
Tivozanib
- Molecular FormulaC22H19ClN4O5
- Average mass454.863 Da
- N-[2-Chloro-4-[(6,7-dimethoxy-4-quinolinyl)oxy]phenyl]-N’-(5-methyl-3-isoxazolyl)urea
- AV 951
- KRN 951
- Kil 8951
- N-[2-Chloro-4-[(6,7-dimethoxy-4-quinolyl)oxy]phenyl]-N’-(5-methyl-3-isoxazolyl)urea
- CAS HCL HYDRATE 682745-41-1
- 682745-43-3 HCL
Tivozanib (AV-951) is an oral VEGF receptor tyrosine kinase inhibitor. It has completed a pivotal Phase 3 investigation for the treatment of first line (treatment naive) patients with renal cell carcinoma.[1] The results from this first line study did not lead to FDA approval, but Tivozanib was approved by the EMA in August 2017[2]
Originally developed at Kirin Brewery, in January 2007 AVEO Pharmaceuticals acquired an exclusive license to develop and commercialize tivozanib in all territories outside of Asia.
In 2010, orphan drug designation was assigned in the E.U. for the treatment of renal cell carcinoma. In 2011, the compound was licensed to Astellas Pharma and AVEO Pharmaceuticals on a worldwide basis for the treatment of cancer
Tivozanib is an orally bioavailable inhibitor of vascular endothelial growth factor receptors (VEGFRs) 1, 2 and 3 with potential antiangiogenic and antineoplastic activities. Tivozanib binds to and inhibits VEGFRs 1, 2 and 3, which may result in the inhibition of endothelial cell migration and proliferation, inhibition of tumor angiogenesis and tumor cell death. VEGFR tyrosine kinases, frequently overexpressed by a variety of tumor cell types, play a key role in angiogenesis.
Tivozanib was originally developed by Kyowa Hakko Kirin and in 2007 AVEO Pharmaceutical acquired all the rights of the compound outside Asia. In December 2015, AVEO reached an agreement with EUSA Pharma, which acquired exclusive rights to tivozanib for advanced renal cell carcinoma in Europe, South America, Asia, parts of the Middle East and South Africa.
Tivozanib is an inhibitor of vascular endothelial growth factor (VEGF) receptors 1, 2, and 3 for first-line treatment of patients with advanced renal cell carcinoma in advanced disease or without VEGFR and mTOR inhibitors and progression after cytokine therapy Advanced renal cell carcinoma patients. Fotivda® is an oral capsule containing 890 μg and 1340 μg of Tivozanib per tablet. The recommended dose is 1 day, each 1340μg, taking three weeks, withdrawal for a week.


- CAS HCL HYDRATE 682745-41-1
ティボザニブ塩酸塩水和物;
Pharmacotherapeutic group
Antineoplastic agents
Therapeutic indication
Fotivda is indicated for the first line treatment of adult patients with advanced renal cell carcinoma (RCC) and for adult patients who are VEGFR and mTOR pathway inhibitor-naïve following disease progression after one prior treatment with cytokine therapy for advanced RCC.
Treatment of advanced renal cell carcinoma
Fotivda : EPAR -Product Information

Tivozanib is synthesized in three main steps using well defined starting materials with acceptable specifications.
Adequate in-process controls are applied during the synthesis. The specifications and control methods for intermediate products, starting materials and reagents have been presented. The critical process parameters are duly justified, methodology is presented and control is adequate.
The characterisation of the active substance and its impurities are in accordance with the EU guideline on chemistry of new active substances. Potential and actual impurities were well discussed with regards to their origin and characterised.
The active substance is packaged in a low-density polyethylene (LDPE) bag which complies with the EC
directive 2002/72/EC and EC 10/2011 as amended.
Product details
| NAME | Fotivda |
|---|---|
| AGENCY PRODUCT NUMBER | EMEA/H/C/004131 |
| ACTIVE SUBSTANCE | tivozanib |
| INTERNATIONAL NON-PROPRIETARY NAME(INN) OR COMMON NAME | tivozanib hydrochloride monohydrate |
| THERAPEUTIC AREA | Carcinoma, Renal Cell |
| ANATOMICAL THERAPEUTIC CHEMICAL (ATC) CODE | L01XE |
Publication details
| MARKETING-AUTHORISATION HOLDER | EUSA Pharma (UK) Limited |
|---|---|
| REVISION | 0 |
| DATE OF ISSUE OF MARKETING AUTHORISATION VALID THROUGHOUT THE EUROPEAN UNION | 24/08/2017 |
Contact address:
EUSA Pharma (UK) Limited
Breakspear Park, Breakspear Way
Hemel Hempstead, HP2 4TZ
United Kingdom
Mechanism
An oral quinoline urea derivative, tivozanib suppresses angiogenesis by being selectively inhibitory against vascular endothelial growth factor.[3] It was developed by AVEO Pharmaceuticals.[4] It is designed to inhibit all three VEGF receptors.[5]
Results
Phase III results on advanced renal cell carcinoma suggested a 30% or 3 months improvement in median PFS compared to sorafenibbut showed an inferior overall survival rate of the experimental arm versus the control arm.[5][6] The Food and Drug Administration‘s Oncologic Drugs Advisory Committee voted in May 2013 13 to 1 against recommending approval of tivozanib for renal cell carcinoma. The committee felt the drug failed to show a favorable risk-benefit ratio and questioned the equipose of the trial design, which allowed control arm patients who used sorafenib to transition to tivozanib following progression disease but not those on the experimental arm using tivozanib to transition to sorafenib. The application was formally rejected by the FDA in June 2013, saying that approval would require additional clinical studies.[6]
In 2016 AVEO Oncology published data in conjunction with the ASCO meeting showing a geographical location effect on Overall Survival in the Pivotal PhIII trial[7]
In 2016 AVEO Oncology announced the start of a second Pivotal PhIII clinical study in Third Line advanced RCC patients. [8]
In 2016 EUSA Pharma and AVEO Oncology announced that Tivozanib had been submitted to the European Medicines Agency for review under the Centralised Procedure. [9]
In June 2017 the EMA Scientific Committee recommended Tivozanib for approval in Europe, with approval expected in September.[10]
In August 2017 the European Commission (EC) formally approved Tivozanib in Europe.[11]
SYNTHESIS
Heterocycles, 92(10), 1882-1887; 2016

CLIP

Paper
Heterocycles (2016), 92(10), 1882-1887
Short Paper | Regular issue | Vol 92, No. 10, 2016, pp. 1882 – 1887
Published online: 5th September, 2016
■ A New and Practical Synthesis of Tivozanib
Chunping Zhu, Yongjun Mao,* Han Wang, and Jingli Xu
*College of Chemistry and Chemical Engineering, Shanghai University of Engineering Science, 333 Longteng Rd., Songjiang, Shanghai, 201620, China
Abstract
New and improved synthetic route of tivozanib is described on a hectogram scale. An reduction cyclization process to prepare the key intermediate 6,7-dimethoxyquinolin-4-ol from the 3-(dimethylamino)-1-(2-nitrophenyl)prop-2- en-1-one compound at H2/Ni condition is adopted in good result. Commercial available materials, simple reaction and operation are used, including nitration, condensation, hydrogenation, chlorination and so on, to give the final product in 28.7% yield over six steps and 98.9% purity (HPLC).
PAPER
https://www.sciencedirect.com/science/article/pii/S0960894X15003054
Bioorganic & Medicinal Chemistry Letters

PAPER
J MED CHEM 2005 48 1359



PATENT
KUBO, Kazuo; (JP).
SAKAI, Teruyuki; (JP).
NAGAO, Rika; (JP).
FUJIWARA, Yasunari; (JP).
ISOE, Toshiyuki; (JP).
HASEGAWA, Kazumasa; (JP)
Scheme 1 and Scheme 2
Skiing


PATENT
MATSUNAGA, Naoki; (JP).
YOSHIDA, Satoshi; (JP).
YOSHINO, Ayako; (JP).
NAKAJIMA, Tatsuo; (JP)
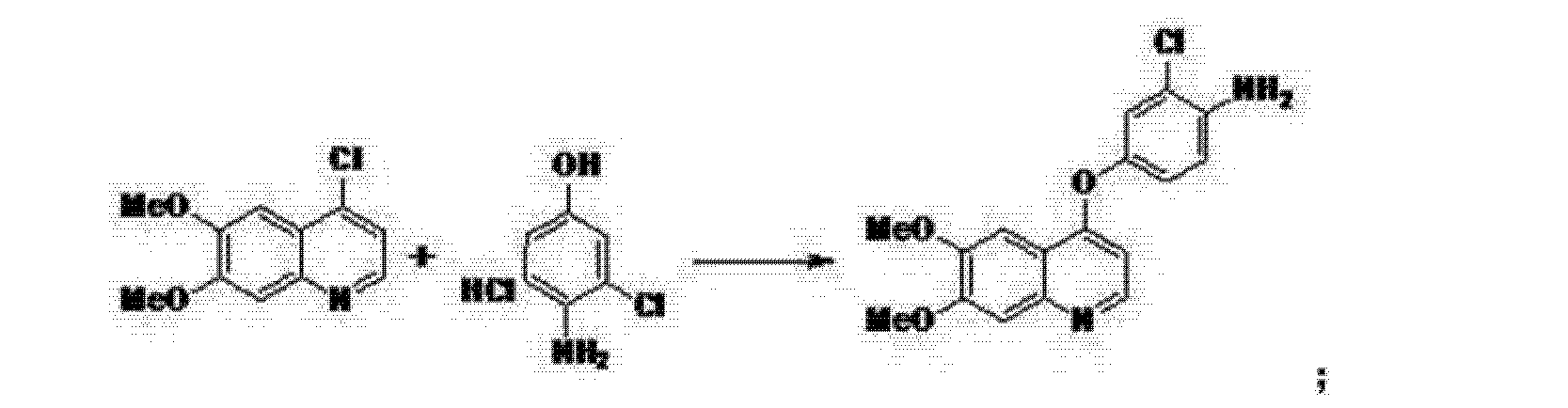
Preparation example: Preparation of N- {2-chloro-1- [(6,7-dimethoxy- 14 1 quinolyl) oxyl] phenyI} – N, – (5-methyl- 3 -isoxazolyl) urea ) Nitration process:

3, 4-Dimethoxyacetophenone (1 500 g) was dissolved in 5:: L 0 ° C of 17% nitric acid (1400 g), and 67% nitric acid (843 0 g) and sodium nitrite g) at a temperature of 5 to 10 ° C. over a period of 2 to 3 hours. After completion of dropping, the mixture was stirred at 5 to 10 ° C. for 1 to 2 hours. Cold water (7. 5 L) was added and after stirring for 30 minutes, filtration and washing with water (30 L). The filtrate was added to water (7. 5 L), neutralized with sodium bicarbonate water, filtered, and washed with water (7 L). The filtrate was dried under reduced pressure to obtain 3, 4-dimethoxy-6-nitroacetophenone (2164 g) (yield = 87.9%).
‘H-NMR (400 MHz, CD C 1 3 / p pm); 62. 5 0 (s, 3 H), 3. 9 7 (s, 3H), 3. 9 9 (s, 3 H), 6. 76 (s, 1 H), 7.6 2 (s, 1 H)
(2) Reduction process:

Methanol (5. 4 L), acetic acid (433 g:), 5% palladium / power monobonn (162 g) was added to 3, 4-dimethoxy-6-nitroacetophenone (1082 g) and hydrogen gas The mixture was stirred for 8 hours under pressure (2 Kg / cm 2, 40 ° C. The reaction solution was filtered, washed with methanol (1 L), and the filtrate was neutralized with aqueous sodium hydroxide solution and concentrated under reduced pressure Water (10 L) was added to the concentrate, stirred overnight, filtered and washed with water (7 L) Toluene (4 L) was added to the filtrate, heated to 80 ° C., 1 After stirring for a while, the residue was concentrated under reduced pressure and the residue was filtered, washed with toluene (300 mL), dried under reduced pressure to give 2-amino-4,5-dimethoxa Cetophenone (576 g) was obtained (yield = 6.1%).
‘H-NM (400 MHz, CD C 1 3 / p pm); 62. 5 6 (s, 3 H), 3. 84 (s, 3H), 3. 88 (s, 3 H), 6. 10 ( s, 1 H), 7.11 (s, 1 H)
(3) Cyclization step:

Tetrahydrofuran (THF) (5. 3 L) and sodium methoxide (3 1 3 g) were added to 2-amino-4, 5-dimethoxyacetophenone (33 7 g) and the mixture was stirred at 20 ° C for 30 minutes. At 0 ° C, ethyl formate (858 g) was added and stirred at 20 ° C for 1 hour. Water (480 mL) was added at 0 ° C. and neutralized with 1 N hydrochloric acid. After filtering the precipitate, the filtrate was washed with slurry with water (2 L). After filtration, the filtrate was dried under reduced pressure to obtain 6, 7-dimethoxy-141 quinolone (3 52 g) (yield = 8.15%).
‘H-NMR (400 MHz, DMS 0 – d 6 / ppm); 63. 8 1 (s, 3 H), 3. 84 (s, 3 H), 5. 94 (d, 1 H), 7. 0 1 (s, 1 H), 7. 43 (s, 1 H), 7. 76 (d, 1 H)
(4) Clovalization process

Toluene (3 L) and phosphorus oxychloride (1300 g) were added to 6, 7-dimethoxy-1-quinolone (105 g), and the mixture was stirred under heating reflux for 1 hour. It was neutralized with aqueous sodium hydroxide solution at 0 ° C. The precipitate was filtered, and then the filtrate was washed with water (10 L) for slurry. After filtering, the filtrate was dried under reduced pressure to obtain 4 1 -chloro- 16, 7-dimethoxyquinoline (928 g) (yield – 87.6 %) c ‘H-NMR (400 MHz, DMS 0 – d 6 / ppm); 63. 9 5 (s, 3 H), 3. 9 6 (s, 3 H), 7. 3 5 (s, 1 H), 7. 43 (s, 1 H) , 7. 54 (d, 1 H), 8. 59 (d, 1 H)
(5) Phenol site introduction step:

4-Amino-3-chlorophenol · HC 1 (990 g) was added to N, N-dimethylacetamide (6. 6 L). Potassium t-butoxide (145 2 g) was added at 0 ° C. and the mixture was stirred at 20 ° C. for 30 minutes. 4-Chloro-6, 7-dimethoxyquinoline (82 5 g) was added thereto, followed by stirring at 115 ° C for 5 hours. After cooling the reaction solution to room temperature, water (8. 3 L) and methanol (8.3 L) were added and the mixture was stirred for 2 hours. After filtration of the precipitate, the filtrate was washed with slurry with water (8. 3 L), filtered, and the filtrate was dried under reduced pressure to give 4- [(4-amino-3-chlorophenol) 6, 7-Dimethoxyquinoline (8 52 g) was obtained (yield = 6 9. 9%).
‘H-NMR (400MH z, DMS 0 – d 6 / ppm); 63. 9 2 (s, 3 H), 3. 93 (s, 3 H), 5. 4 1 (s, 2 H), 6 (D, 1 H), 6. 89 (d, 1 H), 6. 98 (dd, 1 H), 7. 19 (d, 1 H), 7. 36 (s, 1 H) , 7. 48 (s, 1 H), 8. 43 (d, 1 H)
(6) Ureaization process:

To 3 – amino – 5 – methylisoxazole (377 g), pyridine (1 2 1 5:), N, N – dimethylacetamide (4 L) at 0 ° C was added chlorobutyl carbonate phenyl
(60 1 g) was added dropwise and the mixture was stirred at 20 ° C. for 2 hours. 4- [(4-amino-1-chlorophenol) oxy] -6, 7-dimethoxyquinoline (84 7 g) was added to the reaction solution, and the mixture was stirred at 80 ° C. for 5 hours. The reaction solution was cooled to 5 ° C, then added with MeOH (8. 5 L) and water (8. 5 L) and neutralized with aqueous sodium hydroxide solution. After filtering the precipitate, the filtrate was washed with water (8. 5 L) for slurry. After filtration, the filtrate was dried under reduced pressure to give N- {2-chloro-4- [(6,7-dimethoxy-4-quinolyl) oxy] phenyl] – N, 1- -isoxazolyl) urea (1002 g) was obtained (yield = 86.1%).
‘H-NMR (400 MHz, DMS 0 – d 6 / ppm); 62.37 (s, 3 H), 3. 92 (s, 3 H), 3. 94 (s, 3 H), 6. 7 (s, 1 H), 7. 48 (s, 1 H), 7 (s, 1 H), 6. 54 (d, . 5 1 (d, 1 H), 8. 2 3 (d, 1 H), 8. 49
(d, 1 H), 8. 77 (s, 1 H), 1 0.16 (s, 1 H)
PATENT
WO 2011060162
WO 2017037220
CN 106967058
CN 104072492
CN 102532116
CN 102408418
PAPER
Advanced Materials Research Vols. 396-398 (2012) pp 1490-1492

Synthesis of the compounds
The synthesis of 6,7-Dimethoxy-4-quinolinone (2a) The 33.7g (0.173mol) of 2-amino-4,5-dimethoxy acetophenone, 150 ml of methanol and 95.5g (0.69mol) of anhydrous potassium carbonate were added to the 500 ml flask and stirred about 1 h at room temperature. Then, the ethyl formate (75.8g, 0.861mol) was dropped the admixture and reactioned about 2 h in the same temperature. The admixture was filtrated and the 35.2 g white powder compound 2a (C11H11NO3) was obtained with the yield of 81.5% and m.p. 124-125. 1H-NMR (DMSO-d6/ppm): δ 3.81 (s, 3H), 3.84 (s,3H), 5.94 (d,1H), 7.01 (s,1H), 7.43 (s,1H), 7.76 (d,1H). ESI-MS: 206 (M+ +1).
The synthesis of 4-chloro-6,7-dimethoxy-quinoline (2b)The 100 ml of toluene, 15 g (0.103 mol) of phosphorus trichloride and 10.6 g (0.52 mol) compound 2a were added to the 250 ml of three bottles, the obtained mixture was refluxed about 2 h. Then, the reaction mixture was cooled to the room temperature, filtrated and the solid was dried. The 9.3 g similar white powder compound 2b (C11H10ClNO2 ) was obtained with the yield of 96.9% and m.p.138-140 ℃ . 1H-NMR (DMSO-d6/ppm): δ 3.95 (s,3H) , 3.96 (s,3H), 7.35 (s,1H), 7.43 (s,1H), 7.54 (d,1H), 8.59(d,1H). ESI-MS: 225 (M+ +1).
The synthesis of 4-[(4-Amino-3-phenol) oxy]-6,7-dimethoxy-quinoline (2c) The 60 ml of N, N-dimethylformamide, 8.9g (0.05 mol) of 4-amino-3-chlorophenol hydrochloride, 14.5g (0.105 mol) of potassium carbonate and 8.3 g (0.037 mol) compounds 2b were added to the 250 ml of three bottles, the obtained mixture was refluxed about 2 h. Then, the reaction mixture was cooled to the room temperature and the 100 ml of anhydrous ethanol was added. The obtained mixture was stirred about 1 h and filtrated. The filtered product was then dried under the reduced pressure to give the 8.5 g similar white powder compound 2c (C17H15ClN2O3) with the yield of 69.9%. 1H-NMR (DMSO-d6/ppm): δ 3.92 (s,3H), 3.93 (s,3H), 5.41 (s,2H), 6.41 (d,1H), 6.89 (d,1H), 6.98 (dd,1H), 7.19 (d,1H), 7.36 (s,1H), 7.48 (s,1H), 8.43(d,1H). ESI-MS: 331 (M+ +1).
The synthesis of N-{2-chloro-4-[(6,7-dimethoxy-4-quinolyl)oxy]phenyl} -N’- (5-methyl-3- isoxazole-yl) urea (2d) The 100 ml of N,N-dimethylformamide, 5.0g (0.051mol) of 3-amino-5- methylisoxa -zole, 7.98 g (0.051mol) of phenyl chloroformate and 17g (0.051mol) compound 2c were added to the 250 ml of three bottles. The mixture was refluxed about 5 h, cooled to room temperature, added the 100 ml of anhydrous ethanol. The obtained mixture was stirred 1 h and filtrated. The filtered product was slurried in water for washing. The slurry was filtered, and the filtered product was then dried under the reduced pressure to give the 20.0g white crystal compound 2d (C22H19ClN4O5) with the yield of 86.1% and the purity of more than 98.5 %. 1H-NMR (DMSO-d6/ppm): δ 2.37 (s,3H), 3.92 (s,3H), 3.94 (s,3H), 6.50 (s,1H), 6.54 (d,1H), 7.26 (dd,1H), 7.39 (s,1H), 7.48 (s,1H), 7.51 (d,1H), 8.23 (d,1H), 8.49 (d,1H), 8.77 (s,1H), 10.16(s,1H). ESI-MS: 456 (M+ +1).
Conclusions Tivozanib was synthesized through the cyclization, chlorinated, condensation reaction with 2-amino-4,5-dimethoxy acetophenone as the starting material. The total yield was 47.5% and the product purity of more than 98.5 %. The synthetic routs and methods of tivozanib are feasible to industrial production owing to the cheap raw materials, mild reaction conditions, stable technology and high yield.
PATENT
https://patents.google.com/patent/CN102532116B/en
Example

[0035] In 250ml three-neck flask, 80ml of chloroform and 22. 0g (0. 16mol) of anhydrous aluminum chloride at room temperature were successively added dropwise l〇.2g (0. 13mol) acetyl chloride, 13.8g (0. i mole) phthalic dimethyl ether, dropwise, stirred at room temperature until the reaction end point (GLC trace). The reaction solution was poured into 500ml diluted hydrochloric acid, with stirring, the organic phase was separated, the aqueous phase was extracted with chloroform and the combined organic phases were dried over anhydrous sodium sulfate, and concentrated under reduced pressure to give 15. Og of white powder Compound Ia (CltlH12O3), mp 48-52 ° C, 83% yield. HKcnT1): 1673,1585,1515,1418 1H-NMR (CDCl3 / ppm):! S 2. 55 (s, 3H), 3.73 (s, 3H), 3.73 (s, 3H), 6.77 (s, lH) , 7.26 (s, lH), 7.31 (s, lH).
[0036] The two 3 Synthesis of 4-dimethoxy-6-nitroacetophenone (Compound lb) Example
[0037] CN 102532116 B specification 4/6

[0038] In 500ml three-neck flask, was added IOOml formic acid and 18g (0 • lmol) compound la, KTC hereinafter 60ml of concentrated nitric acid was added dropwise, dropwise, warmed to 60-70 ° C, stirred for 30min. The reaction mixture was poured into 500ml ice water bath and stirred, suction filtered to give a pale yellow powder 36.9g Compound lb (CltlH11NO5), mp 135-137 ° C, in 82% yield. 1H-NMR (CDCl3 / ppm): S 2. 50 (s, 3H), 3 97 (s, 3H), 3 99 (s, 3H), 6 76 (s, 1H), 7. 62 (… s, 1H).
Example tri-2-amino-4, Synthesis of 5-dimethoxy acetophenone (Compound Ic), [0039] Embodiment

[0041] In 250ml three-neck flask, 36ml of water was added and 7g (0. 125mol) of reduced iron powder was heated and refluxed for LH, was slowly added 5. 6g (0. 025mol) LB compound, stirred for 3h, filtered off with suction, the filtrate is cooled, to give a yellow powder 7g compound Ic (C10H13NO3), mp 106-108 ° C, in 96% yield.1H-NMR (CDCl3Zppm): S 2. 56 (s, 3H), 3.84 (s, 3H), 3.88 (s, 3H), 6.10 (s, lH), 7.11 (s, lH).
Synthesis of four 6, 7-dimethoxy-4-quinolinone (Compound Id), [0042] Example

[0045] A 33. 7g (0 • 173mol) Compound lc, 150ml methanol and 95. 5g (0 • 69mol) of anhydrous potassium carbonate were added to a 500ml three-necked flask, LH stirred at room temperature, was added dropwise 75. 8g (0. 861mol) ethyl, the reaction incubated 2h. Suction filtration and dried, to give 35. 2g of a white powder compound Id (C11H11NO3), mp 124-125 ° C, yield 81.5%. 1H-NMR (DMSO-Cl6Zppm): 8 3.81 (s, 3H), 3.84 (s, 3H), 5.94 (d, 1H), 7.01 (s, 1H), 7.43 (s, lH), 7.76 (d, lH ).
[0046] Example 4- five-chloro-6, 7-dimethoxy-quinoline (compound Ie) Synthesis of
[0047] CN 102532116 B specification 5/6

[0049] The IOOml toluene, 10. 6g (0 • 52mol) Compound Id and 15g (0 • 103mol) phosphorus trichloride force the opening into a 250ml three-necked flask and heated at reflux for 2h, cooled suction filtration and dried to give 9 . 3g white powder compound Ie (C11H10ClNO2), mp 138-14 (TC, yield 87. 6% .1H-NMR (DMS〇-d6 / ppm): 8 3. 95 (s, 3H), 3.96 ( s, 3H), 7.35 (s, lH), 7.43 (s, lH), 7.54 (d, lH), 8.59 (d, lH).
Six 4 [0050] Example – [(4-amino-phenol) oxy] -6, 7-dimethoxy-quinoline (compound If) Synthesis of

[0053] In 250ml three-neck flask, was added 60ml of N, N- dimethylformamide, 8. 9g (0 • 05mol) 4- amino-3-chlorophenol hydrochloride, 14.5g (0.105mol) of potassium carbonate and (0.037 mol) compound le 8.3g, was heated refluxed for 2h. Cooled to room temperature, IOOml ethanol, stirred, filtered off with suction, and dried to give compound 8. 5g If (C17H15ClN2O3), a yield of 69. gQ / jH-NMlUDMSO-dyppm): S 3.92 (s, 3H), 3.93 ( s, 3H), 5.41 (s, 2H), 6.41 (d, 1H), 6.89 (d, 1H), 6.98 (dd, 1H), 7.19 (d, 1H), 7.36 (s, 1H), 7.48 (s , 1H), 8.43 (d, 1H).
-N’- (5- methyl-3-isobutyl – [0054] Example seven N- {[(6,7- dimethoxy-4-quinolyl) oxy] phenyl} -42- chloro oxazolyl) urea (compound Ig) synthesis of

[0056] The IOOml of N, N- dimethylformamide, 5. Og (0.051mol) of 3-amino-5-methylisoxazole, 7. 98g (0 • 051mol) and phenyl chloroformate 17g (0 • 051mol) If a compound was added to 250ml three-necked flask, the reaction was heated at reflux for 5h, cooled to room temperature, ethanol was added IOOml, stirring, filtration, and dried to give 20. Og compound Ig (C22H19ClN4O5), yield 86 . 1%. 1H-NMR (DMS0-d6 / ppm): S 2.37 (s, 3H), 3.92 (s, 3H), 3.94 (s, 3H), 6.50 (s, lH), 6.54 (d, lH), 7.26 (dd , lH), 7.39 (s, lH), 7.48 (s, lH), 7.51 (d, lH), 8.23 (d, lH), 8.49 (d, lH), 8.77 (s, lH), 10.16 (s, lH).





References
- Tivozanib is currently being evaluated in the pivotal Phase 3 TIVO-3 trial, a randomized, controlled, multi-center, open-label study to compare tivozanib to sorafenib in subjects with refractory advanced RCC. FDA approval is expected in 2018. A Study of Tivozanib (AV-951), an Oral VEGF Receptor Tyrosine Kinase Inhibitor, in the Treatment of Renal Cell Carcinoma, clinicaltrials.gov
- http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/004131/human_med_002146.jsp&mid=WC0b01ac058001d124.
- Campas, C., Bolos, J., Castaner, R (2009). “Tivozanib”. Drugs Fut. 34 (10): 793.
- Aveo Kidney Cancer Drug Shows Success; Shares Up, By John Kell, Dow Jones Newswires[dead link]
- “Phase III Results Lead Aveo and Astellas to Plan Regulatory Submissions for Tivozanib”. 3 Jan 2012.
- “FDA Rejects Renal Cancer Drug Tivozanib”. MedPage Today. June 30, 2013.
- http://meetinglibrary.asco.org/content/165081-176
- http://investor.aveooncology.com/phoenix.zhtml?c=219651&p=irol-newsArticle&ID=2172669
- http://www.eusapharma.com/files/EUSA-Pharma-file-tivozanib-in-EU-March-2016.pdf
- “AVEO Pharma surges 48% on recommendation for European approval of its cancer drug”. Market Watch. June 28, 2017. Retrieved June 28, 2017.
- “AVEO Oncology Announces FOTIVDA® (tivozanib) Approved in the European Union for the Treatment of Advanced Renal Cell Carcinoma” (PDF). AVEO Oncology. August 28, 2017. Retrieved February 9, 2018.
|
|
| Names | |
|---|---|
| IUPAC name
1-{2-Chloro-4-[(6,7-dimethoxyquinolin-4-yl)oxy]phenyl}-3-(5-methylisoxazol-3-yl)urea
|
|
| Other names
AV-951
|
|
| Identifiers | |
|
3D model (JSmol)
|
|
| ChEMBL | |
| ChemSpider | |
| KEGG | |
|
PubChem CID
|
|
| UNII | |
| Properties | |
| C22H19ClN4O5 | |
| Molar mass | 454.87 g·mol−1 |
| Pharmacology | |
| L01XE34 (WHO) | |
|
Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa).
|
|

////////Tivozanib, ema 2017, ASP-4130, AV-951, KRN-951, Kil-8951, Fotivda, Tivopath, orphan drug, ティボザニブ塩酸塩水和物,
CC1=CC(=NO1)NC(=O)NC2=C(C=C(C=C2)OC3=C4C=C(C(=CC4=NC=C3)OC)OC)Cl