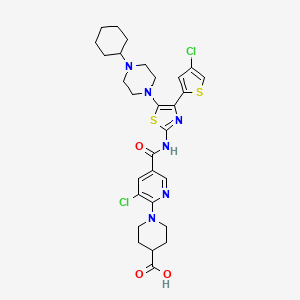
![ChemSpider 2D Image | Roquinimex | C18H16N2O3]()
![CID 55197.png]()
![Roquinimex.svg]()
Roquinimex
- Molecular FormulaC18H16N2O3
- Average mass308.331 Da
4-hydroxy-N,1-dimethyl-2-oxo-N-phenyl-1,2-dihydroquinoline-3-carboxamide
Linomide
N-phenyl-N-methyl-1,2-dihydro-4-hydroxy-1-methyl-2-oxoquinoline-3-carboxamide
1,2-Dihydro-4-hydroxy-N,1-dimethyl-2-oxo-N-phenyl-3-quinolinecarboxamide
372T2944C0, FCF-89
LS-2616
PNU-212616
E. Eriksoo et al., EP 59698; eidem, U.S. Patent 4,738,971 (1982, 1988 both to AB Leo).
Roquinimex (Linomide) is a quinoline derivative immunostimulant which increases NK cell activity and macrophage cytotoxicity. It also inhibits angiogenesis and reduces the secretion of TNF alpha.
Roquinimex (Linomide) is a quinoline derivative immunostimulant which increases NK cell activity and macrophage cytotoxicity. It also inhibits angiogenesis and reduces the secretion of TNF alpha.
Investigated as a treatment for some cancers (including as adjuvant therapy after bone marrow transplantation in acute leukemia) and autoimmune diseases, such as multiple sclerosis and recent-onset type I diabetes.
Roquinimex has been investigated as a treatment for some cancers (including as adjuvant therapy after bone marrow transplantation in acute leukemia) and autoimmune diseases, such as multiple sclerosis and recent-onset type I diabetes. Several trials have been terminated due to serious cardiovascular toxicity.
Synthesis
Ethyl 2-(methylamino)benzoate is condensed with ethyl malonate. Amine-ester ineterchange of that compound with N-methylanilineresults in formation of the amide roquinimex.
PAPER
Using DOE to Achieve Reliable Drug Administration: A Case Study
DuPont Chemoswed, R&D Department, P.O. Box 839, Celciusgatan 35, SE-201 80 Malmö, Sweden
Org. Proc. Res. Dev., 2004, 8 (5), pp 802–807
DOI: 10.1021/op049904l
Design of experiments (DOE), a statistical tool, and mathematical modeling techniques are established and proven methodologies for process and product improvements in the pharmaceutical industry. This contribution presents a case study where an unsatisfactory dissolution capacity for the drug Roquinimex was overcome by investigating the process parameters with the help of an experimental design. By elucidating the detailed effects of temperature, dosing time, and dilution, conformity in the particle size distribution of the active pharmaceutical ingredient (API) from batch to batch in full-scale manufacturing could be ensured. As a direct result the manufactured drug met its specified dissolution capacity, which was a prerequisite for obtaining the desired bioavailability of the pharmaceutical oral formulation. This work demonstrates how the use of DOE in chemical process development adds value by allowing efficient and reliable improvements of a given synthetic step.
1 H NMR (4): δ 12.4 (broad s, 1H, OH), 8.1 (m, 1H, Ar), 7.5 (m, 1H, Ar), 7.1 (m, 7H, Ar), 3.5 (s, 3H, NCH3 ), 3.3 5 (s, 3H, NCH3 ).
SYN
BE 0904431; DE 3609052; GB 2172594; JP 1986221194; US 4672057
![]() By condensation of 4-hydroxy-1-methyl-2-oxo-1,2-dihydroquinoline-3-carboxylic acid ethyl ester (I) with N-methylaniline (II) by heating at 125 C and distillation of the ethanol formed.
By condensation of 4-hydroxy-1-methyl-2-oxo-1,2-dihydroquinoline-3-carboxylic acid ethyl ester (I) with N-methylaniline (II) by heating at 125 C and distillation of the ethanol formed.
CLIP
https://www.sciencedirect.com/science/article/abs/pii/S0731708597001076
![Image result for Roquinimex NMR]() 1H-NMR spectrum of linomide in DMSO at 298 K recorded on a Bruker AC
1H-NMR spectrum of linomide in DMSO at 298 K recorded on a Bruker AC
![Image result for Roquinimex NMR]() 13C-DEPT experiment of linomide in DMSO at 298 K recorded on a Bruker AC
13C-DEPT experiment of linomide in DMSO at 298 K recorded on a Bruker AC
![Image result for Roquinimex NMR]()
![Image result for Linomide NMR]()
A 2D 13C–1H COLOC experiment of linomide in DMSO at 298 K recorded on
![Image result for Linomide NMR]()
![Image result for Linomide NMR]() COSY 45° spectrum of linomide in DMSO at 298 K recorded on a Bruker AC
COSY 45° spectrum of linomide in DMSO at 298 K recorded on a Bruker AC
PATENT
https://patents.google.com/patent/US5912349
U.S. Pat. No. 4,738,971 discloses roquinimex and a method to produce it. The disclosed method starts with N-methylisatoic anhydride (I) and requires three steps. The improved process of the present invention starts with the same N-methylisatoic anhydride (I) and requires fewer steps.
The process of the present invention is practiced according to EXAMPLE 2. It is preferred to perform the claimed process in an aprotic solvent. Suitable aprotic solvents include DMF, THF, glyme, dioxane and ether and mixtures thereof.
The roquinimex produced by the process of the invention (EXAMPLE 2) can be upgraded or purified by the process of EXAMPLE 3.
Roquinimex is known to be useful as a pharmaceutical agent, see U.S. Pat. No. 4,738,971. It is preferably used in treating multiple sclerosis, in particular the treatment of relapsing remitting and secondary progressive multiple sclerosis. In treating multiple sclerosis roquinimex is administered in an oral dose of from about 2.0 to about 5.0 mg/day.
Example 1
N-Methyl-N-Phenyl-α-Carbomethoxyacetamide (V)
Mono-methyl malonate potassium salt also known as potassium methyl malonate (73.32 g, 0.47 mol) and water (50 ml) are cooled to 5° with an ice bath, and concentrated hydrochloric acid (40 ml) is added over a 30 minute period while the temperature is maintained below 10°. The mixture is filtered with suction to remove potassium chloride, and the precipitate washed with methyl t-butylether (75 ml). The aqueous layer of the filtrate is separated and washed with methyl t-butyl ether (3×50 ml). The combined methyl t-butyl ether extracts are dried over anhydrous sodium sulfate; then the solvent was removed under reduced pressure at 45-50° to give carbomethoxy acetic acid. This product was checked by NMR for complete removal of the methyl t-butyl ether solvent.
Carbomethoxy acetic acid (100 g, 0.84 mol) is dissolved in methylene chloride (400 ml). Thionyl chloride (100 g, 0.84 mol) is added via a dropping funnel. It can be added rapidly as there is little, if any, exotherm produced during the addition. After addition, the reaction is refluxed at 40-45° for 1 hr. At the end of the reflux period, 50% of the methylene chloride is removed (200 ml) by distillation at atmospheric pressure and 40-45°. Fresh methylene chloride is added (200 ml) followed by distillation to again remove 50% of the total volume. This add-distillation procedure is repeated two times to give the carbomethoxy acetyl chloride.
The carbomethoxy acetyl chloride mixture is cooled in an ice-salt bath to -5 to 0° and N-methyl aniline (55.64 g, 0.52 mol) in methylene chloride (200 ml) is added at a rate so as to maintain the temperature of the reaction mixture between -5 to 0°. The addition is performed using an addition funnel and can normally be carried out over a 3-5 min time period to control the slight exotherm. Pyridine (66.36 g, 0.84 mol) in methylene chloride (200 ml) is then added to the above mixture. The addition rate is adjusted so as to keep the temperature of the reaction between -5 to 0° during the addition. The addition is performed using an addition funnel and can normally be carried out over a 3-5 min time period to control the slight exotherm. After the addition is complete (as measured by HPLC) the reaction is quenched by pouring the reaction mixture into water (500 ml) and stirring continued for 30 min. The reaction is equilibrated and the methylene chloride layer separated. Additional methylene chloride (400-500 ml) is added and the methylene chloride mixture is washed successively with hydrochloric acid (1N, 2×300 ml), saturated sodium bicarbonate solution (2×300 ml), saline (1×600 ml) and the methylene chloride mixture dried through anhydrous sodium sulfate. Concentration of the mixture under reduced pressure at 40-45° gives the title compound, HPLC (Nucleosil column; acetonitrile/water, 45/55, 1 ml/min, UV=229 nm; Retention times for N-methyl-N-phenyl-α-carbomethoxyacetamide˜6.0 min; N-methyl aniline˜11.0-12.0 min.
Example 2
Preparation of Roquinimex (IV) from N-Methylisotoic anhydride (I) and N-Methyl-α-carbomethoxyacetamide (V)
N-Methyl-N-phenyl-α-carbomethoxyacetamide (V, EXAMPLE 1, 139 g, 0.671 mole) and DMF (695 mL). The mixture is subject to reduced pressure and purged with nitrogen three times. While at room temperature (20-25°), potassium t-butoxide solution (1.714 M in THF, 367 mL, 0.630 mole) is added in one portion. A small exotherm and slight darkening of the mixture followed this addition. The mixture is heated to 80-90° and kept at this temperature for 1.5 hr.
A -78° cooling bath is placed on the receiving flask of the distillation assembly, the nitrogen flow is shut off and the mixture is subject to reduced pressure over 0.5 hr to remove the THF solvent. The pot temperature at the end of the distillation is 72-76°. The amount of distillate collected should be nearly identical to the amount of potassium tert-butoxide reagent used, (367 ml). The mixture is then heated to 80-85° and N-methylisotoic anhydride (I, 70.72 g, 0.400 mole) is added in one portion followed by a 5-10 mL DMF wash. Gas evolution with foaming followed the addition and subsequent wash. The equipment is modified at this point to include a reflux condenser with a vacuum port. With the temperature still at 80-85°, the mixture is placed under reduced pressure and the mixture refluxed for 30 min. After refluxing the temperature is 79°. The reduced pressure and heat source are removed, the system is repressurized with nitrogen and the temperature is allowed to drop to 30° (±2°). Hydrochloric acid (0.6 N, 2.295 L) is added slowly via an addition funnel attached to the claisen head over 2.5 hr, to pH=1.0-1.5, making sure the temperature does not exceed 32°. The temperature control is especially critical at the beginning of the addition when a mild exotherm occurs. The temperature at the end of the addition is nearly room temperature (24-25°). When the acid addition is complete, the resulting slurry is stirred for 30 min and then let stand overnight before filtration. The solids are washed with water (2 ×330 mL) and dried on a nitrogen press to give the title compound, HPLC (Nucleosil column; acetonitrile/water, 45/55, 1 ml/min, UV=229 nm; Retention times 2.29 min.
Example 3
Purification of Roquinimex (IV)
Roquinimex crude is taken up in water (1.5 L) and the slurry is stirred vigorously at 20-25°. The pH is adjusted to 7.5-7.7 with sodium hydroxyde (7%, about 170 mL). (The base can be added as fast as possible but requires longer pH equilibration near the end of the addition (about 1-2 hr total addition time). It is recommended that 85% of the base is initially added to a stable pH and the rest is added dropwise until the pH has stabilized and falls into the desired range of 7.5-7.7.) Nearly all solids should be dissolved (some may remain however). After the base is added and the pH is stabilized for more than 30 min, Darco (charcoal, 15.00 g) is added and the mixture is stirred for 30 min. The mixture is filtered through a 0.45 micron Millipore filter and the filter cake is washed with water (2×175 mL). The filtrate is transferred to a flask.
The mixture is stirred vigorously, heated to 28-32° and hydrochloric acid (6 N, about 120 mL) is added over 30 to 45 min to a pH of 0.5 to 1.0. After the addition is over, the mixture is stirred for 15 min then allowed to stand, without stirring, at the above temperature for 2 hr before filtration. The filter cake is washed with water (2×180 mL) and dried on a nitrogen press to give essentially pure title compound.
PATENT
https://patents.google.com/patent/US6605616
The present invention relates to novel substituted quinoline-3-carboxamide derivatives, to methods for their preparation, to compositions containing them, and to methods and use for clinical treatment of diseases resulting from autoimmunity, such as multiple sclerosis, insulin-dependent diabetes mellitus, systemic lupus erythematosus, rheumatoid arthritis, inflammatory bowel disease and psoriasis and, furthermore, diseases where pathologic inflammation plays a major role, such as asthma, atherosclerosis, stroke and Alzheimer’s disease. More particularly, the present invention relates to novel quinoline derivatives suitable for the treatment of, for example, multiple sclerosis and its manifestations.
BACKGROUND OF THE INVENTION
Autoimmune diseases, e.g., multiple sclerosis (MS), insulin-dependent diabetes mellitus (IDDM), systemic lupuis erythematosus (SLE), rheumatoid arthritis (RA), inflammatory bowel disease (IBD) and psoriasis represent assaults by the body’s immune system which may be systemic in nature, or else directed at individual organs in the body. They appear to be diseases in which the immune system makes mistakes and, instead of mediating protective functions, becomes the aggressor (1).
MS is the most common acquired neurologic disease of young adults in western Europe and North America. It accounts for more disability and financial loss, both in lost income and in medical care, than any other neurologic disease of this age group. There are approximately 250.000 cases of MS in the United States. Although the cause of MS is unknown, advances in brain imaging, immunology, and molecular biology have increased researchers’ understanding of this disease. Several therapies are currently being used to treat MS, but no single treatment has demonstrated dramatic treatment efficacy. Current treatment of MS falls into three categories: treatment of acute exacerbations, modulation of progressive disease, and therapy for specific symptoms.
MS affects the central nervous system and involves a demyelination process, i.e., the myelin sheaths are lost whereas the axons are preserved. Myelin provides the isolating material that enables rapid nerve impulse conduction. Evidently, in demyelination, this property is lost. Although the pathogenic mechanisms responsible for MS are not understood, several lines of evidence indicate that demyelination has an immunopathologic basis. The pathologic lesions, the plaques, are characterized by infiltration of immunologically active cells such as macrophages and activated T cells (2).
In U.S. Pat. No. 4,547,511 and in U.S. Pat. No. 4,738,971 and in EP 59,698 some derivatives of N-aryl-1,2-dihydro-4-substituted-1-alkyl-2-oxo-quinoline-3-carboxamide are claimed as enhancers of cell-mediated immunity. The compound
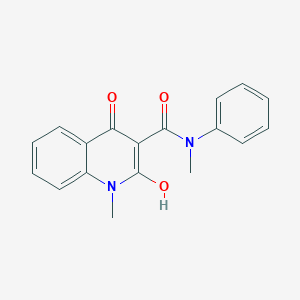
![Figure US06605616-20030812-C00002]()
known as roquinimex (Merck Index 12th Ed., No. 8418; Linomide®, LS2616, N-phenyl-N-methyl-1,2-dihydro-4-hydroxy-1-methyl-2-oxo-quinoline-3-carboxamide) belongs to this series of compounds. Roquinimex has been reported to have multiple immunomodulatory activities not accompanied with general immunosuppression (3-12). Furthermore, in U.S. Pat. No. 5,580,882 quinoline-3-carboxarnide derivatives are claimed to be useful in the treatment of conditions associated with MS. The particular preferred compound is roquinimex. In U.S. Pat. No. 5,594,005 quinoline-3-carboxamide derivatives are claimed to be useful in the treatment of type I diabetes. The particular preferred compound is roquinimex. In WO 95/24195 quinoline-3-carboxamide derivatives are claimed to be useful in the treatment of inflammatory bowel disease. Particularly preferred compounds are roquinimex or a salt thereof. In WO95/24196 quinoline-3-carboxamide derivatives are claimed to be useful in the treatment of psoriasis. Particularly preferred compounds are roquinimex or a salt thereof.
In clinical trials comparing roquinimex to placebo, roquinimex was reported to hold promise in the treatment of conditions associated with MS (13, 14). There are, however, some serious drawbacks connected to roquinimex. For example, it has been found to be teratogenic in the rat, and to induce dose-limiting side effects in man, e.g., a flu-like syndrome, which prevents from using the full clinical potential of the compound.
Further, in WO 92/18483 quinoline derivatives substituted in the 6-position with a RAS (O)n-group (RA=lower alkyl or aryl; n=0−2) are claimed, which possess an immunomodulating, anti-inflammatory and anti-cancer effect.
PAPER
Modified synthesis and antiangiogenic activity of linomide
https://www.sciencedirect.com/science/article/pii/S0960894X00006995?via%3Dihub
![]()
PAPER
https://pubs.acs.org/doi/full/10.1021/jm031044w
1H NMR (CDCl3) δ 3.28 (s, br, 3H, 1-NCH3), 3.50 (s, 3H, 12-NCH3), 7.1−7.3 (m, 7H, 6,8,2‘,3‘,4‘,5‘,6‘-aromatic CH), 7.56 (dt, JHCCH = 7.5 and 8.5 Hz, JHCCCH = 1.5 Hz, 1H, 7-aromatic CH), 8.09 (dd, JHCCH = 8.0 Hz, JHCCCH = 1.5 Hz, 1H, 5-aromatic CH), 12.3 (s, br, 1H, 4-OH). 13C NMR (CDCl3) δ 28.7 (1C, 1-NCH3), 38.3 (1C, br, 12-NCH3), 104.6 (1C, 3-C), 113.5 (1C, 8-CH), 115.3 (1C, 10-C), 121.4 (1C, 6-CH), 124.6 (1C, 5-CH), 125.5 (2C, 2‘,6‘-CH), 126.7 (1C, 4‘-CH), 128.5 (2C, 3‘,5‘-CH), 132.3 (1C, 7-CH), 140.1 (1C, 9-C), 143.8 (1C, 1‘-C), 158.8 (1C, 2-CO), 164.3 (1C, 4-C), 169.4 (1C, 11-CO). MS-ESI: m/z 309 [MH]+. Anal. (C18H16N2O3) C, H, N.
PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2012050500&recNum=1&maxRec=&office=&prevFilter=&sortOption=&queryString=&tab=PCTDescription
1,2-dihydro-4-hydroxy-2-oxo-quinoline-3-carboxanilides have been described in the literature since the 1970s (refs 1-4). The most well-known compound in this class, roquinimex (Linomide), was first described by AB Leo as an immuno-stimulating agent (ref 4) but was later also found to have immuno-modulating effects, as well as anti-angiogenetic effects (refs 5a, b). Roquinimex has been claimed beneficial for the treatment of autoimmune diseases, such as rheumatoid arthritis, multiple sclerosis, systemic lupus erythematosus, inflammatory bowel disease, diabetes type 1, and psoriasis, as well as for the treatment of cancer (refs 6a-d, 9d and refs therein).
![]()
![]()
The compound laquinimod (a 5-Cl, N-Et carboxanilide derivative) has been reported by Active Biotech AB to convey a better therapeutic index compared with roquinimex (refs 7a, b) and is currently in phase III clinical studies for the treatment of multiple sclerosis. Laquinimod has also entered clical trials in Crohn’s disease and
SLE. Two other compounds in the same class under clinical evaluation are tasquinimod (prostate cancer) and paquinimod (systemic sclerosis). Recently, a molecular target for laquinimod was identified as S100A9 (ref 8).
Fujisawa has reported on similar compounds with inhibitory activity on nephritis and on B16 melanoma metastases (refs 9a-d). Also the closely related thieno-pyridone analogs have been described as immunomodulating compounds with anti-inflammatory properties (ref
10).
Another closely related compound class are the corresponding N-pyridyl-carboxamide derivatives, which have been reported to have antitubercular activity as well as anti-inflammatory properties (ref
11). However, according to litterature (ref 10) these derivatives are less active as immunomodulating agents.
The N-hydrogen 3-carboxanilides (“N-H derivatives”) and the N-alkyl 3-carboxanilides (“N-alkyl derivatives”), respectively, are described in the prior art documents relating to inflammation, immunomodulation, and cancer as a homogenous group of compounds in terms of biological effects. Prior art also teaches that the N-alkyl derivatives are the preferred compound derivatives.
![]()
In fact, very few studies (refs 4, 9d) of N-hydrogen derivatives, especially in vivo studies, have been reported. Furthermore, no fundamental biological differences between the N-alkyl derivatives and the N-hydrogen derivatives, respectively, have been described.
However, some chemical properties of the N-hydrogen and the N-alkyl derivatives are different (ref 12). N-Alkyl derivatives adopt a twisted 3D-structure, whereas the N-H derivatives are stabilized by intramolecular hydrogen bonds in a planar structure. The N-alkyl derivatives are more soluble in aqueous media, but also inherently unstable towards nucleophiles, such as amines and alcohols (refs 12, 13).
The N-alkyl derivatives roquinimex (N-Me) and laquinimod (N-Et) have been reported to be metabolized in human microsomes to give the corresponding N-hydrogen derivatives, via N-dealkylation catalyzed mainly by CYP3A4 (refs 14a, b).
bHLH-PAS (basic helix-loop-helix Per-Arnt-Sim) proteins constitute a recently descovered protein family functioning as transcripon factors as homo or hetero protein dimers (refs 15a, b). The N-terminal bHLH domain is responsible for DNA binding and contributes to dimerization with other family members. The PAS region (PAS-A and PAS-B) is also involved in protein-protein interactions determining the choice of dimerization partner and the PAS-B domain harbors a potential ligand binding pocket.
The aryl hydrocarbon receptor (AhR or dioxin receptor) and its dimerization partner ARNT (AhR nuclear translocator) were the first mammalian protein members to be identified. AhR is a cytosolic protein in its non-activated form, associated in a protein complex with Hsp90, p23, and XAP2. Upon ligand activation, typically by chlorinated aromatic hydrocarbons like TCDD, the Ahr enters the nucleus and dimerizes with ARNT. The AhR/ARNT dimer recognizes specific xenobiotic response elements (XREs) to regulate TCDD-responsive genes. The ligand binding domain of AhR (AhR-LBD) resides in the PAS-B domain.
Recently, it has been demonstrated that AhR is involved in Thl7 and Treg cell development and AhR has been proposed as a unique target for therapeutic immuno-modulation (refs 16a-c). The AhR ligand TCDD was shown to induce development of Treg (FoxP3+) cells, essential for controlling auto-immunity, and to suppress symptoms in the EAE model. In addition, activation of AhR has been shown essential for the generation of IL-10 producing regulatory Trl cells (ref 16d), and Ahr ligands have also been proven efficacious in other models of auto-immunity, e.g. diabetes type 1, IBD, and uveitis (refs 16e-h). Apart from controlling autoimmune disorders, AhR activation and Treg cell development have been implicated as a therapeutic strategy for other conditions with an immunological component, such as allergic lung inflammation, food allergy, transplant rejection, bone loss, and type 2 diabetes and other metabolic disorders (refs 17a-e).
Apart from its role as a transcription factor, AhR has been reported to function as a ligand-dependent E3 ubiguitin ligase (ref 18), and ligand-induced degradation of β-catenin has been demonstrated to suppress intestinal cancer in mice (ref 19). In addition, activation of AhR has been implicated to play a protective role in prostate cancer (ref 20).
Other members of the bHLH-PAS family are the HIF-α (hypoxia inducible factor alpha) proteins, which also hetero-dimerize with ARNT. In conditions with normal oxygen levels (normoxia), HIF-α proteins are rapidly degraded by the ubiquitin-proteasome system and they are also inactivated by asparagine hydroxylation. Under hypoxic conditions, however, the proteins are active and upregulate genes as a response to the hypoxic state, e.g. genes for erythropoietin and vascular endothelial growth factor (VEGF). VEGF is essential for blood vessel growth (angiogenesis) and is together with HIF-1α considered as interesting targets for anti-angiogenetic tumour theraphy (ref 21). HIF-α proteins can be negatively and indirectly regulated by AhR ligands, which upon binding with AhR reduce the level of the common dimerization partner ARNT. Anti-angiogenetic effects can possibly also be achieved directly by AhR activity via upregulation of thrombospondin-1 (ref 22).
![Roquinimex]() |
|
|
Title: Roquinimex
CAS Registry Number: 84088-42-6
CAS Name: 1,2-Dihydro-4-hydroxy-N,1-dimethyl-2-oxo-N-phenyl-3-quinolinecarboxamide
Additional Names: N-phenyl-N-methyl-1,2-dihydro-4-hydroxy-1-methyl-2-oxoquinoline-3-carboxamide; 1,2-dihydro-4-hydroxy-N,1-dimethyl-2-oxo-3-quinolinecarboxanilide
Manufacturers’ Codes: LS-2616
Trademarks: Linomide (Pfizer)
Molecular Formula: C18H16N2O3
Molecular Weight: 308.33
Percent Composition: C 70.12%, H 5.23%, N 9.09%, O 15.57%
Literature References: Biological response modifier. Prepn: E. Eriksoo et al., EP 59698; eidem, US 4738971 (1982, 1988 both to AB Leo). Immunopharmacology: A. Tarkowski et al., Immunology 59, 589 (1986). Mechanism of action study: E.-L. Larsson et al.,Int. J. Immunopharmacol. 9, 425 (1987). Clinical evaluation in cancer patients: J. C. S. Bergh et al., Cancer Invest. 15, 204 (1997).
Properties: Crystals from pyridine, mp 200-204°.
Melting point: mp 200-204°
Therap-Cat: Antineoplastic.
Keywords: Antineoplastic; Immunomodulators.
|
Roquinimex (Linomide) is a quinoline derivative immunostimulant which increases NK cell activity and macrophage cytotoxicity. It also inhibits angiogenesis and reduces the secretion of TNF alpha.
/////////////////Roquinimex, Linomide, FCF-89, LS-2616, PNU-212616
CN1C2=CC=CC=C2C(=O)C(=C1O)C(=O)N(C)C3=CC=CC=C3





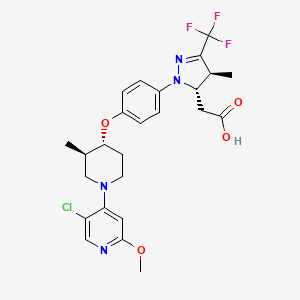






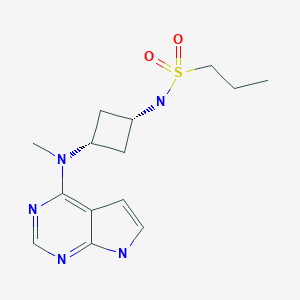
 , Zhengxiang Gu, Elizabeth Anne Jurica
, Zhengxiang Gu, Elizabeth Anne Jurica 


 Rick Ewing, Head, External Partnerships, Discovery Chemistry and Molecular Technologies at Bristol-Myers Squibb
Rick Ewing, Head, External Partnerships, Discovery Chemistry and Molecular Technologies at Bristol-Myers Squibb



 Sundara Lakshmi Kanniah
Sundara Lakshmi Kanniah

 reported that Indoco Remedies was capable of producing commercial quantities of dorzolamide hydrochloride and holds an active US DMF since 2010
reported that Indoco Remedies was capable of producing commercial quantities of dorzolamide hydrochloride and holds an active US DMF since 2010

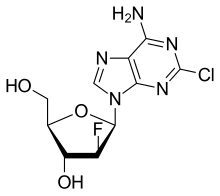
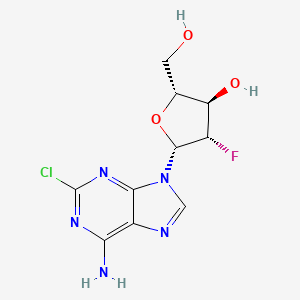























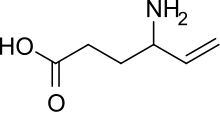



 The enantiocontrolled addition of phthalimide (I) to 1,3-butadiene monoepoxide (II) with a chiral palladium catalyst and Na2CO3 in dichloromethane gives N-(2-hydroxy-1(S)-vinylethyl)phthalimide (III), which is treated with triflic anhydride and TEA in dichloromethane to yield the triflate (IV). The condensation of (IV) with dimethyl malonate (V) by means of NaH in THF affords the alkylated malonate (VI), which is finally decarboxylated and deprotected by a treatment with aqueous refluxing HCl. Note that the synthesis of the biologically active (S)-enantiomer simply requires a change in the chirality of the Pd catalyst used in the first step of the synthesis.
The enantiocontrolled addition of phthalimide (I) to 1,3-butadiene monoepoxide (II) with a chiral palladium catalyst and Na2CO3 in dichloromethane gives N-(2-hydroxy-1(S)-vinylethyl)phthalimide (III), which is treated with triflic anhydride and TEA in dichloromethane to yield the triflate (IV). The condensation of (IV) with dimethyl malonate (V) by means of NaH in THF affords the alkylated malonate (VI), which is finally decarboxylated and deprotected by a treatment with aqueous refluxing HCl. Note that the synthesis of the biologically active (S)-enantiomer simply requires a change in the chirality of the Pd catalyst used in the first step of the synthesis.





 By condensation of 4-hydroxy-1-methyl-2-oxo-1,2-dihydroquinoline-3-carboxylic acid ethyl ester (I) with N-methylaniline (II) by heating at 125 C and distillation of the ethanol formed.
By condensation of 4-hydroxy-1-methyl-2-oxo-1,2-dihydroquinoline-3-carboxylic acid ethyl ester (I) with N-methylaniline (II) by heating at 125 C and distillation of the ethanol formed. 1H-NMR spectrum of linomide in DMSO at 298 K recorded on a Bruker AC
1H-NMR spectrum of linomide in DMSO at 298 K recorded on a Bruker AC 13C-DEPT experiment of linomide in DMSO at 298 K recorded on a Bruker AC
13C-DEPT experiment of linomide in DMSO at 298 K recorded on a Bruker AC


 COSY 45° spectrum of linomide in DMSO at 298 K recorded on a Bruker AC
COSY 45° spectrum of linomide in DMSO at 298 K recorded on a Bruker AC








 +91- 999-997-2051
+91- 999-997-2051 info@synthesiswithcatalysts.com
info@synthesiswithcatalysts.com