

NVP-LXS196
CAS 1874276-76-2
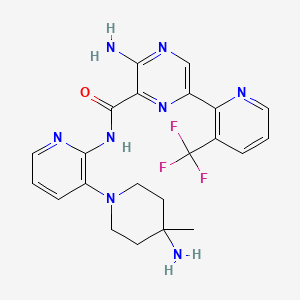
3-amino-N-[3-(4-amino-4-methylpiperidin-1-yl)pyridin-2-yl]-6-[3-(trifluoromethyl)pyridin-2-yl]pyrazine-2-carboxamide
- 3-Amino-N-[3-(4-amino-4-methylpiperidin-1-yl)pyridin-2-yl]-6-[3-(trifluoromethyl)pyridin-2-yl]pyrazine-2-carboxamide
| Molecular Formula: | C22H23F3N8O |
|---|---|
| Molecular Weight: | 472.476 g/mol |
| Inventors | Michael Joseph Luzzio, Julien Papillon,Michael Scott Visser |
| Applicant | Novartis Ag |



SYNTHESIS

Uveal melanoma (UM) is the most common cancer of the eye in adults (Singh AD. et al., Ophthalmology. 201 1 ; 1 18: 1881-5). Most UM patients develop metastases for which no curative treatment has been identified so far. The majority of UM tumors have mutations in the genes GNAQ (guanine nucleotide-binding protein G(q) subunit alpha) and GNA11 (guanine nucleotide-binding protein G(q) subunit 1 1 ), which encode for small GTPases (Harbour JW. Pigment Cell Melanoma Res. 2012;25: 171-81). Both of these mutations lead to activation of the protein kinase C (PKC) pathway. The up-regulation of PKC pathway has downstream effects which leads to constitutive activation of the mitogen-activated protein kinase (MAPK) signaling pathway that has been implicated in causing uncontrolled cell growth in a number of proliferative diseases.
Whilst anti-proliferative effects have been observed with certain PKC pathway inhibitors, no sustained MAPK pathway inhibition has been observed. Thus far, PKC inhibitors (PKCi) have had limited efficacy as single agents in patients (Mochly-Rosen D et al., Nat Rev Drug Discov. 2012 Dec;1 1 (12):937-57). Moreover, inhibition of PKC alone was unable to trigger cell death in vitro and/or tumor regression in vivo (Chen X, et al., Oncogene. 2014;33:4724-34).
The protein p53 is a transcription factor that controls the expression of a multitude of target genes involved in DNA damage repair, apoptosis and cell cycle arrest, which are all important phenomena counteracting the malignant growth of tumors. The TP53 gene is one of the most frequently mutated genes in human cancers, with approximately half of all cancers having inactivated p53. Furthermore, in cancers with a non-mutated TP53 gene, typically the p53 is functionally inactivated at the protein level. One of the mechanisms of p53 inactivation is through its interaction with human homolog of MDM2 (Mouse double minute 2) protein. MDM2 protein functions both as an E3 ubiquitin ligase, that leads to proteasomal degradation of p53, and an inhibitor of p53 transcriptional activation. Therefore, MDM2 is an important negative regulator of the p53 tumor suppressor. MDM2 inhibitors can prevent interaction between MDM2 and p53 and thus allow the p53 protein to exert its effector functions. Whilst TP53 mutations are not common in UM, there are reports suggesting the p53 pathway is inactivated by either high expression of MDM2 protein or downregulation of the PERP protein in UM patients.
A combination of an MDM2 inhibitor (Nutlin-3) has been shown to act synergistically with reactivation of p53 and induction of tumor cell apoptosis (RITA) and Topotecan to cause growth inhibition in UM cell lines (De Lange J. et al., Oncogene. 2012;31 :1 105-16). However, Nutlin-3 and Topotecan delayed in vivo tumor growth only in a limited manner.
PATENT
PATENT
Example 9: 3-amino-N-(3-(4-amino-4-methylpiperidin-l-yl)pyridin-2-yl)- 6-(3-(trifluoromethyl)pyridin-2-yl)pyrazine-2-carboxamide

Synthesis of tert-butyl (4-meth l-l-(2-nitropyridin-3-yl)piperidin-4-yl)carbamate

To a solution of 3-fluoro-2-nitropyridine (11.2 g, 81 mmol) in dioxane (200 mL) was added tert-butyl (4-methylpiperidin-4-yl)carbamate (26 g, 121 mmol). Huenig’s Base (28.3 mL,
162 mmol) was added and the mixture was heated to 85 °C for 18 hrs. The reaction was cooled to RT and concentrated to give a brown solid. The solids were washed with 200 mL of 4: 1 heptane:EtOAc. Slurry was concentrated to half volume and filtered to collect (26.2 g, 78 mmol, 96%) brown solid. LC-MS (Acidic Method): ret.time= 1.46 min, M+H = 337.4
Step 2: Synthesis of tert-butyl (4-meth l-l-(2-nitropyridin-3-yl)piperidin-4-yl)carbamate

To a solution of tert-butyl (4-methyl-l-(2-nitropyridin-3-yl)piperidin-4-yl)carbamate (11.6 g, 37.2 mmol) in ethyl acetate (200 mL). 10% Pd-C (3.48 g) was added and stirred under H2 balloon pressure at RT for 4h. A small amount of MgS04 was added to the reaction and then the reaction mixture was filtered through a pad of cellite, then washed with ethyl acetate (100 mL) and the filtrate was concentrated to afford a brown solid (8.54 g, 27.9 mmol, 85%). LC-MS (Acidic Method): ret.time= 0.91 min, M+H = 307.4.
Step 3: Synthesis of tert-butyl (l-(2-(3-amino-6-(3-(trifluoromethyl)pyridin-2-yl)pyrazine-2-carboxamido)pyridin-3-yl)-4-meth lpiperidin-4-yl)carbamate

To a solution of 3-amino-6-(3-(trifluoromethyl)pyridin-2-yl)pyrazine-2-carboxylic acid in dimethyl formamide (125 mL) was added ((lH-benzo[d][l,2,3]triazol-l- yl) oxy)
tris(dimethylamino) phosphonium hexafluorophosphate(V) (1.8g, 4.24 mmol) and 4-methylmorpholine (1 mL, 9.79 mmol). Reaction stirred at RT for 40 minutes. Tert-butyl (l-(2-aminopyridin-3-yl)-4-methylpiperidin-4-yl) carbamate in dimethylformamide (25 mL) was added and reaction stirred for 16 hrs at RT. The reaction mixture was diluted with EtOAc and was washed with NaHC03(aq) (3 x 200mL) and brine (lx 200mL). The organic phase was dried with Na2S04, filtered and concentrated. The crude product was taken up in acetonitrile (30 mL) and mixture was allowed to stand at RT for a period of time. Yellow solid collected by filtration (1.39g, 74%). LC-MS (Acidic Method): ret.time= 1.13 mm, M+H = 573.3.
Step 4: Synthesis of 3-amino-N-(3-(4-amino-4-methylpiperidin-l-yl)pyridin-2-yl)-6-(3-(trifluoromethyl)pyridin-2-yl)pyrazine-2-carboxamide
A solution of tert-butyl (l-(2-(3-amino-6-(3-(trifluoromethyl)pyridin-2-yl)pyrazine-2-carboxamido)pyridin-3-yl)-4-methylpiperidin-4-yl)carbamate (l -39g, 2.06 mmol) in dichloromethane (10 mL) was cooled to 0 °C. 2,2,2-trifluoroacetic acid (2.4 ml, 31 mmol) was added dropwise to the solution. The mixture was allowed to warm to 22 °C and stirred for 4 hrs. Reaction mixture was concentrated to remove DCM and excess TFA. A red oil was produced, which was taken up in 100 mL CHCI3/IPA 3: 1 and saturated aq. NaHCC was added to neutralize the solution. The mixture was then stirred at 22°C for 16 hrs. The mixture transfered to separatory funnel and aqueous layers were washed with CHCI3/IPA 3: 1 (3X 100 mL). Combined organic phases were dried with Na2S04, filtered and concentrated to afford a yellow solid. The crude product was recrystallized from acetonitrile. A yellow solid was collected by filtration (0.82g, 83%). LC-MS (Acidic Method ): ret.time= 0.75 mm, M+H = 473.2. 1H NMR (400 MHz, Methanol-^) δ 8.92 (dd, J = 5.1, 1.4 Hz, 1H), 8.68 (s, 1H), 8.47 – 8.27 (m, 1H), 8.12 (dd, J = 4.9, 1.6 Hz, 1H), 7.83 – 7.50 (m, 2H), 7.18 (dd, J = 7.9, 4.9 Hz, 1H), 3.02 – 2.65 (m, 4H), 1.54 – 1.24 (m, 4H), 0.74 (s, 3H).
REFERENCES
Visser, M.; Papillon, J.; Fan, J.; et al.
NVP-LXS196, a novel PKC inhibitor for the treatment of uveal melanoma
253rd Am Chem Soc (ACS) Natl Meet (April 2-6, San Francisco) 2017, Abst MEDI 366
| Patent ID | Patent Title | Submitted Date | Granted Date |
|---|---|---|---|
| US2016046605 | PROTEIN KINASE C INHIBITORS AND METHODS OF THEIR USE | 2015-08-05 | 2016-02-18 |
//////////////NVP-LXS196, NVP-LXS 196, 1874276-76-2, Michael Joseph Luzzio, Julien Papillon,Michael Scott Visser, NOVARTIS, PKC inhibitor, uveal melanoma
FC(F)(F)c1cccnc1c2cnc(N)c(n2)C(=O)Nc3ncccc3N4CCC(C)(N)CC4
http://sanfrancisco2017.acs.org/i/803418-253rd-american-chemical-society-national-meeting-expo/289
Michael Visser of @Novartis talking now in 1st time disclosures about a PKC inhibitor to treat uveal melanoma #ACSSanFran
Filed under: Uncategorized Tagged: 1874276-76-2, Julien Papillon, Michael Joseph Luzzio, Michael Scott Visser, novartis, NVP-LXS 196, NVP-LXS196, PKC inhibitor, uveal melanoma







