
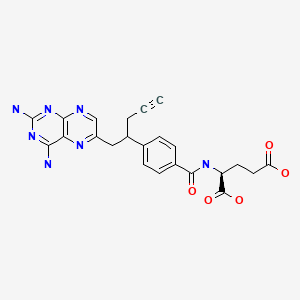
Pralatrexate (JAN/USAN/INN);
10-Propargyl-10-deazaaminopterin;
Folotyn (TN)
Antineoplastic
| Product
CAS:
|
FOLOTYN (Allos Therapeutics)
146464-95-1
|
|---|---|
| Formula |
C23H23N7O5
|
| Exact mass |
477.1761
|

- (2S)-2-((4-((1RS)-1-((2,4-diaminopteridin-6-yl)methyl)but-3-ynyl)benzoyl)amino)pentanedioic acid
- (2S)-2-({4-[1-(2,4-diaminopteridin-6-yl)pent-4-yn-2-yl]benzoyl}amino)pentanedioic acid
- 10-Propargyl-10-deazaaminopterin
- N-(4-(1-((2,4-Diamino-6-pteridinyl)methyl)-3-butynyl)benzoyl)-L-glutamic acid
- PDX
- UNII:A8Q8I19Q20
Japan approved 2017
| 2017/7/3 | PMDA | JAPAN | Pralatrexate | Difolta | Mundipharma | NME |

EMA 2012
The molecule contains two asymmetric carbon centres (C10) and (C19). The C10 position exists in the RS-configuration (approx. 50:50 ratio) on the link between the two aryl groups. The C19 position is contained in the glutamic acid moiety and predominantly exists in the S-configuration. Pralatrexate is an off-white to yellow crystalline material, soluble in aqueous solutions at pH 6.5 or higher and practically insoluble in chloroform, and ethanol. It predominantly exists as a single polymorph (form A).
Pralatrexate, chemically known as “(2S)-2-[[4-[(1RS)-1-[(2,4-diaminopteridin-6-yl)methyl]but-3-ynyl]benzoyl- ]-amino]pentanedioic acid”, also known as “10-Propargyl-10-deazaminopterin” or “PDX”, is a compound which has been tested and found useful in the treatment of cancer. In its racemic form, 2S)-2-[[4-[(1RS)-1-[(2,4-diaminopteridin-6-yl)methyl]but-3-ynyl]benzoyl]a- mino]-pentanedioic acid has been approved by the U.S. Food and Drug Administration (FDA) as a treatment for relapsed and refractory peripheral T-cell lymphoma.
Pralatrexate, was first disclosed in Journal of Medicinal Chemistry. 36: 2228-2231 (1993) by DeGraw et al., and subsequently in U.S. Pat. No. 5,374,726 and U.S. Pat. No. 5,354,741.
Pralatrexate is an antimetabolite for the treatment of relapsed or refractory peripheral T-cell lymphoma. It is more efficiently retained in cancer cells than methotrexate. FDA approved on September 24, 2009.
Pralatrexate (brand name Folotyn) is an anti-cancer therapy.[1] It is the first drug approved as a treatment for patients with relapsed or refractory peripheral T-cell lymphoma, or PTCL[2] — a biologically diverse group of aggressive blood cancers that have a poor prognosis.[2]

Approval
Folotyn was approved by the U.S. Food and Drug Administration (FDA) in September 2009 under the FDA’s accelerated approval,[2] which allows for earlier approval of drugs that meet unmet medical needs.[3] Pralatrexate injection is marketed in the U.S. under the name Folotyn by Spectrum Pharmaceuticals.[2] Clinical trials are currently underway to explore the potential of Folotyn in other blood related cancers and solid tumors.[4]

Mechanism
Pralatrexate is an antifolate (a folate analogue metabolic inhibitor) designed to accumulate preferentially in cancer cells.[1] Based on preclinical studies, researchers believe that pralatrexate selectively enters cells expressing reduced folate carrier type 1 (RFC-1), a protein that is overexpressed on certain cancer cells compared to normal cells.[1]
Antifolates, such as pralatrexate, are part of a group of compounds known as antimetabolites with structural similarity to naturally occurring molecules involved in DNA synthesis.[5] Cancer cells mistake antimetabolites for normal metabolites[5] allowing the compound to stop or slow critical enzymes involved in DNA synthesis which then triggers cell death.[1] Because of their primary effect on DNA synthesis, the antimetabolites are most effective against actively dividing cells and are largely cell-cycle phase specific.[5]
The selectivity of pralatrexate for cancer cells is based upon the observation that cancer cells generally have an overexpression of reduced folate carrier protein-1 (RTC-1) compared to normal somatic cells. This carrier protein allows the entrance of pralatrexate into the cell. Upon entering the cell, folypolyglutamate synthase FPGS catalyzes the polyglutamination of pralatrexate so that it is retained inside the cell.
Once inside, pralatrexate competitively inhibits dihydrofolate reductase (DHFR) and thymidylate synthase. Subsequent depletion of thymidine monophosphate (TMP) occurs so that the cancer cell is unable to synthesize DNA and RNA. As a result, the cancer cell cannot proliferate and is forced to undergo apoptosis. Pralatrexate is more effective against cells that are actively dividing.
Biological Activity
Pralatrexate (Folotyn) is an antifolate, and structurally a folate analog. It acts as an inhibitor of dihydrofolate reductase. It is selective for the reduced folate carrier type 1. Its IC50 is < 300 nM in some cell lines.
Conversion of different model animals based on BSA (Value based on data from FDA Draft Guidelines)
| Species | Mouse | Rat | Rabbit | Guinea pig | Hamster | Dog |
| Weight (kg) | 0.02 | 0.15 | 1.8 | 0.4 | 0.08 | 10 |
| Body Surface Area (m2) | 0.007 | 0.025 | 0.15 | 0.05 | 0.02 | 0.5 |
| Km factor | 3 | 6 | 12 | 8 | 5 | 20 |
| Animal A (mg/kg) = Animal B (mg/kg) multiplied by | Animal B Km |
| Animal A Km |
For example, to modify the dose of resveratrol used for a mouse (22.4 mg/kg) to a dose based on the BSA for a rat, multiply 22.4 mg/kg by the Km factor for a mouse and then divide by the Km factor for a rat. This calculation results in a rat equivalent dose for resveratrol of 11.2 mg/kg.
Discovery
Research on this class of drugs began in the 1950s at SRI International, where scientists were focused on developing new chemotherapies and antifolates that would be effective against tumor cells.[1]
In the late 1970s, researchers at Memorial Sloan Kettering Cancer Center discovered that cancerous cells take in natural folate through a protein identified as plasma membrane transporter (now referred to as “reduced folate carrier type 1” or “RFC-1”). Further research showed that when normal cells evolve into cancerous cells they often overproduce RFC-1 to ensure they get enough folate.[6]
A subsequent scientific collaboration was ultimately formed among SRI International, Memorial Sloan Kettering Cancer Center, and the Southern Research Institute with the intention of developing an antifolate with greater therapeutic selectivity – an agent that could be more effectively internalized into tumors (transported into the cells through RFC-1) and would be more toxic to cancer cells than normal cells.[6]
This collaboration, supported by the National Cancer Institute,[7] led to the identification of pralatrexate in the mid-1990s. Pralatrexate was later licensed to Allos Therapeutics in 2002 for further development.[8] Allos Therapeutics, Inc. was acquired by Spectrum Pharmaceuticals, Inc. on September 5, 2012. Allos is now a wholly owned subsidiary of Spectrum.[9]
Pralatrexate, is a 10-deazaaminopterin derivative which has been developed for the potential treatment of malignancies. Pralatrexate is an antifolate, structurally a folate analog inhibitor of dihydrofolate reductase (DHF ) exhibiting high affinity for reduced folate carrier- 1 (RFC- 1) and iolylpolyglutamate synthetase (FPGS). with antineoplastic and immunosuppressive activities, resulting in extensive internalization and accumulation in tumour cells. Pralatrexate selectively enters cells expressing RFC- 1. Intracellularly, this agent is highly polyglutamylated and competes for the folate binding site of DHFR, blocking tetrahydrofolate synthesis, which may result in depletion of nucleotide precursors; inhibition of DNA. RNA and protein synthesis; and apoptotic tumor cell death. Efficient intracellular polyglutamylation of pralatrexate results in higher intracellular concentrations compared to non-polyglutamylated pralatrexate, which is more readily effuxed by the MRP (multidrug resistance protein) drug efflux pump. RFC- 1, an oncofetal protein expressed at highest levels during embryonic development, may be over expressed on the cell surfaces of various cancer cell types. Pralatrexate is the first and only drug approved by the Food and Drug Administration as a treatment for relapsed or refractory peripheral T-cell lymphoma, demonstrating the ability to reduce tumor size, but not to prolong life.
Pralatrexate is a folate analog metabolic inhibitor that competitively inhibits dihydrofolate reductase. It is also a competitive inhibitor for polyglutamylation by the enzyme folylpolyglutamyl synthetase. This inhibition results in the depletion of thymidine and other biological molecules the synthesis of which depends on single carbon transfer.
US 200510267117 discloses that T cell lymphoma is treated by administering to a patient suffering from T cell lymphoma a therapeutically effective amount of IO-propargyl-10- deazaaminopterin. Remission is observed in human patients, even with drug resistant T cell lymphoma at weekly dosages levels as low as 30 mg/m2. In general, the 10-propargyl-lO- deazaaminopterin is administered in an amount of from 30 to 275 mg/m2 per dose.
US 2011/0190305 discloses diastereomers of 10-propargyl-l 0-deazaminopterin, compositions comprising optically pure diastereomers of 10-propargyl-l 0-deazaminopterin, in particular the two (R,S) diastereomers about the C 10 position, method of preparation of the diastereomers and method of treatment of conditions related to inflammatory disorders and cancer.
US005354751 discloses heteroaroyl-10-deazaaminopterins and 10-alkenyl or 10-alkynyl-lO- deazaaminopterins having pronounced anti-inflammatory activity, anti-leukemic and anti- tumorigenic activity, as well as a method for treatment of inflammatory diseases, leukemia and tumors. Pharmaceutical compositions containing these heteroaroyl-lO-deazaaminopterin compounds are also disclosed. The invention further concerns a process for preparation of these compounds. A method for preparation of I0-propargyl-10-deazaaminopterin compound is also disclosed in this document.
Journal publication Bioorganic and Medicinal Chemistry (19) 2011, page 1151, synthetic approaches to the 2009 new drugs, also discloses a method for synthesis of Pralatrexate. The method comprises alkylating dimethyl homotrephthalate with propargyl bromide in the presence of KH in THF and then with 2,4-diamino-6-(bromomethyl)pteridine hydrobromide in the presence of KH in D F to afford crude product. Subsequent hydrolysis of the diester with aqueous NaOH, followed by acidification with acetic acid to give crude carboxylic acid, followed by thermally induced decarboxylation in D SO to give 10-deazapteroic acid derivative. Activation of carboxylic acid as a mixed anhydride using t-butyl chioroformate prior to coupling with diethyl L-glutamate hydrochloride in the presence of Et3 in DMF to give lO-propargyl-IO-deaza-aminopterin diethyl ester. Finally, saponification of diethyl ester with aqueous NaOH in 2-methoxyethanol, followed by acidifying with AcOH giving Pralatrexate.
Methods of preparing Pralatrexate known in the prior art are not only complicated but preparation of Pralatrexate using the methods disclosed in the prior art also result in very high manufacturing cost. Therefore, there is a need for an improved, simple and cost effective method for preparation of Pralatrexate which can be used for industrial scale preparation of this compound.
PATENT
scheme- 1 .

Scheme-1

Scheme 2

Scheme 3
scheme 4.

https://patents.google.com/patent/WO2013164856A1/en
PATENT
https://patents.google.com/patent/WO2014016740A2/en
Pralatrexate, chemically known as “(25)-2-[[4-[(lR5)-l-[(2,4-diaminopteridin-6- yl)methyl]but-3-ynyl]benzoyl]- amino] pentanedioic acid”, also known as “10-Propargyl- 10-deazaminopterin” or “PDX”, is a compound which has been tested and found useful in the treatment of cancer. In its racemic form, 2S)-2-[[4-[(lRS)-l-[(2,4-diaminopteridin-6- yl)methyl]but-3-ynyl]benzoyl]amino]- pentanedioic acid has been approved by the U.S. Food and Drug Administration (FDA) as a treatment for relapsed and refractory peripheral T-cell lymphoma.
Pralatrexate, represented by Formula (I), was first disclosed in Journal of Medicinal Chemistry. 36: 2228-2231 (1993) by DeGraw et al., and subsequently in US 5374726 and US 5354741.

DeGraw et al, publication, US 5374726 and US 5354741 disclose method for the synthesis of Pralatrexate of Formula (I), comprising alkylation of homoterephthalic acid dimethyl ester with propargyl bromide using Potassium Hydride, which is further coupled with 2,4-diamino-6-bromomethylpteridine in presence of Potassium Hydride followed by hydrolysis in presence of NaOH in 2-methoxyethanol-water mixture and decarboxylation at high temperature in DMSO and subsequent coupling with L-glutamic acid diethyl ester using t-butyl chloroformate and a base, and finally hydrolysis of the product with NaOH in 2-methoxyethanol-water mixture to give Pralatrexate of Formula (I). The process is outlined below as synthetic Scheme- 1.


Scheme- 1
The methods disclosed in DeGraw et al., publication, US 5374726 and US 5354741 suffer from the following disadvantages, which are outlined below:
(i) Use of pyrophoric Potassium hydride in the initial alkylation step and the subsequent coupling step.
(ii) Amide formation in the penultimate step by use of a hazardous chloroformate reagent.
(iii) The final product has a purity of -95% and is contaminated with the 10- deazaminopterin impurity to the level of 4%, which affects the final quality of Active Pharmaceutical ingredient (API) and does not meet the Pharmacopeial specifications. Use of 2-methoxyethanol in the last step which is classified under guideline of International Conference on Harmonisation of Pharmaceutical for Human USE (ICH) as a Class-2 solvent, with a maximum daily exposure limit of 50 ppm. Extensive use of column chromatography during the method adding to the cost of manufacture.
(vi) Low yield of the final Pralatrexate (-5.5 %).
US 6028071 discloses a process for the preparation of Pralatrexate of Formula (I) comprising coupling of homoterephthalic acid dimethyl ester with propargyl bromide using NaH in THF, further coupling of the product with 2,4-diamino-6- bromomethylpteridine using NaH in DMF, followed by hydrolysis with a base in 2- methoxyethanol-water mixture, and decarboxylation at elevated temperatures at 115- 120°C in DMSO, and finally coupling of the product with L-glutamic acid dimethyl ester using benzotriazole-l-yloxytris(dimethylamino) phosphonium hexafluorophosphate (BOP) and triethylamine in DMF, and finally hydrolysis with NaOH in methanol-water mixture to yield Pralatrexate. The process is outlined below as synthetic Scheme-2.

3, R = CH3
4 ■■ H

Scheme-2 The process disclosed in US 6028071 suffer from the following disadvantages outlined below
(i) Use of sodium hydride in the initial alkylation step and the subsequent coupling step.
(ii) Using benzotriazole-l-yloxytris(dimethylamino) phosphonium hexafluoro phosphate (BOP) in coupling reaction that liberates Hexamethylphosphoramide (HMPA), which is carcinogenic
(iii) Extensive column chromatography during the process adding to the cost of manufacture
(iv) Quality of the API obtained by this process is only -98%.
(v) Low yield of Pralatrexate is obtained (2.06%).
(vi) In the propargylation step the ratio of oc-monopropargyl homoterephthalic acid dimethyl ester to oc-monopropargyl homoterephthalic acid dimethyl ester is not less than 75:25.
US 20110190305 relates to optically pure diastereomers of 10-propargyl-lO- deazaminopterin, in particular the two ( ,S) diastereomers about the CIO position. None of the prior art discloses a process for preparing substantially pure Pralatrexate. When the present inventors practiced the invention disclosed in US 6028071 to ascertain the purity of Pralatrexate, they found the content of individual diastereomers at the CIO position to be 50+3.66%.
Example- 11
10-Propar gyl- 10-deazaminopterin (Pralatrexate)
To aqueous NaOH (11.6 g NaOH in 472 mL DM water) and Methanol (944 mL), 10- Propargyl-10-deazaminopterin Dimethyl Ester (59.0 g) was added at 20-25°C and stirred the reaction mass for 8 hours. After completion of reaction which was monitored by HPLC, pH of the reaction mass was adjusted to 6.6 with acetic acid. Excess methanol was evaporated under reduced pressure below 40° C and DM water (1298mL) was added to the residual solution. The pH of the residual solution was adjusted to 4.5 with dilute acetic acid. The reaction mass was stirred for 30 minutes at 20-25° C and filtered the solid precipitated. The solid was furthered purified with DM water (590 mL) by stirring at 20- 25°C for 30-35 minutes. The solid was filtered and dried under vacuum at 35-40° C to give 39 g (70 %) of the title compound.
Purity: 99.56 %
Water content = 4.8 % (w/w)
*H NMR (DMSO-d6; 400MHz): δ 1.91 (m, 1H), 2.05 (m, 1H), 2.33 (t, J=7.2 Hz, 2H), 2.59 (bm, 2H), 2.78 (s, 1H), 3.14-3.20 (bm, 1H), 3.28 (dd, J=14.4 Hz & 6.4 Hz, 1H), 3.64 (quintet, J=7.2 Hz), 4.35 (bm, 1H), 6.30 (bs, 2H, NH2), 7.39 (d, J=8.0 Hz, 2H), 7.61 & 7.63 (2xbs, 2H, NH2), 7.73 (d, J=8.0Hz, 2H), 8.39 (bs, 1H), 8.50 (d, J=7.6 Hz, 1H, NH), 12.20 (bs, 2H, 2xC02H).
13C NMR (DMSO-d6; 100MHz): δ 24.84 (CH2), 25.94 (CH2), 30.46 (CH2), 39.08 (CH2), 43.05 (CH), 51.93 (CH), 72.90 (CH), 82.57 (C), 121.51 (C), 127.35 (2xCH), 127.35 (2xCH), 132.22 (C), 146.69 (C), 147.20 (C), 150.56 (CH), 154.17 (C), 162.41 (C), 162.77 (C), 166.42 & 166.46 (CONH), 173.54 (C02H), 173.94 (C02H).
MS (ES+) m/z: 478 [M+H]+.
IR (KBr, cm-1): 1540, 1557, 1639, 1704, 3300, 3420.
XRD (°2Theta; Cu): 8.47, 10.85, 12.28, 14.34, 15.00, 15.78, 18.90, 21.79, 24.20, 27.5, 28.92, 34.28.
Example- 12 discloses the preparation of Pralatrexate according to US 6028071.
Example-12
To 10-Propargyl-lO-deazaminopterin dimethyl ester (3.0 g) in methanol (181.8 mL), aqueous sodium hydroxide (0.52 g of sodium hydroxide in 13.1 mL demineralized water) was added at 20-25°C accompanied by stirring. The reaction mixture was stirred for 2h at 20-25°C, kept for further 8 hours at the same temperature and diluted with demineralized water (181.8 mL). methanol was recovered under vacuum below 40°C and the residue was left at 20-25°C for 24 hrs. The reaction was monitored by HPLC and acidified with acetic acid (7.5 mL). The solid obtained was filtered, washed with demineralized water (15 mL) and suck-dried for 2-3 hrs. The product was dried under vacuum at 50-55°C for 12 hours.
Weight : 2.5
Yield (%) : 89.2
Purity by HPLC (%) : 99.61
PATENT
Pralatrexate, (2S)-2-[[4-[(1 RS)-1-[(2,4-diaminopteridin-6-yl)methyl]but-3-ynyl]benzoyl]amino]pentandioic acid, also referred to as 10-propargyl-10-deaza-aminopterin, is an anti-cancer drug having the following formula:

Pralatrexate is approved for a treatment for patients with relapsed or refractory peripheral T-cell lymphoma. It is an antifolate and acts as an inhibitor of
dihydrofolate reductase.
[0004] Pralatrexate is disclosed in several documents such as DeGraw et al., (J. Med. Chem, 1993, 36, 2228), US 6,028,071, US 5,354,751, EP 0944389, EP 1891957 and WO 98/02163. US 6,028,071 discloses Pralatrexate and a preparation thereof, as described in the following reaction scheme:

EXAMPLES
Reference examples:
[0099] Pt-MADES (compound 5) and Pt-MADAC (compound 6) may be prepared according to procedures disclosed in US 6,028,071, example 1.
Example 1 : Decarboxylation of Pt-MADAC (compound 6)

[00100] Pt-MADAC (compound 6) (17 g, 43.3 mmol, containing 1.4% of impurity hydro-Pt-MADAC (compound 6a) according to HPLC analysis) was added to N-methyl-2-pyrrolidone (170 mL, 10 Vol.) pre-heated at 120°C. Upon dissolution of the solid, N, N-diisopropylethyl amine (5.6 mL, 32.1 mmol) was added. The reaction mixture was stirred at 120 °C for 0.5 h, then cooled down to room temperature and poured into water (1700 mL, 100 Vol.). The pH was adjusted to 4.5 by addition of aq. HCl (16% w/w). A precipitate formed and was isolated by filtration. The collected solid was dried in a drying oven at 45 °C for 18 h to provide Pt-MADEC (compound 7) as a yellow solid (14.6 g, 97% yield, purity 87.1%), containing 7.5% of Pt-lactone (compound 7b) and 1.4% of hydro-Pt-MADEC (compound 7a) according to HPLC analysis.
Example 2: Decarboxylation of Pt-MADAC (Compound 6) without use of a base
[00101] Pt-MADAC (compound 6) (2 g, 5.10 mmol, containing 1.4% of impurity hydro-Pt-MADAC (compound 6a) according to HPLC analysis) was added to N-methyl-2-pyrrolidone (20 mL, 10 Vol.) pre-heated at 120 °C. The reaction mixture was stirred at 120 °C for 1 h, then cooled down to room temperature and poured into water (200 mL, 100 Vol.). The pH was adjusted to 4.5 by addition of aq. HCl (16% w/w) and the precipitate that formed was isolated by filtration. Drying in a drying oven at 45 °C for 18 h furnished Pt-MADEC (compound 7) as a yellow solid (1.65 g, 93% yield, purity 85.8%), containing 6.9% of Pt-lactone (compound 7b) and 1.4% of hydro-Pt-MADEC (compound 7a) according to HPLC analysis.
Example 3: Purification of Pt-MADEC 7 by precipitation of the corresponding potassium salt

[00102] Pt-MADEC (compound 7) (4 g, corresponding to 9.9 mmol of product considering the residual solvent content, prepared according to example 1) was added to aqueous KOH (12.9 mmol of KOH in 40 mL of water, 10 Vol.). The solid dissolved rapidly, and after 0.5 h, the formation of a precipitate started. After 0.5 h at room temperature the reaction mixture was cooled to 0 °C. After 1 h at 0 °C, the precipitate that had formed was isolated by filtration. Drying in a drying oven at 45 °C for 18 h furnished K-Pt-MADEC (compound 11) as a pale yellow solid (2.5 g, 65% yield, purity 99.5%), containing 0.3% of K-Pt-lactone-open (compound 11b) and 0.1% of hydro-K-Pt-MADEC (compound 11a) according to HPLC analysis.
Example 4: Purification of Pt-MADEC 7 derived from Pt-MADAC containing Pt-NADAC (5%) and Pt-lactone as impurities

[00103] Pt-MADAC (compound 6) (10 g) containing 5% of impurity Pt-NADAC (compound 6c) (according to HPLC analysis) was subjected to the decarboxylation conditions described in Example 1. The isolated Pt-MADEC (compound 7) (8.7 g, 87% yield, purity 83.4%) contained 6.2% of Pt-lactone (compound 7b), 1.6% of Pt- NADEC (compound 7c) and 3.4% of unreacted Pt-NADAC (compound 6c) according to HPLC analysis.
[00104] The compound was subjected to the purification conditions described in Example 3, furnishing K-Pt-MADEC (compound 11) (5.3 g, 55% yield, purity 98.5%), containing 0.3% of K-Pt-lactone-open (compound lib), 0.88% of K-Pt-NADEC (compound 11c) and 0.18% of K-Pt-NADAC (compound 1 Id) according to HPLC analysis.
Example 5: Purification of Pt-MADEC 7 by precipitation of the corresponding sodium salt

[00105] Pt-MADEC (compound 7) (2 g, corresponding to 4.95 mmol of product considering the residual solvent content) was added to aqueous NaOH (6.44 mmol of NaOH in 20 mL of water, 10 Vol.). The solid dissolved rapidly, then after 5 minutes the formation of a precipitate started. After 0.5 h at room temperature, the reaction mixture was cooled to 0 °C. After 1 h at 0 °C the precipitate that had formed was isolated by filtration. The collected solid was then dried in a drying oven at 45 °C for 18 h to provide the Na-Pt-MADEC (compound 12) as a pale yellow solid (1.6 g, 75% yield, purity 96.6%), containing 1.8% of Na-Pt-lactone-open (compound 12b) and 0.3% of hydro-Na-Pt-MADEC (compound 12a) according to HPLC analysis.
Example 6: Purification of Pt-MADEC 7 by precipitation of the corresponding lithium salt

[00106] Pt-MADEC (compound 7) (2 g, corresponding to 4.27 mmol of product, considering the residual solvent content), was added to aqueous LiOH (5.55 mmol of LiOH, in 20 mL of water, 10 Vol.). The solid dissolved rapidly, and after 15 minutes the formation of a precipitate started. After 0.5 h at room temperature the reaction mixture was cooled to 0 °C. After 1 h at 0 °C the precipitate that had formed was isolated by filtration. The collected solid was then dried in a drying oven at 45 °C for 18 h to provide Li-Pt-MADEC (compound 13) as a pale yellow solid (1.1 g, 73% yield, purity 97.7%), containing 0.6% of Li-Pt-lactone-open (compound 13b) and 0.9% of hydro-Li-Pt-MADEC (compound 13a) according to HPLC analysis.
Example 7: Synthesis of Pt-lactone
[00107] To a suspension of Pt-MAD AC (compound 6) (12.2 g, 31.2 mmol) in acetonitrile (122 mL, 10 Vol.), copper (I) iodide (595 mg, 3.12 mmol) and N,N-diisopropylethyl amine (10.9 mL, 62.2 mmol) were added. The reaction mixture was heated to reflux and stirred at reflux for 48 h., and then cooled to room temperature. The resulting precipitate was filtered and washed with acetonitrile (24 mL, 2 Vol.). The collected solid was then dried in a drying oven at 45 °C for 18 h to provide Pt-lactone (compound 7b) as a brown solid (12.3 g, >100% yield, purity 94.1%).
Example 8: Purification of Pt-MADES by formation of the corresponding DMF solvate
[00108] Pt-MADES (compound 5) (40 g, purity 94.7%) was added to DMF (400 mL, 10 Vol.), pre-heated at 120 °C. After dissolution of the product, the reaction mixture was kept at 120°C for 0.5 h, and then cooled to room temperature. The
resulting precipitate was filtered and washed with acetone (2 x 200 mL, 2 x 5 Vol.). Drying in a drying oven at 45 °C for 18 h furnished Pt-MADES (compound 5) as a pale yellow solid (36.4 g, 91% yield, purity 97.5%).
Example 9: purification of Pt-MADES
[00109] Pt-MADES (compound 5) (30 g, purity 94.7%) was suspended in a mixture of MeOH (300 mL, 10 Vol.) and formamide (150 mL, 5 Vol.). The suspension was heated at 60-65 °C for 2 h, then cooled to room temperature. Upon stirring at room temperature for 15 h, the resulting precipitate was filtered and washed with methanol (2 x 30 mL, 2 x 1 Vol.). Drying in a drying oven at 50 °C for 18 h furnished Pt-MADES (compound 5) as a pale yellow solid (22.5 g, 75% yield, purity 98.0%).
Example 10: purification of Pt-MADAC
[00110] Pt-MADAC (compound 6) (50 g, purity 94.0%) was suspended in a mixture of MeOH (150 mL, 3 Vol.) and formamide (50 mL, 1 Vol.). The suspension was heated at 60-65 °C for 2 h, and then cooled to room temperature. Upon stirring at room temperature for 15 h, a precipitate formed and was filtered and washed with methanol (2 x 50 mL, 2 x 1 Vol.). The collected precipitate was then dried in a drying oven at 50°C for 18 h. The thus-produced Pt-MADES was then suspended in MeOH (500 mL, 10 Vol.). This suspension was heated at reflux for 1 h, and then cooled to 0°C. The resulting precipitate was filtered and washed with methanol (2 x 50 mL, 2 x 1 Vol.). Drying in a drying oven at 50 °C for 18 h furnished Pt-MADAC (compound 6) as a pale yellow solid (41 g, 82% yield, purity 96.3%).
Example 11: Preparation of Pralatrexate, sodium salt (PLT-Na) (Compound 10a) by hydrolysis of Pralatrexate ethyl ester (PLT-ES) (Compound 9B)

[00111] A reactor was charged with EtOH (195 mL) and aqueous NaOH (3.75 M, 19.5 mL, 73 mmol). The mixture was then cooled down to 10 °C. 10-Propargyl-10-
deazaaminopterin ethyl ester (PLT-ES, Compound 9B, 13 g, 24.4 mmol) was added, and the temperature was increased to 25 °C over 0.5 h. The resulting suspension was then stirred at 25 °C for 17 h. The solid in the suspension was then isolated by filtration and washed with EtOH (65 mL). The collected solid was then dried in a drying oven at 45 °C for 18 h to provide (2S)-2-[[4-[(1 RS)-1-[(2,4-diaminopteridin-6-yl)methyl]but-3-ynyl]benzoyl]amino]pentanedioic acid disodium salt (Na-PLT; compound 10a) as a pale yellow solid (10.9 g, 86% yield, purity 99.6%).
Example 12: Preparation of Pralatrexate (Compound 10)

[00112] (2S)-2-[[4-[(1 RS)-1-[(2,4-Diaminopteridin-6-yl)methyl]but-3-ynyl]benzo-yl]amino]pentanedioic acid disodium salt (Na-PLT, Compound 10a, 10.9 g, 20.9 mmol) was dissolved in water (109 mL). The pH of the solution was adjusted to 4.5 by addition of aqueous HCl 1N. A precipitate formed and was isolated by filtration and washed with water (54 mL). The collected solid was then dried in a drying oven at 45 °C for 17 h to provide Pralatrexate (Compound 10) as a white solid (9.4 g, 81% yield, and purity 99.7%).
Example 13: Preparation of Pralatrexate ethyl ester (PLT-ES) (compound 9B)

[00113] A reactor was charged with potassium 10-propargyl-4-deoxy-4-amino-10-deazapteroate (K-Pt-MADEC, compound 11, 11.1 g, 28.7 mmol). DCM (82 ml) and 1-methyl-2-pyrrolidinone (16.7 ml) were added to obtain a suspension. 1-Hydroxy-benzotriazole hydrate (HOBt, 0.77 g, 5.74 mmol), N-(3-dimethylaminopropyl)-N’-ethylcarbodiimide hydrochloride (EDC, 6.60 g, 34.4 mmol) and (L)-glutamic acid
diethyl ester hydrochloride (7.98 g, 34.4 mmol) were sequentially added. The resulting mixture was stirred at room temperature until HPLC analysis showed reaction completion. DCM was removed under reduced pressure and MeOH (22.2 mL) was added. The reaction mixture was poured into water pre-acidified with aqueous HCl 16% (w/w, 16.9 mL). The pH was adjusted to 4.5 by addition of aqueous NaOH, resulting in the precipitation of the PLT-ES. The solid precipitate was isolated by filtration and dried in oven at 55 °C for 18 h to provide PLT-ES as a pale yellow solid (13 g, 85% yield, and purity 99.0%).
Example 14: Preparation of crystalline Pralatrexate ethyl ester (compound 9B)
[00114] PLT-ES (Compound 9B, lg) was dissolved in EtOH (15 ml). After a few minutes a precipitate started to form, and the precipitate was isolated by filtration after 45 minutes (0.5g, yellow solid, amorphous form). The mother liquor was left at room temperature overnight, resulting in the precipitation of a yellow solid which was isolated by filtration (0.3g, crystalline form, PXRD is shown on Figure 7).
Example 15: Hydrolysis of Pralatrexate ethyl ester (compound 9B)

[00115] A reactor was charged with MeOH (48 mL), aqueous NaOH (3.75 M, 18.0 mL, 67.5 mmol), and water (6 mL), and the mixture was cooled down to 10 °C. PLT-ES 9B (12 g, 22.5 mmol) was added and the temperature was increased to 25 °C over 1 h. The resulting suspension was stirred at 25 °C for 1 h, and then EtOH (168 mL) was added, resulting in the formation of a precipitate. After stirring for an additional 24 h the solid precipitate was isolated by filtration and washed with EtOH (120 mL).
The collected solid was then dried in a drying oven at 60 °C for 18 h to provide Na-PLT 10a (10.5 g, 90% yield, purity 99.8%) as a pale yellow solid.
PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2014068599
Pralatrexate is chemically, N-(4-{ 1-[(2,4-diaminopteridin-6-yl)methyl]but-3-yn-1 -yl}benzoyl)-L-glutamic acid and has the structural formula:

Pralatrexate is an anti-cancer therapy. It is the first drug approved as a treatment for patients with relapsed or refractory peripheral T-cell lymphoma, or PTCL – a biologically diverse group of aggressive blood cancers. Pralatrexate is currently marketed under the trade name FOLOTYN® by Alios.
Pralatrexate was disclosed in U.S. patent nos. 5,354,751 and 6,028,071.
According to the ‘071 patent, alpha-propargylhomoterephthalic acid dimethyl ester substantially free of homoterephthalic acid dimethyl ester was obtained by chromatographing alpha-propargylhomoterephthalic acid dimethyl ester residue obtained as part of the reaction between homoterephthalic acid dimethyl ester and propargyl bromide in the presence of
tetrahydrofuran and sodium hydride on silica gel using cyclohexane and ethyl acetate (8:1) for the elution.
Pralatrexate was also reported in J. Med. Chem, 1993, 36, 2228-2231. According to the paper, pralatrexate is prepared by crystallizing pralatrexate diethyl ester in a mixture of 2-methoxyethanol and water in the presence of sodium hydroxide.
International patent application publication no. WO 2012/061469 (‘469 patent) disclosed crystalline Form A, Form B and Form C of pralatrexate. According to the ‘469 patent, crystalline pralatrexate Form A can be prepared by crystallizing amorphous pralatrexate in formamide.
According to the ‘469 patent, crystalline pralatrexate Form B can be prepared by crystallizing amorphous pralatrexate in methanol or water.
According to the ‘469 patent, crystalline pralatrexate Form C can be prepared by crystallizing amorphous pralatrexate in a mixture of methanol and water.
Alpha-propargylhomoterephthalic acid dimethyl ester is a key staring material for the preparation of pralatrexate.
Example 1 :
Preparation of Alpha-propargylhomoterephthalic acid dimethyl ester
Sodium hydride (60 gm; 60%) was added to tetrahydrofuran (1500 ml) at room temperature and then cooled to 10 to 15°C. To the solution was added a solution of homoterephthalic acid dimethyl ester (250 gm) in tetrahydrofuran (250 ml) slowly for 15 minutes. The reaction mass was then cooled to 0 to -5°C and then added propargyl bromide (130 gm) in tetrahydrofuran (125 ml) slowly for 15 minutes at 0 to -5°C. The reaction mass was maintained for 2 hours at 0 to -5°C and then added methanol (50 ml). The temperature of the reaction mass was raised to room temperature and then added water (1500 ml) and diisopropyl ether (2500 ml), and then the layers were separated. The organic layer were dried with sodium sulfate and then concentrated to obtain 275 gm of alpha-propargylhomoterephthalic acid dimethyl ester.
Chromatographic purity of alpha-propargylhomoterephthalic acid dimethyl ester: 62.0%; Content of homoterephthalic acid dimethyl ester: 12.0%.
Example 2:
Purification of Alpha-propargylhomoterephthalic acid dimethyl ester
Alpha-propargylhomoterephthalic acid dimethyl ester (275 gm; HPLC Purity: 62.0%) as obtained in example 1 was dissolved in a mixture of hexane (1200 ml) and diisopropyl ether (65 ml) at room temperature. The solution was stirred for 15 hours at room temperature and filtered. The solid obtained was dried to obtain 175 gm of alpha-propargylhomoterephthalic acid dimethyl ester.
Chromatographic purity of alpha-propargylhomoterephthalic acid dimethyl ester: 74.6%; Content of homoterephthalic acid dimethyl ester: 0.4%.
Example 3:
Preparation of 10-propargyl-10-deazaminopterin diethyl ester
Step-I: Preparation oƒ 10-proparsyl-10-carbomethoxy-4-deoxy-4-amino-10-deazapteroic acid methyl ester
Sodium hydride (120 gm; 60%) was added to dimethylformamide (750 ml) at room temperature and then cooled to 0 to -5°C. To the solution was added a solution of alpha-propargylhomoterephthalic acid dimethyl ester (250 gm) in dimethylformamide (750 ml)
slowly for 15 minutes. The reaction mixture was maintained for 30 minutes at 0 to -5 C and then cooled to -20 to -25°C. To the reaction mixture was added 6-bromomethyl-pteridine-2,4-diamine (300 gm) in dimethylformamide (1500 ml) slowly for 30 minutes. The reaction mass was maintained for 2 hours at -20 to -25°C and then added methanol (300 ml). The temperature of the reaction mass was raised to room temperature and then added water (15000 ml) and diisopropyl ether (1500 ml). The contents were stirred for 2 hours at room temperature and filtered. The solid obtained was dried to obtain 198 gm of 10-propargyl-10-carbomethoxy-4-deoxy-4-amino-10-deazapteroic acid methyl ester.
Step-II: Preparation oƒ 10-proparsyl-10-carboxy-4-deoxy-4-amino-10-deazapteroic acid
2-Methoxyethanol (775 gm) was added to 10-propargyI-10-carbomethoxy-4-deoxy-4-amino-10-deazapteroic acid methyl ester (155 gm) at room temperature and then cooled to 15 to 20°C. To the reaction mixture was added a solution of sodium hydroxide (120 gm) in water (930 ml) and maintained for 4 hours at room temperature. The pH of the reaction mass was adjusted to 4.5 to 4.6 with acetic acid (50%) and then added water (3100 ml). The reaction mass was stirred for 2 hours, filtered and then dried to obtain 125 gm of 10-propargyl-10-carboxy-4-deoxy-4-amino-10-deazapteroic acid.
Step-III: Preparation oƒ 10-proparsyl-4-deoxy-4-amino-10-deazapteroic acid
10-Propargyl-10-carboxy-4-deoxy-4-amino-10-deazapteroic acid (135 gm) was added to dimethyl sulfoxide (1350 ml) at 120 to 125°C and maintained for 45 minutes at 120 to 125°C. The reaction mass was poured into water (3000 ml), maintained for 24 hours at room temperature and filtered to obtain a wet solid. To the wet solid was basified and then acetified, and maintained for 2 hours at room temperature. The separated solid was filtered and then dried to obtain 56 gm of 10-propargyl-4-deoxy-4-amino-10-deazapteroic acid.
Step-IV: Preparation oƒ 10-propargyl-10-deazaminopterin diethyl ester
Dimethylformamide (1 12 ml) was added to 10-propargyl-4-deoxy-4-amino-10-deazapteroic acid (14 gm) and stirred for 15 minutes. To the reaction mixture was added triethylamine (14 ml) and then cooled to 0 to -5°C. A solution of (benzotriazol-1-yloxy)tris(dimethylamino)phosphonium hexafluorophosphate (21 gm) in dimethylformamide (28 ml) was added to the reaction mixture and maintained for 1 hour at 0 to -5°C. To the
reaction mixture was added L-glutamic acid diethyl ester (10 gm) in dimethylformamide (28 ml) slowly, maintained for 2 hours at -10 to -15°C and filtered. The pH of the filtrate obtained was adjusted with sodium hydroxide solution and then added water (700 ml) slowly for 45 minutes. The reaction mass was maintained for 2 hours at room temperature, filtered and then dried to obtain 14 gm of 10-propargyl-10-deazaminopterin diethyl ester.
Example 4:
Preparation of pralatrexate
10-Propargyl-10-deazaminopterin diethyl ester (40 gm) was dissolved in tetrahydrofuran (320 ml) at room temperature. The solution was then cooled to 15 to 20°C and added a solution of sodium hydroxide (24 gm) in water (400 ml) slowly for 15 minutes. The reaction mass was maintained for 45 minutes at 15 to 20°C and then added a mixture of tetrahydrofuran (200 ml) and ethyl acetate (200 ml). The layers were separated and to the aqueous layer was added water (80 ml). The separated aqueous layer was then concentrated and pH was adjusted to 4.7 to 4.8 with acetic acid (10%). The contents were stirred for 1 hour at room temperature and filtered. The solid obtained was then dried to obtain 28 gm of pralatrexate.
Chromatographic purity of pralatrexate: 98.5%;
Content of 10-propargyl-4-deoxy-4-amino-10-dezapteroic acid: 0.3%;
Content of 10-deazaaminopterin: 0.5%;
Example 5:
Purification of pralatrexate
The pralatrexate (28 gm: HPLC Purity: 98.5%) as obtained in example 4 was dissolved in tetrahydrofuran (400 ml) and then heated to 60°C. To the contents were added water (200 ml) at 60°C and then cooled to 5 to 10°C. The contents were stirred for 2 hours 30 minutes at 5 to 10°C, filtered and then dried to obtain a solid. The solid was dissolved in dimethyl sulfoxide (138 ml) and then stirred to obtain a clear solution. The solution was filtered through celite bed and then added ethanol (690 ml) slowly for 1 hour. The contents were stirred for 1 hour at room temperature, filtered and then dried to obtain 20 gm of pure pralatrexate.
Chromatographic purity of pralatrexate: 99.5%;
Content of 10-propargyl-4-deoxy-4-amino-10-dezapteroic acid: 0.06%; Content of 10-deazaaminopterin: 0.08%.
PAPER
Nonpolyglutamatable Antifolate N-alpha-(4 amino-4-deoxypteroyl)-Ndelta-hemiphthaloyl-L-ornithine”, JOURNAL OF MEDICINAL CHEMISTRY, AMERICAN CHEMICAL SOCIETY, US, vol. 45, no. 8, 1 January 2002 (2002-01-01), pages 1690 – 1696, XP002291409, ISSN: 0022-2623, DOI: 10.1021/JM010518T *

Details are disclosed for the synthesis of Nα-[4-[2-(2,4-diaminoquinazolin-6-yl)ethyl]benzoyl]-Nδ-hemiphthaloyl-l-ornithine (2) and Nα-[4-[5-(2,4-diaminoteridin-6-yl)pent-1-yn-4-yl]benzoyl]-Nδ-hemiphthaloyl-l-ornithine (6) as analogues of Nα-(4-amino-4-deoxypteroyl)-Nδ-hemiphthaloyl-l-ornithine (1, PT523), a nonpolyglutamatable antifolate currently in advanced preclinical development. In a 72 h growth inhibition assay against cultures of CCRF-CEM human leukemic lymphoblasts, the IC50 of 2 and 6 was 0.69 ± 0.044 nM and 1.3 ± 0.35 nM, respectively, as compared with previously reported values 4.4 ± 0.10 nM for aminopterin (AMT) and 1.5 ± 0.39 nM for PT523. In a spectrophotometric assay of dihydrofolate reductase (DHFR) inhibition using dihydrofolate and NADPH as the cosubstrates, the previously unreported compounds 2 and the mixed 10R and 10S diastereomers of 6 had Ki values of 0.21 ± 0.05 pM and 0.60 ± 0.02 pM, respectively, as compared with previously reported values of 3.70 ± 0.35 pM for AMT and 0.33 ± 0.04 pM for PT523. Thus, while they were comparable to 1 and several of its previously studied analogues in their ability to bind to DHFR and inhibit the growth of CCRF-CEM cells, 2 and the mixed diastereomers of 6 were several times more active than AMT despite the fact that they cannot form γ-polyglutamylated metabolites of the type formed in cells from AMT and other classical antifolates with a glutamate side chain.
Synthesis and In Vitro Antitumor Activity of New Deaza Analogues of the Nonpolyglutamatable Antifolate Nα-(4-Amino-4-deoxypteroyl)-Nδ-hemiphthaloyl-l-ornithine (PT523)

PATENT
https://patents.google.com/patent/CN107488112A/zh
PATENT
US 20150183789
https://patents.google.com/patent/US9440979B2/en
PATENT
Publication numberPriority datePublication dateAssigneeTitle
References
- ^ Jump up to:a b c d e [1], Allos Therapeutics Press Release, “Allos Therapeutics’ Pralatrexate Demonstrates Anticancer Activity in Multiple Cancer Cell Lines”.
- ^ Jump up to:a b c d [2], Allos Therapeutics Press Release, “Allos Therapeutics’ FOLOTYN(TM) First and Only FDA-Approved Therapy for Relapsed or Refractory Peripheral T-cell Lymphoma”.
- Jump up^ [3], FDA, “Fast Track, Accelerated Approval and Priority Review”.
- Jump up^ [4], Allos Therapeutics, “Allos Therapeutics, Inc. Q1 2010 Earnings Call Transcript”.
- ^ Jump up to:a b c [5], Psychiatric Times, “Principles of Oncologic Pharmacotherapy”.
- ^ Jump up to:a b [6], Memorial Sloan Kettering Cancer Center Press Release, “FDA Approves Lymphoma Drug Developed at Memorial Sloan Kettering”.
- Jump up^ [7], National Cancer Institute “NCI Cancer Bulletin: The Next Steps in Drug Development at NCI”.
- Jump up^ “FDA Approves Pralatrexate for Treatment of Peripheral T-Cell Lymphoma” (Press release). SRI International. 2009-09-25. Retrieved 2013-07-10.
- Jump up^ Avery, Greg (2012-09-07). “Purchase of Allos Therapeutics is completed”. Denver Business Journal. Retrieved 2013-07-10.
External links
- Pralatrexate Development Information
- Clinical trials of pralatrexate at ClinicalTrials.gov
- Folotyn website
- FocusonPTCL website
- ASAP Support For Assisting Patients
|
|
 |
|
| Clinical data | |
|---|---|
| Trade names | Folotyn |
| AHFS/Drugs.com | Monograph |
| License data |
|
| Pregnancy category |
|
| Routes of administration |
Intravenous |
| ATC code | |
| Legal status | |
| Legal status |
|
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| IUPHAR/BPS | |
| ChemSpider | |
| UNII | |
| ChEBI | |
| ChEMBL | |
| ECHA InfoCard | 100.205.791 |
| Chemical and physical data | |
| Formula | C23H23N7O5 |
| Molar mass | 477.47 g/mol |
| 3D model (JSmol) | |


////////////プララトレキサート , japan 2017, Pralatrexate
NC1=NC2=NC=C(CC(CC#C)C3=CC=C(C=C3)C(=O)N[C@@H](CCC(O)=O)C(O)=O)N=C2C(N)=N1