WATCH OUT FOR CORRECT FORMATTING TEXT AND STRUCTURE BOTH IN 2-3 DAYS
FDA approves the first drug with an indication for treatment of smallpox
July 13, 2018
Release
The U.S. Food and Drug Administration today approved TPOXX (tecovirimat), the first drug with an indication for treatment of smallpox. Though the World Health Organization declared smallpox, a contagious and sometimes fatal infectious disease, eradicated in 1980, there have been longstanding concerns that smallpox could be used as a bioweapon.
“To address the risk of bioterrorism, Congress has taken steps to enable the development and approval of countermeasures to thwart pathogens that could be employed as weapons. Today’s approval provides an important milestone in these efforts. This new treatment affords us an additional option should smallpox ever be used as a bioweapon,” said FDA Commissioner Scott Gottlieb, M.D. “This is the first product to be awarded a Material Threat Medical Countermeasure priority review voucher. Today’s action reflects the FDA’s commitment to ensuring that the U.S. is prepared for any public health emergency with timely, safe and effective medical products.”
Prior to its eradication in 1980, variola virus, the virus that causes smallpox, was mainly spread by direct contact between people. Symptoms typically began 10 to 14 days after infection and included fever, exhaustion, headache and backache. A rash initially consisting of small, pink bumps progressed to pus-filled sores before finally crusting over and scarring. Complications of smallpox could include encephalitis (inflammation of the brain), corneal ulcerations (an open sore on the clear, front surface of the eye) and blindness.
TPOXX’s effectiveness against smallpox was established by studies conducted in animals infected with viruses that are closely related to the virus that causes smallpox, and was based on measuring survival at the end of the studies. More animals treated with TPOXX lived compared to the animals treated with placebo. TPOXX was approved under the FDA’s Animal Rule, which allows efficacy findings from adequate and well-controlled animal studies to support an FDA approval when it is not feasible or ethical to conduct efficacy trials in humans.
The safety of TPOXX was evaluated in 359 healthy human volunteers without a smallpox infection. The most frequently reported side effects were headache, nausea and abdominal pain.
The FDA granted this application Fast Track and Priority Review designations. TPOXX also received Orphan Drug designation, which provides incentives to assist and encourage the development of drugs for rare diseases and a Material Threat Medical Countermeasure Priority Review Voucher, which provides additional incentives for certain medical products intended to treat or prevent harm from specific chemical, biological, radiological and nuclear threats.
The FDA granted approval of TPOXX to SIGA Technologies Inc.
TPOXX was developed in conjunction with the U.S. Department of Health and Human Services’ Biomedical Advanced Research and Development Authority (BARDA).
Tecovirimat


Tecovirimat
4-trifluoromethyl-N-(3,3a,4,4a,5,5a,6,6a-octahydro-1,3-dioxo-4,6-ethenocycloprop(f)isoindol-2(1H)-yl)-benzamide
N- [(3aR,4R,4aR,5aS,6S, 6aS)- 3,3a,4,4a,5,5a,6,6a- octahydro-1,3-dioxo- 4,6- ethenocycloprop[f]iso- indol-2(1H)-yl]-4- (trifluoromethyl)- benzamide
4 -trifluoromethyl -N- (3, 3a, 4, 4a, 5, 5a, 6, 6a- octahydro-1, 3 -dioxo-4, 6 -ethenocycloprop [f] isoindol -2 ( 1H) -yl ) – benzamide
Details
NDA FILED IN US
2006 ORPHAN DRUG DESIGNATION IN US FOR SMALL POX
2010 ORPHAN DRUG DESIGNATION IN US FOR ORTHOPOX VIRUS
A core protein cysteine protease inhibitor potentially for treatment of smallpox infection.
![]()
SIGA TECHNOLOGIES INNOVATOR
SIGA-246; ST-246
CAS No. 869572-92-9
The Orthopox genus (Orthopoxyiridae) is a member of the Poxyiridae family and the Choropoxivirinae subfamily. The genus consists of numerous viruses that cause significant disease in human and animal populations. Viruses in the orthopox genus include cowpox, monkeypox, vaccina, and variola (smallpox), all of which can infect humans.
The smallpox (variola) virus is of particular importance. Recent concerns over the use of smallpox virus as a biological weapon has underscored the necessity of developing small molecule therapeutics that target orthopoxviruses. Variola virus is highly transmissible and causes severe disease in humans resulting in high mortality rates (Henderson et al. (1999) JAMA. 281:2127-2137). Moreover, there is precedent for use of variola virus as a biological weapon. During the French and Indian wars (1754-1765), British soldiers distributed blankets used by smallpox patients to American Indians in order to establish epidemics (Stern, E. W. and Stern A. E. 1945. The effect of smallpox on the destiny of the Amerindian. Boston). The resulting outbreaks caused 50% mortality in some Indian tribes (Stern, E. W. and Stern A. E.). More recently, the soviet government launched a program to produce highly virulent weaponized forms of variola in aerosolized suspensions (Henderson, supra). Of more concern is the observation that recombinant forms of poxvirus have been developed that have the potential of causing disease in vaccinated animals (Jackson et al. (2001) J. Virol., 75:1205-1210).
The smallpox vaccine program was terminated in 1972; thus, many individuals are no longer immune to smallpox infection. Even vaccinated individuals may no longer be fully protected, especially against highly virulent or recombinant strains of virus (Downie and McCarthy. (1958) J. Hyg. 56:479-487; Jackson, supra). Therefore, mortality rates would be high if variola virus were reintroduced into the human population either deliberately or accidentally.
Variola virus is naturally transmitted via aerosolized droplets to the respiratory mucosa where replication in lymph tissue produces asymptomatic infection that lasts 1-3 days. Virus is disseminated through the lymph to the skin where replication in the small dermal blood vessels and subsequent infection and lysis of adjacent epidermal cells produces skin lesions (Moss, B. (1990) Poxyiridae and Their Replication, 2079-2111. In B. N. Fields and D. M. Knipe (eds.), Fields Virology. Raven Press, Ltd., New York). Two forms of disease are associated with variola virus infection; variola major, the most common form of disease, which produces a 30% mortality rate and variola minor, which is less prevalent and rarely leads to death (<1%). Mortality is the result of disseminated intravascular coagulation, hypotension, and cardiovascular collapse, that can be exacerbated by clotting defects in the rare hemorrhagic type of smallpox (Moss, supra).
A recent outbreak of monkeypox virus underscores the need for developing small molecule therapeutics that target viruses in the orthpox genus. Appearance of monkeypox in the US represents an emerging infection. Monkeypox and smallpox cause similar diseases in humans, however mortality for monkeypox is lower (1%).
Vaccination is the current means for preventing orthopox virus disease, particularly smallpox disease. The smallpox vaccine was developed using attenuated strains of vaccinia virus that replicate locally and provide protective immunity against variola virus in greater than 95% of vaccinated individuals (Modlin (2001) MMWR (Morb Mort Wkly Rep) 50:1-25). Adverse advents associated with vaccination occur frequently (1:5000) and include generalized vaccinia and inadvertent transfer of vaccinia from the vaccination site. More serious complications such as encephalitis occur at a rate of 1:300,000, which is often fatal (Modlin, supra). The risk of adverse events is even more pronounced in immunocompromised individuals (Engler et al. (2002) J Allergy Clin Immunol. 110:357-365). Thus, vaccination is contraindicated for people with AIDS or allergic skin diseases (Engler et al.). While protective immunity lasts for many years, the antibody response to smallpox vaccination is significantly reduced 10 to 15 years post inoculation (Downie, supra). In addition, vaccination may not be protective against recombinant forms of ortho poxvirus. A recent study showed that recombinant forms of mousepox virus that express IL-4 cause death in vaccinated mice (Jackson, supra). Given the side effects associated with vaccination, contraindication of immunocompromised individuals, and inability to protect against recombinant strains of virus, better preventatives and/or new therapeutics for treatment of smallpox virus infection are needed.
Vaccinia virus immunoglobulin (VIG) has been used for the treatment of post-vaccination complications. VIG is an isotonic sterile solution of immunoglobulin fraction of plasma derived from individuals who received the vaccinia virus vaccine. It is used to treat eczema vaccinatum and some forms of progressive vaccinia. Since this product is available in limited quantities and difficult to obtain, it has not been indicated for use in the event of a generalized smallpox outbreak (Modlin, supra).
Cidofovir ([(S)-1-(3-hydroxy-2-phosphonylmethoxypropyl)cytosine][HPMPC]) is a nucleoside analog approved for treatment of CMV retinitis in AIDS patients. Cidofovir has been shown to have activity in vitro against a number of DNA containing viruses including adenovirus, herpesviruses, hepadnaviruses, polyomaviruses, papillomaviruses, and ortho poxviruses (Bronson et al. (1990) Adv. Exp. Med. Biol. 278:277-83; De Clercq et al. (1987) Antiviral Res. 8:261-272; de Oliveira et al. (1996) Antiviral Res. 31:165-172; Snoeck et al. (2001) Clin Infect. Dis. 33:597-602). Cidofovir has also been found to inhibit authentic variola virus replication (Smee et al. (2002) Antimicrob. Agents Chemother. 46:1329-1335).
However, cidofovir administration is associated with a number of issues. Cidofovir is poorly bioavailable and must be administered intravenously (Lalezari et al. (1997) Ann. Intern. Med. 126:257-263). Moreover, cidofovir produces dose-limiting nephrotoxicity upon intravenous administration (Lalezari et al.). In addition, cidofovir-resistance has been noted for multiple viruses. Cidofovir-resistant cowpox, monkeypox, vaccinia, and camelpox virus variants have been isolated in the laboratory by repeated passage in the presence of drug (Smee, supra). Cidofovir-resistance represents a significant limitation for use of this compound to treat orthopoxvirus replication. Thus, the poor bioavailability, need for intravenous administration, and prevalence of resistant virus underscores the need for development of additional and alternative therapies to treat orthopoxvirus infection
In addition to viral polymerase inhibitors such as cidofovir, a number of other compounds have been reported to inhibit orthopoxvirus replication (De Clercq. (2001) Clin Microbiol. Rev. 14:382-397). Historically, methisazone, the prototypical thiosemicarbazone, has been used in the prophylactic treatment of smallpox infections (Bauer et al. (1969) Am. J. Epidemiol. 90:130-145). However, this compound class has not garnered much attention since the eradication of smallpox due to generally unacceptable side effects such as severe nausea and vomiting. Mechanism of action studies suggest that methisazone interferes with translation of L genes (De Clercq (2001), supra). Like cidofovir, methisazone is a relatively non-specific antiviral compound and can inhibit a number of other viruses including adenoviruses, picornaviruses, reoviruses, arboviruses, and myxoviruses (Id.).
Another class of compounds potentially useful for the treatment of poxviruses is represented by inhibitors of S-adenosylhomocysteine hydrolase (SAH). This enzyme is responsible for the conversion of S-adenosylhomocysteine to adenosine and homocysteine, a necessary step in the methylation and maturation of viral mRNA. Inhibitors of this enzyme have shown efficacy at inhibiting vaccinia virus in vitro and in vivo (De Clercq et al. (1998) Nucleosides Nucleotides. 17:625-634.). Structurally, all active inhibitors reported to date are analogues of the nucleoside adenosine. Many are carbocyclic derivatives, exemplified by Neplanacin A and 3-Deazaneplanacin A. While these compounds have shown some efficacy in animal models, like many nucleoside analogues, they suffer from general toxicity and/or poor pharmacokinetic properties (Coulombe et al. (1995) Eur. J. Drug Metab Pharmacokinet. 20:197-202; Obara et al. (1996) J. Med. Chem. 39:3847-3852). It is unlikely that these compounds can be administered orally, and it is currently unclear whether they can act prophylactically against smallpox infections. Identification of non-nucleoside inhibitors of SAH hydrolase, and other chemically tractable variola virus genome targets that are orally bioavailable and possess desirable pharmicokinetic (PK) and absorption, distribution, metabolism, elimination (ADME) properties would be a significant improvement over the reported nucleoside analogues. In summary, currently available compounds that inhibit smallpox virus replication are generally non-specific and suffer from use limiting toxicities and/or questionable efficacies.
In U.S. Pat. No. 6,433,016 (Aug. 13, 2002) and U.S. Application Publication 2002/0193443 A1 (published Dec. 19, 2002) a series of imidodisulfamide derivatives are described as being useful for orthopox virus infections.
Synthesis
![str2]()

RAW MATERIAL
Key RM is, 4,6-Etheno-1H-cycloprop[f]isobenzofuran-1,3(3aH)-dione, 3a,4,4a,5,5a,6-hexahydro-, (3aR,4R,4aR,5aS,6S,6aS)-rel–
cas 944-41-2, [US7655688]
| Molecular Formula: | C11H10O3 |
|---|---|
| Molecular Weight: | 190.1953 g/mol |
- 4,6-Etheno-1H-cycloprop[f]isobenzofuran-1,3(3aH)-dione, 4,4a,5,5a,6,6a-hexahydro-, (3aα,4β,4aα,5aα,6β,6aα)-
- Tricyclo[3.2.2.02,4]non-8-ene-6,7-dicarboxylic anhydride, stereoisomer (8CI)
- 3,6-Cyclopropylene-Δ4-tetrahydrophthalic anhydride
MP 94-96 °C
Ref, Dong, Ming-xin; European Journal of Medicinal Chemistry 2010, V45(9), Pg 4096-4103
SMILES……….
O=C1OC(=O)[C@H]4[C@@H]1[C@H]3C=C[C@@H]4[C@@H]2C[C@@H]23
SYNTHESIS CONTINUED…….![]()

ST-246
Patent
The present invention provides a process for making ST-246 outlined in Scheme 1


P = Boc
Scheme 1
The present invention also provides a process for making ST-246 outlined in, Scheme 2


Scheme 2
The present invention further provides a process for making ST-246 outlined in Scheme 3


ST-246
P = Boc
Scheme 3


P = Boc
Scheme 4
The present invention further provides a process for making ST-246 outlined in
Scheme 5


Scheme 5
Example 1 : Synthetic Route I:


P = Boc
Scheme 1
Step A. Synthesis of Compound 6 (P = Boc)
To a mixture of compound 3 (5.0 g, 26.3 mmol, synthesized according to WO041 12718) in EtOH (80 mL, EMD, AX0441 -3) was added terf-butyl carbazate 5 (3.65 g, 27.6 mmol, Aldrich, 98%). The reaction mixture was heated to reflux for 4 h under nitrogen atmosphere. LC-MS analysis of the reaction mixture showed less than 5% of compound 3 remained. The reaction mixture was evaporated under reduced pressure. The residue was recrystallized from EtOAc – hexanes, the solid was filtered, washed with hexanes (50 mL) and dried under vacuum to afford compound 6 (3.1 g, 39% yield) as a white solid. The filtrate was concentrated and purified by column chromatography eluting with 25% EtOAc in hexanes to give an additional 3.64 g (46% yield) of compound 6 as a white solid. Total yield: 6.74 g (84% yield). 1H NMR in CDCI3: δ 6.30 (br s, 1 H), 5.79 (t, 2H), 3.43 (s, 2H), 3.04 (s, 2H), 1 .46 (s, 9H), 1 .06-1 .16 (m, 2H), 0.18-0.36 (m, 2H); Mass Spec: 327.2 (M+Na)+
Step B. Synthesis of Compound 7 (HCI salt)
Compound 6 (3.6 g, 1 1 .83 mmol) was dissolved in /‘-PrOAc (65 mL, Aldrich, 99.6%). 4M HCI in dioxane (10.4 mL, 41 .4 mmol, Aldrich) was added drop-wise to the above solution keeping the temperature below 20 °C. The reaction mixture was stirred at room temperature overnight (18 h) under nitrogen atmosphere. The resulting solid was filtered, washed with /‘-PrOAc (15 mL) and dried under vacuum to yield HCI salt of compound 7 (1 .9 g, 67% yield) as a white solid. The filtrate was concentrated to 1/3 its volume and stirred at 10 – 15 °C for 30 min. The solid was filtered, washed with minimal volume of /‘-PrOAc and dried to afford additional 0.6 g (21 % yield) of compound 7. Total yield: 2.5 g (88% yield). 1 H NMR in DMSO-d6: δ 6.72 (br s, 3H), 5.68 (m, 2H), 3.20 (s, 2H), 3.01 (s, 2H), 1 .07-1 .17 (m, 2H), 0.18-0.29 (m, 1 H), -0.01 -0.07 (m, 1 H); Mass Spec: 205.1 (M+H)+
Step C. Synthesis of ST-246
To a mixture of compound 7 (0.96 g, 4 mmol) in dry dichloromethane (19 mL) was added triethylamine (1 .17 mL, 8.4 mmol, Aldrich) keeping the temperature below 20 °C. The resulting solution was stirred for 5 minutes at 15 – 20 °C, to it was added drop-wise 4-(trifluoromethyl)benzoyl chloride 8 (0.63 mL, 4.2 mmol, Aldrich, 97%) and the reaction mixture was stirred at room temperature overnight (18 h). LC-MS and TLC analysis showed the correct molecular weight and Rf value of ST-246 but the reaction was not complete. Additional 0.3 mL (2 mmol, 0.5 eq) of 4-(trifluoromethyl)benzoyl chloride 8 was added to the reaction mixture at 15 – 20 °C. The reaction was then stirred at room temperature overnight (19 h). LC-MS analysis indicated ca. 5% of starting material 7 still remained. The reaction was stopped and dichloromethane (30 mL) was added. The organic phase was washed with water (30 mL), saturated aqueous NH CI (30 mL), water (15 mL) and saturated aqueous NaHCO3 (30 mL). The organic phase was separated, dried over Na2SO4, filtered and concentrated to give crude product. The crude product was purified by column chromatography eluting with 30 -50% EtOAc in hexanes to afford ST-246 (0.34 g, 23% yield) as an off-white solid. Analytical data (1H NMR, LC-MS and HPLC by co-injection) were matched with those of ST-246 synthesized according to WO041 12718 and were consistent.
Example 2: Synthetic Route II


Scheme 2
Step A. Synthesis of Compound 9
A mixture of compound 4 (2.0 g, 9.8 mmol) and maleic anhydride 2 (0.96 g, 9.8 mmol, Aldrich powder, 95%) in o-xylene (100 mL, Aldrich anhydrous, 97%) was heated to reflux using a Dean-Stark trap apparatus overnight. After 18 h, LC-MS analysis at 215 nm showed the desired product 9 (86%), an uncyclized product (2.6%) and a dimer by-product (1 1 .6%).

Uncyclized product (MS = 303) Dimer by-product (MS = 489)
The reaction mixture was cooled to 45 °C and evaporated under reduced pressure. The residue was dissolved in EtOAc (50 mL) and the insoluble solid (mostly uncyclized product) was removed by filtration. The filtrate was concentrated and purified by column chromatography eluting with 50% EtOAc in hexanes to yield compound 9 (1 .5 g, 54% yield) as an off-white solid. 1 H NMR in CDCI3: δ 8.44 (s, 1 H), 7.91 (d, 2H), 7.68 (d, 2H), 6.88 (s, 2H); Mass Spec: 285.1 (M+H)+
Step B. Synthesis of ST-246 (Route II)
A mixture of compound 9 (0.97 g, 3.4 mmol) and cycloheptatriene 1 (0.51 mL, 4.42 mmol, distilled before use, Aldrich tech 90%) in toluene (50 mL, Aldrich anhydrous) was heated at 95 °C under nitrogen atmosphere. After 1 .5 h at 95 °C, LC-MS analysis at 254 nm showed 29% conversion to the desired product (endo:exo = 94:6). The resulting solution was continued to be heated at same temperature overnight. After 18 h at 95 °C, LC-MS analysis indicated 75% conversion with an endo:exo ratio of 94:6. The reaction temperature was increased to 1 10 °C and the reaction was monitored. After heating at 1 10 °C for 7 h, LC-MS analysis at 254 nm showed 96.4% conversion to the desired product (endo:exo = 94:6). The volatiles were removed by evaporation under reduced pressure and the reside was purified by column chromatography eluting with 30% EtOAc in hexanes to afford ST-246 (0.29 g, 22.6% yield, HPLC area 99.7% pure and 100% endo isomer) as a white solid. Analytical data (1H NMR, LC-MS and HPLC by co-injection) were matched with those of ST-246 synthesized according to WO041 12718 and were consistent. An additional 0.5 g of ST-246 (38.9% yield, endo:exo = 97: 3) was recovered from column chromatography. Total Yield: 0.84 g (65.4% yield). 1H NMR of ST-246 exo isomer in CDCI3: δ 8.62 (s, 1 H), 7.92 (d, 2H), 7.68 (d, 2H), 5.96 (m, 2H), 3.43 (s, 2H), 2.88 (s, 2H), 1 .17 (s, 2H), 0.24 (q, 1 H), 0.13 (m, 1 H); Mass Spec: 377.1 (M+H)+
Example 3: Synthetic Route III

ST-246 9
P = Boc
Scheme 3
Step A. Synthesis of Compound 10
A mixture of maleic anhydride 2 (15.2 g, 155 mmol, Aldrich powder 95%) and terf-butyl carbazate 5 (20.5 g, 155 mmol, Aldrich, 98%) in anhydrous toluene (150 mL, Aldrich anhydrous) was heated to reflux using a Dean-Stark trap apparatus under nitrogen atmosphere. After refluxing for 2 h, no starting material 2 remained and LC-MS analysis at 254 nm showed the desired product 10 (20% by HPLC area), imine byproduct (18%) and disubstituted by-product (56%). The reaction mixture was concentrated and purified by column chromatography eluting with 25% EtOAc in hexanes to afford compound 10 (5.98 g, 18% yield, HPLC area >99.5% pure) as a white solid. 1 H NMR in DMSO-d6: δ 9.61 (s, 1 H), 7.16 (s, 2H), 1 .42 (s, 9H); Mass Spec: 235.1 (M+Na)+.
duct

C9H12N204 C14H22N405
Mol. Wt.: 212.2 Mol. Wt.: 326.35
Step B. Synthesis of Compound 11 (HCI salt)
Compound 10 (3.82 g, 18 mmol) was dissolved in /‘-PrOAc (57 mL, Aldrich, 99.6%). 4M HCI in dioxane (15.8 mL, 63 mmol, Aldrich) was added drop-wise to the above solution keeping the temperature below 20 °C. The solution was stirred overnight (24 h) at room temperature under nitrogen atmosphere. The resulting solid was filtered, washed with /‘-PrOAc (10 mL) and dried at 45 °C under vacuum for 1 h to afford HCI salt of compound 11 (2.39 g, 89% yield) as a white solid. 1 H NMR in CD3OD: δ 6.98 (s, 2H); Mass Spec: 1 13.0 (M+H)+
Step C. Synthesis of Compound 9 (Route III)
To a mixture of compound 11 (1 .19 g, 8 mmol) in dry dichloromethane (24 mL) was added diisopropylethylannine (2.93 mL, 16.8 mmol, Aldrich redistilled grade) keeping the temperature below 20 °C. The resulting solution was stirred for 5 minute at 15 – 20 °C and to it was added 4-(trifluoromethyl)benzoyl chloride 8 (1 .31 mL, 8.8 mmol, Aldrich, 97%) drop-wise. The reaction was stirred at room temperature for 5 h. LC-MS analysis showed the correct MW but the reaction was not complete. Additional 0.48 mL (0.4 equiv) of 4-(trifluoromethyl)benzoyl chloride 8 was added to the reaction mixture at 15 – 20 °C and the reaction mixture was stirred at room temperature overnight (21 h). The reaction was stopped and dichloromethane (50 mL) was added. The organic phase was washed with water (50 mL), saturated aqueous NH4CI (50 mL), water (30 mL) and saturated aqueous NaHCO3 (30 mL). The organic phase was separated, dried over Na2SO4, filtered and concentrated to give crude product. The crude product was purified by column chromatography eluting with 30 – 35% EtOAc in hexanes to afford compound 9 (0.8 g, 35% yield) as a light pink solid. Analytical data (1H NMR and LC-MS) were consistent with those of compound 9 obtained in Synthetic Route II.
Step D. Synthesis of ST-246 (Route III)
A mixture of compound 9 (0.5 g, 1 .76 mmol) and cycloheptatriene 1 (0.33 mL, 3.17 mmol, distilled before to use, Aldrich tech 90%) in toluene (10 mL, Aldrich anhydrous) was heated at 1 10 – 1 15 °C under nitrogen atmosphere. After 6 h, LC-MS analysis at 254 nm showed 95% conversion to the desired product (endo:exo = 94:6). The resulting solution was heated at same temperature overnight (22 h). LC-MS analysis at 254 nm showed no starting material 9 remained and the desired product (endo:exo = 93:7). The reaction mixture was concentrated and purified by column chromatography eluting with 25 – 35% EtOAc in hexanes to afford ST-246 (0.39 g, HPLC area >99.5% pure with a ratio of endo:exo = 99:1 ) as a white solid. Analytical data (1 H NMR, LC-MS and HPLC by co-injection) were compared with those of ST-246 synthesized according to WO041 12718 and were found to be consistent. An additional 0.18 g of ST-246 (HPLC area >99.5% pure, endo:exo = 91 : 9) was recovered from column chromatography. Total Yield: 0.57 g (86% yield).
Example 4 ; Synthetic Route IV:

P = Boc
Scheme 4
Step A. Synthesis of Compound 10
A mixture of maleic anhydride 2 (3.4 g, 34.67 mmol, Aldrich powder, 95%) and terf-butyl carbazate 5 (4.6 g, 34.67 mmol, Aldrich, 98%) in anhydrous toluene (51 ml_, Aldrich) was heated to reflux using a Dean-Stark trap apparatus under nitrogen atmosphere. After refluxing for 2.5 h, no starting material 2 remained and LC-MS analysis at 254 nm showed the desired product 10 (19% HPLC area), imine by-product (18%) and another by-product (56%). The reaction mixture was concentrated and purified by column chromatography eluting with 30% EtOAc in hexanes to afford compound 10 (1 .0 g, 13.6% yield, HPLC area >99% pure) as a white solid. Analytical data (1H NMR and LC-MS) were consistent with those of compound 10 obtained in Synthetic Route III.
Im ine by-product

Mol. Wt.: 212.2
Step B. Synthesis of Compound 6
A mixture of compound 10 (4.4 g, 20.74 mmol) and cycloheptatriene 1 (3.22 mL, 31 .1 mmol, distilled before to use, Aldrich tech 90%) in toluene (88 mL, 20 volume, Aldrich anhydrous) was heated at 95 °C under nitrogen atmosphere. After 15 h at 95 °C, LC-MS analysis showed 83% conversion to the desired product. The reaction mixture was heated at 105 °C overnight. After total 40 h at 95 – 105 °C, LC-MS analysis at 254 nm showed -99% conversion to the desired product (endo:exo = 93:7). The reaction mixture was concentrated and the crude was purified by column chromatography eluting with 25 – 50 % EtOAc in hexanes to afford compound 6 (2.06 g, 32.6% yield, HPLC area 99.9% pure and 100% endo isomer) as a white solid. 1 H NMR and LC-MS were consistent with those of compound 6 obtained in Synthetic Route I. An additional 4.0 g of 6 (63.4% yield, HPLC area 93% pure with a ratio of endo:exo = 91 : 9) was recovered from column chromatography. Total Yield: 6.06 g (96% yield).
Step C. Synthesis of Compound 7 (HCI salt)
Compound 6 (2.05 g, 6.74 mmol) was dissolved in /‘-PrOAc (26 mL, Aldrich, 99.6%). 4M HCI in dioxane (5.9 mL, 23.58 mmol, Aldrich) was added drop-wise to the above solution keeping the temperature below 20 °C. The solution was stirred overnight (18 h) at room temperature under nitrogen atmosphere. The resulting solid was filtered, washed with /‘-PrOAc (5 mL) and dried under vacuum to yield HCI salt of compound 7 (1 .57 g, 97% yield) as a white solid. Analytical data (1 H NMR and LC-MS) were consistent with those of compound 7 in Synthetic Route I.
Step D. Synthesis of ST-246 (Route IV)
To a mixture of compound 7 (0.84 g, 3.5 mmol) in dichloromethane (13 mL) was added diisopropylethylamine (1 .34 mL, 7.7 mmol) keeping the temperature below 20 °C and the resulting solution was stirred for 5 – 10 minutes. 4-(Trifluoromethyl)benzoyl chloride 8 (0.57 mL, 3.85 mmol, Aldrich, 97%) was added to above solution keeping the temperature below 20 °C. The reaction mixture was stirred at room temperature for 2 h. Additional 0.2 mL (0.4 equiv) of 4-(trifluoromethyl)benzoyl chloride 8 was added to the reaction keeping the temperature below 20 °C. The reaction was stirred at room temperature overnight (24 h). The reaction mixture was diluted with dichloromethane (20 mL). The organic phase was washed with water (20 mL), saturated aqueous NH4CI (20 mL), water (20 mL) and saturated aqueous NaHCO3 (20 mL). The organic phase was separated, dried over Na2SO4, filtered and concentrated to give crude product. The crude product was purified by column chromatography eluting with 30 – 35% EtOAc in hexanes to afford ST-246 (0.25 g, 19% yield, HPLC area >99.5% pure) as a white solid. Analytical data (1H NMR and LC-MS) were consistent with those of ST-246 synthesized according to WO041 12718.
Example 5: Synthetic Route V:

Scheme 5
Step A. Synthesis of Compound 13
To a mixture of compound 7 (1 .6 g, 6.65 mmol, synthesized according to Synthetic Route I) in dichloromethane (80 ml_,) was added triethylamine (2.04 ml_, 14.63 mmol) keeping the temperature below 20 °C and the resulting solution was stirred for 5 – 10 minute. 4-lodobenzoyl chloride 12 (1 .95 g, 7.31 mmol, 1 .1 equiv, Aldrich) was added portion-wise under nitrogen atmosphere to the above solution keeping the temperature below 20 °C. The reaction mixture was stirred at room temperature overnight. After 17 h and 19 h, additional 0.35 g (0.2 equiv) of acid chloride 12 was added to the reaction keeping the temperature below 20 °C. After 24 h, additional 0.18 g (0.1 equiv, used total 1 .6 equiv) of acid chloride 12 was added and the reaction was continued to stir at room temperature overnight (total 43 h). LC-MS analysis at 215 nm showed 43% of the desired product (13) and -5% of compound 7. The reaction was diluted with dichloromethane (100 ml_). The organic phase was washed with saturated aqueous NH4CI (100 ml_), water (100 ml_) and saturated aqueous NaHCO3 (100 ml_). The organic phase was separated, dried over Na2SO4, filtered and concentrated to give crude product. The crude product was purified by column chromatography eluting with 25 – 50% EtOAc in hexanes to afford compound 13 (1 .63 g, 57% yield, HPLC area 93% pure) as a white solid. 1 H NMR in DMSO-d6: δ 1 1 .19 and 10.93 (two singlets with integration ratio of 1 .73:1 , total of 1 H, same proton of two rotamers), 7.93 (d, 2H), 7.66 (d, 2H), 5.80 (s, 2H), 3.36 (s, 2H), 3.27 (s, 2H), 1 .18 (s, 2H), 0.27 (q, 1 H), 0.06 (s,1 H); Mass Spec: 435.0 (M+H)+
Step B. Synthesis of ST-246 (Route V)
Anhydrous DMF (6 ml_) was added to a mixture of compound 13 (0.2 g, 0.46 mmol), methyl 2, 2-difluoro-2-(fluorosulfonyl)acetate (0.44 ml_, 3.45 mmol, Aldrich) and copper (I) iodide (90 mg, 0.47 mmol). The reaction mixture was stirred at -90 °C for 4 h. LC-MS analysis at 254 nm indicated no starting material 13 remained and showed 48% HPLC area of ST-246. The reaction mixture was cooled to 45 °C and DMF was removed under reduced pressure. The residue was slurried in EtOAc (30 mL) and insoluble solid was removed by filtration. The filtrate was concentrated and purified by column chromatography eluting with 25 – 35% EtOAc in hexanes to afford ST-246 (55
mg, 32% yield, 95% pure by HPLC at 254 nm) as off-white solid. Analytical data (1H NMR and LC-MS) were consistent with those of ST-246 synthesized according to WO041 12718.
PAPER
N-(3,3a,4,4a,5,5a,6,6a-Octahydro-1,3-dioxo-4,6- ethenocycloprop[f]isoindol-2-(1H)-yl)carboxamides: Identification of Novel Orthopoxvirus Egress Inhibitors
ViroPharma Incorporated, 397 Eagleview Boulevard, Exton, Pennsylvania 19341, United States Army Medical Research Institute of Infectious Diseases, 1425 Porter Street, Frederick, Maryland 21702, University of Alabama, Birmingham, Alabama 35294, and SIGA Technologies, Inc., 4575 SW Research Way, Corvallis, Oregon 97333
J. Med. Chem., 2007, 50 (7), pp 1442–1444
DOI: 10.1021/jm061484y

A series of novel, potent orthopoxvirus egress inhibitors was identified during high-throughput screening of the ViroPharma small molecule collection. Using structure−activity relationship information inferred from early hits, several compounds were synthesized, and compound 14was identified as a potent, orally bioavailable first-in-class inhibitor of orthopoxvirus egress from infected cells. Compound 14 has shown comparable efficaciousness in three murine orthopoxvirus models and has entered Phase I clinical trials.
http://pubs.acs.org/doi/suppl/10.1021/jm061484y/suppl_file/jm061484ysi20070204_060607.pdf
General Procedure for synthesis of compounds 2-14, 16-18.
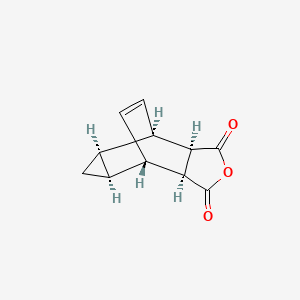
N-(3,3a,4,4a,5,5a,6,6aoctahydro-1,3-dioxo-4,6-ethenocycloprop[f]isoindol-2(1H)-yl)-4- (trifluoromethyl)benzamide (14).
A mixture of 2.00 g (9.8 mmol) of 4-(trifluoromethyl) benzoic acid hydrazide, 1.86 g (9.8 mmol) of 4,4a,5,5a,6,6a-hexahydro-4,6-etheno-1Hcycloprop[f]isobenzofuran-1,3(3aH)-dione, and one drop of diisopropylethylamine in 40 mL of absolute ethanol was refluxed for 4.5 h. Upon cooling to rt, 4 mL of water was added, and the product began to crystallize. The suspension was cooled in an ice bath, and the precipitate collected by filtration. The crystalline solid was air-dried affording 3.20 g (87%) of the product as a white solid;
Mp 194-195 ºC. 1 H NMR, (300 MHz, d6 -DMSO) δ 11.20, 11.09 (2 brs from rotamers, 1H), 8.06 (d, J= 7.8 Hz, 2H), 7.90 (d, J= 7.8 Hz, 2H), 5.78 (m, 2H), 3.26 (m, 4H), 1.15 (m, 2H), 0.24 (dd, J= 7.2, 12.9 Hz, 1H), 0.04 (m, 1H).
Anal. calcd. for C19H15F3N2O3● 0.25H2O: %C, 59.92; %H, 4.10; %F, 14.97; %N, 7.36; %O, 13.65. Found: %C, 59.97; %H, 4.02; %F, 14.94; %N, 7.36; %O, 13.71.

CLICK ON IMAGE
PATENT
US20140316145

CLICK ON IMAGE
http://www.google.com/patents/US8802714
Example 1
Preparation of 4-trifluoromethyl-N-(3,3a,4,4a,5,5a,6,6a-octahydro-1,3-dioxo-4,6-ethenocycloprop[f]isoindol-2(1H)-yl)-benzamide
a. Preparation of Compounds 1(a) and 1(b).

A mixture of cycloheptatriene (5 g, 54.26 mmol) and maleic anhydride (6.13 g, 62.40 mmol) in xylenes (35 mL) was heated at reflux under argon overnight. The reaction was cooled to room temperature and a tan precipitate was collected by filtration and dried to give 2.94 grams (28%) of the desired product, which is a mixture of compounds 1(a) and 1(b). Compound 1(a) is normally predominant in this mixture and is at least 80% by weight. The purity of Compound 1(a) may be further enhanced by recrystallization if necessary. Compound 1(b), an isomer of compound 1(a) is normally less than 20% by weight and varies depending on the conditions of the reaction. Pure Compound 1(b) was obtained by concentrating the mother liquid to dryness and then subjecting the residue to column chromatography. Further purification can be carried out by recrystallization if necessary. 1H NMR (500 MHz) in CDCl3: δ 5.95 (m, 2H), 3.42 (m, 2H), 3.09 (m, 2H), 1.12 (m, 2H), 0.22 (m, 1H), 0.14 (m, 1H).
b. Preparation of N-[(3aR,4R,4aR,5aS,6S,6aS)-3,3a,4,4a,5,5a,6,6a-octahydro-1,3-dioxo-4,6-ethenocycloprop[f]isoindol-2(1H)-yl]-4-(trifluoromethyl)-benzamide. desired
A mixture of compound 1(a) (150 mg, 0.788 mmol) and 4-trifluoromethylbenzhydrazide (169 mg, 0.827 mmol) in ethanol (10 mL) was heated under argon overnight. The solvent was removed by rotary evaporation. Purification by column chromatography on silica gel using 1/1 hexane/ethyl acetate provided 152 mg (51%) of the product as a white solid.
c. Preparation of N-[(3aR,4S,4aS,5aR,6R,6aS)-3,3a,4,4a,5,5a,6,6a-octahydro-1,3-dioxo-4,6-ethenocycloprop[f]isoindol-2(1H)-yl]-4-(trifluoromethyl)-benzamide. UNWANTED
N-[(3aR,4S,4aS,5aR,6R,6aS)-3,3a,4,4a,5,5a,6,6a-octahydro-1,3-dioxo-4,6-ethenocycloprop[f]isoindol-2(1H)-yl]4-(trifluoromethyl)-benzamide was prepared and purified in the same fashion as for N-[(3aR,4R,4aR,5aS,6S,6aS)-3,3a,4,4a,5,5a,6,6a-octahydro-1,3-dioxo-4,6-ethenocycloprop[f]isoindol-2(1H)-yl]-4-(trifluoromethyl)-benzamide by replacing 1(a) with 1(b) and was obtained as a white solid. 1H NMR (300 MHz) in CDCl3: δ 8.62 (s, 1H), 7.92 (d, 2H), 7.68 (d, 2H), 5.96 (m, 2H), 3.43 (s, 2H), 2.88 (s, 2H), 1.17 (s, 2H), 0.24 (q, 1H), 0.13 (m, 1H); Mass Spec: 377.1 (M+H)+.
FINAL COMPD SYNTHESIS
| TABLE 1 | ||||
| Example | **Mass | |||
| Number | R6 | *NMR | Spec | Name |
| 1 |
|
1H NMR in DMSO-d6: δ 11.35 (d, 1H); 11.09 (d, 1H); 8.08 (d, 2H); 7.92 (d, 2H); 5.799 (s, 2H); 3.29 (brs, 4H); 1.17 (m, 2H); 0.26 (m, 1H); 0.078 (s, 1H) | 375 (M − H)− | N-[(3aR,4R,4aR,5aS,6S, 6aS)-3,3a,4,4a,5,5a,6,6a- octahydro-1,3-dioxo- 4,6-ethenocycloprop[f] isoindol-2(1H)-yl]-4- (trifluoromethyl)- benzamide |
TABLE 1 EXAMPLE 1
N- [(3aR,4R,4aR,5aS,6S, 6aS)- 3,3a,4,4a,5,5a,6,6a- octahydro-1,3-dioxo- 4,6- ethenocycloprop[f]iso- indol-2(1H)-yl]-4- (trifluoromethyl)- benzamide
1H NMR in DMSO-d6: δ 11.35 (d, 1H); 11.09 (d, 1H); 8.08 (d, 2H); 7.92 (d, 2H); 5.799 (s, 2H); 3.29 (brs, 4H); 1.17 (m, 2H); 0.26 (m, 1H); 0.078 (s, 1H), 375 (M − H)−
EXAMPLE 42 Characterization of 4-trifluoromethyl-N-(3,3a,4,4a,5,5a,6,6a-octahydro-1,3-dioxo-4,6-ethenocycloprop[f]isoindol-2(1H)-yl)-benzamide (“ ”)
In the present application, ST-246 refers to: N-[(3aR,4R,4aR,5aS,65,6aS)-3,3a,4,4a,5,5a,6,6a-octahydro-1,3-dioxo-4,6-ethenocycloprop[f]isoindol-2(1H)-yl]-4-(trifluoromethyl)-benzamide.
Physico-Chemical Properties
Appearance: ST-246 is a white to off-white powder.
Melting Point: Approximately 196° C. by DSC.
Permeability: The calculated log P is 2.94. Based on the partition coefficient, ST-246 is expected to have good permeability.
Particle Size: The drug substance is micronized to improve its dissolution in the gastrointestinal fluids. The typical particle size of the micronized material is 50% less than 5 microns.
Solubility: The solubility of ST-246 is low in water (0.026 mg/mL) and buffers of the gastric pH range. Surfactant increases its solubility slightly. ST-246 is very soluble in organic solvents. The solubility data are given in Table 5.


CLICK ON IMAGE
PATENT
http://www.google.com/patents/CN101445478A?cl=en
Tecovirimat (ST-246) is an antiviral with activity against orthopoxviruses such as smallpox and is currently undergoing clinical trials. It was previously owned by Viropharma and discovered in collaboration with scientists at USAMRIID. It is currently owned and is synthesized by Siga Technologies, a drug development company in the biodefense arena. It works by blocking cellular transmission of the virus, thus preventing the disease. Tecovirimat has been effective in laboratory testing, with no serious side effects reported to date. Despite not yet having FDA approval for medical use, tecovirimat is stockpiled in the US Strategic National Stockpile as a defense against a smallpox outbreak.[1]

Clinical study
The results of clinical trials involving tecovirimat supports its use against smallpox and other related orthopoxviruses. It has shown potential for a variety of uses including prophylaxis, as a post-exposure therapeutic, as a therapeutic and an adjunct to vaccination.[2]
Tecovirimat can be taken orally and has recently been granted permission to conduct Phase II trials by the U.S. Food and Drug Administration (FDA). In phase I trials tecovirimat was generally well tolerated with no serious adverse events.[3] Due to its importance for biodefense, the FDA has designated tecovirimat for ‘fast-track’ status, creating a path for expedited FDA review and eventual regulatory approval.
Tecovirimat is an orthopoxvirus egress inhibitor. Tecovirimat appears to target the V061 gene in cowpox, which is homologous to the vaccinia virus F13L. By targeting this gene, tecovirimat inhibits the function of a major envelope protein required for the production of extracellar virus. Thus the virus is prevented from leaving the cell, and the spread of the virus within the body is prevented.[4]



References
- Damon, Inger K.; Damaso, Clarissa R.; McFadden, Grant (2014). “Are We There Yet? The Smallpox Research Agenda Using Variola Virus”. PLoS Pathogens 10 (5): e1004108.doi:10.1371/journal.ppat.1004108. PMID 24789223.
- Siga Technologies
- Jordan, R; Tien, D; Bolken, T. C.; Jones, K. F.; Tyavanagimatt, S. R.; Strasser, J; Frimm, A; Corrado, M. L.; Strome, P. G.; Hruby, D. E. (2008). “Single-Dose Safety and Pharmacokinetics of ST-246, a Novel Orthopoxvirus Egress Inhibitor”. Antimicrobial Agents and Chemotherapy 52 (5): 1721–1727. doi:10.1128/AAC.01303-07. PMC 2346641. PMID 18316519.
- Yang, G; Pevear, D. C.; Davies, M. H.; Collett, M. S.; Bailey, T; Rippen, S; Barone, L; Burns, C; Rhodes, G; Tohan, S; Huggins, J. W.; Baker, R. O.; Buller, R. L.; Touchette, E; Waller, K; Schriewer, J; Neyts, J; Declercq, E; Jones, K; Hruby, D; Jordan, R (2005). “An Orally Bioavailable Antipoxvirus Compound (ST-246) Inhibits Extracellular Virus Formation and Protects Mice from Lethal Orthopoxvirus Challenge”. Journal of Virology 79 (20): 13139–13149. doi:10.1128/JVI.79.20.13139-13149.2005. PMC 1235851. PMID 16189015.
Referenced by
Citing Patent Filing date Publication date Applicant Title
CN101912389A * Aug 9, 2010 Dec 15, 2010 中国人民解放军军事医学科学院微生物流行病研究所 Pharmaceutical composition containing ST-246 and preparation method and application thereof
CN102406617A * Nov 30, 2011 Apr 11, 2012 中国人民解放军军事医学科学院生物工程研究所 Tecovirimat dry suspension and preparation method thereof
CN102406617B Nov 30, 2011 Aug 28, 2013 中国人民解放军军事医学科学院生物工程研究所 Tecovirimat dry suspension and preparation method thereof
CN103068232B * Mar 23, 2011 Aug 26, 2015 西佳科技股份有限公司 多晶型物形式st-246和制备方法
US8530509 Jul 29, 2011 Sep 10, 2013 Siga Technologies, Inc. Compounds, compositions and methods for treatment and prevention of orthopoxvirus infections and associated diseases
US8802714 Aug 14, 2013 Aug 12, 2014 Siga Technologies, Inc. Compounds, compositions and methods for treatment and prevention of orthopoxvirus infections and associated diseases
US9045418 Jul 3, 2014 Jun 2, 2015 Siga Technologies, Inc. Compounds, compositions and methods for treatment and prevention of Orthopoxvirus infections and associated diseases
| Cited Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| US20070287735 * | Apr 23, 2007 | Dec 13, 2007 | Siga Technologies, Inc. | Chemicals, compositions, and methods for treatment and prevention of orthopoxvirus infections and associated diseases |
| US20090011037 * | Apr 23, 2008 | Jan 8, 2009 | Cydex Pharmaceuticals, Inc. | Sulfoalkyl Ether Cyclodextrin Compositions and Methods of Preparation Thereof |
| Citing Patent | Filing date | Publication date | Applicant | Title |
| US8530509 | Jul 29, 2011 | Sep 10, 2013 | Siga Technologies, Inc. | Compounds, compositions and methods for treatment and prevention of orthopoxvirus infections and associated diseases |
| US8802714 | Aug 14, 2013 | Aug 12, 2014 | Siga Technologies, Inc. | Compounds, compositions and methods for treatment and prevention of orthopoxvirus infections and associated diseases |
| US9045418 | Jul 3, 2014 | Jun 2, 2015 | Siga Technologies, Inc. | Compounds, compositions and methods for treatment and prevention of Orthopoxvirus infections and associated diseases |
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
N-{3,5-Dioxo-4- azatetracyclo[5.3.2.0{2,6}.0{8,10}]dodec-11-en-4- yl}-4-(trifluoromethyl)benzamide |
|
| Identifiers | |
| UNII | F925RR824R  |
| ChEMBL | CHEMBL1242629 |
| Synonyms | ST-246 |
| Chemical data | |
| Formula | C19H15F3N2O3 |
| Molecular mass | base: 376.3 g/mol |
//////////////////Tecovirimat, FDA 2018, ORPHAN DRUG DESIGNATION, TPOXX, SIGA Technologies Inc, Fast Track, Priority Review
FC(F)(F)c1ccc(cc1)C(=O)NN1C(=O)C2C(C3C=CC2C2CC32)C1=O