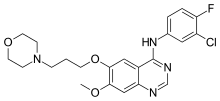
![Gefitinib structure.svg]()
Gefitinib
N-(3-chloro-4-fluorophenyl)-7-methoxy-6-(3-morpholin-4-ylpropoxy)quinazolin-4-amine
| Gefitinib; Iressa; 184475-35-2; ZD1839; N-(3-Chloro-4-fluorophenyl)-7-methoxy-6-(3-morpholinopropoxy)quinazolin-4-amine; Irressat; |
| Molecular Formula: |
C22H24ClFN4O3 |
| Molecular Weight: |
446.902363 g/mol |
![Gefitinib ball-and-stick.png]()
Gefitinib (INN, //, trade name Iressa, marketed by AstraZeneca and Teva), is a drug used for certain breast, lung and other cancers. Gefitinib is an EGFR inhibitor, like erlotinib, which interrupts signaling through the epidermal growth factor receptor(EGFR) in target cells. Therefore, it is only effective in cancers with mutated and overactive EGFR.
Mechanism of action
Gefitinib is the first selective inhibitor of epidermal growth factor receptor‘s (EGFR) tyrosine kinase domain. Thus gefitinib is an EGFR inhibitor. The target protein (EGFR) is a family of receptors which includes Her1(erb-B1), Her2(erb-B2), and Her 3(erb-B3). EGFR is overexpressed in the cells of certain types of human carcinomas – for example in lung and breast cancers. This leads to inappropriate activation of the anti-apoptotic Ras signalling cascade, eventually leading to uncontrolled cell proliferation. Research on gefitinib-sensitive non-small cell lung cancers has shown that a mutation in the EGFR tyrosine kinase domain is responsible for activating anti-apoptotic pathways.[1][2] These mutations tend to confer increased sensitivity to tyrosine kinase inhibitors such as gefitinib and erlotinib. Of the types of non-small cell lung cancer histologies, adenocarcinoma is the type that most often harbors these mutations. These mutations are more commonly seen in Asians, women, and non-smokers (who also tend to more often have adenocarcinoma).
Gefitinib inhibits EGFR tyrosine kinase by binding to the adenosine triphosphate (ATP)-binding site of the enzyme.[3] Thus the function of the EGFR tyrosine kinase in activating the anti-apoptotic Ras signal transduction cascade is inhibited, and malignant cells are inhibited.[4]
Clinical uses
The FDA approved Gefitinib in May 2003 for NSCLC a type of lung cancer,[5] Gefitinib is currently marketed in over 64 countries.
In June 2005 the FDA withdrew approval for use in new patients due to lack of evidence that it extended life.[6]
In Europe gefitinib is indicated since 2009 in advanced NSCLC in all lines of treatment for patients harbouring EGFR mutations. This label was granted after gefitinib demonstrated as a first line treatment to significantly improve progression-free survival vs. a platinum doublet regime in patients harbouring such mutations. IPASS has been the first of four phase III trials to have confirmed gefitinib superiority in this patient population.[7] In most of the other countries where gefitinib is currently marketed it is approved for patients with advanced NSCLC who had received at least one previous chemotherapy regime. However, applications to expand its label as a first line treatment in patients harbouring EGFR mutations is currently in process based on the latest scientific evidence.As at August 2012 New Zealand has approved gefitinib as first line treatment for patients with EGFR mutation for naive locally advanced or metastatic, unresectable NSCLC. This publicly funded for an initial 4 month term and renewal if no progression. [8]
In 2014 in the TRANSCOG study Petty et al., demonstarted gefitinib was effective in esophageal cancer patients whose tumours harboured additional copies of the EGFR gene.[9] While gefitinib has yet to be proven to be effective in other cancers, there is potential for its use in the treatment of other cancers where EGFR overexpression is involved.
Erlotinib is another EGFR tyrosine kinase inhibitor that has a similar mechanism of action to gefitinib.
Experimental Uses
In August 2013, the BBC reported that researchers in Edinburgh and Melbourne found, in a small-scale trial of 12 patients, that the effectiveness of Methotrexate for treating ectopic pregnancy was improved when Gefitinib was also administered.[10]
Studies
IPASS (IRESSA Pan-Asia Study) was a randomized, large-scale, double-blinded study which compared Gefitinib vs. carboplatin/ paclitaxel as a first line treatment in advanced NSCLC.[11] IPASS studied 1,217 patients with confirmed adenocarcinoma histology who were former or never smokers. A pre-planned sub-group analyses showed that progression-free survival (PFS) was significantly longer for Gefitinib than chemotherapy in patients with EGFR mutation positive tumours (HR 0.48, 95 per cent CI 0.36 to 0.64, p less than 0.0001), and significantly longer for chemotherapy than Gefitinib in patients with EGFR mutation negative tumours (HR 2.85, 95 per cent CI 2.05 to 3.98, p less than 0.0001). This, in 2009, was the first time a targeted monotherapy has demonstrated significantly longer PFS than doublet chemotherapy.
EGFR Diagnostic tests
Genzyme, QIAGEN, Argenomics S.A. & other companies make tests to detect EGFR mutations, designed to help predict which lung cancer patients may respond best to some therapies, including Gefitinib and Erlotinib.
The tests examine the genetics of tumors removed for biopsy for mutations that make them susceptible to treatment.
The EGFR mutation test may also help AstraZeneca win regulatory approval for use of their drugs as initial therapies. Currently the TK inhibitors are approved for use only after other drugs fail. In the case of gefitinib, the drug works only in about 10% of patients with advanced non-small cell lung cancer, the most common type of lung cancer.
Adverse effects
As gefitinib is a selective chemotherapeutic agent, its tolerability profile is better than previous cytotoxic agents. Adverse drug reactions (ADRs) are acceptable for a potentially fatal disease.
Acne-like rash is reported very commonly. Other common adverse effects (≥1% of patients) include: diarrhoea, nausea, vomiting, anorexia, stomatitis, dehydration, skin reactions, paronychia, asymptomatic elevations of liver enzymes, asthenia, conjunctivitis, blepharitis.[12]
Infrequent adverse effects (0.1–1% of patients) include: interstitial lung disease, corneal erosion, aberrant eyelash and hair growth.[12]
Iressa was approved and marketed from July 2002 in Japan, making it the first country to import the drug.
Gefitinib is an anilinoquinazoline which is useful in the treatment of a certain type of lung cancer (non-small cell lung cancer or NSCLC) that has not responded to chemotherapy. The chemical name for gefitinib is 4-(3′-chloro-4′-fluoroanilino)-7- methoxy-6-(3-morpholinopropoxy) quinazoline. Its structural formula is :
Gefitinib
The earliest known synthesis of gefitinib was first disclosed in the patent application WO 96/33980. The synthetic method employed is depicted in the following reaction scheme 1.
![Figure imgf000003_0001]()
The process involves selective demethylation of 6,7-dimethoxy quinazoline-4-one using methanesulfonic acid and L-methionine to get its 6-hydroxyl derivative, which is protected by acetylation. The acetoxy compound is chlorinated and condensed with chloro-fluoroaniline. Hydrolysis of the acetoxy compound followed by etherification with 3-morpholinopropyl chloride gives crude gefitinib which is purified by column chromatography. The process suffers from many disadvantages as it involves several protection and deprotection steps. The selective demethylation using methionine results in isomeric impurities and has to be purified or else the impurity carries over to subsequent steps in the preparation of gefitinib making it more difficult to isolate a pure product. The process also leads to formation of an N-alkylated impurity at the final stage which must be separated by column chromatography to obtain gefitinib.
Several other approaches are also described in the literature to make gefitinib.
WO 2004/024703 discloses a process for the preparation of gefitinib starting from 3- hydroxy-4-methoxy benzonitrile which involves condensation of 3-hydroxy-4-methoxy benzonitrile with morpholino propyl chloride, nitration, reduction with sodium dithionite to amino compound, hydrolysis of nitrile to amide, cyclisation in the presence of formamide to obtain quinazoline, chlorination with phosphorous oxychloride and finally condensation with chloro-fluoro aniline to obtain gefitinib. The process involves multiple steps and hence is time consuming.
WO 2005/023783 discloses a process for the manufacture of gefitinib starting from 2- amino-4-methoxy-5-(3-morpholinopropoxy)benzonitrile. The process involves a rearrangement reaction of 3-(3-chloro-4-fluorophenyl)-7-methoxy-6-(3- morpholinopropoxy)3,4-dihydroqunazoline-4-imine. The process is not feasible industrially, as the basic raw material is not readily available on a commercial scale and involves the use of excess 3-chloro-4-fluoroaniline which is expensive. A further draw back of the process is in the isomerization of the 4-imine compound which requires anhydrous conditions at high temperature for a longer duration of 96 hours. All the problems associated with this prior art process are overcome by the novel process of the present invention.
WO2005/070909 discloses a process for the preparation of gefitinib starting from isovanillin as depicted in scheme 2
The WO’ 909 process has disadvantages as it forms cis-trans geometrical isomers of the oxime, which have different reactivities. Furthermore, the process uses a large excess of acetic anhydride to convert the oxime to the nitrile at higher temperature.
The patent applications 901 /CHE/2006 and 903/CHE/2006 disclose another route for preparing gefitinib starting from isovanillin. The process involves formation of a formamido compound [N’-[2-cyano-4-{3-(4-morpholinyl)propoxy}phenyl]-N,N-dimethyl formamide], which is unstable and may result in undesired impurities in the final condensation with 3-chloro-4-fluoro aniline, thereby making the process less feasible on an industrial scale. The processes disclosed in the prior art are cumbersome. Therefore, there exists a need for a more economical and efficient method of making gefitinib which is suitable for industrial scale-up.
The process of the present invention avoids use of reagents such as sodium dithionite, acetic anhydride and allows substantial reduction in the number of problems associated with these reagents.
…………………
CLIP
Gefitinib A mixture of compound 1 starting acid 1 is heated in a closed loop to obtain ammonium 2 in 2 classic resonant3 , the 7 – Bit methoxy given electron, so to 6 – position methoxy-electron density is low, so that the acidic conditions demethylase (with methionine and methanesulfonic acid) may optionally occur in the 6 – position, to give compound 4 , 4 of the phenolic hydroxyl group with an acetyl group after protection thionyl chloride to get five , five and six occurred SNAr reaction 7 , 7deacetylated with ammonia and chloride 8 reaction gefitinib.
 - anti-cancer drugs")
……………..
CLIP
![]()
……………….
\PATENT
http://www.google.com/patents/EP2155656A2?cl=en
Detailed Description of the Invention In an embodiment of the present invention, there is provided an improved synthesis of gefitinib from isovanillin , as depicted below in reaction scheme 3.
isovanillin Formula II Formula in
Formula V Formula IV
W
Gefitinib Formula I
An alternate route for the synthesis of gefitinib from isovanillin according to the present invention is depicted below in reaction scheme 4.
Nitration
Step a Step b
Isovanillin Formula VIII Formula IX
Formula XII Formula XI Formula X
Formula XIII Formula XIV
![Figure imgf000019_0005]()
![Figure imgf000019_0006]()
Example 1 :
Preparation of 4-(3′-chloro-4′-fluoroanilino)-7-methoxy-6-(3- morpholinopropoxy)-quinazoline (gefitinib) (formula I)
Methanol (1200 ml) and 6-(3-morpholino propoxy)-7-methoxy-4-chloro quinazoline (200gm) were stirred for 15 minutes at 25-300C, then a solution of 4-fluoro-3- chloroaniline in methanol (213 gm in 400 ml) was charged and refluxed for 6 hours. The reaction mass was cooled to 15-200C, hydrochloric acid (40 ml) was added drop wise, and stirred at 5-100C for 30 minutes. The solid obtained was filtered and washed with chilled methanol (150ml). The solid was dissolved in a mixture of toluene (30 volume) and methanol (5 volume), the reaction mass was concentrated to half the volume and cooled to 5-10°C. The solid obtained was filtered, washed with toluene (200 ml) and dried at 45-50°C to yield the title compound (183 gm, 70% yield).
Example 2: Preparation of 6-(3-morpholino propoxy)-7-methoxy-4- chloroquinazoline (formula VII)
DMF (3 It), 6-(3-chloropropoxy)-7-methoxy-4-chloro quinazoline (200 gm) and morpholine (210 gm), were heated to 70-750C for 6-8 hours. The reaction mass was cooled to room temperature, and methylene chloride (2.5 It) and water (2.5 It) were charged. The layers separated and the aqueous layer extracted with methylene chloride twice (500 ml). The combined methylene chloride layer was washed with water, dried over sodium sulphate (10 gm) and concentrated completely at 35-40°C to yield the title compound (200 gm, 85% yield).
Example 3: Preparation of 6-(3-chloropropoxy)-7-methoxy-4-chloroquinazoline (formula Vl)
6-(3-chloropropoxy)-7-methoxyquinazoline-4-one (400 gm), thionyl chloride (3.2 It) and DMF (100 ml) were refluxed for 7-8 hours. Thionyl chloride was distilled off completely under reduced pressure below 45°C. Methylene chloride (2.5 It) and water (1.5 It) were charged, stirred for 30 minutes at room temperature and the layers separated. The aqueous layer was extracted twice with methylene chloride (500 ml), the combined methylene chloride layer was washed with 1 % sodium bicarbonate solution (1 It), dried over sodium sulphate (20 gm) and concentrated under reduced pressure at 35-40°C. The residue was stirred with isopropyl alcohol (400 ml) at 40-450C for 1 hour, cooled to 0-50C, the solids filtered, washed with chilled isopropyl alcohol (200 ml) and dried under vacuum at 45°C to yield the title compound (406 gm, 95% yield).
Example 4: Preparation of 6-(3-chloropropoxy)-7-methoxyquinazoline-4-one (formula V)
2-amino-4-methoxy-5-(3-chloropropoxy)benzoic acid (450gm), formamide (2250 ml) and ammonium formate (200 gm) were heated to 170-1800C for 3-4 hours. The reaction mass was concentrated under reduced pressure at 140-1500C. The residue was stirred in methanol (1000 ml) at 45-50°C and cooled to 5-10°C. The solid obtained was filtered to yield the title compound (420 gm, 90% yield).
Example 5: Preparation of 2-amino-4-methoxy-5-(3-chloropropoxy) benzoic acid (formula IV)
a) Preparation of 3-(3-chloropropoxy)-4-methoxy-6-nitrobenzoic acid
Methanol (4 It), 3-(3-chloropropoxy)-4-methoxy-6-nitro benzaldehyde (560 gm) and 30% methanolic NaOH solution (5 ml) were heated to 450C. To this reaction mass 35% of H2O2 solution (1200 ml) was added drop wise in 3-4 hours maintaining a pH of 10.5 – 11.5 with 30% methanolic NaOH solution. The reaction mass was quenched into ice water (10 kg) and the pH adjusted to 2.0-3.0 using hydrochloric acid. The solid obtained was filtered, washed with 50% aqueous methanol (500 ml) and dried at 45-500C to yield the title compound (510 gm, 86% yield).
bi) Preparation of 2-amino-4-methoxy-5-(3-chloropropoxy)benzoic acid – using hydrogen gas
Ethyl acetate (3 It), Pd/C (50 gm) and 3-(3-chloropropoxy)-4-methoxy-6-nitrobenzoic acid (500 gm) were hydrogenated under a hydrogen pressure of 5-6 kg at 35-400C for 3-4 hours. The reaction mass was filtered and the clear filtrate was distilled under reduced pressure at 45-500C. To the residue, hexane (1 It) was charged, stirred at room temperature, the solids filtered and dried at 45-50°C to yield the title compound (403 gm, 90% yield). 5
(bii) Preparation of 2-amino-4-methoxy-5-(3-chloropropoxy)benzoic acid – using hydrazine hydrate
S-p-chloropropoxyH-methoxy-e-nitrobenzoic acid (100 gm), hydrazine hydrate (50 gms), neutral alumina (20gms), charcoal (10 gms), water (50 ml) and methanol (500
10 ml) were mixed together. The reaction mass was heated to 500C. A solution of ferric chloride (2 gms, 0.012M) in 50 ml methanol was introduced slowly at 55-600C. The reaction mass was filtered over hyflo and the clear filtrate evaporated. The residue obtained was dissolved in 1.0-lit ethyl acetate, washed organic extract with water, evaporated to obtain title compound. (75 gms, 83.6%)
15
(biii) Preparation of 2-amino-4-methoxy-5-(3-chloropropoxy)benzoic acid – using ammonium formate
3-(3-chloropropoxy)-4-methoxy-6-nitro benzoic acid (165 gms), 5% Paladium on carbon (16.5 gms) and DMF (0.66 lit) were mixed together. The reaction mass was heated to
20 400C. Ammonium formate (82.5 gms) was charged in lots maintaining temperature below 500C. The temperature of reaction mass slowly raised to 70°Cand maintained for 2 hours. The reaction mass was cooled to 300C and catalyst was removed by filtration and the clear filtrate evaporated. The residue was dissolved in ethyl acetate (0.825 lit), washed with water and evaporated to yield the title compound. (125 gms,
25 84.5%)
Example 6: Preparation of 3-(3-chloropropoxy)-4-methoxy-6-nitro benzaldehyde (formula III)
5-nitro isovanillin (500 gm), acetonitrile (3.5 Its), K2CO3 (750 gm) and 30 chlorobromopropane (780 gm) were refluxed for 4 hours. The reaction mass was filtered hot, washed with acetonitrile (1 It) and the filtrate was distilled off to remove solvent. The residue was dissolved in methylene chloride (4 It) and washed with water. Water (3 It) was charged to the methylene chloride layer, the pH adjusted to 7.0 to 7.5 with acetic acid, the methylene chloride layer separated, dried over sodium sulphate (50 gm) and distilled out completely under reduced pressure below 400C. The residue was stirred with 2 volumes of n-Hexane at 40-450C, cooled slowly to 0-50C, the solids filtered, washed with n-Hexane (250 ml) and dried at 40-450C to yield the title compound (638 gm, 92% yield).
Example 7: Preparation of 5-nitro isovanillin (formula II)
Isovanillin (500 gm) and acetic acid (1750 ml) were cooled to -5 to O0C. To this solution, nitric acid (750 ml) was charged slowly at -5 to O0C with stirring. The temperature of the reaction mass was slowly raised to 25-300C and maintained for 12 hours. The reaction mass was quenched into ice water (4 kg), the solids filtered and washed with water (2 It). The solids were stirred with a 1% sodium bicarbonate solution (1 It), filtered and dried at 45-500C. The solid was dissolved in 6 volumes of ethyl acetate, ethyl acetate was distilled off up to half the volume and 3 volumes of n-Hexane were charged slowly at 45-50°C. The reaction mass was cooled slowly to 0-5°C, maintained for 1 hour, the solids filtered, washed with 0.5 volumes of 1 :1 mixture of ethyl acetate: n-Hexane and dried at 45-500C to yield the title compound (423 gm, 65 % yield) .
Example 8: Preparation of Methyl-2-hydroxy-3-methoxy benzoate (formula VIII)
a) Preparation of 3-hydroxy-4-methoxy benzoic acid
Methanol (350 ml), isovanillin (50 gm) and 30% methanolic sodium hydroxide solution (1 ml), were heated to 450C. To this solution, 35% hydrogen peroxide solution (107 ml) was charged slowly maintaining pH at 10.5 to 11.5 using methanolic sodium hydroxide solution over a period of 2-3 hours. The reaction mass was quenched into chilled water (1 It) and the pH adjusted to 2-3 using hydrochloric acid. The solids were filtered, washed with 50% aqueous methanol (50 ml) and dried at 45-50°C to yield 3-hydroxy-4- methoxy benzoic acid.
b) Preparation of Methyl-2-hydroxy-3-methoxy benzoate
The solid obtained in step a), was refluxed with 10% methanolic hydrochloric acid solution (250 ml) for 6 hours. The reaction mass was quenched into chilled water (1 It) and repeatedly extracted with methylene chloride (250 ml). The combined methylene chloride layer was washed with water (100 ml * 2) and methylene chloride distilled out completely at 35-40°C. The residue was stirred in hexane (150 ml), at 25-300C. The solid obtained was filtered, washed with hexane (25 ml) and dried at 40-45°C to yield the title compound (50 gm, 83% yield).
Example 9: Preparation of Methyl-5-hydroxy-4-methoxy-2-nitro benzoate (formula
IX)
Methyl-2-hydroxy-3-methoxy benzoate (50 gm) and acetic acid (175 ml) were cooled to
0-5°C. To this solution, 70% nitric acid solution (75 ml) was charged slowly at 0-5°C under stirring and the reaction mass was further stirred for 18 hours. The reaction mass was quenched into chilled water (800 ml) and extracted repeatedly with methylene chloride (400 ml). The combined methylene chloride layer was washed with water, followed by 1% potassium carbonate solution (100 ml), dried over sodium sulphate and methylene chloride distilled off completely at 35-40°C. The residue was dissolved in 10% aqueous methanol (250 ml). The filtrate was gradually cooled to 0-5°C and maintained for 1 hour. The solid obtained was filtered, washed with 10% aqueous methanol (100 ml) and dried at 40-450C to yield the title compound (46 gm, 74% yield).
Example 10: Preparation of Methyl-2-amino-5-hydroxy-4-methoxy benzoate (X) Ethyl acetate (300 ml), methyl-5-hydroxy-4-methoxy-2-nitro benzoate (50 gm) and 10% palladium/carbon (5 gm) were hydrogenated under a hydrogen gas pressure of 5-6 kg for 4 hours. The reaction mass was filtered to remove catalyst. The filtrate was distilled off to remove solvent. The residue obtained was stirred in n-hexane (100 ml) at 0-5°C The solid obtained was filtered and washed with n-hexane (25 ml) to yield the title compound (40 gm, 93% yield).
Example 11 : Preparation of 6-hydroxy-7-methoxy-quinazoline-4-one (formula Xl)
Methyl-2-amino-5-hydroxy-4-methoxy benzoate (50 gm), methanol (400 ml) and formamidine acetate (30 gm) were refluxed for 10 hours. The reaction mass was gradually cooled to 5-10°C and stirred for 1 hour. The solid obtained was filtered and washed with methanol (150 ml) and dried at 50-55°C to yield the title compound (45 gm, 92% yield).
Example 12 : Preparation of Gefitinib
Acetonitrile (500 ml), N-(4-fluoro-3-chloro phenyl)-6-hydroxy-7-methoxy quinazoline-4- amine (50 gm), 3-morpholinopropyl chloride (35 gm) and tetrabutyl ammonium bromide (5 gm) were refluxed for 16 hours. The reaction mass was distilled off to remove acetonitrile completely at 40-45°C. To the residue, water (500 ml) was charged and stirred for 15 minutes at 25-300C. The solid obtained was filtered, washed with methanol (50 ml) and dried at 45-50°C. The crude solid was dissolved in a mixture of toluene (1200 ml) and methanol (200 ml). The reaction mass was distilled off under reduced pressure at 40-450C to 400 ml volume, cooled to 10-150C, stirred for 30 minutes, the solid filtered, washed with toluene (40 ml) and dried to yield gefitinib (28 gm, 40% yield).
![]() GEFITINIB
GEFITINIB
![]()
…………
| Indian Patent Number |
236843 |
| Indian Patent Application Number |
http://www.allindianpatents.com/patents/236843
The process of preperation of 4-(3-chloro-4-flurophenylamino)-7-methoxy-6-[3-(4-morpholinyl)propoxy]-quinazoline is substantially as herein described with reference to the foregoing example.
a) 4-methoxy-3-[3-(4-morpholinyl)-propoxy] benzaldehyde:
A mixture of 3-hydroxy -4- methoxy benzaldehyde (20g), 3-
morpholinopropyl chloride (28g), potassium carbonate (50g) and DMF
(140ml) was stirred and heated to 100°C for 3 hours. The reaction mixture
was cooled and filtered. The filtrate was evaporated and the residue
obtained was dissolved in ethyl acetate (200ml). The ethyl acetate layer was
washed with water, dried over anhydrous sodium sulphate. Evaporation of
ethyl acetate yielded 4-methoxy-3-[3-(4-morpholinyl)-propoxy]
benzaldehyde (34.8 gms,95%) of the formula II.
b) 4-methoxy -3[3-(4-morpholinyl)-propoxy] benzonitrile:
The above 34.8 gms of compound was dissolved in 200 ml methanol, and to this added 34.8 g of hydroxylamine hydrochloride and 35 ml of pyridine. The reaction mixture was heated to reflux for 3 hours and then cooled to 10 °C. The material precipitated mass was filtered and the solid mass obtained was washed with chilled methanol (50ml) and dried at room temperature. To this dried material was added 2 volumes of acetic anhydride and heated to 110°C for 4 hours. Then quenched the reaction mass in water and adjusted the pH to 8.0 with sodium bicarbonate and extracted with methylene dichloride. Washed the methylene dichloride layer with water and dried over calcium chloride. On evaporation of the solvent 4-methoxy -3[3-(4-morphoinyl)-propoxy] benzonitrile (31 gms, 90%) of the formula III.
NMR spectrum (CDCI3): 5 2.05 (m, 2H), 2.53 (m, 6H), 3.72 (m, 4H), 3.91 (s, 3H). 4.10(mf 2H), 6.89 (d, 1H), 7.25 (d, 1H), 7.28 (dd, 1H).
c) 4-Methoxy-5-[3-(4-morpholinyl)-propoxy]- 2-nitro benzonitrile: The above compound (31 gms) of the formula III was dissolved in 30 ml of 70% nitric acid and this was added to 55°C preheated 30 ml of 70% nitric acid slowly over a period of 2 hours under stirring. After completion of the addition, continued stirring at the same temperature for further an hour. Cooled the reaction mixture and quenched in cool water and
adjusted the pH to 8.0. The precipitate obtained was filtered and washed with ice cold water and dried the material at 50°C to get 27 gms yellow solid 4-Methoxy-5-[3-(4-morphoiinyl)-propoxy]- 2-nitro benzonitrile (75%) of the formula IV.
NMR spectrum (DMSO-d6): 5 2.18 (m, 2H), 3.26 (m, 4H), 3.53 (m, 4H), 3.99 (s, 3H), 4.04 (m, 2H), 4.29 (m, 2H), 7.73 (s, 1H), 7.91 (s, 1H). d) Synthesis of 2-amino-4-methoxy-5-(3-morpholinopropoxy) benzonitrile: To 4-methoxy-5-[3-(4-morpholinyl) propoxy]-2-nitro benzonitrile (10 g) was added acetic acid (75ml) and water(75ml), stirred the reaction mass for about 10 min, added Iron powder (7g) in portions over a period of 2hrs, Stirred the reaction mixture for about Vi hr at room temperature adjusted PH of the reaction mass to 8 using ammonia solution, extracted the material into ethylacetate, the organic layer was dried over sodium sulfate and concentrated to get product(6g)
1HNMR (CDCI3): 5 2.01 (m, 2H), 2.51 (m. 6H), 3.72 (t, 4H), 3.84 (s, 3H), 3.97 (t, 2H), 4.14 (brs, 2H), 6.23 (s, 1H), 6.85 (s, 1H).
(e) Synthesis of N’-(3-chloro-4-fluorophenyl) N,N-dimethyl formamidine.
To 3-chloro-4-flouro aniline (10g, 0.0687moles) was added Toluene (40ml), N,N-dimethylformamide dimethyl acetal (18.3ml, 0.1374moles) and acetic acid (0.5ml), heated the reaction mixture to 110°C and stirred for about 2hrs, distilled off toluene to yield dark brown liquid (11g)
1HNMR (CDCI3): 5 3.0 (s, 6H), 6.75 (m, 1H) 6.94 (m, 1H), 6.98 (m, 1H), 7.45 (s, 1H).
(f) Synthesis of Gefitinib:
To 2-amino-4-methoxy-5-(3-morpholinopropoxy) benzonitrile (5g, 0.0172moles) was added toluene (30ml), N’-(3-chloro-4-fluorophenyl) N, N-dimethyl formamidine (3.44g, 0.0172moles) and acetic acid (0.5ml) refluxed the reaction mixture for about 4hrs cooled the reaction mass to room temperature, toluene layer was separated washed with water and chilled toluene layer to yield crude gefitinib and which was further recrystallized from
methano! to get pure off-white crystalline compound(3g) having the mp
194-198°C
UV, IR, NMR spectral data together with elemental analysis is in complete
agreement with those of standard substance of Gefitinib
………………
NMR
http://nopr.niscair.res.in/bitstream/123456789/29472/1/IJCB%2053B(10)%201269-1274.pdf
MP 193-95
……………….
PAPER
Org. Process Res. Dev., 2007, 11 (5), pp 813–816
DOI: 10.1021/op700054p
An efficient, economical and large-scale convergent synthesis of epidermal growth factor receptor- tyrosine kinase inhibitors gefitinib (1, Iressa) and erlotinib (2, Tarceva) approved by U.S. FDA for the treatment of non-small-cell lung cancer is described. The formation of 4-anilinoquinazolines are achieved in a simple one-pot reaction of suitable formamidine intermediates and substituted anilines involving Dimroth rearrangement, thereby avoiding the need to make quinazolin-4(3H)-one intermediates, which require a large experimental inputs. Using this process, we have produced drug candidates 1 with overall yield of 66% from 4-methoxy-5-[3-(4-morpholinyl) propoxy]-2-nitrobenzonitrile (3) and 2 with 63% from 4,5-bis(2-methoxyethoxy)-2-nitrobenzonitrile (6) on a multigram scale.
1as off-white solid (304 g, 70% yield). Mp 193–195 °C. HPLC purity >99%.
1H NMR (CDCl3, 200 MHz): δ 2.11 (m, 2H), 2.46–2.59 (m, 6H), 3.74 (dd, J = 4.5 and 4.4Hz, 4H), 3.98 (s, 3H), 4.17 (t,J = 6.5Hz, 2H), 7.09 (s, 1H), 7.16 (t, J = 8.8Hz, 1H), 7.26 (s, 1H), 7.34 (brs, 1H, exchangeable with D2O), 7.50–7.58 (m, 1H), 7.84–7.88 (m, 1H), 8.66 (s, 1H). MS (m/z): 446(M+),128, 100.
Elemental Anal. Calcd for C22H24ClFN4O3: C, 59.19; H, 5.38; N, 12.55. Found: C, 59.17; H, 5.21; N, 12.33.
NMR
![]()
MASS
![]()
………
SEE
http://aerodrive.ccchwc.edu.hk/~lck/1011_7s/10117sgp05ppt.pdf
http://shodhganga.inflibnet.ac.in:8080/jspui/bitstream/10603/18581/4/04_list%20of%20figures.pdf.pdf
http://industrialgreenchem.com/pdf-docs/presentations/igcw%202013/pharma/Dr.%20Manjunatha.pdf
…………..
Synthesis of Gefitinib[J]. CJPH, 2013, 44(11): 1081-1083..
| Synthesis of Gefitinib |
| 1. School of Chemical Engineering, Huaihai Institute of Technology, Lianyungang 222001; 2. Lianyungang Shenghe Biotechnology Limited Company, Lianyungang 222007 |
Gefitinib was synthesized from 3-hydroxy-4-methoxybenzaldehyde via conversion of aldehyde to nitrile, condensation with N-(3-chloropropy1)morpholine, nitration and reduction to give 2-amino-4-methoxy-5-(3-morpholin-4-ylpropoxy)benzonitrile, which was subjected to amidination with 3-chloro-4-fluoroaniline and cyclization in the presence of formic acid with an overall yield of about 44%.
…………..
Synthesis of Gefitinib
LV Tong-jie,OUYANG Gui-ping,MENG Xiang-bing,LIU Xiao-yu(Key Laboratory of Green Pesticide and Agriculture Bioengineering,Ministry of Education,Research and Development Center for fine Chemicals,Guizhou University,Guizhou Guiyang 550025,China)
The 4-(3-chloro-4-fluorophenylamino)-7-methoxy-6-[3-(4-morpholinyl)pro-poxy]quinazoline(Geifitinib,ZD1839) was synthesized from 3-hydroxy-4-methoxybenzaldehyde,through a seven-step procedure of condensation,conversion of aldehyde to nitrile,n-itration,reduction,cyclization,et al.,and the total yield reached 31.81%.The structure of the compound was characterized by IR,1H-NMR,13C-NMR,and MS.
References
- Pao W, Miller V, Zakowski M et al. (September 2004). “EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib”. Proceedings of the National Academy of Sciences of the United States of America 101 (36): 13306–11. doi:10.1073/pnas.0405220101. PMC 516528.PMID 15329413. Retrieved 2009-07-02.
- Sordella R, Bell DW, Haber DA, Settleman J (August 2004). “Gefitinib-sensitizing EGFR mutations in lung cancer activate anti-apoptotic pathways”. Science 305 (5687): 1163–7.doi:10.1126/science.1101637. PMID 15284455.
- Lynch, Thomas J.; Bell, Daphne W.; Sordella, Raffaella; Gurubhagavatula, Sarada; Okimoto, Ross A.; Brannigan, Brain W.; Harris, Patricia L.; Haserlat, Sara M.; Supko, Jeffrey G.; Haluska, Frank G.; Louis, David N.; Christiani, David C.; Settleman, Jeff; Haber, Daniel A (May 20, 2004). “Activating Mutations in the Epidermal Growth Factor Receptor Underlying Responsiveness of Non-Small-Cell Lung Cancer to Gefitinib”. NEJM 350 (21): 2129–39. doi:10.1056/nejmoa040938.
- Takimoto CH, Calvo E. “Principles of Oncologic Pharmacotherapy” in Pazdur R, Wagman LD, Camphausen KA, Hoskins WJ (Eds) Cancer Management: A Multidisciplinary Approach. 11 ed. 2008.
- http://www.accessdata.fda.gov/drugsatfda_docs/label/2003/021399lbl.pdf
- http://www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/ucm110473.htm
- Mok TS, Wu YL, Thongprasert S, et al, Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med 2009; 361: 947–957. Sebastian M, Schmittel A, Reck, M, First-line treatment of EGFR-mutated nonsmall cell lung cancer: critical review on study methodology, European Respiratory Review. 2014 Mar 1;23(131):92-105.
- http://www.pharmac.govt.nz/2012/07/09/2012.07.10%20gefitinib%20funded.pdf
- http://meetinglibrary.asco.org/content/127239-144
- http://www.bbc.co.uk/news/uk-scotland-edinburgh-east-fife-24021956
- Mok TS et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Eng J Med 2009; 361. 10.1056/NEJMoa0810699.
- Rossi S, editor. Australian Medicines Handbook 2004. Adelaide: Australian Medicines Handbook; 2004. ISBN 0-9578521-4-2.
 - anti-cancer drugs")
Updates
H-NMR spectral analysis
![N-(3-chloro-4-fluorophenyl)-7-methoxy-6-(3-morpholin-4-ylpropoxy)quinazolin-4-amine NMR spectra analysis, Chemical CAS NO. 184475-35-2 NMR spectral analysis, N-(3-chloro-4-fluorophenyl)-7-methoxy-6-(3-morpholin-4-ylpropoxy)quinazolin-4-amine H-NMR spectrum]()
CAS NO. 184475-35-2, N-(3-chloro-4-fluorophenyl)-7-methoxy-6-(3-morpholin-4-ylpropoxy)quinazolin-4-amine H-NMR spectral analysis |
C-NMR spectral analysis
![N-(3-chloro-4-fluorophenyl)-7-methoxy-6-(3-morpholin-4-ylpropoxy)quinazolin-4-amine NMR spectra analysis, Chemical CAS NO. 184475-35-2 NMR spectral analysis, N-(3-chloro-4-fluorophenyl)-7-methoxy-6-(3-morpholin-4-ylpropoxy)quinazolin-4-amine C-NMR spectrum]()
CAS NO. 184475-35-2, N-(3-chloro-4-fluorophenyl)-7-methoxy-6-(3-morpholin-4-ylpropoxy)quinazolin-4-amine C-NMR spectral analysis |
SYNTHESIS
Marzaro, Giovanni; Guiotto, Adriano; Pastorini, Giovanni; Chilin, Adriana Tetrahedron, 2010 , vol. 66, 4 pg. 962 – 968
Chandregowda, Venkateshappa; Venkateswara Rao, Gudapati; Chandrasekara Reddy, Goukanapalli Heterocycles, 2007 , vol. 71, 1 p. 39 – 48
Chandregowda, Venkateshappa; Rao, Gudapati Venkateswara; Reddy, Goukanapalli Chandrasekara Synthetic Communications, 2007 , vol. 37, 19 pg. 3409 – 3415
Ming, Dong Li; You, Guang Zheng; Ji, Min Molecules, 2007 , vol. 12, 3 pg. 673 – 678
Knesl, Petr; Roeseling, Dirk; Jordis, Ulrich Molecules, 2006 , vol. 11, 4 pg. 286 – 297
Heterocycles, , vol. 71, 1 p. 39 – 48
Journal of Labelled Compounds and Radiopharmaceuticals, , vol. 48, 11 pg. 829 – 843
Synthetic Communications, , vol. 37, 19 pg. 3409 – 3415
WO2013/180403 A1, ;
…………………..
http://www.google.com/patents/WO2013180403A1?cl=en
N-(3-chloro-4-fluorophenyl)-7-methoxy-6-(3-morpholinopropoxy)quinazoline-4-amine (gefitinib) represented by Chemical Formula 1 below is a quinazoline derivative useful in treatment of non-small cell lung cancer. The structure of gefitinib is shown in the following Chemical Formula 1.
[Chemical Formula 1]
WO 96/33980 discloses the gefitinib synthesis method as represented in Scheme 1 below.
[Scheme 1]
According to the synthesis method of Scheme 1, 6,7-dimethoxy quinazolin-4-one as a starting material is subjected to selective demethylation, condensation with chlorofluoroaniline and then etherification with 4-(3-morpholinopropyl)chloride, thereby synthesizing gefitinib. Because gefitinib thus synthesized contains an excess of an N-alkylated impurity, that is, N-(3-chloro-4-fluorophenyl)-7-methoxy-6-(3-morpholinopropoxy)-N-(3-morpholinopropyl)quinazoline-4-amine in the final step, the impurity should be separated via column chromatography, undesirably lowering the yield and making it difficult to achieve commercial production.
To solve such problems, WO 2004/024703 discloses a method of synthesizing gefitinib from a start material of 3-hydroxy-4-methoxy benzonitrile as shown in Scheme 2 below.
[Scheme 2]
In the synthesis method of Scheme 2, a morpholinopropyl group is introduced before forming a quinazoline ring, thus suppressing the production of N-(3-chloro-4-fluorophenyl)-7-methoxy-6-(3-morpholinopropoxy)-N-(3-morpholinopropyl)quinazoline-4-amine which is the N-alkylated impurity. However, reduction of a nitro compound, formation of a quinazoline ring, and chlorination of the quinazoline ring in the final step to carry out condensation with chlorofluoroaniline are performed in the presence of the morpholinopropyl group, undesirably complicating the reaction process and lengthening the reaction time.
WO 2008/125867 discloses a method of synthesizing gefitinib from a start material of isovanilin as shown in Scheme 3 below.
[Scheme 3]
In the synthesis method of Scheme 3, propoxychloride is introduced before forming the quinazoline ring, thus suppressing the production of the N-alkylated impurity. After a quinazoline ring is formed and a morpholine group is introduced, chlorofluoroaniline is introduced, thus synthesizing gefitinib. However, a chloropropyl group and a morpholine group are separately introduced, instead of the morpholinopropyl group, thus increasing the number of synthesis steps, and also, the nitro reduction and the quinazoline ring reaction are performed in the presence of the chloropropyl group, undesirably causing the production of an impurity.
Additionally, WO 2005/023783 discloses a method of synthesizing gefitinib from imine via a rearrangement reaction, and WO 2005/070909 discloses a method of synthesizing gefitinib by performing nitrilization of oxime and then forming a quinazoline ring.
However, the preparation methods mentioned in the prior techniques produce an excess of impurity or include other routes to suppress the formation of the impurity, undesirably increasing the number of preparation steps and thus resulting in complicated processes and a long synthesis time, and thereby these methods are unsuitable for commercial production.
Therefore, there is required a method of efficiently and simply preparing gefitinib, which may minimize the production of an impurity and be suitable for use in industrial production.
[Citation List]
[Patent Literature]
(Patent Document 1) WO 96/33980
(Patent Document 2) WO 2004/024703
(Patent Document 3) WO 2008/125867
(Patent Document 4) WO 2005/070909
<Example 3-1> Preparation of N-(3-chloro-4-fluorophenyl)-7-methoxy-6-(3-morpholinopropoxy)quinazoline-4-amine (gefitinib)
About 123.0 g of 4-(3-chloro-4-fluorophenylamino)-7-methoxyquinazolin-6-ol and about 1100.0 mL of N,N-dimethylformamide were placed in a flask. The resulting mixture was suspended with stirring while about 186.0 g of potassium carbonate and about 4.7 g of N,N-dimethylaminopyridine were added. The reaction mixture was cooled to about -10℃, slowly added with about 77.0 g of iodotrimethylsilane while paying attention to heat generation, and stirred at about 15℃ for about 1 hr. Then about 75.5 g of 4-(3-chloropropyl)morpholine was diluted with about 130.0 mL of N,N-dimethylformamide and then slowly added. The reaction mixture was heated to about 80℃ and stirred for about 2 hr. The termination of the reaction was confirmed using HPLC and TLC. The reaction product was cooled to about 20℃, slowly added with 2460.0 mL of purified water, and stirred for 30 min, and the produced solid was filtered. The obtained solid was washed with about 490.0 mL of purified water and then dried in a vacuum at about 50℃ for about 3 hr, yielding about 154.7 g of the title compound N-(3-chloro-4-fluorophenyl)-7-methoxy-6-(3-morpholinopropoxy)quinazoline-4-amine (gefitinib) as pale yellow powder.
HPLC purity: 99.21% (N-alkylated impurity, that is, N-(3-chloro-4-fluorophenyl)-7-methoxy-6-(3-morpholinopropoxy)-N-(3-morpholinopropyl)quinazoline-4-amine: 0.3%)
1H-NMR (400MHz, DMSO-d6): 2.0 (m,2H), 2.4 (m,6H), 3.7 (m,4H), 3.9 (s,3H), 4.2 (t,2H), 7.2 (s,1H), 7.4 (t,1H), 7.8 (m,2H), 8.2 (m,1H), 8.5 (s,1H), 9.6 (s,1H)
<Example 3-3> Preparation of N-(3-chloro-4-fluorophenyl)-7-methoxy-6-(3-morpholinopropoxy)quinazoline-4-amine (gefitinib)
About 8.3 g of the title compound N-(3-chloro-4-fluorophenyl)-7-methoxy-6-(3-morpholinopropoxy)quinazoline-4-amine (gefitinib) as pale yellow powder was obtained in the same manner as in Example 3-2, with the exception that dimethylsulfoxide (DMSO) was used as the solvent, instead of N,N-dimethylacetamide (DMAC).
HPLC purity: 98.89% (N-alkylated impurity, that is, N-(3-chloro-4-fluorophenyl)-7-methoxy-6-(3-morpholinopropoxy)-N-(3-morpholinopropyl)quinazoline-4-amine: 0.7%)
<Example 4-2> Purification of N-(3-chloro-4-fluorophenyl)-7-methoxy-6-(3-morpholinopropoxy)quinazoline-4-amine (gefitinib)
About 8.6 g of N-(3-chloro-4-fluorophenyl)-7-methoxy-6-(3-morpholinopropoxy)quinazoline-4-amine obtained in Example 3-2 was suspended with stirring in about 129.0 mL of toluene and about 65.0 mL of anhydrous ethanol, and heated to about 40℃ so as to be thoroughly dissolved. About 1.9 g of neutral activated carbon was added into the resulting solution, stirred for about 1 hr, and filtered to thus remove the activated carbon. The filtrate was concentrated to about 90 mL, and stirred for about 30 min, and the produced solid was filtered. The obtained solid was washed with about 20.0 mL of toluene, and dried at about 40℃ for about 3 hr, thus obtaining about 7.3 g of white gefitinib.
The purified gefitinib was added to about 125.0 mL of anhydrous ethanol to prepare a suspension, which was then refluxed with stirring at about 75℃ so that gefitinib was thoroughly dissolved, and then further stirred for about 1 hr. The solution was gradually cooled to about 20℃ and the produced solid was stirred for about 30 min and then further stirred at about 5℃ for about 1 hr. The obtained solid was filtered, washed with about 7.0 mL of anhydrous ethanol, and dried in a vacuum at about 45℃ for about 5 hr, yielding about 6.5 g of purified white N-(3-chloro-4-fluorophenyl)-7-methoxy-6-(3-morpholinopropoxy)quinazoline-4-amine (gefitinib).
HPLC purity: 99.89% (without N-alkylated impurity, that is, N-(3-chloro-4-fluorophenyl)-7-methoxy-6-(3-morpholinopropoxy)-N-(3-morpholinopropyl)quinazoline-4-amine)
<Example 4-3> Purification of N-(3-chloro-4-fluorophenyl)-7-methoxy-6-(3-morpholinopropoxy)quinazoline-4-amine (gefitinib)
About 6.2 g of purified white N-(3-chloro-4-fluorophenyl)-7-methoxy-6-(3-morpholinopropoxy)quinazoline-4-amine (gefitinib) was obtained in the same manner as in Example 4-1, with the exception that the gefitinib prepared in Example 3-3 was used.
HPLC purity: 99.87% (N-alkylated impurity, that is, N-(3-chloro-4-fluorophenyl)-7-methoxy-6-(3-morpholinopropoxy)-N-(3-morpholinopropyl)quinazoline-4-amine: 0.03%)
| WO1996033980A1 * |
Apr 23, 1996 |
Oct 31, 1996 |
Keith Hopkinson Gibson |
Quinazoline derivatives |
| WO2004024703A1 * |
Sep 9, 2003 |
Mar 25, 2004 |
Astrazeneca Ab |
Process for the preparation of 4- (3’-chloro-4’-fluoroanilino) -7-methoxy-6- (3-morpholinopropoxy) quinazoline |
| WO2005023783A1 * |
Sep 1, 2004 |
Mar 17, 2005 |
Astrazeneca Ab |
Process for the manufacture of gefitinib |
| WO2005070909A1 * |
Jul 27, 2004 |
Aug 4, 2005 |
Prasad Ramanadham Jyothi |
An improved process for the preparation of gefitinib |
| WO2008125867A2 * |
Apr 16, 2008 |
Oct 23, 2008 |
Cipla Ltd |
Process for the preparation of gefitinib |
Filed under:
Uncategorized Tagged:
gefitinib,
ZD 1839 ![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()































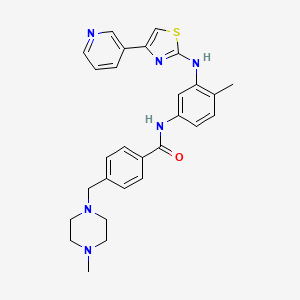












![methanesulfonic acid,4-[(4-methylpiperazin-1-yl)methyl]-N-[4-methyl-3-[(4-pyridin-3-yl-1,3-thiazol-2-yl)amino]phenyl]benzamide NMR spectra analysis, Chemical CAS NO. 1048007-93-7 NMR spectral analysis, methanesulfonic acid,4-[(4-methylpiperazin-1-yl)methyl]-N-[4-methyl-3-[(4-pyridin-3-yl-1,3-thiazol-2-yl)amino]phenyl]benzamide H-NMR spectrum](http://pic11.molbase.net/nmr/nmr_image/2015-01-15/001/603/1603135_1h.png)
![methanesulfonic acid,4-[(4-methylpiperazin-1-yl)methyl]-N-[4-methyl-3-[(4-pyridin-3-yl-1,3-thiazol-2-yl)amino]phenyl]benzamide NMR spectra analysis, Chemical CAS NO. 1048007-93-7 NMR spectral analysis, methanesulfonic acid,4-[(4-methylpiperazin-1-yl)methyl]-N-[4-methyl-3-[(4-pyridin-3-yl-1,3-thiazol-2-yl)amino]phenyl]benzamide C-NMR spectrum](http://pic11.molbase.net/nmr/nmr_image/2015-01-15/001/603/1603135_13c.png)






































 Headquartered in India, Strides Arcolab is a pharmaceutical company with a key focus on development and manufacture of IP-led niche generics and bio-pharmaceuticals. It is also among the world’s largest manufacturers of specialty soft gelatin capsules. With world-class manufacturing facilities, an innovative R&D hub in Bangalore and a strong commercial platform to market branded and commodity generics globally, Strides has earned a reputation for building and scaling profitable businesses in a short span of time.
Headquartered in India, Strides Arcolab is a pharmaceutical company with a key focus on development and manufacture of IP-led niche generics and bio-pharmaceuticals. It is also among the world’s largest manufacturers of specialty soft gelatin capsules. With world-class manufacturing facilities, an innovative R&D hub in Bangalore and a strong commercial platform to market branded and commodity generics globally, Strides has earned a reputation for building and scaling profitable businesses in a short span of time.









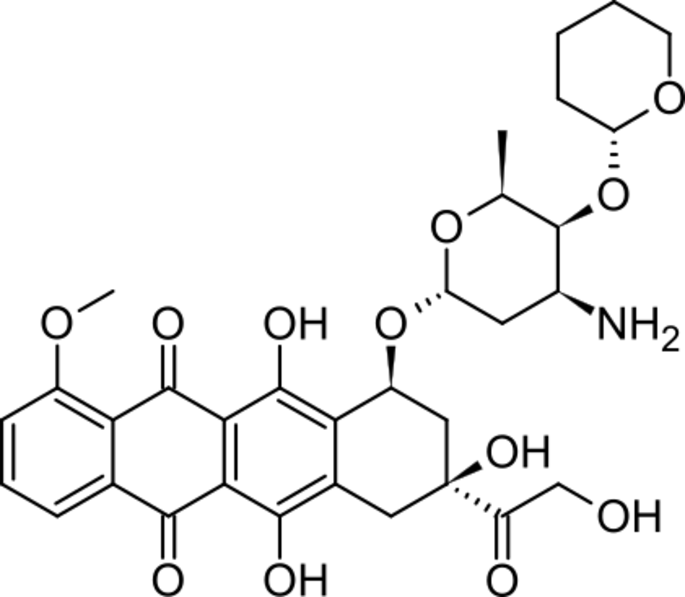
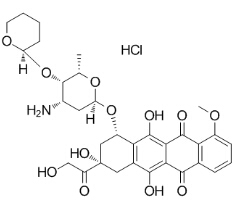 Pirarubicin Hydrochloride
Pirarubicin Hydrochloride 

 .
.")



 .
.






 GOLCONDA
GOLCONDA
















 .
. .
.
 .
. .
. .
. .
. .
.
 .
. .
.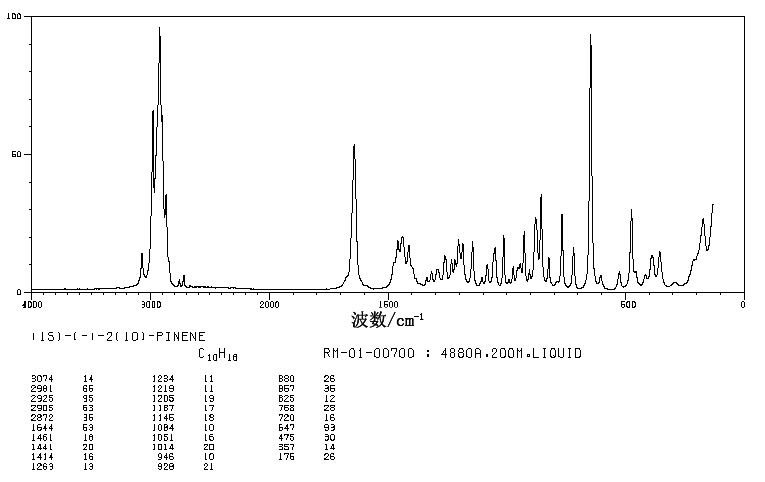









































































 .
. .
.
 .
.
 .
.



 .
.









 .
.





 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..



 amcrasto@gmail.com
amcrasto@gmail.com







 Originally posted on
Originally posted on 








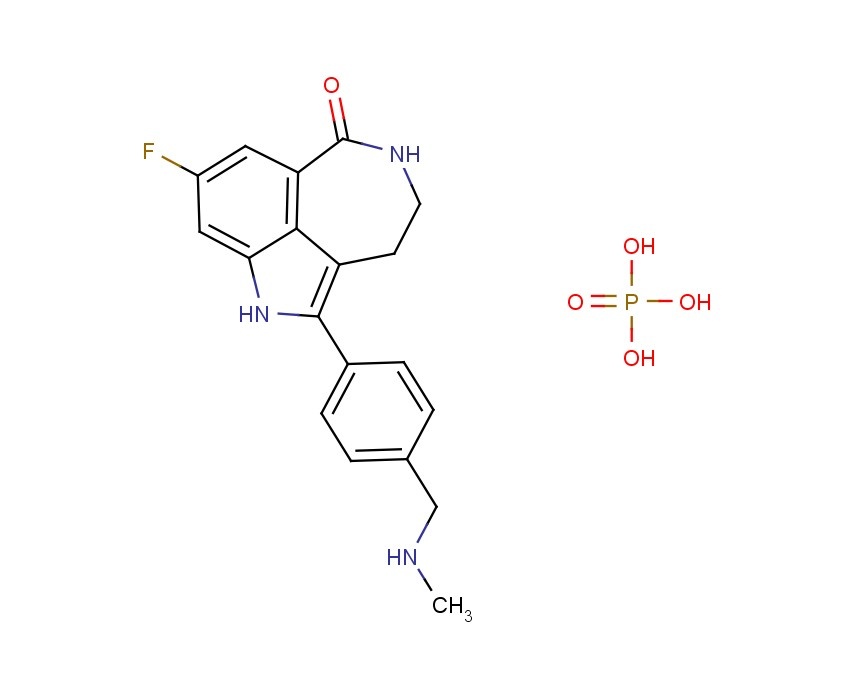




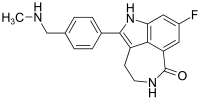
 Novel PARP inhibitor 1 is a promising new candidate for treatment of breast and ovarian cancer. A modified synthetic route to 1 has been developed and demonstrated on 7 kg scale. In order to scale up the synthesis to multikilogram scale, several synthetic challenges needed to be overcome. The key issues included significant thermal hazards present in a Leimgruber–Batcho indole synthesis, a low-yielding side-chain installation, a nonrobust Suzuki coupling and hydrogen cyanide generation during a reductive amination. In addition to these issues, changing from intravenous to oral delivery required a new salt form and therefore a new crystallization procedure. This contribution describes development work to solve these issues and scaling up of the new process in the pilot plant.
Novel PARP inhibitor 1 is a promising new candidate for treatment of breast and ovarian cancer. A modified synthetic route to 1 has been developed and demonstrated on 7 kg scale. In order to scale up the synthesis to multikilogram scale, several synthetic challenges needed to be overcome. The key issues included significant thermal hazards present in a Leimgruber–Batcho indole synthesis, a low-yielding side-chain installation, a nonrobust Suzuki coupling and hydrogen cyanide generation during a reductive amination. In addition to these issues, changing from intravenous to oral delivery required a new salt form and therefore a new crystallization procedure. This contribution describes development work to solve these issues and scaling up of the new process in the pilot plant.



























 AIRPORT
AIRPORT





 FLAG
FLAG























































 GEFITINIB
GEFITINIB
































































 .
.





 Tandoori chicken at Surjit Food Plaza. amritsar
Tandoori chicken at Surjit Food Plaza. amritsar







 Golden Temple – Harmandir Sahib: Free food for everyone
Golden Temple – Harmandir Sahib: Free food for everyone


 tandoori chicken
tandoori chicken





















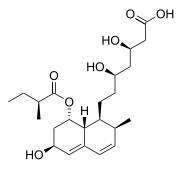
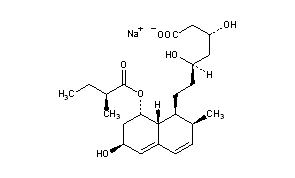

















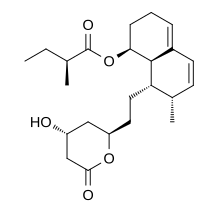





 and
and









Note: Compound name must be entered under “Substance Identification” and then “Names and Synonyms” selected to view synonyms.