![]()



Example 13 WO2015104688
6-(6-aminopyridin-3-yl)-N-(2-morpholin-4-yl-1,3-benzothiazol-6-yl)pyridine-2-carboxamide
| Molecular Formula: | C22H20N6O2S |
|---|---|
| Molecular Weight: | 432.4982 g/mol |
PROBABLE STRUCTURE

Example 1 ……..6′-amino-N-(2-morpholinooxazolo[4,5-b]pyridin-6-yl)-[2,3′-bipyridine]-6-carboxamideWO2015104688

Compound-6: 6′-amino-N-(5-(cyclopropyIamino)-2-morpholinobenzo [d]oxazoI-6-yl)-[2,3′-bipyridine]-6-carboxamide.WO2013042137
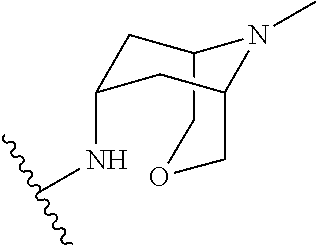
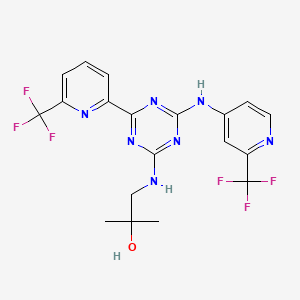
PROBABLE CA 4948, AU 4948,AU-4948, CA-4948
STRUCTURE AND SYNTHESIS COMING……..
| Latest Stage of Development | Preclinical |
| Standard Indication | B cell lymphoma |
| Indication Details | Treat diffuse large B cell lymphoma (DLBCL) |
| Regulatory Designation | |
| Partner | Curis Inc. |
Interleukin-1 Receptor Associated Kinase-4 (IRAK-4) is a serine/threonine protein kinase belonging to tyrosine like kinase (TLK) family. IRAK-4 is one of the important signalling components downstream of IL-1/Toll family of receptors (IL-1R, IL-18R, IL-33R, Toll-like receptors). Recent studies have reported occurrence of oncogenic mutations in MYD88 in 30% of ABC diffuse large B cell lymphomas (ABC DLBCL) and 90% of Waldenstrom’s macroglobulinemia (WM). Most of ABC DLBCLs have a single amino acid substitution of proline for the leucine at position 265 (L265P) in the TIR domain of MYD88 protein resulting in constitutive activation of IRAK-4. Thus, IRAK4 is an attractive therapeutic target for the treatment of B-cell lymphomas with activating MYD88 L265P mutation. We have designed, synthesized and tested small molecule IRAK-4 inhibitors based on hits originating from Aurigene’ s compound library. These novel compounds were profiled for IRAK4 kinase inhibition, anti-proliferative activity, kinase selectivity, and drug-like properties. Furthermore, selected compounds were tested in a proliferation assay and pIRAK1 mechanistic assay using ABC-DLBCL cell lines with activating MYD88 L265P mutation, OCI-lLy10 and OCI-lLy3. We have identified a series of novel bicyclic heterocycles as potent inhibitors of IRAK-4. Aurigene Lead compound exhibited potent inhibitory activity for IRAK-4 with an IC50 of 3nM in biochemical assay. Aurigene Lead compound inhibited pIRAK1 levels, and proliferation of OCI-Ly3 and OCI-Ly10 cells with an IC501of 132nM and 52nM respectively. To the best of our knowledge, Aurigene Lead compound represents the most potent IRAK4 inhibitor reported for target modulation and anti-proliferative activity in DLBCL cell lines with activating MYD88 L265P mutation. Aurigene Lead compound has good oral pharmacokinetic profile in mice and has demonstrated excellent pharmacodynamic effect in an in vivo LPS induced TNF-α model with an ED50 of 3.8 mg/Kg in mice. Preliminary in vitro tox studies indicated clean safety profile. Demonstration of efficacy in OCI-lLy10 mouse tumor model is ongoing. In summary, a series of potent IRAK-4 inhibitors belonging to 3 different chemical series have been discovered and are being evaluated for treatment of B-cell lymphomas.
Curis with the option to exclusively license Aurigene’s orally-available small molecule inhibitor of Interleukin-1 receptor-associated kinase 4 (IRAK4) in the precision oncology field. Curis expects to exercise its option to obtain exclusive licenses to both programs and file IND applications for a development candidate from each in 2015.
Recent studies have also shown that alterations of the MYD88 gene lead to dysregulation of its downstream target IRAK4 in a number of hematologic malignancies, including Waldenström’s Macroglobulinemia and a subset of diffuse large B-cell lymphomas, making IRAK4 an attractive target for the treatment of these cancers.
![]()
Curis and Aurigene Announce Collaboration, License and Option Agreement to Discover, Develop and Commercialize Small Molecule Antagonists for Immuno-Oncology and Precision Oncology Targets
— Agreement Provides Curis with Option to Exclusively License Aurigene’s Antagonists for Immuno-Oncology, Including an Antagonist of PD-L1 and Selected Precision Oncology Targets, Including an IRAK4 Kinase Inhibitor —
— Investigational New Drug (IND) Application Filings for Both Initial Collaboration Programs Expected this Year —
— Curis to issue 17.1M shares of its Common Stock as Up-front Consideration —
— Management to Host Conference Call Today at 8:00 a.m. EST —
LEXINGTON, Mass. and BANGALORE, India, Jan. 21, 2015 (GLOBE NEWSWIRE) — Curis, Inc. (Nasdaq:CRIS), a biotechnology company focused on the development and commercialization of innovative drug candidates for the treatment of human cancers, and Aurigene Discovery Technologies Limited, a specialized, discovery stage biotechnology company developing novel therapies to treat cancer and inflammatory diseases, today announced that they have entered into an exclusive collaboration agreement focused on immuno-oncology and selected precision oncology targets. The collaboration provides for inclusion of multiple programs, with Curis having the option to exclusively license compounds once a development candidate is nominated within each respective program. The partnership draws from each company’s respective areas of expertise, with Aurigene having the responsibility for conducting all discovery and preclinical activities, including IND-enabling studies and providing Phase 1 clinical trial supply, and Curis having responsibility for all clinical development, regulatory and commercialization efforts worldwide, excluding India and Russia, for each program for which it exercises an option to obtain a license.
The first two programs under the collaboration are an orally-available small molecule antagonist of programmed death ligand-1 (PD-L1) in the immuno-oncology field and an orally-available small molecule inhibitor of Interleukin-1 receptor-associated kinase 4 (IRAK4) in the precision oncology field. Curis expects to exercise its option to obtain exclusive licenses to both programs and file IND applications for a development candidate from each in 2015.
“We are thrilled to partner with Aurigene in seeking to discover, develop and commercialize small molecule drug candidates generated from Aurigene’s novel technology and we believe that this collaboration represents a true transformation for Curis that positions the company for continued growth in the development and eventual commercialization of cancer drugs,” said Ali Fattaey, Ph.D., President and Chief Executive Officer of Curis. “The multi-year nature of our collaboration means that the parties have the potential to generate a steady pipeline of novel drug candidates in the coming years. Addressing immune checkpoint pathways is now a well validated strategy to treat human cancers and the ability to target PD-1/PD-L1 and other immune checkpoints with orally available small molecule drugs has the potential to be a distinct and major advancement for patients. Recent studies have also shown that alterations of the MYD88 gene lead to dysregulation of its downstream target IRAK4 in a number of hematologic malignancies, including Waldenström’s Macroglobulinemia and a subset of diffuse large B-cell lymphomas, making IRAK4 an attractive target for the treatment of these cancers. We look forward to advancing these programs into clinical development later this year.”
Dr. Fattaey continued, “Aurigene has a long and well-established track record of generating targeted small molecule drug candidates with bio-pharmaceutical collaborators and we have significantly expanded our drug development capabilities as we advance our proprietary drug candidates in currently ongoing clinical studies. We believe that we are well-positioned to advance compounds from this collaboration into clinical development.”
CSN Murthy, Chief Executive Officer of Aurigene, said, “We are excited to enter into this exclusive collaboration with Curis under which we intend to discover and develop a number of drug candidates from our chemistry innovations in the most exciting fields of cancer therapy. This unique collaboration is an opportunity for Aurigene to participate in advancing our discoveries into clinical development and beyond, and mutually align interests as provided for in our agreement. Our scientists at Aurigene have established a novel strategy to address immune checkpoint targets using small molecule chemical approaches, and have discovered a number of candidates that modulate these checkpoint pathways, including PD-1/PD-L1. We have established a large panel of preclinical tumor models in immunocompetent mice and can show significant in vivo anti-tumor activity using our small molecule PD-L1 antagonists. We are also in the late stages of selecting a candidate that is a potent and selective inhibitor of the IRAK4 kinase, demonstrating excellent in vivo activity in preclinical tumor models.”
In connection with the transaction, Curis has issued to Aurigene approximately 17.1 million shares of its common stock, or 19.9% of its outstanding common stock immediately prior to the transaction, in partial consideration for the rights granted to Curis under the collaboration agreement. The shares issued to Aurigene are subject to a lock-up agreement until January 18, 2017, with a portion of the shares being released from the lock-up in four equal bi-annual installments between now and that date.
The agreement provides that the parties will collaborate exclusively in immuno-oncology for an initial period of approximately two years, with the option for Curis to extend the broad immuno-oncology exclusivity.
In addition Curis has agreed to make payments to Aurigene as follows:
- for the first two programs: up to $52.5 million per program, including $42.5 million per program for approval and commercial milestones, plus specified approval milestone payments for additional indications, if any;
- for the third and fourth programs: up to $50 million per program, including $42.5 million per program for approval and commercial milestones, plus specified approval milestone payments for additional indications, if any; and
- for any program thereafter: up to $140.5 million per program, including $87.5 million per program in approval and commercial milestones, plus specified approval milestone payments for additional indications, if any.
Curis has agreed to pay Aurigene royalties on any net sales ranging from high single digits to 10% in territories where it successfully commercializes products and will also share in amounts that it receives from sublicensees depending upon the stage of development of the respective molecule.
About IRAK4:
Interleukin-1 receptor-associated kinase 4, or IRAK4 is a signaling kinase that becomes inappropriately activated in certain cancers including activated B cell-diffuse large B cell lymphoma (ABC-DLBCL), an aggressive form of lymphoma with poor prognosis. There appears to be a mechanistic link with IRAK4 in ABC-DLBCL where these tumors from approximately 35% of patients harbor oncogenic mutations in the MYD88 gene, which encodes an adaptor protein that interacts directly with IRAK4. MYD88 mutations appear to constitutively activate the IRAK4 kinase complex, driving pro-survival pathways in ABC-DLBCL disease. Oncogenic MYD88 mutations have also been identified in other cancers, including in over 90% of patients with Waldenström’s Macroglobulinemia as well as in a subset of patients with chronic lymphocytic leukemia (CLL).
About Curis, Inc.
Curis is a biotechnology company focused on the development and commercialization of novel drug candidates for the treatment of human cancers. Curis’ pipeline of drug candidates includes CUDC-907, a dual HDAC and PI3K inhibitor, CUDC-427, a small molecule antagonist of IAP proteins, and Debio 0932, an oral HSP90 inhibitor. Curis is also engaged in a collaboration with Genentech, a member of the Roche Group, under which Genentech and Roche are developing and commercializing Erivedge®, the first and only FDA-approved medicine for the treatment of advanced basal cell carcinoma. For more information, visit Curis’ website at www.curis.com.
About Aurigene
Aurigene is a specialized, discovery stage biotechnology company, developing novel and best-in-class therapies to treat cancer and inflammatory diseases. Aurigene’s Programmed Death pathway program is the first of several immune checkpoint programs that are at different stages of discovery and preclinical development. Aurigene has partnered with several large- and mid-pharma companies in the United States and Europe and has delivered multiple clinical compounds through these partnerships. With over 500 scientists, Aurigene has collaborated with 6 of the top 10 pharma companies. Aurigene is an independent, wholly owned subsidiary of Dr. Reddy’s Laboratories Ltd. (NYSE:RDY). For more information, please visit Aurigene’s website at http://aurigene.com/.
Small Molecule IRAK4 Kinase Inhibitor)
Innate immune responses mediated through Toll-like receptors or certain interleukin receptors are important mediators of the body’s initial defense against foreign antigens, while their dysregulation is associated with certain inflammatory conditions. Toll-like receptor and interleukin receptor signaling through the adaptor protein MYD88, results in the assembly and activation of IRAK4, initiating a signaling cascade that induces cytokine and survival factor expression mediated by the transcription factor NFκB. More recently, components of this pathway are recognized to be genetically altered and have important roles in specific human cancers. Toll-like receptor and interleukin receptor signaling through the adaptor protein MYD88, results in the assembly and activation of IRAK4, initiating a signaling cascade that induces cytokine and survival factor expression mediated by the transcription factor NFκB. MYD88 gene mutations are shown to occur in approximately 30% of Activated B-Cell (ABC) subtype of diffuse large B-cell lymphomas (DLBCL)1,2 and in over 90% of the B-cell malignancy Waldenstrom’s macroglobulinemia.3 Due to IRAK4’s central role in these signaling pathways, it is considered an attractive target for generation of therapeutics to treat these B-cell malignancies as well as certain inflammatory diseases.
As part of the collaboration with Aurigene, in October 2015 we exercised our option to exclusively license a program of orally-available, small molecule inhibitors of IRAK4 kinase, including the development candidate, CA-4948. Curis expects to file an IND and initiate clinical testing of CA-4948 in patients with advanced hematologic cancers during the second half of 2016.
1Nature. 2011; 470(7332):115–1192Immunology and Cell Biology. 2011; 89(6):659–6603N Engl J Med. 30, 2012; 367(9):826–833
CLIP
In November 2015, preclinical data were presented at the 2015 AACR-NCI-EORTC Molecular Targets and Cancer Therapeutics Conference in Boston, MA
Aurigene Collaboration (IRAK4 Inhibitor):
In October 2015, Curis exercised its option to exclusively license a program of orally available small molecule inhibitors of IRAK4 kinase, a serine/threonine kinase involved in innate immune responses as well as in certain hematologic cancers. The Company has since designated the development candidate as CA-4948 and expects to file an IND application for this molecule during 2016.
In November 2015, Curis’ collaborator Aurigene presented preclinical data from the IRAK4 program at the 2015 AACR-NCI-EORTC Molecular Targets and Cancer Therapeutics Conference in Boston, MA. This presentation included data from chemically distinct series of small molecule compounds with potent IRAK4 inhibitory activity in biochemical assays as well as in in vivo preclinical models, including MYD88 mutant DLBCL xenograft tumor models as well as a model of inflammatory disease.
CLIP
In April 2014, preclinical data presented at the CHI’s Ninth Drug Discovery Chemistry Conference in San Diego, CA, showed the compounds in vivo to have activity down to 10 mg/kg .
CLIP

 Susanta Samajdar, Ph.D., Research Director, Medicinal Chemistry, Aurigene Discovery Technologies Limited
Susanta Samajdar, Ph.D., Research Director, Medicinal Chemistry, Aurigene Discovery Technologies LimitedApril 24-25 2014
Drug Discovery Chemistry – CHI’s Ninth Annual Conference: Fifth Annual Kinase inhibitor Chemistry, San Diego, CA, USA
Novel IRAK4 inhibitors
Susanta Samajdar from Aurigene Discovery Technologies presented the discovery of new IRAK4 (IL-1 receptor-associated kinase 4) inhibitors. Research began with a HTS campaign using two types of libraries: rationally designed novel scaffolds by hopping and morphing of known IRAK4 inhibitors and novel scaffolds identified by virtual screening of drug-like commercial library. A benzoxazol series was identified and crystallography was used to help their design. Lead optimization culminated in the identification of very potent compounds (AU-2807 and AU-2202) in cell assay (inflammation pathway and oncology pathway, respectively). The compounds were also active against Flt3 and KDR. Some PD in vivo data using LPS and TNFalpha release were presented in which the compound showed activity down to 10 mg/kg: no other in vivo model data were disclosed, but it was mentioned that studies in the CIA (collagen induced arthritis) model was ongoing. Dr Samajdar answered to three questions, one related to IRAK1 selectivity (the answer was that the compound is fully selective against IRAK1 and IRAK2). It was also mentioned that the compounds have a PBB higher than 98%. And the last question was related to the synergetic effect with BTK inhibitor in activated B-cell like diffuse large B-cell lymphoma, and this effect was observed with these compounds.

Research Director at Aurigene Discovery Technologies
PATENT
http://www.google.com/patents/WO2013042137A1?cl=en
Compound-6: Synthesis of 6′-amino-N-(5-(cyclopropyIamino)-2-morpholinobenzo [d]oxazoI-6-yl)-[2,3′-bipyridine]-6-carboxamide.
Step_l^N-cyclopropyl-2-morpholino-6-nitrobenzo[d]oxazol-5-amine.

N-cyclopropyl-2-moφholino-6-nitrobenzo[d]oxazol-5-amine(0.7g,70%) was prepared from 5-fluoro-2-mo holino-6-nitrobenzo[d]oxazole(lg,Intermediate-2) by treating with cyciopropanamine in sealed tube at 100°C for 8-14h. The progress of the reaction was monitored by TLC. After the reaction was completed, it was extracted with water (15ml) and dichioromethane (2x 15ml). The organic layer was collected, washed with brine, dried over sodium sulfate and concentrated under reduced pressure to get the crude. MS (ES) m/e 305(M+1, 50%).
Steg2:6-bromo-N-(5-(cyclopropylamino)-2-morpholinobenzo[d]oxazol-6-yl)
picolinamide.

Step Π and ii):The process of these steps are adopted from step 2 and step 3 of compound- 1.
Step3:6′-amino-N-(5-(cvclopropvlamino)-2-morpholinobenzord]oxazol-6-yl)-r2,3′- bipyridine]-6-carboxamide.

(i) N-(4-methoxybenzyl)-5-(4,4,5,5-tetramethyl-l ,3,2-dioxaborolan-2-yl)pyridin
Na2C03, Pd(dppf)Cl2, ACN, H20, 80-100°C, 8-14h; TFA, 60-70°C, 8-14h.
6′-amino-N-(5-(cyclopropylamino)-2-mo holinobenzo[d]oxazol-6-yl)-[2,3′-bipyridine]-6- carboxamide (0.03g,61%) was prepared from 6-bromo-N-(5-(cyclopropyIamino)-2- moφholinobenzo[d]o azoI-6-yl)picolinamide(0.07g, step-3) by following the same process used in step-1 and 2 of compound-3.
Ή NMR (400 MHz, DMSO-< ):6 1 1.63 (s, IH), 8.90 (s, IH), 8.61 (s, IH), 8.55 (s, IH), 8.37- 8.03 (m, 2H), 7.39 (s, IH), 6.80-6.62 (s, IH), 3.80-3.59 (m, 15H), 2.88-2.64 (m, 2H). MS (ESI): 472 (M+l , 60%).
PATENT
Example 13
6′-amino-N-(2-morphol ne]-6-carboxamide

Step-1: Synthesis of 6-chloro thiazolo[4,5-c]pyridine-2(3H)-thione
Using the same reaction conditions as described in step 1 of example 1, 4,6-dichloropyridin-3-amine (1.3 g, 7 mmol) was cyclised using potassium ethyl xanthate (2.55 g, 15 mmol) in DMF (25mL) at 150°C for 8h to afford the title compound (1.3 g, 86.6 %) as a light brown solid.
1HNMR (400 MHz, DMSO-d6): δ 14.2-14.0 (b, 1H), 8.274 (s, 1H), 7.931 (s, 1H); LCMS: 100%, m/z = 201.3 (M+l)+.
Step-2: Synthesis of 4-(6-chloro thiazolo[4,5-c]pyridin-2-yl) morpholine
To a suspension of 6-chlorothiazolo[4,5-c]pyridine-2(3H)-thione (0.3 g, 1.16 mmol) in
DCM (4 mL), oxalyl chloride (0.2 mL, 2.38 mmol) and DMF (1.5 mL) were added at 0°C. The resulting mixture was slowly allowed to warm to room temperature and stirred there for 1 h. The reaction mixture was again cooled to 0°C and triethyl amine (0.66 mL, 4.76 mmol) and morpholine (0.13 mL, 1.75 mmol) were added. The reaction mixture was stirred at RT for 1 h and quenched with water and extracted with ethyl acetate. The combined organic layers were washed with water, brine, dried over sodium sulphate and concentrated under reduced pressure. The crude material was purified by column chromatography (EtOAc/n-hexanes 3:7) to afford the title compound (0.14 g, 39.6 %) as a light brown solid.
1H NMR (400 MHz, DMSO-d6): δ 8.47 (s, 1H), 8.04 (s, 1H), 3.74-3.72 (m, 4H), 3.61-3.59 (m, 4H); LCMS: m/z = 256.1 (M+l)+.
Step-3: Synthesis of 6′-amino-/V-(2-morpholino thiazolo [4,5-c]pyridin-6-yl)-[2,3′-bipyridine]-6-carboxamide
Using the same reaction conditions as described in step 4 of example 12, 4-(6-chlorothiazolo[4,5-c] pyridin-2-yl) morpholine (0.081 g, 0.32 mmol), was coupled with tert-butyl (6-carbamoyl-[2,3′-bipyridin]-6′-yl)carbamate (intermediate 2) (0.1 g, 0.32 mmol) using cesium carbonate (0.21 g, 0.64 mmol), XantPhos (0.028g, 0.047mmol) and Pd2(dba)3 (0.015 mg, 0.015 mmol) in toluene : dioxane (2:2mL) to get the crude product. The resultant crude was purified by 60-120 silica gel column chromatography using 2% methanol in DCM as eluent. Further the resultant crude was purified by prep HPLC to afford title compound (0.01 g, 6 %) as an off-white solid.
1H NMR (400 MHz, DMSO-d6): δ 10.65 (s, 1H), 8.88 (d, 1H), 8.85 (dd, 1H), 8.71 (s, 1H), 8.55 (s, 1H), 8.22-8.13 (m, 4 H), 7.09 (d, 1H), 3.73 (t, 4H), 3.58 (t, 4H). LCMS: 100%, m/z = 434.2 (M+l)+.
Example 11
(S)-2-(2-methylpyridin-4-yl)-N-(2-morpholino-5-(pyrrolidin-3-ylamino)oxazolo[4,5-b]pyridin-6-yl)oxazole-4-carboxamide

Step l:Preparation of (S)-tert-butyl 3-((2-morpholino-6-nitrooxazolo[4,5-b]pyridin-5-yl)amino)pyrrolidine- 1 -carboxylate
A solution of 5-chloro-2-morpholino-6-nitrooxazolo[4,5-b]pyridine (300mg, 1.0563 mmol) (S)-tert-butyl 3 -aminopyrrolidine- 1 -carboxylate (237mg, 1.267 mmol) and potassium carbonate (292mg, 2.112 mmol) in DMF (2mL) was heated at 100°C for 2h. Reaction was quenched with ice water and filtered the solid. The resultant crude was purified by 60-120 silica gel column chromatography using 1 % methanol in DCM as eluent to obtain the title compound (350mg, 76.25%). LCMS: m/z: 435.4 (M+l)+.
Step 2:Preparation of (S)-tert-butyl 3-((6-amino-2-morpholinooxazolo[4,5-b]pyridin-5-yl)amino)pyrrolidine- 1 -carboxylate
Using the same reaction conditions as described in step 5 of example 1, (S)-tert-butyl 3- ((2-morpholino-6-nitrooxazolo[4,5-b]pyridin-5-yl)amino)pyrrolidine-l -carboxylate (350mg, 0.806 mmol) was reduced with zinc dust (422mg, 6.451 mmol) and ammonium chloride (691mg, 12.903 mmol) in THF/methanol/H20 (10mL/2mL/lmL) to get the title compound (240mg, 71.8%). LCMS: m/z: 405.2 (M+l)+.
Step 3:Preparation of (S)-tert-butyl 3-((6-(2-(2-methylpyridin-4-yl)oxazole-4-carboxamido)-2-morpholinooxazolo[4,5-b]pyridin-5-yl)amino)pyrrolidine-l-carboxylate
Using the same reaction conditions as described in step 6 of example 1, (S)-tert-butyl 3-((6-amino-2-morpholinooxazolo[4,5-b]pyridin-5-yl)amino)pyrrolidine-l -carboxylate (115mg, 0.284 mmol), was coupled with 2-(2-methylpyridin-4-yl)oxazole-4-carboxylic acid (70mg, 0.341 mmol) using EDCI.HCl (82mg, 0.426 mmol), HOBt (58mg, 0.426 mmol), DIPEA (0.199mL, 1.138 mmol) in DMF (2mL) to afford the title compound (lOOmg, 59.52%). LCMS: m/z: 591.4 (M+l)+.
Step 4: Preparation of (S)-2-(2-methylpyridin-4-yl)-N-(2-morpholino-5-(pyrrolidin-3-ylamino)oxazolo[4,5-b]pyridin-6-yl)oxazole-4-carboxamide
Using the same reaction conditions as described in step 8 of example 1, (S)-tert-butyl 3- ((6-(2-(2-methylpyridin-4-yl)oxazole-4-carboxamido)-2-morpholinooxazolo[4,5-b]pyridin-5-yl)amino)pyrrolidine-l -carboxylate (lOOmg, 0.169 mmol) was deprotected using methanolic HC1 (5mL) to get the crude product. This was then purified by prep HPLC to get the title compound (9mg, 10.84%).
1HNMR (CDCI3, 400MHz): δ 9.91 (s, 1H), 8.78 (s, 1H), 8.74-8.73 (d, 1H), 8.45 (s, 1H), 7.82 (s, 1H), 7.76-7.74 (d, 1H), 4.50 (s, 1H), 4.04-4.03 (d, 4H), 3.30-3.00 (m, 7H), 2.70 (s, 3H), 2.40-1.80 (m, 4H), 1.00-0.08 (m, 1H). LCMS: 100%, m/z = 491.3 (M+l)+.
REFERENCES
http://www.curis.com/images/stories/pdfs/posters/Aurigene_IRAK4_AACR-NCI-EORTC_2015.pdf
http://www.curis.com/images/stories/pdfs/posters/Aurigene_IRAK4_AACR_20150421.pdf
1Nature. 2011; 470(7332):115–119
2Immunology and Cell Biology. 2011; 89(6):659–660
3N Engl J Med. 30, 2012; 367(9):826–833
April 2014, preclinical data presented at the CHI’s Ninth Drug Discovery Chemistry Conference in San Diego, CA
November 2015, preclinical data were presented at the 2015 AACR-NCI-EORTC Molecular Targets and Cancer Therapeutics Conference in Boston, MA
http://pubs.acs.org/doi/abs/10.1021/jm5016044
http://cancerres.aacrjournals.org/content/75/15_Supplement/3646
2015 Apr 18-22; Philadelphia, PA. Philadelphia (PA): AACR; Cancer Res 2015;75(15 Suppl):Abstract nr 3646. doi:10.1158/1538-7445.AM2015-3646
////////IRAK4 Kinase Inhibitor, Curis, Aurigene, CA 4948, AU 4948, CA-4948, AU-4948, 1428335-77-6
c21ccc(cc1sc(n2)N3CCOCC3)NC(c4nc(ccc4)c5ccc(nc5)N)=O
सुकून उतना ही देना प्रभू, जितने से जिंदगी चल जाये। औकात बस इतनी देना, कि औरों का भला हो जाये।
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE

Join me on Facebook 


 amcrasto@gmail.com
amcrasto@gmail.com
 LIONEL MY SON
LIONEL MY SON
सुकून उतना ही देना प्रभू, जितने से
जिंदगी चल जाये।
औकात बस इतनी देना,
कि औरों का भला हो जाये।
Read all about Organic Spectroscopy on ORGANIC SPECTROSCOPY INTERNATIONAL 
Filed under: Uncategorized Tagged: 1428335-77-6, AU 4948, Aurigene, CA 4948, curis, IRAK4 Kinase Inhibitor



































































 L. Nathan Tumey, Ph.D., Principal Research Scientist, Pfizer Global R&D
L. Nathan Tumey, Ph.D., Principal Research Scientist, Pfizer Global R&D

















 \
\























































































































 Drug Authorization Procedures in the EU
Drug Authorization Procedures in the EU