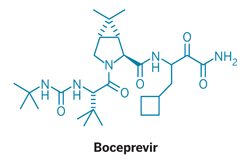
BOCEPREVIR
Handelsname: Victrelis®,
Patentnummer: WO2002008244
CAS394730-60-0
N-[3-amino-1-(cyclobutylmethyl)-2,3-dioxopropyl]-3-{N-[(tert-butylamino)carbonyl]-3-methyl-L-valyl}-6,6-dimethyl-3-azabicyclo[3.1.0]hexane-2-carboxamide
Hepatitis C virus (HCV) chronically infects more than 200 million people worldwide, and current treatment options have been very limited. Boceprevir, a protease inhibitor, which is a drug molecule approved in 2011, is useful for the treatment of human hepatitis C virus infections. It is an amorphous mixture of two diastereomers in the ratio 1.15:1, which differ in their stereochemical configuration at the third carbon atom from the ketoamide end of the molecule. Boceprevir is used in combination with interferon α-2b and ribavirin in the treatment of chronic HCV genotype 1 infection.
PAPER
Org. Process Res. Dev., Article ASAP
DOI: 10.1021/op500065t
Efforts toward the synthesis and process optimization of boceprevir 1 are described. Boceprevir synthesis was optimized by telescoping the first three steps and last two steps of the five-step process. Optimization of oxidation, which is one of the critical steps in the total synthesis, is discussed. A control strategy for the three impurities is described. A novel process for the synthesis of fragment A (2) has been developed, which is the key starting material for the synthesis of boceprevir.
…………………
![]()
![]()
WO 2015004685
( 1 R,5S)-N-[3-Amino- 1 -(cyclobutylmethyl)-2,3-dioxopropyl]-3-[2(S)-[[[( 1 , 1 -dimethylethyl) amino]carbonyl]amino]-3,3-dimethyl-l-oxobutyl]-6,6-dimethyl-3-azabicyclo [3.1.0]hexan-2(S)-carboxamide (Boceprevir); having formula I. It is a hepatitis C virus (“HCV”) protease inhibitor, developed by Merck & Co and marketed under the brand name of VICTRELIS.
![]()
Formula I
U.S. patent number 6,992,220, U.S. patent application numbers 201 1034705, U.S. 20050249702 and U.S. 201001 13821 are disclosed process for the preparation of Boceprevir.
U.S. patent number 7,326,795 claims Boceprevir bisulfate adduct as a product. Advanced Organic Chemistry, 4th ed., Jerry March Ed., John Wiley and Sons, 1972 disclosed purification methods from bisulfate adduct to provide the compound in a pure form.
U.S. patent number 8,222,427 claims a process for the purification of Boceprevir through a corresponding bisulfite adduct, wherein the compound of Formula I is dissolved in organic solvent, which is treated with an aqueous phase comprising bisulfite, thereby forming an aqueous solution of the bisulfite adduct of the compound of Formula I, which is subsequently regenerated from the aqueous phase without isolating the bisulfite adduct.
![]()
Examples:
Example 1:
183.7 gm of l-Dimethylaminopropyl-3-ethylcarbodiimide hydrochloride and 500 ml of dimethylsulfoxide were taken at 23-25 °C and to this 500 ml of ethyl acetate was added then cooled to 2-8 °C. 3-[2-(3-Tert-butylureido)-3,3-dimethyl-butyryl]-6,6-dimethyl-3-azabicyclo[3.1.0] hexane-2 carboxylic acid(2-carbamoyl-l-cyclobutyl-(methyl-2-hydroxy-ethyl)amide (Hydroxy Boceprevir) 100 gm was added to the reaction mixture under stirring at same temperature followed by 86.5 gm of dichloroacetic acid and continued stirring for 1-2 hrs. After completion of the reaction, 2500 mL of water was added to the reaction mixture at 2-10 °C and the reaction mixture temperature was raised to 15-20 °C. Ethyl acetate 600 ml was added to the reaction mass and the organic layer was separated. The product was extracted from aqueous layer with ethyl acetate. The organic layer was washed with 5% w/w hydrochloric acid followed by water. To the organic layer, aqueous solution of sodium bisulfite (300 gm in 600 ml) was added and stirred for 2 hrs. The layers were separated and organic layer was extracted with water. Thereafter, extracted aqueous layer was washed with ethyl acetate. To the aqueous layer sodium bisulfite (5.1 gm in 17 ml of water) was added and stirred for 30 min. The obtained solution was degassed and the pH was adjusted to 1.0 to 2.5 with dilute hydrochloric acid (15 ml of 35% w/w hydrochloric acid and 15 ml of water) and cooled to 10-15 °C. The obtained solid was filtered and washed with water to yield pure Boceprevir.
Exam le 2:
202 gm of l-Dimethylaminopropyl-3-ethylcarbodiimide hydrochloride and 500 ml of dimethylsulfoxide were taken at 23-25 °C and stirred, to this reaction mixture 500 ml of ethyl acetate was added; stirred and cooled to 2-8 °C. Hydroxy Boceprevir 100 gm was added under stirring at same temperature followed by 92.7 gm of dichloroacetic acid and continued stirring for 2-4 hrs. After completion of the reaction, 2500 mL of water was added to the reaction mixture at 2-10 °C and temperature was raised to 20-25 °C. Ethyl acetate 600 ml was added to the reaction mass and the organic layer was separated. The product was extracted from aqueous layer with ethyl acetate. The both organic layers were combined and stirred with dilute hydrochloric acid solution (prepared by mixing 50 ml of ~35% w/w of hydrochloric acid and 950 mL of water). The organic layer containing the product was separated and washed with water. The organic layer was cooled to 1-5 °C. To the organic layer, aqueous solution of sodium bisulfite (300 gm in 600 ml) was added and stirred for 2 hrs at 5- 9 °C. The organic layer was cooled without agitation and added precooled water at 5-10 °C. The aqueous layer containing the product was collected. The aqueous layer filtered through hyflo and washed with precooled water. Further the aqueous layer was diluted with precooled water, and adjusted the pH to 2 – 2.8 with dilute hydrochloric acid. Vacuum was applied to the aqueous layer and the temperature was slowly raised to less than 23 °C under reduced pressure. The separated solid was filtered at 22-30 °C and washed with water. Further, the filtered solid was washed with water having pH 1.8-2.4 (The pH of the water was adjusted with HC1). The product was dried at 24-28 °C under reduced pressure to yield pure Boceprevir.
Example 7:
100 gm of Crude Boceprevir was added to 300 mL of ethanol-isopropyl alcohol (1 : 1) at 22-30 °C and contents were stirred for about 40 minutes. The resulting solution was added to water slowly at 5-10 °C and stirred for 2-4 hrs at the same temperature. The product was filtered, washed with water and dried at 25-30°C under reduced pressure.
![]()
…………………
SCHERING CORPORATION Patent: WO2008/76316 A2, 2008 ; Location in patent: Page/Page column 27 ;
or eq https://www.google.co.in/patents/EP2121604A2?cl=en
Hepatitis C virus (HCV) is a (+)-sense single-stranded RNA virus that has been implicated as the major causative agent in non-A, non-B hepatitis; an HCV protease necessary for polypeptide processing and viral replication has been identified. U.S. Patent No. 7,012,066 discloses a genus of HCV protease inhibitor compounds that includes the compound of Formula I, (1 R,5S)-N-[3-amino-1-(cyclobutylmethyl)-2,3- dioxopropyl]-3-[2(S)-[[[(1 , 1 -dimethylethyl)amino]-carbonyl]amino]-3,3-dimethyl-1 – oxobutyl]-6,6-dimethyl-3-azabicyclo[3.1.0]hexan-2(S)-carboxamide.
Formula I
US2005/0059800, published March 17, 2005, discloses a process for preparing the compound of Formula I and discloses a bisulfite adduct of Formula I which can be used to provide the compound in a pure form in accordance with the methods taught in Advanced Organic Chemistry, 4th ed., Jerry March Ed., John Wiley and Sons, 1972.
US2005/0020689, filed January 27, 2005, discloses processes for preparing an intermediate useful in preparing the compound of Formula I. Methods for preparing diastereomers of the compound of Formula I are disclosed in US2005/0249702, filed November 10, 2005. Published US Patent Application No. 2007/0149459, filed November 13, 2006, discloses oxidation processes for preparing the compound of Formula I.
Purification of the compound of Formula I is difficult for several reasons. The compound Formula I is an alpha-keto amide that is unstable and forms dimers, especially under basic conditions. Also, the compound of Formula I is amorphous, thus it does not crystallize and precipitation does not improve the purity of the solid —
Previously published procedures for preparing the compound of Formula I resulted in about 63 to about 98.5% purity.
Historically, aldehydes and ketones have been purified by preparing their bisulfite adduct. Bisulfite purification of these types of compounds was performed through isolation of a solid bisulfite adduct intermediate from aqueous alcoholic solution by filtration. Regeneration of an aldehyde or ketone from an isolated bisulfite adduct is accomplished using a base or a strong acid. Examples appearing in the literature of regeneration using bases includes: Na2Cθ3 in Org. Synthesis Coll. Vol. 4, 903 (1963); NaOH in WO 2006/074270 A2; and K2CO3 in Tetrahedron Lett., 45, 3219 (2004). Examples of regeneration using acids include: H2SO4 in J. Am. Chem. Soc, 70, 1748 (1948); and HCI in WO 99/57123.
For the preparation of a purified product, isolation of an intermediate solid bisulfite adduct is not preferred since filtration of the adduct is required. In addition, base regeneration of the adduct to yield the substrate is not appropriate in those cases wherein the regenerated product is unstable in basic conditions, for example, where the regenerated product is the compound of Formula I. When acid conditions are used to regenerate the substrate compound from a bisulfite adduct, generally strongly acidic conditions and heating are necessary (see references above).
Published international application no. WO 99/57123 reports using non- alcoholic solvent in a process for forming a bisulfite adduct, however the process required isolation of a solid bisulfite adduct and regeneration the substrate from the adduct using NaOH.
A non-aqueous method for regeneration of a substrate from the corresponding bisulfite adduct was reported in J. Org. Chem., 64, 5722 (1999) as a means to overcome side-reactions such as degradation and hydrolysis during regeneration of aldehyde/ketone with a base or an acid. In this method, trimethylsilyl chloride (TMSCI) or its equivalent was employed in acetonitrile. During the process TMS2O, NaCI1 SO2 and HCI were generated as co-products when TMSCI was used.
Removal of the co-products required the process steps of filtration (for NaCI), aqueous work-up (for NaCI and excess TMSCI) and distillation (for TMS2O), which requires use of a high boiling solvent. Regeneration of aldehydes from the corresponding bisulfite adducts with ammonium acetate in solvent-free conditions was reported in J. of Chem. Research, 237 (2004), however this process requires microwave irradiation.
Published international application no. WO 2006/076415 describes regeneration of an aldehyde from a corresponding bisulfite adduct isolated from an alcoholic solvent system using a carbonate base with a lower alkyl carbonyl compound, for example, acetone and glyoxylic acid.
SCHEME Il
solv
Bisulfite Adduct
Formula I in water Formula I
SCHEME III
Formula I
Published U.S. patent application no. 2007/0149459, published June 28, 2007, discloses several alternate procedures for oxidizing the intermediate compound of the Formula II:
Formula II, to obtain the compound of Formula I.
HPLC Determination of Purity
The purity of the compound of Formula I is determined by HPLC according to the methods described below:
alternatively, the following equipment and conditions are used:
![Figure imgf000029_0001]()
Example 1
(Purification Process of Scheme III, Regeneration Option “a”)
Preparation of Compound: To a reactor was charged (16.5 kg) of the compound of Formula II,
Formula Il24.3 Kg of EDCI1 and 190 L of EtOAc. The batch temperature was adjusted between 15 and 250C. At the same temperature, Et3N (9.60 kg, 3 eq) followed by EtOAc rinse (8 L) was charged. To the resultant mixture was charged DMSO (83 L) while maintaining the temperature of the batch between 150C and 250C. CH3SO3H (10.89 kg) was charged while maintaining the reaction mixture between 150C and 30° C. After agitating at the reaction mixture for 1.5 hours while maintaining the reaction mixture between 200C and 300C, the reaction mixture was cooled to a temperature between -50C and 50C.
Purification of the Compound of Formula I
In a separate reactor was charged 165 L of water and 33 L of EtOAc, and the mixture was cooled below 50C. The reaction mixture containing the compound was transferred into the mixture of cold water/EtOAc at 0 to 100C. The organic layer was separated and washed with water (99 L) three times. Step 1 : To the resulting organic solution was added NaHSθ3 aqueous solution
(prepared from 49.5 kg of NaHSO3 and 109 L of water). The whole was agitated for 3 h at 20-300C. The aqueous NaHSO3 layer was separated and saved. The organic layer was concentrated to about 116 L of volume and diluted with MTBE (220 L). The separated aqueous NaHSO3 layer was added to the organic layer. The resultant mixture was agitated for 3 h at 20-30 0C. The organic layer was separated and cooled to 0-10 0C.
Step 2: To the cooled organic layer of Step 1 was added cold water (165 L, 0-100C) without agitation, and the whole was agitated for 5 min. The aqueous layer was separated, and a solution of water (2 L) containing NaHSO3 (0.71 kg) was added to the water layer. The water layer was distilled to the final volume of about 171 L under vacuum below 25 0C to remove volatiles.
Step 3: (Regeneration method a): The resultant water layer of Step 2 was added into a slurry of NaCI (49.5 kg) in acetone (83 L) at 20-300C. The separated acetone layer followed by acetone rinse (8 L) was added through a 0.2 micron filter to water (347 L) over 20 min at 15-25 0C. After agitation for about 1 h, the precipitate was filtered and washed with water (83 L). The wet cake was dried under vacuum at 30-400C to produce 13.0 kg (79%) of the purified compound as a white solid.
![]()
………………..
US2007/149459
http://www.google.co.in/patents/US20070149459
EXAMPLESPreparation of (1R,2S,5S)-N-[3-amino-1-(cyclobutylmethyl)-2,3-dioxopropyl]-3-{N-[(tert-butylamino)carbonyl]-3-methyl-L-valyl}-6,6-dimethyl-3-azabicyclo-[3.1.0]hexane-2-carboxamide (the Compound of Structure 2 in Scheme A, Below)
Example 1Preparation of Compound 2 Using Aqueous Acetic Acid in the Reaction Mixture
Into a 1 L, three necked flask is placed KBr (10 g, 84 mmol), NaOAc (10 g, 122 mmol), Compound 1 (50 g, 96 mmol), and TEMPO (15 g, 96 mmol), followed by 500 mL of MTBE. The reaction mixture is stirred at 350-400 rpm and the temperature is maintained at a temperature of from 10° C. to 20° C. Acetic acid (50 mL, 874 mmol), and water (5 mL) are added to the reaction mixture and the two phase mixture is agitated for 15 minutes. Continuously, over a two hour period, to the reaction mixture is added 158 mL of a 0.82 M solution of NaOCl (130 mmol). When all of the NaOCl solution is added, the reaction mixture is stirred for an additional 3 hours while maintaining the temperature. Water (50 mL) is added.
The layers are separated and the organic layer is washed twice with water (2×250 mL). A solution of ascorbic acid, which is prepared from 50 g of sodium ascorbate, 200 mL of water, and 50 mL of 4N HCl, is added to the organic layer and the mixture is stirred for about 1 hour. After the layers are separated, the organic layer is washed twice with water (2×250 mL). The organic layer is concentrated by distilling off solvent at low temperature (0-5° C.) until the total volume is about 350 mL. The concentrated organic layer is added dropwise over 30 minutes into a 3 L flask containing 2 L of n-heptane at about 0° C. providing a white precipitate. The white precipitate is collected by filtration, washed with n-heptane (400 mL) and dried in a vacuum oven (2 hr at 25° C., 8 hr at 350, and 8° C. at 45° C.). The product is obtained as a white powder (typically 94-96% yield).
1H NMR, δ 0.84 (d, J=2.3 Hz, 3H), 0.90-1.02 (m, 9H), 0.99 (d, J=4.0 Hz, 3H), 1.24 (s, 9H), 1.40-1.86 (m, 7H), 1.90-2.10 (m, 3H), 2.25-2.40 (m, 1H), 3.75 (dd, J=5.3 and 10.4 Hz, 1H), 4.10 (dd, J=6.8 and 10.4 Hz, 1H), 4.4 (dd, J=3.0 and 5.3 Hz, 2H), 5.17 (dddd, J=4.6, 8.1, 8.1, and 10.4 Hz, 1H), 5.3 (br s, 2H), 6.71 (d, J=14.7 Hz, 1H), 6.90 (dd, J=2.3 and 19.0 Hz, 1H), and 7.34 (dd, J=7.1 and 20.2 Hz, 1H).
Example 2Preparation of Compound 2 Using Glacial Acetic Acid in the Reaction Mixture
Into a 2 L, three necked flask was charged KBr (20 g, 168 mmol), NaOAc (20 g, 243 mmol), Compound 1 (100 g, 192 mmol), and TEMPO (30 g, 192 mmol), followed by 800 mL of MTBE. The reaction mixture was stirred at 350400 rpm while the temperature of the reaction mixture was maintained at a temperature of from 10° C. to 20° C. Acetic acid (70 mL, 1223 mmol, used as received), was added and the mixture was agitated for 15 minutes additional. Continuously, over a two hour period, 315 ml of a 0.73M solution of NaOCl (230 mmol) was added to the reaction mixture. When all of the NaOCl solution had been added, agitation was continued for an additional 3 hours. Water (100 mL) was added to the reaction mixture at the end of 3 hours. The layers were separated and the organic layer was washed once with water (500 mL).
A solution of ascorbic acid, which was prepared from 100 g of sodium ascorbate, 456 mL of water, and 44 mL of 36% HCl, was added to the organic layer and the mixture was stirred for about 2 hours. The layers were separated and then a solution of 3.5N HCL was added and stirred about 30 minutes. After the layers were separated, the organic layer was washed three times with water (3×500 mL). This organic layer was then added drop-wise over 30 minutes into a 5 L flask containing 3 L of n-heptane at about −10 to about 0° C. The white precipitate was filtered, washed with n-heptane (600 mL) and dried in a vacuum oven (2 hr at 25° C., 8 hr at 350, and 8° C. at 45° C.). The product was obtained as a white powder (93% yield).
1H NMR, δ 0.84 (d, J=2.3 Hz, 3H), 0.90-1.02 (m, 9H), 0.99 (d, J=4.0 Hz, 3H), 1.24 (s, 9H), 1.40-1.86 (m, 7H), 1.90-2.10 (m, 3H), 2.25-2.40 (m, 1H), 3.75 (dd, J=5.3 and 10.4 Hz, 1H), 4.10 (dd, J=6.8 and 10.4 Hz, 1H), 4.4 (dd, J=3.0 and 5.3 Hz, 2H), 5.17 (dddd, J=4.6, 8.1, 8.1, and 10.4 Hz, 1H), 5.3 (br s, 2H), 6.71 (d, J=14.7 Hz, 1H), 6.90 (dd, J=2.3 and 19.0 Hz, 1H), and 7.34 (dd, J=7.1 and 20.2 Hz, 1H).
![]() Boceprevir
Boceprevir
…………………
![]()
![]()
Chinese journal of medicinal chemistry 2011, 21, 5 , pg 409-10
![screenshot-wenku baidu com 2015-04-23 09-24-00]()
![]()
……………………
J Med Chem,2006,49(20):6074-6086.
……………………………………….
WO2004/113294 A1
…………..
MSN LABORATORIES LIMITED; THIRUMALAI RAJAN, Srinivasan; ESWARAIAH, Sajja; VENKAT REDDY, Ghojala; SAHADEVA REDDY, Maramreddy Patent: WO2014/61034 A1, 2014 ;
………………………..
WO2013066734A1
MERCK SHARP and DOHME CORP.; WU, George, G.; ITOH, Tetsuji; MCLAUGHLIN, Mark; LIU, Zhijian; QIAN, Gang Patent: WO2013/66734 A1, 2013 ;
Example 1: Cyclobutylacetonitrile
Step 1 : Cyclobutylmethyl methanesulfonate
![Figure imgf000029_0002]()
A 50-L jacket vessel was charged with DCM (20 L) (KF 34 ppm), and cyclobutylmethyl alcohol (5.0 kg, 58.0 mol) followed by TEA (8850 mL, 63.5 mol). The reaction mixture was cooled to approximately -10°C, and MsCl (4735 mL, 60.8 mol) was added via an addition funnel dropwise over approximately 3 hours, while the temperature was maintained below -5°C. The reaction resulted in a yellow slurry after 70 minutes of aging. H20 (8 L) was added to give a clear solution, which was agitated for 15 minutes. Then, the organic layer was separated. H20 (8 L) was charged to the organic layer. The mixture was agitated for 20 minutes, and then the organic layer was separated. Brine (10% solution, 4 L) was charged to the organic layer. The mixture was agitated for 20 minutes, and then the organic layer was separated. The organic phase was concentrated by vacuum distillation at approximately 30°C to 40°C and 28 inches Hg, resulting in a light brown residue (10.0 kg crude, approximately 9.5 kg product assumed, 58.0 mol, approximately 100% yield). A portion of the material was purified by distillation for characterization.
1H NMR (CDC13, 400 MHz): δ 4.18 (d, J = 6.8 Hz, 2H), 3.00 (s, 3H), 2.71 (m, 1H), 2.11 (m, 2H), 2.00-1.80 (m, 4H).
Step 2: Cyclobutylacetonitrile
![Figure imgf000030_0001]()
A 100-L RB flask was set up with a mechanical stirrer, a thermocouple, an addition funnel, a N2 inlet, and a condenser that is connected to a scrubber (11 L bleach and 5 L 2N NaOH). DMSO (30.3 L) (KF approximately 680 ppm) and NaCN (3030 g, 61.8 mol) were charged to the flask. The mixture was heated to approximately 75 °C by steam to dissolve most chunks of NaCN, resulting in a turbid solution. The product of Step 1 (9476 g, 57.7 mol) in DMSO (4 L) was added dropwise in 1 hour, 40 minutes while the temperature was maintained below approximately 87°C. The reaction was aged at approximately 85°C for 3 hours and cooled down to RT. H20 (24 L) and MTBE (24 L) were charged. The mixture was agitated, and the organic layer was separated. The aqueous layer was extracted with MTBE (18 L), and the combined organic layer was agitated with H20 (12 L) and separated. The organic layer was washed with 10% brine (4 L and 2 L), and concentrated by vacuum distillation at approximately 45°C and approximately 20 inches Hg, giving a light brown liquid (7.235 kg crude, 73.3% by GC assay, 5.30 kg product assay, 55.7 mol, 96.5% for two steps).
Ή NMR (CDCI3, 400 MHz): δ 2.65 (m, 1H), 2.41 (d, J – 5.2 Hz , 2H), 2.18 (t, J = 6.8 Hz, 2H), 2.00-1.80 (m, 4H).
Example 2: Ethyl 4-cyclobutyl-3-oxobutanoate
![Figure imgf000030_0002]()
THF (20 L) and zinc dust (2.75 kg, 42.0 mol) were charged under N2 to a 50-L jacketed vessel with a thermocouple, an addition funnel and a condenser. The mixture was stirred, and chlorotrimethylsilane (0.571 kg, 5.26 mol) was added at RT. The mixture was heated at 67°C for 30 minutes. Cyclobutylacetonitrile (2.5 kg, 26.3 mol, product of Example 1) was added at 67°C. Ethyl bromoacetate (6.108 kg, 36.6 mol) was added to the mixture at approximately 67°C to 70°C for over 3 hours. After the addition, the mixture was heated at approximately 70°C for 1 hour and then cooled to approximately 0°C to 5°C. 10% H2S04 (aq.) (35 L, 33.9 mol, approximately 1.3 eq.) was added slowly. The mixture was aged at RT for 1 hour. The organic layer was separated and subsequently washed with 10% aqueous citric acid (15 L, 7.88 mol, 0.3 eq.), 10% aqueous Na2S205 (25 L), 10% Na2S205 (aq.) (10 L), and 10% brine (10 L). The organic layer was concentrated in vacuo to afford the crude product (4.08 kg assay, 22.15 mol) in 84% yield. A part of the material was purified by distillation for characterization (with NMR in CDC13, approximately 10-15% enol-form of the compound was observed, major keto-form as shown.)
1H NMR (CDC13, 400 MHz): δ 4.19 (q, J = 7.1 Hz, 2 H), 3.38 (s, 2 H), 2.75-2.65 (m, 1H), 2.65-2.63 (m, 2 H), 2.19-2.08 (m, 2 H), 1.95-1.79 (m, 2 H), 1.73-1.60 (m, 2 H), 1.27 (t, J = 7.1 Hz, 3 H).
13C NMR (CDC13, 400 MHz): δ 202.2, 167.2, 61.3, 50.0, 49.3, 31.1, 28.4, 18.7,
14.1.
Example 3: Ethyl 2-chloro-4-c clobut l-3-oxobutanoate
Methyl t-butyl ether (30.2 L), and the crude product of Example 2 (3.78 kg assay,
20.52 mol) were charged to a 100-L RB flask with an overhead stirrer, an addition funnel, a thermometer, and an acid scrubber (with 2N NaOH at RT under N2). Sulfuryl chloride (2.98 kg,
22.06 mol) was added at approximately 20°C to 23 °C over 1.5 hours. After addition, the mixture was cooled to approximately 5°C and then quenched with 1M K3P04 (aq.) (23.6 L). The organic layer was separated and concentrated under vacuum to afford the crude chloride (4.487 kg, assume 100% yield, 20.52 mol), which was used in the next reaction without purification. A part of the material was purified by distillation for characterization (with NMR in CDC13,
approximately 10% enol-form of the compound was observed, major keto-form was shown below).
1H NMR (CDCI3, 400 MHz): δ 4.73 (s, 1 H), 4.29 (q, J = 7.1 Hz, 2 H), 2.89-2.79 (m, 2 H), 2.79-2.69 (m, 1 H), 2.20-2.07 (m, 2 H), 1.98-1.78 (m, 2 H), 17.3-1.61 (m, 2 H), 1.32 (t, J = 7.1 Hz, 3 H).
13C NMR (CDC13, 400 MHz): δ 198.1, 165.0, 63.1, 60.9, 45.7, 31.0, 28.3, 18.7, 13.9. Example 4: -C clobut l-l-ethox -l,3-dioxobutan-2-yl 4-methoxybenzoate
The crude chloride product of Example 3 (4.487 kg assumed, 20.52 mol) and Ν,Ν-dimethylformamide (11.2 L) were charged to a 50-L jacketed vessel with a thermocouple and a condenser at RT under N2. -Methoxybenzoic acid (3.75 kg, 24.62 mol) and TEA (2.285 kg, 22.57 mol) were added to the mixture. The mixture was heated at 55°C for 14 hours. The mixture was cooled to approximately 10°C, diluted with methyl tert-butyl ether (24 L), quenched with ¾0 (24 L). The organic layer was separated and subsequently washed with IN NaHC03 (20 L), then H20 (18 L) with NaCl (0.90 kg) and NaHC03 (0.45 kg). The organic layer was separated and concentrated in vacuo to afford the product (6.07 kg, 18.15 mol) in 88% assay yield. A part of the material was purified by distillation for characterization.
1H NMR (CDCI3, 400 MHz): δ 8.09 (dt, J = 2.1, 9.0 Hz, 2 H), 6.96 (dt, J = 2.1, 9.0 Hz, 2 H), 5.66 (s, 1 H), 4.31 (q, J = 7.1 Hz, 2 H), 3.88 (s, 3 H), 2.86 (dd, J = 5.7, 7.6 Hz, 2 H, 2.83-2.74 (m, 1 H), 2.23-2.12 (m, 2H), 1.98-1.80 (m, 2 H), 1.74-1.65 (m, 2 H), 1.32 (t, J = 7.1 Hz, 3 H).
Example 5: (2 -3-Amino-4-cyclobutyl-l-ethoxy-l-oxobut-2-en-2-yl 4-methoxybenzoate
The crude product of Example 4 (5.97 kg, 17.85 mol), 1-propanol (12 L), and EtOH (12 L) were charged to a 100-L RB flask with an overhead stirrer and a thermometer at RT under N2. NH4OAc (4.82 kg, 62.5 mol) was added to the mixture. The mixture was heated at 50°C for 1 hour. The mixture was concentrated in vacuo to remove H20 azeotropically with continuous addition of 1-propanol (total approximately 24 L). The mixture was solvent-switched to i‘PrOAc (24 L) under vacuum. The mixture was quenched with 2M K3P04 (aq.) (17.85 L). The organic layer was separated and washed with 15% brine (18 L) twice. The organic layer was concentrated in vacuo to afford crude enamine product (5.95 kg, assume 100% yield, 17.85 mol).
1H NMR (CDC13, 400 MHz): δ 8.12 (d, J= 8.0 Hz, 2H), 6.98 (d, J= 8.0 Hz, 2H),
6.02 (s, 2H), 4.15 (q, J= 8 Hz, 2H), 3.89 (s, 3H), 2.60-2.53 (m, 1H), 2.33 (s, 2H), 2.13-2.06 (m,
2H), 1.91-169 (m, 4H), 1.20 (t, J = 8 Hz, 3H).
13C NMR (CDC13, 400 MHz): δ 165.7, 167.6, 163.6, 153.9, 132.1, 122.2, 113.9,
113.7, 112.5, 59.6, 44.5, 37.8, 33.9, 28.5, 28.4, 18.5, 14.4.
Example 6A: 3-[(tert-Butoxycarbonyl)amino]-4-cyclobutyl-l-ethoxy-l-oxobut-2-yl 4- methoxybenzoate
The crude product of Example 5 (5.92 kg, 17.75 mol) and MeOH (23.7 L) were charged to a 100-L RB flask with an overhead stirrer, a thermocouple, and an addition funnel at RT under N2. Di-tert-butyl dicarbonate (5.81 kg, 26.6 mol) and sodium cyanoborohydride
(1.171 kg, 18.64 mol) were charged to the mixture. A solution of glycolic acid (1.485 kg, 19.53 mol) in MeOH (3.55 L) was added to the mixture drop wise at a rate to maintain the temperature at approximately 15°C to 22°C. The mixture was aged at approximately 20°C for approximately 8-10 hours. EtOAc (3.49 L, 35.5 mol) and a solution of glycine (0.866 kg, 11.4 mol) in H20 (11 L) were added to the mixture at RT. Then, 2M K3P04 (aq ) solution (17.75 L) was added. The mixture was aged for 20 minutes. The mixture was extracted with methyl tert-butyl ether (28 L). The organic layer was separated and washed subsequently with 2M K3P04 (aq.) solution (17.75 L), 10% brine (17.75 L, twice). The organic layer was concentrated under vacuum to afford the desired two diastereoisomers in almost 1 : 1 ratio (7.30 kg, 16.76 mol) in 94% assay yield.
1H NMR (CDCI3, 400 MHz): δ 8.02 (d, J= 8.0 Hz, 2H), 6.94 (d, J= 8.0 Hz, 1H),
6.93 (d, J= 8.0 Hz, 1H), 5.30 (d, J= 4.0 Hz, 0.5H), 5.17 (d, J= 4.0 Hz, 0.5H), 4.80 (d, J= 8.0 Hz, 0.5H), 4.63 (d, J = 8.0 Hz, 0.5H), 4.27-4.18 (m, 3H), 3.86 (s, 3H), 2.50-2.30 (m, 1H), 2.15- 2.00 (m, 2H), 1.89-1.60 (m, 6H), 1.43 -1.42 (m, 9H), 1.27 (t, J= 8.0 Hz, 3H).
Example 6B: 3-[(tert-Butoxycarbonyl)amino]-4-cyclobutyl-l-ethoxy-l-oxobut-2-yl 4- methoxybenzoate (First alternate procedure)
The crude product of Example 5 (19.2 g, 58.0 mmol) and MeOH (100 mL) were charged to an autoclave with a thermocouple at RT. Di-tert-butyl dicarbonate (19.0 g, 87.0 mmol) and 5% Ir/CaC03 (10.0 g) were charged to the mixture. The mixture was heated to 40°C under sealed conditions, where H2 was transferred until the internal pressure became
approximately 200 psig. The mixture was heated at 40°C at approximately 200 psig for 20 hours. The reaction mixture was cooled to RT and filtered to remove the solid to afford a clear solution. EtOAc (5.7 mL, 58 mmol) and a solution of glycine (2.8 g, 38 mmol) in H20 (37 mL) were added to the mixture at RT. Then, 2M K3P04 (aq ) solution (58 mL) was added. The mixture was aged for 20 minutes. The mixture was extracted with methyl tert-butyl ether (130 mL). The organic layer was separated and washed subsequently with 2M 3P04 (aq.) solution (58 mL), 10% brine (58 mL, twice). The organic layer was concentrated under vacuum to afford the desired two diastereoisomers in almost 1 :1 ratio (23 g, 52 mmol) in a 90% assay yield.
1H NMR (CDC13, 400 MHz): δ 8.02 (d, J= 8.0 Hz, 2H), 6.94 (d, J= 8.0 Hz, 1H), 6.93 (d, J= 8.0 Hz, 1H), 5.30 (d, J= 4.0 Hz, 0.5H), 5.17 (d, J- 4.0 Hz, 0.5H), 4.80 (d, J= 8.0 Hz, 0.5H), 4.63 (d, J= 8.0 Hz, 0.5H), 4.27-4.18 (m, 3H), 3.86 (s, 3H), 2.50-2.30 (m, 1H), 2.15- 2.00 (m, 2H), 1.89-1.60 (m, 6H), 1.43 -1.42 (m, 9H), 1.27 (t, J= 8.0 Hz, 3H). Example 6C: 3-[(tert-Butoxycarbonyl)amino]-4-cyclobutyl-l-ethoxy-l-oxobut-2-yl 4- methoxybenzoate (Second alternate procedure)
![Figure imgf000035_0001]()
NaBH4 (0.23 g, 6 mmol) and THF (5 mL) were charged to a 100-ml RB flask. The mixture was cooled to -10°C. Methanesulfonic acid (0.78 mL, 12 mmol) was charged slowly into the mixture at less than -8°C and the mixture was agitated for 15 minutes. A 0.3M solution of the crude product of Example 5 (1 g, 3 mmol) in THF was charged slowly into the mixture at below -8°C. The mixture was agitated for 16 hours. H20 (1 ml) was charged slowly into the mixture at 0°C, and the mixture was warmed to RT. Di-tert-butyl dicarbonate (1.31 g, 6 mmol) and 2M aqueous NaOH (3.75 ml) were charged into the mixture. The mixture was agitated for 2 hours at RT. An assay of the reaction mixture gave the product (1.23 g, 94%). Example 7A: Ethyl 3-f(tert-buyoxycarbonyl)aminoJ-4-cyclobutyl-2-hydroxybutanoate
![Figure imgf000035_0002]()
The crude product of Example 6A (6.0 kg, 13.78 mol) and MeOH (24 L) were charged into a 10-gallon autoclave at RT. The mixture was heated to 70°C under sealed conditions, where NH4 was transferred until the internal pressure became approximately 80 psig. The mixture was heated at 70°C at approximately 80 psig for 22 hours. The mixture was cooled to RT. NH4 was vented at RT. DMSO (5.4 L) was added to the mixture, and the mixture was aged at RT for 1 hour. The mixture was transferred into a 100-L RB flask with an overhead stirrer and a thermometer. The autoclave was rinsed with MeOH, and the mixture and rinse liquid were combined. This combined mixture was concentrated to remove MeOH under vacuum. Then, the flask was rinsed with DMSO (2.6 L) to wash the walls. Total DMSO volume was 8.0 L. The mixture was heated to 70°C to dissolve the solid to afford a clear solution, which was cooled to RT slowly to afford a slurry. ¾0 (32.0 L) was charged for approximately 1.5 hours at 20°C to 27°C. After addition of H20, the mixture was aged at RT overnight and then cooled to 0°C to 5°C for 4 more hours. The mixture was filtered to collect the solid, which was washed with cold H20 (12 L). The solid was dried at 40°C in a vacuum oven with N2 sweep (approximately 150 torr) to afford the crude product 5.63 kg (3.75 kg).
1H NMR (DMSO-d6, 400 MHz): δ 7.20-7.15 (m, 2H), 7.25 (d, J= 12.0 Hz, 0.5H), 5.92 (d, J= 12.0 Hz, 0.44H), 5.52-5.44 (m, 1H), 3.83-3.81 (m, 0.5H), 3.74-3.62 (m, 1.5H), 2.29- 2.22 (m, 1H), 2.03-1.92 (m, 2H), 1.83-1.70 (m, 2H), 1.62-1.24 (m, 13H).
13C NMR (DMSO-d6, 400 MHz) δ 175.2, 174.6, 155.5, 155.4, 78.0, 77.9, 74.4, 72.7, 51.9, 51.8, 38.8, 35.8, 33.3, 33.2, 33.0, 28.8, 28.7, 28.6, 28.5, 28.4, 28.2, 18.6, 18.5.
Example 7B: Ethyl 3-[(tert-buyoxycarbonyl)amino]-4-cyclobutyl-2-hydroxybutanoate
The crude product of Example 6A (6.0 g, 84 wt%, 11.57 mmol) and CaCl2 (1.413 g, 12.73 mmol) and 7N NH3 in MeOH (60 mL, 420 mmol) were charged into a 40 mL vial. The mixture was aged at approximately 33°C for 3 hours. The mixture was concentrated under reduced pressure to afford the product (7.8 g crude, assume 100% yield) as a tan solid. Example 8: Ethyl 3-amino-4-cyclobutyl-2-hydroxybutanoate hydrochloride
![Figure imgf000037_0001]()
IP A (13.8 L) was charged into a 100-L RB flask with a mechanical stirrer, dry and clean with a thermometer and an addition funnel, followed by addition of the product of Example 7 (3.46 kg assay, 12.70 mol). HCI in IPA (5-6 M 13.8 L, 69 mol) was slowly added into the reaction mixture. The reaction mixture was heated at 50°C for 4 hours. The mixture was cooled to RT. Then, MTBE (28 L) was added to the mixture over 30 minutes. The reaction mixture was cooled to 0°C to 5°C by MeOH/ice bath for 1.5 hour. The mixture was filtered to collect the solid, which was washed with MTBE (7 L) twice. The wet cake was dried under vacuum with N2 and sweep overnight to afford the product as an off-white solid (2.15 kg, 10.30 mol) in 76.6% overall yield for Examples 5-8.
1H NMR (DMSO-d6, 400 MHz): δ 8.20-7.95 (m, 3H), 7.54-7.44 (m, 2H), 6.46 (d, J= 4.0 Hz, 0.5H), 6.26 (d, J= 8.0 Hz, 0.5H), 4.22 (s, 0.5H), 3.98 (s, 0.5H), 3.26 (s, 0.5H), 3.10 (d, J= 4.0 Hz, 0.5H), 2.45-2.36 (m, 1H), 2.00-1.96 (m, 2H), 1.81-1.39 (m, 6H).
13C NMR (DMSO-d6, 400 MHz) δ 174.1, 173.6, 71.2, 69.8, 51.7, 51.5, 36.0, 34.6,
31.7, 31.5, 28.0, 27.8, 27.7, 18.3, 18.1.
Exam le 9: Ethyl 3-amino-4-cyclobutyl-2-hydroxybutanoate hydrochloride (Recrystallization)
H20 (3.0 L), CH3CN (6 L) and the product of Example 8 (2.00 kg, 9.58 mol) were charged to a 100-L RB flask with an overhead stirrer, a thermocouple and a condenser at RT under N2. The mixture was heated to 65°C to get a clear solution. The mixture was cooled to 50°C to get a thin slurry. CH3CN (6.0 L) was added at 50°C for over 1 hour. The mixture was cooled to 40°C. CH3CN (9.0 L) was added at 40°C for over 1 hour. The mixture was cooled to 30°C. CH3CN (18 L) was added at 30°C. The mixture was cooled to approximately 0°C to 5°C and stirred for 1 hour before filtration. The mixture was filtered, washed with CH3CN (4 L) twice, and dried with N2 stream to afford the recrystallized product as a white solid (1.887 kg, 9.04 mol, 94% isolated yield).
Ή NMR (DMSO-d6, 400 MHz): δ 8.20-7.95 (m, 3H), 7.54-7.44 (m, 2H), 6.46 (d, J= 4.0 Hz, 0.5H), 6.26 (d, J= 8.0 Hz, 0.5H), 4.22 (s, 0.5H), 3.98 (s, 0.5H), 3.26 (s, 0.5H), 3.10 (d, J= 4.0 Hz, 0.5H), 2.45-2.36 (m, 1H), 2.00-1.96 (m, 2H), 1.81-1.39 (m, 6H).
13C NMR (DMSO-d6, 400 MHz): δ 174.1, 173.6, 71.2, 69.8, 51.7, 51.5, 36.0, 34.6, 31.7, 31.5, 28.0, 27.8, 27.7, 18.3, 18.1.
Example 10: (lR,2S,5S)-N-(4-amino-l-cyclobutyl-3-hydroxy-4-oxobutan-2-yl)-3-[N-(t rt- butylcarbamoyl)-3-methyl-I^valyl]-6,6-dimethyl-3-azabicyclo[3A ]h
Hydroxybenzotiazole (HOBT, 4.83 g, 31.5 mmol), water (4.5 mL), (1R,2S,5S)-N- (4-amino- 1 -cyclobutyl-3 -hydroxy-4-oxobutan-2-yl)-3- [N-(tertbutylcarbamoyl)-3 -methylvalyl] – 6,6-dimethyl-3-azabicyclo[3.1.0]hexane-2-carboxamide (30 g, 60.6 mmol), HCl salt product of Example 9 (13.79 g, 66.1 mmol), ethyl acetate (120 mL) and N-methyl-2-pyrrolidone (NMP, 30 mL) were added at 19°C to a three-necked 500mL RB flask equipped with an overhead stirrer and a thermocouple. N-methylmorpholine (13.3 mL, 121 mmol) was added to the mixture at 19°C. l-Ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDCI, 15.0 g, 78.0 mmol) was added to the mixture at 21°C. Ethyl acetate (30 mL) was then added to the mixture at 18°C.
The mixture was agitated at approximately 20°C to 24°C for about 16 hours. After the reaction was complete, ethyl acetate (120 mL) was added at 23°C. The mixture was washed with 10% aqueous potassium carbonate solution (180 mL) twice at approximately 20°C to 24°C. Then, the organic layer was washed with 3.3% aqueous HCl (180 mL) twice at approximately 12°C to 18°C. The organic layer then was washed with 10% aqueous potassium carbonate solution (180 mL) and water (180 mL). The organic layer was concentrated to approximately 100 mL volume and was added to heptane (900 mL) dropwise at approximately -10°C to -5°C to precipitate the product. The mixture was filtered and washed with heptane. The solid was dried in vacuo at approximately 50°C to 60°C overnight. 31.3 g of the product compound was obtained as a white solid in 99% yield. The above procedure is in accordance with the processes disclosed in U.S. Patent Application Publication No. US2010/519485 Al, the disclosures of which are herein
incorporated by reference. It will be appreciated that the processes disclosed therein can be modified without undue experimentation to prepare specifically desired materials. The results of H NMR and C NMR for the above procedure were consistent with those reported in U.S. Patent Application Publication No. US2010/519485 Al .
Example 11: (lR,5S)-N-[3-Amino-l-(cyclobutylmethyl)-2,3-dioxopropyl]-3-[2(S)-[[[(l,l- dimethylethyl)amino]carbonyl]amino]-3,3-dimethyl-l-oxobutyl]-6,6-dimazabicyclo[3.1.0]hexan-2(S)-carboxamide
Acetic acid (27.0 mL, 472 mmol) and MTBE (240 mL) at RT were added to a three-necked 1L RB flask equipped with an overhead stirrer, a thermocouple and a chiller. The mixture was cooled to approximately 14°C, then the product from Example 10 (30.0 g, 57.5 mmol) was charged at approximately 14°C. The mixture was cooled to approximately 11°C. 2,2,6,6-Tetramethylpiperidin-l-yl)oxyl (TEMPO, 9.97 g, 63.8 mmol) was added to the mixture. A pre-mixed solution containing 40% aqueous sodium permanganate (17.02 g, 48.0 mmol) and water (99 mL) at approximately 12°C to 14°C was added to the reaction mixture over about 2 hours. The mixture was agitated at approximately 12°C until completion.
After the reaction was complete, the mixture was cooled to approximately 1°C. Water (30 mL) was added, then aqueous layer was separated. The organic layer was then washed with water (150 mL) at approximately 0°C to 10°C, and then washed with a pre-mixed solution of sodium ascorbate (30.0 g, 151 mmol) in water (150 mL) and concentrated HCl (12.42 mL, 151 mmol) at approximately 5°C to 15°C. The mixture was agitated at approximately 5°C to 10°C for 2 hours; then aqueous layer was separated. The organic layer was further washed with 2.5 N HCl (120 mL) at approximately 0°C to 10°C and with water (150 mL) at
approximately 0°C to 10°C four times. The organic layer (approximately 170 mL) was then added dropwise to heptane (720 mL) at approximately -20°C to -15°C to precipitate the product. The mixture was then warmed to -5°C and filtered to collect the solid. The solid was washed with heptane, dried in a vacuum oven with nitrogen sweep at room temperature to afford 27.1 g of desired product of Formula II as a white solid in 91% yield.
The above procedure is in accordance with the processes disclosed in U.S.
Provisional Patent Application No.61/482,592 (unpublished), the disclosures of which are herein incorporated by reference. It will be appreciated that the processes disclosed therein can be modified without undue experimentation to prepare specifically desired materials. The results of 1H NMR and 13C NMR for the above procedure were consistent with those reported in U.S. Provisional Patent Application No.61/482,592 (unpublished).
……………………………………………………………….
![[14C]-Boceprevir NMR spectra analysis, Chemical CAS NO. 394730-60-0 NMR spectral analysis, [14C]-Boceprevir H-NMR spectrum]()
13C NMR PREDICT
![[14C]-Boceprevir NMR spectra analysis, Chemical CAS NO. 394730-60-0 NMR spectral analysis, [14C]-Boceprevir C-NMR spectrum]()
| WO2010138889A1 * |
28 May 2010 |
2 Dec 2010 |
Concert Pharmaceuticals, Inc. |
Peptides for the treatment of hcv infections |
| WO2011125006A2 * |
31 Mar 2011 |
13 Oct 2011 |
Pfizer Inc. |
Novel sultam compounds |
| US20110034705 * |
17 Dec 2008 |
10 Feb 2011 |
Schering-Plough Corporation |
Process For the Synthesis of 3- Amino-3-Cyclobuthylmethyl-2-Hydroxypropionamide or Salts Thereof |
| US8188137 |
14 Aug 2009 |
29 May 2012 |
Avila Therapeutics, Inc. |
HCV protease inhibitors and uses thereof |
| US8524760 |
10 Apr 2012 |
3 Sep 2013 |
Celgene Avilomics Research, Inc. |
HCV protease inhibitors and uses thereof |
| EP2704570A1 * |
2 May 2012 |
12 Mar 2014 |
Merck Sharp & Dohme Corp. |
Drug substances, pharmeceutical compositions and methods for preparing the same |
| WO2014061034A1 * |
17 Oct 2013 |
24 Apr 2014 |
Msn Laboratories Limited |
Process for preparation of boceprevir and intermediates thereof |
References
- 1 Kiser JJ, Burton JR, Anderson PL, Everson GT (May 2012). “Review and Management of Drug Interactions with Boceprevir and Telaprevir”. Hepatology 55 (5): 1620–8. doi:10.1002/hep.25653. PMC 3345276. PMID 22331658.
- 2
- Degertekin B, Lok AS (May 2008). “Update on viral hepatitis: 2007″. Curr. Opin. Gastroenterol. 24 (3): 306–11. doi:10.1097/MOG.0b013e3282f70285. PMID 18408458.
- 3
- Njoroge FG, Chen KX, Shih NY, Piwinski JJ (January 2008). “Challenges in modern drug discovery: a case study of boceprevir, an HCV protease inhibitor for the treatment of hepatitis C virus infection”. Acc. Chem. Res. 41 (1): 50–9. doi:10.1021/ar700109k. PMID 18193821.
- 4
- “Boceprevir – FDA Antiviral Drugs”. FDA. FDA. April 2011. Retrieved April 2014.
- 5
- “Interim Results from Boceprevir Phase II Study in Genotype 1 Treatment-Naive Hepatitis C Patients Presented At EASL – Forbes.com” (Press release). Forbes.com. Retrieved 2008-05-19.
- 6
- “FDA Approves Merck’s VICTRELIS™ (boceprevir), First-in-Class Oral Hepatitis C Virus (HCV) Protease Inhibitor” (Press release). Merck & Co. Retrieved 2011-05-14.
- 7
- http://www.hcvadvocate.org/news/reports/EASL_2009/Advocate_EASL_2009_Coverage.htm
- 8
- Poordad, F et al. (March 2011). “Boceprevir for Untreated Chronic HCV Genotype 1 Infection”. N Engl J Med. 364 (13): 1195–206. doi:10.1056/NEJMoa1010494. PMID 21449783.
- 9
- Jensen, D (March 2011). “A New Era of Hepatitis C Therapy Begins”. N Engl J Med. 364 (13): 1272–1273. doi:10.1056/NEJMe1100829. PMID 21449791.
- 10
Bacon, B et al. (March 2011). “Boceprevir for Previously Treated Chronic HCV Genotype 1 Infection”. N Engl J Med. 364 (13): 1207–17. doi:10.1056/NEJMoa1009482. PMC 3153125. PMID 21449784.
Filed under:
GENERICS,
Uncategorized Tagged:
BOCEPREVIR,
FDA 2011,
msd,
schering,
victrelis ![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()

 :
:







 Originally posted on
Originally posted on 






























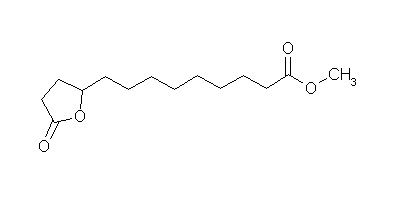







nonanoic acid methyl ester")

 Instructions
Instructions


























































 A Rationale for Changing the Synthetic Route
A Rationale for Changing the Synthetic Route











 , such as genotoxins, biocatalysis, green solvents, predicting effective solvent combinations, and process validation. Almost 85% of the references cited were published after the first edition was published, and virtually all examples in the Figures are new. Trevor Laird kindly wrote a forward for this edition.
, such as genotoxins, biocatalysis, green solvents, predicting effective solvent combinations, and process validation. Almost 85% of the references cited were published after the first edition was published, and virtually all examples in the Figures are new. Trevor Laird kindly wrote a forward for this edition.")

")

















































![[14C]-Boceprevir NMR spectra analysis, Chemical CAS NO. 394730-60-0 NMR spectral analysis, [14C]-Boceprevir H-NMR spectrum](http://pic11.molbase.net/nmr/nmr_image/2014-11-29/002/493/2493725_1h.png)
![[14C]-Boceprevir NMR spectra analysis, Chemical CAS NO. 394730-60-0 NMR spectral analysis, [14C]-Boceprevir C-NMR spectrum](http://pic11.molbase.net/nmr/nmr_image/2014-11-29/002/493/2493725_13c.png)









































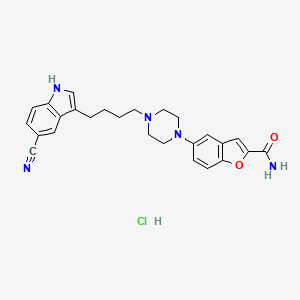




















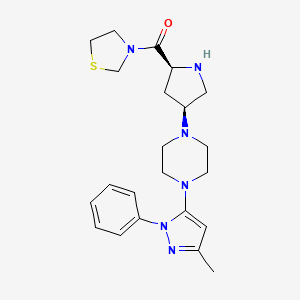







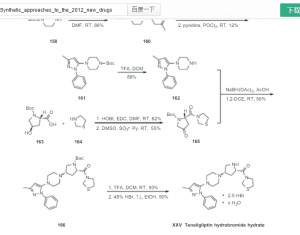





















































 trelagliptin succinate
trelagliptin succinate






























































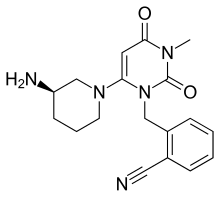

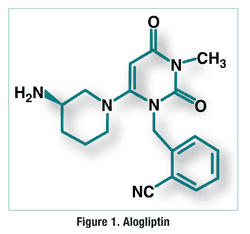




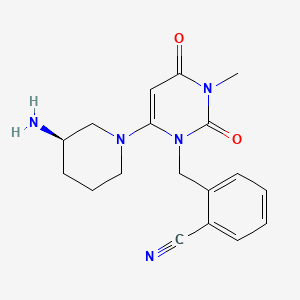


 Alogliptin benzoate
Alogliptin benzoate LIONEL MY SON
LIONEL MY SON
















