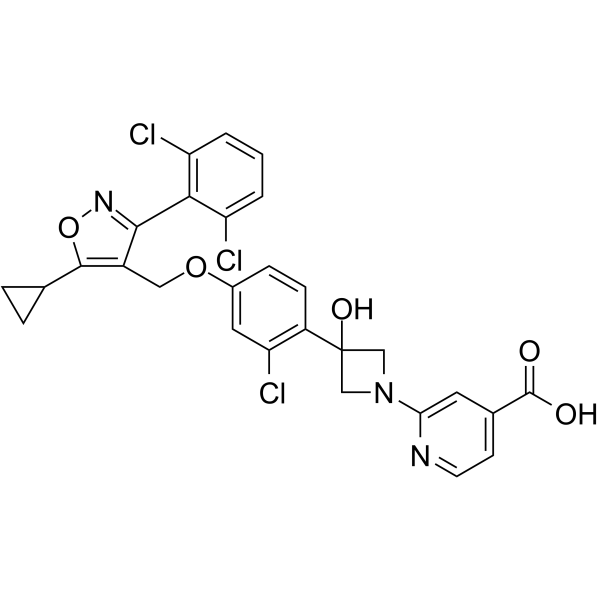
![Voclosporin.svg]()
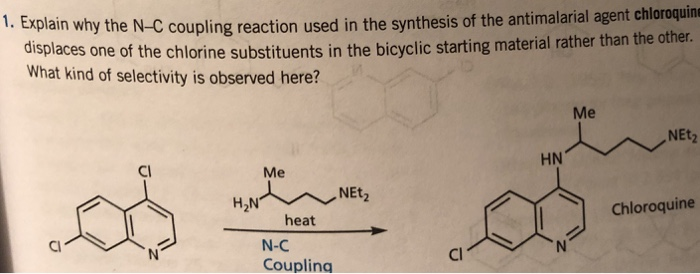
![ChemSpider 2D Image | Voclosporin | C63H111N11O12]()
![Voclosporin | C63H111N11O12 - PubChem]()
![Structure of VOCLOSPORIN]()
Voclosporin
- Molecular FormulaC63H111N11O12
- Average mass1214.622 Da
VOCLOSPORIN
(3S,6S,9S,12R,15S,18S,21S,24S,30S,33S)-30-Ethyl-33-[(1R,2R,4E)-1-hydroxy-2-methyl-4,6-heptadien-1-yl]-6,9,18,24-tetraisobutyl-3,21-diisopropyl-1,4,7,10,12,15,19,25,28-nonamethyl-1,4,7,10,13,16,19,22,2 5,28,31-undecaazacyclotritriacontane-2,5,8,11,14,17,20,23,26,29,32-undecone
1,4,7,10,13,16,19,22,25,28,31-Undecaazacyclotritriacontane-2,5,8,11,14,17,20,23,26,29,32-undecone, 30-ethyl-33-[(1R,2R,4E)-1-hydroxy-2-methyl-4,6-heptadien-1-yl]-1,4,7,10,12,15,19,25,28-nonamethyl-3,2 1-bis(1-methylethyl)-6,9,18,24-tetrakis(2-methylpropyl)-, (3S,6S,9S,12R,15S,18S,21S,24S,30S,33S)-
2PN063X6B1
8889
SA247, ISAtx 247, ISAtx-247, ISAtx247, Luveniq, LX211,
Aurinia Pharmaceuticals (following its merger with Isotechnika ), in collaboration with licensee Paladin Labs (a subsidiary of Endo International plc ), 3SBio ,and ILJIN , is developing a capsule formulation of the immunosuppressant calcineurin inhibitor peptide voclosporin for the treatment of psoriasis, the prevention of organ rejection after transplantation, autoimmune disease including systemic lupus erythematosus and lupus nephritis, and nephrotic syndrome including focal segmental glomerulosclerosis;
Voclosporin is an experimental immunosuppressant drug being developed by Aurinia Pharmaceuticals. It is being studied as a potential treatment for lupus nephritis (LN) and uveitis.[1] It is an analog of ciclosporin that has enhanced action against calcineurin and greater metabolic stability.[2] Voclosporin was discovered by Robert T. Foster and his team at Isotechnika in the mid 1990s.[3] Isotechnika was founded in 1993 and merged with Aurinia Pharmaceuticals in 2013.
Initially, voclosporin was a mixture of equal proporations of cis and trans geometric isomers of amino acid-1 modified cyclosporin. Later, in collaboration with Roche in Basel, Switzerland, voclosporin’s manufacturing was changed to yield the predominantly trans isomer which possesses most of the beneficial effect of the drug (immunosuppression) in the treatment of organ transplantation and autoimmune diseases.
Patent
WO-2020082061
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2020082061&_cid=P12-K9MDK8-59382-1
Novel crystalline forms of voclosporin which is a structural analog of cyclosporine A as calcineurin signal-transduction pathway inhibitor useful for treating lupus nephritis.
Voclosporin is a structural analog of cyclosporine A, with an additional single carbon extension that has a double-bond on one side chain. Voclosporin has the chemical name (3S,6S,9S,l2R,l5S,l8S,2lS,24S,30S,33S)-30-Ethyl-33-[(lR,2R,4E)-l-hydroxy-2-methyl-4,6-heptadien-l-yl]-6,9,l8,24-tetraisobutyl-3,2l-diisopropyl-l,4,7,l0,l2,l5,l9,25,28-nonamethyl-l,4,7,l0,l3,l6,l9,22,25,28,3 l-undecaazacyclotritriacontane-2,5,8,l l,l4,l7,20,23,26,29,32-undecone and the following chemical structure:
![]()
Voclosporin is reported to be a semisynthetic structural analogue of cyclosporine that exerts its immunosuppressant effects by inhibition of the calcineurin signal-transduction pathway and is in Phase 3 Clinical Development for Lupus Nephritis.
[0003] Voclosporin and process for preparation thereof are known from International Patent Application No. WO 1999/18120.
[0004] Certain mixtures of cis and trans-isomers of cyclosporin A analogs referred to as
ISATX247 in different ratios are known from U.S. Patent No. 6,998,385, U.S. Patent No. 7,332,472 and U.S. Patent No. 9,765,119.
[0005] Polymorphism, the occurrence of different crystal forms, is a property of some molecules and molecular complexes. A single compound, like Voclosporin, may give rise to a variety of polymorphs having distinct crystal structures and physical properties like melting point, thermal behaviors (e.g. measured by thermogravimetric analysis – “TGA”, or differential scanning calorimetry – “DSC”), powder X-ray diffraction (PXRD) pattern, infrared absorption fingerprint, Raman absorption fingerprint, and solid state (13C-) NMR spectrum. One or more of these techniques may be used to distinguish different polymorphic forms of a compound.
[0006] Different salts and solid state forms (including solvated forms) of an active
pharmaceutical ingredient may possess different properties. Such variations in the properties of different salts and solid state forms and solvates may provide a basis for improving formulation, for example, by facilitating better processing or handling characteristics, improving the dissolution profile, or improving stability (polymorph as well as chemical stability) and shelf-life. These variations in the properties of different salts and solid state forms may also provide improvements to the final dosage form, for instance, if they serve to improve bioavailability. Different salts and solid state forms and solvates of an active pharmaceutical ingredient may also give rise to a variety of polymorphs or crystalline forms, which may in turn provide additional opportunities to use variations in the properties and characteristics of a solid active pharmaceutical ingredient for providing an improved product.
[0007] Discovering new salts, solid state forms and solvates of a pharmaceutical product can provide materials having desirable processing properties, such as ease of handling, ease of processing, storage stability, and ease of purification or as desirable intermediate crystal forms that facilitate conversion to other salts or polymorphic forms. New salts, polymorphic forms and solvates of a pharmaceutically useful compound can also provide an opportunity to improve the performance characteristics of a pharmaceutical product (dissolution profile, bioavailability, etc.). It enlarges the repertoire of materials that a formulation scientist has available for formulation optimization, for example by providing a product with different properties, e.g., a different crystal habit, higher crystallinity or polymorphic stability which may offer better processing or handling characteristics, improved dissolution profile, or improved shelf-life.
[0008] For at least these reasons, there is a need for solid state forms (including solvated forms) of Voclosporin and salts thereof.
HPLC method:
Method description
Column: Zorbax SB C18, 1.8 pm, 100×2.1 mm
Mobile phase: A: 38 ACN : 7 TBME : 55 voda : 0.02 H3P04 (V/V/V/V)
B: 70 ACN : 7 TBME : 23 voda : 0.02 H P04 (V/V/V/V)
Flow rate: 0.5 mL/min
Gradient
![]()
Analysis time: 26 minutes + 3 minutes equilibration
Injection volume: 3.0 pL
Column temperature: 90 °C
Diluent: Ethanol
Detection: UV, 210 nm
EXAMPLES
[0095] The starting material Voclosporin crude may be obtained according to ET.S. Patent No. 6,998,385 ETnless otherwise indicated, the purity is determined by HPLC (area percent). The crude product contained according to HPLC analysis 42.6 % trans-Voclosporin (further only Voclosporin), 40.2 % cis-Voclosporin and 2.9 % Cyclosporin A. The crude Voclosporin was purified by column chromatography on silica gel using a mixture of toluene and acetone 82 : 18 (v/v) as mobile phase. The fractions were monitored by HPLC. The appropriate fractions were joined and evaporated, obtaining purified Voclosporin as a white foam. According to HPLC analysis it contained 85.7 % Voclosporin, 3.6 % cis-Voclosporin and 2.6 % Cyclosporin A (further only purified Voclosporin).
[0096] The Voclosporin crude (containing about 42.6 % of Voclosporin) was used for further optimization of the chromatographic separation of cis-Voclosporin and Voclosporin and the effort resulted in improved process for chromatographic separation which includes purification by column chromatography on silica gel using a mixture of toluene and methylisobutylketone 38 : 62 as mobile phase. The fractions were monitored by HPLC. The appropriate fractions were joined and evaporated to a dry residue, weighing 31.0 grams. This residue was not analyzed. The material was dissolved in 25 ml of acetone and then 50 ml of water was added and the solution was let to crystallize for 2 hours in the refrigerator. Then the crystalline product was separated by filtration and dried in vacuum dryer (40 °C, 50 mbar, 12 hours), obtaining 29.6 g of dry product containing 90.6 % of Voclosporin, 0.4 % cis-Voclosporin and 3.7 % Cyclosporin A (further mentioned as final Voclosporin).
Example 1: Preparation of Voclosporin Form A
4.1 grams of Purified Voclosporin was dissolved in acetone and the solution was evaporated to 8.0 grams and the concentrate was diluted by 6 ml of water. The solution was let to crystallize in refrigerator at about 2 °C for 12 hours. The crystalline product was filtered off, washed by a mixture of acetone and water 1 : 1 (v/v) and dried on open air obtaining 2.6 grams of crystalline product Form A. Voclosporin form A was confirmed by PXRD as presented in Figure 1.
Example 2: Preparation of Voclosporin Form B
[0097] 1.0 gram of Purified Voclosporin was dissolved in a mixture of 1.5 ml acetone and 3.0 ml n-hexane. The solution was let to crystallize in refrigerator at about 2 °C for 12 hours. The crystalline product was filtered off, washed by a mixture of acetone and hexane 1 : 2 (v/v) and dried on open air obtaining 0.5 grams of crystalline product Form B. Voclosporin form B was confirmed by PXRD as presented in Figure 2.
Example 3: Preparation of Amorphous Voclosporin
[0098] 2.0 grams of Purified Voclosporin was dissolved in 40 ml of hot cyclohexane and the solution was stirred for 12 hours at room temperature. Then the crystalline product was filtered off and washed with 5 ml of cyclohexane and dried on open air, obtaining 1.3 grams of amorphous powder. Amorphous Voclosporin was confirmed by PXRD as presented in Figure 3
Example 4: Preparation of Voclosporin Form C
[0099] Final Voclosporin (2 grams) was dissolved in acetonitrile (20 ml) at 50 °C, water (6 ml) was added with stirring, and the clear solution was allowed to crystallize 5 days at 20 °C. Colorless needle crystals were directly mounted to the goniometer head in order to define the crystal structure. Voclosporin form C was confirmed by X-ray crystal structure determination.
References
External links
Voclosporin
![Voclosporin.svg]() |
| Names |
| IUPAC name
(3S,6S,9S,12R,15S,18S,21S,24S,30S,33S)-30-Ethyl-33-[(1R,2R,4E)-1-hydroxy-2-methyl-4,6-heptadien-1-yl]-6,9,18,24-tetraisobutyl-3,21-diisopropyl-1,4,7,10,12,15,19,25,28-nonamethyl-1,4,7,10,13,16,19,22,25,28,31-undecaazacyclotritriacontane-2,5,8,11,14,17,20,23,26,29,32-undecone
|
| Other names
VCS, ISA247, Luveniq
|
| Identifiers |
|
|
|
|
|
|
| ChemSpider |
|
|
|
|
|
|
|
|
| Properties |
|
|
C63H111N11O12 |
| Molar mass |
1214.646 g·mol−1 |
Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa).
|
REFERENCES
1: Mok CC. Calcineurin inhibitors in systemic lupus erythematosus. Best Pract Res Clin Rheumatol. 2017 Jun;31(3):429-438. doi: 10.1016/j.berh.2017.09.010. Epub 2017 Oct 11. Review. PubMed PMID: 29224682.
2: Dang W, Yin Y, Wang Y, Wang W, Su J, Sprengers D, van der Laan LJW, Felczak K, Pankiewicz KW, Chang KO, Koopmans MPG, Metselaar HJ, Peppelenbosch MP, Pan Q. Inhibition of Calcineurin or IMP Dehydrogenase Exerts Moderate to Potent Antiviral Activity against Norovirus Replication. Antimicrob Agents Chemother. 2017 Oct 24;61(11). pii: e01095-17. doi: 10.1128/AAC.01095-17. Print 2017 Nov. PubMed PMID: 28807916; PubMed Central PMCID: PMC5655111.
3: Wong TC, Lo CM, Fung JY. Emerging drugs for prevention of T-cell mediated rejection in liver and kidney transplantation. Expert Opin Emerg Drugs. 2017 Jun;22(2):123-136. doi: 10.1080/14728214.2017.1330884. Epub 2017 May 22. Review. PubMed PMID: 28503959.
4: Chow C, Simpson MJ, Luger TA, Chubb H, Ellis CN. Comparison of three methods for measuring psoriasis severity in clinical studies (Part 1 of 2): change during therapy in Psoriasis Area and Severity Index, Static Physician’s Global Assessment and Lattice System Physician’s Global Assessment. J Eur Acad Dermatol Venereol. 2015 Jul;29(7):1406-14. doi: 10.1111/jdv.13132. Epub 2015 Apr 27. PubMed PMID: 25917315.
5: Simpson MJ, Chow C, Morgenstern H, Luger TA, Ellis CN. Comparison of three methods for measuring psoriasis severity in clinical studies (Part 2 of 2): use of quality of life to assess construct validity of the Lattice System Physician’s Global Assessment, Psoriasis Area and Severity Index and Static Physician’s Global Assessment. J Eur Acad Dermatol Venereol. 2015 Jul;29(7):1415-20. doi: 10.1111/jdv.12861. Epub 2015 Apr 27. PubMed PMID: 25917214.
6: Maya JR, Sadiq MA, Zapata LJ, Hanout M, Sarwar S, Rajagopalan N, Guinn KE, Sepah YJ, Nguyen QD. Emerging therapies for noninfectious uveitis: what may be coming to the clinics. J Ophthalmol. 2014;2014:310329. doi: 10.1155/2014/310329. Epub 2014 Apr 24. Review. PubMed PMID: 24868451; PubMed Central PMCID: PMC4020293.
7: Hardinger KL, Brennan DC. Novel immunosuppressive agents in kidney transplantation. World J Transplant. 2013 Dec 24;3(4):68-77. doi: 10.5500/wjt.v3.i4.68. Review. PubMed PMID: 24392311; PubMed Central PMCID: PMC3879526.
8: Ling SY, Huizinga RB, Mayo PR, Larouche R, Freitag DG, Aspeslet LJ, Foster RT. Cytochrome P450 3A and P-glycoprotein drug-drug interactions with voclosporin. Br J Clin Pharmacol. 2014 Jun;77(6):1039-50. doi: 10.1111/bcp.12309. PubMed PMID: 24330024; PubMed Central PMCID: PMC4093929.
9: Mayo PR, Ling SY, Huizinga RB, Freitag DG, Aspeslet LJ, Foster RT. Population PKPD of voclosporin in renal allograft patients. J Clin Pharmacol. 2014 May;54(5):537-45. doi: 10.1002/jcph.237. Epub 2013 Nov 30. PubMed PMID: 24243422.
10: Gubskaya AV, Khan IJ, Valenzuela LM, Lisnyak YV, Kohn J. Investigating the Release of a Hydrophobic Peptide from Matrices of Biodegradable Polymers: An Integrated Method Approach. Polymer (Guildf). 2013 Jul 8;54(15):3806-3820. PubMed PMID: 24039300; PubMed Central PMCID: PMC3770487.
11: Ling SY, Huizinga RB, Mayo PR, Freitag DG, Aspeslet LJ, Foster RT. Pharmacokinetics of voclosporin in renal impairment and hepatic impairment. J Clin Pharmacol. 2013 Dec;53(12):1303-12. doi: 10.1002/jcph.166. Epub 2013 Oct 8. PubMed PMID: 23996158.
12: Mayo PR, Huizinga RB, Ling SY, Freitag DG, Aspeslet LJ, Foster RT. Voclosporin food effect and single oral ascending dose pharmacokinetic and pharmacodynamic studies in healthy human subjects. J Clin Pharmacol. 2013 Aug;53(8):819-26. doi: 10.1002/jcph.114. Epub 2013 Jun 4. PubMed PMID: 23736966.
13: Schultz C. Voclosporin as a treatment for noninfectious uveitis. Ophthalmol Eye Dis. 2013 May 5;5:5-10. doi: 10.4137/OED.S7995. Print 2013. PubMed PMID: 23700374; PubMed Central PMCID: PMC3653814.
14: Gomes Bittencourt M, Sepah YJ, Do DV, Agbedia O, Akhtar A, Liu H, Akhlaq A, Annam R, Ibrahim M, Nguyen QD. New treatment options for noninfectious uveitis. Dev Ophthalmol. 2012;51:134-61. doi: 10.1159/000336338. Epub 2012 Apr 17. Review. PubMed PMID: 22517211.
15: Khan IJ, Murthy NS, Kohn J. Hydration-induced phase separation in amphiphilic polymer matrices and its influence on voclosporin release. J Funct Biomater. 2012 Oct 30;3(4):745-59. doi: 10.3390/jfb3040745. PubMed PMID: 24955746; PubMed Central PMCID: PMC4030927.
16: Roesel M, Tappeiner C, Heiligenhaus A, Heinz C. Oral voclosporin: novel calcineurin inhibitor for treatment of noninfectious uveitis. Clin Ophthalmol. 2011;5:1309-13. doi: 10.2147/OPTH.S11125. Epub 2011 Sep 13. PubMed PMID: 21966207; PubMed Central PMCID: PMC3180504.
17: Busque S, Cantarovich M, Mulgaonkar S, Gaston R, Gaber AO, Mayo PR, Ling S, Huizinga RB, Meier-Kriesche HU; PROMISE Investigators. The PROMISE study: a phase 2b multicenter study of voclosporin (ISA247) versus tacrolimus in de novo kidney transplantation. Am J Transplant. 2011 Dec;11(12):2675-84. doi: 10.1111/j.1600-6143.2011.03763.x. Epub 2011 Sep 22. PubMed PMID: 21943027.
18: Kuglstatter A, Mueller F, Kusznir E, Gsell B, Stihle M, Thoma R, Benz J, Aspeslet L, Freitag D, Hennig M. Structural basis for the cyclophilin A binding affinity and immunosuppressive potency of E-ISA247 (voclosporin). Acta Crystallogr D Biol Crystallogr. 2011 Feb;67(Pt 2):119-23. doi: 10.1107/S0907444910051905. Epub 2011 Jan 15. PubMed PMID: 21245533; PubMed Central PMCID: PMC3045272.
19: Kunynetz R, Carey W, Thomas R, Toth D, Trafford T, Vender R. Quality of life in plaque psoriasis patients treated with voclosporin: a Canadian phase III, randomized, multicenter, double-blind, placebo-controlled study. Eur J Dermatol. 2011 Jan-Feb;21(1):89-94. doi: 10.1684/ejd.2010.1185. PubMed PMID: 21227890.
20: Deuter CM. [Systemic voclosporin for uveitis treatment]. Ophthalmologe. 2010 Jul;107(7):672-5. doi: 10.1007/s00347-010-2217-5. German. PubMed PMID: 20571806.
//////////VOCLOSPORIN, Voclosporin, ISA247, ISAtx 247, ISAtx-247, ISAtx247, Luveniq, LX211,
CC[C@@H]1NC([C@@H](N(C([C@@H](N(C([C@@H](N(C([C@@H](N(C([C@H](NC([C@@H](NC([C@@H](N(C([C@H](C(C)C)NC([C@@H](N(C(CN(C1=O)C)=O)C)CC(C)C)=O)=O)C)CC(C)C)=O)C)=O)C)=O)C)CC(C)C)=O)C)CC(C)C)=O)C)C(C)C)=O)C)[C@@H]([C@@H](C/C=C/C=C)C)O)=O




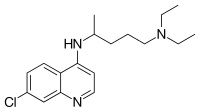
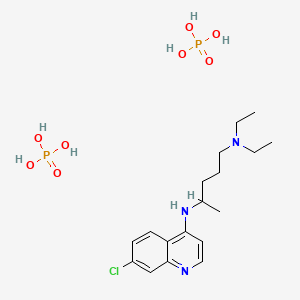

 Elevated occurrence of chloroquine- or multi-resistant malaria
Elevated occurrence of chloroquine- or multi-resistant malaria



























![(1R,5S,6R,7R,10R,11R,14R,15S,20R,21R)-21-[(2R)-2-Amino-2,3,3-trimethylbutoxy]-20-(5-carbamoyl-1,2,4-triazol-1-yl)-5,7,10,15-tetramethyl-7-[(2R)-3-methylbutan-2-yl]-17-oxapentacyclo[13.3.3.01,14.02,11.05,10]henicos-2-ene-6-carboxylic acid.png](http://pubchem.ncbi.nlm.nih.gov/image/imgsrv.fcgi?cid=59312774&t=l)



















.gif)