholin-4-ium chloride (Compound (I), diHCI salt).
[000244 ] To a stirred suspension of compound (I) (free base, 60.00 g) in absolute EtOH (600 mL) was added 170 mL of 2.5 M HCl (in ethanol), 25 mL EtOH being added to wash down the sides of the flask. The resulting homogeneous solution was stirred at room temperature (20 min) and then evaporated to near dryness (to frothing). After chasing with EtOH (2 x 150 mL), the residue was taken up again in EtOH (150 mL) and then was followed by the slow addition of heptane until the mixture appeared saturated (33 mL required for cloudiness to remain). After sitting overnight, two layers had formed. After adding additional heptane (250 mL) crystallization still could not be induced and so the reaction mixture was concentrated to a volume of -200 mL at which time the mixture was homogeneous. This thick homogeneous solution was added dropwise to very rapidly stirred (mechanical) EtOAc (2 L). After the addition was complete, a 25 mL EtOH rinse of the original flask and addition funnel was added to the rapidly stirred mixture. The rapid stirring was continued for another ~1 h and then the mixture was filtered and the solid (partly gummy) was washed with EtOAc (300 mL) and then heptane. As soon as the heptane wash began, the solid got much gummier. The fritted Buchner funnel and its contents were covered (paper towel/rubber band) and immediately placed in the vacuum oven. After overnight vacuum at -45 0C, the vacuum was released under nitrogen, and the Buchner funnel containing the product (foamy solid) was immediately placed in a zip-lock back and then, under nitrogen (glove bag), transferred to a bottle and the foamy solid broken up (spatula) to a powder. A second night under high vacuum (-45 0C) resulted in only 1.3 g of additional weight loss. Constant weight was essentially attained with the third night of high vacuum (-45 0C) where only 0.2 g of weight was lost. The final weight of material was 68.05 g (yield of 97%), containing 0.29 eq (4.8% w/w) of EtOAc, 0.035 eq (0.3% w/w) EtOH, and 0.03 eq (0.6% w/w) heptane. The purity was 99.6%.
[ 000247] Calculated FW: 534.63 (does not take into account the 0.8 H2O which probably arose during handling of this very hygroscopic powder, since 1H NMR shows no evidence for H2O).
[ 000248] The ethyl chloride level in this material was measured and found to be 98 ppm. The sample was also analyzed and found to contain 5,800 ppm of heptane.
[000249] Analysis of another portion of this sample yielded the following results: 99.6% AUC, 1640 ppm ethanol, 41,480 ppm ethyl acetate, 5600 ppm heptane, no anisole detected, and 120 ppm ethyl chloride.
[000250] A procedure for recrystallizing the salt was also developed using the above dried salt. This procedure would work just was well on the highly pure crude salt (containing residual EtOH) obtained from concentrating the HCl salt-forming reaction mixture:
[000251] The salt (575 mg) was dissolved in twice the mass of absolute EtOH (1.157 g) and then heated under nitrogen. To this hot solution (stirred) was added 1.6 g of 25% EtOH (in EtOAc) followed by the addition of EtOAc (0.25 mL) resulting in a cloudiness that remained. The cloudy hot solution was allowed to cool to room temperature during which time crystallization occurred. After crystallization was complete (2 h), the crystalline solid was filtered, washed with anhydrous EtOAc (~40 mL), and vacuum dried to give 424 mg of the dihydrochloride salt of compound (I) as a free-flowing solid (tiny beads, 99.8% AUC) containing only 0.05 eq (0.45% w/w) of EtOH and 0.015 eq (0.26% w/w) of EtOAc. Slightly better recovery (460 mg from 586 mg) was attained using isopropanol/EtOAc but the level of solvent entrapment was higher [0.085 eq (1.0% w/w) of isopropanol and 0.023 eq (0.4% w/w) ofEtOAc].
Example 3: Large Scale Synthesis of Compound (I) di-HCl
[000252 ] Reagents and solvents were used as received from commercial suppliers. Progress of the reactions was monitored by HPLC, GC/MS, or 1H NMR. Thin-layer chromatography (TLC) was performed using Analtech silica gel plates and visualized by UV light (254 nm). High pressure liquid chromatography (HPLC) was performed on an Agilent 1100 Series instruments. Proton and carbon nuclear magnetic resonance spectra were obtained using a Bruker AV 300 at 300 MHz for proton and 75 MHz for carbon. The solvent peak was used as the reference peak for proton and carbon spectra. Preparation of 4-(2-(4-Bromophenoxy)ethyl)morpholine (2)
[000253 ] A 50 L jacketed reactor equipped with a reflux condenser and temperature probe was charged with 4-(3-chloropropyl)morpholine (2.44 kg, 0.54 mol), 4-bromophenol (2.27 kg, 0.54 mol, 1.0 equiv.), powdered potassium carbonate (6.331 kg, 1.88 mol, 3.50 equiv.), and DMF (12.2 L) and stirred. The reaction mixture was then heated to 60-65 0C and stirred overnight. After 17.5 h, the reaction mixture was cooled to 20-25 °C. The reaction mixture was charged to a different reactor equipped with bottom valve for the work-up. While maintaining a temperature between 20-30 0C, DI water (48.7 L) was charged to the reactor. The phases were separated. The aqueous layer was extracted with MTBE (3 x 24.4 L). To the combined organics, DI water (18.3 L) and then 6M sodium hydroxide (18.2 L) were added. The mixture was stirred for 2-5 minutes and the phases were separated. The organic phase was washed with water (24.4 L) and brine (24.4 L), dried over magnesium sulfate, filtered, and concentrated to give 337Og of a yellow oil (89% crude yield, 99.4% AUC by HPLC).
Preparation of 6-fluoropyridin-3-ylboronic acid (4)
[000254] A 72 L reactor equipped with reflux condenser, and temperature probe. To the reactor 5-bromo-2-fluoropyridine (1.17 L, 0.568 mol), toluene (18.2 L), and triisopropyl borate (3.13 L, 0.68 mol, 1.2 equiv.) were charged and stirred. Tetrahydrofuran (4.4 L) was added to the reactor and the reaction mixture was cooled to between —35 to -50 0C. While maintaining a temperature between -35 to —45 0C, n-butyl lithium (2.5 M solution of hexanes, 5.44 L, 0.68 mol, 1.2 equiv.) was cautiously added to the reactor. After 5 h, the reaction was deemed complete and the reaction mixture was warmed to between -15 to -20 0C. To the reaction was added 2M HCl (11.80L) to the reactor while maintaining a temperature between -15 0C and 0 0C. The reaction mixture was stirred at 18 to 23 0C for (16 h) and the phases were separated. The organics were then extracted with 6 M sodium hydroxide (6.0 L). The acidic anbasic aqueous phases were mixed in the reactor and 6 M HCl (2.5 L) was added until pH 7.5 was achieved. Sodium chloride (6.0 kg) was then added to the aqueous phase. The aqueous phase was then extracted with THF (3 * 20 L). The combined organics were dried with magnesium sulfate and concentrated to give 1300 g of a tan solid (81% crude yield).
Preparation of 4-(2-(4-(6-fluoropyridin-3-yl)phenoxy)ethyl)morpholine (5) [000255] A 72 L reactor equipped with reflux condenser, sparging tube, bubbler, and temperature probe was charged with 6-fluoropyridin-3-ylboric acid (2.84 kg, 1.24 equiv.), 4- (2-(4-bromophenoxy)ethyl)morpholine (4.27 kg, 1.0 equiv.), and DME (27 L). Agitation was started and sodium carbonate (4.74 kg, 3.0 equiv.) as a solution in DI water (17.1 L) was then charged to the reaction mixture. Argon was bubbled through the reaction mixture for 50 minutes. Under an argon atmosphere, tetrakis(triphenylphosphine)palladium (750 g, 0.04 equiv.) was added to the reaction mixture as a slurry in DME (1.0 L). The reaction mixture was heated to 75 – 85 0C and stirred overnight (17 h). The reaction mixture was cooled to between 18 – 22°C. DI water (26.681kg) and MTBE (26.681 L) were charged to the reactor and stirred for 5 minutes. The phases were separated and the aqueous phase was extracted with MTBE (2 x 26.7 L). The combined organics were extracted with 2M HCl (1 x 15.0 L, 3 x 21.8 L). The aqueous phase was then charged back to the reactor and ethyl acetate was added (26.7 L). The pH was adjusted to 6.2 using 6 M sodium hydroxide (26.7 L) while maintaining a temperature between 15 – 25 0C. The phases were separated and the aqueous phase was extracted with ethyl acetate (2 x 26.7 L). The combined organics were dried with magnesium sulfate and concentrated to give 4555 g of a residue (101% crude yield, 67.1% AUC by HPLC).
Purification of 4-(2-(4-(6-fluoropyridin-3-yl)phenoxy)ethyl)morpholine (5)
[000256] The crude product (575 g) was purified by silica gel chromatography by eluting with methanol/ethyl acetate/heptane (30% ethyl acetate/heptane, 50% ethyl acetate/heptane, 75% ethyl acetate/heptane, 100% ethyl acetate, and 5% methanol/ethyl acetate). Concentration of the pure fractions by TLC (10% methanol/dichloromethane, Rf = 0.3) provided 420 g of a light brown solid (73% recovery, >99.9% AUC by HPLC).
Preparation of 2-(5-(4-(2-morpholinoethoxy)phenyl)pyridin-2-yl)acetonitrile (6)
[ 000257] A 1 M solution of NaHMDS (2.0 L, 5.0 equiv.) in THF was charged to a 5-L flask and cooled to -20 to -15 0C. While maintaining a temperature below -10 0C, fluoride (119.7g, 1.0 equiv.) in THF (500 mL) was charged to the flask over 20 minutes. Acetonitrile (82.5 mL, 4.0 equiv.) in THF (170 mL) was added to the flask over 20 minutes, while maintaining a temperature below —100C. The reaction mixture was then stirred for 1 h. To the reaction was added brine (1.5 L, 12.6 vol.) at a rate as to maintain a temperature below 10 0C. The solution was then warmed to room temperature and the layers were allowed to separate. The mixture was filtered over Celite and washed with THF (I x 200 mL, 1 x 100 mL). The aqueous phase was extracted with toluene (750 mL). The combined organics were dried with magnesium sulfate, filtered, washed with toluene (2 * 25OmL), and concentrated to dryness. Toluene (IL) was added and the solution was concentrated to dryness again to give 169.8 g of an oil. MTBE (1190 mL, 7 vol.) was added to the oil at 50 0C and stirred for 15 minutes. Heptane (850 mL, 5vol.) was added over ten minutes at 50 0C. The mixture was then cooled to room temperature over 1.5 h and stirred for 2 h. The slurry was filtered, washed with 1 :4 MBTE/heptane (2 x 100 mL), and dried in an oven overnight at 45 0C to give 102.3 g of an off-white solid (80% yield, 98.8% AUC by HPLC).
Preparation of methyl 2-(5-(4-(2-morpholinoethoxy)phenyl)pyridin-2-yl)acetate (7)
[000258] Nitrile 6 (101 g) and methanol (1.01 L, 10 vol.) were charged to a 3-L flask equipped with stir bar and thermocouple. Concentrated H2SO4 (175 mL, 10.0 equiv.) was added drop wise to the solution over 15 minutes while maintaining a temperature below 60 0C. Followed by 30% fuming sulfuric acid (124 mL) was added drop wise to the solution while maintaining a temperature below 60 0C. The solution was then heated to reflux with a heating mantle and stirred overnight. When the reaction was deemed complete, it was cooled to 20 0C. In a second flask (22 L), saturated sodium bicarbonate (10.7 L) and dichloromethane (1.1 L) were charged and cooled to 15 0C. While maintaining a temperature below 20 0C, the reaction mixture was added to the sodium bicarbonate/dichloromethane mixture. The quench was stirred for 15 minutes and the phases were separated. The aqueous phase was extracted with dichloromethane (I x 55OmL, 1 x 30OmL). The combined organics were dried with magnesium sulfate and concentrated to dryness to give 105 g of an orange solid (94% crude yield, 97.7% AUC by HPLC).
Preparation of 2-(5-(4-(2-morpholinoethoxy)phenyl)pyridin-2-yl)-N-benzylacetamide (Compound (I))
[ 000259] Ester 7 (103 g), anisole (513 mL, 5 vol.), and benzylamine (94 mL, 3.0 equiv.) were charged to a 3 L flask equipped with thermocouple and overhead stirrer. The reaction mixture was then heated to 142 0C and stirred for two days. The reaction mixture was cooled to 45-50 0C and stirred for 2 hours. To the mixture was added n-heptane (1.5 L) dropwise over an hour. The solution was cooled to room temperature over three hours and then stirred overnight. The resulting slurry was filtered, washed with 4: 1 Anisole/n-heptane (200 mL) and n-heptane (3 x 100 mL). Drying in the oven overnight, the resulting product was 112. Ig of a tan solid (90% yield, 99.6% AUC by HPLC). The use of a single isomer of heptane was essential to adequately quantitate the residual solvent.
Preparation of 2-(5-(4-(2-morpholinoethoxy)phenyl)pyridin-2-yl)-N-benzylacetamide dihydrochloride salt (Compound (I) 2HC1)
[000260 ] EtOH (1.0 L) was charged to a 2-L flask and acetyl chloride (62.5 raL, 3.0 equiv.) was added slowly to the flask and stirred for 40 minutes. The resulting solution was added to compound (I) (100 g) over 30 minutes while maintaining a temperature of 30 0C. The solution was concentrated to a mass of 270 g. The concentrated solution was added to ethyl acetate (2 L) over 20 minutes with rapid stirring. The mixture was stirred overnight and then filtered under nitrogen to give two distinct solid products, tan solids (73.5 g) and darker solids (42.2 g). The solids were dry blended to give a combined yield of 99%. The HPLC analysis indicated 99.0% purity (AUC).
Analysis indicated that ethanol was present at 2530 ppm, ethyl acetate at 48,110 ppm, ethyl chloride at 170 ppm, and no heptane and anisole were detected. Palladium content was assayed three times and measured to be 29 ppm, 2 ppm, and less than 1 ppm.
Crystallization Study of Compound (I) 2HCl
Preparation of 2-(5-(4-(2-morpholinoethoxy)phenyl)pyridin-2-yl)-N-benzylacetamide (Compound (I))
[000268] To a 22-L reactor was charged compound 7 (650 g, 1.82 mol), anisole (3.25
L, 5 vol, anhydrous) and benzylamine (600 mL, 0.92 vol, 3 equiv). The batch (approximately 18 °C) was heated to 142 ± 5 °C over 1 hour 44 minutes, with dissolution occurring at 30 0C. The batch was maintained at 142 ± 5 0C for 69 hours 30 minutes at which point HPLC analysis indicated that compound 7 was 0.9% by conversion (specification <1.7% by conversion). The batch was cooled to 45-50 0C over 5 hours 12 minutes (to aid cooling the nitrogen flow was increased once the batch was approximately 72 0C). At that temperature range, the batch was poorly stirring and on mixing, the batch temperature increased to 52 0C. It was >50 °C for <15 minutes. The batch was aged for 2 hours 2 minutes once initially <50 0C, then n-heptane (9.75 L, 15 vol, 99%) was added to the batch over 1 hour 56 minutes, maintaining the batch temperature at 45-50 °C. The heating was then discontinued and the batch cooled to 25 0C over 10 hours 32 minutes and then to approximately 20 °C over 20 minutes. The total time the batch was maintained <25 0C was 4 hours 50 minutes (2 hours 47 minutes at approximately 20 0C). The batch was filtered under suction via a 24-inch polypropylene filter funnel (fitted with a PTFE cloth) and the reactor rinsed with anisole/n- heptane (1.3 L, 4: 1) and the rinse transferred to the cake. The cake was then washed successively with two portions of /i-heptane (1.3 L, 0.65 L). The total filtration time was 39 minutes. The batch (net wet weight 1004 g of KX2391) was transferred to three glass trays and placed into a vacuum oven set at 50 0C and dried to constant weight over 96 hours 26 minutes.
Preparation of 2-(5-(4-(2-morpholinoethoxy)phenyl)pyridin-2-yl)-N-benzylacetamide mesylate (Compound (I)-MSA)
[000269] Compound (I) (520 g, 1.21 mol) was transferred to reactor 1 using acetone (41.6 vol, 80 vol, ACS) to facilitate the transfer. The batch was heated to 50 ± 5 0C over 33 minutes with dissolution occurring at 30 0C . The batch was clarified into a second reactor via a transfer pump fitted with an inline filter (Pall P/N 12077, 10 micron) and reheated from 46 0C to 50 ± 5 0C. Methanesulfonic acid (121.4 g, 1.05 equiv, 99% extra pure) was added to the pale yellow batch over 12 minutes and the heating then discontinued. After fourteen minutes, white solids were observed, which increased in number to give after 59 minutes a white suspension. The batch was in the range of 25 ± 5 0C after 7 hours 51 minutes and aged for a further 19 hours 21 minutes (10 hours 30 minutes at <27 0C). The batch was filtered under suction via a 24-inch polypropylene filter (PTFE cloth) and the reactor rinsed with acetone (2.0 L, clarified, ACS) and the rinse transferred to the cake. The cake was covered with a stainless steel cover and sucked dry under a flow of nitrogen. The total filtration time was 21 minutes. The batch (net wet weight 764 g) was transferred to three glass drying trays and dried in a vacuum oven to constant weight at 25 ± 5 °C over 21 hours 54 minutes (565 g, 89% of theory). A sample was removed for analysis and the batch maintained in vacuo at 25 ± 5 °C. The batch was then transferred to two 80-oz amber glass bottles (Teflon lined polypropylene closure), blanketed with argon and stored at -10 to -20 °C.
[00045] The synthesis of 4-(2-(4-(6-fluoropyridin-3-yl)phenoxy)ethyl)morpholine is shown in the scheme below:
[00046] 4-(2-(4-(6-fluoropyridin-3-yl)phenoxy)ethyl)morpholine (5) was synthesized in 3 steps. Intermediate 2 was synthesized using an ether coupling reaction e.g., using Williamson ether synthesis. Ether formation between 4-(2-chloroethyl)morpholine (1) and A- bromophenol was carried out in the presence of potassium carbonate and DMF to afford 4-(2- (4-bromophenoxy)ethyl)morpholine (2). Rigorously dry conditions were not essential for this reaction and a basic wash with sodium hydroxide was used to remove any remaining A- bromophenol. In another aspect of the invention, intermediate 2 is synthesized using any ether formation reaction. Intermediate 2 is synthesized starting from compound 1 containing any leaving group. For example, the skilled chemist would start with compounds of the
general formula
wherein the leaving group “LG” includes but is not limited to halogen, tosylate, mesylate, trifluate, etc.
[00047] Compound 5 was formed using a Suzuki reaction. Formation of the aryl borate, 6-fluoropyridin-3-yl-3-boronic acid (4), was carried out by forming the aryl anion using n-BuLi followed by in situ quenching with triisopropylborate (Li, et ah, J. Org. Chem. 2002, 67, 5394-5397). The resulting 6-fluoropyridin-3-yl-3-boronic acid (4) was coupled to 4-(2-(4-bromophenoxy)ethyl)morpholine (2) in a solution of DME and aqueous sodium carbonate using tetrakis(triphenylphosphine)palladium to afford 4-(2-(4-(6-fluoropyridin-3- yl)phenoxy)ethyl)morpholine (5), which was purified using silica gel chromatography. The skilled chemist would know that other transition metal coupling reaction are used to prepare compound 5.
[00048] The synthesis of 2-(5-(4-(2-morpholinoethoxy)phenyl)pyridin-2-yl)-JV- benzylacetamide dihydro chloride is shown below:
[00049] 2-(5-(4-(2-morpholinoethoxy)phenyl)pyridin-2-yl)-N-benzylacetamide dihydrochloride (KX2-391 HCl) was synthesized in four linear steps. The fluoride of 4-(2-(4- (6-fluoropyridin-3-yl)phenoxy)ethyl)morpholine (5) was displaced by the anion of acetonitrile formed using commercially available NaHMDS. Acetonitrile was added slowly to a cooled mixture of compound 5 and base to form 2-(5-(4-(2- morpholinoethoxy)phenyl)pyridin-2-yl)acetonitrile (6). In another aspect of the invention, intermediate 5 may have a leaving group other than fluorine. Thus, compounds of the general formula:
would be pursued where LG includes other leaving groups known to the skilled chemist.
[00050] Acid catalyzed methanolysis of 2-(5-(4-(2-morpholinoethoxy)phenyl)pyridin-
2-yl)acetonitrile (6) was carried out using a mixture of concentrated sulfuric and fuming sulfuric acid. The use of fuming sulfuric acid removed residual water from the reaction mixture and reduced the amount of carboxylic acid by-product formed. The reaction mixture was quenched by adding the reaction mixture to a solution of saturated sodium bicarbonate and dichloromethane while maintaining the temperature below 20 ºC. Any carboxylic acid contaminant was readily removed with aqueous work-up. In another aspect of the invention, other acid catalyzed conditions are used by the skilled artisan for alcoho lysis of the nitrile of compound 6 to produce compound 7.
[00051] The resulting methyl 2-(5-(4-(2-morpholinoethoxy)phenyl)pyridin-2- yl)acetate (7) and benzyl amine were coupled in anisole at high temperature to afford 2-(5-(4- (2-morpholinoethoxy)phenyl)pyridin-2-yl)-N-benzylacetamide (KX2-391). An HCl solution formed by adding acetyl chloride to absolute ethanol was added to KX2-391 to form the bis- HCl salt, 2-(5-(4-(2-morpholinoethoxy)phenyl)pyridin-2-yl)-N-benzylacetamide dihydrochloride, (KX2-di-HCl).
[00052] The synthesis of the mesylate salt of KX2-391 (KX2-391 -MSA) is depicted in the scheme below:
[00053] 2-(5-(4-(2-morpholinoethoxy)phenyl)pyridin-2-yl)-N-benzylacetamide mesylate (KX2-391 MSA) was synthesized in four linear steps starting from compound 5.
The first 3 steps were carried out similar to the procedure discussed above for KX2-391 2HCl to afford methyl 2-(5-(4-(2-morpholinoethoxy)phenyl)pyridin-2-yl)acetate (KX2-391). KX2-
391 was converted to the methanesulfonate salt by treatment with methanesulfonic acid
(MSA) in acetone at 50 ºC to afford 2-(5-(4-(2-morpholinoethoxy)phenyl)pyridin-2-yl)-JV- benzylacetamide mesylate (KX2-391 MSA).
EXAMPLES Example 1: Small Scale Synthesis of KX2-391
[000343] The preliminary synthesis described below was illustrated in
US20060160800A1. This procedure is useful for small scale reactions, for example, reactions that produce up to 50 g of product.
[000344] For the following synthesis, unless otherwise noted, reagents and solvents were used as received from commercial suppliers. Proton and carbon nuclear magnetic resonance spectra were obtained on a Bruker AC 300 or a Bruker AV 300 spectrometer at 300 MHz for proton and 75 MHz for carbon. Spectra are given in ppm (δ) and coupling constants, J, are reported in Hertz. Tetramethylsilane was used as an internal standard for proton spectra and the solvent peak was used as the reference peak for carbon spectra. Mass spectra and LC-MS mass data were obtained on a Perkin Elmer Sciex 100 atmospheric pressure ionization (APCI) mass spectrometer. LC-MS analyses were obtained using a Luna C8(2) Column (100 x 4.6 mm, Phenomenex) with UV detection at 254 nm using a standard solvent gradient program (Method B). Thin-layer chromatography (TLC) was performed using Analtech silica gel plates and visualized by ultraviolet (UV) light, iodine, or 20 wt % phosphomolybdic acid in ethanol. HPLC analyses were obtained using a Prevail Cl 8 column (53 x 7 mm, Alltech) with UV detection at 254 nm using a standard solvent gradient program (Method A or B). Method A:
A = Water with 0.1 v/v Trifluoroacetic Acid
B = Acetonitrile with 0.1 v/v Trifluoroacetic Acid
Method B:
A = Water with 0.02 v/v Trifluoroacetic Acid
B = Acetonitrile with 0.02 v/v Trifluoroacetic Acid
Synthesis of Η-benzyl-2- (5-bromopyridin-2-yl)acetamide :
[000345] A flask was charged with 5-(5-bromopyridin-2(lH)-ylidene)-2,2-dimethyl- l,3-dioxane-4,6-dione (1.039 g, 3.46 mmol), benzylamine (0.50 mL, 4.58 mmol), and toluene (20 mL). The reaction was brought to reflux under nitrogen for 18 hours, then cooled and placed in a freezer until cold. The product was collected by filtration and washed with hexanes to yield a mass of bright white crystals (1.018 g, 96%).
[000346] To a stirring solution of 4-(4,4,5,5-tetramethyl[l,3,2]dioxaborolan-2-yl)- phenol (2.55 g, 11.58 mmol), 2-morpholin-4-ylethanol (1.60 mL, 1.73 g, 13.2 mmol) and triphenyl phosphine (3.64 g, 13.9 mmol) in methylene chloride (60 mL) at 0 ºC was added dropwise DIAD (2.82 g, 13.9 mmol). The reaction was allowed to warm to room temperature and stir overnight. After 18 hours, additional portions of triphenyl phosphine (1.51 g, 5.8 mmol), 2-morpholin-4-ylethanol (0.70 mL, 5.8 mmol), and DIAD (1.17 g, 5.8 mmol) were added. After stirring an additional 2 hours at room temperature the reaction was concentrated and the residue purified by flash chromatography (5% to 25% EtOAc in CHCI3) to provide the product as a white solid (2.855 g, 74%).
Synthesis of 2-(5-(4-(2-morpholinoethoxy)phenyl)pyridin-2-yl)-N-benzylacetamide KX2-391
[000347] A lO rnL reaction tube with a septum closure and stir bar was charged with N- benzyl-2-(5-bromopyridin-2-yl)acetamide (123 mg, 0.403 mmol), 4-(2-(4-(4,4,5,5- tetramethyl[l,3,2]dioxaborolan-2-yl)-phenoxy)ethyl)morpholine (171 mg, 0.513 mmol), and FibreCat 1007 (30 mg, 0.015 mmol). Ethanol (3 mL) was added, followed by aqueous potassium carbonate solution (0.60 mL, 1.0 M, 0.60 mmol). The tube was sealed and heated under microwave conditions at 150 ºC for 10 minutes. The reaction was cooled and concentrated to remove the majority of the ethanol, and then taken up in 10 mL of ethyl acetate and washed successively with water and saturated sodium chloride solution. The organic layer was dried with MgSO4, filtered and concentrated to a white solid. This white solid was triturated with ethyl ether to give KX2-391 as a white solid (137 mg, 79%): mp 135-137 ºC; 1H NMR (300 MHz,CDCl3) δ 8.70 (d, IH, J=2.0 Hz), 7.81 (dd, IH, J=2.4 Hz, J=8.0Hz), 7.65 (br s, IH), 7.49 (d, 2H, J=8.8 Hz), 7.37-7.20 (m, 6H), 7.01 (d, 2H, J=8.8 Hz), 4.49 (d, 2H, J=5.8 Hz), 4.16 (t, 2H, J=5.7 Hz, 3.82 (s, 2H), 3.78-3.72 (m, 4H), 2.84 (t, 2H, J=5.7 Hz), 2.62-2.58 (m, 4H); HPLC (Method B) 98.0% (AUC), tR = 1.834 min.; APCI MS m/z 432 [M+H]+.
Example 2: Intermediate Scale Synthesis of KX2-391 di-hydrochloride
[000348] The synthesis outlined in this example can be used on intermediate-scale reactions. The preparation of batches of at least 50 g of the dihydrochloride salt of KX2-391 is shown in Scheme 1. The linear synthesis consisted of 6 steps, a seventh step being the preparation of one of the reagents, 6-fluoropyridin-3-ylboronic acid (which is also available commercially). The overall yield of the sequence was 35% with an average yield of 83%, with the lowest yielding step being that of 68%. Of the seven steps only one required chromatography. The procedure listed below was performed on a 70 g scale.
[000349] The first step is a Williamson ether synthesis between 4-bromophenol (131 g) and N-chloroethylmorpholine (1 as the HCl salt; 141 g) using K2CO3 powder (3 to 3.5 equivalents) as the base and having acetonitrile as the solvent. The ingredients were mixed and stirred at reflux overnight with high conversion (96.3-99.1%). After dilution with dichloromethane and heptane, the reaction mixture was filtered and evaporated to give the desired product 2 in essentially a quantitative yield (216 g). Note that with similar substrates (e.g., 4-bromo-3-fluorophenol), conversions (even with extensive heating) were not always so high (e.g., 59.9-98.3%). Both the alkyl chloride and the K2CO3 are preferably purchased from Aldrich. If continued heating does not drive reaction to completion, unreacted bromophenol can readily be removed by dissolving the crude reaction mixture in 4 parts toluene and washing out the phenol with 4 parts 15% aqueous NaOH. [000350] One of the reagents required for the second step (Suzuki coupling) was 6- fluoropyridin-3-ylboronic acid (4). Although available commercially, this reagent was readily prepared by lithium-bromide exchange of 5-bromo-2-fluoropyridine (3, 102 g) with n- butyllithium (1.2 eq) at low temperatures (<-60 ºC) in TBME followed by the addition of triisopropylborate (1.65 eq). Both stages of the reaction are brief, with an overall reaction time (including addition times) of ~3 h. Quenching is achieved with aqueous 24% NaOH, which also extracts the product leaving impurities in the organic layer. Once the aqueous layer is removed, it is then neutralized with HCl and extracted with EtOAc. After drying the organics and diluting with some heptane, concentration leads to precipitation/ crystallization of the product. Filtration gave the boronic acid 4 in relatively high purity (96.4% AUC) and good yield (69 g, 79-90%; see note on estimation of yield in the experimental section), which can be used without further purification.
[000351] The second reaction step in the linear sequence (a Suzuki coupling) is a simple reaction to set up; all the reagents [2 (111 g), aqueous Na2CO3, DME, and Pd(PPh3)4 (0.04 eq)] were charged to the reaction flask and the mixture heated at reflux; note that the reaction mixture was degassed to remove oxygen. Once the reaction is complete (within 7 h), the work-up involved decanting (or siphoning off) of reaction solution from the organic salts on the side of the flask (there was no visible aqueous layer), the flask was rinsed, and dried, and the solvent was removed from the combined organics. Crystallization of crude 5 from isopropanol/heptane provided material of improved purity compared to the crude, but still required chromatography (ratio of silica gel to crude was -8.5:1) to obtain material of adequate purity (>98%); the yield was 68% (79.5 g). Use of clean 5 prevented the need for chromatography in the next step, acetonitrile displacement of the fluorine atom. [000352] The replacement of fluoride with acetonitrile was also a simple reaction, and a simple room temperature crystallization of the crude product provided clean 6 in high yield and purity. The reaction involved initial formation of the “enolate” from acetonitrile (6.5 eq) using potassium hexamethyldisilane KHMDS (8 eq)/THF at -10 ºC followed immediately by the addition of fluoride 5 (79 g). The reaction was quick and after one hour quenching was achieved with saturated brine. After drying and evaporation of solvent of the organics, the resulting crude mixture consisted of only two components, the desired product and a much less polar product from apparent self-condensation of acetonitrile. The crude mixture was swirled in isopropanol/heptane and allowed to sit overnight, which resulted in complete crystallization of the product, which was filtered off and washed to provide high purity 6 (99.3% AUC) in good yield (64 g, 76%).
[000353] Methanolysis of 6 (64 g) was accomplished by heating in 40% H2SO4 (in
MeOH) until the reaction was complete (25 h). The reaction was then cooled, stirred with MgSO4 to convert traces of hydro lyzed product (ArCH2-CO2Me) back to product, and then added to cooled, aqueous K2CO3, with simultaneous extraction into dichloromethane. Drying and evaporation of most of the DCM followed by addition of 5% EtOAc (in heptane) and further concentration resulted in the crystallization of the product. Filtration of the solid and washing gave high purity (98.9% AUC) 7 in good yield (82%), additional high purity product (4 g) being obtained from the mother liquors for a total yield of 61.7 g (87%). [000354] The amidation step also involved charging of the reaction vessel with the ingredients (7 (61 g), benzyl amine (3 eq), and high boiling anisole) and then heating at reflux until the reaction was complete. Cooling of the reaction mixture resulted in complete crystallization of the target compound with high purity (98.9%) and good yield (81%). [000355] The final step was the formation of the dihydro chloric salt of the target compound. In order to ensure complete protonation at both basic sites, the reaction was conducted in absolute ethanol, which freely dissolved the dihydrochloride salt. After evaporation to near dryness, the reaction mixture was “chased” with ethanol twice to remove excess hydrogen chloride. The resulting viscous oil was dissolved in ethanol (2 parts) and then added, with rapid stirring, to a large volume (20 parts) EtOAc (ethyl acetate). Filtration, washing with ethyl acetate (no heptane) and vacuum drying provided the dihydrochloride salt of KX2-391 as a creamy-white powder. A total of 68 g (yield of 97%) was obtained of the final salt in high purity (99.6% AUC), which contained traces of EtOAc (4.8% w/w), EtOH (0.3% w/w), and heptane (0.6% w/w; from a final wash with heptane prior to vacuum drying). This salt was also crystallized (instead of the precipitation method described above) from hot EtOH/EtOAc to afford crystalline beads that had much lower entrapped solvent levels (only 0.26% w/w of EtOAc and 0.45% w/w of EtOH) and was free-flowing.
Preparation of 4-(2-(4-bromophenoxy)ethyl)morpholine (2):
[000356] A 5 L three-necked round-bottomed flask, equipped with mechanical stirrer, thermometer with adapter, condenser, and nitrogen inlet (on top of condenser), was charged with 1 (140.7 g, 0.756 mol), 4-bromophenol (130.6 g, 0.755 mol), anhydrous K2CO3 powder (367.6 g, 2.66 mol, 3.5 eq), and acetonitrile (1.3 L). The mixture was vigorously stirred (blade touching bottom of flask) at 80 ºC (overnight), followed by dilution with DCM (500 mL) and heptane (200 mL) and filtration through Celite. Evaporation to dryness (rotovap, then high vac) gave 2 as a light yellow oil (216.00 g, yield of 100%, 96.3% AUC, contains 3.7% unreacted bromophenol). This material was used successfully without further purification.
[000358] That the bromophenol can be readily removed was demonstrated on a 2 g sample by first dissolving the sample in toluene (8 g) and washing with 8 g of 15% aqueous NaOH; liquid chromatography showed no trace of unreacted bromophenol in the recovered product (1.97 g; 98.5% recovery).
Preparation of 6-fluoropyridin-3-ylboronic acid (4):
[000359] To stirred and cooled (dry ice-acetone bath) anhydrous [TBME] (620 mL; in a
3 L three-necked round-bottomed flask equipped with mechanical stirrer, temperature probe with adapter, and nitrogen inlet) was added (via syringe) 2 M BuLi (352 mL, 0.704 mol, 1.2 eq). To this rapidly stirred and cooled (< -75 ºC) mixture was added a solution of 3 (102.2 g, 0.581 mol) in anhydrous TBME (100 mL) over a period of 13 min during which time the internal temperature rose to -62 ºC. The reaction was stirred for another 45 min (the temperature was maintained between -62 ºC and -80 ºC), followed by the rapid and sequential addition of four portions of triisopropylborate (total of 180 g, 0.957 mol, 1.65 eq). At the end of the addition the internal temperature had risen to -33 ºC. After stirring an additional 45 min over the cold bath (internal temperature lowered from -33 ºC to -65 ºC), the cold bath was removed and the stirred mixture on its own rose to -22 ºC over a period of 50 min. After warming (via water bath) to 6 ºC over a period of 15 min, the stirred reaction mixture was placed in an ice-water bath and then quenched under nitrogen with a cooled solution of NaOH (160 g) in water (500 mL). Once the addition was complete, the internal temperature was 20 ºC. This mixture was stirred at room temperature for 1.5 h. The aqueous layer was removed, neutralized to pH 7 with -350 mL concentrated HCl, and then extracted with EtOAc (3 x 1 L). Because the pH was now 8-9, the aqueous layer was adjusted to pH 7 using ~15 mL concentrated HCl and extracted further (2 x 1 L) with ethyl acetate. The combined EtOAc extracts were dried (Na2SO4), filtered, and concentrated to a volume of -150 mL. With swirling of the concentrate, heptane was added in portions (total volume of 300 mL) resulting in the precipitation/crystallization of the product. Filtration, washing of the solid with heptane (100 mL, 300 mL, then another 300 mL), and air drying gave the title product as an off-white solid (68.6 g, yield of 79-90%*; LC purity of 96.4%, NMR showed an estimated 5.5% w/w of heptane), which was used successfully without further purification. LC/MS showed it to be a mixture of the two following entities, the intensity of the higher molecular weight entity being major (*Note: yield of reaction is 79% if the boronic acid is assumed to be the only constituent and is 90% if it is assumed that the cyclic borate is the only constituent):
[000360] A 2 L three-necked round-bottomed flask equipped with mechanical stirrer, thermometer and adapter, condenser, and nitrogen inlet (at top of condenser) was charged with 2 (110.7 g, 0.387 mol), 4 (71.05 g, 0.477 mol, 1.23 eq) and DME (700 mL). The resulting stirred solution was degassed by passing a rapid stream of nitrogen through the stirred solution over a period of 5 min followed by the addition of a degassed solution of Na2CO3 (121.06 g, 1.142 mol, 3 eq) in H2O (250 mL) and also solid Pd(PPh3)4 (19.8 g, 0.044 eq). Immediately after the last addition, the head space above the reaction mixture was purged with nitrogen and the mixture then stirred at 80-85 ºC (internal temperature) for 7 h, followed by cooling to room temperature. Because of the lack of an aqueous layer, the supernatant was decanted, leaving behind the inorganic salts (with adsorbed water). The reaction flask with the inorganic salts was washed with 50% dichloromethane/ethyl acetate (2 x 250 mL), the washes being added to the decanted supernatant. These combined organics were dried (Na2SO4), filtered, and evaporated to dryness to a dark brown oil (148 g). To this oil was added 15O g of 50% heptane/isopropyl alcohol (IPA) and after swirling and cooling (via ice water bath), crystallization began. Additional heptane (50 g) was added and the resulting solid was filtered, washed, and air dried to give 48 g of a light brown solid. After evaporating the filtrate to dryness, the resulting mixture was swirled in 100 mL of 50% heptane/IPA followed by the addition of more heptane (-100 mL), stoppering and placing in the freezer for crystallization. The resulting solid was filtered, washed with heptane, and air dried to give 61 g of a gummy solid. Evaporation of the resulting filtrate gave an oil (34 g) which contained significant less polar impurities including Ph3P=O and so it was partitioned between 2 N HCl (240 mL) and EtOAc (220 mL). The bottom aqueous layer was removed and then stirred with EtOAc while neutralizing with K2CO3 to a pH of 7-8. The EtOAc layer was dried, filtered, and evaporated to dryness (22 g). The 48 g, 61 g, and 22 g portions were chromato graphed over silica gel (1.1 Kg) packed in DCM. Elution with DCM (400 mL), 50% DCM/EtOAc (5 L), and then 50% DCM/EtOAc (8 L) containing increasing amounts of MeOH/Et3N (beginning with 1.5% MeOH/1% Et3N and ending with 5% MeOH/3% Et3N) gave 77.68 g of a viscous oil (purity 98.0%) which immediately crystallized upon swirling in heptane (300 mL). Filtration, washing with heptane and air drying gave 75.55 g (98.7% AUC) of solid 5. Additional pure 5 (total of 3.9 g, 98.6-99.3% AUC) was obtained from earlier chromatographic fractions containing Ph3P=O by cleaning them up as done for the above 34 g sample, followed by evaporative crystallization. The total yield of 5 was 79.5 g (68%). 1H NMR (CDCl3) δ 2.59 (t, 4 H), 2.84 (t, 2 H), 3.75 (t, 4 H), 4.16 (t, 2 H), 6.97 (dd, 1 H), 7.01 (d, 2 H), 7.46 (d, 2 H), 7.92 (ddd, 1 H), 8.37 (fine d, 1 H). MS (from LC/MS): m/z 303.2 [M + I].
Preparation of 2-(5-(4-(2-morpholinoethoxy)phenyl)pyridin-2-yl)acetonitrile (6):
[000361] A 3 L three-necked round-bottomed flask was equipped with mechanical stirrer, thermometer and adapter, additional funnel, and nitrogen inlet (on top of addition funnel, positive pressure through a bubbler). With a rapid stream of nitrogen going through the bubbler, the stopper was removed and the flask was charged with KHMDS (415.8 g, 2.08 mol) and then anhydrous THF (1 L). To the stirred and cooled (ice/methanol bath, internal temperature of solution was -8 ºC) KHMDS/THF solution was added dropwise a solution of MeCN (70 g) in THF (110 mL) over a period of 22 min followed immediately by the relatively rapid (4 min) addition of a solution of 5 (79.06 g, 0.262 mol) in THF (400 mL), after which time the internal temperature of the reaction mixture had reached 10 ºC. With continued cooling (1 h) the internal temperature was -6 ºC and by TLC the reaction appeared complete. After an additional 30 min (internal temperature of -3 ºC), the reaction mixture was quenched with saturated brine (1 L) and diluted with EtOAc (500 mL). After removing the aqueous layer, the organic solution was dried (Na2SO4), filtered, and evaporated to dryness (to an oil) followed by completely dissolving in IPA (150 mL), diluting with heptane (300 mL), adding seed crystals (prepared by dissolving -100 mg of crude oil in IPA (-150 mg) and diluting with heptane (-2.5 mL)), and allowing to stand overnight. After stirring to break up the crystalline solid, the solid was filtered, washed with 250 mL 2:1 heptane/IP A and then multiple washes with heptane and air dried to give 64.38 g (yield of 76%) of title product 6 as a crystalline tan solid (LC purity of 99.3%). Another 5.88 g of less pure material was obtained from the filtrate.
Preparation of methyl 2-(5-(4-(2-morpholinoethoxy)phenyl)pyridin-2-yl)acetate (7): [000363] A 2 L single-necked round-bottomed flask was charged with 6 (64.00 g, 0.198 mol) and MeOH (360 g) followed by the slow, careful, and dropwise addition OfH2SO4 (240 g) and the resulting homogeneous solution stirred at reflux (115 ºC oil bath) until the reaction was complete (25 h with 0.8% unreacted starting material) with 3.5% ArCH2CO2H. After brief cooling, MgSO4 (75 g) was added and the mixture swirled and allowed to stand an additional 45 min (composition now 96.3% product, 0.8% unreacted starting material, and 2.5% ArCH2CO2H). The reaction mixture was then added slowly to a rapidly stirred and cooled (ice-water bath) mixture of DCM (2 L) and a solution OfK2CO3 (450 g) in H2O (600 mL). The resulting emulsion was allowed to stand overnight. The clear portions of organic solution were siphoned off and the remainder portions were treated iteratively with water and DCM, the clear organics being combined with the original portion that was siphoned off. The combined organics were dried (Na2SO4), filtered, and concentrated to a volume of ~1.2 L followed by the addition of 300 mL of 5% EtOAc (in heptane) and then heptane (300 mL) and the mixture concentrated (rotovap with heat) again to remove the DCM. At this point 15 mL EtOAc was added and the hot mixture swirled until crystallization had begun, swirling continued until crystallization was near complete, and then allowed to stand and cool to room temperature for complete crystallization. The solid was then filtered, washed with 300 mL 5% EtOAc (in heptane) and heptane (100 mL) and then fully air dried to give 57.74 g (yield of 82%) of 7 as a light yellow solid (98.9% AUC). Another 3.94 g of clean product (97.9% AUC) was obtained from the filtrate (total yield of 87%).
[000364] 1H NMR (CDCl3) δ 2.60 (t, 4 H), 2.84 (t, 2 H), 3.74 (overlapping t and s, 6 H),
Preparation of 2-(5-(4-(2-morpholinoethoxy)phenyl)pyridin-2-yl)-N-benzylacetamide (KX2-391 free base).
[000365] A l L single-necked round-bottomed flask was charged with 7 (61.4 g, 0.172 mol), benzyl amine (55.6 g, 0.519 mol, 3 eq), and anhydrous anisole (300 g) and then stirred at reflux until reaction was essentially complete (23 h, 165 ºC oil bath temperature; internal temperature was 147 ºC) and then allowed to cool to near room temperature. A portion (1 mL) of the reaction mixture was diluted with toluene (1 mL) resulting in the complete crystallization of that portion. This seed was then added to the reaction mixture and allowed to stand until the whole reaction mixture had crystallized to a single block. Toluene (150 mL) was added and the mixture swirled to break up the solid. Heptane/toluene (1 :1, 100 mL) was added and the solid mixture broken up further. Finally, heptane (50 mL, then 25 mL) was added and the mixture broken up even further, allowing to stand an additional 30 min before filtering the solid. Filtration of the solid, washing with 2:1 toluene/heptane (300 mL), 1 :2 toluene/heptane (300 mL), and then heptane (2 x 300 mL), and then drying (air, then high vac) gave 60.16 g (yield of 81%) of title product as a white solid (≥98.9% AUC). Another 2.5 g of less pure (97.4%) material was obtained from the mother liquors. 1H NMR (CDCl3) δ 2.60 (t, 4 H), 2.83 (t, 2 H), 3.74 (t, 4 H), 3.82 (s, 2 H), 4.18 (t, 2 H), 4.49 (d, 2 H), 7.01 (d, 2 H), 7.2-7.35 (m, 6 H), 7.49 (d, 2 H), 7.64 (br t, 1 H), 7.81 (dd, 1 H), 8.69 (fine d, 1 H). MS (from LC/MS): m/z 432.5 [M + I].
Preparation of 4-(2-(4-(6-(2-(benzylamino)-2-oxoethyl)pyridinium-3-yl)phenoxy)ethyl)- morpholin-4-ium chloride (KX2-391, diHCl salt).
[000366] To a stirred suspension of KX2-391 (free base, 60.00 g) in absolute EtOH (600 niL) was added 170 niL of 2.5 M HCl (in ethanol), 25 niL EtOH being added to wash down the sides of the flask. The resulting homogeneous solution was stirred at room temperature (20 min) and then evaporated to near dryness (to frothing). After chasing with EtOH (2 x 150 mL), the residue was taken up again in EtOH (150 mL) and then was followed by the slow addition of heptane until the mixture appeared saturated (33 mL required for cloudiness to remain). After sitting overnight, two layers had formed. After adding additional heptane (250 mL) crystallization still could not be induced and so the reaction mixture was concentrated to a volume of -200 mL at which time the mixture was homogeneous. This thick homogeneous solution was added dropwise to very rapidly stirred (mechanical) EtOAc (2 L). After the addition was complete, a 25 mL EtOH rinse of the original flask and addition funnel was added to the rapidly stirred mixture. The rapid stirring was continued for another ~1 h and then the mixture was filtered and the solid (partly gummy) was washed with EtOAc (300 mL) and then heptane. As soon as the heptane wash began, the solid got much gummier. The fritted Buchner funnel and its contents were covered (paper towel/rubber band) and immediately placed in the vacuum oven. After overnight vacuum at ~45 ºC, the vacuum was released under nitrogen, and the Buchner funnel containing the product (foamy solid) was immediately placed in a zip-lock back and then, under nitrogen (glove bag), transferred to a bottle and the foamy solid broken up (spatula) to a powder. A second night under high vacuum (-45 ºC) resulted in only 1.3 g of additional weight loss. Constant weight was essentially attained with the third night of high vacuum (~45 ºC) where only 0.2 g of weight was lost. The final weight of material was 68.05 g (yield of 97%), containing 0.29 eq (4.8% w/w) of EtOAc, 0.035 eq (0.3% w/w) EtOH, and 0.03 eq (0.6% w/w) heptane. The purity was 99.6%.
Elemental analysis (for C26H29N3O3 • 2 HCl • 0.035 EtOH • 0.29 EtOAc • 0.03 heptane • 0.8 H2O): Calculated (%): C, 60.03; H, 6.54; N, 7.65; Cl, 12.91 Observed (%):C, 59.85/59.97; H, 6.54/6.47; N, 7.67/7.67; Cl, 13.10/13.24 Calculated FW: 534.63 (does not take into account the 0.8 H2O which probably arose during handling of this very hygroscopic powder, since 1H NMR shows no evidence for H2O). [000367] The ethyl chloride level in this material was measured and found to be 98 ppm. The sample was also analyzed and found to contain 5,800 ppm of heptane. [000368] Analysis of another portion of this sample yielded the following results: 99.6% AUC, 1640 ppm ethanol, 41,480 ppm ethyl acetate, 5600 ppm heptane, no anisole detected, and 120 ppm ethyl chloride.
[000369] A procedure for recrystallizing the salt was also developed using the above dried salt. This procedure would work just was well on the highly pure crude salt (containing residual EtOH) obtained from concentrating the HCl salt-forming reaction mixture: [000370] The salt (575 mg) was dissolved in twice the mass of absolute EtOH (1.157 g) and then heated under nitrogen. To this hot solution (stirred) was added 1.6 g of 25% EtOH (in EtOAc) followed by the addition of EtOAc (0.25 mL) resulting in a cloudiness that remained. The cloudy hot solution was allowed to cool to room temperature during which time crystallization occurred. After crystallization was complete (2 h), the crystalline solid was filtered, washed with anhydrous EtOAc (~40 mL), and vacuum dried to give 424 mg of the dihydrochloride salt of KX2-391 as a free-flowing solid (tiny beads, 99.8% AUC) containing only 0.05 eq (0.45% w/w) of EtOH and 0.015 eq (0.26% w/w) of EtOAc. Slightly better recovery (460 mg from 586 mg) was attained using isopropanol/EtOAc but the level of solvent entrapment was higher [0.085 eq (1.0% w/w) of isopropanol and 0.023 eq (0.4% w/w) OfEtOAc].
Example 3: Large Scale Synthesis of KX2-391 di-HCl
[000371] Reagents and solvents were used as received from commercial suppliers.
Progress of the reactions was monitored by HPLC, GC/MS, or 1H NMR. Thin-layer chromatography (TLC) was performed using Analtech silica gel plates and visualized by UV light (254 nm). High pressure liquid chromatography (HPLC) was performed on an Agilent 1100 Series instruments. Proton and carbon nuclear magnetic resonance spectra were obtained using a Bruker AV 300 at 300 MHz for proton and 75 MHz for carbon. The solvent peak was used as the reference peak for proton and carbon spectra.
Preparation of 4-(2-(4-Bromophenoxy)ethyl)morpholine (2) [000372] A 50 L jacketed reactor equipped with a reflux condenser and temperature probe was charged with 4-(3-chloropropyl)morpholine (2.44 kg, 0.54 mol), 4-bromophenol (2.27 kg, 0.54 mol, 1.0 equiv.), powdered potassium carbonate (6.331 kg, 1.88 mol, 3.50 equiv.), and DMF (12.2 L) and stirred. The reaction mixture was then heated to 60-65 ºC and stirred overnight. After 17.5 h, the reaction mixture was cooled to 20-25 ºC. The reaction mixture was charged to a different reactor equipped with bottom valve for the work-up. While maintaining a temperature between 20-30 ºC, DI water (48.7 L) was charged to the reactor. The phases were separated. The aqueous layer was extracted with MTBE (3 x 24.4 L). To the combined organics, DI water (18.3 L) and then 6M sodium hydroxide (18.2 L) were added. The mixture was stirred for 2-5 minutes and the phases were separated. The organic phase was washed with water (24.4 L) and brine (24.4 L), dried over magnesium sulfate, filtered, and concentrated to give 337Og of a yellow oil (89% crude yield, 99.4% AUC by HPLC).
Preparation of 6-fluoropyridin-3-ylboronic acid (4)
[000373] A 72 L reactor equipped with reflux condenser, and temperature probe. To the reactor 5-bromo-2-fluoropyridine (1.17 L, 0.568 mol), toluene (18.2 L), and triisopropyl borate (3.13 L, 0.68 mol, 1.2 equiv.) were charged and stirred. Tetrahydrofuran (4.4 L) was added to the reactor and the reaction mixture was cooled to between -35 to -50 ºC. While maintaining a temperature between -35 to -45 ºC, n-butyl lithium (2.5 M solution of hexanes, 5.44 L, 0.68 mol, 1.2 equiv.) was cautiously added to the reactor. After 5 h, the reaction was deemed complete and the reaction mixture was warmed to between -15 to -20 ºC. To the reaction was added 2M HCl (11.80L) to the reactor while maintaining a temperature between -15 ºC and 0 ºC. The reaction mixture was stirred at 18 to 23 ºC for (16 h) and the phases were separated. The organics were then extracted with 6 M sodium hydroxide (6.0 L). The acidic anbasic aqueous phases were mixed in the reactor and 6 M HCl (2.5 L) was added until pH 7.5 was achieved. Sodium chloride (6.0 kg) was then added to the aqueous phase. The aqueous phase was then extracted with THF (3 x 20 L). The combined organics were dried with magnesium sulfate and concentrated to give 1300 g of a tan solid (81% crude yield).
Preparation of 4-(2-(4-(6-fluoropyridin-3-yl)phenoxy)ethyl)morpholine (5)
[000374] A 72 L reactor equipped with reflux condenser, sparging tube, bubbler, and temperature probe was charged with 6-fluoropyridin-3-ylboric acid (2.84 kg, 1.24 equiv.), A- (2-(4-bromophenoxy)ethyl)morpholine (4.27 kg, 1.0 equiv.), and DME (27 L). Agitation was started and sodium carbonate (4.74 kg, 3.0 equiv.) as a solution in DI water (17.1 L) was then charged to the reaction mixture. Argon was bubbled through the reaction mixture for 50 minutes. Under an argon atmosphere, tetrakis(triphenylphosphine)palladium (750 g, 0.04 equiv.) was added to the reaction mixture as a slurry in DME (1.0 L). The reaction mixture was heated to 75 – 85 ºC and stirred overnight (17 h). The reaction mixture was cooled to between 18 – 22ºC. DI water (26.681kg) and MTBE (26.681 L) were charged to the reactor and stirred for 5 minutes. The phases were separated and the aqueous phase was extracted with MTBE (2 x 26.7 L). The combined organics were extracted with 2M HCl (1 x 15.0 L, 3 x 21.8 L). The aqueous phase was then charged back to the reactor and ethyl acetate was added (26.7 L). The pH was adjusted to 6.2 using 6 M sodium hydroxide (26.7 L) while maintaining a temperature between 15 – 25 ºC. The phases were separated and the aqueous phase was extracted with ethyl acetate (2 x 26.7 L). The combined organics were dried with magnesium sulfate and concentrated to give 4555 g of a residue (101% crude yield, 67.1% AUC by HPLC).
Purification of 4-(2-(4-(6-fluoropyridin-3-yl)phenoxy)ethyl)morpholine (5)
[000375] The crude product (575 g) was purified by silica gel chromatography by eluting with methanol/ethyl acetate/heptane (30% ethyl acetate/heptane, 50% ethyl acetate/heptane, 75% ethyl acetate/heptane, 100% ethyl acetate, and 5% methanol/ethyl acetate). Concentration of the pure fractions by TLC (10% methanol/dichloromethane, Rf = 0.3) provided 420 g of a light brown solid (73% recovery, >99.9% AUC by HPLC).
Preparation of 2-(5-(4-(2-morpholinoethoxy)phenyl)pyridin-2-yl)acetonitrile (6)
[000376] A 1 M solution of NaHMDS (2.0 L, 5.0 equiv.) in THF was charged to a 5-L flask and cooled to -20 to -15 ºC. While maintaining a temperature below -10 ºC, fluoride (119.7g, 1.0 equiv.) in THF (500 mL) was charged to the flask over 20 minutes. Acetonitrile (82.5 mL, 4.0 equiv.) in THF (170 mL) was added to the flask over 20 minutes, while maintaining a temperature below -10 ºC. The reaction mixture was then stirred for 1 h. To the reaction was added brine (1.5 L, 12.6 vol.) at a rate as to maintain a temperature below 10 ºC. The solution was then warmed to room temperature and the layers were allowed to separate. The mixture was filtered over Celite and washed with THF (1 x 200 mL, 1 x 100 mL). The aqueous phase was extracted with toluene (750 mL). The combined organics were dried with magnesium sulfate, filtered, washed with toluene (2 x 25OmL), and concentrated to dryness. Toluene (IL) was added and the solution was concentrated to dryness again to give 169.8 g of an oil. MTBE (1190 niL, 7 vol.) was added to the oil at 50 ºC and stirred for 15 minutes. Heptane (850 rnL, 5vol.) was added over ten minutes at 50 ºC. The mixture was then cooled to room temperature over 1.5 h and stirred for 2 h. The slurry was filtered, washed with 1 :4 MBTE/heptane (2 x 100 mL), and dried in an oven overnight at 45 ºC to give 102.3 g of an off-white solid (80% yield, 98.8% AUC by HPLC).
Preparation of methyl 2-(5-(4-(2-morpholinoethoxy)phenyl)pyridin-2-yl)acetate (7)
[000377] Nitrile 6 (101 g) and methanol (1.01 L, 10 vol.) were charged to a 3-L flask equipped with stir bar and thermocouple. Concentrated H2SO4 (175 mL, 10.0 equiv.) was added drop wise to the solution over 15 minutes while maintaining a temperature below 60 ºC. Followed by 30% fuming sulfuric acid (124 mL) was added drop wise to the solution while maintaining a temperature below 60 ºC. The solution was then heated to reflux with a heating mantle and stirred overnight. When the reaction was deemed complete, it was cooled to 20 ºC. In a second flask (22 L), saturated sodium bicarbonate (10.7 L) and dichloromethane (1.1 L) were charged and cooled to 15 ºC. While maintaining a temperature below 20 ºC, the reaction mixture was added to the sodium bicarbonate/dichloromethane mixture. The quench was stirred for 15 minutes and the phases were separated. The aqueous phase was extracted with dichloromethane (I x 55OmL, 1 x 30OmL). The combined organics were dried with magnesium sulfate and concentrated to dryness to give 105 g of an orange solid (94% crude yield, 97.7% AUC by HPLC).
Preparation of 2-(5-(4-(2-morpholinoethoxy)phenyl)pyridin-2-yl)-N-benzylacetamide (KX2-391)
[000378] Ester 7 (103 g), anisole (513 mL, 5 vol.), and benzylamine (94 mL, 3.0 equiv.) were charged to a 3 L flask equipped with thermocouple and overhead stirrer. The reaction mixture was then heated to 142 ºC and stirred for two days. The reaction mixture was cooled to 45-50 ºC and stirred for 2 hours. To the mixture was added n-heptane (1.5 L) dropwise over an hour. The solution was cooled to room temperature over three hours and then stirred overnight. The resulting slurry was filtered, washed with 4:1 Anisole/n-heptane (200 mL) and n-heptane (3 χ100 mL). Drying in the oven overnight, the resulting product was 112. Ig of a tan solid (90% yield, 99.6% AUC by HPLC). The use of a single isomer of heptane was essential to adequately quantitate the residual solvent. See Figure 5 for 1H NMR of KX2- 391. Preparation of 2-(5-(4-(2-morpholinoethoxy)phenyl)pyridin-2-yl)-N-benzylacetamide dihydrochloride salt (KX2-391 2HC1)
[000379] EtOH (1.0 L) was charged to a 2-L flask and acetyl chloride (62.5 niL, 3.0 equiv.) was added slowly to the flask and stirred for 40 minutes. The resulting solution was added to KX2-391 (100 g) over 30 minutes while maintaining a temperature of 30 ºC. The solution was concentrated to a mass of 270 g. The concentrated solution was added to ethyl acetate (2 L) over 20 minutes with rapid stirring. The mixture was stirred overnight and then filtered under nitrogen to give two distinct solid products, tan solids (73.5 g) and darker solids (42.2 g). The solids were dry blended to give a combined yield of 99%. The HPLC analysis indicated 99.0% purity (AUC). Analysis indicated that ethanol was present at 2530 ppm, ethyl acetate at 48,110 ppm, ethyl chloride at 170 ppm, and no heptane and anisole were detected. Palladium content was assayed three times and measured to be 29 ppm, 2 ppm, and less than 1 ppm.
2-(5-(4-(2-morpholinylethoxy)phenyl)pyridine-2-yl)-N-benzyl-acetamide, development code KX -01, KX2-391, have the structure shown in formula I.
Patent CN10118473B and US7300931B disclose compound KX2-391, and disclose its application in the treatment of cell proliferative disorders. KX2-391 and its pharmaceutically acceptable salts are effective Src tyrosine kinase inhibitors, which can effectively treat diseases and disorders regulated by Src kinase. KX2-391 has a GI50 of 9-60 nM in cancer cell lines and is currently in clinical phase II.
KX2-391 has polymorphism. Polymorphism refers to the phenomenon that the same compound can form two or more molecular spatial arrangements by controlling its different production conditions to produce different solid crystals. Different crystal forms of the same compound have the same chemical composition. , But the microscopic crystal structure is different, which leads to differences in their appearance, physical and chemical properties and biological activity. The phenomenon of polymorphism directly affects the processing performance of the drug formulation, and affects the stability, solubility, and bioavailability of the drug, and further affects the quality, safety, effectiveness and application of the drug. Therefore, in drug research and development, the polymorphism of drugs should be fully considered. At present, KX2-391 is still in the research and development stage, and a comprehensive study of its solid form is of great significance to the research and development of KX2-391 and the approval of the market.
Example 1
2-(5-(4-(2-morpholinylethoxy)phenyl)pyridin-2-yl)-N-benzyl-acetamide (KX2-391) crystal form (i.e. having formula (I) The structure of the crystalline diaryl compound, the subsequent examples are referred to as the preparation of KX2-391 crystal form B)
Put KX2-391 (5.0g) in a 500ml round bottom flask, add 150ml methanol to dissolve KX2-391 completely, and place it at 50°C and stir. 300ml of purified water was gradually added dropwise. After the addition, the resulting slurry was stirred at room temperature for 1 hour to crystallize, filtered with suction, and dried under vacuum at 50°C. The resulting solid was KX2-391 crystal form B. The purity detected by HPLC is ≥99.83%.
Example 2 Preparation of KX2-391 crystal form B
Put KX2-391 (5.0g) in a 100ml round bottom flask, add 25ml of DMSO to dissolve KX2-391 completely, and stir at room temperature. Gradually add 50ml of purified water dropwise. After the dropwise addition, the resulting slurry was stirred at 0°C for 1h to crystallize, filtered with suction, and dried under vacuum at 50°C. The resulting solid was KX2-391 crystal form B. HPLC detection purity ≥99.81%.
Example 3 Preparation of KX2-391 crystal form B
Put KX2-391 (5.0g) in a 250ml round bottom flask, add 15ml of dichloromethane to dissolve KX2-391 completely, and stir at 30°C. Gradually add 100ml of n-heptane dropwise. After the dropwise addition, the resulting slurry was stirred at room temperature for 0.5h to crystallize, filtered with suction, and dried under vacuum at 50°C. The resulting solid was KX2-391 crystal form B. HPLC detection purity ≥99.80%.
Example 4 Preparation of KX2-391 crystal form B
Put KX2-391 (2.0g) in a 500ml round bottom flask, add 100ml of acetone to completely dissolve KX2-391, and stir at room temperature. Gradually add 150 ml of n-hexane, and after the addition is complete, the resulting slurry is stirred at 0°C for 1 h to crystallize, filtered with suction, and dried in vacuum at 50°C. The obtained solid is KX2-391 crystal form B. HPLC detection purity ≥99.79%.
Example 5 Preparation of KX2-391 crystal form B
Put KX2-391 (2.0g) in a 250ml round bottom flask, add 50ml of THF to dissolve KX2-391 completely, and place it at 40°C and stir. Gradually add 100 ml of methyl tert-butyl ether dropwise. After the dropwise addition, the resulting slurry was stirred at room temperature for 2 hours to crystallize, filtered with suction, and dried under vacuum at 50°C. The resulting solid was KX2-391 crystal form B. HPLC detection purity ≥99.81%.
Example 6 Detection of KX2-391 crystal form B
The KX2-391 crystal form B prepared in Example 1 was tested by XRPD method. The equipment used is RIGAKU TTR III X-ray powder diffractometer, measurement conditions and methods: Cu (target), 40KV-30mA (working voltage and current), 2θ=2~50 degrees (scanning range), 4.0deg /min. (scanning speed), the obtained spectrum is shown in Figure 1. It can be seen from Figure 1 that the XRPD spectrum of KX2-391 crystal form B provided in Example 1 is 2.10, 3.68, 4.16, 6.24, 8.33, There are peaks at 12.53, 16.26, 16.75, 18.33, 19.05, 19.85, 21.00, 21.50, 21.92, 22.50, 23.16, 25.08, 25.35, 25.70, 27.49, 29.67, 33.97, and 38.43.
The invention also adopts the DSC-TGA method to detect the crystal form B of KX2-391 provided by the invention. The equipment used is METTLER TOLEDO’s TGA-DSC, testing environment conditions 22℃, relative humidity RH68%, temperature range 0-400℃, heating rate 12℃/min, protective gas N 2 , The resulting maps are shown in Figure 2 and Figure 3. It can be seen from Figure 2 that the DSC spectrum of KX2-391 crystal form B provided in Example 1 has endothermic peaks at 126.9°C and 137.4°C. It can be seen from Figure 3 that the TGA pattern of KX2-391 crystal form B provided in Example 1 has no significant weight loss before 200°C.
PAPER
Journal of Medicinal Chemistry (2018), 61(11), 4704-4719.
The discovery of potent, peptide site directed, tyrosine kinase inhibitors has remained an elusive goal. Herein we describe the discovery of two such clinical candidates that inhibit the tyrosine kinase Src. Compound 1 is a phase 3 clinical trial candidate that is likely to provide a first in class topical treatment for actinic keratosis (AK) with good efficacy and dramatically less toxicity compared to existing standard therapy. Compound 2 is a phase 1 clinical trial candidate that is likely to provide a first in class treatment of malignant glioblastoma and induces 30% long-term complete tumor remission in animal models. The discovery strategy for these compounds iteratively utilized molecular modeling, along with the synthesis and testing of increasingly elaborated proof of concept compounds, until the final clinical candidates were arrived at. This was followed with mechanism of action (MOA) studies that revealed tubulin polymerization inhibition as the second MOA.
(2S)-4-amino-N-[(1R,2S,3S,4R,5S)-5-amino-2-[(2S,3R,4S,5S,6R)-4-amino-3,5-dihydroxy-6-(hydroxymethyl)oxan-2-yl]oxy-4-[(2R,3R,4S,5S,6R)-6-(aminomethyl)-3,4,5-trihydroxyoxan-2-yl]oxy-3-hydroxycyclohexyl]-2-hydroxybutanamide;sulfuric acid AmikacinCAS Registry Number: 37517-28-5 CAS Name:O-3-Amino-3-deoxy-a-D-glucopyranosyl-(1®6)-O-[6-amino-6-deoxy-a-D-glucopyranosyl-(1®4)]-N1-[(2S)-4-amino-2-hydroxy-1-oxobutyl]-2-deoxy-D-streptamine Additional Names: 1-N-[L(-)-4-amino-2-hydroxybutyryl]kanamycin AMolecular Formula: C22H43N5O13Molecular Weight: 585.60Percent Composition: C 45.12%, H 7.40%, N 11.96%, O 35.52% Literature References: Semisynthetic aminoglycoside antibiotic derived from kanamycin A. Prepn: Kawaguchi et al.,J. Antibiot.25, 695 (1972); H. Kawaguchi, T. Naito, DE2234315; H. Kawaguchi et al.,US3781268 (both 1973 to Bristol-Myers). Biological formation from kanamycin A: L. M. Cappelletti, R. Spagnoli, J. Antibiot.36, 328 (1983). Microbiological evaluation: Price et al.,ibid.25, 709 (1972). Pharmacokinetics: Cabana, Taggart, Antimicrob. Agents Chemother.3, 478 (1973). In vitro studies: Yu, Washington, ibid.4, 133 (1973); Bodey, Stewart, ibid. 186. Pharmacology in humans: Bodey et al.,ibid.5, 508 (1974). Toxicity studies: Fujisawa et al.,J. Antibiot.27, 677 (1974). Review: K. A. Kerridge in Pharmacological and Biochemical Properties of Drug Substancesvol. 1, M. E. Goldberg, Ed. (Am. Pharm. Assoc., Washington, DC, 1977) pp 125-153. Comprehensive description: P. M. Monteleone et al.,Anal. Profiles Drug Subs.12, 37-71 (1983).Properties: White crystalline powder from methanol-isopropanol, mp 203-204° (sesquihydrate). [a]D23 +99° (c = 1.0 in water). LD50 in mice of solns pH 6.6, pH 7.4 (mg/kg): 340, 560 i.v. (Kawaguchi).Melting point: mp 203-204° (sesquihydrate)Optical Rotation: [a]D23 +99° (c = 1.0 in water)Toxicity data: LD50 in mice of solns pH 6.6, pH 7.4 (mg/kg): 340, 560 i.v. (Kawaguchi) Derivative Type: SulfateCAS Registry Number: 39831-55-5Trademarks: Amiglyde-V (Fort Dodge); Amikin (BMS); Amiklin (BMS); BB-K8 (BMS); Biklin (BMS); Lukadin (San Carlo); Mikavir (Salus); Novamin (BMS); Pierami (Fournier)Molecular Formula: C22H43N5O13.2H2SO4Molecular Weight: 781.76Percent Composition: C 33.80%, H 6.06%, N 8.96%, O 42.98%, S 8.20%Properties: Amorphous form, dec 220-230°. [a]D22 +74.75° (water).Optical Rotation: [a]D22 +74.75° (water) Therap-Cat: Antibacterial.Therap-Cat-Vet: Antibacterial.Keywords: Antibacterial (Antibiotics); Aminoglycosides.
Amikacin Sulfate is the sulfate salt of amikacin, a broad-spectrum semi-synthetic aminoglycoside antibiotic, derived from kanamycin with antimicrobial property. Amikacin irreversibly binds to the bacterial 30S ribosomal subunit, specifically in contact with 16S rRNA and S12 protein within the 30S subunit. This leads to interference with translational initiation complex and misreading of mRNA, thereby hampering protein synthesis and resulting in bactericidal effect. This agent is usually used in short-term treatment of serious infections due to susceptible strains of Gram-negative bacteria.Amikacin disulfate is an aminoglycoside sulfate salt obtained by combining amikacin with two molar equivalents of sulfuric acid. It has a role as an antibacterial drug, an antimicrobial agent and a nephrotoxin. It contains an amikacin(4+).
Amikacin sulfate is semi-synthetic aminoglycoside antibiotic derived from kanamycin. It is C22H43N5O13•2H2SO4•O-3-amino-3-deoxy-α-D-glucopyranosyl-(1→4)-O-[6-amino-6-deoxy-α-Dglucopyranosyl-( 1→6)]-N3-(4-amino-L-2-hydroxybutyryl)-2-deoxy-L-streptamine sulfate (1:2)
M.W. 585.61The dosage form is supplied as a sterile, colorless to light straw colored solution for intramuscular or intravenous use. Each mL contains 250 mg amikacin (as the sulfate), 0.66% sodium metabisulfite, 2.5% sodium citrate dihydrate with pH adjusted to 4.5 with sulfuric acid.
Amikacin may be administered once or twice a day and is usually given by the intravenous or intramuscular route, though it can be given via nebulization. There is no oral form available, as amikacin is not absorbed orally. In people with kidney failure, dosage must be adjusted according to the creatinine clearance, usually by reducing the dosing frequency.[9] In people with a CNS infection such as meningitis, amikacin can be given intrathecally (by direct injection into the spine) or intraventricularly (by injection into the ventricles of brain).[4]
An liposome inhalation suspension is also available and approved to treat Mycobacterium avium complex (MAC) in the United States.[15][16] The application for Arikayce was withdrawn in the European Union because the Committee for Medicinal Products for Human Use (CHMP) was of the opinion that the benefits of Arikayce did not outweigh its risks.[17]
Special populations
Amikacin should be used in smaller doses in the elderly, who often have age-related decreases in kidney function, and children, whose kidneys are not fully developed yet. It is considered pregnancy category D in both the United States and Australia, meaning they have a probability of harming the fetus.[4] Around 16% of amikacin crosses the placenta; while the half-life of amikacin in the mother is 2 hours, it is 3.7 hours in the fetus.[9] A pregnant woman taking amikacin with another aminoglycoside has a possibility of causing congenital deafness in her child. While it is known to cross the placenta, amikacin is only partially secreted in breast milk.[4]
In general, amikacin should be avoided in infants.[18] Infants also tend to have a larger volume of distribution due to their higher concentration of extracellular fluid, where aminoglycosides reside.[3]
The elderly tend to have amikacin stay longer in their system; while the average clearance of amikacin in a 20-year-old is 6 L/hr, it is 3 L/hr in an 80-year-old.[19]
Clearance is even higher in people with cystic fibrosis.[20]
In people with muscular disorders such as myasthenia gravis or Parkinson’s disease, amikacin’s paralytic effect on neuromuscular junctions can worsen muscle weakness.[4]
Adverse effects
Side-effects of amikacin are similar to those of other aminoglycosides. Kidney damage and ototoxicity (which can lead to hearing loss) are the most important effects, occurring in 1–10% of users.[12] The nephro- and ototoxicity are thought to be due to aminoglycosides’ tendency to accumulate in the kidneys and inner ear.[3]
Diagram of the inner ear. Amikacin causes damage to the cochlea and vestibules.
Amikacin can cause neurotoxicity if used at a higher dose or for longer than recommended. The resulting effects of neurotoxicity include vertigo, numbness, tingling of the skin (paresthesia), muscle twitching, and seizures.[4] Its toxic effect on the 8th cranial nerve causes ototoxicity, resulting in loss of balance and, more commonly, hearing loss.[3] Damage to the cochlea, caused by the forced apoptosis of the hair cells, leads to the loss of high-frequency hearing and happens before any clinical hearing loss can be detected.[9][21] Damage to the ear vestibules, most likely by creating excessive oxidative free radicals. It does so in a time-dependent rather than dose-dependent manner, meaning that risk can be minimized by reducing the duration of use.[22]
The amikacin liposome inhalation suspension prescribing information includes a boxed warning regarding the increased risk of respiratory conditions including hypersensitivity pneumonitis (inflamed lungs), bronchospasm (tightening of the airway), exacerbation of underlying lung disease and hemoptysis (spitting up blood) that have led to hospitalizations in some cases.[15][16] Other common side effects in patients taking amikacin liposome inhalation suspension are dysphonia (difficulty speaking), cough, ototoxicity (damaged hearing), upper airway irritation, musculoskeletal pain, fatigue, diarrhea and nausea.[15][16]
Contraindications
Amikacin should be avoided in those who are sensitive to any aminoglycoside, as they are cross-allergenic (that is, an allergy to one aminoglycoside also confers hypersensitivity to other aminoglycosides). It should also be avoided in those sensitive to sulfite (seen more among people with asthma),[9] since most amikacin usually comes with sodium metabisulfite, which can cause an allergic reaction.[4]
In general, amikacin should not be used with or just before/after another drug that can cause neurotoxicity, ototoxicity, or nephrotoxicity. Such drugs include other aminoglycosides; the antiviral acyclovir; the antifungal amphotericin B; the antibiotics bacitracin, capreomycin, colistin, polymyxin B, and vancomycin; and cisplatin, which is used in chemotherapy.[4]
Amikacin can be inactivated by other beta-lactams, though not to the extent as other aminoglycosides, and is still often used with penicillins (a type of beta-lactam) to create an additive effect against certain bacteria, and carbapenems, which can have a synergistic against some Gram-positive bacteria. Another group of beta-lactams, the cephalosporins, can increase the nephrotoxicity of aminoglycoside as well as randomly elevating creatinine levels. The antibiotics chloramphenicol, clindamycin, and tetracycline have been known to inactivate aminoglycosides in general by pharmacological antagonism.[4]
Potent diuretics not only cause ototoxicity themselves, but they can also increase the concentration of amikacin in the serum and tissue, making the ototoxicity even more likely.[4]Quinidine also increases levels of amikacin in the body.[12] The NSAIDindomethacin can increase serum aminoglycoside levels in premature infants.[4] Contrast mediums such as ioversol increases the nephrotoxicity and otoxicity caused by amikacin.[12]
Amikacin can decrease the effect certain vaccines, such as the live BCG vaccine (used for tuberculosis), the cholera vaccine, and the live typhoid vaccine by acting as a pharmacological antagonist.[12]
Pharmacology
Mechanism of action
The 30S subunit of the prokaryotic ribosome. The orange represents the 16S rRNA, and the blue represents the various proteins attached.
Amikacin irreversibly binds to 16S rRNA and the RNA-binding S12 protein of the 30S subunit of prokaryotic ribosome and inhibits protein synthesis by changing the ribosome’s shape so that it cannot read the mRNAcodons correctly.[9][23] It also interferes with the region that interacts with the wobble base of the tRNA anticodon.[24] It works in a concentration-dependent manner, and has better action in an alkaline environment.[3]
At normal doses, amikacin-sensitive bacteria respond within 24–48 hours.[9]
Resistance
Amikacin evades attacks by all antibiotic-inactivating enzymes that are responsible for antibiotic resistance in bacteria, except for aminoacetyltransferase and nucleotidyltransferase.[25] This is accomplished by the L-hydroxyaminobuteroyl amide (L-HABA) moiety attached to N-1 (compare to kanamycin, which simply has a hydrogen), which blocks the access and decreases the affinity of aminoglycoside-inactivating enzymes.[25][26][27] Amikacin ends up with only one site where these enzymes can attack, while gentamicin and tobramycin have six.[11]
Bacteria that are resistant to streptomycin and capreomycin are still susceptible to amikacin; bacteria that are resistant to kanamycin have varying susceptibility to amikacin. Resistance to amikacin also confers resistance to kanamycin and capreomycin.[28]
Resistance to amikacin and kanamycin in Mycobacterium, the causative agent of tuberculosis, is due to a mutation in the rrs gene, which codes for the 16S rRNA. Mutations such as these reduce the binding affinity of amikacin to the bacteria’s ribosome.[29] Variations of aminoglycoside acetyltransferase (AAC) and aminoglycoside adenylyltransferase (AAD) also confer resistance: resistance in Pseudomonas aeruginosa is caused by AAC(6′)-IV, which also confers resistance to kanamycin, gentamicin, and tobramycin, and resistance in Staphylococcus aureus and S. epidermidis is caused by AAD(4′,4), which also confers resistance to kanamycin, tobramycin, and apramycin.[26] Some strains of S. aureus can also inactivate amikacin by phosphorylating it.[13]
Pharmacokinetics
Amikacin is not absorbed orally and thus must be administered parenterally. It reaches peak serum concentrations in 0.5–2 hours when administered intramuscularly. Less than 11% of the amikacin actually binds to plasma proteins. It is distributed into the heart, gallbladder, lungs, and bones, as well as in bile, sputum, interstitial fluid, pleural fluid, and synovial fluids. It is usually found at low concentrations in the cerebrospinal fluid, except when administered intraventricularly.[4] In infants, amikacin is normally found at 10–20% of plasma levels in the spinal fluid, but the amount reaches 50% in cases of meningitis.[9] It does not easily cross the blood-brain barrier or enter ocular tissue.[3]
While the half-life of amikacin is normally two hours, it is 50 hours in those with end-stage renal disease.[11]
The vast majority (95%) of amikacin from an IM or IV dose is secreted unchanged via glomerular filtration and into the urine within 24 hours.[4][11] Factors that cause amikacin to be excreted via urine include its relatively low molecular weight, high water solubility, and unmetabolized state.[18]
In dogs and cats, amikacin is commonly used as a topical antibiotic for ear infections and for corneal ulcers, especially those that are caused by Pseudomonas aeruginosa. The ears are often cleaned before administering the medication, since pus and cellular debris lessen the activity of amikacin.[32] Amikacin is administered to the eye when prepared as an ophthalmic ointment or solution, or when injected subconjunctivally.[35] Amikacin in the eye can be accompanied by cephazolin. Despite its use there amikacin (and all aminoglycosides) are toxic to intraocular structures.[36]
In horses, amikacin is FDA-approved for uterine infections (such as endometriosis and pyometra) when caused by susceptible bacteria.[37] It is also used in topical medication for the eyes and arthroscopic lavage; when combined with a cephalosporin, is used to treat subcutaneous infections that are caused by Staphylococcus. For infections in the limbs or joints, it is often administered with a cephalosporin via limb perfusion directly into the limb or injected into the joint.[32][38] Amikacin is also injected into the joints with the anti-arthritic medication Adequan in order to prevent infection.[39]
Side effects in animals include nephrotoxicity, ototoxicity, and allergic reactions at IM injection sites. Cats tend to be more sensitive to the vestibular damage caused by ototoxicity. Less frequent side effects include neuromuscular blockade, facial edema, and peripheral neuropathy.[3][32]
The half-life in most animals is one to two hours.[40]
Treating overdoses of amikacin requires kidney dialysis or peritoneal dialysis, which reduce serum concentrations of amikacin, and/or penicillins, some of which can form complexes with amikacin that deactivate it.[3]
The safety and efficacy of amikacin liposome inhalation suspension, an inhaled treatment taken through a nebulizer, was demonstrated in a randomized, controlled clinical trial where patients were assigned to one of two treatment groups.[15] One group of patients received amikacin liposome inhalation suspension plus a background multi-drug antibacterial regimen, while the other treatment group received a background multi-drug antibacterial regimen alone.[15] By the sixth month of treatment, 29 percent of patients treated with amikacin liposome inhalation suspension had no growth of mycobacteria in their sputum cultures for three consecutive months compared to 9 percent of patients who were not treated with amikacin liposome inhalation suspension.[15]
Amikacin is a semi-synthetic aminoglycoside antibiotic with a broad antibacterial spectrum and strong antibacterial activity against a variety of bacteria; its sulfate has become a clinically commonly used first-line anti-infective drug in the world and continues to Develop new dosage forms and uses.
[0003] Amikacin sulfate is suitable for Pseudomonas aeruginosa and other Pseudomonas, Escherichia coli, Proteus, Klebsiella, Enterobacter, Serratia, Acinetobacter Severe infections caused by other sensitive gram-negative bacilli and Staphylococcus (methicillin-sensitive strains), such as bacteremia or sepsis, bacterial endocarditis, lower respiratory tract infections, bone and joint infections, biliary tract infections, abdominal infections, Complex urinary tract infections, skin and soft tissue infections, etc. Because it is stable to most aminoglycoside inactivating enzymes, it is especially suitable for the treatment of serious infections caused by gram-negative bacilli against kanamycin, gentamicin or tobramycin-resistant strains.
[0004] Amikacin, also known as amikacin, has a molecular weight of 585. The most commonly used synthetic route is a silyl protecting routes, such as the document “amikacin by New Method” (Author: Jiangzhong Liang, Wang Yu; Fine & Specialty Chemicals, 2004, 12 (10), 26- 28) The main process recorded is: (1) Using kanamycin A (KMA) as a raw material to protect the 11 amino groups and hydroxyl groups of kanamycin to obtain methylsilyl kanamycin; (2) ) Using YN-phthalimido-α-hydroxybutyric acid (PHBA) and N-hydroxy-phthalimide (NOP) as raw materials in dicyclohexylcarbodiimide (DCC) The active ester compound is prepared in the presence; (3) acylation (transesterification reaction) with methylsilyl kanamycin and active ester, and then acidolysis and hydrazinolysis reactions to obtain amikacin. As shown in the following route:
[0005] 1. Silanization protection reaction:
[0006]
[0007] 2. Preparation of Living King®:
[0008]
[0009] 3. Acylation reaction:
U
[0011] 4. Acidolysis reaction:
[0012]
[0013] 5. Hydrazine reaction:
[0014]
[0015] The acylation reaction in the above route adopts a transesterification reaction between a silyl group protection reactant and an independently prepared active ester. Due to the active transesterification reaction, a large excess of reactant active ester is needed to improve the reaction yield, and there is an independent unit reaction for preparing active ester, and the raw material N-hydroxy-phthalimide is used. (NOP), increasing the usage amount of reaction solvent, the solvent in the process is volatile, the loss is large, the environment is affected, and the production cost is increased.
[0016] How to find a direct one-step acylation reaction between the silyl group protection and YN-phthalimido-α-hydroxybutyric acid (PHBA), which can not only ensure the synthesis yield, but also reduce the synthesis The steps are easy to operate, and the N-hydroxy-phthalimide (NOP), the raw material for preparing active esters, is no longer used, and the acylation reaction conditions that reduce solvent consumption are a very beneficial synthetic process line.
Example 1
[0046] 600mL of acetonitrile was put into the silanization reaction flask, and 0.1 billion kanamycin A (KMA) was added. After the feeding port was closed and stirred for 10 minutes, hexamethyldisilazane (HMDS) was added. 400mL, heated to reflux, refluxed at 75~80°C for 7hr. Use drinking water to cool the outside of the reaction flask to lower the temperature to below 35°C, and let it stand for natural layering. Separate and collect the lower layer to obtain a silyl group protected product.
[0047] Add 1000mL acetone to the silyl group protection product, start stirring, add 60g γ-N-phthalimido-α-hydroxybutyric acid (PHBA), and then add 2.5g catalyst 4-N, N -Lutidine (DMAP), cooled to -15~-1 (TC〇
[0048] Dissolve 60gN, N-bicyclohexylcarbodiimide with 300mL of acetone, add its flow to the above-mentioned reactant, control the flow rate of 5mL/min, and control the temperature of the reactant to -15~-10°C; the flow is completed Continue the reaction for 1 hour.
[0049] After the completion of the acylation reaction, the material was transferred to the acidolysis bottle, the stirring was turned on, and 400mL of 4.0mol/L hydrochloric acid was added for acidolysis, and the feed solution was pH 3.0 and allowed to stand for 60 minutes. The lower acid hydrolysis solution was collected by suction filtration, and the filter cake (DCU) was washed three times with 150 mL of deionized water, and the washing water was incorporated into the acid hydrolysis solution.
[0050] The acid hydrolysate was transferred to a distillation flask. Turn on the vacuum, the degree of vacuum: <0.07Mpa, the distillation temperature is controlled at 40~68°C, the distillation time: 2.5 hours after the distillation is complete; transfer the PKS concentrate in the distillation flask into the hydrazinolysis flask, and add 7.Omol/ L ammonia water 200mL, so that the pH of the material solution reaches 8.0; add 180mL hydrazine hydrate, increase the temperature, the temperature is 85~95°C, hydrazinolysis 3.5 hours, use drinking water to cool outside the hydrazinolysis bottle, and cool to 40 °C.
[0051] Add 4.0111〇1/1 hydrochloric acid 12001^ to the hydrazinolysis bottle, adjust? !1 is 4.0. Turn on the vacuum filtration. With 5001 ^ deionized water top washing filter, 1510mL of amikacin synthetic solution, amikacin content 5.8% (g/mL), relative to the synthetic yield of kanamycin A is 72.5 %.
[0052] Example 2
[0053] 600mL of acetonitrile was put into the silanization reaction flask, 0.1 billion kanamycin A (KMA) was added, the feeding port was closed and stirred for 10 minutes, and hexamethyldisilazane (HMDS) was added 500mL, heated to reflux, refluxed at 75~80°C for 8hr. After the reaction is completed, cool down to 40°C with drinking water and let stand for natural layering. Separate and collect the lower layer to obtain a silyl group protected product.
[0054] Add 1000mL acetone to the silyl group protection product, start stirring, add 70g Y-N-phthalimido-α-hydroxybutyric acid (PHBA), and add 3.0g catalyst 1-hydroxybenzo Triazole (HOBT), after the material is dissolved, the temperature is reduced to -15~-10°C.
[0055] Dissolve 70g of N,N-bicyclohexylcarbodiimide with 300mL of acetone, add its flow to the above-mentioned reactants, control the flow rate of 6mL/min, and control the temperature of the reactants from -15 to -10°C; the flow is completed Continue the reaction for 1.5 hours.
[0056] After the acylation reaction is completed, the material is transferred to the acidolysis bottle, the stirring is turned on, and 6.0m〇l/L hydrochloric acid 300mL is added for acidolysis, the feed solution is pH 2.0, and the acidolysis is completed, and it is allowed to stand for 50 minutes. The lower acid hydrolysis solution was collected by suction filtration, the filter cake (DCU) was washed three times with 200 mL of deionized water, and the washing water was incorporated into the acid hydrolysis solution.
[0057] Transfer the acid hydrolysate into a distillation flask. Turn on the vacuum, vacuum degree: <-0.07Mpa, the distillation temperature is controlled at 40~68°C, the distillation time is 3.0 hours, except for acetone. After the distillation is completed, transfer the PKS concentrate in the distillation flask into the hydrazinolysis flask, add 150 mL of 10.0 mol/L ammonia water, the pH of the feed solution is 8.5; add 200 mL of hydrazine hydrate, increase the temperature at 85~95 °C, hydrazinolysis 4 After hours, use drinking water to cool down outside the hydrazinolysis bottle to 45°C.
[0058] Add 6.0111〇1/1 hydrochloric acid 10001^ to the hydrazinolysis bottle, adjust? !1 is 3.0. Turn on the vacuum filtration, use 8001^ deionized water to wash and filter the fish, to obtain 1620 mL of amikacin synthetic solution, and the amikacin content is 5.5% (g/mL). The synthetic yield relative to kanamycin A was 73.7%.
^ Jump up to:abcdefghijklmn Plumb, Donald C. (2011). “Amikacin Sulfate”. Plumb’s Veterinary Drug Handbook (7th ed.). Stockholm, Wisconsin; Ames, Iowa: Wiley. pp. 39–43. ISBN978-0-470-95964-0.
^World Health Organization (2019). World Health Organization model list of essential medicines: 21st list 2019. Geneva: World Health Organization. hdl:10665/325771. WHO/MVP/EMP/IAU/2019.06. License: CC BY-NC-SA 3.0 IGO.
^ Jump up to:ab Aronson J. K., ed. (2016). “Amikacin”. Meyler’s Side Effects of Drugs (16th ed.). Oxford: Elsevier. pp. 207–209. ISBN978-0-444-53716-4.
^ Vardanyan, Ruben; Hruby, Victor (2016). “Chapter 32: Antimicobacterial Drugs”. Synthesis of Best-Seller Drugs. Boston: Academic Press. pp. 669–675. ISBN978-0-12-411492-0.
^ Maire, P.; Bourguignon, L.; Goutelle, S.; Ducher, M.; Jelliffe, R. (2017). “Chapter 20 – Individualizing Drug Therapy in the Elderly”. Individualized Drug Therapy for Patients. Boston: Academic Press. pp. 373–382. ISBN978-0-12-803348-7.
^ Bauman, Robert W. (2015). Microbiology: with diseases by body system (4th ed.). Boston: Pearson. ISBN978-0-321-91855-0.
^“Amikacin”. DrugBank. 2 August 2017. Archived from the original on 16 August 2017. Retrieved 10 August 2017.
^ Jump up to:ab Mudd, Efrain (7 August 2017). “O Aminoglycosides”. Pharmacological Sciences. Archived from the original on 16 August 2017. Retrieved 14 August 2017.
^ Park, Je Won; Ban, Yeon Hee; Nam, Sang-Jip; Cha, Sun-Shin; Yoon, Yeo Joon (1 December 2017). “Biosynthetic pathways of aminoglycosides and their engineering”. Current Opinion in Biotechnology. Chemical biotechnology: Pharmaceutical biotechnology. 48: 33–41. doi:10.1016/j.copbio.2017.03.019. ISSN0958-1669. PMID28365471.
^ Monteleone, Peter M.; Muhammad, Naseem; Brown, Robert D.; McGrory, John P.; Hanna, Samir A. (1 January 1983). Amikacin Sulfate. Analytical Profiles of Drug Substances. 12. pp. 37–71. doi:10.1016/S0099-5428(08)60163-X. ISBN9780122608124. ISSN0099-5428.
^ Orsini, James A. (1 August 2017). “Update on Managing Serious Wound Infections in Horses: Wounds Involving Joints and Other Synovial Structures”. Journal of Equine Veterinary Science. 55: 115–122. doi:10.1016/j.jevs.2017.01.016. ISSN0737-0806.
^ Wanamaker, Boyce P.; Massey, Kathy (25 March 2014). Applied Pharmacology for Veterinary Technicians – E-Book. Elsevier Health Sciences. p. 392. ISBN978-0-323-29170-5.
^ Papich, Mark G. (October 2015). “Amikacin”. Saunders Handbook of Veterinary Drugs: Small and Large Animal (4th ed.). Elsevier Health Sciences. pp. 25–27. ISBN978-0-323-24485-5. Archived from the original on 10 September 2017.
External links
“Amikacin”. Drug Information Portal. U.S. National Library of Medicine.
“Amikacin sulfate”. Drug Information Portal. U.S. National Library of Medicine.
Arcus Biosciences is developing etrumadenant, the lead from the small molecule adenosine (A2a/A2b) dual receptor antagonist program, for treating cancer. In November 2020, preliminary data from ARC-7 in metastatic NSCLC were expected to report in the first half of 2021.
OriginatorArcus Biosciences
ClassAmines; Antineoplastics; Nitriles; Pyridines; Pyrimidines; Small molecules; Triazoles
Mechanism of ActionAdenosine A2A receptor antagonists; Adenosine A2B receptor antagonists
Phase IINon-small cell lung cancer
Phase I/IIProstate cancer
Phase IBladder cancer; Breast cancer; Cancer; Colorectal cancer; Endometrial cancer; Gastrointestinal cancer; Head and neck cancer; Malignant melanoma; Merkel cell carcinoma; Oesophageal cancer; Ovarian cancer; Renal cancer
19 Sep 2020Updated efficacy and adverse events data from a phase I/Ib trial in Non-small cell lung cancer presented at the 45th European Society for Medical Oncology Congress (ESMO-2020)
06 Aug 2020Efficacy data from a phase I trial in Colorectal cancer presented at the American Association for Cancer Research Meeting (AACR-2020)
13 Jul 2020Arcus Biosciences and Gilead Sciences complete closing of partnership agreement to co-develop and co-promote AB 928 in USA
PAPER
Organic Process Research & Development (2020), 24(7), 1254-1261.
AB928 is a potent and selective dual antagonist of the A2a and A2b receptors, which is currently in clinical trials. Here, we report the development of two scalable and practical syntheses of AB928. The first-generation synthesis was used to successfully obtain AB928 in excellent yield and purity to support our preclinical and initial clinical studies. Recently, we have developed a second-generation synthesis of AB928 featuring a palladium-free protocol to access 3-(2-amino-6-chloropyrimidin-4-yl)-2-methylbenzonitrile, a key intermediate in the AB928 synthesis. The new method is scalable, practical, and significantly more cost-effective.
Example 1: Synthesis of 3-[2-amino-6-(l-{[6-(2-hydroxypropan-2-yl)pyridin-2-yl]methyl}-lH-l,2,3-triazol-4-yl)pyrimidin-4-yl]-2-methylbenzonitrile (Compound I)
[0208] Step 1 : In a 250mL round bottom flask equipped with a magnetic stir bar was successively charged the boronic ester (3.89 g, 16 mmol) and the 2-amino-4,6-dichloropyrimidine (3.67 g, 22,4 mmol). Absolute ethanol (100 mL) was added followed by a solution of KHCO3 (4.81 g, 48 mmol) in deionized water (19 mL). The resulting suspension was degassed with nitrogen for 5 minutes. PdChiPPluk (112 mg, 1 mol%) was then added and the mixture was heated to 78 °C for 3 hours under a nitrogen atmosphere. Ethanol was evaporated under reduced pressure and deionized water (150 mL) was added. The suspension was filtered and the solid was washed with additional water (100 mL). The solid was then dissolved in acetone (220 mL) and collected in a 500 mL round bottom flask. A mixture of silica and celite (1 : 1, 150 g) was added and the solvent was removed under reduced pressure. The resulting crude material was purified by flash chromatography over silica gel (dichloromethane/ethyl acetate gradient 0% to 15%). The desired product was obtained as a white solid (1.91 g, 49%). LCMS: Method A, retention time = 2.93 min, ESI MS [M+H]+ for C12H9CIN4, calcd 245.7, found 245.2
[0209] Step 2 : In a round-bottom flask 5.1 g (20.8 mmol) of chloro-pyrimidine was suspended in 42 mL of degassed THF. To this suspension was added 8.68 mL (62.4 mmol) of Et3N and 5.95 mL (25.0 mmol) of TIPS-acetylene. The reaction mixture was stirred for 5 min, followed by addition of 219 mg (0.312 mmol) of PdCl2(PPh3)2 and 119 mg (0.624 mmol) of Cul. The reaction mixture was stirred at 50 °C for 5h under N2. After cooling the reaction to room temp., solvent was removed and the crude material was resuspended in 100 mL EtOAc from which insoluble solid was filtered off. The filtrate was washed with (1 : 1) NH4CI/NH4OH (2 x 100 mL) and 10% Na2S204 (1 x 100 mL). The organic layer was dried using Na2S04, concentrated and taken to next step without further purification.
[0210] Step 3 : In a round-bottom flask the crude TIPS product from previous step was dissolved in 42 mL dry THF and cooled to 0 °C. To this was added 25 mL (25.0 mmol) of TBAF (1.0 M in THF). The reaction was stirred at 0 °C for 15 min. Saturated NH4CI (100 mL) was added to quench the reaction. The organics were extracted from the aqueous layer with EtOAc (2 x 100 mL). The combined organic layer was washed with (1 : 1) NH4CI/NH4OH (2 x 100 mL) and 10% Na2S204 (1 x 100 mL). The organic layer was dried using Na2S04, concentrated and the pure product 5 was obtained by triturating with 40% CH2Cl2/Hexane as a light brown solid. Yield: 3.71 g (76%, 2-steps).
[0211] Step 4 : To a solution of methylmagnesium bromide (3 M in Et20, 40 mL, 120 mmol, 4.0 equiv) at 0 °C under N2 was added a solution of methyl 2-(hydroxymethyl)pyridine-2-carboxylate (5.0 g, 29.9 mmol) in THF (70 mL, 0.4 M) over the course of 30 minutes. The resulting mixture was allowed to warm to room temperature and stirred for 3 h. The reaction mixture was quenched with NH4CI aq (55 mL) and EtOAc (50 mL) was added. The organic phase was separated, and the aqueous phase was extracted with EtOAc (3 x 40 mL). The combined organic extracts were washed with saturated aqueous sodium bisulfite (7 x 20 mL), then dried (Ni^SCh), filtered and concentrated in vacuo to give the title compound (3.45 g, 69% yield; 96% purity as judged by LCMS) as a pale yellow liquid. LCMS: Method A, retention time = 0.722 and 1.06 min, ESI MS [M+H]+ for C9H13NO2, calcd 167.09, found 167.2
[0212] Step 5 : To a solution of 2-hydroxymethyl-6-(l -hydroxy- 1 -methyl ethyljpyri dine (5 g,
29.9 mmol, 1.0 equiv) in PhMe (33 mL, 0.9 M) at 0 °C under N2 was added diphenylphosphoryl azide (7.73 mL, 35.9 mmol, 1.2 equiv.), followed by l,8-diazabicyclo[5.4.0]undec-7-ene (5.37 mL, 35.9 mmol, 1.2 equiv.). The resulting mixture was to warm to room temperature and stirred for 14 h. Upon completion, diluted with ethyl acetate and washed with water, the organic layer was dried (Na2S04), filtered and concentrated. The residue was dissolved in 1N aq HC1 (2 eq, 60 mmol) and extracted with MTBE in hexanes (3:7, 100 mL), the organic layer was washed with water (50 mL) and the combined aqueous layer was neutralized with 2N aqueous NaOH and extracted with ethyl acetate (3X75 mL), dried the organic layer (Na2S04), filtered through a plug of cotton and concentrated the filtrate to afford the pure compound as pale yellow color liquid (3.75 g, 75%). LCMS: Method A, retention time = 2.67 min, ESI MS [M+H]+ for C9H12N4O, calcd 193.1, found 193.2
[0213] Step 6: A mixture of azide (3.34 g, 17.4 mmol), alkyne (3.71 g, 15.8 mmol), copper(II) sulfate (39 mg; 0.158 mmol), and sodium ascorbate (156 mg, 0.790 mmol) in 2: 1 /-BuOH/EbO (158 mL) was heated at 60 °C for 13 h. The solvent was removed in vacuo, the residue dry loaded onto silica gel, and purified by silica gel chromatography (0-100% EtOAc in hexanes) to afford the desired product as an off-white solid (6.08 g, 90%). ‘H NMR (400 MHz, DMSO-cfc) d 8.69 (s, 1H), 7.90 (d, J= 7.8 Hz, 1H), 7.80 (t, J= 7.8 Hz, 1H), 7.76 (d, J= 7.8 Hz, 1H), 7.61 (d, J= 8.0 Hz, 1H), 7.51 (t, /= 7.8 Hz, 1H), 7.28 (s, 1H), 7.10 (d, J= 7.6 Hz, 2H), 6.90 (s, 2H), 5.81 (s, 2H), 5.23 (s, 1H), 2.55 (s, 3H), 1.38 (s, 6H). ESI MS [M+H]+ for C23H23N8O, calcd 427.2, found 427.3.
Example 2: Preparation of Crystalline Solid Form of 3-[2-amino-6-(l-{[6-(2-hydroxypropan-2-yl)pyridin-2-yl]methyl}-lH-l,2,3-triazol-4-yl)pyrimidin-4-yl]-2-methylbenzonitrile
[0214] The product from Example 1, Step 6 (7.53 g) was dissolved in acetone (109 mL) by heating to reflux at which point water (218 mL) was added at a rate of 10 mL/min to initiate crystallization. The mixture was cooled and the solids were collected by filtration, washed with 1 :2 acetone/water (109 mL), and dried under vacuum to afford Form I of Compound I as a white solid (7.08 g; 94%).
PATENT
WO 2019161054
PATENT
WO2020185859 , claiming method for treating a subject identified as having an oncogene driven cancer comprising an agent (eg AB-928) targeting the extracellular production of adenosine and/or antagonizing the activation by adenosine of one of its receptors.
Processes for preparing aminopyrimidine compounds, particularly etrumadenant (AB-928).
Example 1: Trifluoroethanol Assisted Condensation of B-Ketoesters to Provide a
Hydroxypyrimidine (and Chloropyrimidine).
bromo-2-methylaniline (18.6 g, 100 mmol) dropwise so that a fine white suspension forms. The mixture was cooled to 0 °C and a solution of sodium nitrite (7.31 g, 106 mmol) in water (15.1 mL) was added dropwise. The mixture was stirred at 0 °C for 30 minutes. To the resultant homogeneous mixture at 0 °C was added sodium bicarbonate (17.8 g, 212 mmol) at such a rate to avoid excessive gas evolution. The aqueous phase of the resultant brown suspension was found to have pH ~7. This suspension was maintained at 0 °C.
[0070] In a separate flask, copper cyanide (9.85 g, 110 mmol), potassium cyanide (13.0 g, 200 mmol), and water (31 mL) were heated to 60 °C to form a homogeneous solution. To this solution at 60 °C with stirring was added the above suspension dropwise to avoid excessive gas evolution. After addition, the mixture was stirred at 100 °C for 30 minutes. The mixture was cooled, MTBE (200 mL) was added, the mixture agitated, and filtered to remove any solids, washing with MTBE. The organic phase was dried over Na2SO4 and concentrated. The resultant crude product was purified by vacuum distillation to afford the desired product as a light orange solid (13.6 g, 69%).
[0071] Step 2: In a two liter two-necked flask, aryl bromide (101.9 g, 520 mmol, 1.0 equiv.) was dissolved in THF (520 mL) under an atmosphere of N2, and the mixture was cooled in an
ice-water bath. iPrMgClLiCl (400 mL, 1.3 M in THF, 520 mmol, 1.0 equiv.) was added by cannula. Upon completion of the addition, the ice bath was removed. After four hours, the flask was cooled in an ice-water bath and dry ice (~ 230 g, 5.2 mol, 10 equiv.) was added portionwise to prevent overheating or bubbling over (note: CO2 gas can be bubbled through the solution in place of solid dry ice). When bubbling from the addition was complete, the mixture was diluted with MTBE (500 mL) and 2M HC1 (250 mL). The layers were separated, and the aqueous layer was washed with additional MTBE (500 mL). The organic layer was extracted with 10% NaOH (190 mL x 2), and the combined aqueous layers were cooled in an ice-water bath and acidified with concentrated HC1 until a white precipitate formed. The precipitate was isolated by filtration and washed with water before being dried overnight in a vacuum oven at 80° C to afford the benzoic acid as a white solid (64.1 g, 76% yield).
[0072] Step 3: The benzoic acid (50 g, 311 mmol, 1.0 equiv.) was suspended in CH2CI2, and oxalyl chloride (40 mL, 466 mmol, 1.5 equiv.) was added, followed by DMF (~ 30 drops). Off gassing was observed immediately, and the reaction flask was open to the atmosphere under positive pressure of N2. Upon complete consumption of the starting acid as determined by LCMS and visual inspection (complete dissolution of starting material), the reaction mixture was concentrated. Excess oxalyl chloride was removed by azeotropic distillation with toluene to afford the corresponding acid chloride as a tannish-brown solid.
[0073] In a separate two-necked flask equipped with an overhead stirrer, potassium ethyl malonate (66.1 g, 388 mmol, 1.25 equiv.), triethylamine (108 mL, 777 mmol, 2.5 equiv.) and MeCN (777 mL) were cooled in a salt/ice-brine bath. Solid MgCl2 (74 g, 777 mmol, 2.5 equiv.) was added, and the resulting suspension was vigorously stirred at ~ -10° C. After one hour, the solid acid chloride was added at a rate to ensure dissolution into the thick suspension. The suspension rapidly became homogenous, and the stirring rate was reduced to avoid splashing.
The ice bath was removed. Upon complete consumption of the starting material as determined by TLC analysis, the reaction mixture was cooled in an ice-water bath, and 2M HC1 (971 mL, 1.9 mol, 6.25 equiv.) was added, and the ice bath was removed. After 30 minutes, the layers were separated, and the aqueous layer was extracted with MTBE. The combined organic layers were washed with saturated NaHCO3 and brine, dried over sodium sulfate, filtered, and concentrated to afford the keto-ester as a tannish-brown solid (67 g, 93% yield).
[0074] Step 4: A round-bottom flask was charged with 42.0 g (181.8 mmol) of the b-keto-ester, 32.7 g (181.8 mmol) of guanidinium carbonate and 227 mL of trifluoroethanol. The suspension was then heated to reflux under N2 for 16 h.
[0075] Work-up: The reaction was cooled to room temperature and solvent was evaporated under reduced pressure to obtain a viscus red oil. The oil was re-dissolved in 250 mL H2O and the aqueous solution was extracted with dichloromethane (2 x 250 mL). The aqueous phase is then acidified to pH ~2-3 using 1.0 M HCl(aq ). The precipitated product was collected by filtration, washed thoroughly with H2O and dried in a vacuum oven at 70 °C. Yield 30.81 g (75%), Purity >99%.
[0076] Step 5: A round-bottom flask was charged with 50.0 g (221.2 mmol) pyrimidone from step 4 and 100.8 g (442.2 mmol) of benzyltriethylammonium chloride. The mixture was suspended in 442.2 mL of dry acetonitrile and 31.0 mL (331.8 mmol) of POCI3 was added. The suspension thus obtained was then heated to reflux under N2 for 4 h.
[0077] Work-up: The reaction was cooled to room temperature and ~200 g crushed ice was added. The mixture was then stirred for 30 min flowed by dropwise addition of ice-cold 15% aqueous NH4OH to ~ pH 10 -11. {Note: Slow addition of cold NH4OH is recommended to avoid sudden exotherm due to quenching of excess POCI3). The suspension was then stirred at room temperature for an additional 1.5 h. The precipitated product was collected by filtration, washed thoroughly with H2O and dried in a vacuum oven at 70 °C. Yield 48.2 g (89%), Purity >99%.
[0078] The synthetic route for preparing 3-[2-amino-6-(l- {[6-(2-hydroxypropan-2-yl)pyridin-2-yl]methyl}-1H-1 ,2,3-triazol-4-yl)pyrimidin-4-yl]-2-methylbenzonitrile utilizing boronic ester benzonitrile to linked the phenyl and pyrimidine rings is shown below and is also provided in WO2018/136700.
[0079] The scheme below displays the synthetic route used to prepare the boronic ester benzonitrile used in the process above and subsequent reaction with pyrimidine to form a compound of Formula (I). Notably, the desired linkage between the pyrimidine and the phenyl provides a yield of less than 50%.
[0080] The below scheme displays the synthetic route used to prepare a compound of Formula (I) that utilized a conversion of a b-diketoester to a pyrimidine using guanidine. The route provides a 75% yield.
Example 1: Synthesis of 3-[2-amino-6-(1-{[6-(2-hydroxypropan-2-yl)pyridin-2-yl]methyl}-1H-1,2,3-triazol-4-yl)pyrimidin-4-yl]-2-methylbenzonitrile
[0269] Step 1: In a 250mL round bottom flask equipped with a magnetic stir bar was successively charged the boronic ester (3.89 g, 16 mmol) and the 2-amino-4,6- dichloropyrimidine (3.67 g, 22,4 mmol). Absolute ethanol (100 mL) was added followed by a solution of KHCO3 (4.81 g, 48 mmol) in deionized water (19 mL). The resulting suspension was degassed with nitrogen for 5 minutes. PdCl2(PPh3)2 (112 mg, 1 mol%) was then added and the mixture was heated to 78 °C for 3 hours under a nitrogen atmosphere. Ethanol was evaporated under reduced pressure and deionized water (150 mL) was added. The suspension was filtered and the solid was washed with additional water (100 mL). The solid was then dissolved in acetone (220 mL) and collected in a 500 mL round bottom flask. A mixture of silica and celite (1:1, 150 g) was added and the solvent was removed under reduced pressure. The resulting crude material was purified by flash chromatography over silica gel (dichloromethane/ethyl acetate gradient 0% to 15%). The desired product was obtained as a white solid (1.91 g, 49%). LCMS: Method A, retention time = 2.93 mm, ESI MS [M+H]+ for C12H9ClN4, calcd 245.7, found 245.2
[0270] Step 2: In a round-bottom flask 5.1 g (20.8 mmol) of chloro-pyrimidine was suspended in 42 mL of degassed THF. To this suspension was added 8.68 mL (62.4 mmol) of Et3Ν and 5.95 mL (25.0 mmol) of TIPS -acetylene. The reaction mixture was stirred for 5 min, followed by addition of 219 mg (0.312 mmol) of PdCl2(PPh3)2 and 119 mg (0.624 mmol) of Cul. The reaction mixture was stirred at 50 °C for 5h under N2. After cooling the reaction to room temp., solvent was removed and the crude material was resuspended in 100 mL EtOAc from which insoluble solid was filtered off. The filtrate was washed with (1:1) NH4C1/NH4OH (2 × 100 mL) and 10% Na2S2O4 (1 × 100 mL). The organic layer was dried using Na2SO4, concentrated and taken to next step without further purification.
[0271] Step 3: In a round-bottom flask the crude TIPS product from previous step was dissolved in 42 mL dry THF and cooled to 0 °C. To this was added 25 mL (25.0 mmol) of TBAF (1.0 M in THF). The reaction was stirred at 0 °C for 15 mm. Saturated NH4Cl (100 mL) was added to quench the reaction. The organics were extracted from the aqueous layer with EtOAc (2 x 100 mL). The combined organic layer was washed with (1:1) NH4Cl/NH4OH (2 x 100 mL) and 10% Na2S2O4 (1 x 100 mL). The organic layer was dried using Na2SO4, concentrated and the pure product 5 was obtained by triturating with 40% CH2Cl2/Hexane as a light brown solid. Yield: 3.71 g (76%, 2-steps).
[0272] Step 4: To a solution of methylmagnesium bromide (3 M in Et2O, 40 mL, 120 mmol, 4.0 equiv) at 0 °C under N2 was added a solution of methyl 2-(hydroxymethyl)pyridine-2-carboxylate (5.0 g, 29.9 mmol) in THF (70 mL, 0.4 M) over the course of 30 minutes. The resulting mixture was allowed to warm to room temperature and stirred for 3 h. The reaction mixture was quenched with NH4Cl aq (55 mL) and EtOAc (50 mL) was added. The organic phase was separated, and the aqueous phase was extracted with EtOAc (3 x 40 mL). The combined organic extracts were washed with saturated aqueous sodium bisulfite (7 x 20 mL), then dried (Na2SO4), filtered and concentrated in vacuo to give the title compound (3.45 g, 69% yield; 96% purity as judged by LCMS) as a pale yellow liquid. LCMS: Method A, retention time = 0.722 and 1.06 mm, ESI MS [M+H]+ for C9H13NO2, calcd 167.09, found 167.2
[0273] Step 5: To a solution of 2-hydroxymethyl-6-(1-hydroxy-1-methylethyl)pyridine (5 g, 29.9 mmol, 1.0 equiv) in PhMe (33 mL, 0.9 M) at 0 °C under N2 was added diphenylphosphoryl azide (7.73 mL, 35.9 mmol, 1.2 equiv.), followed by l,8-diazabicyclo[5.4.0]undec-7-ene (5.37 mL, 35.9 mmol, 1.2 equiv.). The resulting mixture was to warm to room temperature and stirred for 14 h. Upon completion, diluted with ethyl acetate and washed with water, the organic layer was dried (Na2SO4), filtered and concentrated. The residue was dissolved in 1N aq HCl (2 eq, 60 mmol) and extracted with MTBE in hexanes (3:7, 100 mL), the organic layer was washed with water (50 mL) and the combined aqueous layer was neutralized with 2N aqueous NaOH and extracted with ethyl acetate (3×75 mL), dried the organic layer (Na2SO4), filtered through a plug of cotton and concentrated the filtrate to afford the pure compound as pale yellow color liquid (3.75 g, 75%). LCMS: Method A, retention time = 2.67 mm, ESI MS [M+H]+ for C9H12N4O, calcd 193.1, found 193.2
[0274] Step 6: A mixture of azide (3.34 g, 17.4 mmol), alkyne (3.71 g, 15.8 mmol), copper(II) sulfate (39 mg; 0.158 mmol), and sodium ascorbate (156 mg, 0.790 mmol) in 2:1 t-BuOH/H2O (158 mL) was heated at 60 °C for 13 h. The solvent was removed in vacuo, the residue dry loaded onto silica gel, and purified by silica gel chromatography (0-100% EtOAc in hexanes) to afford the desired product as an off-white solid (6.08 g, 90%). 1H NMR (400 MHz, DMSO-d6) δ 8.69 (s, 1H), 7.90 (d, J = 7.8 Hz, 1H), 7.80 (t, J = 7.8 Hz, 1H), 7.76 (d, J = 7.8 Hz, 1H), 7.61 (d, J= 8.0 Hz, 1H), 7.51 (t, J = 7.8 Hz, 1H), 7.28 (s, 1H), 7.10 (d, J = 7.6 Hz, 2H), 6.90 (s, 2H), 5.81 (s, 2H), 5.23 (s, 1H), 2.55 (s, 3H), 1.38 (s, 6H). ESI MS [M+H]+ for C23H23N8O, calcd 427.2, found 427.3.
CitrullineCAS Registry Number: 372-75-8 CAS Name:N5-(Aminocarbonyl)-L-ornithine Additional Names: d-ureidonorvaline; a-amino-d-ureidovaleric acid; Nd-carbamylornithine Molecular Formula: C6H13N3O3Molecular Weight: 175.19 Percent Composition: C 41.13%, H 7.48%, N 23.99%, O 27.40%Line Formula: H2NCONH(CH2)3CH(NH2)COOH Literature References: An amino acid, first isolated from the juice of watermelon, Citrullus vulgaris Schrad., Cucurbitaceae: Wada, Biochem. Z.224, 420 (1930); isoln from casein: Wada, ibid.257, 1 (1933). Synthesis from ornithine through copper complexes: Kurtz, J. Biol. Chem.122, 477 (1938); by alkaline hydrolysis of arginine: Fox, ibid.123, 687 (1938); from cyclopentanone oxime: Fox et al.,J. Org. Chem.6, 410 (1941). Crystallization: Matsuda et al.,JP71 174 (1971 to Ajinomoto), C.A.74, 126056u (1971). Crystal and molecular structure: Naganathan, Venkatesan, Acta Crystallogr.27B, 1079 (1971); Ashida et al.,ibid.28B, 1367 (1972). Use in asthenia and hepatic insufficiency: FR2198739 (1974 to Hublot & Vallet), C.A.82, 144952c (1975). Clinical trial in treatment of lysinuric protein intolerance: J. Rajantie et al.,J. Pediatr.97, 927 (1980); T. O. Carpenter et al.,N. Engl. J. Med.312, 290 (1985).Properties: Prisms from methanol + water, mp 222°. [a]D20 +3.7° (c = 2). pK1 2.43; pK2 9.41. Sol in water. Insol in methanol, ethanol.Melting point: mp 222°pKa: pK1 2.43; pK2 9.41Optical Rotation: [a]D20 +3.7° (c = 2) Derivative Type: HydrochlorideCAS Registry Number: 34312-10-2Molecular Formula: C6H13N3O3.HClMolecular Weight: 211.65Percent Composition: C 34.05%, H 6.67%, N 19.85%, O 22.68%, Cl 16.75%Properties: Crystals, dec 185°. [a]D22 +17.9° (c = 2).Optical Rotation: [a]D22 +17.9° (c = 2) Derivative Type: Malate (salt)CAS Registry Number: 54940-97-5Trademarks: Stimol (Biocodex)Molecular Formula: C6H13N3O3.C4H6O5Molecular Weight: 309.27Percent Composition: C 38.84%, H 6.19%, N 13.59%, O 41.39% Therap-Cat: Treatment of asthenia.
Asklepion is developing an iv formulation of citrulline, Citrupress, for the potential treatment of pulmonary hypertension and for the potential prevention of clinical sequelae of acute lung injury complicating congenital heart repair surgery in pediatric patients, and also investigating the drug for the potential treatment of acute sickle cell crisis. In August 2016, a phase III study was initiated for preventing clinical sequelae of acute lung injury?in pediatric patients undergoing cardiopulmonary bypass (CPB) for heart defects; in July 2019, results were expected in October 2019.
Citrulline is an amino acid. It is made from ornithine and carbamoyl phosphate in one of the central reactions in the urea cycle. It is also produced from arginine as a by-product of the reaction catalyzed by NOS family. Its name is derived from citrullus, the Latin word for watermelon, from which it was first isolated.
Citrulline is made from ornithine and carbamoyl phosphate in one of the central reactions in the urea cycle. It is also produced from arginine as a byproduct of the reaction catalyzed by NOS family (NOS; EC 1.14.13.39).[6] It is made from arginine by the enzymetrichohyalin at the inner root sheath and medulla of hair follicles.[7]Arginine is first oxidized into N-hydroxyl-arginine, which is then further oxidized to citrulline concomitant with release of nitric oxide.
Citrulline is also made by enterocytes of the small intestine.[2][8]
Function
Several proteins contain citrulline as a result of a posttranslational modification. These citrulline residues are generated by a family of enzymes called peptidylarginine deiminases (PADs), which convert arginine into citrulline in a process called citrullination or deimination with the help of calcium ion. Proteins that normally contain citrulline residues include myelin basic protein (MBP), filaggrin, and several histone proteins, whereas other proteins, such as fibrin and vimentin are susceptible to citrullination during cell death and tissue inflammation.
Circulating citrulline concentration is a biomarker of intestinal functionality.[9][10
PAPER
Biochemistry, 53(41), 6511-6519; 2014
PAPER
Journal of the Chemical Society of Pakistan, 34(2), 451-454; 2012
PAPER
Journal of Agricultural and Food Chemistry, 66(33), 8841-8850; 2018
l-Citrulline is a nonessential amino acid with a variety of physiological functions and can be enzymatically produced by arginine deiminase (ADI, EC 3.5.3.6). The enzymatic-production approach is of immense interest because of its mild conditions, high yield, low cost, and environmental benignity. However, the major hindrances of l-citrulline industrialization are the poor thermostability and enzyme activity of ADI. Hence, in this work, directed evolution and site-directed mutagenesis aided with in silico screening, including the use of b-factor values and HoTMuSiC, were applied to a previously identified ADI from Enterococcus faecalis SK23.001 (EfADI), and a triple-site variant R15K–F269Y–G292P was obtained. The triple-site variant displays a 2.5-fold higher specific enzyme activity (333 U mg–1), a lower Km value of 6.4 mM, and a 6.1-fold longer half-life (t1/2,45°C = 86.7 min) than wild-type EfADI. This work provides a protein-engineering strategy to improve enzyme activity and thermostability, which might be transferrable to other ADIs and enzymes.
Biocatalytic transformation of carbamate formed readily from CO2 and NH3 provides attractive green routes for mitigation of these important environmental pollutants. Accordingly, a coupled-enzyme system was developed for the one-pot production of citrulline through carbamoylation of ornithine in aqueous solutions of CO2 and NH3. Hyperthermophilic ornithine carbamoyltransferases are produced recombinantly in E. coli with carbamate kinases known to have a propensity for carbamoyl phosphate synthesis. Importantly, in vitro biocatalysis is carried out by E. coli cell lysate prepared through coexpression of the required recombinant enzymes in a single bacterial culture, greatly reducing limitations normally associated with protein production and purification. Acetate kinase that is endogenous in the lysate also recycles the required ATP cofactor, which would otherwise have been required in costly stoichiometric amounts. Recombinant lysates catalyze the production of carbamoyl phosphate with substoichiometric ATP (>300 turnovers) as well as its in situ reaction with ornithine to give citrulline in high yield (>95%) and g L–1 h–1 titers. The system is active over a wide range of NH3 concentrations (2.5 mM – 2 M), and >90% conversions of NH3 may be reached within 1.5 h. Aqueous NH3 used to sequester CO2 gas (10% v/v) may be directly used as the biocatalyst feedstock. In preliminary studies, citrulline is found to be an effective organic nitrogen fertilizer of the wheat grass Brachypodium distachyon. Therefore, lysates described here constitute a cost-effective biocatalytic platform for one-pot production of a promising organic nitrogen fertilizer, under mild reaction conditions, from environmental pollutants as feedstock.
Process for preparing citrulline from a transition metal complex of ornithine using cyanate useful to reduce the incidence or severity of cardiopulmonary bypass-induced pulmonary injury due to free radical formation in a patient during cardiopulmonary bypass.
Ornithine is an alpha amino acid with a terminal amino group opposite the alpha carbon.
Citrulline is an alpha amino acid with a terminal carbamido group in the same position as the terminal amino group of ornithine. Dr. A. Kurtz described synthesis of racemic citrulline from racemic ornithine in 1938 (J. Biol. Chem., 122:477-484), and that disclosure was followed up by synthesis of optically active /-citrulline from /-ornithine in 1949 (J. Biol. Chem., 180: 1253-1267). Optical activity was preserved by complexing the starting material (/-ornithine) in a transition metal complex via the alpha amino and carboxyl groups, then reacting the terminal amino group with urea to from a carbamido derivative (see Figure 1). Kurth 1949 describes numerous other syntheses, all depending on the transition metal complex to preserve the alpha amino acid character of the starting compound while derivatizing other parts of the molecule. An example of this synthesis is described in Example 1 below.
Details of various steps in the improved processes developed by the present inventors for producing pharmaceutical grade citrulline are discussed below.
Synthesis of Citrulline from Ornithine
[00014] The present inventors preserved the stereochemical structure around the alpha carbon of the alpha amino acid during reaction of amino groups elsewhere on the compound by complexing the alpha end of the molecule with a transition metal atom, as reported by
Kurth 1938 and 1949. The initial production of the /-ornithine-copper complex is carried out as described by Kurtz. Kurtz describes a variety of transition metals as the complexing metal in the 1949 paper, but the preferred metal is copper (II), based on the ease of forming stable complexes and the ease with which copper (II) may subsequently be removed from the product. The copper is typically supplied as cupric sulfate, although complex formation from copper (II) acetate, cupric carbonate, or cupric oxide have also been reported.
[00015] The present inventors have discovered an alternative method of derivatizing the terminal amino group of the complexed alpha amino acid using cyanate rather than the urea reaction reported by Kurth. An example of this improved synthesis is shown in Figure 2A and described in Example 3 below. Use of cyanate as the derivatizing agent has been found to produce fewer distinct product compounds, which simplifies purification of the desired citrulline product. Kurth carried out urea derivatization by refluxing the copper complex in the presence of excess urea. Cyanate derivatization may be carried out at lower temperatures (e.g. 55°C-65°C) which may contribute to higher yield of citrulline, based on the initial amount of ornithine. Cyanate is preferably provided in excess, and the reaction is driven by precipitation of the citrulline: copper complex. The precipitated complex is washed with water to remove unreacted copper (e.g., wash until no blue coloration persists in the filtrate). The precipitated copper complex of citrulline may be recovered and dried.
Enriching Citrulline as a Copper Complex
[00016] The inventors have discovered that the relative citrulline content of the reaction
product(s) can be enhanced by reprecipitation of the citrulline: copper complex. Precipitated copper complex of citrulline (produced, for example, by reaction of a ornithine: copper complex with urea or cyanate in water) may be dried. The
citrulline: copper complex may be redissolved by suspending the precipitate in water and acidifying the suspension until the complex dissolves. Acidification may be
accomplished by adding concentrated acid, preferably hydrogen chloride, to the suspension while stirring. Once the copper: citrulline complex solution is clear, base (typically sodium hydroxide) is added to bring the pH up to 7-10. Both the acidification and subsequent neutralization steps are actively cooled (temperature not more than 45°C) to protect the citrulline product from hydrolysis or reaction to produce side products. The precipitate is washed with water (e.g., until the filtrate is free of chloride by checking the filtrate for turbidity with silver nitrate), and then the precipitate is dried. Reprecipitation under these conditions is selective for citrulline: copper complex over ornithine: copper complex, because the ornithine complex is more soluble in water. If the dried complex contains higher than the desired level of ornithine contamination (e.g., greater than 10 mole% ornithine – as measured by NMR, for example), the complex may be redissolved and reprecipitated as necessary to further lower the relative amount of ornithine.
Recovering Citrulline from Its Copper Complex
[00017] Once the ornithine content in the copper: citrulline complex precipitate is sufficiently low
(preferably less than 10 mole% ornithine), the precipitate is resuspended in water and citrulline is freed from the complex by removing the copper as an inorganic precipitate, typically copper sulfide (See Figure 2B). Sulfide may be introduced in a variety of salt forms, but the inventors have found it preferable to use hydrogen sulfide gas as the sulfide source. In a preferred mode, the aqueous suspension is placed in a stirred, pressure vessel. The air is then pumped out of the reactor’s head space to form an under pressure. The reactor is then repressurized with hydrogen sulfide gas over the aqueous suspension (preferably at low temperature, e.g., 0°C-5°C, to maximize the solubility of hydrogen sulfide). Hydrogen sulfide is continuously added to the reactor to maintain parity with ambient pressure during consumption of this gas. Copper salts will precipitate, leaving citrulline in solution. As hydrogen sulfide is consumed, the pressure in the vessel decreases; the reaction is complete when the pressure stabilizes. Reaction of hydrogen sulfide with residual copper salts (for example chloride or sulfate) will lower the pH; typically the pH will be below 4, preferably pH~3. Copper salts typically include copper (II) sulfide, but may also include copper (I) sulfide and copper oxide. The solution temperature is elevated for filtration, typically to about 30°C, to promote solubility of the citrulline and drive off excess hydrogen sulfide gas, while precipitated copper salts are removed by filtration.
Purifying Citrulline
[00018] For pharmaceutical use, the active compound must be substantially free of contaminants, and further purification steps are necessary to produce a pharmaceutical grade product. For the purposes of this invention, substantially free of contaminants is considered to include: ornithine not more than (NMT) 0.8%, individual specified impurities NMT 0.15%, individual unspecified (unknown) impurities NMT 0.1%; total related substances NMT 1.3%, and Cu not more than lOppm. For citrulline manufactured from ornithine using copper complex to protect the alpha amino acid functions, the inventors have found that desired purification after citrulline is released from the copper complex can be achieved by activated carbon adsorption of contaminants and solvent/anti- solvent crystallization of the active pharmaceutical component.
[00019] The citrulline-containing aqueous solution remaining after removal of precipitated copper salts is neutralized to stabilize the citrulline against hydrolysis, to enhance adsorption of residual copper to activated carbon, and to facilitate solvent/anti-solvent precipitation of citrulline; pH is preferably adjusted to 5.9 ± 0.2, the isoelectric point of citrulline. The neutralized citrulline solution may be passed through a nano-filter to remove any bacteria and/or bacterial cell wall fragments that contaminate the solution. The nano-filtered solution may be held in a semi-sterile reservoir for staging purposes between the subsequent purification steps. The neutralized citrulline solution is treated with activated carbon, either by mixing with carbon dust or passing the solution through an activated carbon adsorber bed. The aqueous citrulline-containing effluent from the activated carbon is mixed with an anti-solvent to induce anti-solvent crystallization. Suitable anti solvents are miscible with water, including aliphatic alcohols, such as 2-propanol, ethanol or methanol, as well as acetone. A preferred antisolvent for citrulline is acetone, when mixed with approximately two volumes of water (e.g., 1 volume of water to 1.8 volumes of acetone). Acetone is preferably pre-cooled so that the resultant suspension is 0°C- 10°C. The cooled suspension may be collected in a reservoir or processed by filtration immediately to recover the citrulline precipitate.
Microbial control:
[00020] Because citrulline synthesis and purification occur in aqueous solution, there is increased risk of microbial contamination and endotoxin accumulation in the product. Washing the citrulline: copper precipitate, and addition of H2S to acid solution minimize any accumulation of microbes. From the exposure of the complex to FES until treatment with acetone the aqueous solutions of citrulline are preferably kept in sealed vessels to limit microbial contamination and growth. Enclosing the purification steps to minimize contact with the environment and use of sterile filters to capture potential microbial contamination allows the manufacturing to be performed in an ISO 8 cleanroom. Alternatively, the final purification steps can be carried out in a sterile GMP environment of the sort used for aseptic filling of sterile dosage products (e.g., ISO Class 5/6).
[00021] If examination of the solution prior to the anti-solvent precipitation shows the amounts of microbes or endotoxin levels exceed those aceptable for injectable therapeutic compositions (e.g., 50 EU/g API, more preferably 20 EU/g), the product may be subjected to nano-filtration to remove microbes and endotoxin, before being recovered by anti-solvent precipitation and drying. The citrulline and water molecules pass through the nano-filtration membrane, but the larger bacteria and bacterial cell wall fragments are retained by the filter.
Filter press
[00022] The reaction mixtures may be pumped through a filter press to collect / remove the
suspended solids. See the general picture in Figure 3, and the attached photograph in Figure 4. The press is composed of a series of plates 1 which are then hydraulically pressed together. The hydraulic pressure ensures that the system is sealed. The suspension is then pumped through a central tube 2 where it spreads-out across several chambers 3 between the plates. The walls of the plates have a filter sheet, which allows the filtrate to flow past and exit via an internal cavity 4.
[00023] The general advantage of a filter press is that it allows a high surface area for filtration.
This effect greatly accelerates the portion-wise collection and washing of the complex and API. This system may be used to collect the copper salts after exposure to hydrogen sulfide. In the latter case, the suspension is pumped from the reactor into the press, and the filtrate may then be passed through an in-line 5 pm filter to catch any residual particulate copper, then an in-line sterile 0.2 pm filter at the entry port of a semi-sterile container for holding.
The press may be used to collect:
• Crude citrulline copper complex
• The complex after the pH-driven re-precipitation
• Precipitated copper salts (where citrulline leaves as solution in the filtrate)
• Precipitated citrulline from anti-solvent precipitation prior to drying
Semi-sterile containers
[00024] A useful semi-sterile container is basically a closed vessel equipped with a stirrer and ports for the addition and removal of liquid, and a pH meter. The container should be sterilized (e.g., treated with isopropyl alcohol solution and rinsed with water) directly prior to use and not opened during use. A sterile, air filter attached to the lid allows air to flow into the container as the liquid is being pumped out. The pH adjustment may be performed in this container, before treatment with activated carbon. The container is not particularly suitable for the long-term storage of the solutions.
Activated carbon adsorber bed
[00025] The solution may be pumped from the semi sterile container through the activated carbon bed (a column packed with granulated activated carbon) pre-flushed with argon. The liquid is then returned to the semi-sterile container via an in-line 5 pm filter and the 0.2 pm sterile filter at the entry port. If the solution is pumped in a cyclic manner with the stirrer activated for not less than 6 hours, the sterile filter acts as a“microbial scrubber” continually collecting any microbes in the solution. The activated carbon primarily removes any organic impurities and will also remove any residual dissolved copper ions. The 5 pm filter catches any carbon particles which detach from the bed.
Sterile bags
[00026] After processing in the activated carbon adsorber bed, the solution may be passed into a single use sterile bag via another sterile filter. The solution may be stored longer in the bag than in the semi-sterile container. At this point, a test for the presence of microbes and/or bacterial endotoxins can be carried out. If endotoxins are observed, then the cut off (nano-filtration) membrane may be employed. If not, the citrulline is ready to be
recovered from the solution by anti-solvent precipitation. Collection of the solution in a sterile bag allows the citrulline solution to be processed batch-wise, where conveniently sized portions of citrulline are precipitated and recovered in the filter press.
Solvent/Anti-solvent Mixing
[00027] The aqueous citrulline solution is mixed with pre-cooled anti-solvent to precipitate the citrulline from solution. After mixing with anti-solvent, the threat posed by bacterial growth is not higher than that for other APIs. The addition of the organic solvent makes the resulting solution bacteriostatic at a minimum. This precipitation improves the purity of citrulline, reducing, in particular, the ornithine levels, and allows for the rapid extraction of citrulline from solution.
Final drying
[00028] The precipitate is dried to remove residual acetone and water. Drying may be carried-out in a conical dryer, firstly to drive off the acetone anti-solvent, then moisture and finally the water of crystallization. The conical dryer can also be used to homogenize the product. The final, dry product of anti-solvent precipitation may be stored, and ultimately dissolved in sterile aqueous diluent for therapeutic administration.
[00029] On dissolution in sterile aqueous media, citrulline prepared as described herein may be used to treat pulmonary hypertension (WO/2000/073322), bronchopulmonary dysplasia (WO/2009/099998), sickle cell crisis (WO/2018/157137), cardiac surgery patients (WO/2005/082042), cardiopulmonary bypass patients (WO/2018/125999), and vasospasm as a complication of subarachnoid hemorrhage (WO/2009/099999), by parenteral administration as described in these documents, incorporated herein by reference.
EXAMPLES
Example 1. Synthesis of citrulline from ornithine using urea.
[00030] L-Citrulline is synthesized from L-omithine and urea. A flow chart of the reaction is shown in Figure 1 A.
[00031] L-Citrulline is prepared synthetically starting from L-ornithine hydrochloride. Into a 120- L reactor containing approximately 50 liters of water, 10 kilograms of L-omithine hydrochloride is added and dissolved. The solution is neutralized with potassium hydroxide and then converted to its copper complex by the addition of 15kg copper sulfate (molar equivalent amount). The copper complex protects the 2-amino carboxylic acid functionality in the molecule while chemistry is performed on the terminal amino group. The L-ornithine copper complex is then exposed to an excess of urea at reflux, which promotes its conversion to the copper complex of L-citrulline. The resulting copper complex of L-citrulline then is precipitated and collected by filtration.
[00032] The isolated copper complex of L-citrulline is dried and testing is performed. The
appearance is verified, and an in-use performance test is done to determine suitability to proceed.
Example 2. Purification of citrulline from copper-citrulline complex.
[00033] L-Citrulline synthesized from L-ornithine and urea is purified by resin-based purification and recrystallization. A flow chart of the reaction is shown in Figure IB.
[00034] In a 120-L reactor, ~13 kilograms of the L-citrulline copper complex prepared in
Example 1 is added to a stirring solution of sodium sulfide (Na2S) in water
(approximately 8 kilograms Na2S in 50 liters of water), causing the precipitation of copper sulfide and the freeing of L-citrulline. The solution is filtered to remove the copper salts. The pH of the resulting aqueous solution containing the sodium salt of L- citrulline and residual sodium sulfide is lowered to 4 by the addition of an acidic ion exchange resin (such as Amberlite). A constant stream of argon gas is passed through the solution to remove the residual sulfide as hydrogen disulfide. The pH of the solution is then raised to 5.9 ± 0.2 using sodium hydroxide to form isoelectric L-citrulline.
Activated carbon is then added to the reaction mixture to remove residual impurities, in particular residual copper ions. The solids (Amberlite and activated carbon) are then removed by filtration, and the filtrate is concentrated to approximately 50 liters (either by evaporation or reverse osmosis). L-citrulline is then precipitated from the aqueous solution by the addition of an equal part of acetone, and the mixture is cooled to near 0°C. The precipitate is collected by filtration and dried in a vacuum oven.
[00035] The non-sterile bulk powder is then reconstituted and processed for endotoxin reduction and sterile filtration steps followed by crystallization, drying and micronization in an aseptic environment. The sterile bulk powder is then used as the“raw material” for aseptic filling into glass vials to produce the finished drug product which may be reconstituted with a sterile diluent prior to use.
Example 3. Synthesis of citrulline from ornithine using cyanate
[00036] L-Citrulline was prepared synthetically starting from L-omithine hydrochloride. Into a reactor containing sodium hydroxide (11 kg) in water (170 kg), L-ornithine hydrochloride (44 kg) was added and dissolved. The temperature was maintained at no more than 40°C by active cooling. The ornithine was then converted to its copper complex by the addition of 0.5 molar equivalents of copper sulfate (33 kg) and stirring at ambient temperature for more than 15 minutes. The copper complex protects the 2-amino carboxylic acid functionality of the molecule while chemistry is performed on the terminal amino group. A molar excess of potassium cyanate (32 kg) is then added to the L-ornithine copper complex, and the solution is held at 55°C-65°C for 4.0-4.5 hours, which promotes its conversion to the copper complex of L-citrulline. The resulting copper complex of L-citrulline precipitates during the reaction, and it is collected by filtration.
Example 4. Purification of therapeutic grade citrulline.
[00037] The dry copper: citrulline complex produced in Example 3 is added to a reactor
containing water, which is stirred to resuspend the complex. Concentrated hydrogen chloride solution is added to convert the complex into a solution of copper (II) chloride and citrulline hydrochloride, while the temperature of the reactor is maintained at no more than 45°C by active cooling. Once the contents of the reactor are in solution, sodium hydroxide is added to raise the pH to 7-10, while the temperature is maintained at no more than 40°C. The copper complex of citrulline then precipitates. The precipitate is collected and washed with water until no blue coloration persists in the filtrate.
[00038] The washed precipitate is tested to determine the relative ornithine content. If ornithine is greater than 10 mole%, the precipitate is redissolved and resuspended as described above, until the ornithine content is lowered to not more than 10 mole%.
[00039] Once the precipitate achieves the desired ornithine content, it is resuspended in water in a stirred reactor, and hydrogen sulfide gas is introduced into the suspension to precipitate copper sulfide and dissolve citrulline. The solution is warmed to 30°C ± 2°C to ensure citrulline is fully solubilized, and precipitated copper salts are removed by filtration. The citrulline-containing filtrate is passed thorough micro- and sterile-filtrations and collected in a semi-sterile reactor.
[00040] Activated carbon is used to remove residual impurities, in particular an organic
component and residual copper ions. The pH of the resulting aqueous solution containing L-citrulline and residual copper is adjusted to 5.9 ± 0.2 with sodium hydroxide to form isoelectric citrulline solution. The isoelectric citrulline solution is treated with active carbon granules, preferably by passing the solution through an active carbon adsorber bed, and passed through micro and sterile filters after the active carbon treatment.
[00041] L-citrulline is then precipitated from the aqueous solution by the addition of acetone anti solvent, and the mixture is cooled to near 0°C. Addition of 1.5 to 2 volume equivalents of acetone produce dihydrate crystals of citrulline. The precipitate is collected by filtration. The crystals are dried in a vacuum in a conical dryer at temperature of no more than 45°C to remove acetone and water, resulting in an anhydrous crystalline solid. This solid citrulline corresponds to the orthorhombic d form anhydrous crystals reported by Allouchi, et al., 2014 ( Cryst . Growth Des., 14: 1279-1286).
[00042] Either the dihydrate crystals or the anhydrous crystals may be used therapeutically. The solid or an aqueous solution/suspension may be administered enterally, or the solid may be redissolved for parenteral administration. To produce a final therapeutic product, the non-sterile bulk powder was reconstituted and underwent endotoxin reduction and sterile filtration steps followed by crystallization, drying and micronization in an aseptic environment. The sterile bulk powder was then used as the“raw material” for aseptic filling into glass vials to produce the finished drug product which was reconstituted with a sterile diluent prior to use.
References
^“Citrulline – Compound Summary”. PubChem Compound. USA: National Center for Biotechnology Information. 16 September 2004. Identification. Retrieved 1 May 2012.
^ Rogers, G. E.; Rothnagel, J. A. (1983). “A sensitive assay for the enzyme activity in hair follicles and epidermis that catalyses the peptidyl-arginine-citrulline post-translational modification”. Current Problems in Dermatology. 11: 171–184. doi:10.1159/000408673. ISBN978-3-8055-3752-0. PMID6653155.
^ Crenn, P.; et al. (2000). “Post-absorptive plasma citrulline concentration is a marker of intestinal failure in short bowel syndrome patients”. Gastroenterology. 119 (6): 1496–505. doi:10.1053/gast.2000.20227. PMID11
The U.S. Food and Drug Administration approved Ebanga (Ansuvimab-zykl), a human monoclonal antibody, for the treatment for Zaire ebolavirus (Ebolavirus) infection in adults and children. Ebanga blocks binding of the virus to the cell receptor, preventing its entry into the cell.
Zaire ebolavirus is one of four Ebolavirus species that can cause a potentially fatal human disease. It is transmitted through blood, body fluids, and tissues of infected people or wild animals, and through surfaces and materials, such as bedding and clothing, contaminated with these fluids. Individuals who care for people with the disease, including health care workers who do not use correct infection control precautions, are at the highest risk for infection.
During an Ebola outbreak in the Democratic Republic of the Congo (DRC) in 2018-2019, Ebanga was evaluated in a clinical trial (the PALM trial). The PALM trial was led by the U.S. National Institutes of Health and the DRC’s Institut National de Recherche Biomédicale with contributions from several other international organizations and agencies.
In the PALM trial, the safety and efficacy of Ebanga was evaluated in a multi-center, open-label, randomized controlled trial. 174 participants (120 adults and 54 pediatric patients) with confirmed Ebolavirus infection received Ebanga intravenously as a single 50 mg/kg infusion and 168 participants (135 adults and 33 pediatric patients) received an investigational control. The primary efficacy endpoint was 28-day mortality. The primary analysis population was all patients who were randomized and concurrently eligible to receive either Ebanga or the investigational control during the same time period of the trial. Of the 174 patients who received Ebanga, 35.1% died after 28 days, compared to 49.4% of the 168 patients who received a control.
The most common symptoms experienced while receiving Ebanga include: fever, tachycardia (fast heart rate), diarrhea, vomiting, hypotension (low blood pressure), tachypnea (fast breathing) and chills; however, these are also common symptoms of Ebolavirus infection. Hypersensitivity, including infusion-related events, can occur in patients taking Ebanga, and treatment should be discontinued in the event of a hypersensitivity reaction.
Patients who receive Ebanga should avoid the concurrent administration of a live virus vaccine against Ebolavirus. There is the potential for Ebanga to inhibit replication of a live vaccine virus and possibly reduce the efficacy of this vaccine.
FDA granted the approval to Ridgeback Biotherapeutics, LP.
Ansuvimab, sold under the brand name Ebanga, is a monoclonal antibody medication for the treatment of Zaire ebolavirus (Ebolavirus) infection.[1][2]
The most common symptoms include fever, tachycardia (fast heart rate), diarrhea, vomiting, hypotension (low blood pressure), tachypnea (fast breathing) and chills; however, these are also common symptoms of Ebolavirus infection.[1]
Ansuvimab was approved for medical use in the United States in December 2020.[1][2]
Ansuvimab is a monoclonal antibody therapy that is infused intravenously into patients with Ebola virus disease. Ansuvimab is a neutralizing antibody,[3] meaning it binds to a protein on the surface of Ebola virus that is required to infect cells. Specifically, ansuvimab neutralizes infection by binding to a region of the Ebola virus envelope glycoprotein that, in the absence of ansuvimab, would interact with virus’s cell receptor protein, Niemann-Pick C1 (NPC1).[6][7][8] This “competition” by ansuvimab prevents Ebola virus from binding to NPC1 and “neutralizes” the virus’s ability to infect the targeted cell.[6]
Effector function
Antibodies have antigen-binding fragment (Fab) regions and constant fragment (Fc) regions. The Neutralization of virus infection occurs when the Fab regions of antibodies binds to virus antigen(s) in a manner that blocks infection. Antibodies are also able to “kill” virus particles directly and/or kill infected cells using antibody-mediated “effector functions” such as opsonization, complement-dependent cytotoxicity, antibody-dependent cell-mediated cytotoxicity and antibody-dependent phagocytosis. These effector functions are contained in the Fc region of antibodies, but is also dependent on binding of the Fab region to antigen. Effector functions also require the use of complement proteins in serum or Fc-receptor on cell membranes. Ansuvimab has been found to be capable of killing cells by antibody-dependent cell-mediated cytotoxicity.[3] Other functional killing tests have not been performed.
Ansuvimab has also shown success with lowering the mortality rate from ~70% to about 34%. In August 2019, Congolese health authorities, the World Health Organization, and the U.S. National Institutes of Health promoted the use of ansuvimab, alongside REGN-EB3, a similar Regeneron-produced monoclonal antibody treatment, over other treatments yielding higher mortality rates, after ending clinical trials during the outbreak.[13][14]
In an experiment described in the 2016 paper, rhesus macaques were infected with Ebola virus and treated with a combination of ansuvimab and another antibody isolated from the same subject, mAb100. Three doses of the combination were given once a day starting 1 day after the animals were infected. The control animal died and the treated animals all survived.[3]
Ansuvimab monotherapy
In a second experiment described in the 2016 paper, rhesus macaques were infected with Ebola virus and only treated with ansuvimab. Three doses of ansuvimab were given once a day starting 1 day or 5 days after the animals were infected. The control animals died and the treated animals all survived.[3] Unpublished data referred to in a publication of the 2018 Phase I clinical trial results of ansuvimab, reported that a single infusion of ansuvimab provided full protection of rhesus macaques and was the basis of the dosing used for human studies.[5][4]
Experimental use in the Democratic Republic of Congo
During the 2018 Équateur province Ebola outbreak, ansuvimab was requested by the Democratic Republic of Congo (DRC) Ministry of Public Health. Ansuvimab was approved for compassionate use by the World Health OrganizationMEURI ethical protocol and at DRC ethics board. Ansuvimab was sent along with other therapeutic agents to the outbreak sites.[19][20][11] However, the outbreak came to a conclusion before any therapeutic agents were given to patients.[11]
Approximately one month following the conclusion of the Équateur province outbreak, a distinct outbreak was noted in Kivu in the DRC (2018–20 Kivu Ebola outbreak). Once again, ansuvimab received approval for compassionate use by WHO MEURI and DRC ethic boards and has been given to many patients under these protocols.[11] In November 2018, the Pamoja Tulinde Maisha (PALM [together save lives]) open-label randomized clinical control trial was begun at multiple treatment units testing ansuvimab, REGN-EB3 and remdesivir to ZMapp. Despite the difficulty of running a clinical trial in a conflict zone, investigators have enrolled 681 patients towards their goal of 725. An interim analysis by the Data Safety and Monitoring Board (DSMB) of the first 499 patient found that ansuvimab and REGN-EB3 were superior to the comparator ZMapp. Overall mortality of patients in the ZMapp and remdesivir groups were 49% and 53% compared to 34% and 29% for ansuvimab and REGN-EB3. When looking at patients who arrived early after disease symptoms appeared, survival was 89% for ansuvimab and 94% for REGN-EB3. While the study was not powered to determine whether there is any difference between REGN-EB3 and ansuvimab, the survival difference between those two therapies and ZMapp was significant. This led to the DSMB halting the study and PALM investigators dropping the remdesivir and ZMapp arms from the clinical trial. All patients in the outbreak who elect to participate in the trial will now be given either ansuvimab or REGN-EB3.[21][22][13][12]
On December 21, 2020, the US Food and Drug Administration approved Ebanga (ansuvimab-zykl) for the treatment for Zaire ebolavirus (Ebolavirus) infection in adults and children. Ebanga had been granted US Orphan Drug designation and Breakthrough Therapy designations. Ansuvimab is a human IgG1 monoclonal antibody that binds and neutralizes the virus.
The safety and efficacy of Ebanga were evaluated in the multi-center, open-label, randomized controlled PALM trial. In this study, 174 participants (120 adults and 54 pediatric patients) with confirmed Ebolavirus infection received Ebanga intravenously as a single 50 mg/kg infusion and 168 participants (135 adults and 33 pediatric patients) received an investigational control. The primary efficacy endpoint was 28-day mortality. Of the 174 patients who received Ebanga, 35.1% died after 28 days, compared to 49.4% of the 168 patients who received a control.
Ebanga is the 12th antibody therapeutic to be granted a first approval in the US or EU during 2020.
^ Jump up to:abcdef Clinical trial number NCT03478891 for “Safety and Pharmacokinetics of a Human Monoclonal Antibody, VRC-EBOMAB092-00-AB (MAb114), Administered Intravenously to Healthy Adults” at ClinicalTrials.gov
On November 25, 2020, the Food and Drug Administration granted accelerated approval to naxitamab (DANYELZA, Y-mAbs Therapeutics, Inc.) in combination with granulocyte-macrophage colony-stimulating factor (GM-CSF) for pediatric patients one year of age and older and adult patients with relapsed or refractory high-risk neuroblastoma in the bone or bone marrow demonstrating a partial response, minor response, or stable disease to prior therapy.
Efficacy was evaluated in patients with relapsed or refractory neuroblastoma in the bone or bone marrow enrolled in two single-arm, open-label trials: Study 201 (NCT 03363373) and Study 12-230 (NCT 01757626). Patients with progressive disease following their most recent therapy were excluded. Patients received 3 mg/kg naxitamab administered as an intravenous infusion on days 1, 3, and 5 of each 4-week cycle in combination with GM-CSF subcutaneously at 250 µg/m2/day on days -4 to 0 and at 500 µg/m2/day on days 1 to 5. At the investigator’s discretion, patients were permitted to receive pre-planned radiation to the primary disease site in Study 201 and radiation therapy to non-target bony lesions or soft tissue disease in Study 12-230.
The main efficacy outcome measures were confirmed overall response rate (ORR) per the revised International Neuroblastoma Response Criteria (INRC) and duration of response (DOR). Among 22 patients treated in the multicenter Study 201, the ORR was 45% (95% CI: 24%, 68%) and 30% of responders had a DOR greater or equal to 6 months. Among 38 patients treated in the single-center Study 12-230, the ORR was 34% (95% CI: 20%, 51%) with 23% of patients having a DOR greater or equal to 6 months. For both trials, responses were observed in either the bone, bone marrow or both.
The prescribing information contains a Boxed Warning stating that naxitamab can cause serious infusion-related reactions and neurotoxicity, including severe neuropathic pain, transverse myelitis and reversible posterior leukoencephalopathy syndrome (RPLS). To mitigate these risks, patients should receive premedication prior to each naxitamab infusion and be closely monitored during and for at least two hours following completion of each infusion.
The most common adverse reactions (incidence ≥25% in either trial) in patients receiving naxitamab were infusion-related reactions, pain, tachycardia, vomiting, cough, nausea, diarrhea, decreased appetite, hypertension, fatigue, erythema multiforme, peripheral neuropathy, urticaria, pyrexia, headache, injection site reaction, edema, anxiety, localized edema, and irritability. The most common Grade 3 or 4 laboratory abnormalities (≥5% in either trial) were decreased lymphocytes, decreased neutrophils, decreased hemoglobin, decreased platelet count, decreased potassium, increased alanine aminotransferase, decreased glucose, decreased calcium, decreased albumin, decreased sodium and decreased phosphate.
The recommended naxitamab dose is 3 mg/kg/day (up to 150 mg/day) on days 1, 3, and 5 of each treatment cycle, administered after dilution as an intravenous infusion in combination with GM-CSF, subcutaneously at 250 µg/m2/day on days -4 to 0 and at 500 µg/m2/day on days 1 to 5. Treatment cycles are repeated every 4 to 8 weeks.
This review used the Real-Time Oncology Review (RTOR) pilot program and the Assessment Aid, a voluntary submission from the applicant to facilitate the FDA’s assessment.
This application was granted accelerated approval based on overall response rate and duration of response. Continued approval may be contingent upon verification and description of clinical benefit in confirmatory trials.
Indication:Ovarian cancer; Breast cancer; Non small cell lung cancer (NSCLC)лурбинектединلوربينيكتيدين芦比替定(1R,1’R,2’R,3’R,11’S,12’S,14’R)-5′,12′-Dihydroxy-6,6′-dimethoxy-7′,21′,30′-trimethyl-27′-oxo-2,3,4,9-tetrahydrospiro[β-carboline-1,26′-[17,19,28]trioxa[24]thia[13,30]diazaheptacyclo[12.9.6.13,11. 02,13.04,9.015,23.016,20]triaconta[4,6,8,15,20,22]hexaen]-22′-yl acetate [ACD/IUPAC Name]2CN60TN6ZS497871-47-3[RN]9397
Lurbinectedin is in phase III clinical development for the treatment of platinum refractory/resistant ovarian cancer.
Phase II clinical trials are also ongoing for several oncology indications: non-small cell lung cancer, breast cancer, small cell lung cancer, head and neck carcinoma, neuroendocrine tumors, biliary tract carcinoma, endometrial carcinoma, germ cell tumors and Ewing’s family of tumors.
Lurbinectedin, sold under the brand name Zepzelca, is a medication for the treatment of adults with metastatic small cell lung cancer (SCLC) with disease progression on or after platinum-based chemotherapy.[1][2][3]
The most common side effects include leukopenia, lymphopenia, fatigue, anemia, neutropenia, increased creatinine, increased alanine aminotransferase, increased glucose, thrombocytopenia, nausea, decreased appetite, musculoskeletal pain, decreased albumin, constipation, dyspnea, decreased sodium, increased aspartate aminotransferase, vomiting, cough, decreased magnesium and diarrhea.[1][2][3]
Lurbinectedin is a synthetic tetrahydropyrrolo [4, 3, 2-de]quinolin-8(1H)-one alkaloid analogue with potential antineoplastic activity.[4] Lurbinectedin covalently binds to residues lying in the minor groove of DNA, which may result in delayed progression through S phase, cell cycle arrest in the G2/M phase and cell death.[4]
Lurbinectedin was approved for medical use in the United States in June 2020.[5][1][2][3][6]
Structure
Lurbinectedin is structurally similar to trabectedin, although the tetrahydroisoquinoline present in trabectedin is replaced with a tetrahydro β-carboline which enables lurbinectedin to exhibit increased antitumor activity compared with trabectedin.[7]
Biosynthesis
Lurbinectedin a marine agent isolated from the sea squirt species Ecteinascidia turbinata. Synthetic production is necessary because very small amounts can be obtained from sea organisms. For example, one ton (1000 kg) of sea squirts are required to produce one gram of trabectedin, which is analogue of lurbinectedin. Complex synthesis of lurbinectedin starts from small, common starting materials that require twenty-six individual steps to produce the drug with overall yield of 1.6%.[8][9]
Mechanism of action
According to PharmaMar,[10] lurbinectedin inhibits the active transcription of the encoding genes. This has two consequences. On one hand, it promotes tumor cell death, and on the other it normalizes tumor microenvironment. Active transcription is the process by which there are specific signal where information contained in the DNA sequence is transferred to an RNA molecule. This activity depends on the activity of an enzyme called RNA polymerase II. Lurbinectedin inhibits transcription through a very precise mechanism. Firstly, lurbinectedin binds to specific DNA sequences. It is at these precise spots that slides down the DNA to produce RNA polymerase II that is blocked and degraded by lurbinectedin. Lurbinectedin also has important role in tumor microenvironment. The tumor cells act upon macrophages to avoid them from behaving like an activator of the immune system. Literally, macrophages work in any tumor’s favor. Macrophages can contribute to tumor growth and progression by promoting tumor cell proliferation and invasion, fostering tumor angiogenesis and suppressing antitumor immune cells.[11][12] Attracted to oxygen-starved (hypoxic) and necrotic tumor cells they promote chronic inflammation. So, not only that macrophages inhibit immune system avoiding the destruction of tumor cells, but they also create tumor tissue that allows tumor growth. However, macrophages associated with tumors are cells that are addicted to the transcription process. Lurbinectedin acts specifically on the macrophages associated with tumors in two ways: firstly, by inhibiting the transcription of macrophages that leads to cell death and secondly, inhibiting the production of tumor growth factors. In this way, lurbinectedin normalizes the tumor microenvironment.
History
Lurbinectedin was approved for medical use in the United States in June 2020.[5][1][2][3][6]
Efficacy was demonstrated in the PM1183-B-005-14 trial (Study B-005; NCT02454972), a multicenter open-label, multi-cohort study enrolling 105 participants with metastatic SCLC who had disease progression on or after platinum-based chemotherapy.[3][6] Participants received lurbinectedin 3.2 mg/m2 by intravenous infusion every 21 days until disease progression or unacceptable toxicity.[3] The trial was conducted at 26 sites in the United States, Great Britain, Belgium, France, Italy, Spain and Czech Republic.[6]
The U.S. Food and Drug Administration (FDA) granted the application for lurbinectedin priority review and orphan drug designations and granted the approval of Zepzelca to Pharma Mar S.A.[3][13]
Research
Clinical Trials
Lurbinectedin can be used as monotherapy in the treatment of SCLC. Lurbinectedin monotherapy demonstrated the following clinical results in relapsed extensive stage SCLC:
For sensitive disease (chemotherapy-free interval of ≥ 90 days) overall response rate (ORR) was 46.6% with 79.3% disease control rate and median overall survival (OS) being increased to 15.2 months.[14]
For resistant disease (chemotherapy-free interval of < 90 days) overall response rate (ORR) was 21.3% with 46.8% disease control rate and 5.1 months median overall survival (OS).[14]
Lurbinectedin is also being investigated in combination with doxorubicin as second-line therapy in a randomized Phase III trial.[medical citation needed] While overall survival in this trial is not yet known, response rates at second line were
91.7% in sensitive disease with median progression-free survival of 5.8 months, and
33.3% in resistant disease with median progression-free of 3.5 months.[15]
Lurbinectedin is available in the U.S. under Expanded Access Program (EAP).[15][16]
The ecteinascidins are exceedingly potent antitumour agents isolated from the marine tunicate Ecteinascidia turbinata. Several ecteinascidins have been reported previously in the patent and scientific literature. See, for example:
U.S. Patent No 5.256.663, which describes pharmaceutical compositions comprising matter extracted from the tropical marine invertebrate, Ecteinascidia turbinata, and designated therein as ecteinascidins, and the use of such compositions as antibacterial, antiviral, and/ or antitumour agents in mammals.
U.S. Patent No 5.089.273, which describes novel compositions of matter extracted from the tropical marine invertebrate, Ecteinascidia turbinata, and designated therein as ecteinascidins 729, 743, 745, 759A, 759B and 770. These compounds are useful as antibacterial and/or antitumour agents in mammals.
U.S. Patent No 5.149.804 which describes Ecteinascidins 722 and 736 (Et’s 722 and 736) isolated from the Caribbean tunicate Ecteinascidia turbinata and their structures. Et’s 722 and 736 protect mice in vivo at very low concentrations against P388 lymphoma, B 16 melanoma, and Lewis lung carcinoma.
U.S. Patent No 5.478.932, which describes ecteinascidins isolated from the Caribbean tunicate Ecteinascidia turbinata, which provide in vivo protection against P388 lymphoma, B 16 melanoma, M5076 ovarian sarcoma, Lewis lung carcinoma, and the LX- 1 human lung and MX- 1 human mammary carcinoma xenografts.
U.S. Patent No 5.654.426, which describes several ecteinascidins isolated from the Caribbean tunicate Ecteinascidia turbinata, which provide in vivo protection against P388 lymphoma, B 16 melanoma, M5076 ovarian sarcoma, Lewis lung carcinoma, and the LX-1 human lung and MX- 1 human mammary carcinoma xenografts.
U.S. Patent No 5.721.362 which describes a synthetic process for the formation of ecteinascidin compounds and related structures.
U.S. Patent No 6.124.292 which describes a series of new ecteinascidin- like compounds.
WO 0177115, WO 0187894 and WO 0187895, which describe new synthetic compounds of the ecteinascidin series, their synthesis and biological properties.
See also: Corey, E.J., J. Am. Chem. Soc, 1996, 118 pp. 9202-9203; Rinehart, et al., Journal of Natural Products, 1990, “Bioactive Compounds from Aquatic and Terrestrial Sources”, vol. 53, pp. 771- 792; Rinehart et al., Pure and Appl. Chem., 1990, “Biologically active natural products”, vol 62, pp. 1277- 1280; Rinehart, et al., J. Org. Chem., 1990, “Ecteinascidins 729, 743, 745, 759A, 759B, and 770: potent Antitumour Agents from the Caribbean Tunicate Ecteinascidia tuminata”, vol. 55, pp. 4512-4515; Wright et al., J. Org. Chem., 1990, “Antitumour Tetrahydroisoquinoline Alkaloids from the Colonial ascidian Ecteinascidia turbinata”, vol. 55, pp. 4508-4512; Sakai et al., Proc. Natl. Acad. Sci. USA 1992, “Additional anitumor ecteinascidins from a Caribbean tunicate: Crystal structures and activities in vivo”, vol. 89, 1 1456- 1 1460; Science 1994, “Chemical Prospectors Scour the Seas for Promising Drugs”, vol. 266, pp.1324; Koenig, K.E., “Asymmetric Synthesis”, ed. Morrison, Academic Press, Inc., Orlando, FL, vol. 5, 1985, p. 71; Barton, et al., J. Chem Soc. Perkin Trans., 1 , 1982, “Synthesis and Properties of a Series of Sterically Hindered Guanidine bases”, pp. 2085; Fukuyama et al., J. Am. Chem. Soc, 1982, “Stereocontrolled Total Synthesis of (+)-Saframycin B”, vol. 104, pp. 4957; Fukuyama et al., J. Am. Chem. Soc, 1990, “Total Synthesis of (+) – Saframycin A”, vol. 112, p. 3712; Saito, et al., J. Org. Chem., 1989, “Synthesis of Saframycins. Preparation of a Key tricyclic Lactam Intermediate to Saframycin A”, vol. 54, 5391; Still, et al., J Org. Chem., 1978, “Rapid Chromatographic Technique for Preparative Separations with Moderate Resolution”, vol. 43, p. 2923; Kofron, W.G.; Baclawski, L.M., J. Org. Chem., 1976, vol. 41, 1879; Guan et al., J. Biomolec Struc & Dynam., vol. 10, pp. 793-817 (1993); Shamma et al., “Carbon- 13 NMR Shift Assignments of Amines and Alkaloids”, p. 206 (1979); Lown et al., Biochemistry, 21, 419-428 (1982); Zmijewski et al., Chem. Biol. Interactions, 52, 361-375 (1985); Ito, CRC Crit. Rev. Anal. Chem., 17, 65- 143 (1986); Rinehart et al., “Topics in Pharmaceutical Sciences 1989”, pp. 613-626, D. D. Breimer, D. J. A. Cromwelin, K. K. Midha, Eds., Amsterdam Medical Press B. V., Noordwijk, The Netherlands (1989); Rinehart et al., “Biological Mass Spectrometry”, 233-258 eds. Burlingame et al., Elsevier Amsterdam (1990); Guan et al., Jour. Biomolec. Struct. & Dynam., vol. 10 pp. 793-817 (1993); Nakagawa et al., J. Amer. Chem. Soc, 11 1 : 2721-2722 (1989);; Lichter et al., “Food and Drugs from the Sea Proceedings” (1972), Marine Technology Society, Washington, D.C. 1973, 117- 127; Sakai et al., J. Amer. Chem. Soc, 1996, 1 18, 9017; Garcϊa-Rocha et al., Brit. J. Cancer, 1996, 73: 875-883; and pommier et al., Biochemistry, 1996, 35: 13303- 13309;
In 2000, a hemisynthetic process for the formation of ecteinascidin compounds and related structures such as phthalascidin starting from natural bis(tetrahydroisoquinoline) alkaloids such as the saframycin and safracin antibiotics available from different culture broths was reported; See Manzanares et al., Org. Lett., 2000, “Synthesis of Ecteinascidin ET-743 and Phthalascidin Pt-650 from Cyanosafracin B”, Vol. 2, No 16, pp. 2545-2548; and International Patent Application WO 00 69862.
Ecteinascidin 736 was first discovered by Rinehart and features a tetrahydro-β-carboline unit in place of the tetrahydroisoquinoline unit more usually found in the ecteinascidin compounds isolated from natural sources; See for example Sakai et al., Proc. Natl. Acad. Sci. USA 1992, “Additional antitumor ecteinascidins from a Caribbean tunicate: Crystal structures and activities in vivo”, vol. 89, 11456-11460.
Et-736
WO 9209607 claims ecteinascidin 736, as well as ecteinascidin 722 with hydrogen in place of methyl on the nitrogen common to rings C and D of ecteinascidin 736 and O-methylecteinascidin 736 with methoxy in place of hydroxy on ring C of ecteinascidin 736.
Despite the positive results obtained in clinical applications in chemotherapy, the search in the field of ecteinascidin compounds is still open to the identification of new compounds with optimal features of cytotoxicity and selectivity toward the tumour and with a reduced systemic toxicity and improved pharmacokinetic properties.
Ecteinascidins is a group of naturally occurring marine compounds and analogs thereof, which are well identified and structurally characterized, and are disclosed to have antibacterial and cytotoxic properties. See for example, European Patent 309.477; WO 03/66638; WO 03/08423; WO 01/77115; WO 03/014127; R. Sakai et al., 1992, Proc. Natl. Acad. Sci. USA 89, pages 11456-11460; R. Menchaca et al., 2003, J. Org. Chem. 68(23), pages 8859-8866; and I. Manzanares et al., 2001, Curr. Med. Chem. Anti–Cancer Agents, 1, pages 257-276; and references therein. Examples of ecteinascidins are provided by ET-743, ET-729, ET-745, ET-759A, ET-759B, ET-759C, ET-770, ET-815, ET-731, ET-745B, ET-722, ET-736, ET-738, ET-808, ET-752, ET-594, ET-552, ET-637, ET-652, ET-583, ET-597, ET-596, ET-639, ET-641, and derivatives thereof, such as acetylated forms, formylated forms, methylated forms, and oxide forms.
[0003] The structural characterizations of such ecteinascidins are not given again explicitly herein because from the detailed description provided in such references and citations any person of ordinary skill in this technology is capable of obtaining such information directly from the sources cited here and related sources.
[0004] At least one of the ecteinascidin compounds, ecteinascidin 743 (ET-743), has been extensively studied, and it will be referred to specifically herein to illustrate features of this invention. ET-743 is being employed as an anticancer medicament, under the international nonproprietary name (INN) trabectedin, for the treatment of patients with advanced and metastatic soft tissue sarcoma (STS), after failure of anthracyclines and ifosfamide, or who are unsuited to receive such agents, and for the treatment of relapsed platinum-sensitive ovarian cancer in combination with pegylated liposomal doxorubicin.
[0005] ET-743 has a complex tris(tetrahydroisoquinoline) structure of formula
[0006] It was originally prepared by isolation from extracts of the marine tunicate Ecteinascidia turbinata. The yield was low, and alternative preparative processes had been sought.
[0007] The first synthetic process for producing ecteinascidin compounds was described in U.S. Pat. No. 5,721,362. This process employed sesamol as starting material and yielded ET-743 after a long and complicated sequence of 38 examples each describing one or more steps in the synthetic sequence.
[0008] An improvement in the preparation of one intermediate used in such process was disclosed in U.S. Pat. No. 6,815,544. Even with this improvement, the total synthesis was not suitable for manufacturing ET-743 at an industrial scale.
[0009] A hemisynthetic process for producing ecteinascidin compounds was described in EP 1.185.536. This process employs cyanosafracin B as starting material to provide ET-743. Cyanosafracin B is a pentacyclic antibiotic obtained by fermentation from the bacteria Pseudomonas fluorescens.
[0010] An improvement in such hemisynthetic process was disclosed in EP 1.287.004.
[0011] To date four additional synthetic process (2 total and 2 formal synthesis) have been disclosed in patent applications JP 2003221395, WO 2007/045686, and WO 2007/087220 and in J. Org. Chem. 2008, 73, pages 9594-9600.
[0012] WO 2007/045686 also relates to the synthesis of Ecteinascidins-583 and 597 using intermediate compounds of formula:
[0013] Total synthesis strategies for the synthesis of the pentacyclic core of ET-743 are overviewed in FIG. 1.
PAPER
Angewandte Chemie, International Edition (2019), 58(12), 3972-3975.
An efficient and scalable approach is described for the total synthesis of the marine natural product Et‐743 and its derivative lubinectedin, which are valuable antitumor compounds. The method delivers 1.6 % overall yield in 26 total steps from Cbz‐protected (S)‐tyrosine. It features the use of a common advanced intermediate to create the right and left parts of these compounds, and a light‐mediated remote C−H bond activation to assemble a benzo[1,3]dioxole‐containing intermediate.
“Lurbinectedin”. Drug Information Portal. U.S. National Library of Medicine.
“Lurbinectedin”. NCI Dictionary of Cancer Terms. National Cancer Institute.
Clinical trial number NCT02454972 for “Clinical Trial of Lurbinectedin (PM01183) in Selected Advanced Solid Tumors” at ClinicalTrials.gov
FDA grants accelerated approval to lurbinectedin for metastatic small cell lung cancer
On June 15, 2020, the Food and Drug Administration granted accelerated approval to lurbinectedin(ZEPZELCA, Pharma Mar S.A.) for adult patients with metastatic small cell lung cancer (SCLC) with disease progression on or after platinum-based chemotherapy.
Efficacy was demonstrated in the PM1183-B-005-14 trial (Study B-005; NCT02454972), a multicenter open-label, multi-cohort study enrolling 105 patients with metastatic SCLC who had disease progression on or after platinum-based chemotherapy. Patients received lurbinectedin 3.2 mg/m2 by intravenous infusion every 21 days until disease progression or unacceptable toxicity.
The main efficacy outcome measures were confirmed overall response rate (ORR) determined by investigator assessment using RECIST 1.1 and response duration. Among the 105 patients, the ORR was 35% (95% CI: 26%, 45%), with a median response duration of 5.3 months (95% CI: 4.1, 6.4). The ORR as per independent review committee was 30% (95% CI: 22%, 40%) with a median response duration of 5.1 months (95% CI: 4.9, 6.4).
The most common adverse reactions (≥20%), including laboratory abnormalities, were myelosuppression, fatigue, increased creatinine, increased alanine aminotransferase, increased glucose, nausea, decreased appetite, musculoskeletal pain, decreased albumin, constipation, dyspnea, decreased sodium, increased aspartate aminotransferase, vomiting, cough, decreased magnesium and diarrhea.
The recommended lurbinectedin dose is 3.2 mg/m2 every 21 days.
This indication is approved under accelerated approval based on overall response rate and duration of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in confirmatory trials.
This review was conducted under Project Orbis, an initiative of the FDA Oncology Center of Excellence. Project Orbis provides a framework for concurrent submission and review of oncology drugs among international partners. For this application, a modified Project Orbis was undertaken because of the timing of submission to other regulatory agencies. FDA is collaborating with the Australian Therapeutic Goods Administration (TGA). FDA approved this application 2 months ahead of the goal date. The review is ongoing for the Australian TGA.
1: Calvo E, Moreno V, Flynn M, Holgado E, Olmedo ME, Lopez Criado MP, Kahatt C, Lopez-Vilariño JA, Siguero M, Fernandez-Teruel C, Cullell-Young M, Soto Matos-Pita A, Forster M. Antitumor activity of lurbinectedin (PM01183) and doxorubicin in relapsed small-cell lung cancer: results from a phase I study. Ann Oncol. 2017 Oct 1;28(10):2559-2566. doi: 10.1093/annonc/mdx357. PubMed PMID: 28961837.
2: Erba E, Romano M, Gobbi M, Zucchetti M, Ferrari M, Matteo C, Panini N, Colmegna B, Caratti G, Porcu L, Fruscio R, Perlangeli MV, Mezzanzanica D, Lorusso D, Raspagliesi F, D’Incalci M. Ascites interferes with the activity of lurbinectedin and trabectedin: Potential role of their binding to alpha 1-acid glycoprotein. Biochem Pharmacol. 2017 Nov 15;144:52-62. doi: 10.1016/j.bcp.2017.08.001. Epub 2017 Aug 4. PubMed PMID: 28782526.
3: Belgiovine C, Bello E, Liguori M, Craparotta I, Mannarino L, Paracchini L, Beltrame L, Marchini S, Galmarini CM, Mantovani A, Frapolli R, Allavena P, D’Incalci M. Lurbinectedin reduces tumour-associated macrophages and the inflammatory tumour microenvironment in preclinical models. Br J Cancer. 2017 Aug 22;117(5):628-638. doi: 10.1038/bjc.2017.205. Epub 2017 Jul 6. PubMed PMID: 28683469; PubMed Central PMCID: PMC5572168.
4: Jimeno A, Sharma MR, Szyldergemajn S, Gore L, Geary D, Diamond JR, Fernandez Teruel C, Soto Matos-Pita A, Iglesias JL, Cullell-Young M, Ratain MJ. Phase I study of lurbinectedin, a synthetic tetrahydroisoquinoline that inhibits activated transcription, induces DNA single- and double-strand breaks, on a weekly × 2 every-3-week schedule. Invest New Drugs. 2017 Aug;35(4):471-477. doi: 10.1007/s10637-017-0427-2. Epub 2017 Jan 20. PubMed PMID: 28105566.
5: Paz-Ares L, Forster M, Boni V, Szyldergemajn S, Corral J, Turnbull S, Cubillo A, Teruel CF, Calderero IL, Siguero M, Bohan P, Calvo E. Phase I clinical and pharmacokinetic study of PM01183 (a tetrahydroisoquinoline, Lurbinectedin) in combination with gemcitabine in patients with advanced solid tumors. Invest New Drugs. 2017 Apr;35(2):198-206. doi: 10.1007/s10637-016-0410-3. Epub 2016 Nov 21. PubMed PMID: 27873130.
6: Harlow ML, Maloney N, Roland J, Guillen Navarro MJ, Easton MK, Kitchen-Goosen SM, Boguslawski EA, Madaj ZB, Johnson BK, Bowman MJ, D’Incalci M, Winn ME, Turner L, Hostetter G, Galmarini CM, Aviles PM, Grohar PJ. Lurbinectedin Inactivates the Ewing Sarcoma Oncoprotein EWS-FLI1 by Redistributing It within the Nucleus. Cancer Res. 2016 Nov 15;76(22):6657-6668. doi: 10.1158/0008-5472.CAN-16-0568. Epub 2016 Oct 3. PubMed PMID: 27697767; PubMed Central PMCID: PMC5567825.
7: Céspedes MV, Guillén MJ, López-Casas PP, Sarno F, Gallardo A, Álamo P, Cuevas C, Hidalgo M, Galmarini CM, Allavena P, Avilés P, Mangues R. Lurbinectedin induces depletion of tumor-associated macrophages, an essential component of its in vivo synergism with gemcitabine, in pancreatic adenocarcinoma mouse models. Dis Model Mech. 2016 Dec 1;9(12):1461-1471. Epub 2016 Oct 20. PubMed PMID: 27780828; PubMed Central PMCID: PMC5200894.
8: Santamaría Nuñez G, Robles CM, Giraudon C, Martínez-Leal JF, Compe E, Coin F, Aviles P, Galmarini CM, Egly JM. Lurbinectedin Specifically Triggers the Degradation of Phosphorylated RNA Polymerase II and the Formation of DNA Breaks in Cancer Cells. Mol Cancer Ther. 2016 Oct;15(10):2399-2412. Epub 2016 Sep 14. PubMed PMID: 27630271.
9: Metaxas Y, Cathomas R, Mark M, von Moos R. Combination of cisplatin and lurbinectedin as palliative chemotherapy in progressive malignant pleural mesothelioma: Report of two cases. Lung Cancer. 2016 Dec;102:136-138. doi: 10.1016/j.lungcan.2016.07.012. Epub 2016 Jul 14. PubMed PMID: 27440191.
10: Lima M, Bouzid H, Soares DG, Selle F, Morel C, Galmarini CM, Henriques JA, Larsen AK, Escargueil AE. Dual inhibition of ATR and ATM potentiates the activity of trabectedin and lurbinectedin by perturbing the DNA damage response and homologous recombination repair. Oncotarget. 2016 May 3;7(18):25885-901. doi: 10.18632/oncotarget.8292. PubMed PMID: 27029031; PubMed Central PMCID: PMC5041952.
11: Takahashi R, Mabuchi S, Kawano M, Sasano T, Matsumoto Y, Kuroda H, Kozasa K, Hashimoto K, Sawada K, Kimura T. Preclinical Investigations of PM01183 (Lurbinectedin) as a Single Agent or in Combination with Other Anticancer Agents for Clear Cell Carcinoma of the Ovary. PLoS One. 2016 Mar 17;11(3):e0151050. doi: 10.1371/journal.pone.0151050. eCollection 2016. PubMed PMID: 26986199; PubMed Central PMCID: PMC4795692.
12: Pernice T, Bishop AG, Guillen MJ, Cuevas C, Aviles P. Development of a liquid chromatography/tandem mass spectrometry assay for the quantification of PM01183 (lurbinectedin), a novel antineoplastic agent, in mouse, rat, dog, Cynomolgus monkey and mini-pig plasma. J Pharm Biomed Anal. 2016 May 10;123:37-41. doi: 10.1016/j.jpba.2016.01.043. Epub 2016 Jan 21. PubMed PMID: 26871278.
13: Elez ME, Tabernero J, Geary D, Macarulla T, Kang SP, Kahatt C, Pita AS, Teruel CF, Siguero M, Cullell-Young M, Szyldergemajn S, Ratain MJ. First-in-human phase I study of Lurbinectedin (PM01183) in patients with advanced solid tumors. Clin Cancer Res. 2014 Apr 15;20(8):2205-14. doi: 10.1158/1078-0432.CCR-13-1880. Epub 2014 Feb 21. PubMed PMID: 24563480.
14: Romano M, Frapolli R, Zangarini M, Bello E, Porcu L, Galmarini CM, García-Fernández LF, Cuevas C, Allavena P, Erba E, D’Incalci M. Comparison of in vitro and in vivo biological effects of trabectedin, lurbinectedin (PM01183) and Zalypsis® (PM00104). Int J Cancer. 2013 Nov;133(9):2024-33. doi: 10.1002/ijc.28213. Epub 2013 May 25. PubMed PMID: 23588839.
15: Vidal A, Muñoz C, Guillén MJ, Moretó J, Puertas S, Martínez-Iniesta M, Figueras A, Padullés L, García-Rodriguez FJ, Berdiel-Acer M, Pujana MA, Salazar R, Gil-Martin M, Martí L, Ponce J, Molleví DG, Capella G, Condom E, Viñals F, Huertas D, Cuevas C, Esteller M, Avilés P, Villanueva A. Lurbinectedin (PM01183), a new DNA minor groove binder, inhibits growth of orthotopic primary graft of cisplatin-resistant epithelial ovarian cancer. Clin Cancer Res. 2012 Oct 1;18(19):5399-411. doi: 10.1158/1078-0432.CCR-12-1513. Epub 2012 Aug 15. PubMed PMID: 22896654.
Launched (Metastatic non small cell lung cancer – China – May-2018)
Orphan Drug; Priority Review
MOA:VEGFR inhibitor
Indication:advanced gastric adenocarcinoma; Advanced renal cell carcinoma (RCC); Medullary thyroid cancer (MTC); Metastatic colorectal cancer (CRC); Non small cell lung cancer (NSCLC); Soft tissue sarcoma; Ovarian cancerStatus:Phase III (Active)
Anlotinib (AL3818) is a highly potent and selective VEGFR2 inhibitor with IC50 less than 1 nM. It has broad-spectrum antitumor potential in clinical trials.
Anlotinib dihydrochloride is in phase II/III clinical trials for the treatment of metastatic colorectal cancer and advanced gastric adenocarcinoma. The compound was co-developed by CTTQ Pharmaceutical (正大天晴) and Advenchen Laboratory.
It is also in phase II clinical trials for the treatment of ovarian cancer, endometrial cancer, non small cell lung cancer (NSCLC), medullary thyroid cancer (MTC), soft tissue sarcoma and advanced renal cell carcinoma (RCC).
In 2015, orphan drug designation was received in the U.S. for the treatment of ovarian cancer.
new process to synthesize l-((4-(4-Fluoro-2-methyl- lH- indol-5-yloxy)-6-methoxyquinolin-7-yloxy)methyl)cyclopropanamine (AL3818) by condensing intermediate (XI) with (Yl) in a solvent at the presence of KI or Nal, or intermediate (X2) with (Y2) in a solvent to form intermediate (Z) which is deprotected to give the final compound (AL3818) in Scheme I. A stable crystalline form of l-((4-(4-Fluoro-2 -methyl- lH-indol-5-yloxy)-6- methoxyquinolin-7-yloxy)-methyl)cyclopropanamine and its salts as well as crystalline forms of salts have also been prepared.
Wherein, R is selected from H and Ci-Cealkoxy.
Process A
R is selected from H and C1 -C6 alkoxy
The final compound (AL3818) was prepared according to Process Al when R is H by deprotecting intermediate (Z-l) with HCOONH4 (ammonium formate) and Pd/C in an alcoholic solvent, such as MeOH, at 25°C-80°C for 0.1-4 hours. (Z-l) was prepared by reacting intermediate (XI) with (Yl-1) at the presence of KI or Nal with K2CO3 in a solvent, such as acetone or DMF, at a temperature of 60°C-160°C for 2-24 hours.
Process Al (R=H)
The final compound (AL3818) was prepared according to Process A2 when R is 4-OMe by deprotecting intermediate (Z-2) with TFA in DCM at 0°C-30°C for 1-24 hours. (Z-2) was prepared by reacting intermediate (XI) with (Y 1-2) at the presence of KI or Nal with K2C03 in a solvent, such as acetone or DMF, at a temperature of 60°C -160°C for 2-24 hours.
Process A2 (R=4-OMe)
The present invention relates a new process to synthesize l-((4-(4-Fluoro-2 -methyl- 1H- indol-5-yloxy)-6-methoxyquinolin-7-yloxy)methyl)cyclopropanamine (AL3818) by reacting intermediate (X2) with (Y2) in a solvent to form intermediate (Z) which is deprotected to give the final compound (AL3818) according to Process B. Proce B
R is selected from H and C1-C6 alkoxy
The final compound (AL3818) was prepared according to Process Bl when R is H by deprotecting intermediate (Z-1) with HCOONH4 (ammonium formate) and Pd/C in an alcoholic solvent, such as MeOH, at 25°C-80°C for 0.1-4 hours. (Z-1) was prepared by reacting intermediate (X2-1) with (Y2) in a solvent, such as pyridine or lutidine, at a temperature of 60°C – 160°C for 1-12 hours.
Process Bl R=H)
The final compound (AL3818) was prepared according to Process B2 when R is 4-OMe by deprotecting intermediate (Z-2) with TFA in DCM at 0°C-30°C for 1-24 hours. (Z-2) was prepared by reacting intermediate (X2-2) with (Y2) in a solvent, such as pyridine or lutidine, at a temperature of 60°C -160°C for 1-12 hours.
Process B2 (R=4-OMe)
The following examples further illustrate the present invention, but should not be construed as in any way to limit its scope.
Example 1
Representation of Process A, Process Al
Process for preparation of l-((4-(4-Fluoro-2 -methyl- lH-indol-5-yloxy)-6-methoxy- quinolin-7-yloxy)methyl)cyclopropanamine (AL3818)
To a stirred mixture of benzyl l-(hydroxymethyl)cyclopropylcarbamate (50 g) and DCM (200 ml) was added DIPEA (39g). The result solution was cooled to 0-5 °C with ice/water and further stirred under this temperature for 15 min. MsCl (30g) was added via an addition funnel dropwise keeping temperature below 5°C for about 1.5 hours. After completion of addition, the reaction mixture was allowed stirring at 0-5°C for 30 min and quenched with saturated NaHC03 (150 ml). The solution was extracted with 150 ml DCM twice. The combined DCM layer was washed with 0.1 N HCl (400 ml) followed by brine. It was dried over Na2S04 and concentrated to obtain an off-white solid 60 gram as (l-(benzyloxycarbonylamino)cyclopropyl)methyl methanesulfonate (Yl-1), MS: (M+l) 300.
To a stirred mixture of (Yl-1) (16 g), XI [(4-(4-fluoro-2-methyl-lH-indol-5-yloxy)-6- methoxy-7-hydroxyquinoline, 12 g] , K2CO3 (21 g) and KI (21 g) was added DMF (100 ml), the reaction suspension was heated at 80°C for 10 hours and (Yl-l) (10 g) was added to continuously heated 80°C for 10 hours. The reaction then was quenched with water (150 ml) and extracted with 150 ml DCM twice. The combined DCM layer was washed with 2 N NaOH (100 ml) followed by water and brine. It was dried over Na2SC>4 and concentrated, further recrystallized from EtOH to obtain a yellow solid as benzyl l-((4-(4-fluoro-2-methyl-lH-indol-5-yloxy)-6-methoxyquinolin- 7-yloxy)methyl)cyclopropylcarbamate (Z-l) 9.5 g. MS: (M+l) 542.
To a stirred mixture of (Z-l) (9.5 g), HCOONH4 (4.7 g) and Pd/C (10%, wet 50%, 4.7g) was added MeOH, the reaction mixture was heated at 45°C for 1.5 hours. It was then cooled and filtered through Celite, further evaporated. 2N HCl (200 ml) was added and extracted with DCM/MeOH (10/1, 100 ml) twice. The aqueous layer was basified with 3N NaOH to adjust pH 11-12 to generate a solid precipitation. The solid was filtered and washed with water to neutral, further suction dry. The solid was dissolved into a mixture of DCM/MeOH (250 ml, 10/1) and further washed with water and brine. It was dried with MgS04 and filtered, further evaporated to give a light yellow solid 5.5 g crude product. Further purification was conducted by dissolving the crude product into DCM/MeOH (40 ml, 10/1) to triturate with petroleum ether (40 ml) for 2 hours slow stirring. The precipitate was filtered and dried in an oven to give the final crystalline product 4.4 g (MP: 203-208 C) and it can be further purified by recrystallizing from EtOH to give purer final product as a same crystalline form. MS: (M+l) 408; ¾ NMR(DMSO-dg) δ 0.60- 0.63(d, 4H), 2.41(s, 1H), 2.42-2.5 l(t, 2H), 3.3 l(s, 2H), 3.96(s, 3H), 4.04(s, 2H), 6.27(s, 1H), 6.31-6.32(m, 1H), 6.97-7.02(t, 1H), 7.20-7.22(d, 1H), 7.36(s, 1H), 7.60(s, 1H), 8.40-8.42(d, 1H), 1 1.41(s, 1H). MP: 208-210°C; DSC Melting Range (Endo): 207-220°C with Peak Temp=216°CPATENTWO 2019154273https://patentscope.wipo.int/search/en/detail.jsf;jsessionid=11C1DF5485B11ADA40E45C9488AB5679.wapp1nB?docId=WO2019154273&tab=FULLTEXT Tyrosine kinases are a group of enzymes that catalyze the phosphorylation of protein tyrosine residues. They play an important role in intracellular signal transduction. They are involved in the regulation, signal transmission and development of normal cells, and are also related to tumor cells. Proliferation, differentiation, migration and apoptosis are closely related. Many receptor tyrosine kinases are related to the formation of tumors, and can be divided into epidermal growth factor receptor (EGFR), platelet-derived growth factor receptor (PDGFR), and vascular endothelial cell growth factor receptor according to the structure of their extracellular region. Body (VEGFR), Fibroblast Growth Factor Receptor (FGFR), etc.[0003]WO2008112407 discloses the compound 1-((4-(4-fluoro-2-methyl-1H-indol-5-yloxy)-6-methoxyquinolin-7-yloxy in Example 24 )Methyl)cyclopropylamine and its preparation method, its structural formula is shown in formula I:[0004]
[0005]It is a multi-target receptor tyrosine kinase inhibitor that can inhibit the activity of vascular endothelial cell growth factor receptors (VEGFR1, VEGFR2/KDR and VEGFR3), stem cell factor receptors, platelet-derived growth factor receptors and other kinase activities. Inhibit the downstream signal transduction mediated by VEGFR2, thereby inhibiting tumor angiogenesis.[0006]Solid drugs generally have multiple crystal forms, such as polymorphs, solvates (hydrates), salts, and co-crystals. The change in the crystal form of the same drug usually results in different melting points, solubility, stability, biological activity, etc., which are important factors that affect the difficulty of drug preparation, storage stability, preparation difficulty, and bioavailability. . When the compound has multiple crystal forms, due to the specific thermodynamic properties and stability of the specific crystal form of the drug, it is important to understand the crystal form of the compound used in each dosage form during the preparation process to ensure the production process Use the same form of medicine. Therefore, it is necessary to ensure that the compound is a single crystal form or a known mixture of some crystal forms.[0007]WO2016179123 discloses the crystalline form 1 of the free base anhydrate of the compound of formula I and a preparation method thereof. CN201010245688.1 discloses the anhydrate and dihydrate crystals of quinoline derivative dihydrochloride and the preparation method thereof.[0008]The discovery of a variety of new crystal forms of medicinal compounds provides an opportunity to improve the physical properties of the drug, that is, to expand all the properties of the substance, which can better guide the research of the compound and its preparation. Therefore, the quinoline derivative provided in this application The crystals and pharmaceutical compositions containing the crystals have commercial value in the manufacture of medicines and other applications.Example 1 1-((4-(4-Fluoro-2-methyl-1H-indol-5-yloxy)-6-methoxyquinolin-7-yloxy)methyl)cyclopropylamine (Formula I compound) preparation[0081]
[0082]Put intermediate 1 (its chemical name is (1-((4-(4-fluoro-2-methyl-1H-indol-5-yl)oxy-6-methoxy Quinolin-7-yl)oxy)methyl)cyclopropyl)benzyl carbamate) 100g, 10% palladium on carbon 30g, ammonium formate 50g and methanol 800ml. Incubate the reaction at 45-55°C, TLC tracking showed that the reaction was complete, filtered, the filter cake was washed with a small amount of methanol, the filtrate was concentrated to dryness under reduced pressure, ethyl acetate and 2mol/L hydrochloric acid were added, stirred for 10 minutes, and then stood for 10 minutes. Separate the aqueous phase, adjust the pH to above 12 with 4N sodium hydroxide, and a large amount of solids will precipitate out. After washing with water until neutral, the aqueous phase is filtered to obtain the crude product of the title compound.[0083]Example 2 Preparation of amorphous compound of formula I[0084]According to the preparation method disclosed in Example 24 of WO2008112407, 1-((4-(4-fluoro-2-methyl-1H-indol-5-yloxy)-6-methoxyquinolin-7-yl (Oxy)methyl)cyclopropylamine is composed of (1-(((4-(4-fluoro-2-methyl-1H-indol-5-yl)oxy-6-methoxyquinolin-7-yl )Oxy)methyl)cyclopropyl)benzyl carbamate (Intermediate 1) was prepared according to the following methods 2.1 and 2.2.[0085]2.1 Take 100 mg of Intermediate 1 and Pd/C (10%, 40 mg) into ethanol (20 ml), and hydrogenate at 50 psi for 12 hours. The reaction solution was filtered with diatomaceous earth, and evaporated to obtain an amorphous compound of formula I, and its X-ray powder diffraction (XRD) pattern was obtained as shown in FIG. 11.[0086] 2.2 Take 100 mg of Intermediate 1, acetic acid (1ml) and 33% hydrobromic acid/acetic acid (1ml) and mix. The reaction was stirred for 1 hour at room temperature, diluted with ethyl acetate/water, and then basified with sodium carbonate. The organic layer is dried, concentrated, and purified by silica gel column to obtain the amorphous compound of formula I.PATENTUS 20160326138https://patents.google.com/patent/US20160326138A1/enNew process has been outlined in Scheme I.
The present invention relates a new process to synthesize 1-((4-(4-Fluoro-2-methyl-1H-indol-5-yloxy)-6-methoxyquinolin-7-yloxy)methyl)cyclopropanamine (AL3818) by condensing intermediate (X1) with (Y1) in a solvent at the presence of KI or NaI to form intermediate (Z) which is deprotected to give the final compound (AL3818) according to Process A.
[0040] The final compound (AL3818) was prepared according to Process A1 when R is H by deprotecting intermediate (Z-1) with HCOONH4 (ammonium formate) and Pd/C in an alcoholic solvent, such as MeOH, at 25° C.-80° C. for 0.1-4 hours. (Z-1) was prepared by reacting intermediate (X1) with (Y1-1) at the presence of KI or NaI with K2CO3 in a solvent, such as acetone or DMF, at a temperature of 60° C.-160° C. for 2-24 hours.
[0041] The final compound (AL3818) was prepared according to Process A2 when R is 4-OMe by deprotecting intermediate (Z-2) with TFA in DCM at 0° C.-30° C. for 1-24 hours. (Z-2) was prepared by reacting intermediate (X1) with (Y1-2) at the presence of KI or NaI with K2CO3 in a solvent, such as acetone or DMF, at a temperature of 60° C.-160° C. for 2-24 hours.
[0042] The present invention relates a new process to synthesize 1-((4-(4-Fluoro-2-methyl-1H-indol-5-yloxy)-6-methoxyquinolin-7-yloxy)methyl)cyclopropanamine (AL3818) by reacting intermediate (X2) with (Y2) in a solvent to form intermediate (Z) which is deprotected to give the final compound (AL3818) according to Process B.
[0043] The final compound (AL3818) was prepared according to Process B1 when R is H by deprotecting intermediate (Z-1) with HCOONH4 (ammonium formate) and Pd/C in an alcoholic solvent, such as MeOH, at 25° C.-80° C. for 0.1-4 hours. (Z-1) was prepared by reacting intermediate (X2-1) with (Y2) in a solvent, such as pyridine or lutidine, at a temperature of 60° C.-160° C. for 1-12 hours.
[0044] The final compound (AL3818) was prepared according to Process B2 when R is 4-OMe by deprotecting intermediate (Z-2) with TFA in DCM at 0° C.-30° C. for 1-24 hours. (Z-2) was prepared by reacting intermediate (X2-2) with (Y2) in a solvent, such as pyridine or lutidine, at a temperature of 60° C.-160° C. for 1-12 hours.
[0045] The following examples further illustrate the present invention, but should not be construed as in any way to limit its scope.
Example 1Representation of Process A, Process A1Process for preparation of 1-((4-(4-Fluoro-2-methyl-1H-indol-5-yloxy)-6-methoxy-quinolin-7-yloxy)methyl)cyclopropanamine (AL3818)
[0046] To a stirred mixture of benzyl 1-(hydroxymethyl)cyclopropylcarbamate (50 g) and DCM (200 ml) was added DIPEA (39 g). The result solution was cooled to 0-5° C. with ice/water and further stirred under this temperature for 15 min. MsCl (30 g) was added via an addition funnel dropwise keeping temperature below 5° C. for about 1.5 hours. After completion of addition, the reaction mixture was allowed stirring at 0-5° C. for 30 min and quenched with saturated NaHCO3 (150 ml). The solution was extracted with 150 ml DCM twice. The combined DCM layer was washed with 0.1 N HCl (400 ml) followed by brine. It was dried over Na2SO4 and concentrated to obtain an off-white solid 60 gram as (1-(benzyloxycarbonylamino)cyclopropyl)methyl methanesulfonate (Y1-1), MS: (M+1) 300.
[0047] To a stirred mixture of (Y1-1) (16 g), X1 [(4-(4-fluoro-2-methyl-1H-indol-5-yloxy)-6-methoxy-7-hydroxyquinoline, 12 g], K2CO3 (21 g) and KI (21 g) was added DMF (100 ml), the reaction suspension was heated at 80° C. for 10 hours and (Y1-1) (10 g) was added to continuously heated 80° C. for 10 hours. The reaction then was quenched with water (150 ml) and extracted with 150 ml DCM twice. The combined DCM layer was washed with 2 N NaOH (100 ml) followed by water and brine. It was dried over Na2SO4 and concentrated, further recrystallized from EtOH to obtain a yellow solid as benzyl 1-((4-(4-fluoro-2-methyl-1H-indol-5-yloxy)-6-methoxyquinolin-7-yloxy)methyl)cyclopropylcarbamate (Z-1) 9.5 g. MS: (M+1) 542.
[0048] To a stirred mixture of (Z-1) (9.5 g), HCOONH4 (4.7 g) and Pd/C (10%, wet 50%, 4.7 g) was added MeOH, the reaction mixture was heated at 45° C. for 1.5 hours. It was then cooled and filtered through Celite, further evaporated. 2N HCl (200 ml) was added and extracted with DCM/MeOH (10/1, 100 ml) twice. The aqueous layer was basified with 3N NaOH to adjust pH 11-12 to generate a solid precipitation. The solid was filtered and washed with water to neutral, further suction dry. The solid was dissolved into a mixture of DCM/MeOH (250 ml, 10/1) and further washed with water and brine. It was dried with MgSO4 and filtered, further evaporated to give a light yellow solid 5.5 g crude product. Further purification was conducted by dissolving the crude product into DCM/MeOH (40 ml, 10/1) to triturate with petroleum ether (40 ml) for 2 hours slow stirring. The precipitate was filtered and dried in an oven to give the final crystalline product 4.4 g (MP: 203-208° C.) and it can be further purified by recrystallizing from EtOH to give purer final product as a same crystalline form. MS: (M+1) 408; 1H NMR (DMSO-d6) δ 0.60-0.63 (d, 4H), 2.41 (s, 1H), 2.42-2.51 (t, 2H), 3.31 (s, 2H), 3.96 (s, 3H), 4.04 (s, 2H), 6.27 (s, 1H), 6.31-6.32 (m, 1H), 6.97-7.02 (t, 1H), 7.20-7.22 (d, 1H), 7.36 (s, 1H), 7.60 (s, 1H), 8.40-8.42 (d, 1H), 11.41 (s, 1H). MP: 208-210° C.; DSC Melting Range (Endo): 207-220° C. with Peak Temp=216° C. TGA demonstrating as an unsolvated material with weight loss at about 210° C. (between 205-215° C.). XRPD having pattern comprising characteristic 10 peaks with intensity % greater than 10% expressed in d values and angles as follows:
[0049] It was similar prepared according to the preparation procedures of (Z-1) described in Example 1 by using 4-methoxybenzyl 1-(hydroxymethyl)cyclopropylcarbamate to first generate (1-((4-methoxybenzyloxy)carbonylamino)cyclopropyl)methyl methanesulfonate (Y1-2) then to give 4-methoxybenzyl 1-((4-(4-fluoro-2-methyl-1H-indol-5-yloxy)-6-methoxyquinolin-7-yloxy)-methyl)cyclopropylcarbamate (Z-2), MS: (M+1) 572
[0050] To a stirred mixture of (Z-2) (1.5 g) in DCM (15 ml) at 0° C. was added TFA (1.5 ml) for about 30 min and warmed up to RT. The reaction was stirred at RT for 2 hours and added into water (30 ml). The aqueous layer was extracted with DCM twice (100 ml×2) and basified with 2N NaOH to adjust pH 11-12. The mixture was extracted with DCM (100 ml×3) and further washed with brine (100 ml). It was dried with MgSO4 and filtered. The solution was evaporated to give 1.05 g crude final product. Further purification was conducted to dissolve the crude product into DCM/MeOH and triturated with petroleum ether and dried in an oven to give the final pure product 0.8 g AL3818 with the same crystalline form.
Example 3Representation of Process A, Process B1Process for preparation of 1-((4-(4-Fluoro-2-methyl-1H-indol-5-yloxy)-6-methoxy-quinolin-7-yloxy)methyl)cyclopropanamine (AL3818)
[0051] To a mixture of benzyl 1-((4-chloro-6-methoxyquinolin-7-yloxy)methyl)cyclopropyl-carbamate (X2-1) (5 g), 4-fluoro-2-methyl-1H-indol-5-ol (Y2) (5 g) and DMAP (4 g) was added 1,6-lutidine (15 ml). The reaction was stirred and heated at 135° C. for 5 hours and was cooled followed by adding IPA with slow stirring for 2 hours at RT. The solid was filtered and further washed with IPA, dried to give (Z-1) 5.2 g as a solid. It was then similarly prepared according to deprotection procedures described of (Z-1) in Example 1 to give the final compound AL3818 with the same crystalline form.
Example 4Representation of Process A, Process B2Process for preparation of 1-((4-(4-Fluoro-2-methyl-1H-indol-5-yloxy)-6-methoxy-quinolin-7-yloxy)methyl)cyclopropanamine (AL3818)
[0052] (Z-2) was similarly prepared according to the procedures described in Example 3 by using 4-methoxybenzyl 1-((4-chloro-6-methoxyquinolin-7-yloxy)methyl)cyclopropylcarbamate (X2-2) and (Y2). It was then similarly prepared according to deprotection procedures of (Z-2) described in Example 2 to give the final compound AL3818 with the same crystalline form.
Example 5
[0053] Preparation of 1-((4-(4-Fluoro-2-methyl-1H-indol-5-yloxy)-6-methoxy-quinolin-7-yloxy)-methyl)cyclopropanamine bishydrochloride acid salt and its crystalline
[0054] To a 25 ml flask was added 250 mg free base (AL3818), 4N HCl in dioxane 0.625 mL (2.5 mmol, 4 eq.) in 10 ml EtOH, the reaction was heated at 75° C. for 30 minutes, cooled to RT and stirred for O.N. The solid was filtered and rinsed with acetone twice. It was dried in oven at 50° C. for 4 hours to give 126 mg white solid as the bishydrochloride salt as a crystalline and further recrystallized from EtOH to give a purer product as a same crystalline form. 1H NMR (DMSO-d6) δ 1.09-1.24 (m, 4H), 2.43 (s, 3H), 4.08 (s, 3H), 4.40 (s, 2H), 6.32 (s, 1H), 6.76 (s, 1H), 7.05-7.11 (t, 1H), 7.27-7.30 (d, 1H), 7.65 (s, 1H), 7.82 (s, 1H), 8.64 (s, 2H), 8.70-8.73 (m, 1H), 11.51 (s, 1H). Chloride ion chromatography showed 2 molecular ratio ions (16.1%). DSC Melting Range (Exo): 249-280 with Peak Temp=268° C.
[0055] To a 10 mL flask, charged 140 mg of 3818-2HCl salt from above Example 4 and 0.7 mL (×5 with salt volume) of 80% MeOH in H2O. The result suspension was heated to 70° C. to form a solution and cooled to RT and further stirred for O.N. The solid was filtered and rinsed with acetone twice. It was dried in oven at 50° C. for 4 hours to obtain off-white solid 110 mg as the crystalline bishydrochloride hydrate salt. 1H NMR (DMSO-d6) δ 1.09 (s, 2H), 1.22 (s, 2H), 2.44 (s, 1H), 2.52 (s, 2H), 4.09 (s, 3H), 4.44 (s, 2H), 6.32 (s, 1H), 6.81-6.82 (d, 1H), 7.08-7.14 (t, 1H), 7.29-7.32 (d, 1H), 7.79 (s, 1H), 7.85 (s, 1H), 8.75-8.78 (d, 1H), 8.85 (s, 2H), 11.66 (s. 1H). Chloride ion chromatography showed 2 molecular ratio ions (17.8%). DSC Melting Range (Exo): 207-260° C. with Peak Temp=226° C. TGA demonstrating 2.68% (˜3%, 1 water) weight loss till 120° C. (between 115-125° C.) and further weight loss at about 170° C. (between 165-175° C.).
Vildagliptin was approved by the European Medicines Agency (EMA) on Sep 26, 2007, and approved by Pharmaceuticals and Medical Devices Agency of Japan (PMDA) on Jan 20, 2010, following by China Food and Drug Administration (CFDA) on Aug 15, 2011. It was developed and marketed as Galvus® by Novartis in EU.
Vildagliptin is a potent selective inhibitor of dipeptidyl peptidase-4 (DPP-4) that improves glycaemic control by increasing islet α-cell and β-cell responsiveness to glucose. It is used to reduce hyperglycemia in type 2 diabete.
Galvus®is available as film-coated tablet for oral use, containing 50 mg free Vildagliptin. The recommended dose of vildagliptin is 100 mg, administered as one dose of 50 mg in the morning and one dose of 50 mg in the evening.Drug Name:VildagliptinResearch Code:LAF-237; DSP-7238; NVP-LAF-237Trade Name:Galvus® / Jalra® / Xiliarx® / Equa®MOA:Dipeptidyl peptidase-4 (DPP-4) inhibitorIndication:Type 2 diabetesStatus:ApprovedCompany:Novartis (Originator)Sales:$1,140 Million (Y2015); $1,224 Million (Y2014); $1,200 Million (Y2013); $910 Million (Y2012); $677 Million (Y2011);ATC Code:A10BH02
Vildagliptin, previously identified as LAF237, is a new oral anti-hyperglycemic agent (anti-diabetic drug) of the new dipeptidyl peptidase-4 (DPP-4) inhibitor class of drugs. Vidagliptin subsequently acts by inhibiting the inactivation of glucagon-like peptide-1 (GLP-1) and gastric inhibitory polypeptide (GIP) by DPP-4. This inhibitory activity ultimately results in a two-fold action where GLP-1 and GIP are present to potentiate the secretion of insulin by beta cells and suppress glucagon secretion by alpha cells in the islets of Langerhans in the pancreas. It is currently in clinical trials in the U.S. and has been shown to reduce hyperglycemia in type 2 diabetes mellitus. While the drug is still not approved for use in the US, it was approved in Feb 2008 by European Medicines Agency for use within the EU and is listed on the Australian PBS with certain restrictions.
Vildagliptin, sold under the brand name Galvus among others, is an oral anti-hyperglycemic agent (anti-diabetic drug) of the dipeptidyl peptidase-4 (DPP-4) inhibitor class of drugs. Vildagliptin inhibits the inactivation of GLP-1[2][3] and GIP[3] by DPP-4, allowing GLP-1 and GIP to potentiate the secretion of insulin in the beta cells and suppress glucagon release by the alpha cells of the islets of Langerhans in the pancreas.
Adverse effects observed in clinical trials include nausea, hypoglycemia, tremor, headache and dizziness. Rare cases of hepatoxicity have been reported.[5]
There have been case reports of pancreatitis associated with DPP-4 inhibitors. A group at UCLA reported increased pre-cancerous pancreatic changes in rats and in human organ donors who had been treated with DPP-4 inhibitors.[6][7] In response to these reports, the United States FDA and the European Medicines Agency each undertook independent reviews of all clinical and preclinical data related to the possible association of DPP-4 inhibitors with pancreatic cancer. In a joint letter to the New England Journal of Medicines, the agencies stated that “Both agencies agree that assertions concerning a causal association between incretin-based drugs and pancreatitis or pancreatic cancer, as expressed recently in the scientific literature and in the media, are inconsistent with the current data. The FDA and the EMA have not reached a final conclusion at this time regarding such a causal relationship. Although the totality of the data that have been reviewed provides reassurance, pancreatitis will continue to be considered a risk associated with these drugs until more data are available; both agencies continue to investigate this safety signal.”[8]
Vildagliptin is an active pharmaceutical substance with an empirical formula of C17H25N3O2 and a molecular weight of 303.40 g/mol. Vildagliptin is the international common accepted name for (2S)-1-[[(3-hydroxytricyclo[3.3.1.13,7]dec-1-yl)amino]acetyl]-2-pyrrolidine carbonitrile and has the structure of formula (I).
[0003]Vildagliptin is a dipeptidyl peptidase IV (DPP-IV) inhibitor and is disclosed in U.S. Pat. No. 6,166,063 (“the ‘063 patent”), the disclosure of which is incorporated herein by reference. The ‘063 patent discloses a synthesis of vildagliptin using the synthetic process represented in Scheme 1.
[0004]Vildagliptin can exist as the (2S) and (2R) enantiomers. The stereoisomer with the desired biological activity is the (2S) enantiomer. Accordingly, it is desirable to synthesize (2S)-vildagliptin with high stereochemical purity. A process that yields vildagliptin with a high enantiomeric purity is disclosed in International Patent Publication WO 2004/092127, the disclosure of which is incorporated herein by reference. This reference discloses compositions containing from 95% to 99.99% of (2S)-vildagliptin.
[0069]This example illustrates the synthesis of the compound of formula (I) in accordance with embodiments of the invention.
[0070]Into a 100 mL rounded reaction vessel were charged 3 g (17.37 mmol) of 1-chloroacetyl-2-cyanopyrrolidine, 3.22 g (19.82 mmol) of 1-amino-3-adamantanol, 2.78 g (20.1 mmol) of potassium carbonate, and 30 mL isopropyl acetate. The mixture was refluxed for 4 h, cooled to room temperature, and the salts were filtered and washed with acetonitrile. The mother liquors were evaporated to dryness to obtain an oil which was aged in MEK from which a white solid crystallizes at 0-5° C. The solid was filtered washing the cake with MEK and dried at 40° C. in a vacuum oven until constant weight.
[0071]Yield: 36%. Assay: 99.21%. HPLC purity: 97.55% of vildagliptin (measured according to Example 2). HPLC chiral purity: more than 99.99% of vildagliptin (measured according to Example 7).
[0072]These results demonstrate that a compound of formula (I) comprising less than 0.01% of (2R)-1-[N-(3-hydroxytricyclo[3.3.1.13,7]dec-1-yl)glycyl]-2-pyrrolidinecarbonitrile (i.e., (2R)-vildagliptin).
Vildagliptin is chemically known as (S)-l-[2-(3-Hydroxyadamantan-l-ylamino) acetyl] pyrrolidine-2-carbonitrile and exist as (2S) and (2R) enantiomers. The stereoisomer with the desired biological activity is the (2S) enantiomer, represented by the following structure:
U.S. Patent No. 6,166,063 (“the Ό63 patent”) discloses new class of Dipeptidyl peptidase 4 (DPP-4) inhibitors such as vildagliptin. The ‘063 patent further discloses a process for the preparation of vildagliptin by acylation of L-prolinamide with chloroacetyl chloride in the presence of a base in dichloromethane or tetrahydrofuran as solvent, filtration and subsequent dehydration with trifluoroacetic anhydride (TFAA) to provide (S) -1- (2- chloroacetyl) pyrrolidin-2-carbonitrile. The carbonitrile intermediate is isolated by distilling out the solvent, co-distillation with ethyl acetate, partitioning between water and ethyl acetate, extraction of the resulting aqueous layer with ethyl acetate followed by aqueous washings of the organic layer and concentrating to obtain carbonitrile intermediate as yellow solid. This is later reacted with about 2 moles of l-aminoadamantane-3-ol in the presence of about 4 moles of potassium carbonate in dichloromethane (DCM) or tetrahydrofuran (THF) for 6 days. Finally, the obtained crude vildagliptin is subjected to chromatography employing SIMS/Biotage Flash chromatography system providing vildagliptin with melting point of 138°C-140°C. The disclosed process is schematically represented as follows:
Amide Carbonitrile
A similar process is described in J. Med. Chem. 2003, 46, 2774-2789, where acylation of L-prolinamide with chloroacetyl chloride is carried out in the presence of potassium carbonate in tetrahydrofuran as solvent and subsequent dehydration with TFAA to provide (S) -1- (2-chloroacetyl) pyrrolidin-2 -carbonitrile. The carbonitrile intermediate was isolated by adding ethyl acetate, distillation of the solvent, partitioning between water and aqueous sodium bicarbonate, extraction of the resulting aqueous layer with ethyl acetate followed by aqueous washings of the organic layer and concentrating to obtain carbonitrile intermediate as yellow- white solid which was reacted with about 2-3 moles of 1- aminoadamantane-3-ol in the presence of about 3 moles of potassium carbonate in DCM or THF for 1-3 days followed by purification from a mixture of ethyl acetate and isopropanol provided Vildagliptin as a white solid.
U.S. Patent No. 6,011,155 discloses a process for the preparation of (S) -1- (2- bromooacetyl) pyrrolidin-2-carbonitrile by acylation of L-prolinamide with bromoacetyl bromide in the presence of triethyl amine and catalytic amount of DMAP in DCM as solvent wherein the resulting (S)-l -(2 -bromoacetyl) pyrrolidin-2-carboxamide is isolated and subsequently dehydrated with TFAA to obtain the carbonitrile intermediate as dark yellow solid.
U.S. Patent application No. 2008/0167479 discloses preparation of Vildagliptin with high chemical and enantiomeric purities wherein (S) -1- (2-chloroacetyl) pyrrolidin-2- carbonitrile is prepared in one step process by acylation of prolinamide with chloroacetyl chloride in a mixture of isopropyl acetate and DMF followed by dehydration with cyanuric chloride to obtain the carbonitrile intermediate as an oil which was crystallized from isopropanol. The resulting carbonitrile intermediate is reacted with l-aminoadamantane-3- ol in the presence of alkali metal carbonates such as potassium carbonate and an optional additive such as I in a solvent comprising at least an ester or ether or nitrile solvent and purification of vildagliptin from methyl ethyl ketone or from a mixture of isopropanol and methyl t-butyl ether.
PCT Publication No. 2010/022690 discloses a process for the preparation of vildagliptin wherein (S)-l -(2-chloroacetyl) pyrrolidin-2-carboxamide intermediate is isolated as a trialkylamine hydrohalide salt in two fractions and. dehydrated with TFAA to obtain (S)-l- (2-chloroacetyl) pyrrolidin-2-carbonitrile as light yellow powder after crystallization from heptane. The resulting carbonitrile intermediate is then reacted with 3-amino-l- adamantanol in the presence of alkali metal carbonate base and an alkali metal iodide as a catalyst in a mixture of organic ketones, ester and polar aprotic solvents. The crude product was subjected to multiple crystallizations in order to achieve high chemical purity of vildagliptin. This publication also disclosed final crystallization of vildagliptin from 2- butanone, toluene, 2-methyl tetrahydrofuran, isopropyl acetate, dimethyl carbonate, isopropanol. This process adds an extra step of isolation of the said carboxamide intermediate, uses mixture of solvents in the preparation of vildagliptin and to multiple crystallizations which makes the process uneconomical on large scale.
PCT Publication No. 2011/101861 discloses a process for the preparation of vildagliptin wherein (S)-l-(2-chloroacetyl) pyrrolidin-2-carboxamide and (S)-l-(2-chloroacetyl) pyrrolidin-2-carbonitrile intermediates are isolated as solids after purification and drying. Further, (S)-l-(2-chloroacetyl) pyrrolidin-2-carbonitrile is then converted to vildagliptin by reacting it with l-aminoadamantane-3-ol in the presence of potassium carbonate and KI in a suitable ether solvent like THF and purifying the obtained vildagliptin from a mixture of ethyl acetate and methanol. This publication also provided an alternate process for the preparation of vildagliptin by reacting 2-(3-hydroxyadamantan-l-yl amino) acid or derivative thereof with pyrrolidine-2-carbonitrile and various solvents from which vildagliptin may be crystallized such as ethyl acetate, 2-butanone, or mixture of ethyl acetate-methanol, ethyl acetate-isopropanol, methanol-DCM, ethyl acetate-cyclohexane and 2-butanone-methyl t-butyl ether.
U.S. Patent No. 7,375,238 discloses a one-pot process for the preparation of vildagliptin without isolation of the carboxamide and carbonitrile intermediates and further involves preparation of Vildagliptin by using potassium carbonate and potassium iodide (KI) as catalysts in 2-butanone solvent. Purification of the crude vildagliptin was carried out from a mixture of isopropanol and methyl t-butyl ether in the presence of 1,8- diazabicyclo[5.4.0]undec-7-ene (DBU) base and final recrystallization from 2-butanone afforded pure vildagliptin. This process suffers from certain draw backs such as use of mixture of solvents for the acylation and condensation reactions; use of base and expensive additive such as KI in the condensation reaction.
PCT Publication No. 2011/012322 discloses a process wherein the (S) -1- (2-chloroacetyl) pyrrolidin-2-carbonitrile intermediate is isolated, purified and reacted with 1- aminoadamantane-3-ol in the presence of a phase transfer catalyst, optionally an inorganic base and a solvent selected from nitrile, ketone, ether, ester and mixtures thereof in a two phase reaction system wherein the first phase consist of a liquid phase and the second phase consists of an inorganic base. The final purification of vildagliptin was carried out in 2- butanone solvent.
PCT Publication No. 2013/179300 discloses preparation of vildagliptin from organic solvents such as aromatic hydrocarbons, aliphatic hydrocarbons, halogenated hydrocarbons, ethers, nitrile, dialkyl formamides, dialkylacetamides, dialkyl sulfoxides in the presence of organic or inorganic base. The resulting crude vildagliptin was purified by acid-base treatment and crystallization from a solvent selected from aliphatic hydrocarbons, aromatic hydrocarbons, ketones, esters, nitrile, ether, cyclic ether and alcohol or mixtures thereof.
PCT Publication No. 2012/022994 involves conversion of racemic vildagliptin to (S)- enantiomer via formation of vildagliptin adducts and final purification from ethyl acetate or mixture of ethyl acetate with 1% water.
U.S. Application No. 2006/0210627 discloses crystalline Form A of vildagliptin and its preparation from 2-butanone, isopropanol, acetone or a mixture of isopropanol-ethyl acetate in the presence of DBU base. This publication also discloses amorphous vildagliptin and its preparation by lyophilization from a water solution.
PCT Publication No. 2014/102815 disclosed a process for the preparation of vildagliptin by isolating the carboxamide and carbonitrile intermediates after crystallization and drying. The resulting carbonitrile intermediate is reacted with l-aminoadamantane-3-ol in the presence of organic base or inorganic base in nitrile, ester or alcohol solvent.
IN 3965 MUM/2013 publication discloses a process for the preparation of vildagliptin by preparing and crystallizing (S) -1- (2-chloroacetyl) pyrrolidin-2-carbonitrile intermediate and reacting it with l-aminoadamantane-3-ol in the presence of a potassium carbonate, optionally in presence of suitable catalyst such as KI in ketone solvent or in mixture of ketone with non polar solvents.
C.N. publication No. 102617434 discloses a one pot process for the preparation of Vildagliptin by reacting salt of pyrrolidine carbonitrile such as TFA salt with haloacetyl halide in the presence of a base followed by insiru reaction with l-aminoadamantane-3-ol in the presence of tertrabutyl ammonium iodide in halogenated hydrocarbon or ether as solvent to get vildagliptin which is further crystallized from ethyl acetate-petroleum ether.
C.N. publication No. 103804267 discloses a process for the preparation of vildagliptin by reacting (S)-l -(2 -haloacetyl) pyrrolidin-2-carbonitrile with l-aminoadamantane-3-ol in a mixed system of an organic solvent and water in the presence of a base and phase transfer catalyst followed by crystallization of the obtained crude vildagliptin.
C.N. publication No. 103787944 disclosed dehydration of-1- (2-chloroacetyl) -2- (S) – pyrrolidine carboxamide in the presence of a dehydrating agent and an acid-binding agent in an organic solvent followed by crystallization from mixture of isopropyl ether and ethyl acetate to provide l-(2-chloroacetyl)-2-(S)-pyrrolidine carbonitrile as white or pale yellow solid powder.
Furthermore, several techniques are known in the art for the purification of vildagliptin such as chromatography (US 6,166,063); or acid-base purification (IN 61 /MUM/2012 publication) or via formation of inorganic salt complexes (WO 2011/042765); or by solvent crystallizations such as mixture of ethyl acetate and isopropanol (J. Med. Chem. 2003, 46, 2774-2789); isopropanol and MTBE in the presence of DBU base and final recrystallization from 2-butanone (US 7,375,238); methyl ethyl ketone or from a mixture of isopropanol and MTBE (US 2008/0167479); acetone, 2-butanone, cyclohexanone, ethyl acetate, isopropyl acetate or dimethyl carbonate (IN 61 /MUM/2012 publication); 2- butanone (WO 2011/012322); aliphatic hydrocarbons, aromatic hydrocarbons, ketones, esters, nitrile, ether, cyclic ether and alcohol or mixtures thereof (WO 2013/179300); or from ethyl acetate or mixture of ethyl acetate with 1% water (WO 2012/022994).
Most of the processes known in the art for synthesizing vildagliptin are associated with one or more of the following disadvantages:
a) use of toxic TFAA for dehydration which is costly and environmentally harmful, b) lengthy and time consuming condensation process,
c) conventional solvents used in the condensation stage are costly, volatile, flammable, toxic, causing adverse health effects, in, addition to this potentially unsafe peroxide forming solvents such as THF were used, which process is more costlier than the process not having such elements,
d) purification of vildagliptin by chromatographic purification or by formation of inorganic salt complexes or by multiple crystallizations which are tedious, labor intensive, uses high amounts of solvents, require precise monitoring and time consuming and hence not viable for commercial scale operations.
Therefore, the present invention fulfills the need in the art and provides simple, industrially feasible and scalable processes for the preparation and purification of vildagliptin that circumvent disadvantages associated with the prior art process, proved to be advantageous from environmental and industrial point of view and also fulfill purity criteria. These processes allow the final product to be produced in a higher yield and purity by minimizing number of processing steps and reducing the number of solvent usage which is very practical for scale-up production, especially in terms of operating efficiency.
The new processes has a further advantage in recovering the expensive 1- aminoadamantane-3-ol from the reaction mixture and recycling in a simple manner that avoids use of inorganic salt complexes, which is economical and applicable on an industrial scale.
EXAMPLE 1: Preparation of (2S)- 1 -(Chloroacetyl)-2-pyrrolidinecarbonitrile.
To a solution of L-Prolinamide (100 gms) dissolved in DCM (1000 mL) was added triethyl amine (88.6 gms) and DMAP (1.07 gms) at 25-30°C under N2 atmosphere and stirred for 15 min at 25-30°C. This solution was added to a solution of chloroacetyl chloride (98.9 gms) in DCM (500 mL) under N2 atmosphere at -5 to 0°C over 2-3 hr. Raised the reaction mass temperature to 0-5°C and stirred for lhr. After reaction completion, charged phosphorus oxy chloride (201.5 gms) to the reaction mass at 0-5 °C, heated the reaction mass temperature to reflux and stirred for 6hr at same temperature. After reaction completion, allowed to cool to 10-20°C and added DM water (500 mL). Aqueous layer was separated and the organic layer was washed with DM water. To the organic layer DM water (300 mL) was added at 25-30°C and adjusted the reaction mass pH to 6.5-7.5 with -500 mL of sodium bicarbonate solution (-40 g of NaHC03 dissolved in 500 mL of DM Water). Separated the aqueous layer and concentrated the organic layer under vacuum at temperature of 30-40°C to get residual mass. Charged isopropanol (100 mL) and distilled out solvent completely under vacuum at <50°C. The resulting residue was allowed to cool to 30-40°C and charged isopropanol (500 mL). Heated the reaction mass temperature to 40- 45°C, stirred for 30 min at 40-45°C, allowed to cool to 0-5°C, stirred for 2 hr, filtered and washed wet cake with chilled isopropanol (100 mL), dried at 40-45°C for 6 hr to provide 115 gms of (2S)-l-(CMoroace1yl)-2-pyrrolidinecarbonitrile.
HPLC Purity: 99.86%.
Example 2: Preparation of Vildagliptin
To (2S)-l-(Chloroacetyl)-2 -Pyrrolidine carbonitrile (100 gms) dissolved in DM Water (500 mL), charged l-aminoadamantane-3-ol (242.2 g) at 25-35°C. Heated the reaction mass temperature to 40-45°C and stirred for 8-10 hr at 40-45°C. After reaction completion, allowed to cool to 25-30°C and charged DM water (700 mL) and DCM (600 mL). Separated the organic layer and extracted the aqueous layer with DCM. The total organic layer was concentrated under vacuum at temperature 30-40°C to get residual mass. Ethyl acetate (100 mL) was added to the residual mass and distilled completely under vacuum at <50°C. Charged ethyl acetate (500 mL) and refluxed for 1 hr. Allowed to cool to 25-30°C and stirred for 2 hr. Filtered the reaction mass and washed with ethyl acetate (100 mL) then dried at 50-55°C for 6 hr to provide 130 gms of crude vildagliptin.
HPLC Purity: 99.56%.
Dimer impurity content: <0.32%;
R-isomer content (by chiral HPLC): <0.2%;
l-aminoadamantane-3-ol content (by GC): 0.56%.
EXAMPLE 3: Preparation of Vildagliptin (using K2C03 and KI)
To l-aminoadamantane-3-ol (19.4 g) taken in DM Water (50 mL), added potassium carbonate (8.0 gms), potassium iodide (0.1 gm) and stirred for 15 mins at 25-35°C. (2S)-1- (Chloroacetyl)-2-Pyrrolidine carbonitrile (10 gms) was added at 25-35°C and stirred for 15 mins at 25-35°C. Raised the reaction mass temperature to 40-45°C and stirred for 4 hr at 40-45°C. After reaction completion, cooled to 25-30°C and charged DCM (50 mL). Separated the organic layer and extracted the aqueous layer with DCM. The total organic layer was washed with DM water and the resulting organic layer was concentrated under vacuum at temperature <40°C to get residual mass. Charged ethyl acetate (70 mL) to above residual mass and refluxed for 1 hr. Cooled to 25-30°C and stirred for 2 hr. Filtered the reaction mass and wash wet cake with ethyl acetate (10 mL). Suck dried for 30 min, dried initially at 25-35°C for 1 hr and then at 50-55°C for 6 hr to provide 12 gms of crude vildagliptin.
EXAMPLE 4; Preparation of Vildagliptin (using K2HP04 buffer and KI)
·
To l-aminoadamantane-3-ol (19.4 g) taken in DM Water (100 mL), added K2HP04 (10.1 gms), potassium iodide (0.1 gm) and stirred for 15 rnins at 25-35°C. (2S)-l-(Chloroacetyl)-
2- Pyrrolidine carbonitrile (10 gms) was added at 25-35°C and stirred for 15 mins at 25- 35°C. Raised the reaction mass temperature to 40-45°C and stirred for 8-10 hr at 40-45°C. After reaction completion, cooled to 25-30°C and filtered the reaction mass to remove salts. The resulting filtrate was extracted with DCM, and the resulting organic layer was concentrated initially by atmospheric distillation and later under vacuum at temperature 30- 40°C to get residual mass. Charged ethyl acetate (50 mL) to above residual mass and refluxed for 1 hr. Cooled to 25-30°C and stirred for 2 hr. Filtered the reaction mass and washed the wet cake with ethyl acetate (10 mL). Suck dried for 30 min, dried initially at 25-35°C for 1 hr and then at 50-55°C for 6 hr to provide 12 gms of crude vildagliptin.
HPLC Purity: 96.54%
Dimer impurity content: 2.55%;
R-isomer content (by chiral HPLC): not detected
l-aminoadamantane-3-ol content (by GC): 0.86%.
Example 5: Purification of Vildagliptin.
Vildagliptin crude (100 gms) was dissolved in isopropanol (900 mL) by heating to 50-55°C and stirred for 30 min. Filtered the reaction mass over hyflo bed (10 gms) at 50-55°C and washed the hyflo bed with hot isopropanol (100 mL). Distilled out solvent under vacuum at
35-40°C up to 4 volumes remains and allowed to cool to 20-25°C and stirred for 1 hr at same temperature. Further, allowed to cool to 5-10°C, stirred for 2 hrs, filtered and washed with isopropanol (100 mL). The wet product was dried at 50-55°C under vacuum for 8 hr to provide 80 gms of pure vildagliptin.
HPLC Purity: 99.89%;
Dimer impurity content: <0.1 %;
R-isomer content (by chiral HPLC): not detected
l-aminoadarnantane-3-ol content (by GC): 0.06%.
The purified vildagliptin (I) was analyzed by powder X-ray diffraction (PXRD) and is set forth in Figure. 01.
EXAMPLE 6: Preparation of Vildagliptin To a solution of L-Prolinamide (100 gms) dissolved in DCM (1000 mL) was added triethyl amine (88.6 gms) and DMAP (1.07 gms) at 25-30°C under N2 atmosphere and stirred for 15 min at 25-30°C. This solution was added to a solution of chloroacetyl chloride (118.7 gms) in DCM (500 mL) under N2 atmosphere at -5 to 0°C over 2-4 hr. Heated the reaction mass temperature to 10-15°C and stirred until reaction completion, charged phosphorus oxychloride (201.5 gms) to the reaction mass at 0-5°C, heated the reaction mass temperature to reflux and stirred for 6hr at same temperature. After reaction completion, allowed to cool to 5-15°C and slowly added DM water (500 mL). Aqueous layer was separated and the organic layer was washed with DM water. To the organic layer, DM water (300 mL) was added at 25-30°C and adjusted the reaction mass pH to 6.5-7.5 with -200 mL of sodium bicarbonate solution (-16 g of NaHC03 dissolved in 200 mL of DM Water). Separated the aqueous layer and concentrated the organic layer under vacuum at temperature of 30-40°C to get residual mass. The residual mass was dissolved in DM Water (640 mL), charged l-aminoadamantane-3-ol (310.6 g) at 25-35°C. Heated the reaction mass temperature to 40-45 °C and stirred for 9 hr at the same temperature. After reaction completion, allowed to cool to 25-30°C and charged DM water (900 mL) and DCM (1280 mL). Separated the organic layer and extracted the aqueous layer with DCM. The aqueous layer was separated and kept aside for l-aminoadamantane-3-ol recovery. The total organic layer was treated with P.S. 133 carbon, stirred for 30 rnins at 25-30°C and filtered over hyflo bed. The resulting filtrate was concentrated under, vacuum at temperature 30-40°C to get residual mass. To the residual mass, charged ethyl acetate (128 mL) and distilled completely under vacuum at 30-40°C to get semi solid mass. Charged ethyl acetate (640 mL) to the obtained semi solid and refluxed for 1 hr. The reaction mass was allowed to cool to 25-30°C and stirred for 2 hr. Filtered the reaction mass and washed with ethyl acetate (128 mL) to obtain wet cake. Again charged ethyl acetate (512 mL) to the obtained wet cake and refluxed for 1 hr. The reaction mass was allowed to cool to 25- 30°C and stirred for 2 hr. Filtered the reaction mass and washed with ethyl acetate (128 mL) and then dried at 50-55°C for 6 hr to provide 175 gms of crude vildagliptin.
HPLC Purity: 99.66%.
Dimer impurity content: <0.2%;
R-isomer content (by chiral HPLC) : <0.1 %;
l-aminoadamantane-3-ol content (by GC): <0.7%.
DSC: 150.12°C.
EXAMPLE 7: Purification of Vildagliptin. Vildagliptin crude (100 gms) was dissolved in isopropanol (1100 mL) by heating to 50- 55°C and stirred for 30 min. Filtered the solution over hyflo bed at 50-55°C and wash with hot isopropanol (100 mL). Distilled out solvent under vacuum at <55°C up to 5 volumes remains and allowed to cool to 20-25 °C and stirred for 1 hr at same temperature. Further allowed to cool to 10-15 °C, stirred for 2 hrs, filtered and washed with chilled isopropanol (100 mL). The wet product was dried at 50-55°C under vacuum for 8 hr to provide 80 gms of pure vildagliptin. HPLC Purity: >99.8%;
Dimer impurity content: <0.1%;
R-isomeri content (by chir‘al HPLC) : <0.1%;
l-aminoadamantane-3-ol content (by GC): <0.1%.
DSC: 151.92°C.
Example 8: Recovery of l-aminoadamantane-3-ol of formula (IV).
To aqueous layer (1700 mL) from example 1, 50% C.S.lye (435 mL) was added to adjust the pH to 13.0-14.0 at 25-35°C and stirred for 15 mins at 25-35°C. Raised the reaction mass temperature to 60-70°C and stirred for 3 hrs. Cooled to 25-35°C and added DCM (1700 mL), stirred for 15 min and separated the organic layer. The aqueous layer was extracted with DCM and the total organic layer was distilled out completely under vacuum at <40°C to get semisolid mass. Charged ethyl acetate (150 mL) and distilled out solvent completely under vacuum at <50°C to get semisolid material. Charged ethyl acetate (400 mL), stirred for 30 min at 40-45°C and cooled to 25-35°C. Further allowed to cool to 0- 5°C, stirred for 2hr, filtered the reaction mass at 5-10°C and washed with ethyl acetate (100 mL). The wet product was dried at 50-55°C under vacuum for 8 hr to obtain 140 gms of 1- aminoadamantane-3-ol.
An original synthesis of vildagliptin ((S)-1-[2-(3-hydroxyadamantan-1-ylamino)acetyl]pyrrolidine-2-carbonitrile), a powerful DPP-4 inhibitor, was developed. Vildagliptin was assembled from 3-amino-1-adamantanol, glyoxylic acid and l-prolinamide in a 4-step reaction sequence with the isolation of only two intermediates. The procedure is competitive with existing protocols, leading to vildagliptin in 63% overall yield.
PAPER
A Facile and Economical Method to Synthesize Vildagliptin
A mild and economical method to prepare vildagliptin had been reported with a good yield. In this paper, vildagliptin was synthesized from L-proline and 3-amino-1-adamantanol through chloride acetylation, amination, dehydration and substitution. The total yield of the target compound was 59%.
Dofetilide was first approved by the U.S. Food and Drug Administration (FDA) on Oct 1, 1999, then approved by European Medicine Agency (EMA) on Nov 29, 1999. It was developed and marketed as Tikosyn® by Pfizer.
Dofetilide is a selective blocker of delayed rectifier outward potassium current (IKr). It is indicated for the maintenance of normal sinus rhythm (delay in time to recurrence of atrial fibrillation/atrial flutter [AF/AFl]) in patients with atrial fibrillation/atrial flutter of greater than one week duration who have been converted to normal sinus rhythm.
Tikosyn® is available capsule for oral use, containing 0.125, 0.25 or 0.5 mg of free Dofetilide. The recommended dose is 500 µg orally twice daily.
Dofetilide is a class III antiarrhythmic agent.[1] It is marketed under the trade name Tikosyn by Pfizer, and is available in the United States in capsules containing 125, 250, and 500 µg of dofetilide. It is not available in Europe[2] or Australia.[3] In the United States it is only available by mail order or through specially trained local pharmacies.[4]
Based on the results of the Danish Investigations of Arrhythmias and Mortality on Dofetilide (“DIAMOND”) study,[7] dofetilide does not affect mortality in the treatment of patients post-myocardial infarction with left ventricular dysfunction, however it was shown to decrease all-cause readmissions as well as CHF-related readmissions.[8] Because of the results of the DIAMOND study, some physicians use dofetilide in the suppression of atrial fibrillation in individuals with LV dysfunction, however use appears limited: After initially receiving marketing approval in Europe in 1999, Pfizer voluntarily withdrew this approval in 2004 for commercial reasons[2] and it is not registered in other first world countries.
It has clinical advantages over other class III antiarrhythmics in chemical cardioversion of atrial fibrillation, and maintenance of sinus rhythm, and does not have the pulmonary or hepatotoxicity of amiodarone, however atrial fibrillation is not generally considered life-threatening, and dofetilide causes an increased rate of potentially life-threatening arrhythmias in comparison to other therapies.[9]
Contraindications
Prior to administration of the first dose, the corrected QT (QTc) must be determined. If the QTc is greater than 440 msec (or 500 msec in patients with ventricular conduction abnormalities), dofetilide is contraindicated. If heart rate is less than 60 bpm, the uncorrected QT interval should be used. After each subsequent dose of dofetilide, QTc should be determined and dosing should be adjusted. If at any time after the second dose of dofetilide the QTc is greater than 500 msec (550 msec in patients with ventricular conduction abnormalities), dofetilide should be discontinued. [4]
Adverse effects
Torsades de pointes is the most serious side effect of dofetilide therapy. The incidence of torsades de pointes is 0.3-10.5% and is dose-related, with increased incidence associated with higher doses. The majority of episodes of torsades de pointes have occurred within the first three days of initial dosing. Patients should be hospitalized and monitored for the first three days after starting dofetilide.[10]
The risk of inducing torsades de pointes can be decreased by taking precautions when initiating therapy, such as hospitalizing individuals for a minimum of three days for serial creatinine measurement, continuous telemetry monitoring and availability of cardiac resuscitation.
This causes the refractory period of atrial tissue to increase, hence its effectiveness in the treatment of atrial fibrillation and atrial flutter.
Dofetilide does not affect dV/dTmax (the slope of the upstroke of phase 0 depolarization), conduction velocity, or the resting membrane potential.
Dofetilide synthesis
There is a dose-dependent increase in the QT interval and the corrected QT interval (QTc). Because of this, many practitioners will initiate dofetilide therapy only on individuals under telemetry monitoring or if serial EKG measurements of QT and QTc can be performed.
Pharmacokinetics
Peak plasma concentrations are seen two to three hours after oral dosing when fasting. Dofetilide is well absorbed in its oral form, with a bioavailability of >90%. Intravenous administration of dofetilide is not available in the United States. [12]
The elimination half-life of dofetilide is roughly 10 hours; however, this varies based on many physiologic factors (most significantly creatinine clearance), and ranges from 4.8 to 13.5 hours. Due to the significant level of renal elimination (80% unchanged, 20% metabolites), the dose of dofetilide must be adjusted to prevent toxicity due to impaired renal function.[13]
Dofetilide is metabolized predominantly by CYP3A4enzymes predominantly in the liver and GI tract. This means that it is likely to interact with drugs that inhibit CYP3A4, such as erythromycin, clarithromycin, or ketoconazole, resulting in higher and potentially toxic levels of dofetilide. [14]
Metabolism
A steady-state plasma level of dofetilide is achieved in 2–3 days.
About 20 percent of dofetilide is metabolized in the liver via the CYP3A4 isoenzyme of the cytochrome P450 enzyme system. Drugs that interfere with the activity of the CYP3A4 isoenzyme can increase serum dofetilide levels. If the renal cation exchange system is interfered with (as with the medications listed above), a larger percentage of dofetilide is cleared via the CYP3A4 isoenzyme system.
History
After its initial US FDA approval, due to the pro-arrhythmic potential it was only made available to hospitals and prescribers that had received education and undergone specific training in the risks of treatment with dofetilide; however, this restriction was subsequently removed in 2016. [15
SYN
REF
Route 1
Reference:1. US5079248A / US4959366A.
2. J. Med. Chem.1990, 33, 1151-1155.
SYN
SYN
SYN
SYN
EP 0245997; JP 1987267250; US 4959366; US 5079248
This compound can be prepared by several related ways: 1) The condensation of N-methyl-2-(4-nitrophenyl)ethylamine (I) with 4-(2-chloroethoxy)nitrobenzene (II) by means of NaI and K2CO3 in refluxing acetonitrile gives 1-(4-nitrophenoxy)-5-(4-nitrophenyl)-3-methyl-3-azapentane (III), which is reduced with H2 over Pd/C in ethanol, yielding the corresponding diamino derivative (IV). Finally, this compound is acylated with methanesulfonyl anhydride in dichloromethane. 2) The condensation of (I) with N-[4-(2-chloroethoxy)phenyl]methanesulfonamide (V) with NaI and K2CO3 as before gives 1-[4-(methanesulfonamide)phenoxy]-3-methyl-5-(4-nitrophenyl)-3-azapentane (VI), which is reduced with H2 over Pd/C as before, yielding the corresponding amino derivative (VII). Finally, this compound is acylated with methanesulfonyl anhydride as usual. 3) The condensation of (II) with N-[4-[2-(methylamino)ethyl]phenyl]methanesulfonamide (VIII) with NaI and K2CO3 as usual gives 1-[4-(methanesulfonamido)phenyl]-3-methyl-5-(4-nitrophenoxy)-3-azapentane (IX), which is reduced with H2 and RaNi to the corresponding amino derivative (X). Finally, this compound is acylated with methanesulfonyl chloride and pyridine. 4) By condensation of N-[4-[2-(methanesulfonyloxy)ethyl]phenyl]methanesulfonamide (XI) with N-[4-[2-(methylamino)ethoxy]phenyl]methanesulfonamide (XII) in refluxing ethanol. 5) By condensation of (V) with (VIII) by means of NaHCO3.
^ Banchs JE; Wolbrette DL; Samii SM; et al. (November 2008). “Efficacy and safety of dofetilide in patients with atrial fibrillation and atrial flutter”. J Interv Card Electrophysiol. 23(2): 111–5. doi:10.1007/s10840-008-9290-6. PMID18688699. S2CID25162347.
^ Lenz TL; Hilleman DE (November 2000). “Dofetilide: A new antiarrhythmic agent approved for conversion and/or maintenance of atrial fibrillation/atrial flutter”. Drugs Today. 36 (11): 759–71. doi:10.1358/dot.2000.36.11.601530. PMID12845335.
^ Torp-Pedersen C, Møller M, Bloch-Thomsen PE, et al. (September 1999). “Dofetilide in patients with congestive heart failure and left ventricular dysfunction. Danish Investigations of Arrhythmia and Mortality on Dofetilide Study Group”. The New England Journal of Medicine. 341 (12): 857–65. doi:10.1056/NEJM199909163411201. PMID10486417.
^ Torp-Pedersen C; ller M; Mø Bloch-Thomsen PE; et al. (September 1999). “Dofetilide in patients with congestive heart failure and left ventricular dysfunction. Danish Investigations of Arrhythmia and Mortality on Dofetilide Study Group”. N. Engl. J. Med. 341 (12): 857–65. doi:10.1056/NEJM199909163411201. PMID10486417.
^ Micromedex Drugdex drug evaluations micromedex.com
^ Torp-Pedersen C, Møller M, Bloch-Thomsen PE, et al. Dofetilide in patients with congestive heart failure and left ventricular dysfunction. Danish Investigations of Arrhythmia and Mortality on Dofetilide Study Group. N Engl J Med 1999; 341:857.
^ Walker DK, Alabaster CT, Congrave GS, et al. Significance of metabolism in the disposition and action of the antidysrhythmic drug, dofetilide. In vitro studies and correlation with in vivo data. Drug Metab Dispos 1996; 24:447.
DofetilideCAS Registry Number: 115256-11-6CAS Name:N-[4-[2-[Methyl[2-[4-[(methylsulfonyl)amino]phenoxy]ethyl]amino]ethyl]phenyl]methanesulfonamideAdditional Names: 1-(4-methanesulfonamidophenoxy)-2-[N-(4-methanesulfonamidophenethyl)-N-methylamino]ethaneManufacturers’ Codes: UK-68798Trademarks: Tikosyn (Pfizer)Molecular Formula: C19H27N3O5S2Molecular Weight: 441.56Percent Composition: C 51.68%, H 6.16%, N 9.52%, O 18.12%, S 14.52%Literature References: Potassium channel blocker. Prepn: J. E. Arrowsmith et al.,EP245997; P. E. Cross et al.,US4959366 (1987, 1990 both to Pfizer); idemet al.,J. Med. Chem.33, 1151 (1990). HPLC determn in urine: D. K. Walker et al.,J. Chromatogr.568, 475 (1991). Mechanism of action study: D. Carmeliet, J. Pharmacol. Exp. Ther.262, 809 (1992). Review of pharmacology and pharmacokinetics: H. S. Rasmussen et al.,ibid.20, Suppl. 2, S96-S105 (1992). Clinical trial in atrial fibrillation and flutter: B. L. Norgaard et al.,Am. Heart J.137, 1062 (1999); in congestive heart failure: C. Torp-Pedersen et al.,N. Engl. J. Med.341, 857 (1999).Properties: Crystals from ethyl acetate/methanol (10:1), mp 147-149° (Cross); from hexane/ethyl acetate, mp 151-152° (Arrowsmith). Also reported as white crystalline solid, mp 161° (Rasmussen). pKa 7.0, 9.0, 9.6. Distribution coefficient (pH 7.4): 0.96. Sol in 0.1M NaOH, acetone, 0.1M HCl; very slightly sol in water, propan-2-ol.Melting point: mp 147-149° (Cross); mp 151-152° (Arrowsmith); mp 161° (Rasmussen)pKa: pKa 7.0, 9.0, 9.6Therap-Cat: Antiarrhythmic (class III).Keywords: Antiarrhythmic; Potassium Channel Blocker.
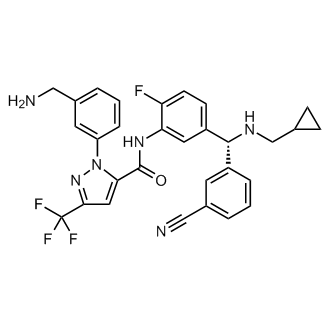

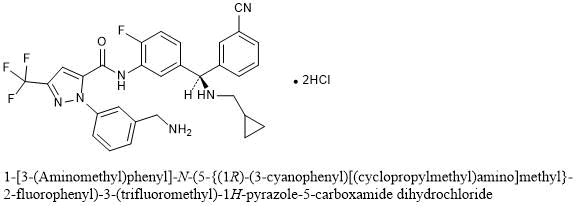
 2HCl : 635.48
2HCl : 635.48





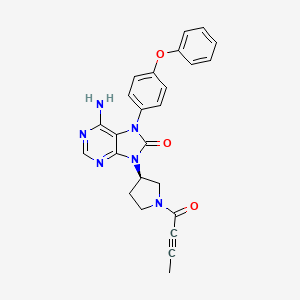





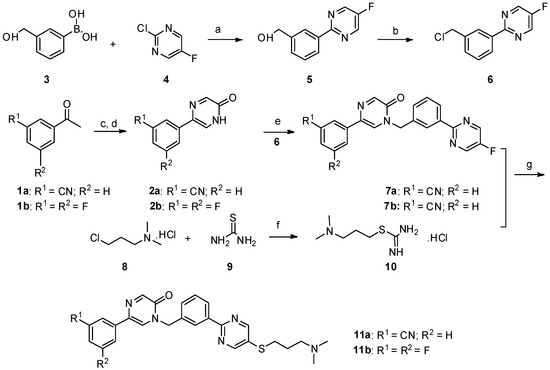
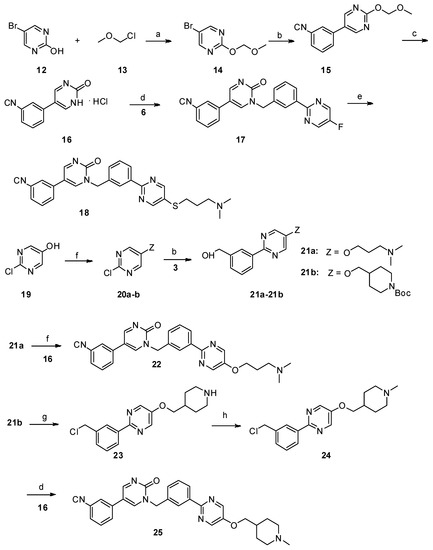
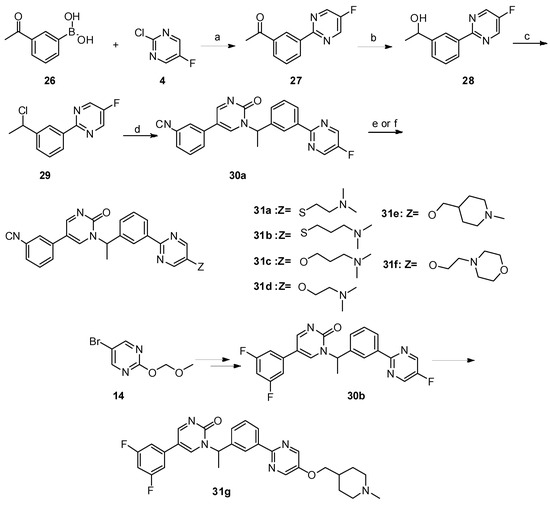
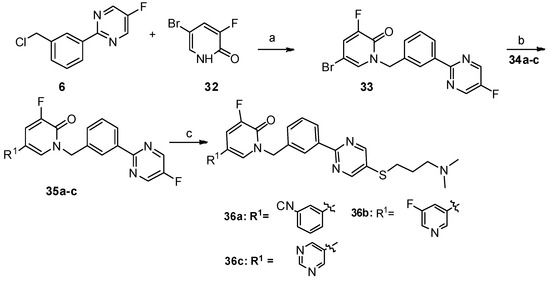
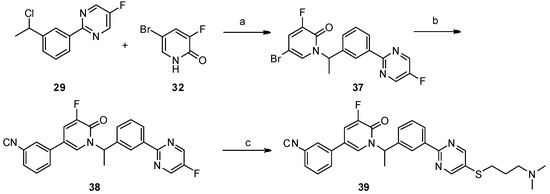

.png)










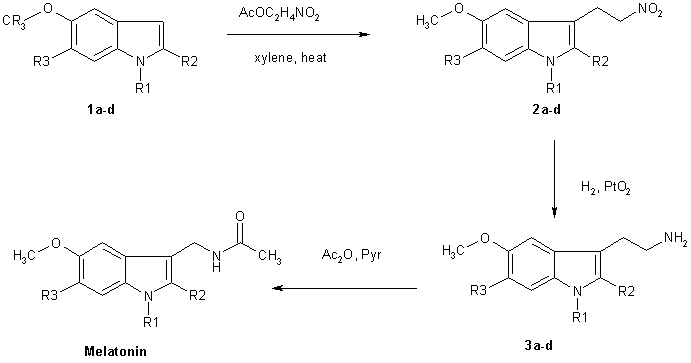
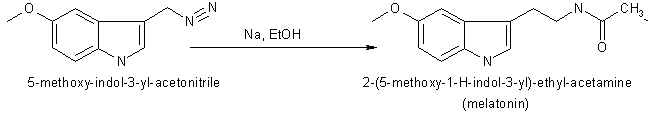
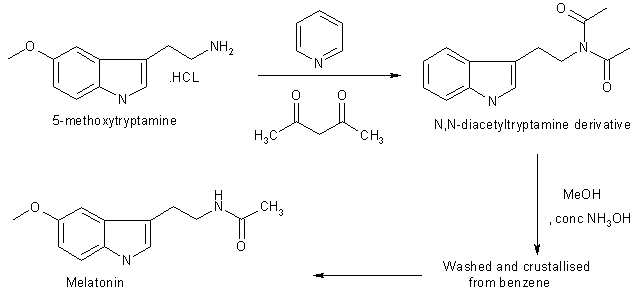
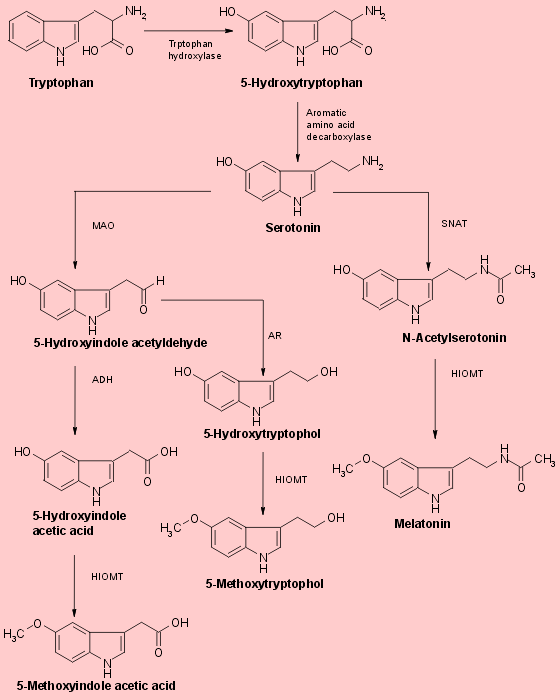 Fig.1 Biosynthesis of pineal methoxyindoles from serotonin
Fig.1 Biosynthesis of pineal methoxyindoles from serotonin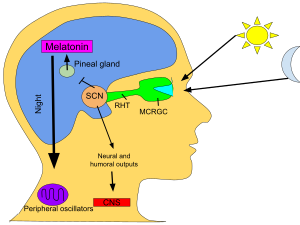

















 This is the first drug approved for PET imaging of prostate-specific membrane antigen positive lesions in men with prostate cancer.
This is the first drug approved for PET imaging of prostate-specific membrane antigen positive lesions in men with prostate cancer.
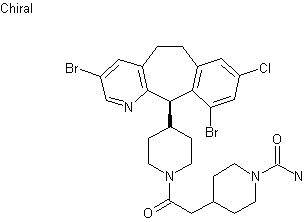

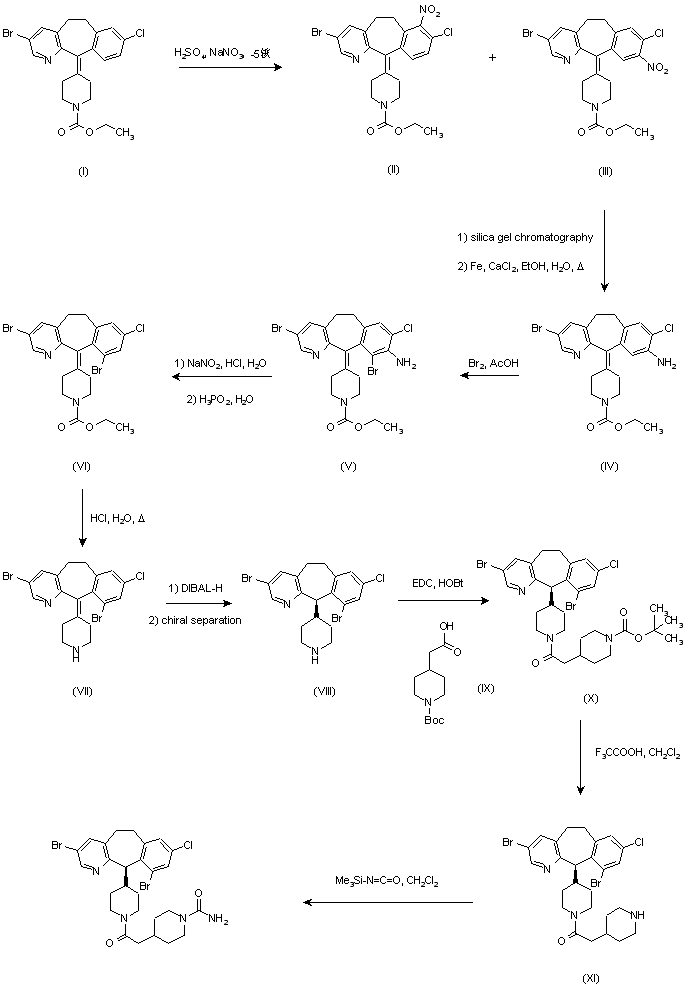
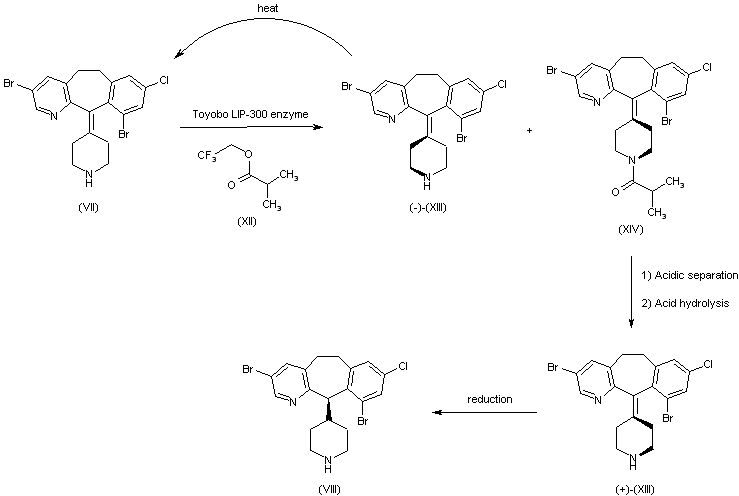
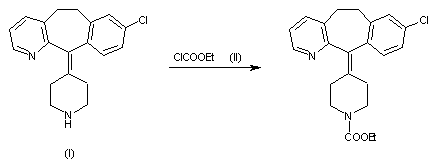
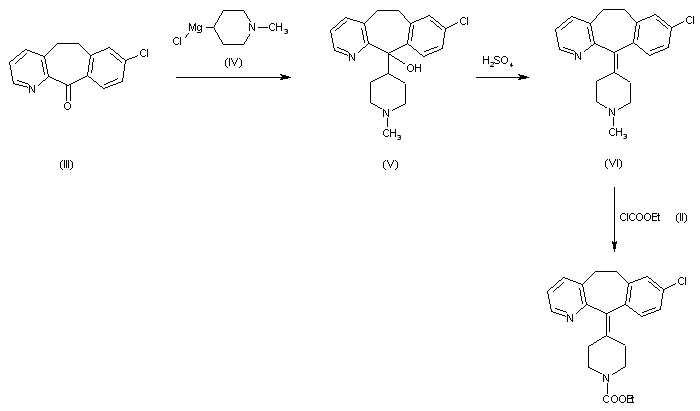
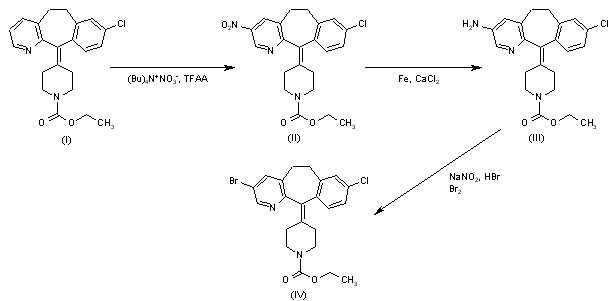
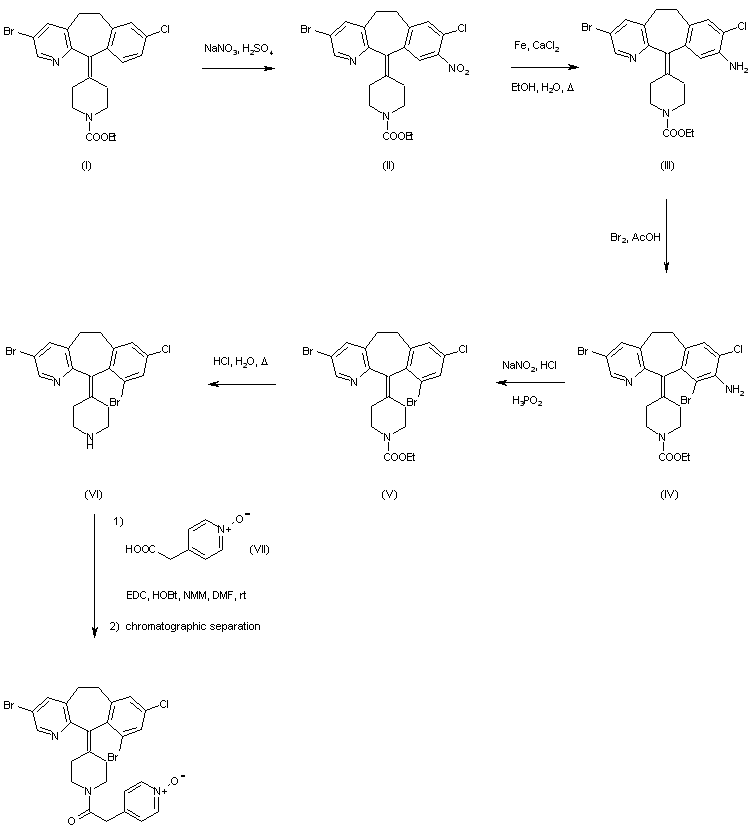
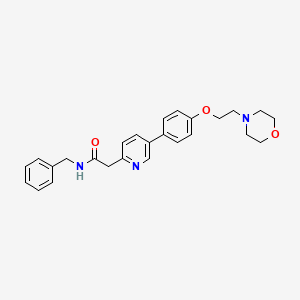















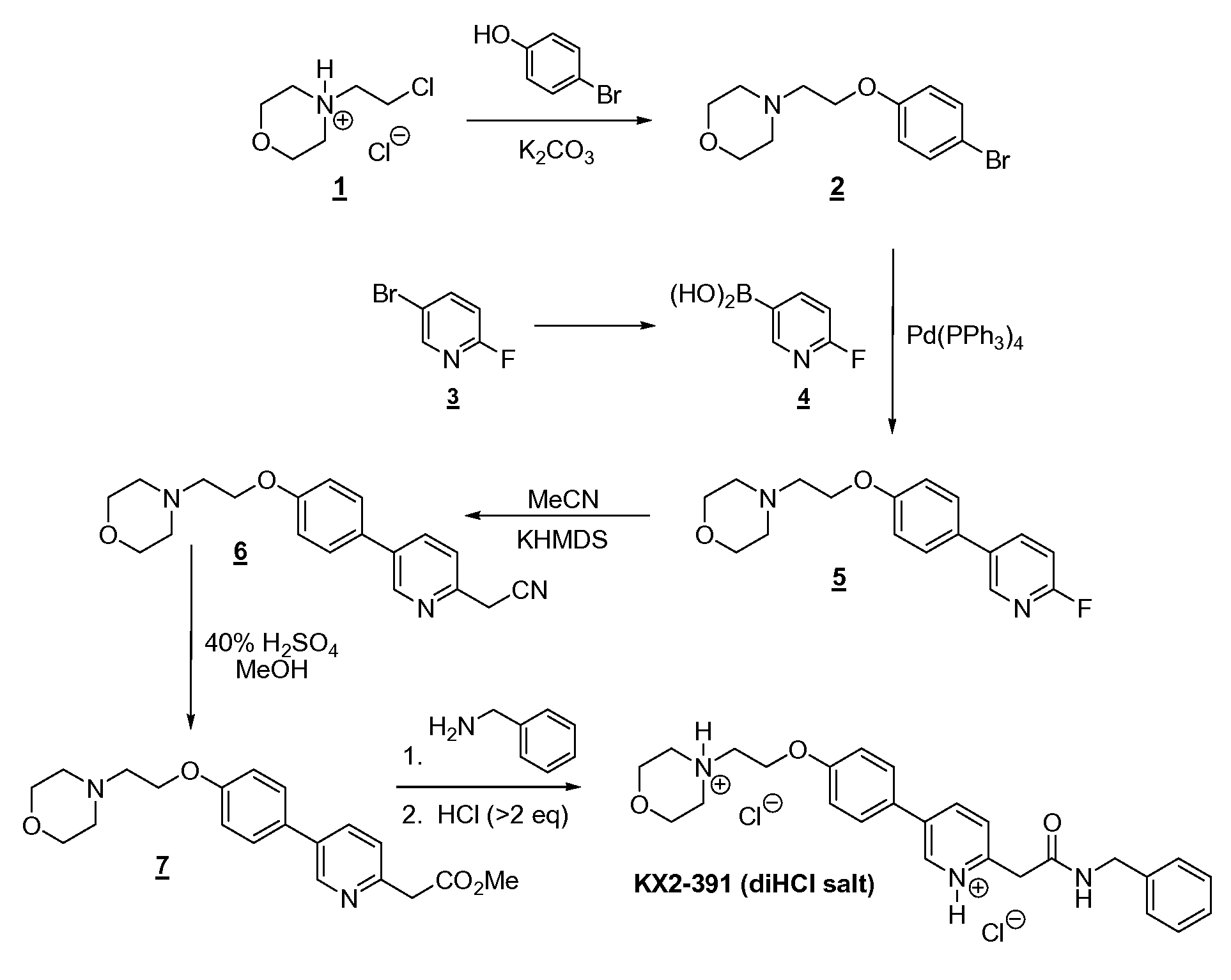







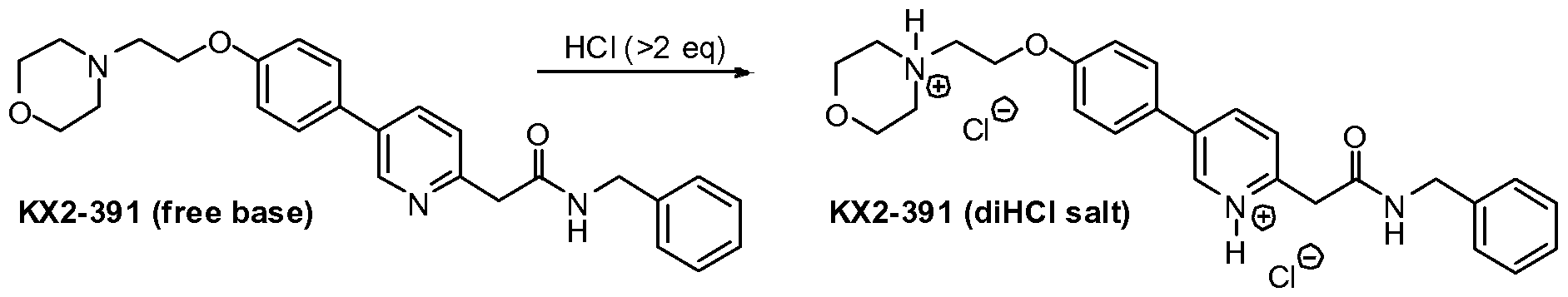



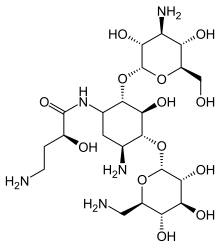


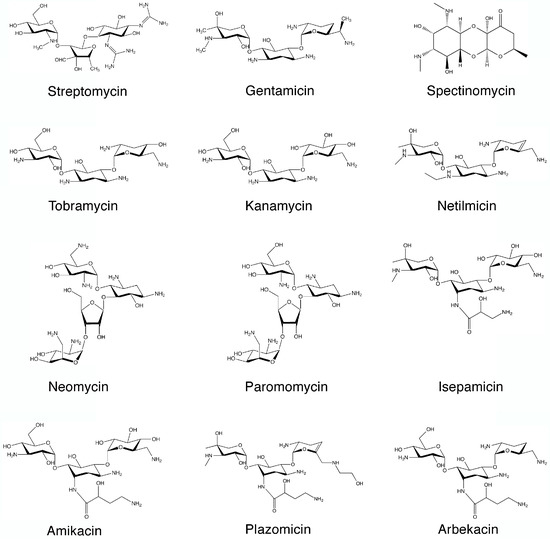


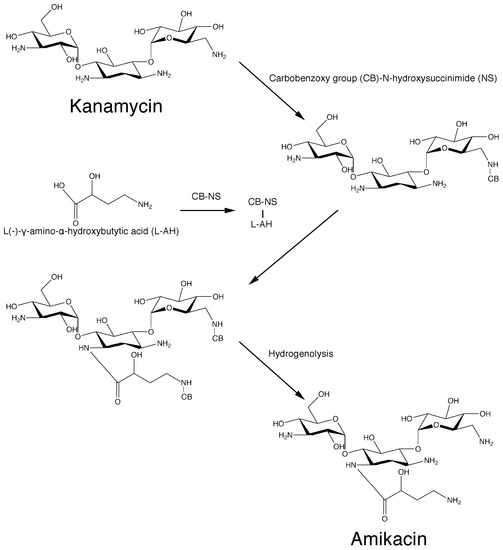


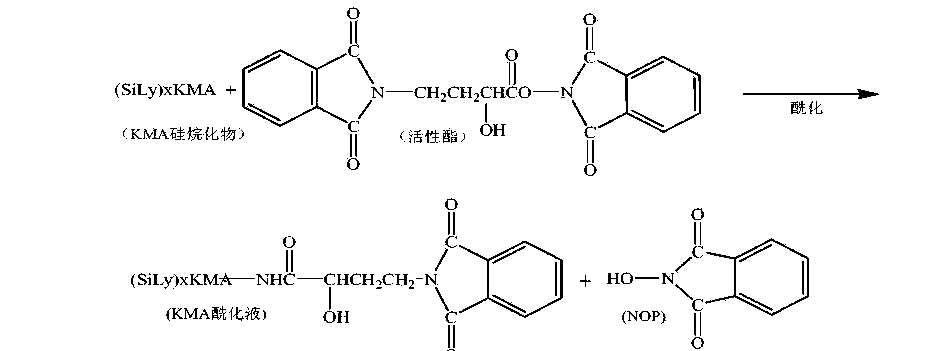









 ). A constant stream of argon gas is passed through the solution to remove the residual sulfide as hydrogen disulfide. The pH of the solution is then raised to 5.9 ± 0.2 using sodium hydroxide to form isoelectric L-citrulline.
). A constant stream of argon gas is passed through the solution to remove the residual sulfide as hydrogen disulfide. The pH of the solution is then raised to 5.9 ± 0.2 using sodium hydroxide to form isoelectric L-citrulline.