
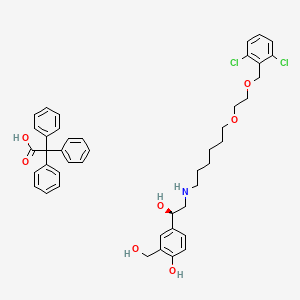


Vilanterol trifenatate, ビランテロールトリフェニル酢酸塩
ビランテロールトリフェナテート
UNII-40AHO2C6DG; GW642444M; CAS 503070-58-4
free form, 503068-34-6
444
642444
GSK-642444
GW-642444
GW-642444M
4-[(1R)-2-[6-[2-[(2,6-dichlorophenyl)methoxy]ethoxy]hexylamino]-1-hydroxyethyl]-2-(hydroxymethyl)phenol;2,2,2-triphenylacetic acid
| Molecular Formula: | C44H49Cl2NO7 |
|---|---|
| Molecular Weight: | 774.776 g/mol |
4-[(1R)-2-({6-[(2-{[(2,6-Dichlorophenyl)methyl]oxy}ethyl)oxy]hexyl}-amino)-1-hydroxyethyl]-2-(hydroxymethyl)phenol Acetate Salt
white crystalline solid: mp (DSC) 131.9−134.2 °C, [α]D 20 −14.6 (c 1.034 in MeOH). MS ES +ve m/z 289, 486/488 (M + H)+. 1H NMR δ (500 MHz, CD3OD) 7.47 (2H, m), 7.38 (8H, m), 7.28 (6H, tt, J 7.1, 1.8 Hz), 7.22 (4H, m), 6.86 (1H, d, J 7.9 Hz), 4.94 (1H, dd, J 9.7, 4.6 Hz), 4.91 (2H, s), 4.74 (2H, s), 3.79 (2H, m), 3.69 (2H, m), 3.56 (2H, t, J 6.1 Hz), 3.10 (2H, m), 2.99 (2H, m), 1.72 (2H, m), 1.65 (2H, m), 1.45 (4H, m). 13C NMR δ (125 MHz, CD3OD) 180.1, 156.2, 147.7, 140.3, 137.9, 134.5, 133.0, 131.9, 131.6, 129.6, 128.9, 128.1, 127.1, 127.0, 126.7, 116.0, 72.1, 71.4, 71.3, 71.1, 70.1, 68.4, 60.9, 55.4, 48.9, 30.5, 27.4, 27.1, 26.8. Anal. found: C, H, N, Cl.
Vilanterol is a selective long-acting beta2-adrenergic agonist (LABA) with inherent 24-hour activity for once daily treatment of COPD and asthma. Its pharmacological effect is attributable to stimulation of intracellular adenylyl cyclase which catalyzes the conversion of adenosine triphosphate (ATP) to cyclic-3′,5′-adenosine monophosphate (cAMP). Increases in cyclic AMP are associated with relaxation of bronchial smooth muscle and inhibition of release of hypersensitivity mediators from mast cells in the lungs.
Vilanterol is approved for use in several combination products such as with fluticasone furoate under the tradename Breo Ellipta and in combination with umeclidinium bromide as Anoro Ellipta. Approved by the FDA in 2013, use of Breo Ellipta is indicated for the long-term, once-daily maintenance treatment of airflow obstruction in patients with COPD, including chronic bronchitis and emphysema. It is also indicated for once-daily maintenance treatment of asthma in patients aged 18 or older with reversible obstructive airways disease.
Vilanterol is approved for use in several combination products such as with fluticasone furoate under the tradename Breo Ellipta and in combination with umeclidinium bromide as Anoro Ellipta. Approved by the FDA in 2013, use of Breo Ellipta is indicated for the long-term, once-daily maintenance treatment of airflow obstruction in patients with COPD, including chronic bronchitis and emphysema. It is also indicated for once-daily maintenance treatment of asthma in patients aged 18 or older with reversible obstructive airways disease.
Vilanterol (INN, USAN) is an ultra-long-acting β2 adrenoreceptor agonist (ultra-LABA), which was approved in May 2013 in combination with fluticasone furoate for sale as Breo Ellipta by GlaxoSmithKline for the treatment of chronic obstructive pulmonary disease (COPD).[1]
Vilanterol is available in following combinations:
- with inhaled corticosteroid fluticasone furoate — fluticasone furoate/vilanterol (trade names Breo Ellipta (U.S.), Relvar Ellipta (EU,RU, JPN))
- with muscarinic antagonist umeclidinium bromide — umeclidinium bromide/vilanterol (trade name Anoro Ellipta)
The other active component of BREO ELLIPTA is vilanterol trifenatate, a LABA with the chemical name triphenylacetic acid-4-{(1R)-2-[(6-{2-[2,6-dicholorobenzyl)oxy]ethoxy} hexyl)amino]-1-hydroxyethyl}-2-(hydroxymethyl)phenol (1:1) and the following chemical structure:
 |
Vilanterol trifenatate is a white powder with a molecular weight of 774.8, and the empirical formula is C24H33Cl2NO5•C20H16O2. It is practically insoluble in water.


https://www.accessdata.fda.gov/drugsatfda_docs/nda/2013/203975Orig1s000ChemR.pdf
PATENT
WO 2003024439
https://patents.google.com/patent/WO2003024439A1/ru
PAPER
Journal of Medicinal Chemistry (2010), 53(11), 4522-4530

A series of saligenin β2 adrenoceptor agonist antedrugs having high clearance were prepared by reacting a protected saligenin oxazolidinone with protected hydroxyethoxyalkoxyalkyl bromides, followed by removal of the hydroxy-protecting group, alkylation, and final deprotection. The compounds were screened for β2, β1, and β3 agonist activity in CHO cells. The onset and duration of action in vitro of selected compounds were assessed on isolated superfused guinea pig trachea. Compound 13f had high potency, selectivity, fast onset, and long duration of action in vitro and was found to have long duration in vivo, low oral bioavailability in the rat, and to be rapidly metabolized. Crystalline salts of 13f (vilanterol) were identified that had suitable properties for inhaled administration. A proposed binding mode for 13f to the β2-receptor is presented.
Synthesis and Structure−Activity Relationships of Long-acting β2Adrenergic Receptor Agonists Incorporating Metabolic Inactivation: An Antedrug Approach
4-[(1R)-2-({6-[(2-{[(2,6-Dichlorophenyl)methyl]oxy}ethyl)oxy]hexyl}-amino)-1-hydroxyethyl]-2-(hydroxymethyl)phenol (13f) Triphenylacetate Salt
β 2- adrenergic receptor agonist is most widely used in clinical treatment of asthma and chronic obstructive pulmonary disease drugs. Currently available on the market β2_ adrenoceptor agonists longest duration of action of 12 hours, which resulted in the need twice daily dosing. Over the last decade, the development of high potency, high selectivity, rapid onset, long duration of action, is administered once daily β2- adrenoreceptor agonists caused great concern in the pharmaceutical industry. Triflate vilanterol by Glaxo Group Limited to develop a new type of ultra-long-acting β 2- adrenergic receptor agonist, on 18 December 2013 by the US FDA clearance to market its drugs name Anoro Ellipta0
vilanterol chemical name is 4 – {(lR) -2 – [(6- {2 _ [(2,6- dichlorobenzyl) oxy] ethoxy} hexyl) amino] -1 – hydroxyethyl} -2_ (hydroxymethyl) phenol, having the formula as follows:

At present the synthesis of chiral vilanterol reported mainly in the following two ways:
1, and references J.Med.Chem.2010,53,4522-4530 Patent W02003024439, synthetic routes such as
under:

1.2, and references J.Med.Chem.2010,53,4522-4530 Patent W02003024439, synthetic routes such as
under:

Two or more routes are carried over a key intermediate in the alkylation of the amine compound X and then deprotecting to give the target compound I. Use of highly toxic chiral oxazaborolidine key intermediate in the process for preparing a compound X as a catalyst is expensive, and serious environmental pollution can not be recycled, high production costs; while boron reducing agent used in the process alkoxy – tetrahydrofuran solution of dimethyl sulfide have high reactivity shortcomings need to use special equipment. Further, throughout the synthesis process used in amounts of sodium hydride, sodium hydride in the reaction process will emit a lot of heat, and the use of sodium hydride and stored under harsh conditions, there are security risks in industrial production, is not suitable for industrial production.
Laurus Labs Limited was improved synthesis process described above, Patent W02014041565, which scheme is as follows:

While this synthesis will replace potassium t-butoxide, sodium hydride, to reduce the security risks in industrial production, but the process for preparing a key intermediate compound using X is still toxic as chiral oxazaborolidine catalyst, and environmental pollution high production cost issues remain unresolved.
An epoxy compound IV (preparation described in Bioorganic & Medicinal Chemistry Letters, 23 (5), 2013,1548-1552 and Patent CN101684074A) amine VI with a chiral auxiliary to give the chiral compound V.

Wherein the amine is a chiral auxiliary or S- S- phenylethylamine naphthylethyl amine, amine chiral auxiliary used has S- (a) – methylbenzylamine, (S) -2_ A -1-phenylethylamine, S – (-) _ N- benzyl-1-phenylethylamine, S – (-) – l_ (l- naphthyl) ethylamine
Example a
(R) -1- (2,2- dimethyl–4H- benzo [d] [I, 3] dioxin-6-yl) _2_ (⑶-1- phenyl-ethylamino) ethanol, and the step of preparing a salt of I): 2, 2- dimethyl-6- ethylene prepared -4H- benzo [d] [I, 3] dioxane (compound of formula IV) burning
Was added to a three neck round bottom flask, 12.8 g of 2-bromo-1- (2,2-dimethyl -4H-1,3- benzodioxin-6-yl) (Compound of formula II) ethanone and 100 ml of methanol, stirred and dissolved it was cooled to -10 ° C, followed by the slow addition of 2.4 g of sodium borohydride addition was completed, the reaction at room temperature for 90 minutes. Was added to the reaction mixture quenched with 50 ml aqueous ammonium chloride solution, stirred and concentrated to remove most of the methanol for 10 minutes, then extracted with 50 ml of methylene chloride, the aqueous phase was repeatedly extracted three times with 50 ml dichloromethane and the combined organic phases . The organic phase was washed with 20 ml of distilled water and once with 20 ml of saturated brine once, dried over anhydrous sodium sulfate, filtered, and concentrated. Then a mixture of tetrahydrofuran and methanol in this step the resulting compound (about 12 g) was dissolved in a total volume of 200 ml (volume ratio of tetrahydrofuran to methanol is 1: 1), 20.8 g of potassium carbonate was added, and the reaction at room temperature for 18 hour. The reaction was concentrated to remove most of the organic solvent, 100 ml of distilled water was added to the concentrate, and then 60 ml of methylene chloride was separated out and the aqueous phase repeatedly extracted three times with 30 ml of methylene chloride, the organic phase was washed with 20 ml of distilled water once with 20 ml saturated brine once, dried over anhydrous sodium sulfate, and concentrated to give a white solid. Compound IV obtained in this step without further purification was used directly in the next reaction.
. [0012] Step 2): (R) -1- (2, 2 ~ _ methyl -4H- benzo [d] [I, 3] dioxo TK 6-yl) -2 – ((S preparation) -1-phenyl-ethylamino) ethanol
The 8.24 g of the epoxy compound IV dissolved in 30 ml dimethyl sulfoxide at room temperature was slowly added 5.8 g S- (a) – methylbenzylamine, and then controlling the reaction temperature at 60 ° C 3 hours, by TLC monitoring the reaction is complete. Wait until the reaction mixture was cooled, added to 90 ml saturated aqueous sodium bicarbonate, and extracted with ethyl acetate (3 x 50 mL), the organic phase was dried over anhydrous sodium sulfate, then filtered and concentrated to give (R) -1- (2, 2-methyl–4H- benzo [d] [1,3] dioxin-6-yl) -2 – ((S) -1- phenylethyl) ethanol The crude product was 10.3 g, yield rate of 73%. The crude product obtained in this step without further purification was used directly in the next salt-forming reaction. [0013] 1H-NMR (500 MHz, CDCl3) δ 1.27 (d, J = 12.2 Hz, 3H), 1.49 (s, 6H), 2.94 (dd, J = 24.8 and 11.4 Hz, 1H), 3.21 (dd, J = 24.8 and 11.4 Hz, 1H), 4.32-4.39 (m, 1H), 4.59 (s, 2H), 4.84 – 4.89 (m, 1H), 6.82 (d, J = 15.0 Hz, 1H), 7.06 (d , J = 3.1 Hz, 1H), 7.25 – 7.35 (m, 6H).
[0014] LC-MS: m / z = 328.1 (C20H25NO3 + H +).
[0015] Chiral HPLC: R- configuration: 96.4%, S- configuration: 3.6%.
[0016] Step 3) (! R) -1- (2,2- dimethyl-benzo -41- [(1] [1,3] dioxin-6-yl) -2 – (( preparation of different salts of 1-phenyl-ethylamino) ethanol 5)
Step 2) The obtained crude product was equally divided into four parts, each of 20 ml of methanol are added to the solvent, stirring at 40 ° C under conditions to dissolve and camphorsulfonic acid were added to a solution of four parts, methanesulfonic acid , oxalic acid and benzoic acid is added in an amount of 1.5 equivalent of the crude product, after the addition was complete, stirring was continued for 2 hours, allowed to stand overnight and cooled at 0 ° C, filtered, to give the corresponding salt. The results shown in the following table.

[0017] Second Embodiment
(R) -1- (2,2- dimethyl–4H- benzo [d] [I, 3] dioxane _6_ yl) _2_ (⑶-2- methoxy-1-phenyl ethanol and salts thereof ethylamino)
Step I): (R) -1- (2, 2- dimethyl -4H- benzo [d] [I, 3] dioxin-6-yl) ~ 2 ~ (⑶-2- methoxy preparation of 1-phenyl-ethylamino) ethanol
The method of preparation of a Compound IV The procedure of Example I) the same embodiment.
[0018] The epoxy compound IV was added 8.24 g to 50 ml of acetonitrile solvent, stirring and dissolved slowly added
9.06 g S-2- methoxy-1-phenylethylamine, followed by stirring at 80 ° C for 6 hours. After completion of the reaction was monitored by TLC, the reaction mixture was concentrated. 30 ml of saturated aqueous sodium bicarbonate, and extracted with ethyl acetate (3×30 mL), the organic phase was dried over anhydrous sodium sulfate, filtered, and concentrated to give (R) -1- (2, 2- dimethyl -4H- benzo [d] [1,3] dioxin-6-yl) -2 – ((S) -2_ gas-methoxy-1-phenylethyl-yl) ethanol 9.8 g crude was wide, wide rate of 68%. The crude product obtained in this step without further purification was used directly in the next salt-forming reaction.
[0019] 1H-NMR (500 MHz, CDCl3) δ 1.49 (s, 6H), 2.98 – 3.21 (m, 2H), 3.34 (s, 3H), 3.55 – 3.80 (m, 2H), 4.02 (dd, J = 12.4 and 2.3 Hz, 1H), 4.59 (s, 2H), 4.86 – 4.88 (m, 1H), 6.82 (d, J = 7.5 Hz, 1H), 7.06 (d, J = 1.4 Hz, 1H), 7.28 –
7.37 (m, 6H).
[0020] LC-MS: m / z = 358.0 (C21H27NO4 + H +).
[0021] Chiral HPLC: R- configuration: 97.1%, S- configuration: 2.9%.
[0022] Step 2)  R) -l_ (2,2- dimethyl–4H- benzo [d] [l, 3] dioxin-6-yl) -2 – ((S) _2 preparation of different salts methoxy-1-phenyl-ethylamino) ethanol –
R) -l_ (2,2- dimethyl–4H- benzo [d] [l, 3] dioxin-6-yl) -2 – ((S) _2 preparation of different salts methoxy-1-phenyl-ethylamino) ethanol –
The procedure of Example I) thus-obtained crude product is equally divided into four parts, each mixed solvent was added 25 ml of ethanol and water (Vis: V # 1: 1) and stirred at 60 ° C under conditions so dissolved, then four solutions are each selected fumaric acid, malic acid, maleic acid and tartaric acid, acid is added in an amount 1.2 equivalents of crude product, after the addition was complete, stirring continued for 2 hours, allowed to stand between 5 ° C cooled overnight and filtered to give the corresponding salt. The results shown in the following table.

[0023] Example three
(R) -2- (benzyl ((S) -1-phenylethyl) amino) -1- (2, 2 – dimethyl – -4H- benzo [d] [I, 3] dioxane ethanol and salts of 6-yl)
Step I): (R) _2_ (benzyl ((S) -1-phenylethyl) atmosphere yl) -1- (2, 2 – dimethyl – -4H- benzo [d] [I, 3] preparation dioxin-6-yl) ethanol
The method of preparation of a Compound IV The procedure of Example I) the same embodiment.
[0024] 8.24 g of the epoxy compound IV were added to 50 ml of tetrahydrofuran solvent, and stirred to dissolve slowly added
10.97 g (i) S-benzyl-1-N- phenethylamine, the reaction was refluxed for 4 hours, the reaction was complete by TLC monitoring. Wait until the reaction solution was cooled, 30 ml of saturated aqueous ammonium chloride was added, stirred at room temperature for 10 minutes, then add 3 g of sodium chloride, stirring was continued for 30 minutes standing layer, the aqueous phase was extracted with ethyl acetate (3×30 mL), the organic phase was dried over anhydrous sodium sulfate, filtered, and concentrated to give (R) -2_ (benzyl ((S) -1-phenylethyl) amino) -1- (2, 2 – dimethyl -4H- benzo [d] [1,3] dioxin-6-yl) ethanol The crude product was 9.3 g, 56% yield. The crude product obtained in this step without further purification was used directly in the next salt-forming reaction.
[0025] 1H-NMR (500 MHz, CDCl3) δ 1.27 (d, J = 12.4 Hz, 3H), 1.49 (s, 6H), 2.78 – 3.21 (m, 2H), 3.46 (s, 1H), 4.00 – 4.08 (m, 2H), 4.59 (s, 2H), 4.85 – 4.88 (m, 1H), 6.81 (d, J = 14.9 Hz, 1H), 7.05 – 7.37 (m, 12H).
[0026] LC-MS: m / z = 418.1 (C27H31NO3 + H +).
[0027] Chiral HPLC: R- configuration: 95.8%, S- configuration: 4.2%.
[0028] Step 2): (R) _2- (benzyl ((S) -1-phenylethyl) gas-yl) -1- (2, 2 – dimethyl – -4H- benzo [d] [ preparation I 3] dioxin-6-yl) ethanol of different salts
A mixed solvent of water -.V The procedure of Example I embodiment) of the obtained crude product was equally divided into four parts, each of which shall propanol and 30 ml of water is 3: 2) at 80 ° C for dissolution while stirring, and then was added to four parts, respectively, fumaric acid, citric acid, maleic acid and tartaric acid, the acid is added in an amount 1.2 equivalents of crude product, after the addition was complete, stirring continued for 2 hours, allowed to stand at 5 ° C for cooling overnight and filtered, to give the corresponding salt. The results shown in the following table.

[0029] Fourth Embodiment
(R) -1- (2,2- dimethyl–4Η- benzo [d] [I, 3] dioxane _6_-yl) -2- (S) -1- (naphthyl _1_ yl) ethanol and salts thereof ethylamino)
Step I): (R) -1- (2,2_ dimethyl -4H- benzo [d] [1,3] dioxin-6-yl) -2_
Preparation (S) _1_ (naphthalen-1-yl) ethylamino) ethanol Preparation of Compound IV in a procedure as in Example I) the same embodiment.
[0030] The 8.24 g of the epoxy compound IV were added to 40 ml _2_ N- methyl pyrrolidone was slowly added with stirring so that after dissolution 9.58 g S – (-) – 1- (1- naphthyl) ethylamine, temperature was controlled at 100 ° C for 6 hours, the reaction was complete by TLC monitoring. After the reaction was cooled, 60 ml of saturated aqueous sodium bicarbonate, and extracted with ethyl acetate (3X 50 ml), the organic phase was dried over anhydrous sodium sulfate, filtered, and concentrated to give 00-1- (2,2-bis methyl-4! l-benzo [d] [l, 3] dioxin-6-yl) -2- (S) -1- (naphthalen-1-yl) ethylamino) ethanol The crude product 9.5 g, yield 63%. The crude product obtained in this step without further purification was used directly in the next salt-forming reaction.
[0031] 1H NMR (500 MHz, CDCl3) δ 1.40 (d, J = 11.9 Hz, 3H), 1.49 (s, 6H), 2.95 (dd, J = 24.7 and 11.0 Hz, 1H), 3.21 (dd, J = 24.9 and 11.0 Hz, 1H), 4.59 (s, 2H), 4.89 – 4.95 (m, 2H), 6.80 – 8.01 (m, I OH).
[0032] LC-MS: m / z = 378.2 (C24H27NO3 + H +).
[0033] Chiral HPLC: R- configuration: 97.8%, S- configuration: 2.3%.
[0034] Step 2): (R) -l_ (2,2- dimethyl–4H- benzo [d] [1,3] dioxin-6-yl) -2- (S) -1 preparation of (naphthalene-1-yl) ethylamino) ethanol salt of different –
The procedure of Example I embodiment) of the obtained crude product was equally divided into four parts, each of which shall solvent was added 25 ml of butanol was stirred at 80 ° C for the condition to be dissolved and then the mixture was four respective selection naphthalenesulfonic acid, camphorsulfonic acid, methanesulfonic acid and benzoic acid treatment, acid is added in an amount 1.5 equivalents crude product, after completion, stirring was continued for 2 hours, allowed to stand overnight and cooled at 0 ° C, filtered, to give the corresponding salt. The results shown in the following table.

[0035] Embodiment V
(S) – (2- (tert-butoxy quasi-yl) ((R) -2- (2, 2- dimethyl-benzo [d] [I, 3] dioxin-6-yl) – 2 preparation amino) phenylacetate -2_ their salts light ~ ethyl)
The I step) (2S) – Preparation of [(tert-butoxycarbonyl) amino] (phenyl) acetate Patent Documents US8455514 and CN102120724A prepared (2S) according to – [(tert-butoxycarbonyl) amino] (phenyl) acetic acid methyl ester.
[0036] 1H-NMR (500 MHz, CDCl3) δ 1.42 (s, 9H), 3.67 (s, 3H), 6.19 (s, 1H), 7.20 – 7.38 (m, 5H).
[0037] Step 2): (S) – (2- (tert-butoxy quasi-yl) ((R) -2- (2,2- dimethyl-benzo [d] [I, 3] dioxane ) -2-6-yl) -2-hydroxyethyl) aminophenyl acetate
The 8.24 g of the epoxy compound IV were added to 30 ml of dimethyl sulfoxide, added slowly with stirring to dissolve after
12.72 g (2S) – [(tert-butoxycarbonyl) amino] (phenyl) acetate, the reaction temperature is controlled at 70 ° C 4 h, monitoring by TLC the reaction was complete. Wait until the reaction solution was cooled, added 60 mL of saturated aqueous sodium bicarbonate, and extracted with ethyl acetate (3 x 50 mL), the organic phase was dried over anhydrous sodium sulfate, filtered, and concentrated to give (S) – (2- (tert oxycarbonyl group) ((R) -2- (2, 2- dimethyl-benzo [d] [l, 3] dioxin-6-yl) -2-hydroxyethyl) amino) phenyl _2_ acetate The crude product was 11.2 g, yield 59%. The crude product obtained in this step without further purification was used directly in the next salt-forming reaction.
[0038] 1H-NMR (500 MHz, CDCl3) δ 1.42 (s, 9H), 1.49 (s, 6H), 3.48 (dd, J = 23.7and 7.5 Hz, 1H), 3.67 (s, 3H), 3.78 ( dd, J = 24.8 and 7.6 Hz, 1H), 4.59 (s, 2H), 5.52 – 5.55 (m, 1H), 6.41 (s, 1H), 6.80 – 7.32 (m, 8H).
[0039] LC-MS: m / z = 472.1 (C26H33NO7 + H +).
[0040] Chiral HPLC: R- configuration: 96.1%, S- configuration: 4.0%.
[0041] Step 3) (S) – (2_ (tert quasi-yl) ((R) _2_ (2,2_-dimethyl-benzo [d] [1,3] dioxin-6-yl) preparation of amino group) of different salts of methyl-2-phenyl-2-hydroxyethyl)
Step 2) The obtained crude product was equally divided into four parts, each solvent were added 20 ml of methanol was stirred at 40 ° C under conditions to dissolve, then the mixture was four respective selection acid, hydrochloric acid, naphthalenesulfonic acid, and methanesulfonic acid treatment, acid is added in an amount 1.5 equivalents crude product, after completion, stirring was continued for 2 hours, allowed to stand overnight and cooled at 0 ° C, filtered, to give the corresponding salt. The results shown in the following table.

Vilanterol is chemically described as 4-{(lR)-2-[6-{2-(2, 6-dichlorobenzyl) oxy] ethoxy} hexyl) amino]- l-hydroxyethyl}-2-(hydroxymethyl) phenol as represented by Formula I.

Formula I The compound 4-{(lR)-2-[(6-{2-[(2,6-dicUorobenzyl)oxy]emoxy}hexyl)amino]-l- hydroxy ethyl} -2-(hydroxymethyl)phenol is specifically described in WO2003/024439, as are pharmaceutically acceptable salts thereof, in particular the acetate, triphenylacetate, a-phenylcinnamate, 1-naphthoate and (R)-mandelate salts. More specifically the preferred pharmaceutically acceptable salt is triphenylacetate salt.
The PCT publication WO 2003/024439, the corresponding US equivalent US 7,361,787 (herein after the ‘787 patent) and J.Med.Chem, 2010, 53, 4522-4530 discloses the process for preparation of vilanterol along with pharmaceutically acceptable salt. The ‘787 patent reaction sequence is schematically represented as follows:

The process described in the ‘787 patent uses alcoholic solvent during the acetonide cleavage of Formula XIV, which tends to result in the formation of the corresponding ether impurities. This requires repetitive purifications, which can be tedious to practice during scale up process. Moreover the dibromo hexane used in the process contains the corresponding 1, 5-dibromo alkanes which tends to react in the same sequential manner to generate the corresponding analogues, which requires repetitive purifications to separate out from the final API. The ‘787 patent imply the use of column chromatographic procedures which are not feasible on the commercial scale.
The ‘787 patent further elucidates the process for preparing (5R)-5-(2, 2-dimethyl-4H-l,

isomeric impurities for the chiral intermediate would carry forward during the process 2013/000556
which results in the formation of various isomeric impurities which are difficult to separate and need more tedious procedures. Moreover reagents like sodium hydride are difficult to handle during the scale up process as it tends to generate high exothermicity, which can affect the yield and purity of the said compound.
The purity and the yield of vilanterol trifenatate as per the disclosed process are not satisfactory and also the said process involves chromatography techniques to isolate the intermediate compounds. The said techniques are tedious, labor intensive, time consuming process not suitable for industrial scale and which in turn result to an increase in the manufacturing cost. Moreover the said process involves the use of vilanterol trifenatate which degrades to form certain impurities and results in the formation of the final compound with a lesser purity.
In view of intrinsic fragility there is a need in the art to develop a simple, industrially feasible and scalable process for the synthesis of vilanterol that would avoid the aforementioned difficulties. Moreover it becomes necessary to prepare highly chiral pure oxazolidinone intermediate to prepare chirally pure vilanterol.
Examplel2: Preparation of 4-((R)-2-{6-[2-(2, 6-Dichlorobenzyloxy)-ethoxy]- hexylamino}-l-hydroxy ethyl)-2-hydroxymethyI-phenol (I-Vilanterol)
Compound XTV (1.0 eqt) was dissolved in acetone (10V) under nitrogen at ambient temperature. The reaction mass was cooled to 0-5°C and 0.5N HCl (12V) was added slowly. The reaction mass was allowed to stir for completion over one hour period. The reaction mass was diluted with dichloromethane and water, followed by addition of saturated sodium bicarbonate solution (lOv) at 0-5°C. The organic layer was separated then washed successively with water/saturated brine and dried over sodium sulfate the solution was concentrated to dryness under vacuum to obtain the residue, followed by column chromatography (MeOH-DCM as eluent). The pure fractions were concentrated under vacuum to afford the title compound as pale yellow color oil.
Yield: 77%; purity by HPLC: 99.15%; Chiral purity: R-isomer: 99.97%; S-isomer: 0.03%
Examplel3: Preparation of 4-((R)-2-{6-[2-(2, 6-Dichlorobenzyloxy)-ethoxy]- hexylamino}-l-hydroxy ethyI)-2-hydroxymethyl-phenol triphenyl acetate (IA: Vilanterol trifenatate)
Triphenyl acetic acid (l.Oeqt) was added to a solution of compound I (l.Oeqt) in acetone (20V) at ambient temperature and the mixture heated to 50-55°C to obtain a homogenous solution. The mixture was allowed to cool to ambient temperature; the resultant product was filtered, washed with chilled acetone, dried under vacuum at 50°C to afford the title compound as a white solid.
Yield: 69%; purity by HPLC: 99.79%; chiral purity-R-isomer: 99.96%; S-isomer: 0.049%
β 2- adrenergic receptor agonist is most widely used in clinical treatment of asthma and chronic obstructive pulmonary disease drugs. Currently available on the market β 2- adrenoreceptor agonist longest duration of action of 12 hours, which resulted in the need twice daily dosing. Over the last decade, the development of high potency, high selectivity, rapid onset, long duration of action, once daily dosing of β 2- adrenoreceptor agonists caused great concern in the pharmaceutical industry. Three acid vilanterol by Glaxo Group Limited development of a new Ultralente β 2- adrenergic receptor agonists, having bronchodilatory action.
[0003] vilanterol chemical name is 4 – {(lR) -2 – [(6- {2 – [(2,6- dichlorobenzyl) oxy] ethoxy} hexyl) amino] – 1-hydroxyethyl} -2_ (hydroxymethyl) phenol, having the formula as follows:

[0005] vilanterol synthetic routes are:

[0007] (5R) -5- (2, 2- dimethyl -4H-1,3- benzodioxin-6-yl) -1,3-oxazolidin-2-one was prepared an important intermediate Whelan Castro. The synthesis of this intermediate are currently two main ways:
[0008] 1: Reference Laurus Labs Limited published patent W02014041565, its main synthetic routes are as follows:
[0009]

[0010] obvious drawback of this method, the starting material is 4-bromo-2-hydroxymethyl-phenol, expensive, the next two steps harsh reaction conditions, where low temperature -75 ° C, and the yield rate is not high. Obviously not suitable for large-scale industrial production.
[0011] 2: Reference J. Med Chem 2010, 53, 4522-4530, and patent W02003024439, scheme is as follows:
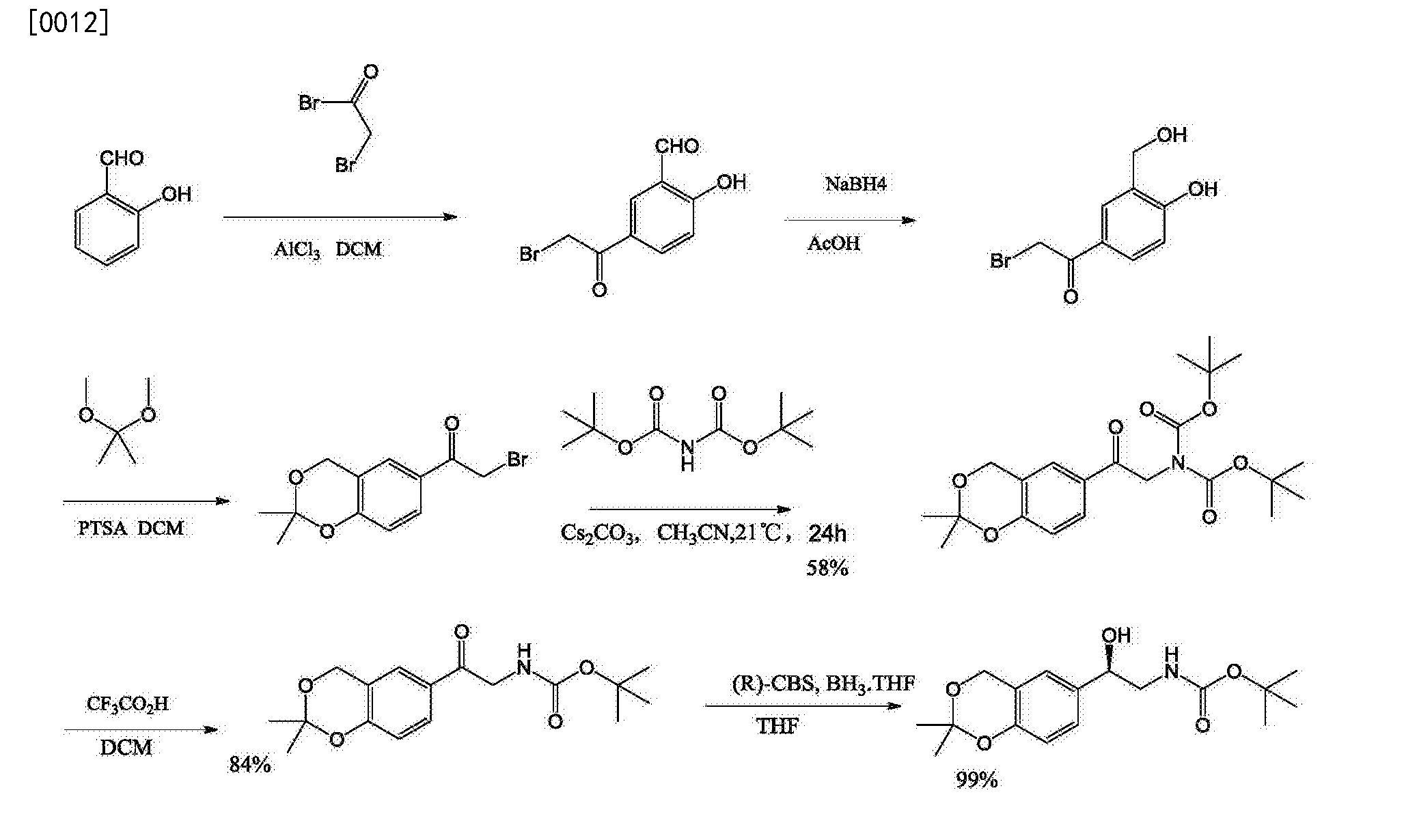
[0013]

The route salicylaldehyde as raw material, the final seven-step synthesis intermediates, but the reaction step, 2-bromo-1- (2,2-dimethyl -4H-1,3- benzodioxin en-6-yl) ethanone di-t-butyl imine and a dicarboxylic acid, a lower yield, only 58%; while the imine dicarboxylate and cesium carbonate expensive, more cost high; the next step and also acidolysis out a tert-butoxycarbonyl group, relatively low utilization atoms.
Synthetic Route [0046] The reaction is as follows:
[0047]

Preparation of 5- (2-bromoacetyl) -2-hydroxyphenyl 4-carbaldehyde: [0048] Example 1
[0049] Under nitrogen, the ice bath, the aluminum trichloride 164g (5eq) dispersed into 600mL (20-fold amount) in DCM was slowly added dropwise bromoacetyl bromide 99. 4g (2eq), 20min After completion of the dropwise addition, the temperature warmed to room temperature, the reaction LH, salicylaldehyde to this mixture was added dropwise 30g, 20min dropwise addition, dropwise, the reaction overnight at 35 ° C. To the reaction mixture was added ice-water, the organic layer was separated, washed with water, dried and concentrated to dryness in vacuo.With DCM and recrystallized from n-hexane, the product was filtered to give 36. 5g, about 61% yield. 4 bandit 1 (4001 hold, 0)? (: 13): Sll.52 (s, lH), 9.99 (s, lH), 8.30 (s, lH), 8.17 (d, lH, J = 8Hz), 7.10 (d, lH, J = 8Hz), 4.39 (s, 2H); MS (-ESI) m / z 240 [MH]
– 5 -phenyl-1-one Preparation of 2-bromo-1- [4-hydroxy-3- (hydroxymethyl): [0050] Example 2
[0051] 40. 0g of the compound 4 dissolved in 400mL of acetic acid (10 times the amount), under ice-cooling, sodium borohydride was added portionwise 6. 8g (1. leq), was added stirred at rt for lh, TLC showed the reaction complete.Concentrated in vacuo to remove most of acetic acid, diluted with water and neutralized with sodium bicarbonate, extracted with EA, the organic phase washed with water and brine, dried over anhydrous sodium sulfate, and concentrated in vacuo to crude off-white powder did. After laundering refluxed with DCM to give a white powder 32g, 80% yield.
[0052] ^ NMR (400MHz, DMS0-d6): δ 10. 53 (s, 1H), 7. 99 (s, 1H), 7. 79 (d, 1H, J = 8Hz), 6.87 (d, lH , J = 8Hz), 4.75 (s, 2H), 4.50 (s, 2H); MS (+ ESI) m / z 267 [m + Na] +
[0053] Example 3: 2-amino-1- [4-hydroxy-3- (hydroxymethyl) – phenyl-1-one hydrochloride (6)
[0054] 10. 0g of the compound 5 was added to 200mL of ethyl acetate, was added hexamethylenetetramine (1. leq) 6. 2g, room temperature lh, TLC showed complete reaction. After filtration the filter cake was dried in vacuo as a white powder 15. 6g.The above white powder was dissolved in 150mL of ethanol, concentrated hydrochloric acid (5eq) 17. 5mL, room temperature overnight, the reaction was concentrated to dryness in vacuo to give an off-white powder 16. 0g (mixture) administered directly in the next step.
[0055] ^ NMR (400MHz, DMS0-d6): δ 10. 89 (s, 1H), 8. 40 (s, 2H), 7. 98 (d, 1H, J = 2Hz), 7 · 70 (dd , 1H, J = 8Hz and 2Hz), 7 · 02 (d, 1H, J = 8Hz), 4 · 49 (s, 2H), 4 · 43 (s, 2H); MS (+ ESI) m / z 182 [M + H] +
Preparation of 2- (3-hydroxymethyl-4-hydroxyphenyl) -2-carbonyl-ethyl carbamate ⑵ of: [0056] Example 4
[0057] The product from the previous step, compound 6 (hydrochloride) 16. 0g added to 150mL of THF and 150mL water was added 20. 6gNaHC03 (5eq), dissolved 30mL THF was added dropwise to a solution of 9. 8g Boc20, 20min After dropping. Reaction at room temperature lh, TLC showed complete reaction. Water was added, extracted with EA, the organic phase was washed successively with water and brine, dried over anhydrous sodium sulfate, and concentrated in vacuo to a crude solid powder did, then after 1-2 times the amount of reflux in DCM starched white powder 8. 7g, two step yield 76%.
[0058] ^ NMRQOOMHz, DMS0-d6):.. Δ 10. 35 (dr, 1H), 7 94 (s, 1H), 7 75 (d, 1H, J = 8Hz), 6 · 95 (t, 1H , J = 4Hz), 6 · 85 (t, 1H, J = 8Hz), 4 · 49 (s, 2H), 4 · 35 (d, 1H, J = 4Hz), L 39 (s, 9H); MS (ES +) m / z 304 [m + Na] +
[0059] Example 5: 2- (2,2-dimethyl -4H-1,3- benzodioxin-6-yl) -2-carbonyl-ethyl carbamate (7 ) preparation of
[0060] 7. 0g of the compound 2 was dissolved in 70mL of DCM (10-fold amount) was added a catalytic amount of p-toluenesulfonic acid (0. 05eq), was added dropwise 2-dimethoxyethane at reflux propane (2eq) was dissolved in 2-fold amount of DCM, 40min addition was complete, the reaction lh, TLC showed complete reaction. The reaction mixture was washed with saturated NaHC (V Sin three times, the organic phase was dried over anhydrous sodium sulfate, and concentrated in vacuo to give a yellow oil. Of isopropyl ether and recrystallized from n-heptane to obtain a white powder 6. 7g, 83% yield.
[0061] iHNMRGOOMHz, CDC13):. Δ 7. 77 (dd, 1H, J = 8Hz and 2Hz), 7 65 (s, 1H), 6 86 (d, 1H, J = 8Hz), 5 51 (.. dr, 1H), 4 87 (s, 2H), 4 56 (d, 2H, J = 4Hz), 1 56 (s, 6H), 1 47 (s, 9H);…. MS (ES +) m / z 344 [M + Na] +
[0062] Example 6: (2R) -2- (2, 2- dimethyl -4H-1,3- benzodioxin-6-yl) -2-hydroxyethyl carbamate butyl ester (8)
[0063] The catalyst was added 0. 78mL to 10mL of anhydrous THF under nitrogen was added dropwise BH3 ice bath. THF, 20min addition was complete. Was added dropwise under ice-cooling 2. 5g of compound 7 was dissolved in 20mL of anhydrous THF, 50min dropwise addition, reaction was warmed to room temperature 0. 5h, TLC indicated complete reaction. After quenched with methanol under ice-cooling the reaction, the reaction solution was concentrated in vacuo, water was added, extracted with EA, the organic phase washed with water and brine, dried over anhydrous sodium sulfate, and concentrated in vacuo to give a pale yellow oil 2. 8g. After petroleum ether starched white powder 2. 2g, 88% yield.
[0064] iHNMRGOOMHz, CDC13):… Δ 7. 13 (dd, 1H, J = 8Hz and 2Hz), 6 99 (s, 1H), 6 79 (d, 1H, J = 8Hz), 4 92 ( dr, 1H), 4. 71-4. 74 (m, 1H), 3. 42 (d, 1H, J = 12Hz), 3. 20-3. 25 (m, 1H), 1.53 (s, 6H) , 1.44 (s, 9H); MS (+ ESI) m / z 346 [m + Na] +
[0065] Example 7: (5R) -5- (2, 2- dimethyl -4H-1,3- benzodioxin-6-yl) -1, 3 oxazolidin -2 – preparation of ⑴ -one
[0066] Under nitrogen, 8 dissolved in 15mL of DMF 1. 8g compound, at 10-15 ° C, potassium tert-butoxide was added 0. 7g (l. Leq), After completion of the reaction at room temperature lh, TLC the reaction was complete. Ice water was added, a white solid was precipitated, stirring at room temperature after 3h, filtered off with suction, the filter cake was dried to obtain a white powder l.Og, 72% yield (purity 99.6%, ee 99.2%).
[0067] iHNMRGOOMHz, CDC13): δ 7. 15 (dd, 1H, J = 8Hz and 4Hz), 7 · 02 (s, 1H), 6 · 83 (d, 1H, J = 8Hz), 6.09 (br, lH), 5.52 (t, lH, J = 8Hz), 4.84 (s, 2H), 3.92 (t, lH, J = 8Hz), 3.53 (t, lH, J = 8Hz), 1.53 (s, 6H); MS (+ ESI) m / z 250 [m + H] +.
PATENT
https://patents.google.com/patent/WO2017001907A1/en
onverting the formed alcohol, preferably Compound II, to Vilanterol trifenatate, according to the below scheme:


timarate

VII L-tait rate


Example 16: Vilanterol base
Compound VII (5 g, obtained by procedure in Example 10) was dissolved in 5 EtOH (50 mL), followed by addition of 1M HCI solution (50 mL). The mixture was
stirred at room temp, for 90 minutes. Afterwards, pH of the mixture was adjusted to
~9 by addition of 20 % K2C03 solution (25 mL). The mixture was then extracted to dichloromethane (100 mL). Organic phase was washed with water (2 x 25 mL), dried over MgS04 and evaporated to dryness. The residue was purified by column 10 chromatography, elution with mixture of dichloromethane/ethanol/ammonia (50/8/1 ) to give title compound as brownish slightly yellowish oil .
Example 17: Vilanterol trifenatate
Vilanterol base (0.620 g) was dissolved in EtOH (6 mL). Triphenylacetic acid
(0.370 g) was added and the mixture was heated to 50° C and stirred at the same 15 temp, for 15 min. The mixture was then cooled to room temp., followed by cooling in ice-water bath for 90 minutes. The formed suspension was filtered, the filtration cake was washed with cold EtOH and dried at room temp, overnight.
Example 18: Preparation of Vilanterol base 20
( l/ )-2-[(6-{2-[(2,6-dichlorobenzyl)oxy]ethoxy}hexyl)amino]-l-(2,2-dimethyl- 4H-l,3-benzodioxin-6-yl)ethanol (15.5 g, obtained according to the procedure in US
2005/0075394, Example 77(iv)) was dissolved in EtOH (50 mL), followed by addition of 1M HCI solution (50 mL). The mixture was stirred at room temperature for 90 minutes.
Afterwards, the pH of the mixture was adjusted to ~9 by addition of 20 % K2C03 25 solution (25 mL). The mixture was then extracted to dichloromethane ( 100 mL). The organic phase was washed with water (2 x 25 mL), dried over MgS04 and evaporated to dryness.
The crude vilanterol base ( 14.5 g, 90.9 % purity) was dissolved in
dichloromethane and the solution was loaded on a column packed with 300 g Diol-silica 30 in dichloromethane. The column was eluted with dichloromethane with gradient of ethanol (2 – 20 %) . The chromatographic fractions were monitored by TLC. The
fractions containing relatively pure vilanterol were joined and evaporated to dryness, obtaining 11.0 g of vilanterol with purity 97.1 %.
Example 21: Preparation of Vilanterol L-tartrate
EtOH (700 mL) was mixed with 1 M aq. HCI acid (700 mL), the formed mixture 25 was cooled to 5 °C, followed by addition of compound VII L-tartrate ( 100 g, obtained by procedure in Example 15). The mixture was stirred at 5 °C for 15 hours. Afterwards, DCM (500 mL) was added, the mixture was cooled to 0 °C and aq. Solution of K2C03 ( 130g of K2C03 in 200 mL of water) was then added drop wise to the stirred reaction mixture until pH 9 – 9.5 was obtained. Temp, during the addition was kept below 5 °C. 30 The water phase was separated, and extracted with additional DCM (300 mL).
Combined organic extracts were warmed to temp. 20-25 °C and washed with water (2 x 500 mL), 1% brine (500 mL) and 24% brine (500 mL). Afterwards, organic extract was mixed with solution of L-Tartaric acid (26.6 g) in EtOH (210 mL). The mixture was stirred for 10 min. at temp. 20-25°C and then heated by setting the temp, of the 35 reactor jacket to 40°C. All DCM solvent was distilled off under vacuum to residual approximate 350 mL. The mixture was then cooled to 25°C, followed by addition of
EtOAc ( 1.5 L) . The mixture was stirred at 20-25 °C for 1 hour then cooled to -5 °C and stirred overnight. The product was separated by filtration, washed with cold EtOAc and dried under inert gas and room temp. Isolated yield 85%, chemical purity 99.8%, 5 optical purity 99.93%. The sample was analyzed by PXRD, the PXRD pattern is
presented in Figure 5.
Example 22: Preparation of Vilanterol trifenatate
Dichloromethane (256 mL) was mixed with water (256 mL), the formed mixture was cooled to 0 °C, followed by addition of Vilanterol L-tartrate (32 g, obtained by 10 procedure in Example 21 ) and EtOH (64 mL). Afterwards, 25% aq. solution of ammonia (34 mL) was then added drop wise to the stirred mixture. Temp, during the addition was kept below 5 °C. The water phase was separated, and extracted with additional
DCM (128 mL) . Combined organic extracts were warmed to temp. 20-25 °C mixed with MTBE (220 mL), EtOH (64 mL). The obtained mixture was then washed with water (3 x 15 220 mL). Afterwards, the obtained organic extract was mixed with triphenylacetic acid ( 14.5 g) and stirred until complete dissolution at temp. 20-25°C. Then EtOH (96 mL) was added and the mixture was heated by setting the temp, of the reactor jacket to
40°C. Part of DCM solvent was distilled off under vacuum to residual approximate volume 220 mL, The mixture was then cooled to 25°C, followed by addition of MTBE 20 (256 mL). The mixture was stirred at 20-25 °C for 1 hour then cooled to -5 °C and for additional 2 hours. The product was separated by filtration, washed with cold MTBE and dried under inert gas and room temp. Isolated yield 93%, chemical purity 99.8%, optical purity 99.93%.
References
- Jump up^ “FDA approves Breo Ellipta to treat chronic obstructive pulmonary disease”. Retrieved 10 May 2013.
- Harrell AW, Siederer SK, Bal J, Patel NH, Young GC, Felgate CC, Pearce SJ, Roberts AD, Beaumont C, Emmons AJ, Pereira AI, Kempsford RD: Metabolism and disposition of vilanterol, a long-acting beta(2)-adrenoceptor agonist for inhalation use in humans. Drug Metab Dispos. 2013 Jan;41(1):89-100. doi: 10.1124/dmd.112.048603. Epub 2012 Oct 4. [PubMed:23043183]
- Spyratos D, Sichletidis L: Umeclidinium bromide/vilanterol combination in the treatment of chronic obstructive pulmonary disease: a review. Ther Clin Risk Manag. 2015 Mar 25;11:481-7. doi: 10.2147/TCRM.S67491. eCollection 2015. [PubMed:25848294]



| Patent ID | Title | Submitted Date | Granted Date |
|---|---|---|---|
| US2012309725 | COMBINATIONS OF A MUSCARINIC RECEPTOR ANTAGONIST AND A BETA-2 ADRENORECEPTOR AGONIST |
2010-11-29
|
2012-12-06
|
| US2014116434 | Dry Powder Inhaler Compositions |
2012-06-01
|
2014-05-01
|
| US2013157991 | Dry Powder Inhalation Drug Products Exhibiting Moisture Control Properties and Methods of Administering the Same |
2011-08-31
|
2013-06-20
|
| US2017189424 | FLUTICASONE FUROATE IN THE TREATMENT OF COPD |
2015-05-27
|
|
| US9763965 | AGGREGATE PARTICLES |
2013-04-11
|
2015-03-26
|
| Patent ID | Title | Submitted Date | Granted Date |
|---|---|---|---|
| US8309572 | Muscarinic acetylcholine receptor antagonists |
2012-02-22
|
2012-11-13
|
| US8534281 | Manifold for use in medicament dispenser |
2006-12-11
|
2013-09-17
|
| US8161968 | Medicament dispenser |
2004-07-21
|
2012-04-24
|
| US8511304 | Medicament dispenser |
2003-01-22
|
2013-08-20
|
| US2011319371 | PHARMACEUTICAL FORMULATIONS COMPRISING 4-HEXYL)AMINO]-1-HYDROXYETHYL}-2-(HYDROXYMETHYL)PHENOL |
2011-12-29
|
| Patent ID | Title | Submitted Date | Granted Date |
|---|---|---|---|
| US6878698 | Anti-inflammatory androstane derivatives |
2003-05-15
|
2005-04-12
|
| US6537983 | Anti-inflammatory androstane derivatives |
2003-03-06
|
2003-03-25
|
| US7488827 | Muscarinic Acetylcholine Receptor Antagonists |
2007-10-25
|
2009-02-10
|
| US6759398 | Anti-inflammatory androstane derivative |
2002-11-28
|
2004-07-06
|
| US9750726 | COMBINATIONS OF A MUSCARINIC RECEPTOR ANTAGONIST AND A BETA-2 ADRENORECEPTOR AGONIST |
2015-12-16
|
2016-04-07
|
| Patent ID | Title | Submitted Date | Granted Date |
|---|---|---|---|
| US8183257 | Muscarinic Acetylcholine Receptor Antagonists |
2009-05-14
|
2012-05-22
|
| US7776895 | Inhalation devices for delivering phenethanolamine derivatives for the treatment of respiratory diseases |
2009-03-12
|
2010-08-17
|
| US7439393 | Phenethanolamine Derivatives for Treatment of Respiratory Diseases |
2008-01-03
|
2008-10-21
|
| US7498440 | Muscarinic acetylcholine receptor antagonists |
2007-08-09
|
2009-03-03
|
| US7629335 | Anti-inflammatory androstane derivative |
2007-02-01
|
2009-12-08
|
 |
|
| Clinical data | |
|---|---|
| License data | |
| ATC code |
|
| Legal status | |
| Legal status | |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEBI | |
| ECHA InfoCard | 100.217.751  |
| Chemical and physical data | |
| Formula | C24H33Cl2NO5 |
| Molar mass | 486.43 g/mol |
| 3D model (JSmol) | |
/////////////Vilanterol trifenatate, HY-14300A, CS-1679, fda 2013, Breo Ellipta, Relvar Ellipta, 444 , 642444 , GSK-642444 , GW-642444 , GW-642444M , ビランテロール , ビランテロールトリフェニル酢酸塩 , ビランテロールトリフェナテート
C1=CC=C(C=C1)C(C2=CC=CC=C2)(C3=CC=CC=C3)C(=O)O.C1=CC(=C(C(=C1)Cl)COCCOCCCCCCNCC(C2=CC(=C(C=C2)O)CO)O)Cl