
 Aurobindo Pharma MD and CEO N. Govindarajan at a company research centre. “It [the transition] is purely driven by the need to get more into areas where there is scope for better profit margins,
Aurobindo Pharma MD and CEO N. Govindarajan at a company research centre. “It [the transition] is purely driven by the need to get more into areas where there is scope for better profit margins,
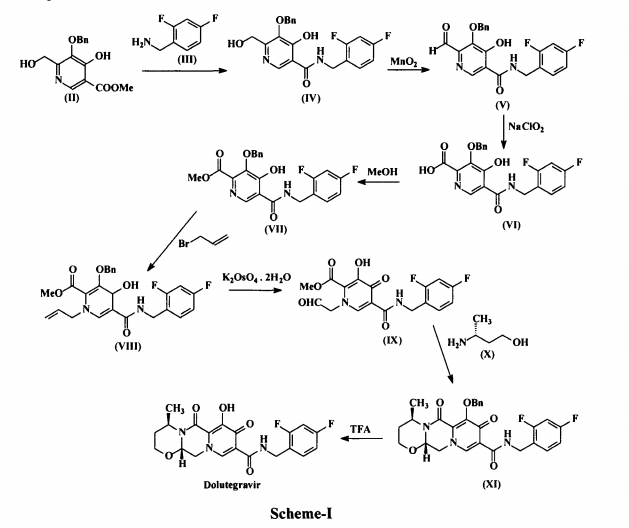
Dolutegravir (I) is chemically known as (4/?,12aS)-N-[(2,4-difluorophenyl)methyl]-3,4,6,8,12,12a-hexahydro-7-hydroxy-4-methyl-6,8-dioxo-2//-pyrido[r,2′:4,5]pyrazino[2,l-b][l,3]oxazine-9-carboxamide. Dolutegravir is a human immunodeficiency virus type 1 (HIV-1) integrase strand transfer inhibitor (INSTI) indicated in combination with other antiretroviral agents for the treatment of HIV-1 infection. Dolutegravir is being marketed under the trade name Tivicay®. US 8,129,385 disclosed Dolutegravir or its pharmaceutically acceptable salts thereof. US ‘385 also discloses a process for the preparation of Dolutegravir (I). The process involves the condensation of 5-benzyloxy-4-hydroxy-6-hydroxymethyl nicotinic acid (II) with 2,4-difluorobenzylamine (III) to produce 5-benzyloxy-N-(2,4-difluorobenzyl)-4-hydroxy-6-hydroxymethyl nicotinic acid amide (IV), which is further under goes oxidation using manganese dioxide (Mn02) to produce 5-benzyloxy-N-(2,4-difluorobenzyl)-6-formyl-4-hydroxy-nicotinic acid amide (V). This amide compound (V) is reacted with sodium chlorite (NaClCh) to produce 3-benzyloxy-5-(2,4-difluorobenzylcarbamoyl)-4- hydroxy-pyridine-2-carboxylic acid (VI), which is further treated with methanol (MeOH) to produce 3-benzyloxy-5-(2,4-difluorobenzyl)-4-hydroxy-pyridine-2-carboxylic acid methyl ester (VII).

The methyl ester compound (VII) is reacted with 3-bromopropene to produce l-allyl-3-benzyloxy-5-(2,4-difluorobenzyl)-4-oxo-l,4-dihydro-pyridine-2- carboxylic acid methyl ester (VIII), which is further reacted with potassium osmate dihydrate (K2OSO4.2H2O) to produce 3-benzyloxy-5-(2,4-difluorobenzylcarbamoyl)-4-oxo-l-(2-oxo-ethyl)-l,4-dihydropyridine-2-carboxylic acid methyl ester (IX). The compound (IX) is reacted with (R)-3-amino-l-butanol (X) to produce benzyloxy Dolutegravir (XI), which is deprotected by treating with TFA to produce Dolutegravir (I). The process is as shown in scheme-I below:
The major disadvantage with the above prior-art process is that it involves large no of steps and tedious work-up procedures to isolate the required product. This results a longer period of time cycle is required to produce Dolutegravir (I), which in turn renders the process more costly and less eco friendly. Further the above processes are low yielding and with less purity. US 8,217,034 discloses variant process for the preparation of Dolutegravir.
This process involves the reaction of methyl l-(2,2-dihydroxyethyl)-4-oxo-3-[(phenylmethyl)oxy]-l,4-dihydro-2-pyridine carboxylate (XII) with (R)-3-amino-l-butanol (X) to produce (4R, 12o5)-4-methyl-7-[(phenylmethyl)oxy]-3,4,12,12a-tetrahydro-2//-pyrido[ 1 \2′,4,5] pyrazino[2,l-b][l,3]oxazine-6,8-dione (XIII), which is further undergoes bromination using NBS to produce (4R,12aS)-9-bromo-4-methyl-7-[(phenylmethyl)oxy]-3,4,12,12a-tetrahydro-2H-pyrido[r,2′:4,5]pyrazino[2,l-b][l,3]oxazine-6,8-dione (XIV). The bromo Compound (XIV) is condensed with 2,4-difluorobenzylamine (III) in the presence of Tetrakis(triphenylphosphine)palladium (Pd(PPh3)4) to produce benzyloxy Dolutegravir (XI), which is hydrogenated in the presence of Pd/C to produce Dolutegravir (I). The process is as shown in Scheme-II below:

The major disadvantage with the above prior art process of preparing Dolutegravir is the use of expensive reagent tetrakis(triphenylphosphine)palladium (Pd(PPh3)4> in coupling step. Use of this reagent on industrial scale is not preferred, which makes the process more expensive. WO 2011/119566 discloses another variant process for the preparation of Dolutegravir.
This process involves the reaction of l-(2,2-dimethoxyethyl)-5-methoxy-6-(methoxycarbonyl)-4-oxo-l,4-dihydropyridine-3-carboxylic acid (XV) with acetic acid in presence of methane sulfonic acid to produce 5-methoxy-6-(methoxycarbonyl)-4-oxo-l-(2-oxoethyl)-l,4-dihydropyridine-3-carboxylic acid (XVI), which is further condensed with (R)-3-amino-l-butanol (X) to produce (4R,12aS)-7-methoxy-4-methyl-6,8-dioxo-3,4,6,8,12,12a-hexahydro-2//-pyrido[ 1 ‘,2’:4,5]pyrazino[2,1 -b] [ 1,3]-oxazine-9-carboxylic acid (XVII). This acid Compound XVII is acylated with 2,4-difluorobenzylamine (III) in the presence of carbonyldiimidazole (CDI) to produce methoxy Dolutegravir (XVIII), which is demethylated in the presence of lithium bromide (LiBr) to produce Dolutegravir (I).
The process is as shown in Scheme-3 below:

The major disadvantage of the above prior art process of preparing Dolutegravir is the use of expensive and highly moisture sensitive reagent, 1,1-carbonyldiimidazole (CDI), during acylation. Use of this reagent on industrial scale is not preferred due to anhydrous conditions required in the process. However, there is always a need for alternative preparative routes, which for example, involve fewer steps, use reagents that are less expensive and/or easier to handle, consume smaller amounts of reagents, provide a higher yield of product, have smaller and/or more eco-friendly waste products, and/or provide a product of higher purity. Hence, there is a need to develop cost effective and commercially viable process for the preparation of Dolutegravir of formula (I). The present invention is related to a process for the preparation of pure Dolutegravir of formula (I), wherein optically active acid addition salt of (R)-3-amino-l-butanol (X) is directly condensed with 5-methoxy-6-(methoxycarbonyl)-4-oxo-l-(2-oxoethyl)-l,4-dihydropyridine-3-carboxylic acid (XVI) instead of condensing with free base of (R)-3-amino-1-butanol (X). The present invention is also related to a process for the preparation of pure Dolutegravir of formula (I), wherein, inexpensive and easily handling condensing reagents in the condensation of (4R, 12aS)-7-methoxy-4-methyl-6,8-dioxo-3,4,6,8,12,12a-hexahydro-2//-pyrido[l’,2′:4,5]pyrazino [2,l-b][l,3]oxazine-9-carboxylic acid (XVII) with 2,4-difluorobenzylamine (III).


AN IMPROVED PROCESS FOR THE PREPARATION OF DOLUTEGRAVIR
| APPLICATION NUMBER | 1361/CHE/2013 |
| APPLICANT NAME | AUROBINDO PHARMA LTD |
| DATE OF FILING | 27/03/2013 |
| PUBLICATION DATE (U/S 11A) | 16/01/2015 |
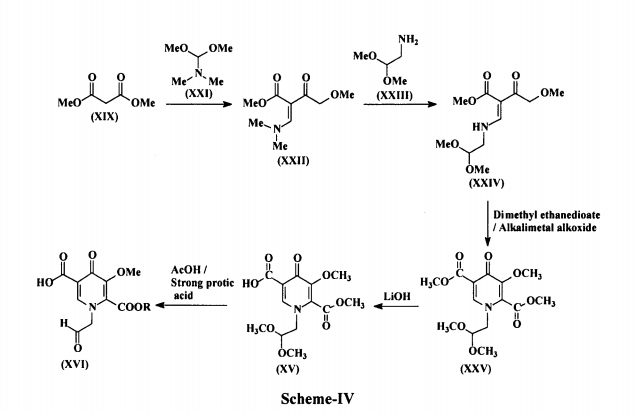
In another embodiment, 5-methoxy-6-(methoxycarbonyl)-4-oxo-l-(2-oxoethyl)-l,4- dihydropyridine-3-carboxylic acid (XVI) used in the present invention is prepared by reacting 4-methoxyacetoacetate (XIX) with N,N-dimethyl-l,l- bis(methyloxy)methanamine (DMF-DMA) (XX) to produce methyl-2- (dimethylaminomethylene)-4-methoxy-3-oxo-butanoate(methyl-3-(dimethylamino)-2 [(methyloxy)acetyl]-2-propenoate) (XXI), which is reacted with aminoacetaldehyde dimethyl acetal (XXII) to produce methyl-2-(2,2-dimethoxyethylaminomethylene)-4-methoxy-3-oxo-butanoate(methyl-3-{[2,2-bis(methyloxy)ethyl]amino}-2-[(methyloxy) acetyl]-2-propenoate) (XXIII).
The compound (XXIII) is contacted with dimethyl ethanedioate in presence of alkali metal alkoxide to produce dimethyl-1-(2,2-dimethoxyethyl)-3-methoxy-4-oxo-l ,4-dihydropyridine-2,5-dicarboxylate (XXIV), which is selectively hydrolyzed with a base to produce l-[2,2-bis(methyloxy)ethyl]-5-(methyloxy)-6-[(methyloxy)carbonyl]-4-oxo-l ,4-dihydro-3-pyridinecarboxylic acid (XV). The compound (XV) is treated with a catalytic amount of a strong protic acid in the presence of acetic acid in an organic solvent to produce a reaction mixture containing 5- methoxy-6-(methoxycarbonyl)-4-oxo-l-(2-oxoethyl)-l,4-dihydropyridine-3-carboxylic acid (XVI), The process is as shown in Scheme-IV below:
The following examples illustrate the nature of the invention and are provided for illustrative purposes only and should not be construed to limit the scope of the invention.
Example-1:
EXAMPLES: Example-1: Process for the preparation of Dolutegravir
Step-i: Preparation of (/?)-3-amino-l-butanol tartarate salt: D-(+) Tartaric acid (12.7 g, 0.085 mol) was added in to a solution of (i?,5)-3-amino-l-butnaol (7.5 g, 0.084 mol) in methanol (100 ml) at 40 °C. The reaction mixture was stirred for about 1 hour at 35-40 °C and the reaction mass was cooled to 0-5°C and maintained for 30-40 minutes. The obtained solid was filtered and washed with chilled methanol (10 ml) at 0-5 °C. The solid was dried to get (i?)-3-amino-l-butanol tartarate salt (8.0 g, 40%).
Step-ii: Preparation of (4rt,12a£)-7-methoxy-4-methyl-6,8-dioxo-3,4,6,8,12,12a-hexahydro-2H-pyrido[l’,2′;4,5]pyrazino[2,l-b][l,3]oxazine-9-carboxylic acid (XVII): l-[2,2-Bis(methyloxy)ethyl]-5-(methyloxy)-6-[(methyloxy)carbonyl]-4-oxo-l,4-dihydro-3-pyridinecarboxylic acid (XV) (lOOg; 0.3175 moles) was suspended in acetonitrile (800 ml) and heated to 80-82°C. A mixture of acetic acid (95.25 g), methanesulfonic acid (9.14 g; 0.09525 moles) and acetonitrile (200 ml) were added to the slurry at 80-82°C. The reaction mass was continued at 80-82°C to complete the reaction. After completion of the reaction, anhydrous sodium acetate (65 g) and (/?)-3-amino-l-butanol tartrate salt (79.68g; 0.3334 moles) were added at 20-25°C and stirred at 60-65°C to complete the reaction. The reaction mass was concentrated and acidified with IN aqueous hydrochloric acid (750 ml) and extracted with methylene chloride (1500 ml) at ice cold temperature. The organic layer was separated, concentrated, treated with hot methanol (350 ml) for 2 h, filtered, washed with methanol and dried to yield (4R,12aS)-7-methoxy-4-methyl-6,8-dioxo-3,4,6,8,12,12a-hexahydro-2H-pyrido[ 1′ ,2′ :4,5]pyrazino[2,1 -b] [ 1,3]oxazine-9-carboxylic acid (XVII) (72 g; HPLC purity: 99.07%).
Step-iii: Process for the preparation of Dolutegravir (I). Method A: Triethylamine (3.61 g; 0.0357 moles) was added to the suspension of (4R,12aS)-7- methoxy-4-methyl-6,8-dioxo-3,4,6,8,12,12a-hexahydro-2H-pyrido[ 1′ ,2′ :4,5]pyrazino[2,1 – b][l,3]oxazine-9-carboxylic acid (XVII) (10 g; 0.0325 moles) in methylene chloride (50 ml), and cooled to 10-15°C. Pivaloyl chloride (4.3 g; 0.0357 moles) was added to the reaction mass, and stirred at 10-15°C for 1 h. Thereafter, 2,4-difiuorobenzylamine (5.58 g; 0.0389 moles) was added at 10-15°C and then warmed to 20-25°C to complete the reaction. After completion of the reaction, IN aqueous hydrochloric acid (20 ml) was added, organic layer was separated, washed with 5% w/w aqueous sodium bicarbonate solution (10 ml) followed by 15% w/w aqueous sodium chloride solution (10 ml) and concentrated. To the concentrated mass, acetonitrile (100 ml) and Lithium bromide (5.08 g; 0.0584 moles) were added and heated to 65-70°C for 3 h to complete the reaction. After completion of the reaction, the reaction mass was acidified with 5N aqueous hydrochloric acid (40 ml), concentrated to about 50 ml and DM water was added to crystallize the product at 20-25°C. The slurry was stirred for 2 h, filtered, washed with DM water and dried to yield (4R,12aS)-N-(2,4-difluorobenzyl)-7-hydroxy-4-methyl-6,8-dioxo-3,4,6,8,12,12a,-hexahydro-2H-pyrido[ 1′ ,2′ :4,5]pyrazino[2,1 -b] [ 1,3]oxazine-9-carboxamide (I) (11.5 g, HPLC purity: 99.63%).
Method B: Isobutyl chloroformate (4.65 gm, 0.03404 moles) in methylene chloride (10 ml) was added to the solution of N-methylmorpholine (3.45 gm, 0.03410 moles) and (4R,12aS)-7-methoxy-4-methyl-6,8-dioxo-3,4,6,8,12,12a-hexahydro-2H-pyrido[ 1′ ,2′ :4,5]pyrazino-[2,1 -b][l,3]oxazine-9-carboxy!ic acid (XVII) (10.0 gm, 0.03245 moles) in methylene chloride (60 ml) at -10 to 0°C in about 1 h. 2,4-Difloro benzyl amine (4.88 gm, 0.03409 moles) in methylene chloride (10 ml) was added to the cold reaction mass, and stirred at 20-30°C for completion of reaction. After completion of reaction, the reaction mass was washed with 5%w/w aqueous sodium bicarbonate solution (20 ml), IN hydrochloric acid (20 ml), DM water (20 ml) and concentrated. Acetonitrile (120 ml) and lithium bromide (4.8 gm, 0.05516 moles) were added to the concentrated mass, and stirred at 70-80°C for 3 h to complete the reaction. After completion of reaction, the reaction mass was acidified with 5N aqueous hydrochloric acid (40 ml) and concentrated to about 50 ml. DM Water (100 ml) was added to the concentrated reaction mass and stirred for 2 h at 25-30°C to crystallize the product. The product was filtered, washed with DM Water (50 ml) and dried to yield Dolutegravir (I) (10.7 gm, HPLC purity: 99.60%).
Example-2: Process for the preparation of Dolutegravir (I) (4R, 12aS)-N-(2,4-difluorobenzyl)-7-methoxy-4-methyl-6,8-dioxo-3,4,6,8,12,12a,-hexahydro-2H-pyrido[r,2′:4,5]pyrazino[2,l-b][l,3]oxazine-9-carboxamide (XVIII) (2 g, 0.0046 moles) was suspended in isopropyl alcohol (20 ml) and lithium bromide (0.8 g, 0.00924 moles) was added and stirred at 70-80°C for 15 h to complete the reaction. After completion of reaction the reaction mass was acidified with 5N aqueous hydrochloric acid (5 ml) and concentrated. DM Water (20 ml) was added to the concentrated mass and stirred at 25-30°C to crystallize the product. The product was filtered, washed with DM Water and dried to yield Dolutegravir (I) (1.5 g, HPLC purity: 97.93%).


/////
Filed under: Uncategorized Tagged: AUROBINDO PHARMA, dolutegravir, INDIA







