Image may be NSFW.
Clik here to view. Image may be NSFW.
Image may be NSFW.
Clik here to view.Image may be NSFW.
Clik here to view.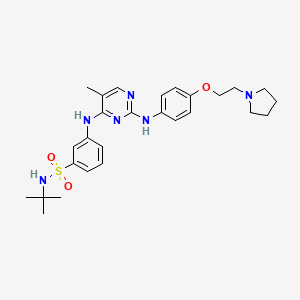
Image may be NSFW.
Clik here to view.Image may be NSFW.
Clik here to view.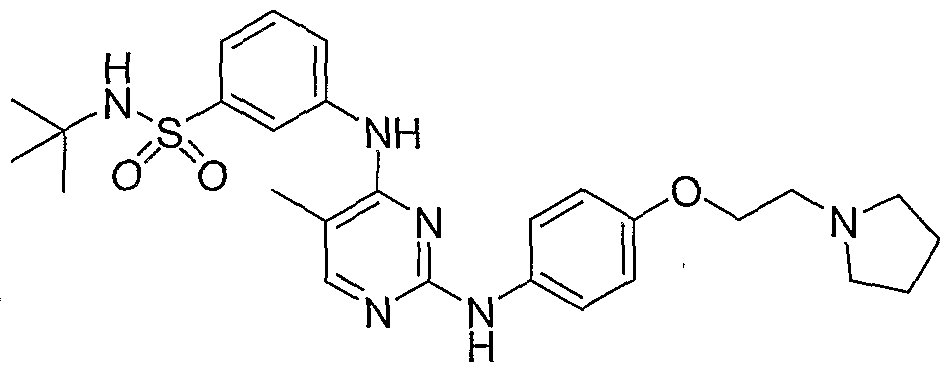
FEDRATINIB
SAR-302503; TG-101348, 6L1XP550I6, 936091-26-8 [RN], WHO 9707
| Molecular Formula: | C27H36N6O3S |
|---|---|
| Molecular Weight: | 524.684 g/mol |
FLT3, JAK2
http://www.ama-assn.org//resources/doc/usan/fedratinib.pdf
Fedratinib had been in phase III clincial trials by Sanofi for the treatment of myelofibrosis.
However, Sanofi had discontinued this research because of the safety issues. Orphan drug designation was assigned in the U.S. and in Japan for this indication. In 2017, the clinical hold was lifted in the U.S. by Impact Biomedicines.
MYELOFIBROSIS (MF), SANOFI , phase 3
Benzenesulfonamide, N-(1,1-dimethylethyl)-3-[[5-methyl-2-[[4-[2-(1-pyrrolidinyl)ethoxy]phenyl]amino]-4-pyrimidinyl]amino]-
N-tert-butyl-3-{[5-methyl-2-({4-[2-(pyrrolidin-1-yl)ethoxy]phenyl}amino)pyrimidin-4-yl]amino}benzenesulfonamide
N-tert-butyl-3-[[5-methyl-2-[4-(2-pyrrolidin-1-ylethoxy)anilino]pyrimidin-4-yl]amino]benzenesulfonamide
USAN (AB-104) FEDRATINIB
THERAPEUTIC CLAIM Antineoplastic
CHEMICAL NAMES
1. Benzenesulfonamide, N-(1,1-dimethylethyl)-3-[[5-methyl-2-[[4-[2-(1-
pyrrolidinyl)ethoxy]phenyl]amino]-4-pyrimidinyl]amino]-
2. N-tert-butyl-3-[(5-methyl-2-{4-[2-(pyrrolidin-1-yl)ethoxy]anilino}pyrimidin-4-
yl)amino]benzenesulfonamide
MOLECULAR FORMULA C27H36N6O3S
MOLECULAR WEIGHT 524.7
SPONSOR Sanofi
CODE DESIGNATIONS SAR302503; TG101348
CAS REGISTRY NUMBER……….936091-26-8
WHO 9707
Image may be NSFW.
Clik here to view.
TG-101348 , a dual-acting JAK2/FLT3 small molecule kinase inhibitor, has been evaluated in phase III clinical development at Sanofi (formerly known as sanofi-aventis) for the oral treatment of intermediate-2 or high risk primary myelofibrosis, post-polycythemia vera myelofibrosis or post-essential thrombocythemia myelofibrosis with splenomegaly. However, development of the compound has been discontinued due to safety issues.
In preclinical models of myeloproliferative diseases, TG-101348, administered orally, was shown to reduce V617F-expressing cell populations in a dose-dependent manner without adversely impacting normal hematopoiesis. The reduction of V617F- expressing cell populations correlated with improved survival and reduced morbidity. Orphan drug designation was assigned in the U.S. and in Japan for the treatment of secondary and primary myelofibrosis. In July 2010, TargeGen was acquired by Sanofi. In 2013, orphan drug designation was assigned by the FDA for the treatment of polycythemia vera.
Fedratinib is an orally bioavailable, small-molecule, ATP-competitive inhibitor of Janus-associated kinase 2 (JAK2) with potential antineoplastic activity. Fedratinib competes with JAK2 as well as the mutated form AK2V617F for ATP binding, which may result in inhibition of JAK2 activation, inhibition of the JAK-STAT signaling pathway, and the induction of tumor cell apoptosis. JAK2 is the most common mutated gene in bcr-abl-negative myeloproliferative disorders (MPDs); the mutated form JAK2V617F has a valine-to-phenylalanine modification at position 617 and plays a key role in tumor cell proliferation and survival.
Fedratinib (TG101348; SAR302503) is an orally available inhibitor of Janus kinase 2 (JAK-2) developed for the treatment of patients with myeloproliferative diseases including myelofibrosis. Fedratinib acts as a competitive inhibitor of protein kinase JAK-2 with IC50=6 nM; related kinases FLT3 and RET are also sensitive, with IC50=25 nM and IC50=17 nM, respectively. Significantly less activity was observed against other tyrosine kinases including JAK3 (IC50=169 nM).[1] In treated cells the inhibitor blocks downstream cellular signalling (JAK-STAT) leading to suppression of proliferation and induction of apoptosis.
Myelofibrosis is a myeloid malignancy associated with anemia, splenomegaly, and constitutional symptoms. Patients with myelofibrosis frequently harbor JAK-STAT activating mutations that are sensitive to TG101348. Phase I trial results focused on safety and efficacy of Fedratinib in patients with high- or intermediate-risk primary or post–polycythemia vera/essential thrombocythemia myelofibrosis have been published in 2011.[2]
Fedratinib was originally discovered at TargeGen. In 2010, Sanofi-Aventis acquired TargeGen and continued development of fedratinib until 2013. In 2016, Impact Biomedicines acquired the rights to fedratinib from Sanofi and continued its development for the treatment of myelofibrosis and polycythemia vera. In January 2018, Celgene acquired Impact Biomedicines.[3]
Image may be NSFW.
Clik here to view.
SYN
Image may be NSFW.
Clik here to view.
WO2007053452A1. +Bioorganic & Medicinal Chemistry Letters, 27(12), 2668-2673; 2017
Condensation of 3-bromo-N-tertbutylbenzylsulfonamide with 2-chloro-5-methyl-pyrimidin-4-ylamine in the presence of Pd2(dba)3, Xantphos, Cs2CO3 in refluxing dioxane gives sulfonamide derivative , which is coupled with 4-[2-pyrrolidin-1-yl-ethoxy]phenylamine in AcOH at 150°C to provide the title compound
PRODUCT PATENT
WO2007053452A1.
| Inventors | Jon Jianguo Cao, John Hood, Dan Lohse, Chi Ching Mak, Pherson Andrew Mc, Glenn Noronha, Ved Pathak, Joel Renick, Richard M. Soll, Binqi Zeng, Less « |
| Applicant | Targegen, Inc. |
https://encrypted.google.com/patents/WO2007053452A1?cl=en
EXAMPLE 90. 7V-fe^-Butyl-3-{5-methyl-2-14-(2-pyrrolidm-l-yl-ethoxy)-phenylaminol- pyrimidin-4-ylaminol-benzenesuIfonamide (Compound LVII)
LVII
[0203] A mixture of intermediate 33 (0.10 g, 0.28 mmol) and 4-(2-pyrrolidin-l-yl- ethoxy)-phenylamine (0.10 g, 0.49 mmol) in acetic acid (3 mL) was sealed in a microwave reaction tube and irradiated with microwave at 150 °C for 20 min. After cooling to room temperature, the cap was removed and the mixture concentrated. The residue was purified by HPLC and the corrected fractions combined and poured into saturated NaHCO3 solution (30 mL). The combined aqueous layers were extracted with EtOAc (2 x 30 mL) and the combined organic layers washed with brine, dried over anhydrous Na2SO4and filtered. The filtrate was concentrated and the resulting solid dissolved in minimum atnount of EtOAc and hexanes added until solid precipitated. After filtration, the title compound was obtained as a white solid (40 mg, 27%).
[0204] 1H NMR (500 MHz, DMSO-d6): δ 1.12 (s, 9H), 1.65-1.70 (m, 4H), 2.12 (s, 3H), 2.45-2.55 (m, 4H), 2.76 (t, J= 5.8 Hz, 2H), 3.99 (t, J= 6.0 Hz, 2H), 6.79 (d, J= 9.0 Hz, 2H), 7.46-7.53 (m, 4H), 7.56 (s, IH), 7.90 (s, IH), 8.10-8.15 (m, 2H), 8.53 (s, IH), 8.77 (s, IH). MS (ES+): m/z 525 (M+H)+. it •ιr
Image may be NSFW.
Clik here to view.
PATENTS
PAPER
Bioorganic & Medicinal Chemistry Letters, 27(12), 2668-2673; 2017
PATENT
The compound and the pharmaceutical compositions described herein can be used for treating or delaying development of myelofibrosis in a subject. N-teft-Butyl-3-[(5-methyl-2-{ [4- (2-pyrrolidin-l-ylethoxy)phenyl]amino}pyrimidin-4-yl)amino]benzenesulfonamide has the following chemical structure:

Example 4. Synthesis of TG101348
Example 4.1 N-fer^-Butyl-3-(2-chloro-5-methyl-pyrimidin-4-ylamino)-benzenesulfonamide
(Intermediate)
Example 4.1(a)

1 2 Intermediate
[0162] A mixture of 2-chloro-5-methyl-pyrimidin-4-ylamine (1) (0.4 g, 2.8 mmol), 3-bromo-N- teft-butyl-benzenesulfonamide (2) (1.0 g, 3.4 mmol), Pd2(dba¾ (0.17 g, 0.19 mmol), Xantphos (0.2 g, 3.5 mmol) and cesium carbonate (2.0 g, 6.1 mmol) was suspended in dioxane (25 mL) and heated at reflux under the argon atmosphere for 3 h. The reaction mixture was cooled to room temperature and diluted with DCM (30 mL). The mixture was filtered and the filtrate
concentrated in vacuo. The residue was dissolved in EtOAc and hexanes added until solid precipitated. After filtration, the title compound (1.2 g, 98%) was obtained as a light brown solid. It was used in the next step without purification. MS (ES+): m/z 355 (M+H)+.
Example 4.1(b)

SM2 Intermediate[0163] The Intermediate was synthesized from 2,4-dichloro-5-methylpyrimidine (SMI) and N-t- butyl-3-aminobenzenesulfonamide (SM2) in the following steps: (1) Mix MeOH (6.7UOa) and SMI (Combi Blocks) (UOa); (2) Add SM2 (1.15UOa, 082eq) and H20 (8.5UOa); (3) Heat 45°C, 20h, N2, IPC CPL SM2<2%; (4) Cool 20°C; (5) Centrifuge, N2; (6) Wash H20 (2.1UOa) + MeOH (1.7UOa); (7) Mix solid in H20 (4.3UOa) + MeOH (3.4UOa); (8) Centrifuge, N2; (9) Wash H20 (2.1UOa) + MeOH (1.7UOa); and (10) Dry 45°C, vacuum, 15h. Obtained
Intermediate, mass 49.6kg (UOb); Yield 79%; OP: 99.6%.
Example 4.2 N-½ri-Butyl-3-[(5-methyl-2-{ [4-(2-pyrrolidin-l- ylethoxy)phenyl]amino}pyrimidin-4-yl)amino]benzenesulfonamide

Intermediate TG101348
Example 4.2(a)
[0164] A mixture of N-ieri-Butyl-3-(2-chloro-5-methyl-pyrimidin-4-ylamino)- benzenesulfonamide (Intermediate) (0.10 g, 0.28 mmol) and 4-(2-pyrrolidin-l-yl-ethoxy)- phenylamine (3) (0.10 g, 0.49 mmol) in acetic acid (3 mL) was sealed in a microwave reaction tube and irradiated with microwave at 150 °C for 20 min. After cooling to room temperature, the cap was removed and the mixture concentrated. The residue was purified by HPLC and the corrected fractions combined and poured into saturated NaHCC^ solution (30 mL). The combined aqueous layers were extracted with EtOAc (2 x 30 mL) and the combined organic layers washed with brine, dried over anhydrous Na2S04 and filtered. The filtrate was concentrated and the resulting solid dissolved in minimum amount of EtOAc and hexanes added until solid precipitated. After filtration, the title compound was obtained as a white solid (40 mg, 27%). ]H NMR (500 MHz, DMSO-d6): δ 1.12 (s, 9H), 1.65-1.70 (m, 4H), 2.12 (s, 3H), 2.45-2.55 (m, 4H), 2.76 (t, /=5.8 Hz, 2H), 3.99 (t, 7=6.0 Hz, 2H), 6.79 (d, 7=9.0 Hz, 2H), 7.46-7.53 (m, 4H), 7.56 (s, 1H), 7.90 (s, 1H), 8.10-8.15 (m, 2H), 8.53 (s, 1H), 8.77 (s, 1H). MS (ES+): m/z 525 (M+H)+.
Example 4.2(b)
[0165] N-½ri-Butyl-3-[(5-methyl-2-{ [4-(2-pyrrolidin-l-ylethoxy)phenyl]amino}pyrimidin-4- yl)amino]benzenesulfonamide dihydrochloride monohydrate was prepared from 4-[2-(l- pyrrolidinyl)ethoxy] aniline dihydrochloride (SM3) and Intermediate following steps (A) and (B).
[0166] Step (A), preparation of free base of SM3 (3) from SM3, comprised steps (1) – (9): (1) Solubilize NaOH (0.42UOb) in H20 (9UOb); (2) Cool <20°C, N2; (3) Add TBME (6UOb) then SM3 (Malladi Drugs) (1.06UOb); (4) Mix >20mn then stop; (5) Drain Aq Ph then extract by TBME (3UOb); (6) Combine Or Ph; (7) Concentrate, vacuum, T<40°C, to an Oil; (8) Solubilize in IPA (2.5UOb); and (9) Calculate dry extract 23%.
[0167] Step (B) comprised the steps (1) – (6): (1) Mix IPA (10.5UOb) and Intermediate (UOb); (2) Add free base of SM3 (0.75UOb, 1.33eq/ interm); (3) add HC1 cone (0.413UOb); (4) Heat 70°C, 20h, N2, IPC CPL Interm<2%; (5) Cool <20°C; (2) Centrifuge, N2; (3) Wash IPA (3UOb); (4) Dry 50°C, vacuum, 26h; (5) De-lump in Fitzmill; and (6) polybag (x2) / poly drum. Obtained TG101348 dihydrochloride monohydrate, mass 83.8kg; Yield 98%; OP: 99.5%. Example 5 Capsule Form of TG101348 and Process of Making TG101348
PATENT
WO 2010017122
US 2007259904
WO 2007053452
Paper
JAK inhibitors: pharmacology and clinical activity in chronic myeloprolipherative neoplasms.
Treliński J, Robak T.
Curr Med Chem. 2013;20(9):1147-61.
Santos FP, Verstovsek S.
Anticancer Agents Med Chem. 2012 Nov;12(9):1098-109. Review.
Looi CY, Imanishi M, Takaki S, Sato M, Chiba N, Sasahara Y, Futaki S, Tsuchiya S, Kumaki S.
PLoS One. 2011;6(8):e23640. doi: 10.1371/journal.pone.0023640. Epub 2011 Aug 10
PATENT
Example 90 N-tert-Butyl-3-{5-methyl-2-[4-(2-pyrrolidin-1-yl-ethoxy)-phenylamino]-pyrimidin-4-ylamino}-benzenesulfonamide (Compound LVII)

A mixture of intermediate 33 (0.10 g, 0.28 mmol) and 4-(2-pyrrolidin-1-yl-ethoxy)-phenylamine (0.10 g, 0.49 mmol) in acetic acid (3 mL) was sealed in a microwave reaction tube and irradiated with microwave at 150° C. for 20 min. After cooling to room temperature, the cap was removed and the mixture concentrated. The residue was purified by HPLC and the corrected fractions combined and poured into saturated NaHCO3 solution (30 mL). The combined aqueous layers were extracted with EtOAc (2×30 mL) and the combined organic layers washed with brine, dried over anhydrous Na2SO4 and filtered. The filtrate was concentrated and the resulting solid dissolved in minimum amount of EtOAc and hexanes added until solid precipitated. After filtration, the title compound was obtained as a white solid (40 mg, 27%).
1H NMR (500 MHz, DMSO-d6): δ 1.12 (s, 9H), 1.65-1.70 (m, 4H), 2.12 (s, 3H), 2.45-2.55 (m, 4H), 2.76 (t, J=5.8 Hz, 2H), 3.99 (t, J=6.0 Hz, 2H), 6.79 (d, J=9.0 Hz, 2H), 7.46-7.53 (m, 4H), 7.56 (s, 1H), 7.90 (s, 1H), 8.10-8.15 (m, 2H), 8.53 (s, 1H), 8.77 (s, 1H). MS (ES+): m/z 525 (M+H)+.
Example 76 N-tert-Butyl-3-(2-chloro-5-methyl-pyrimidin-4-ylamino)-benzenesulfonamide (Intermediate 33)

A mixture of 2-chloro-5-methyl-pyrimidin-4-ylamine (0.4 g, 2.8 mmol), 3-bromo-N-tert-butyl-benzenesulfonamide (1.0 g, 3.4 mmol), Pd2(dba)3 (0.17 g, 0.19 mmol), Xantphos (0.2 g, 3.5 mmol) and cesium carbonate (2.0 g, 6.1 mmol) was suspended in dioxane (25 mL) and heated at reflux under the argon atmosphere for 3 h. The reaction mixture was cooled to room temperature and diluted with DCM (30 mL). The mixture was filtered and the filtrate concentrated in vacuo. The residue was dissolved in EtOAc and hexanes added until solid precipitated. After filtration, the title compound (1.2 g, 98%) was obtained as a light brown solid. It was used in the next step without purification. MS (ES+): m/z 355 (M+H)+.
PATENT
https://encrypted.google.com/patents/US20090286789
- Example 90N-tert-Butyl-3-{5-methyl-2-[4-(2-pyrrolidin-1-yl-ethoxy)-phenylamino]-pyrimidin-4-ylamino}-benenesulfonamide (Compound LVII)
-
[0308]
-
[0309]A mixture of intermediate 33 (0.10 g, 0.28 mmol) and 4-(2-pyrrolidin-1-yl-ethoxy)-phenylamine (0.10 g, 0.49 mmol) in aeetie acid (3 mL) was sealed in a microwave reaction tube and irradiated with microwave at 150° C. for 20 min. After cooling to room temperature, the cap was removed and the mixture concentrated. The residue was purified by HPLC and the corrected fractions combined and poured into saturated NaIICO3 solution (30 mL). The combined aqueous layers were extracted with EtOAc (2×30 mL) and the combined organic layers washed with brine, dried over anhydrous Na2SO4 and filtered. The filtrate was concentrated and the resulting solid dissolved in minimum amount of EtOAc and hexanes added until solid precipitated. After filtration, the title compound was obtained as a white solid (40 mg, 27%).
-
[0310]1H NMR (500 MHz, DMSO-d6): δ 1.12 (s, 9H), 1.65-1.70 (m, 4H), 2.12 (s, 3H), 2.45-2.55 (m, 4H), 2.76 (t, J=5.8 Hz, 2H), 3.99 (t, J=6.0 Hz, 2H), 6.79 (d, J=9.0 Hz, 2H), 7.46-7.53 (m, 4H), 7.56 (s, 1H), 7.90 (s, 1H), 8.10-8.15 (m, 2H), 8.53 (s, 1H), 8.77 (s, 1H). MS (ES+): m/z 525 (M+H)+.

PATENT
WO 2015117053
References
- Jump up^ Pardanani, A.; Hood, J.; Lasho, T.; Levine, R. L.; Martin, M. B.; Noronha, G.; Finke, C.; Mak, C. C.; Mesa, R.; Zhu, H.; Soll, R.; Gilliland, D. G.; Tefferi, A. (2007). “TG101209, a small molecule JAK2-selective kinase inhibitor potently inhibits myeloproliferative disorder-associated JAK2V617F and MPLW515L/K mutations”. Leukemia. 21 (8): 1658–1668. doi:10.1038/sj.leu.2404750. PMID 17541402.
- Jump up^ Pardanani, A.; Gotlib, J. R.; Jamieson, C.; Cortes, J. E.; Talpaz, M.; Stone, R. M.; Silverman, M. H.; Gilliland, D. G.; Shorr, J.; Tefferi, A. (2011). “Safety and Efficacy of TG101348, a Selective JAK2 Inhibitor, in Myelofibrosis”. Journal of Clinical Oncology. 29 (7): 789–796. doi:10.1200/JCO.2010.32.8021. PMC 4979099 Image may be NSFW.
Clik here to view. . PMID 21220608.
. PMID 21220608. - Jump up^ “Celgene to Acquire Impact Biomedicines, Adding Fedratinib to Its Pipeline of Novel Therapies for Hematologic Malignancies (NASDAQ:CELG)”. ir.celgene.com. Retrieved 2018-01-18.

External links
- Fedratinib, Impact Biomedicines
| Image may be NSFW. Clik here to view.  |
|
| Names | |
|---|---|
| IUPAC name
N–tert-Butyl-3-{5-methyl-2-[4-(2-pyrrolidin-1-yl-ethoxy)-phenylamino]-pyrimidin-4-ylamino}-benzenesulfonamide
|
|
| Other names
SAR302503; TG101348
|
|
| Identifiers | |
|
|
|
3D model (JSmol)
|
|
| Properties | |
| C27H36N6O3S | |
| Molar mass | 524.68 g·mol−1 |
| Density | 1.247 ± 0.06 g/cm3 |
|
Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa).
|
|
| Infobox references | |

////////////////FEDRATINIB, SAR-302503, TG-101348, SANOFI, PHASE 3, TG101348, SAR302503, TG 101348, SAR 302503, Orphan drug designation
CC1=CN=C(N=C1NC2=CC(=CC=C2)S(=O)(=O)NC(C)(C)C)NC3=CC=C(C=C3)OCCN4CCCC4