Image may be NSFW.
Clik here to view. Image may be NSFW.
Image may be NSFW.
Clik here to view.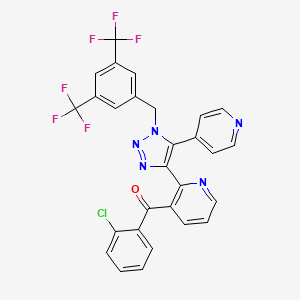
Tradipitant
VLY-686, LY686017
- Molecular Formula C28H16ClF6N5O
- Average mass 587.903 Da
PHASE 2, Gastroparesis; Pruritus
pyridine-containing NK-1 receptor antagonist ie tradipitant, useful for treating anxiety, pruritus and alcoholism.
Vanda Pharmaceuticals, under license from Eli Lilly, was developing tradipitant, a NK1 antagonist, for treating anxiety disorder, pruritus and alcohol dependence. The company was also investigating the drug for treating gastroparesis. In February 2017, tradipitant was reported to be in phase 2 clinical development for treating anxiety and pruritus.
- Originator Eli Lilly
- Developer Eli Lilly; National Institute on Alcohol Abuse and Alcoholism; Vanda Pharmaceuticals
- Class Antipruritics; Anxiolytics; Chlorobenzenes; Pyridines; Small molecules; Triazoles
- Mechanism of Action Neurokinin 1 receptor antagonists; Substance P inhibitors
Highest Development Phases
- Phase II Gastroparesis; Pruritus
- Discontinued Alcoholism; Social phobia
- The drug had been in phase II clinical trials at Lilly and the National Institute on Alcohol Abuse and Alcoholism for the treatment of alcoholism; however, no recent development has been reported for this research.
- A phase II clinical trial for the treatment of social phobia has been completed by Lilly.
PATENT WO 2003091226
Image may be NSFW.
Clik here to view.
Image may be NSFW.
Clik here to view.
SYNTHESIS
Image may be NSFW.
Clik here to view.
Condensation of 2-chloropyridine with thiophenol in the presence of K2CO3 in DMF at 110ºC yields sulfide intermediate,
which is then oxidized by means of NaOCl in AcOH to give 2-(benzenesulfonyl)pyridine.
This is treated with (iPr)2NH and n-BuLi in THF at -60 to -70°C and subsequently couples with 2-chlorobenzaldehyde in THF at -60 to -70°C to furnish (2-(phenylsulfonyl)pyridin-3-yl)-(2-chlorophenyl)methanone.
Ketone couples with the enolate of 4-acetylpyridine (formed by treating 4-acetylpyridine (VII) with t-BuOK in DMSO) in the presence of LiOH in DMSO and subsequently is treated with PhCOOH in iPrOAc to give rise to pyridine benzoate derivative.
This finally couples with 1-azidomethyl-3,5-bistrifluoromethylbenzene (obtained by treating 3,5-bis(trifluoromethyl)benzylchloride with NaN3 ini DMSO) in the presence of K2CO3 in t-BuOH to afford the title compound Tradipitant.
Tradipitant (VLY-686 or LY686017) is an experimental drug that is a neurokinin 1 antagonist. It works by blocking substance P, a small signaling molecule. Originally, this compound was owned by Eli Lilly and named LY686017. VLY-686 was purchased by Vanda Pharmaceuticals from Eli Lilly and Company in 2012.[1] Vanda Pharmaceuticals is a U.S. pharmaceutical company that as of November 2015 only has 3 drugs in their product pipeline: tasimelteon, VLY-686, and iloperidone.[2]
Tachykinins are a family of peptides that are widely distributed in both the central and peripheral nervous systems. These peptides exert a number of biological effects through actions at tachykinin receptors. To date, three such receptors have been characterized, including the NK-1 , NK-2, and NK-3 subtypes of tachykinin receptor.
The role of the NK-1 receptor subtype in numerous disorders of the central nervous system and the periphery has been thoroughly demonstrated in the art. For instance, NK-1 receptors are believed to play a role in depression, anxiety, and central regulation of various autonomic, as well as cardiovascular and respiratory functions. NK- 1 receptors in the spinal cord are believed to play a role in pain transmission, especially the pain associated with migraine and arthritis. In the periphery, NK-1 receptor activation has been implicated in numerous disorders, including various inflammatory disorders, asthma, and disorders of the gastrointestinal and genitourinary tract.
There is an increasingly wide recognition that selective NK-1 receptor antagonists would prove useful in the treatment of many diseases of the central nervous system and the periphery. While many of these disorders are being treated by new medicines, there are still many shortcomings associated with existing treatments. For example, the newest class of anti-depressants, selective serotonin reuptake inhibitors (SSRIs), are increasingly prescribed for the treatment of depression; however, SSRIs have numerous side effects, including nausea, insomnia, anxiety, and sexual dysfunction. This could significantly affect patient compliance rate. As another example, current treatments for chemotherapy- induced nausea and emesis, such as the 5-HT3receptor antagonists, are ineffective in managing delayed emesis. The development of NK-1 receptor antagonists will therefore greatly enhance the ability to treat such disorders more effectively. Thus, the present invention provides a class of potent, non-peptide NK-1 receptor antagonists, compositions comprising these compounds, and methods of using the compounds.
Indications
Pruritus
It is being investigated by Vanda Pharmaceuticals for chronic pruritus (itchiness) in atopic dermatitis. In March 2015, Vanda announced positive results from a Phase II proof of concept study.[3] A proof of concept study is done in early stage clinical trials after there have been promising preclinical results. It provides preliminary evidence that the drug is active in humans and has some efficacy.[4]
Alcoholism
VLY-686 reduced alcohol craving in recently detoxified alcoholic patients as measured by the Alcohol Urge Questionnaire.[5] In a placebo controlled clinical trial of recently detoxified alcoholic patients, VLY-686 significantly reduced alcohol craving as measured by the Alcohol Urge Questionnaire. It also reduced the cortisol increase seen after a stress test compared to placebo. The dose given was 50 mg per day.
Social anxiety disorder
In a 12-week randomized trial of LY68017 in 189 patients with social anxiety disorder, 50 mg of LY68017 did not provide any statistically significant improvement over placebo.[6]
PATENT
WO03091226,
https://www.google.com/patents/WO2003091226A1?cl=en
PATENT
The compound {2-[l-(3,5-bis-trifluoromethyl-benzyl)-5-pyridin-4-yl-lH-[l,2,3]triazol-4-yl]- pyridin-3-yl}-(2-chlorophenyl)-methanone, depicted below as the compound of Formula I, was first described in PCT published application WO2003/091226.

(I)
Because the compound of Formula I is an antagonist of the NK-I subtype of tachykinin receptor, it is useful for the treatment of disorders associated with an excess of tachykinins. Such disorders include depression, including major depressive disorder; anxiety, including generalized anxiety disorder, panic disorder, obsessive compulsive disorder, and social phobia or social anxiety disorder; schizophrenia and other psychotic disorders, including bipolar disorder; neurodegenerative disorders such as dementia, including senile dementia of the Alzheimer’s type or Alzheimer’s disease; disorders of bladder function such as bladder detrusor hyper-reflexia and incontinence, including urge incontinence; emesis, including chemotherapy-induced nausea and acute or delayed emesis; pain or nociception; disorders associated with blood pressure, such as hypertension; disorders of blood flow caused by vasodilation and vasospastic diseases, such as angina, migraine, and Reynaud’s disease; hot flushes; acute and chronic obstructive airway diseases such as adult respiratory distress syndrome, bronchopneumonia, bronchospasm, chronic bronchitis, drivercough, and asthma; inflammatory diseases such as inflammatory bowel disease; gastrointestinal disorders or diseases associated with the neuronal control of viscera such as ulcerative colitis, Crohn’s disease, functional dyspepsia, and irritable bowel syndrome (including constipation-predominant, diarrhea- -?-
predominant, and mixed irritable bowel syndrome); and cutaneous diseases such as contact dermatitis, atopic dermatitis, urticaria, and other eczematoid dermatitis.
In PCT published application, WO2005/042515, novel crystalline forms of the compound of Formula I, identified as Form IV and Form V, are identified. Also described in WO2005/042515 is a process for preparation of the compound of Formula I, comprising reacting (2-chlorophenyl)-[2-(2- hydroxy-2-pyridin-4-yl-vinyl)pyridin-3-yl]methanone or a phosphate salt thereof with l-azidomethyl-3,5- bistrifluoromethylbenzene in the presence of a suitable base and a solvent. Use of this procedure results in several shortcomings for synthesis on a commercial scale. For example, use of the solvent DMSO, with (2- chlorophenyl)-[2-(2-hydroxy-2-pyridin-4-yl-vinyl)pyridin-3-yl]methanone phosphate, requires a complex work-up that has a propensity to emulsify. This process also requires extraction with CH2CI2, the use of which is discouraged due to its potential as an occupational carcinogen, as well as the use of MgSC>4 and acid-washed carbon, which can generate large volumes of waste on a commercial scale. Conducting the reaction with (2-chlorophenyl)-[2-(2-hydroxy-2-pyridin-4-yl-vinyl)pyridin-3-yl]methanone in isopropyl alcohol, as also described in WO2005/042515, is also undesirable due to the need to incorporate a free base step. Furthermore, variable levels of residual l-azidomethyl-3,5-bistrifluoromethylbenzene, a known mutagen, are obtained from use of the procedures described in WO2005/042515.
An improved process for preparing the compound of Formula I would control the level of 1- azidomethyl-3,5-bistrifluoromethylbenzene impurity, and improve the yield. We have discovered that use of the novel salt, (2-chlorophenyl)-[2-(2-hydroxy-2-pyridin-4-yl-vinyl)pyridin-3-yl]methanone benzoate, as well as use of tert-butanol as the reaction solvent, improves reaction times and final yield, and decreases impurities in the final product. In addition, a novel process for the preparation of (2-chlorophenyl)- [2-(2- hydroxy-2-pyridin-4-yl-vinyl)pyridin-3-yl]methanone benzoate, in which a pre-formed enolate of 4-acetyl pyridine is added to (2-phenylsulfonyl-pyridine-3-yl)-(2-chlorophenyl)methanone, results in an overall improved yield and improved purity, and is useful on a commercial scale.
EXAMPLES
Example 1 {2-[l-(3,5-bistrifluoromethylbenzyl)-5-pyridin-4-yl-lH-[l,2,3]triazol-4-yl]-pyridin-3-yl}-(2-chlorophenyl)- methanone (Form IV)

Suspend (2-chlorophenyl)-[2-(2-hydroxy-2-pyridin-4-yl-vinyl)pyridin-3-yl] methanone benzoate (204.7 g; 1.04 equiv; 445 mmoles) in t-butanol (614 mL) and treat the slurry with potassium carbonate (124.2 g; 898.6 mmoles). Heat to 7O0C with mechanical stirring for 1 hour. Add l-azidomethyl-3,5- bistrifluoromethylbenzene (115.6 g; 1.00 equiv; 429.4 mmoles) in a single portion, then heat the mixture to reflux. A circulating bath is used to maintain a condenser temperature of 3O0C. After 18 hours at reflux, HPLC reveals that the reaction is complete (<2% l-azidomethyl-3,5-bistrifluoromethylbenzene remaining). The mixture is cooled to 7O0C, isopropanol (818 mL) is added, then the mixture is stirred at 7O0C for 1 hour. The mixture is filtered, and the waste filter cake is rinsed with isopropanol (409 mL). The combined filtrate and washes are transferred to a reactor, and the mechanically stirred contents are heated to 7O0C. To the dark purple solution, water (1.84 L) is added slowly over 35 minutes. The solution is cooled to 6O0C, then stirred for 1 hour, during which time a thin precipitate forms. The mixture is slowly cooled to RT, then the solid is filtered, washed with 1 : 1 isopropanol/water (614 mL), subsequently washed with isopropanol (410 mL), then dried in vacuo at 450C to produce 200.3 g of crude {2-[l-(3,5- bistrifluoromethylbenzyl)-5-pyridin-4-yl-lH-[l,2,3]triazol-4-yl]-pyridin-3-yl}-(2-chlorophenyl)-methanone as a white solid. Crude {2-[l-(3,5-bistrifluoromethylbenzyl)-5-pyridin-4-yl-lH-[l,2,3]triazol-4-yl]-pyridin- 3-yl}-(2-chlorophenyl)-methanone (200.3 g) and isopropyl acetate (600 mL) are charged to a 5L 3-neck jacketed flask, then the contents heated to 750C. After dissolution is achieved, the vessel contents are cooled to 550C, then the solution polish filtered through a 5 micron filter, and the filter rinsed with a volume of isopropyl acetate (200 mL). After the polish filtration operation is complete, the filtrates are combined, and the vessel contents are adjusted to 5O0C. After stirring for at least 15 minutes at 5O0C, 0.21 grams of {2-[l-(3,5-bistrifluoromethylbenzyl)-5-pyridin-4-yl-lH-[l,2,3]triazol-4-yl]-pyridin-3-yl}-(2- chlorophenyl)-methanone Form IV seed (d90 = 40 microns) is added, and the mixture stirred at 5O0C for at least 2 h. Heptanes (1.90 L) are then added over at least 2 h. After the heptanes addition is completed, the slurry is stirred for an hour at 5O0C, cooled to 230C at a rate less then 2O0C per hour, then aged at 230C for an hour prior to isolation. The mixture is then filtered in portions through the bottom outlet valve in the reactor into a 600 mL filter. The resulting wetcake is washed portionwise with a solution containing heptanes (420 mL) and isopropyl acetate (180 mL), which is passed directly through the 5L crystallization vessel. The wetcake is blown dry for 5 minutes with nitrogen, then transferred to a 500 mL plastic bottle. The product is dried at 5O0C for 4 h. to produce 190.3g of pure {2-[l-(3,5- bistrifluoromethylbenzyl)-5-pyridin-4-yl-lH-[l,2,3]triazol-4-yl]-pyridin-3-yl}-(2-chlorophenyl)- methanone, Form IV in 75.0% yield with 100% purity, as determined by HPLC analysis. Particle size is reduced via pin or jet mill. 1H NMR (400 MHz, CDCl3): 5.46 (s, 2H); 7.19 (m, 5H); 7.36 (dd, IH, J = 4.9, 7.8); 7.45 (s, 2H); 7.59 (m, IH); 7.83 (s, IH); 7.93 (dd, IH, J = 1.5, 7.8); 8.56 (dd, IH, J= 1.5, 4.9); 8.70 (d, 2H, J= 5.9).
Preparation 1-A (2-chlorophenyl)-[2-(2-hydroxy-2-pyridin-4-yl-vinyl)pyridin-3-yl]methanone benzoate Charge powdered KOfBu (221.1 g, 1.93 moles, 1.40 eq.) to Reactor A, then charge DMSO (2 L) at
250C over 10 min. The KOfBu/DMSO solution is stirred for 30 min at 230C, then a solution of 4-acetyl pyridine (92 mL, 2.07 moles, 1.50 eq) in DMSO (250 mL) is prepared in reactor B. The contents of reactor B are added to Reactor A over 10 minutes, then the Reactor A enolate solution is stirred at 230C for Ih. In a separate 12-L flask (Reactor C), solid LiOH (84.26 g, 3.45 moles, 2.0 eq) is poured into a mixture of (2- phenylsulfonyl-pyridin-3-yl)-(2-chlorophenyl)methanone (500.0 g, 1.34 moles, 1.0 eq) and DMSO (2L), with stirring, at 230C. The enolate solution in reactor A is then added to Reactor C over a period of at least 15 minutes, and the red suspension warmed to 4O0C. The reaction is stirred for 3h, after which time HPLC analysis reveals less than 2% (2-phenylsulfonyl-pyridin-3-yl)-(2-chlorophenyl)methanone. Toluene (2.5 L) is charged, and the reactor temperature cooled to 3O0C. The mixture is quenched by addition of glacial acetic acid (316 mL, 5.52 moles, 4.0 eq), followed by 10 % NaCl (2.5 L). The biphasic mixture is transferred to a 22-L bottom-outlet Morton flask, and the aqueous layer is removed. The aqueous layer is then extracted with toluene (750 mL). The combined organic layers are washed with 10 % NaCl (750 mL), then concentrated to 4 volumes and transferred to a 12-L Morton flask and rinsed with isopropyl acetate (4 vol, 2 L). The opaque amber solution is warmed to 75 degrees to 750C over 40 min. Benzoic acid (171. Ig, 1.34 moles, 1.0 eq) is dissolved in hot isopropyl acetate (1.5 L), and charged to the crude free base solution over at least 30 min. The crude solution containing benzoate salt is stirred for 0.5 h at 750C then cooled to 23 0C. When solids are first observed, the cooling is stopped and the mixture is aged for an hour at the temperature at which crystals are first observed. Alternatively, if seed crystal is available, the mixture may be seeded with (2-chlorophenyl)-[2-(2-hydroxy-2-pyridin-4-yl-vinyl)pyridin-3-yl]methanone benzoate (2.25g) at 750C, followed by stirring for 0.5 h at 750C, then cooling to 230C over at least 1.5 h. The mixture is then cooled to <5 0C, then filtered through paper on a 24cm single-plate filter. The filtercake is then rinsed with cold z‘-PrOAc (750 mL) to produce granular crystals of bright orange-red color. The wet solid is dried at 550C to produce 527.3 g (83% yield) with 99.9% purity. (2-chlorophenyl)-[2-(2-hydroxy-2- pyridin-4-yl-vinyl)pyridin-3-yl]methanone benzoate. Anal. Calcd. for C26Hi9N2ClO4: C, 68.05; H, 4.17; N, 7.13. Found: C, 67.89; H, 4.15; N 6.05. HRMS: calcd for C19H13ClN2O2, 336.0666; found 336.0673.
The synthesis of(2-chlorophenyl)-[2-(2-hydroxy-2-pyridin-4-yl-vinyl)pyridin-3-yl]methanone benzoate proceeds optimally when the potassium enolate of 4-acetyl pyridine is pre-formed using KOfBu in DMSO. Pre-formation of the enolate allows the SNAR (nucleophilic aromatic substitution) reaction to be performed between room temperature and 4O0C, which minimizes the amount of degradation. Under these conditions, the SNAR is highly regioselective, resulting in a ratio of approximately 95:5 preferential C – acylation. In all cases, less polar solvents such as THF or toluene, or co-solvents of these solvents mixed with DMSO, results in a substantial increase of acylation at the oxygen in the SNAR, and leads to a lower yield of product. This is a substantial improvement over the procedures described in WO2005/042515 for synthesis of the free base or the phosphate salt, in which the SNAR is performed at 60-700C, resulting in a substantial increase in chemical impurity. Using the conditions described in WO2005/042515, when scaled to 2kg, results in maximum yields of 55%, with sub-optimal potency. In comparison, the improved conditions described herein can be run reproducibly from 0.4 to 2kg scale to give yields of 77-83%, with >99% purity. In addition, the reaction can be held overnight at 4O0C with minimal degradation, whereas holding the reaction for 1 h past completion at 60-70°C results in substantial aromatized impurity. The reaction may also be performed using sodium tert-amylate as the base, in combination with an aprotic solvent, such as DMSO or DMF.
The title compound exists as a mixture of tautomers and geometric isomers. It is understood that each of these forms is encompassed within the scope of the invention.

Preparation 1-B
(2-chlorophenyl)-[2-(2-hydroxy-2-pyridin-4-yl-vinyl)pyridin-3-yl]methanone toluate The procedure described in Preparation 1-A is followed, with the following exception. Solid toluic acid (1.0 eq) is added to the crude free base solution at 550C, then the solution cooled to 45 0C. The solution is stirred for one hour at 45 0C, then slowly cooled to 23 0C. When solids are first observed, the cooling is stopped and the mixture is aged for an hour at the temperature at which crystals are first observed. Alternatively, if seed crystal is available, the mixture may be seeded, aged for 3 h at 450C , then cooled to O0C over 4 h. The isolation slurry is filtered, and the wetcake washed with MeOH (3 volumes). The wetcake is dried at 5O0C to provide 14.0 g (76.4%) of (2-chlorophenyl)-[2-(2-hydroxy-2-pyridin-4-yl- vinyl)pyridin-3-yl]methanone toluate as a light red powder.
As with the benzoate salt, the toluate salt can also exist as a mixture of tautomers and geometric isomers, each of which is encompassed within the scope of the invention. (2-chlorophenyl)-[2-(2-hydroxy- 2-pyridin-4-yl-vinyl)pyridin-3-yl]methanone toluate . 13C NMR (125 MHz,DMS0-d6) δ 194.5, 167.8, 167.4, 155.5, 150.7 (2C), 147.4, 144.0, 143.4, 142.7, 138.6, 133.0, 130.8, 130.7, 130.5, 129.8(2C), 129.5(2C), 128.5, 128.0, 127.9, 119.9 (2C), 118.6, 92.6, 21.5.
Preparation 1-C
(2-phenylsulfonyl-pyridin-3-yl)-(2-chlorophenyl)methanone
A solution of 1.3 eq of diisopropylamine (based on 2-benzenesulfonyl pyridine) in 5 volumes of THF in a mechanically stirred 3 -necked flask is cooled to -70 to -75 0C. To this solution is added 1.05 eq of w-butyllithium (1.6M in hexanes) at such a rate as to maintain the temperature below -6O0C. The light yellow solution is stirred at -60 to -70 0C for 30 minutes. Once the temperature has cooled back down to – 60 to -650C, 1.0 eq of 2-benzene-sulfonyl pyridine, as a solution in 3 volumes of THF, is added at the fastest rate that will maintain the reaction temperature under -6O0C. A yellow suspension forms during the addition that becomes yellow-orange upon longer stirring. This mixture is stirred for 3 hours at -60 to – 750C, and then 1.06 eq of 2-chlorobenzaldehyde, as a solution in 1 volume of THF, is added dropwise at a sufficient rate to keep the temperature under -55 0C. The suspension gradually turns orange-red, thins out, and then becomes a clear red solution. The reaction mixture is allowed to stir at -60 to -7O0C for 1 hour, 3N aqueous HCl (7 volumes) is added over 20-30 minutes, and the temperature is allowed to exotherm to 0-100C. The color largely disappears, leaving a biphasic yellow solution. The solution is warmed to at least 1O0C, the layers are separated, and the aqueous layer is back-extracted with 10 volumes of ethyl acetate. The combined organic layers are washed with 10 volumes of saturated sodium bicarbonate solution and concentrated to about 2 volumes. Ethyl acetate (10 volumes) is added, and the solution is once again concentrated to 2 volumes. The thick solution is allowed to stand overnight and is taken to the next step with no purification of the crude alcohol intermediate. The crude alcohol intermediate is transferred to a 3 -necked flask with enough ethyl acetate to make the total solution about 10 volumes. The yellow solution is treated with 3.2 volumes of 10% aqueous (w/w) potassium bromide, followed by 0.07 eq of 2,2,6,6-Tetramethylpiperidine-N-oxide (TEMPO). The orange mixture is cooled to 0-50C and treated with a solution of 1.25 eq of sodium bicarbonate in 12% w/w sodium hypochlorite (9 volumes) and 5 volumes of water over 30-60 minutes while allowing the temperature to exotherm to a maximum of 2O0C. The mixture turns dark brown during the addition, but becomes yellow, and a thick precipitate forms. The biphasic light yellow mixture is allowed to stir at ambient temperature for 1-3 hours, at which time the reaction is generally completed. The biphasic mixture is cooled to 0-50C and stirred for 3 hours at that temperature. The solid is filtered off, washed with 4 volumes of cold ethyl acetate, followed by 4 volumes of water, and dried in vacuo at 450C to constant weight. Typical yield is 80-83% with a purity of greater than 98%. 1H NMR (600 MHz, CDCl3-^) δ ppm 7.38 (td, ./=7.52, 1.28 Hz, 1 H) 7.47 (dd, ./=7.80, 1.30 Hz, 1 H) 7.51 (td, ./=7.79, 1.60 Hz, 1 H) 7.51 (t, ./=7.89 Hz, 2 H) 7.50 – 7.54 (m, J=7.75, 4.63 Hz, 1 H) 7.60 (t, J=7.43 Hz, 1 H) 7.73 (dd, J=7.75, 1.60 Hz, 1 H) 7.81 (dd, J=7.79, 1.56 Hz, 1 H) 8.00 (dd, ./=8.44, 1.10 Hz, 2 H) 8.76 (dd, ./=4.63, 1.61 Hz, 1 H).
Preparation 1-D 1 -azidomethyl-3,5-bistrifluoromethyl-benzene
Sodium azide (74.3 g, 1.14 mol) is suspended in water (125 mL), then DMSO (625 mL) is added. After stirring for 30 minutes, a solution consisting of 3,5-Bis(trifluoromethyl)benzyl chloride (255.3 g, 0.97 moles) and DMSO (500 mL) is added over 30 minutes. (The 3,5-Bis(trifluoromethyl)benzyl chloride is heated to 350C to liquefy prior to dispensing (MP = 30-320C)). The benzyl chloride feed vessel is rinsed with DMSO (50 mL) into the sodium azide solution, the mixture is heated to 4O0C, and then maintained for an hour at 4O0C, then cooled to 230C.
In Process Analysis: A drop of the reaction mixture is dissolved in d6-DMSO and the relative intensities of the methylene signals are integrated (NMR verified as a 0.35% limit test for 3,5- Bis(trifluoromethyl)benzyl Chloride). Work-up: After the mixture reaches 230C , it is diluted with heptanes (1500 mL), then water (1000 mL) is added, and the mixture exotherms to 350C against a jacket setpoint of 230C. The aqueous layer is removed (-2200 mL), then the organic layer (approximately 1700 mL) is washed with water (2 X 750 mL). The combined aqueous layers (-3700 mL) are analyzed and discarded.
The solvent is then partially removed via vacuum distillation with a jacket set point of 850C, pot temperature of 60-650C and distillate head temperature of 50-550C to produce 485g (94.5% yield) of 51 Wt% solution title compound as a clear liquid. Heptanes can be either further removed by vacuum distillation or wiped film evaporation technology. 1H NMR (400 MHz, CDCl3): 4.58 (s, 2H); 7.81 (s, 2H); 7.90 (s, IH).
Preparation 1-E 2-benzene-sulfonyl pyridine Charge 2-chloropyridine (75 mL, 790 mmol), thiophenol (90 mL, 852 mmol), and DMF (450 mL) to a 2L flask. Add K2CO3 (134.6 g, 962 mmol), then heat to HO0C and stir for 18 hours. Filter the mixture, then rinse the waste cake with DMF (195 mL). The combined crude sulfide solution and rinses are transferred to a 5-L flask, and the waste filtercake is discarded. Glacial acetic acid (57 mL, 995 mmol) is added to the filtrate, then the solution is heated to 4O0C, and 13 wt % NaOCl solution (850 mL, 1.7 mol) is added over 2 hours. After the reaction is complete, water (150 mL) is added, then the pH of the mixture adjusted to 9 with 20 % (w/v) NaOH solution (250 mL). The resulting slurry is cooled to <5 0C, stirred for 1.5 h, then filtered, and the cake washed with water (3 x 200 mL). The product wetcake is dried in a 550C vacuum oven to provide 2-benzene-sulfonyl pyridine (149 g, 676 mmol) in 86 % yield: 1H NMR (500 MHz, CDCl3) δ 8.66 (d, J = 5.5 Hz, IH), 8.19 (d, J = 1.1 Hz, IH), 8.05 (m, 2H), 7.92 (ddd, J= 9.3, 7.7, 1.6 Hz, IH), 7.60 (m, IH), 7.54 (m, 2H), 7.44 (m, IH); IR (KBr) 788, 984, 1124, 1166, 1306, 1424, 1446, 1575, 3085 cm“1; MS (TOF) mlz 220.0439 (220.0427 calcd for C11H10NO2S, MH); Anal, calcd for C11H9NO2S: C, 60.26; H, 4.14; N, 6.39; S, 14.62. Found: C, 60.40; H, 4.02; N, 6.40; S, 14.76.
As noted above, use of the improved process of the present invention results in an improved habit of the crystalline Form IV compound of Formula I. The improved habit reduces surface area of the crystal, improves the filtration, and washing, and improves the efficiency of azide mutagen rejection. These improvements are described in greater detail below.
In patent application WO2005/042515, the polish filtration is carried out in 7 volumes (L/kg) of isopropanol near its boiling point (65-83 0C), a process that is difficult and hazardous to execute in commercial manufacturing because of the high risk of crystallization on the filter and/or vessel transfer lines due to supersaturation. In the preferred crystallization solvent, isopropyl acetate, the polish filtration is conducted in four volumes of isopropyl acetate at temperatures from 45 to 55 0C. This temperature range is 35 to 45 0C lower than the boiling point of isopropyl acetate, which provides a key safety advantage.
PATENT
PATENT
WO 2017031215
EXAMPLES
Example 1: Preparation of Compound (I) via Negishi Coupling Route
Example 1 provides a scheme including preparations 1A-1D, described below, for the synthesis of the compound of Formula (I) and intermediates used in the route. An overview of the scheme is as follows:
Image may be NSFW.
Clik here to view.
80 on ma s ale
Example 1A: Preparation of Compound (I)
Image may be NSFW.
Clik here to view.
Zinc dust (200 mg, 3.06 mmol) combined with 2.0 mL of dimethylformamide was treated with 0.010 mL of 1,2-dibromoethane and heated to 65°C for 3 minutes. The mixture was cooled to ambient temperature and treated with 0.010 mL of trimethylsilyl chloride. After 5 minutes, 1.26 mL of 1M zinc chloride in diethyl ether was added to the mixture followed by Compound (Ila) (600 mg, 1.20 mmol). The mixture was heated to 65°C and further treated with 0.020 mL each of 1,2-dibromoethane and trimethylsilyl chloride. After 2.5 hours, via HPLC chromatogram, the reaction showed some formation of the zincate and was allowed to stir at ambient temperature for 16 hours. At this time
tetrakis(triphenylphosphine)palladium(0) (70 mg, 0.06 mmol), Compound (Ilia) (357 mg, 1.20 mmol) were added to the reaction and the mixture heated to 65°C. HPLC analysis showed the formation of Compound (I) in the reaction.
IB: Preparation of Comp
Image may be NSFW.
Clik here to view.
To a solution of Compound (IV) (8.00 g, 18 mmol) in 40 mL of 1,2-dichloroethane was added a solution of iodine monochloride (10.7 g, 65.9 mmol) in 40 mL of 1,2-dichloroethane resulting in a slurry. The slurry was heated to 75°C for 4 hours then cooled to ambient temperature. The solids were collected by filtration, washed with heptane, then combined with 90 mL of ethyl acetate and 80 mL of saturated sodium thiosulfate solution. The organic phase was washed with saturated sodium chloride solution and dried with sodium sulfate. The mixture was concentrated to yield 7.80 g (87%) of Compound (Ila) as a yellow solid. The product could be further purified by silica gel chromatography. Thus 2.0 g of yellow solid was dissolved in dichloromethane and charged onto a silica gel column. The product was eluted using tert-butyl methyl ether to provide 1.87 g (93% recovery) of Compound (Ila) as a white powder. Analytical data: Iodine monochloride complex: ¾ NMR (500 MHz, DMSO-de) δ 8.80 (2 H), 8.05 (1 H), 7.77 (2 H), 7.59 (2 H), 5.86 (2 H).
Uncomplexed: ¾ NMR (500 MHz, DMSO-de) δ 8.71 (2 H), 8.03 (1 H), 7.74 (2 H), 7.44 (2 H), 5.86 (2 H).
It was observed that the iodination proceeded smoothly as a suspension in 1,2-dichloroethane with IC1 (4.0 equiv) at 75°C. An ICl-Compound (Ila) complex was initially isolated by filtration. Compound (Ila) was then obtained in approximately 85% yield by treatment of the ICl-Compound (Ila) complex with sodium thiosulfate. This protocol provided a viable means of isolation of Compound (Ila) without the use of DMF.
Example 1C: Preparation of silyl substituted triazole (Compound IV)
Image may be NSFW.
Clik here to view.
A mixture of Compound (V) (8.07 g, 30.0 mmol) and Compound (VI) (5.12 g, 29.2 mmol) was heated to 100°C for 18 hours. To the mixture was added 40 mL of heptane and the reaction was allowed to cool with rapid stirring. After 1 hour the solids were collected by filtration and washed with heptane then dried to 9.30 g (72%) of Compound (IV) as a tan solid. Analytical data: ¾ NMR (500 MHz, DMSO-de) δ 8.66 (2 H), 8.04 (1 H), 7.67 (2 H), 7.32 (2 H), 5.72 (2 H), 0.08 (9 H).
It was further found that combining Compound (V) and Compound (VI) (neat) and heating at 95 – 105°C afforded a 92: 8 mixture of regioisomers as shown below:
Image may be NSFW.
Clik here to view.
Crystallization of the mixture from heptane afforded Compound (IV) in 62-72% yield, thus obviating the need for chromatography to isolate Compound (IV).
Example ID: Preparation of starting material Compound (VI)
Image may be NSFW.
Clik here to view.
Zinc bromide (502 g, 2.23 mole) was added in approximately 100 g portions to 2.0 L of tetrahydrofuran cooled to between 0 and 10°C. To this cooled solution was added 4-bromopyridine hydrochloride (200 g, 1.02 mol), triphenylphosphine (54 g, 0.206 mol), and palladium (II) chloride (9.00 g, 0.0508 mol). Triethylamine (813 g, 8.03 mol) was then added at a rate to maintain the reaction temperature at less than 10°C, and finally
trimethylsilylacetylene (202 g, 2.05 mol) was added. The mixture was heated to 60°C for 4.5 hours. The reaction was cooled to -5°C and combined with 2.0 L of hexanes and treated with 2 L of 7.4 M NH4OH. Some solids were formed and were removed as much as possible with the aqueous phase. The organic phase was again washed with 2.0 L of 7.4 M NH4OH, followed by 2 washes with 500 mL of water, neutralized with 1.7 L of 3 M hydrochloric acid, dried with sodium sulfate, and concentrate to a thick slurry. The slurry was combined with 1.0 L of hexanes to give a precipitate. The precipitate was removed by filtration and the filtrate was concentrated to 209 g of dark oil. The product was purified by distillation (0.2 torr, 68°C) to give 172 g (96%) of Compound (VI) as colorless oil. Analytical data: ¾ NMR (500 MHz, DMDO-de) δ 8.57 (2 H), 7.40 (2 H), 0.23 (9 H).
EXAMPLE 2 – Preparation of Compound (Ilia)
Example 2 provides a morpholine amide route for the synthesis of Compound (Ilia). In this approach, morpholine amide (Compound VII) was prepared from 2-chlorobenzoyl chloride (Preparation 2A). Metallation of 2-bromopyridine with LDA (1.09 equiv.) in THF at -70°C followed by addition of (Compound VII) afforded Compound (Ilia) in 37% yield after crystallization from IP A/heptane (Preparation 2B). This sequence provides a direct route to Compound (Ilia), and a means to isolate Compound (Ilia) without the use of
chromatography. Compound (Ilia) may then be used to form Compound (I) as shown in Example 1A above (Preparation 2C).
Preparation 2A: Preparation of Compound (VII)
Image may be NSFW.
Clik here to view.
Toluene (1.5 L) was added to Compound (IX) (150 g, 0.86 mol) and cooled to 10°C. Morpholine (82 mL, 0.94 mol) was added to the clear solution over 10 minutes. The resulting white slurry was stirred for 20 minutes then pyridine (92 mL, 1.2 mol) was added dropwise over 20 minutes. The cloudy white mixture was stirred in a cold bath for 1 hour. Water (600 mL) was added in a single portion and the cold bath removed. The mixture was stirred for 20 minutes and the layers are separated. The organic layer was washed with a mixture of 1 N HC1 and water (2: 1, 500 mL:250 mL). The pH of the aqueous layer was ~ 2. The organic layer was washed with a mixture of saturated NaHCCb and water (1 : 1, 100 mL: 100 mL). The pH of the aqueous layer was ~ 9. The layers were separated. The organic layer was concentrated in vacuo to an oil. The oil was dissolved in IPA (70 mL) and heated at 60°C for 30 min. The clear solution was allowed to cool to 30°C, then heptane (700 mL, 4.7 v) was added dropwise. The resulting slurry was stirred at RT for 2 hours then cooled to 0°C for 1 hour. The slurry was filtered at RT, washed with heptane then dried under vacuum at 30°C overnight. Compound (VII) (156.2 g, 81%) was obtained as a white solid. Analytical data: ¾ NMR (500 MHz, CDCh) δ 7.42-7.40 (m, 1 H), 7.35-7.29 (m, 3 H), 3.91-3.87 (m, 1 H), 3.80-3.76 (m, 3 H), 3.71 (ddd, J= 11.5, 6.8, 3.3 Hz, 1 H), 3.60 (ddd, J = 11.2, 6.4, 3.4 Hz, 1 H), 3.28 (ddd, J= 13.4, 6.3, 3.2 Hz, 1 H), 3.22 (ddd, J= 13.7, 6.8, 3.3 Hz, 1 H); LRMS (ES+) calcd for CnHi3F6ClN02 (M+H)+ 226.1, found 225.9 m/z.
Preparation 2B: Preparation of Compound (Ilia)
Image may be NSFW.
Clik here to view.
THF (75 mL) was added to diisopropyl amine (4.9 mL, 34.8 mmol) and cooled to a
temperature of -70°C under N2 atmosphere. 2.5 M w-BuLi in hexanes (13.9 mL, 34.8 mmol) was added in a single portion (a 30-40°C exotherm) to the clear solution and cooled back to -70°C. Compound (VIII) (5.0 g, 31.6 mmol) was added neat to the LDA solution (a 2 to 5°C exotherm) followed by a THF (10 mL) rinse, keeping T< -65°C. This clear yellow solution was stirred at -70°C for 15 min. Compound (VII) (7.1 g, 31.6 mmol) in THF (30 mL) was added keeping T< -65°C. The resulting clear orange solution was stirred at -70°C for 3 hours. MeOH (3 mL) was added to quench reaction mixture and the cold bath was removed. 5 N HC1 (25 mL) was added to the reaction solution. MTBE (25 mL) was added, and the layers were separated. The organic layer was washed with water (25 mL X 2). The organic layer was dried over MgS04 and filtered. The organic layer was concentrated in vacuo to an orange oil. The oil was dissolved in IPA (15 mL, 3 vol) at ambient temperature. Heptane (25 mL) was added dropwise and the resulting slurry was stirred at RT for 1 hour. The slurry was cooled to 0°C for 1 hour and filtered. The filter cake was rinsed with chilled heptane (20 mL) and dried under vacuum at 30°C overnight. Compound (Ilia) (4.25 g, 45%) was obtained as a yellow solid.
Several reactions were run at different temperatures and with different addition rates of Compound (VII). If the reaction temperature was maintained below -65°C and Compound (VII) was added in <5 min, it was found that the reaction worked well. If the temperature was increased and/or the addition time of Compound (VII) was increased, then yields suffered, and the work-up was complicated by emulsions.
Preparation 2C: Preparation of Compound (I)
Compound (Ilia) may then reacted with Compound (Ila) to produce Compound (I) as shown in Preparation 1A.
EXAMPLE 3
Example 3 describes a new route for the synthesis of an intermediate free base, which may be used to form Compound (I) as described further below.
Example 3A: Preparation of starting material (Compound X) from 2-Chloronicotinonitrile
Image may be NSFW.
Clik here to view.
A mixture of NaH (40.0 g, 1 mol, 60% dispersion in mineral oil) and 2-chloronicotinonitrile (69.3 g, 500 mmol) in THF (1 L) was heated to reflux. A solution of 4-acetylpyridine (60.6 g, 500 mmol) in THF (400 mL) was added over a period of 40 min. The resulting dark brown mixture was stirred at reflux for ~ 2 h. The heating mantle was then removed, and AcOH (58 mL, 1 mol) was added. EtOAc (1 L) and H2O (1 L) were then added, and the layers were separated. The organic layer was concentrated to afford an oily solid. CH3CN (500 mL) was added, and the mixture was stirred for 30 min. H2O (1 L) was then added. The mixture was stirred for 1 h then filtered. The solid was rinsed with 2: 1
CH3CN-H2O (900 mL) and hexanes (400 mL) then dried under vacuum at 45°C overnight to afford 61.4 g (55% yield) of Compound (X) as yellow solid. Compound (X) exists as an approximate 95:5 enol-ketone mixture in CDCI3. Analytical data for enol: IR (CHCI3): 3024, 2973, 2229, 1631, 1597, 1579, 1550, 1497; ¾ NMR (500 MHz, CDCI3) δ 8.69 (dd, J= 4.4,
1.7 Hz, 2H), 8.55 (dd, J = 5.2, 1.8 Hz, 1H), 7.97 (dd, J= 7.9, 1.8 Hz, 1H), 7.70 (dd, J= 4.6, 1.5 Hz, 2H, 7.17 (dd, J = 7.8, 5.0 Hz, 1H), 6.59 (s, 1H); LRMS (ES+) calcd for C13H10N3O (M+H)+ 224.1, found 224.0 m/z.
Preparation 3B: Preparation of Compound (XI)
Preparation 3B(1):
Image may be NSFW.
Clik here to view.
(X) (XI)
Compound (XI) may be prepared using Compound (X).
Preparation 3B(2):
Alternatively, the following procedure for the conversion of nitrile into an acid which may also yield compound (XI). A mixture of Compound (X) (1 eq) and NaOH (1.5 eq) in 1 : 1 fhO-EtOH (3.5 mL/g of Compound (X)) was heated at 65°C overnight. The reaction mixture was cooled to RT then added to CH2C12 (12.5 mL/g of Compound (X)) and H20 (12.5 mL/g of Compound (X)). Cone. HC1 (2.5 mL/g of Compound (X)) was then added, and the layers were separated. The aqueous layer was extracted with CH2CI2 (10 mL/g of Compound (X)). The combined organic extracts were washed with H2O (12.5 ml/g of Compound (X)), dried (MgS04), filtered and concentrated to afford Compound (XI).
Preparation 3C
Compound Compound (XI) may then be converted into a Stage C intermediate free base, with observed 87% conversion in Grignard reaction as shown above. A complete synthesis route for Com ound (I) starting from compound Compound (XI) is depicted below.
Image may be NSFW.
Clik here to view.
Detailed experimental procedures for the synthesis of benzoate salt and final step are given in
International Patent Application Publication WO 2008/079600 Al .
References
- “Company Overview of Eli Lilly & Co., Worldwide License to Develop and Commercialize VLY-686”. Bloomberg Business. Retrieved 16 November 2015.
- [1]
- “Vanda Pharmaceuticals Announces Tradipitant Phase II Proof of Concept Study Results for Chronic Pruritus in Atopic Dermatitis”. PR Newswire. Retrieved 16 November 2015.
- Schmidt, B (2006). “Proof of principle studies”. Epilepsy Res. 68 (1): 48–52. doi:10.1016/j.eplepsyres.2005.09.019. PMID 16377153.
- George, DT; Gilman, J; Hersh, J; et al. (2008). “Neurokinin 1 receptor antagonism as a possible therapy for alcoholism.”. Science. 6: 1536–1539. doi:10.2147/SAR.S70350. PMC 4567173Image may be NSFW.
Clik here to view. . PMID 26379454.
. PMID 26379454. - Tauscher, J; Kielbasa, W; Iyengar, S; et al. (2010). “Development of the 2nd generation neurokinin-1 receptor antagonist LY686017 for social anxiety disorder”. European Neuropsychopharmacology. 20 (2): 80–87. doi:10.1016/j.euroneuro.2009.10.005. PMID 20018493.

George, D.T.; Gilman, J.; Hersh, J.; Thorsell, A.; Herion, D.; Geyer, C.; Peng, X.; Kielbasa, W.; Rawlings, R.; Brandt, J.E.; Gehlert, D.R.; Tauscher, J.T.; Hunt, S.P.; Hommer, D.; Heilig, M. Neurokinin 1 receptor antagonism as a possible therapy for alcoholism, Science 2008, 319(5869): 1536
Gackenheimer, S.L.; Gehlert, D.R.In vitro and in vivo autoradiography of the NK-1 antagonist (3H)-LY686017 in guinea pig brain39th Annu Meet Soc Neurosci (October 17-21, Chicago) 2009, Abst 418.16
Tonnoscj, K.; Zopey, R.; Labus, J.S.; Naliboff, B.D.; Mayer, E.A.
The effect of chronic neurokinin-1 receptor antagonism on sympathetic nervous system activity in irritable bowel syndrome (IBS) Dig Dis Week (DDW) (May 30-June 4, Chicago) 2009, Abst T1261
Kopach, M.E.; Kobierski, M.E.; Coffey, D.S.; et al.
Process development and pilot-plant synthesis of (2-chlorophenyl)[2-(phenylsulfonyl)pyridin-3-yl]methanone
Org Process Res Dev 2010, 14(5): 1229
| Patent ID | Patent Title | Submitted Date | Granted Date |
|---|---|---|---|
| US2016060250 | NOVEL INTERMEDIATE AND PROCESS USEFUL IN THE PREPARATION OF -(2-CHLOROPHENYL)-METHANONE | 2015-11-10 | 2016-03-03 |
| US2015320866 | PHARMACEUTICAL COMPOSITION COMPRISING ANTIEMETIC COMPOUNDS AND POLYORTHOESTER | 2013-12-13 | 2015-11-12 |
| US2014206877 | NOVEL INTERMEDIATE AND PROCESS USEFUL IN THE PREPARATION OF -(2-CHLOROPHENYL)-METHANONE | 2014-03-27 | 2014-07-24 |
| US2012225904 | New 7-Phenyl-[1, 2, 4]triazolo[4, 3-a]Pyridin-3(2H)-One Derivatives | 2010-11-09 | 2012-09-06 |
| US2010056795 | NOVEL INTERMEDIATE AND PROCESS USEFUL IN THE PREPARATION OF -(2-CHLOROPHENYL)-METHANONE | 2010-03-04 | |
| US7381826 | Crystalline forms of {2-[1-(3, 5-bis-trifluoromethyl-benzyl)-5-pyridin-4-yl-1H-[1, 2, 3]triazol-4-yl]-pyridin-3-yl}-(2-chlorophenyl)-methanone | 2007-04-05 | 2008-06-03 |
| US7320994 | Triazole derivatives as tachykinin receptor antagonists | 2005-10-27 | 2008-01-22 |
| Image may be NSFW. Clik here to view. |
|
| Legal status | |
|---|---|
| Legal status |
|
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| ChemSpider | |
| Chemical and physical data | |
| Formula | C28H16ClF6N5O |
| Molar mass | 587.90 g/mol |
| 3D model (Jmol) | |
Tradipitant
Tradipitant is being evaluated in a Phase II study in treatment resistant pruritus in atopic dermatitis.
Tradipitant is an NK-1 receptor antagonist licensed from Eli Lilly in 2012. Tradipitant has demonstrated proof-of-concept in alcohol dependence in a study published by the NIH1. In that study tradipitant was shown to reduce alcohol cravings and voluntary alcohol consumption among patients with alcohol dependence. NK-1R antagonists have been evaluated in a number of indications including chemotherapy-induced nausea and vomiting (CINV), post-operative nausea and vomiting (PONV), alcohol dependence, anxiety, depression, and pruritus.
The NK-1R is expressed throughout different tissues of the body, with major activity found in neuronal tissue. Substance P (SP) and NK-1R interactions in neuronal tissue regulate neurogenic inflammation locally and the pain perception pathway through the central nervous system. Other tissues, including endothelial cells and immune cells, have also exhibited SP and NK-1R activity2. The activation of NK-1R by the natural ligand SP is involved in numerous physiological processes, including the perception of pain, behavioral stressors, cravings, and the processes of nausea and vomiting1,2,3. An inappropriate over-expression of SP either in nervous tissue or peripherally could result in pathological conditions such as substance dependence, anxiety, nausea/vomiting, and pruritus1,2,3,4. An NK-1R antagonist may possess the ability to reduce this over-stimulation of the NK-1R, and as a result address the underlying pathophysiology of the symptoms in these conditions.
References
- George DT, Gilman J, Hersh J, Thorsell A, Herion D, Geyer C, Peng X, Keilbasa W, Rawlings R, Brandt JE, Gehlert DR, Tauscher JT, Hunt SP, Hommer D, Heilig M. Neurokinin 1 receptor antagonism as a possible therapy for alcoholism. Science. 2008; 319(5869):1536-9
- Almeida TA, Rojo J, Nieto PM, Pinto FM, Hernandez M, et al. Tachykinins and tachykinin receptors: structure and activity relationships. Current Medicinal Chemistry. 2004;11:2045-2081.
- Hargreaves R, Ferreira JC, Hughes D, Brands J, Hale J, Mattson B, Mill S. Development of aprepitant, the first neurokinin-1 receptor antagonist for the prevention of chemotherapy-induced nausea and vomiting. Annals of the New York Academy of Sciences. 2011; 1222:40-48.
- Stander S, Weisshaar E, Luger A. Neurophysiological and neurochemical basis of modern pruritus treatment. Experimental Dermatology. 2007;17:161-69.
///////////////////tradipitant, PHASE 2, VLY-686, LY686017, традипитант , تراديبيتانت , 曲地匹坦 , VANDA, ELI LILLY, Gastroparesis Pruritus
Filed under: Phase2 drugs, Uncategorized Tagged: eli lilly, Gastroparesis Pruritus, LY686017, традипитант, phase 2, tradipitant, VANDA, VLY-686, 曲地匹坦, تراديبيتانت Image may be NSFW.
Clik here to view.
 Image may be NSFW.
Image may be NSFW.Clik here to view.
 Image may be NSFW.
Image may be NSFW.Clik here to view.
 Image may be NSFW.
Image may be NSFW.Clik here to view.
 Image may be NSFW.
Image may be NSFW.Clik here to view.
 Image may be NSFW.
Image may be NSFW.Clik here to view.
 Image may be NSFW.
Image may be NSFW.Clik here to view.
 Image may be NSFW.
Image may be NSFW.Clik here to view.
